Introduction
Gastric cancer (GC) is the fifth most common type of
cancer and the third leading cause of cancer-associated mortality
worldwide (1). Since it is
commonly diagnosed at advanced stages with lymphatic metastasis and
extensive invasion, the majority of patients with GC have a poor
prognosis. However, the average 5-year survival rate of patients
with early GC can reach >90% (2). Gastric carcinogenesis, particularly
the intestinal type, is considered to be a multistep and sequential
process that includes chronic superficial gastritis, atrophic
gastritis (AG), intestinal metaplasia (IM), dysplasia, intramucosal
carcinoma and invasive neoplasia (3). Among these stages, IM is defined as a
precancerous lesion of the gastric mucosa and is considered a risk
factor in gastric tumourigenesis. However, the molecular mechanism
underlying IM formation in the human stomach remains unclear.
Recently, endoscopic techniques have achieved great progress in the
treatment of precancerous lesions of the stomach. However,
endoscopic techniques remain unsatisfactory for detecting early GC
and IM due to a lack of sensitivity and high costs (4). Therefore, it is necessary to
elucidate the precise mechanism underlying IM formation, and to
identify valuable biomarkers for early GC and IM diagnosis and
prognosis.
Caudal-related homeobox transcription factor 2
(CDX2) is an intestinal-specific transcription factor, which has
been strongly implicated in the development of intestinal
epithelial cells, where it regulates intestinal markers, including
mucin 2 (MUC2) (5) and trefoil
factor 3 (TFF3) (6). Normally,
CDX2 expression is limited to the intestines, and CDX2 is absent in
normal gastric mucosa. However, CDX2 exhibits a high expression
level in gastric IM tissues, whereas its expression is decreased in
dysplasia and GC stages (7,8). In
transgenic mice expressing CDX2 in parietal cells (9), normal gastric epithelial cells are
completely replaced by IM with the induction of MUC2, and these
mice further develop intestinal-type adenocarcinomas, thus
indicating that ectopic CDX2 expression may serve an important role
in gastric tumourigenesis by inducing IM. Farnesoid X receptor
(FXR) is a ligand-activated transcription factor that has a high
affinity for physiological bile acids, such as chenodeoxycholic
acid (CDCA) and deoxycholic acid (DCA) (10). FXR primarily regulates the
homoeostasis of bile acids, including bile acid synthesis,
transport and intestinal reabsorption (11,12).
FXR deficiency in mice results in increased intestinal epithelial
cell proliferation and intestinal tumourigenesis (13,14).
In addition, upregulation of FXR has been reported in gastric IM
(15); however, the function of
FXR in IM formation in the stomach remains to be determined.
Clinical and experimental studies have reported that
AG and IM are associated with the phenomenon of duodenogastric
reflux (DGR) (16,17). Bile acids, which are the primary
duodenal components of DGR, are generally considered to be
carcinogens in gastric IM formation and gastric tumourigenesis
(18,19). Bile acids, particularly CDCA and
DCA, have been reported to be associated with the induction of CDX2
and MUC2 expression. It has previously been demonstrated that bile
acid treatment can lead to an increase in CDX2 and MUC2 expression
in Barrett's oesophageal cells (20). Xu et al also reported that
CDCA stimulates the upregulation of CDX2 and MUC2 by activating FXR
in normal rat gastric epithelial cells in a dose-dependent manner
(21). However, the exact
molecular mechanisms whereby bile acids promote human gastric IM
formation remain unclear. Therefore, the present study aimed to
investigate the effect of bile acids on molecular alterations in
gastric IM formation and the molecular mechanisms involved.
Materials and methods
Clinical samples and cell culture
A total of 40 human gastric IM tissues and paired
normal gastric mucosa tissues were obtained from patients (27 males
and 13 females; age, 37-74 years) with DGR who had undergone
endoscopic biopsy at the First Affiliated Hospital of Xi'an
Jiaotong University (Xi'an, China) between March 2014 and July
2016. Biopsy specimens from the antrum and corpus were fixed in a
solution of 4% paraformaldehyde for 24 h at room temperature and
were embedded in paraffin. The 4-µm tissues were then
deparaffinized using xylene and ethanol, and were stained with 0.5%
hematoxylin for 5 min, followed by 0.5% eosin for 1 min at room
temperature. Images were acquired using a Nikon ECLIPSE Ti-S
microscope mounted with a Nikon digital camera (Nikon Corporation,
Tokyo, Japan) and were assessed for the presence of intestinal
metaplasia, which was recognized morphologically by the presence of
goblet cells, absorptive cells and cells resembling colonocytes.
Patients diagnosed with GC were excluded from this study. All
tissue samples were obtained from patients that had provided
informed consent, and the study protocol was approved by the Ethics
Committee of the First Affiliated Hospital of Xi'an Jiaotong
University. Normal human gastric epithelial cells (GES-1) were
purchased from the Shanghai Institute of Cell Biology, Chinese
Academy of Sciences (Shanghai, China). The cells were maintained at
37°C in RPMI-1640 medium supplemented with 10% foetal bovine serum
(both from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
in a humidified incubator containing 5% CO2. Once GES-1
cells reached 70% confluence, they were serum-deprived for 24 h
prior to treatment with various doses of DCA or CDCA (50, 100, 150,
200, 400 and 600 µmol/l) along with the FXR agonist GW4064
(1 µmol/l), the FXR antagonist Z-guggulsterone (20
µmol/l) or the NF-κB inhibitor pyrrolidine dithiocarbamate
(PDTC; 50 µmol/l) for different durations (6, 12, 24 and 48
h). CDCA and DCA were purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). GW4064, Z-guggulsterone and PDTC were
purchased from Selleck Chemicals LLC (Houston, TX, USA).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cell lines and tissue
samples using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to a standard protocol. The
PrimeScript® RT Reagent kit (Takara Biotechnology Co.,
Ltd., Dalian, China) was used to convert RNA into cDNA at 37°C for
15 min and 85°C for 5 sec, followed by maintenance at 4°C,
according to the manufacturer's protocol. The PCR primer sequences
were as follows: CDX2, forward, 5′-GAACCTGTGCGAGTGGATG-3′ and
reverse, 5′-GGATGGTGATGTAGCGACTG-3′; MUC2, forward,
5′-CAACGATTCCTACGCTCTCC-3′ and reverse, 5′-CTTCTTCTTGTCAGCCAGCA-3′;
FXR, forward, 5′-TGCAGATCAGACCGTGAATGA-3′ and reverse,
5′-TTGGTTGCCATTTCCGTCAAA-3′; and GAPDH, forward,
5′-TGCACCACCAACTGCTTAGC-3′ and reverse, 5′-GGCATGGACTGTGGTCATGAG-3′
(all from Sangon Biotech Co., Ltd., Shanghai, China). qPCR was
conducted using SYBR Premix Ex Taq II (Takara Biotechnology Co.,
Ltd.) on a CFX96™ Real-Time PCR Detection system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The real-time cycler
conditions (two-step method) were as follows: i) Initial
denaturation at 95°C for 30 sec; ii) 40 cycles of denaturation at
95°C for 5 sec and annealing/extension at 60°C for 1 min. The
relative mRNA expression levels were quantified using the
2−ΔΔCq method (22).
Each experiment was repeated three times.
Vector construction and transfection
Fragments of the CDX2 and MUC2 5′-flanking sequence
(2,581 and 2,665 bp) were amplified using PCR from the DNA of GES-1
cells and cloned into the luciferase reporter vector pGL3.0-Basic
(Promega Corporation, Madison, WI, USA). The sequences of the
primers for CDX2 were forward 5′-CGGGGTACCAGAGCCACGTCTTCAGG-3′ and
reverse 5′-GGAAGATCTGCACGGAGCTAGGGTAC-3′. The sequences of the
primers for MUC2 were forward 5′-GAGGCTAGCCCGGGCTTCCTGGTGAGTC-3′
and reverse 5′-GAGCTCGAGCATGGTGGCTGGCAGGGGC-3′. Potential NF-κB or
CDX2-binding sites in the CDX2 or MUC2 promoter were identified
using bioinformatics analysis (http://jaspar.genereg.net/). Mutagenesis of the CDX2
and MUC2 promoters was performed using a site-directed mutagenesis
kit (Takara Biotechnology Co., Ltd.). All constructs were verified
by sequencing (data not shown). The pNF-κB-Luc plasmid was
purchased from Clontech Laboratories, Inc. (Mountainview, CA, USA).
The phU6-EGFP-short hairpin (sh) RNA-CDX2 lentiviral vector (target
sequence, 5′-ACAAATATCGAGTGGTGTA-3′) and control vector (target
sequence, 5′-TTCTCCGAACGTGTCACGT-3′) were purchased from Shanghai
GeneChem Co., Ltd. (Shanghai, China). Small interfering (si)RNA-FXR
(target sequence, 5′-GTAGCAGAGATGCCTGTAA-3′) and scrambled control
(target sequence, 5′-TTCTCCGAACGTGTCACGT-3′) were purchased from
Shanghai GenePharma Co., Ltd. For siRNA transfection, cells were
seeded at a density of 1×105 cells/well in 6-well plates
and were transfected with 50 µM siRNA-FXR or scrambled
control for 24 h using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. For lentiviral infection, cells were seeded in 24-well
plates overnight at a density of 5×104 cells/well and
were infected with the phU6-EGFP-shRNA-CDX2 lentiviral vector and
the control vector at a multiplicity of infection of 10 for 12 h,
according to the manufacturer's protocol.
Dual-luciferase reporter assay
GES-1 cells were plated on 24-well plates at a
density of 5×104 cells/well the day prior to
transfection. The CDX2 or MUC2 promoter luciferase constructs or
the pNF-κB-Luc plasmid were co-transfected with a pRL-TK construct
(Promega Corporation) into the cells using
Lipofectamine® 2000. A total of 24 h post-transfection,
cells were incubated in medium containing 200 µmol/l bile
acids for an additional 48 h and were harvested for analysis with
the Dual-Luciferase Reporter Assay system (Promega Corporation),
according to the manufacturer's protocol. Luciferase activity was
measured using a PerkinElmer EnSpire Multilabel Reader 2300
(PerkinElmer, Inc., Waltham, MA, USA). Luciferase intensity was
normalized to Renilla luciferase activity to normalize for
transfection efficiency. Each experiment was repeated three
times.
Cell growth assay
The cells were trypsinized and seeded into 96-well
culture plates (Corning, Inc., Corning, NY, USA) at a density of
5×103 cells/well. After 24 h incubation, the cells were
treated with dimethyl sulfoxide (DMSO), or 100, 200, 400 or 600
µmol/l of DCA or CDCA. The cells were then harvested at 6,
12, 24 and 48 h and cell growth was assessed using the Cell
Counting Kit 8 (CCK8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan), according to the manufacturer's protocol.
Absorbance was measured at 450 nm. Each experiment was repeated
three times.
Preparation of nuclear extracts
Nuclear extracts were prepared using the Nuclear
Extraction kit (Abcam, Cambridge, MA, USA). Briefly, cells were
grown to 70-80% confluence on a 100-mm plate. Subsequently, culture
medium was removed and cells were washed twice with PBS. After
removal of the PBS, 3 ml fresh PBS was added to the plate and the
cells were scraped into a 15-ml conical tube. The cells were
centrifuged for 5 min at 179 × g and the supernatant was discarded;
subsequently, the cell pellet was resuspended in 300 µl 1X
Pre-Extraction Buffer, transferred to a microcentrifuge vial and
incubated on ice for 10 min. The preparation was then vigorously
vortexed for 10 sec, centrifuged for 1 min at 12,400 × g and the
cytoplasmic extract was carefully removed from the nuclear pellet.
Subsequently, 2 volumes of Extraction Buffer containing
dithiothreitol and protease inhibitor cocktail was added to the
nuclear pellet, which was incubated on ice for 15 min; the sample
was vortexed for 5 sec every 3 min. Finally, the suspension was
centrifuged at 16,900 × g for 10 min at 4°C and the supernatant was
transferred into a new microcentrifuge vial. The nuclear protein
was then quantified using Bradford reagent (Thermo Fisher
Scientific, Inc.) and used for western blotting.
Western blotting
The cells were lysed with radioimmunoprecipitation
assay buffer (Beyotime Institute of Biotechnology, Shanghai,
China). Protein quantification of each sample was performed using
Bradford reagent (Thermo Fisher Scientific, Inc.). Subsequently,
cell lysates containing 50 µg total protein were subjected
to 10% SDS-PAGE (Beyotime Institute of Biotechnology) and proteins
were transferred to polyvinylidene fluoride membranes (EMD
Millipore, Billerica, MA, USA). After blocking with 5% fat-free dry
milk at room temperature for 2 h, the membranes were incubated
overnight at 4°C with primary antibodies at 1:1,000 dilution. The
membranes were then washed four times with Tris-buffered
saline-0.1% Tween-20 (8 min//wash) and were incubated with a
horseradish peroxidase (HRP)-conjugated secondary antibody at
1:5,000 dilution at room temperature for 2 h. Chemiluminescent HRP
substrate (EMD Millipore) was used to visualize the protein bands
and protein expression was semi-quantified using ImageJ version
1.46 software (National Institutes of Health, Bethesda, MD, USA).
The antibodies against MUC2 (cat. no. sc-13312), FXR (cat. no.
sc-13063) and GAPDH (cat. no. sc-47724) were purchased from Santa
Cruz Biotechnology, Inc. (Dallas, TX, USA), and the antibodies
against CDX2 (cat. no. 12306), Histone H3 (cat. no. 4499), p50
(cat. no. 13586) and p65 (cat. no. 4764) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The HRP-conjugated
anti-rabbit (cat. no. 7074) and anti-mouse (cat. no. 7076)
secondary antibodies were purchased from Cell Signaling Technology,
Inc., and the HRP-conjugated anti-goat (cat. no. sc-2354) secondary
antibody was purchased from Santa Cruz Biotechnology, Inc.
Immunohistochemistry
Tissues were fixed with 10% neutral formalin at room
temperature for 24 h and embedded in paraffin. Subsequently,
4-µm tissue sections were prepared. The immunohistochemical
staining procedure was performed using the standard
Streptavidin-Biotin Complex (SABC) staining method (23). The sections were incubated with
primary antibodies against CDX2 (cat. no. 12306; Cell Signaling
Technology, Inc.), MUC2 (cat. no. sc-13312) and FXR (cat. no.
sc-13063) (both from Santa Cruz Biotechnology, Inc.) at 1:100
dilution overnight at 4°C, followed by incubation with biotinylated
secondary antibodies (1:100 dilution) for 30 min and treatment with
HRP-conjugated SABC (cat. no. SA1022 and SA1023; Wuhan Boster
Biological Technology, Ltd., Wuhan, China) for 30 min at room
temperature. Images were acquired using a Nikon ECLIPSE Ti-S
microscope mounted with a Nikon digital camera (Nikon Corporation).
The CDX2-, MUC2- and FXR-stained sections were divided into two
groups (negative and positive) based on the extent and intensity of
staining. The extent of positively stained cells was categorized as
follows: 0 (<5%), 1 (5-25%), 2 (25-50%), 3 (50–75%) and 4
(>75%). Staining intensity was categorized as follows: 0
(negative), 1 (weakly positive), 2 (moderately positive) and 3
(strongly positive). The immunoreactivity score (IRS) was defined
as the product of the extent and intensity scores. An IRS of ≤3 was
defined as negative, and a score of >3 was defined as positive.
Two pathologists evaluated all specimens in a blinded manner.
Quantitative chromatin
immunoprecipitation (qChIP)
qChIP assays were conducted using an EZ-ChIP™ Assay
kit (EMD Millipore), as previously described (24). Chromatin-protein complexes were
immunoprecipitated with 5 µg anti-CDX2, anti-p50 and
anti-p65 antibodies, or 1 µg rabbit immunoglobulin G (cat.
no. ab171870; Abcam) as a negative control. qPCR was performed to
amplify the regions of interest or internal negative control
regions. The real-time cycler conditions (two-step method) were as
follows: i) Initial denaturation at 95°C for 30 sec; ii) 40 cycles
of denaturation at 95°C for 5 sec and annealing/extension at 60°C
for 1 min. Fold enrichment ratio was calculated as the value of the
ChIP sample versus the negative control. Fold enrichment =
E(Input Cq-ChIP Cq)/E(Input
Cq-Negative Control Cq), where E refers to primer
efficiency. The primer sequences of the CDX2 promoter region (−403
to −186 bp) were forward, 5′-TTCGAGGGGTTGTGCGTAGAGTGCG-3′ and
reverse, 5′-AGGCGGTCCCTCCCTCTGGCCT-3′. The primer sequences of the
MUC2 promoter region (−251 to −168 bp) were forward,
5′-CTACAGGGCTGCCTCATCCT-3′ and reverse,
5′-AATATTGATTCAGGTTATCGGAGGT-3′. Each sample was assessed in
triplicate.
Animal model
A mixture of bile acids at molar concentrations,
which have previously been described as near 'physiologic'
(25–27), was used in this study. A total of
25 female mice (age, 7 weeks; weight, ~21 g) were purchased from
Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China) and were
housed in the Laboratory Animal Centre of Xi'an Jiaotong
University. Mice had free access to water and Purina 5L79 rodent
chow (Nestlé Purina PetCare Company, St. Louis, MO, USA) and were
maintained under the following conditions: Temperature, 22-25°C;
humidity, 50-60%; 12-h light/dark cycle. Bile acids (0.15 ml) were
administered to the stomach of C57BL/6J mice via a plastic feeding
tube (20 g), two times per day, for 45 days. The animals were
separated into three experimental and two control groups (n=5
mice/group). The experimental groups were treated with i) DCA (10
mmol/l in 0.01 mol/l PBS), ii) CDCA (10 mmol/l in 0.01 mol/l PBS)
or iii) a mixture of DCA and CDCA (10 mmol/l in 0.01 mol/l PBS).
The control groups included i) a PBS-treated group (0.01 mol/l) and
ii) an untreated group. After 45 days administration, the mice were
sacrificed by cervical dislocation and gastric mucosa samples from
each group were obtained for western blotting. The experimental
protocols were evaluated and approved by the Animal Care and Use
Committee of the Medical School of Xi'an Jiaotong University.
Statistical analysis
All data are presented as the means ± standard
deviation. The χ2 test or one-way analysis of variance
with Dunnett's post hoc test were used to analyse the differences
among the groups. The correlation between CDX2 and FXR or MUC2 mRNA
expression was explored using the Pearson Correlation test. All
statistical analyses were performed using SPSS 18.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Bile acids affect the viability of GES-1
normal human gastric epithelial cells
To assess the toxic effect of bile acids on GES-1
cell viability, the cells were treated with various doses of DCA or
CDCA (100, 200, 400 and 600 µmol/l) for different durations
(6, 12, 24 and 48 h). Cell viability was then detected using the
CCK8 assay. As shown in Fig. 1A and
B, compared with in the blank control group, treatment with DCA
or CDCA at 100 or 200 µmol/l for 6, 12 or 24 h did not have
a toxic effect. However, when cells were treated for 48 h, the
viability of the cells cultured with 100 and 200 µmol/l DCA
or CDCA was significantly inhibited. Treatment with 400 or 600
µmol/l DCA or CDCA, even for a short period of time, induced
a severe toxic effect on cell viability. The maximum toxic effect
of bile acids was detected in the group treated with 600
µmol/l for 48 h. These data demonstrated that long-term
treatment with high-dose bile acids was toxic to GES-1 cells. To
avoid these toxic effects, GES-1 cells were treated with
physiological concentrations of bile acids (<200 µmol/l)
for 24 h in subsequent experiments.
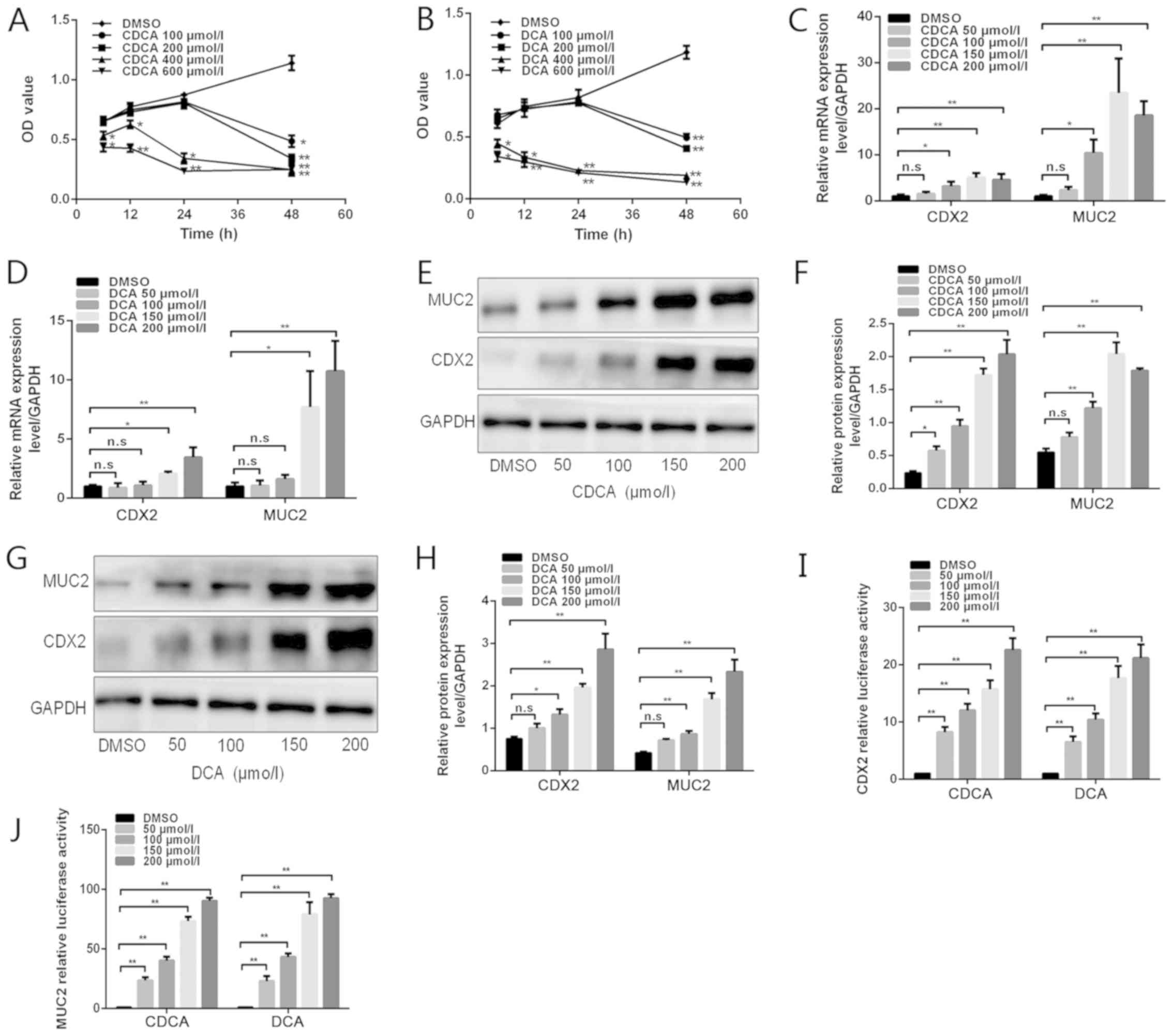 | Figure 1Effects of bile acids on CDX2 and
MUC2 expression in GES-1 cells. (A and B) GES-1 cells were treated
with DMSO or various doses of bile acids (100, 200, 400 and 600
µmol/l) for different periods of time (6, 12, 24 and 48 h).
Cell viability was then determined using a Cell Counting Kit 8
assay. (C and D) GES-1 cells were treated with DMSO or various
concentrations of bile acids (50, 100, 150 and 200 µmol/l)
for 24 h. Reverse transcription-quantitative polymerase chain
reaction was performed to determine the mRNA expression levels of
CDX2 and MUC2 in GES-1 cells treated with bile acids. (E) Western
blot analysis of CDX2 and MUC2 protein expression in each group of
GES-1 cells stimulated with CDCA. (F) Comparison of CDX2 and MUC2
protein levels in each group of cells stimulated with CDCA. (G)
Western blot analysis of CDX2 and MUC2 protein expression in each
group of GES-1 cells stimulated with DCA. (H) Comparison of CDX2
and MUC2 protein levels in each group of cells stimulated with DCA.
(I and J) GES-1 cells treated with various concentrations of bile
acids were transfected with 2.5 kb CDX2- or 2.6 kb MUC2-luc
promoter constructs. A dual-luciferase reporter assay was conducted
to determine (I) CDX2 and (J) MUC2 promoter activity. Data are
presented as the means ± standard deviation from three independent
experiments. *P<0.05, **P<0.01. CDCA,
chenodeoxycholic acid; CDX2, caudal-related homeobox transcription
factor 2; DCA, deoxycholic acid; DMSO, dimethyl sulfoxide; MUC2,
mucin 2; n.s., not significant; OD, optical density. |
Bile acids upregulate CDX2 and MUC2
expression in GES-1 cells
To determine the effects of bile acids on the
formation of gastric IM, RT-qPCR and western blotting were
performed to detect CDX2 and MUC2 expression levels in cells
treated with bile acids (Fig.
1C–J). Treatment with CDCA at 50, 100, 150 or 200 µmol/l
for 24 h increased CDX2 and MUC2 expression, at both the mRNA and
protein levels, in a dose-dependent manner (Fig. 1C, E and F). Similarly, DCA
treatment led to an increase in CDX2 and MUC2 expression in a
dose-dependent manner (Fig. 1D, G and
H). Subsequently, GES-1 cells were transfected with 2.5 kb
CDX2- and 2.6 kb MUC2-luc promoter constructs and were treated with
various concentrations of DCA and CDCA. As shown in Fig. 1 I and J, bile acids significantly
increased CDX2 and MUC2 transcriptional activity in a
dose-dependent manner. These results indicated that treatment of
GES-1 cells with bile acids may lead to a dose-dependent
upregulation of CDX2 and MUC2 expression.
CDX2 regulates bile acid-induced
expression of MUC2
The intestinal-specific transcription factor CDX2
can directly bind to the MUC2 promoter and promote its
transcription (5). This study
aimed to investigate whether CDX2 has a pivotal role in bile
acid-induced upregulation of MUC2 transcription. MUC2 promoter
activity and protein expression levels were detected in GES-1 cells
transfected with shRNA-CDX2 lentiviral vectors following treatment
with 200 µmol/l bile acids (Fig. 2A–C). As shown in Fig. 2B, the shRNA-CDX2 construct
successfully knocked down CDX2 expression. Knockdown of CDX2
expression attenuated bile acid-induced MUC2 promoter activity and
protein expression (Fig.
2A–C).
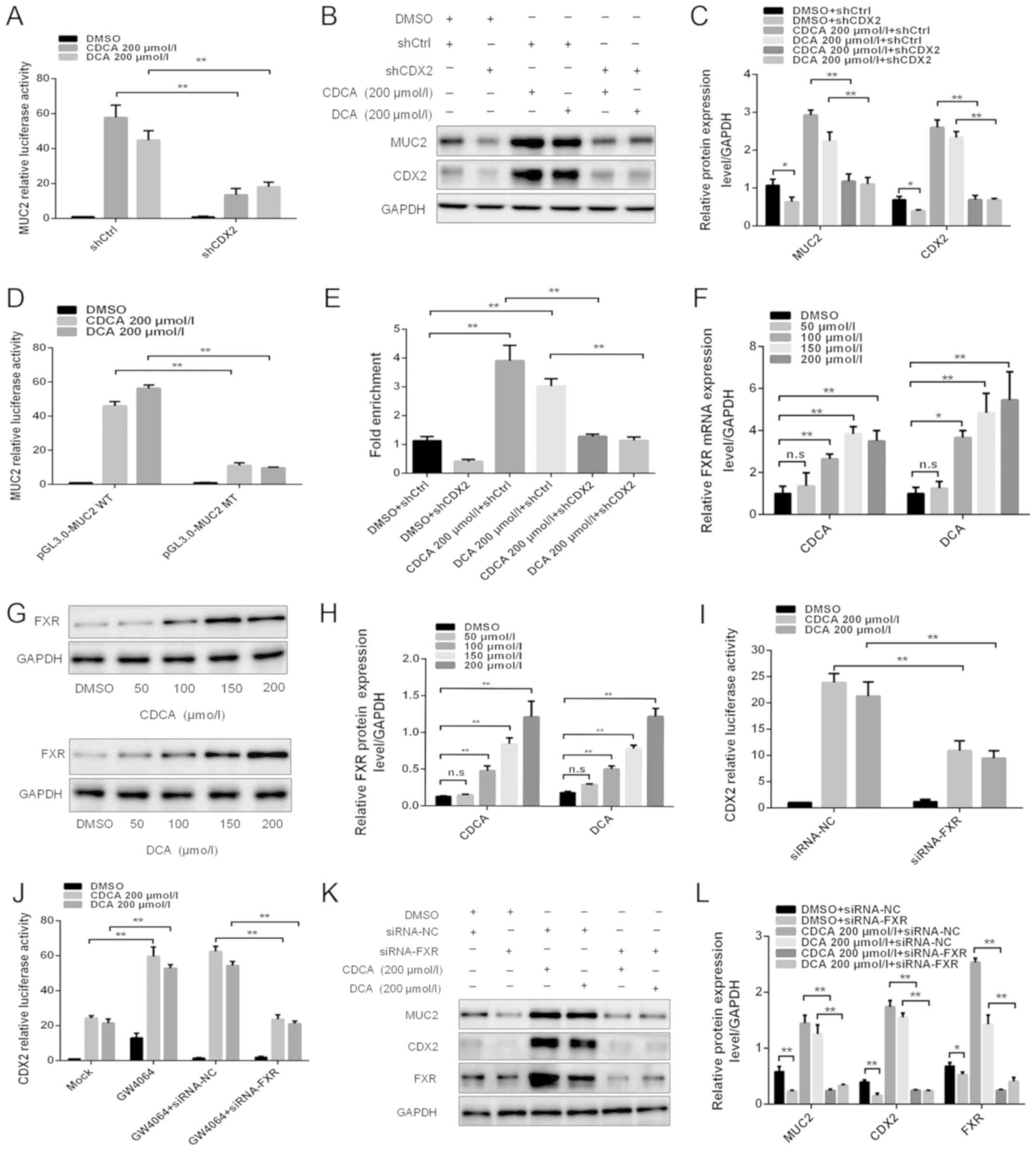 | Figure 2FXR is involved in the regulation of
bile acid-induced CDX2 and MUC2 expression. (A) Dual-luciferase
reporter assay was conducted to determine the effect of knockdown
of CDX2 expression on bile acid-enhanced MUC2 promoter activity.
(B) Western blotting was conducted to determine the effect of
knockdown of CDX2 on bile acid-enhanced MUC2 expression. (C)
Comparison of CDX2 and MUC2 protein levels in each group of GES-1
cells. (D) Mutagenesis of the MUC2 promoter was performed using a
site-directed mutagenesis kit. A dual-luciferase reporter assay was
conducted to detect the luciferase activities in GES-1 cells
transfected with a MUC2 promoter reporter construct or mutated
construct. (E) A quantitative chromatin immunopre-cipitation assay
was performed to investigate the effect of bile acids on the
binding of CDX2 to the MUC2 promoter. (F) Reverse
transcription-quantitative polymerase chain reaction was conducted
to detect the mRNA expression levels of FXR in cells treated with
DMSO, or CDCA or DCA at various concentrations (0, 100, 150 and 200
µmol/l) for 24 h. (G) Western blot analysis of FXR protein
expression in each group of bile acid-treated GES-1 cells. (H)
Comparison of FXR protein levels in each group of GES-1 cells
stimulated with CDCA or DCA. (I) siRNA-FXR was transfected into
cells with the CDX2-luc promoter construct. A dual-luciferase
reporter assay was conducted to determine the effect of the
downregulation of FXR on bile acid-enhanced CDX2 promoter activity.
(J) Effects of treatment with the FXR agonist GW4064 on bile
acid-enhanced CDX2 promoter activity. (K) Western blotting was
performed to investigate the effect of FXR downregulation on bile
acid-induced CDX2 and MUC2 expression. (L) Comparison of CDX2, MUC2
and FXR protein expression in each group of GES-1 cells. Data are
presented as the means ± standard deviation from three independent
experiments. *P<0.05, **P<0.01. CDCA,
chenodeoxycholic acid; CDX2, caudal-related homeobox transcription
factor 2; DCA, deoxycholic acid; DMSO, dimethyl sulfoxide; FXR,
farnesoid X receptor; MT, mutated type; MUC2, mucin 2; NC, negative
control; n.s., not significant; sh, short hairpin RNA; si/siRNA,
small interfering RNA; WT, wild-type. |
Potential CDX2-binding sites were identified in the
MUC2 promoter using bioinformatics analysis. To further demonstrate
that CDX2 transcriptionally activated MUC2 expression by binding to
the ATAAA motif in the MUC2 promoter, the MUC2 promoter luciferase
construct was used to produce a mutant construct. The present study
indicated that transfection with the MUC2 mutant construct led to a
significantly lower level of luciferase activity in GES-1 cells
after bile acids treatment compared with transfection with the
wild-type construct (Fig. 2D).
Furthermore, a qChIP assay was performed to investigate the effects
of bile acids on the binding of CDX2 to the MUC2 promoter. The
results revealed that bile acids increased the binding of CDX2 to
the MUC2 promoter, whereas knockdown of CDX2 abolished enhanced
binding (Fig. 2E). These results
suggested that CDX2 may regulate bile acid-induced expression of
MUC2 in GES-1 cells.
FXR is involved in bile acid-induced CDX2
and MUC2 expression
FXR is a bile acid nuclear receptor, which has been
reported to be upregulated by bile acids in GC (10). This study aimed to investigate
whether FXR is involved in bile acid-induced CDX2 and MUC2
expression in GES-1 cells. A significant increase in FXR expression
at the mRNA and protein levels was observed in cells treated with
bile acids (Fig. 2F–H).
The present study then assessed whether FXR
participated in the regulation of bile acid-induced CDX2
transcription. siRNA-FXR was co-transfected into GES-1 cells with
the CDX2-luc promoter construct. As shown in Fig. 2I, downregulation of FXR expression
attenuated bile acid-enhanced CDX2 transcriptional activity.
Conversely, treatment with the FXR agonist GW4064 synergistically
enhanced bile acid-induced CDX2 transcriptional activity (Fig. 2J). In addition, downregulation of
FXR abolished the synergistic effect of bile acids and GW4064 on
bile acid-induced CDX2 promoter activity (Fig. 2J). Western blotting revealed that
downregulation of FXR expression abolished the bile acid-induced
upregulation of CDX2 and MUC2 expression (Fig. 2K and L). These results indicated
that FXR participated in the transactivation of CDX2 induced by
bile acids. Taken together, these results suggested that FXR may
participate in the regulatory effects of bile acid-induced CDX2 and
MUC2 expression on gastric IM formation.
NF-κB is involved in regulation of the
bile acid-induced FXR/CDX2/MUC2 signalling pathway
The bile acid-induced NF-κB signalling pathway
serves an important role in gastric tumourigenesis (28,29).
This study assessed whether NF-κB is involved in regulating the
bile acid-induced FXR/CDX2/MUC2 signalling pathway in GES-1 cells.
The NF-κB luciferase plasmid was transfected into GES-1 cells that
were treated with 200 µmol/l bile acids. An increase in
NF-κB activity was detected following treatment with bile acids
(Fig. 3A). An increase in p50 and
p65 protein levels was also detected in response to treatment with
bile acids (Fig. 3B and C). These
results indicated that NF-κB was activated in GES-1 cells treated
with bile acids.
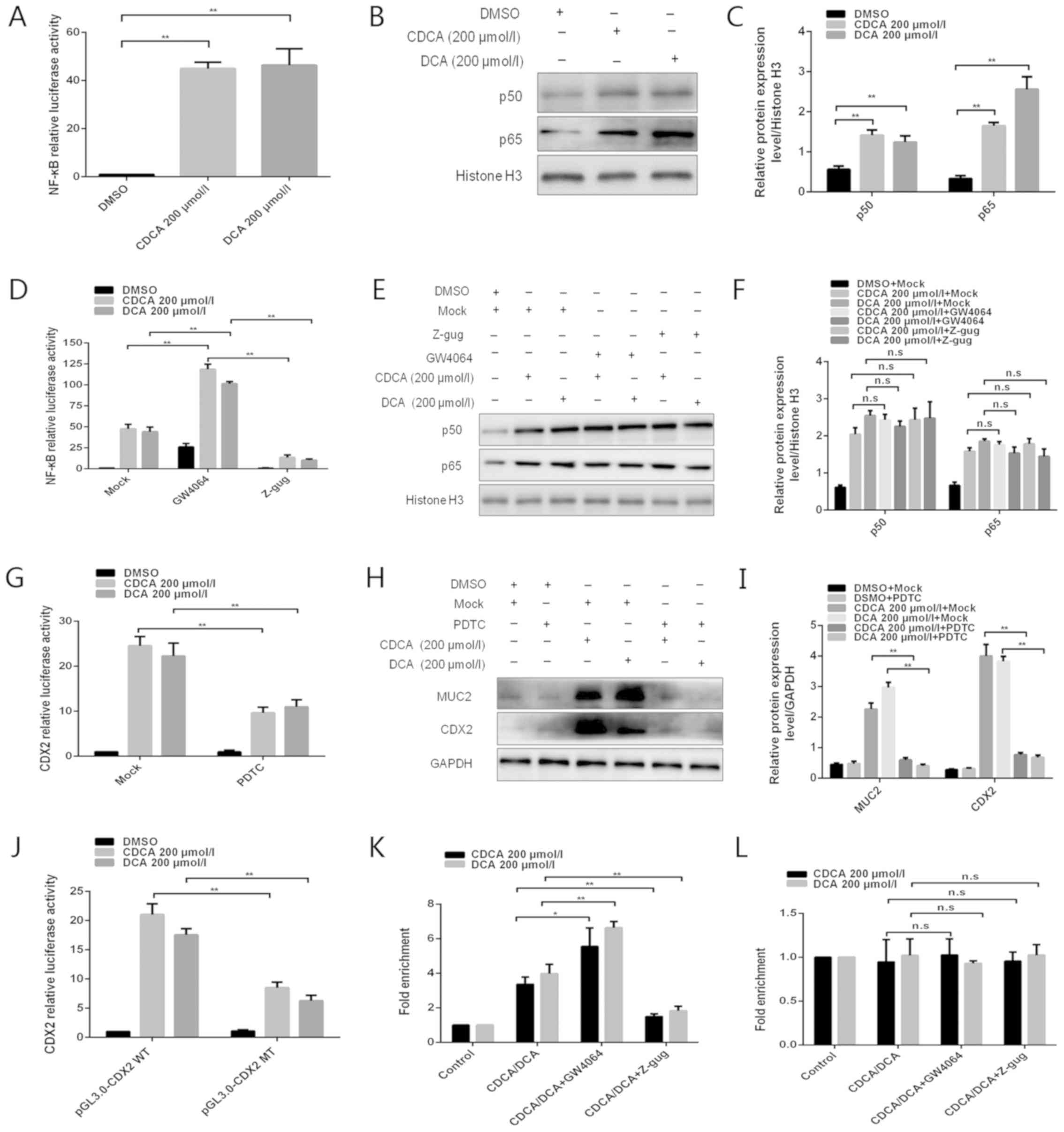 | Figure 3NF-κB is involved in the regulation
of bile acid-induced FXR/CDX2/MUC2 signalling pathway activation.
(A) Dual-luciferase reporter assay was conducted to determine the
effect of bile acids on NF-κB activity. (B) Western blotting was
conducted to detect the effects of bile acids on p50/p65 protein
expression. (C) Comparison of p50/p65 protein levels in each group
of GES-1 cells. (D) Dual-luciferase reporter assay was performed to
detect the effect of the FXR agonist GW4064 or the antagonist Z-gug
on bile acid-enhanced NF-κB activity. (E) Western blotting was
conducted to determine the effect of the FXR agonist GW4064 or the
antagonist Z-gug on bile acid-induced alterations in p50 and p65
protein levels. (F) Comparison of p50 and p65 protein levels in
each group of GES-1 cells. (G) Dual-luciferase reporter assay was
conducted to determine the effect of the NF-κB inhibitor PDTC on
bile acid-enhanced CDX2 promoter activity. (H) Western blotting was
performed to determine the effect of the NF-κB inhibitor PDTC on
bile acid-induced alterations in CDX2 and MUC2 protein expression
levels. (I) Comparison of CDX2 and MUC2 protein levels in each
group of GES-1 cells. (J) Dual-luciferase reporter assay was
conducted to detect the luciferase activities in GES-1 cells
transfected with CDX2 promoter reporter construct or mutated
construct. (K and L) Quantitative chromatin immunoprecipitation
assay was performed to investigate the effect of the FXR agonist
GW4064 or the antagonist Z-gug on the binding of NF-κB (K) p50 or
(L) p65 protein to the CDX2 promoter. Data are presented as the
means ± standard deviation from three independent experiments.
*P<0.05, **P<0.01. CDCA,
chenodeoxycholic acid; CDX2, caudal-related homeobox transcription
factor 2; DCA, deoxycholic acid; DMSO, dimethyl sulfoxide; MT,
mutated type; MUC2, mucin 2; NF-κB, nuclear factor-κB; n.s., not
significant; PDTC, pyrrolidine dithiocarbamate; WT, wild-type;
Z-gug, Z-guggulsterone. |
The interaction between FXR and NF-κB has been
reported in the hepatic inflammatory response (30). This study aimed to investigate
whether FXR is involved in regulating bile acid-induced NF-κB
activity. Compared with in the control group, treatment with GW4064
enhanced bile acid-induced NF-κB activity, whereas Z-guggulsterone
attenuated this activity (Fig.
3D), indicating that NF-κB activity was regulated by FXR
following treatment of GES-1 cells with bile acids. However,
treatment with GW4064 or Z-guggulsterone did not affect nuclear p50
and p65 protein expression (Fig. 3E
and F).
A previous study demonstrated that the CDX2 promoter
contains an NF-κB binding site and can be bound by p50/p65
(31). Recently, Chen et al
(32) reported that CDX2 can be
activated by the NF-κB signalling pathway in gastric IM. Therefore,
the present study assessed whether NF-κB was involved in regulating
bile acid-induced CDX2 expression in GES-1 cells. Treatment with
PDTC, an inhibitor of NF-κB activation, strongly attenuated bile
acid-induced CDX2 promoter-driven luciferase activity (Fig. 3G). Furthermore, PDTC attenuated
bile acid-induced protein expression of CDX2 and MUC2 (Fig. 3H and I). These results suggested
that bile acid-induced transactivation of CDX2 may be regulated by
NF-κB.
In order to determine whether NF-κB
transcriptionally activated CDX2 expression by binding to the
special motif in the CDX2 promoter, mutagenesis of the predicted
NF-κB binding site was performed in the CDX2 promoter using a
site-directed mutagenesis kit. Transfection with the CDX2 mutant
construct led to a decrease in luciferase activity in GES-1 cells
following treatment with bile acids (Fig. 3J). Furthermore, a qChIP assay was
conducted to investigate the effect of bile acids on the binding of
NF-κB to the CDX2 promoter. The present study indicated that bile
acid treatment enhanced the binding of p50, but not p65, to the
CDX2 promoter (Fig. 3K and L). The
present study also explored whether FXR participated in the
regulation of the binding of NF-κB to the CDX2 promoter. Treatment
with GW4064 or Z-guggulsterone enhanced or attenuated bile
acid-induced binding of p50 to the CDX2 promoter, respectively.
However, the binding activity of p65 to the CDX2 promoter exhibited
no obvious alterations in response to treatment with GW4064 or
Z-guggulsterone. These results demonstrated that bile acid-induced
transactivation of CDX2 may be mediated by enhanced p50 binding to
the CDX2 promoter and that this effect is regulated by FXR. Taken
together, these data indicated that NF-κB may be involved in
regulation of the bile acid-induced FXR/CDX2/MUC2 signalling
pathway.
High expression levels of CDX2 are
correlated with FXR and MUC2 in gastric IM tissues
To further elucidate the expression pattern of MUC2,
CDX2 and FXR in the formation of gastric IM, immunohistochemistry
was conducted using paraffin-embedded gastric IM and paired normal
gastric mucosa tissues (Fig.
4A–G). Representative CDX2, MUC2 and FXR staining images in
normal mucosa and IM tissues are shown in Fig. 4A. Positive CDX2, MUC2 and FXR
staining was increased in IM tissues compared with in normal
gastric mucosa to (Fig. 4B, D and
F). Analysis of the IHC scores also revealed that the IRS of
CDX2, MUC2 and FXR staining was higher in IM tissues than in normal
tissues (Fig. 4C, E and G).
RT-qPCR was further conducted to detect the mRNA expression levels
of CDX2, FXR and MUC2 in gastric IM tissues and paired normal
gastric tissues. As shown in Fig.
4H–J, the mRNA expression levels of CDX2, FXR and MUC2 were
significantly upregulated in IM tissues compared with in normal
gastric mucosa. Furthermore, CDX2 mRNA levels were positively
correlated with FXR and MUC2 mRNA levels in 40 gastric IM tissues
(Fig. 4K and L). Taken together,
these data suggested that the expression levels of CDX2, FXR and
MUC2 were upregulated in IM tissues relative to normal tissues, and
were positively correlated with one another.
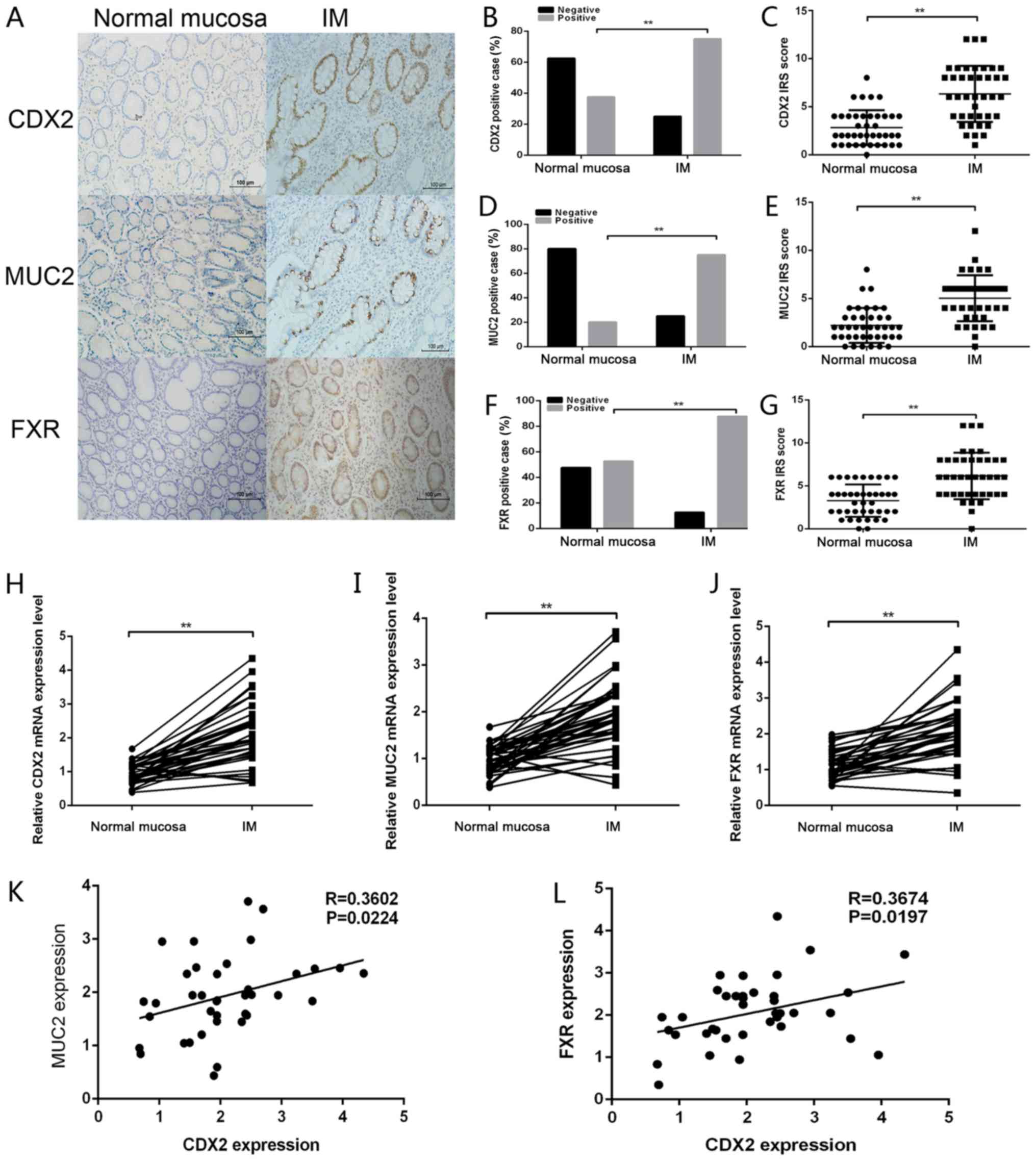 | Figure 4CDX2 expression is correlated with
FXR and MUC2 expression in gastric IM tissues. (A)
Immunohistochemical staining of CDX2, MUC2 and FXR in gastric
mucosal tissues with or without IM. Scale bar, 100 µm. (B)
CDX2, (D) MUC2 and (F) FXR staining was classified into negative
and positive, and the percentage of tissues in each group was
shown. IRS of (C) CDX2, (E) MUC2 and (G) FXR staining in normal
mucosa and IM tissues. Reverse transcription-quantitative
polymerase chain reaction was performed to determine the mRNA
expression levels of (H) CDX2, (I) MUC2 and (J) FXR in mucosal
tissues with or without IM. Correlations between the mRNA
expression levels of CDX2 and (K) MUC2 (r=0.3602; P<0.05) and
(L) FXR (r=0.3674; P<0.05) in gastric IM tissues. Data are
presented as the means ± standard deviation from three independent
experiments. **P<0.01. CDX2, caudal-related homeobox
transcription factor 2; FXR, farnesoid X receptor; IM, intestinal
metaplasia; IRS, immunoreactivity score; MUC2, mucin 2. |
Bile acids induce molecular alterations
linked to premalignant lesions in murine gastric mucosa in
vivo
To further investigate the chronic effects of bile
acids on gastric IM formation in vivo, an animal model of
DGR was generated by administering bile acids to the stomach of
mice (33). Western blot analyses
revealed that, compared with in mice exposed to PBS alone, the
murine gastric mucosa exposed to DCA, CDCA, or a mixture of DCA and
CDCA, exhibited significant increases in the expression levels of
FXR, p50, p65, CDX2 and MUC2 (Fig.
5A–F). However, H&E-stained murine mucosa revealed no
obvious IM-like alterations in mice treated with DCA, CDCA or a
mixture of both (data not shown). These results further confirmed
the pivotal role of bile acids in gastric IM formation via the
induction of molecular alterations linked to premalignant
lesions.
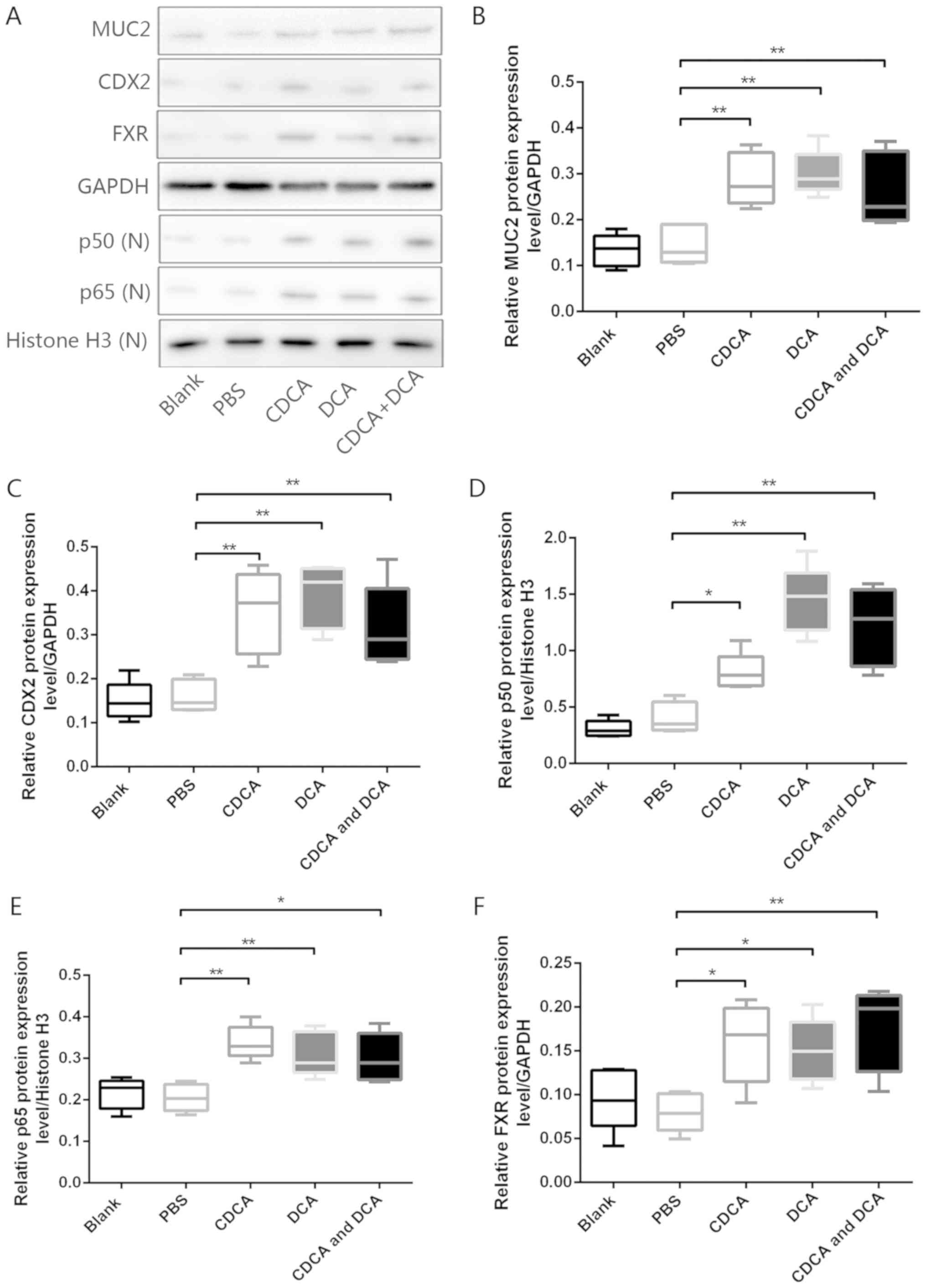 | Figure 5Bile acids induce molecular
alterations linked to premalignant lesions in murine gastric mucosa
in vivo. (A) Western blot analysis of MUC2, CDX2, p50, p65
and FXR protein expression in murine gastric mucosa samples from
each group. Comparison of (B) MUC2, (C) CDX2, (D) p50, (E) p65 and
(F) FXR protein expression levels in each group.
*P<0.05, **P<0.01. CDCA,
chenodeoxycholic acid; CDX2, caudal-related homeobox transcription
factor 2; DCA, deoxycholic acid; FXR, farnesoid X receptor; MUC2,
mucin 2; N, nuclear. |
Discussion
GC is one of the leading causes of cancer-associated
morbidity and mortality worldwide; however, the exact molecular
mechanisms that underlie GC tumourigenesis remain elusive. IM is
considered to be a precancerous lesion of the gastric mucosa. IM
places patients at a higher risk of developing gastric
adenocarcinoma than patients without IM (34). In addition, mounting
epidemiological evidence has demonstrated that exposure to bile
acids can induce IM in the stomach (19). Tatsugami et al reported that
grades of atrophy and IM are significantly positively correlated
with bile acid concentrations in patients with Helicobacter
pylori infections (18).
According to a multi-centric and large-scale cross-sectional study
in Japan (35), the risk of
developing IM is significantly higher in patients with high bile
acid concentrations, regardless of H. pylori infection
status. The critical role of bile acids in IM formation in the
stomach is widely accepted; however, the molecular mechanisms by
which bile acids promote IM formation in the human stomach remain
to be characterized.
Increasing evidence has indicated that CDX2 has a
crucial role in gastric IM formation and gastric carcinogenesis.
Silberg et al (36)
observed that overexpression of CDX2 in transgenic mice leads to IM
formation in the stomach, alongside the presence of intestinal-type
goblet cells and the expression of intestine-specific genes. This
result indicated that activated CDX2 is involved in the initiation
of IM formation, which can lead to intestinal neoplasia of the
gastric mucosa. As an intestinal-specific transcription factor,
CDX2 directly regulates several genes involved in proliferation,
differentiation, and intestinal cell fate specification, including
sucrose-isomaltase (37), MUC2
(5), TFF3 (6) and guanylyl cyclase C (38). This study revealed that treatment
with bile acids increased the mRNA and protein expression levels of
CDX2 and MUC2 in GES-1 cells. Xu et al reported that RGM-1
normal rat gastric epithelial cells exposed to bile acids exhibit
upregulated CDX2 and MUC2 expression (21), which is consistent with the present
findings. In addition, the transcriptional activity of CDX2 and
MUC2 was increased in GES-1 cells treated with bile acids in a
dose-dependent manner, thus indicating that treatment with bile
acids increased CDX2 and MUC2 expression at the transcriptional
level. Furthermore, this study revealed that CDX2 regulated bile
acid-induced MUC2 transcription in GES-1 cells by binding to the
MUC2 promoter, which was consistent with the results of a previous
study (5). However, several
studies have indicated that bile acids stimulate MUC2 transcription
via activation of NF-κB, but not CDX2, in oesophageal
adenocarcinoma (39) and
colorectal cancer (40), thus
suggesting that the pathogenesis of different tumour types may
differ. In addition, immunohistochemistry demonstrated that CDX2
and MUC2 expression levels were significantly upregulated in IM
tissue compared with in normal gastric mucosa tissue, and CDX2
expression was positively correlated with that of MUC2 in IM
tissues. This result supported the present in vitro
findings. Taken together, these results suggested that treatment of
GES-1 cells with bile acids may lead to the upregulation of CDX2
and MUC2 expression at the mRNA and protein levels in a
dose-dependent manner.
FXR is a potent nuclear receptor of bile acid, which
exerts important effects on the homeostasis of bile acids,
cholesterol and lipids (11,41,42).
Aberrant expression of FXR has been associated with human
colorectal cancer (43), prostate
cancer (44) and breast cancer
(45). FXR is slightly detectable
in normal human stomach tissue. In a previous study, high
expression levels of FXR were reported to be associated with
gastric IM formation (15). The
present study revealed that treatment of GES-1 cells with bile acid
induced overexpression of FXR. The immunohistochemistry staining
results indicated that FXR expression was significantly increased
in IM tissues compared with in normal mucosa. Consistent with this
study, De Gottardi et al (46) reported that Barrett's oesophagus, a
precancerous lesion of the oesophageal mucosa, also exhibits high
expression levels of FXR, implying that this protein may be
involved in the pathogenesis of Barrett's oesophagus. The role of
FXR in bile acid-induced CDX2 and MUC2 expression in GES-1 cells
was also investigated. The present results indicated that FXR
participated in bile acid-induced transactivation of CDX2 in GES-1
cells, a result that was consistent with the work of Li et
al (47). In oesophageal
adenocarcinoma, the FXR antagonist Z-guggulsterone has been
reported to suppress bile acid-induced constitutive CDX2
expression, implying that FXR may regulate bile acid-induced CDX2
expression (48). In the present
study, FXR expression was positively correlated with CDX2
expression in gastric IM tissues. Taken together, these results
indicated that FXR may be involved in the regulation of bile
acid-induced CDX2 and MUC2 expression in gastric IM formation. Lian
et al reported that proper activation of FXR is a protective
mechanism by which gastric mucosa resists inflammation-mediated
damage (49). In addition, colon
cells with decreased FXR expression fail to prevent intracellular
bile acid accumulation to cytotoxic levels, thus predisposing these
cells to tumourigenesis (50). It
is possible that the upregulation of FXR in response to bile acids
is a protective mechanism by which the gastric mucosa resists bile
acid-induced damage. However, further studies are required to
investigate the role of FXR in gastric IM formation.
NF-κB is involved in several important biological
phenomena, including proliferation, inflammation, apoptosis and
differentiation (51). Bile acids
can induce the activation of NF-κB in oesophageal cancer (52) and colon cancer (39), triggering a wide array of
pro-inflammatory cascades and disturbing cellular homeostasis. The
present study demonstrated that exposure of GES-1 cells to bile
acids increased NF-κB activity, as well as p50 and p65 protein
expression. The potential role of FXR in bile acid-induced NF-κB
activity was subsequently investigated. Our preliminary data
demonstrated that treatment with GW4064 or Z-guggulsterone enhanced
or attenuated bile acid-induced NF-κB activity, respectively. This
result suggested that FXR may be a positive modulator of bile
acid-induced NF-κB activity in GES-1 cells. However, bile
acid-induced increases in nuclear p50 and p65 protein levels were
not significantly affected by treatment with GW4064 or
Z-guggulsterone, indicating that FXR may affect the activity of
NF-κB but not its nuclear expression. Consistent with these
results, Lee et al (53)
reported that downregulation of FXR in pancreatic carcinoma cells
decreases NF-κB activity and consequently inhibits its target
genes. However, in inflammatory bowel disease, overexpression of
NF-κB results in suppression of FXR activity, indicating that NF-κB
may participate in the regulation of FXR activity (54). Wang et al (30) also described a negative crosstalk
between the FXR and NF-κB signalling pathways in the hepatic
inflammatory response. Therefore, the exact molecular mechanism
underlying the effects of FXR and NF-κB on gastric IM formation
remains to be further explored.
This study aimed to investigate whether NF-κB was
involved in the regulation of bile acid-induced CDX2 expression.
The results indicated that PDTC, an inhibitor of NF-κB activation,
attenuated CDX2 promoter-driven luciferase activity, as well as
CDX2 and MUC2 protein expression, following bile acid treatment,
thus preliminarily verifying our hypothesis. A previous study
demonstrated that bile acids activate CDX2 transcription via NF-κB
and upregulate CDX2 expression in oesophageal keratinocytes
(55). Different NF-κB subunits
exert different functions on CDX2 transcriptional activity.
Debruyne et al (38)
reported that DCA induces p50 nuclear translocation and enhances
the binding activity of p50 to the CDX2 promoter, whereas p65
remains in the cytoplasm in human oesophageal cells. Kim et
al (31) also reported that
the transcriptional activity of CDX2 depends on the balance between
the p50 and p65 subunits. The present study observed that
mutagenesis of the predicted NF-κB binding site in the CDX2
promoter abolished enhanced luciferase activity in GES-1 cells
following bile acid treatment. A qChIP assay indicated that bile
acid treatment led to increased binding of p50, but not p65, to the
CDX2 promoter. Furthermore, treatment with GW4064 or
Z-guggulsterone enhanced or attenuated the binding activity of p50
to the CDX2 promoter, respectively. Yamada et al
demonstrated that CDCA enhances p50 protein binding to the CDX2
promoter in oesophageal adenocarcinoma. However, treatment with
Z-guggulsterone does not reduce CDCA-induced binding activity of
p65 to the CDX2 promoter (48).
Taken together, these results indicated that bile acid-induced
transcriptional activation of CDX2 in GES-1 cells may be increased
via the enhancement of p50 binding to the CDX2 promoter; this
effect may be regulated by FXR.
The results of the DGR animal model experiment
demonstrated an increase in the protein expression levels of FXR,
p50, p65, CDX2 and MUC2 in gastric mucosa exposed to DCA, CDCA, or
a mixture of both, providing further evidence to verify the present
findings. However, H&E-stained murine mucosa revealed no
obvious IM-like alterations in mucosa treated with DCA, CDCA or a
mixture of both. There are three potential reasons for this
phenomenon: i) The time period over which the animals were treated
with bile acids was not sufficient to stimulate gastric IM
formation. ii) Gastric IM formation is a complex process that
depends on multifactorial interactions, such as H. pylori
infection, bile acids and acid. iii) The animal model of DGR used
in this study did not wholly mimic the complex dynamic environment
of DGR in vivo, including the time interval of DGR and the
fluctuating concentration of bile acids. In our future in
vivo studies, we aim to mimic the actual DGR environment and
thereby clarify the molecular mechanisms underlying IM formation in
the human stomach.
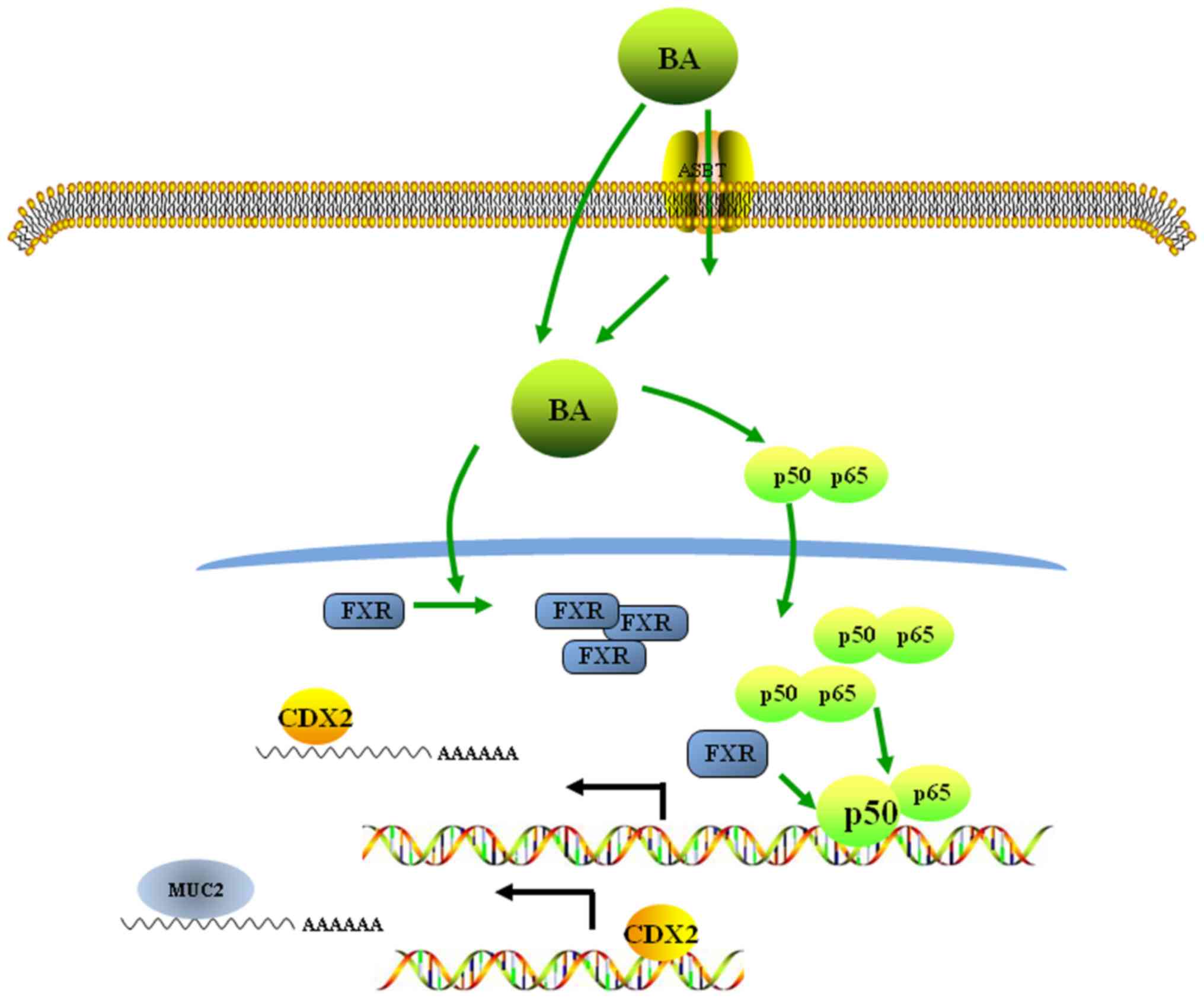
In conclusion, the results of the present study
suggested a molecular mechanism underlying the bile acid-induced
acquisition of an intestinal phenotype in normal gastric epithelial
cells. These data revealed that bile acids may activate the
FXR/NF-κB signalling pathway, inducing the ectopic expression of
CDX2, an intestinal-specific transcription factor, and MUC2, an
intestinal metaplasia marker, in normal gastric epithelial cells
(Fig. 6). These observations of IM
formation in the human stomach may contribute to our understanding
of the pathogenic mechanisms underlying neoplastic transformation
in the stomach.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 81101874 and
81172362), The Science and Technology Project of Shaanxi Province
(grant nos. 2016SF-015 and 2016SF-157), and The Coordinative and
Innovative Plan Projects of the Science and Technology Program in
Shaanxi Province (grant no. 2013KTCQ03-08).
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
JHY and JBZ performed the experiments, acquired the
data and drafted the manuscript. KY, JQ and CBW collected human
gastric IM tissues, and assisted with qPCR and western blotting.
YHW and KW analysed and interpreted data. XJS and JHY substantially
contributed to the study conception and design. All authors read
and approved the final manuscript and agreed to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients, and the study protocol was approved by the Ethics
Committee of The First Affiliated Hospital of Xi'an Jiaotong
University. All animal research was carried out following approval
by the Institutional Animal Care and Use Committee of The First
Affiliated Hospital of Xi'an Jiaotong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Costamagna G and Cesaro P: Early gastric
cancer: Detection and endoscopic treatment. Ann Ital Chir.
83:183–191. 2012.PubMed/NCBI
|
|
3
|
Correa P and Piazuelo MB: The gastric
precancerous cascade. J Dig Dis. 13:2–9. 2012. View Article : Google Scholar :
|
|
4
|
Compare D, Rocco A and Nardone G:
Screening for and surveillance of gastric cancer. World J
Gastroenterol. 20:13681–13691. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yamamoto H, Bai YQ and Yuasa Y:
Homeodomain protein CDX2 regulates goblet-specific MUC2 gene
expression. Biochem Biophys Res Commun. 300:813–818. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shimada T, Koike T, Yamagata M, Yoneda M
and Hiraishi H: Regulation of TFF3 expression by homeodomain
protein CDX2. Regul Pept. 140:81–87. 2007. View Article : Google Scholar
|
|
7
|
Liu Q, Teh M, Ito K, Shah N, Ito Y and
Yeoh KG: CDX2 expression is progressively decreased in human
gastric intestinal metaplasia, dysplasia and cancer. Modern
pathology: An official journal of the United States and Canadian
Academy of Pathology Inc. 20:1286–1297. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Eda A, Osawa H, Yanaka I, Satoh K, Mutoh
H, Kihira K and Sugano K: Expression of homeobox gene CDX2 precedes
that of CDX1 during the progression of intestinal metaplasia. J
Gastroenterol. 37:94–100. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mutoh H, Hakamata Y, Sato K, Eda A, Yanaka
I, Honda S, Osawa H, Kaneko Y and Sugano K: Conversion of gastric
mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice.
Biochem Biophys Res Commun. 294:470–479. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lefebvre P, Cariou B, Lien F, Kuipers F
and Staels B: Role of bile acids and bile acid receptors in
metabolic regulation. Physiol Rev. 89:147–191. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Makishima M: Nuclear receptors as targets
for drug development: Regulation of cholesterol and bile acid
metabolism by nuclear receptors. J Pharmacol Sci. 97:177–183. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Redinger RN: The role of the enterohepatic
circulation of bile salts and nuclear hormone receptors in the
regulation of cholesterol homeostasis: Bile salts as ligands for
nuclear hormone receptors. Can J Gastroenterol. 17:265–271. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Modica S, Murzilli S, Salvatore L, Schmidt
DR and Moschetta A: Nuclear bile acid receptor FXR protects against
intestinal tumorigenesis. Cancer Res. 68:9589–9594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maran RR, Thomas A, Roth M, Sheng Z,
Esterly N, Pinson D, Gao X, Zhang Y, Ganapathy V, Gonzalez FJ, et
al: Farnesoid X receptor deficiency in mice leads to increased
intestinal epithelial cell proliferation and tumor development. J
Pharmacol Exp Ther. 328:469–477. 2009. View Article : Google Scholar :
|
|
15
|
Ku HJ, Kim HY, Kim HH, Park HJ and Cheong
JH: Bile acid increases expression of the histamine-producing
enzyme, histidine decarboxylase, in gastric cells. World J
Gastroenterol. 20:175–182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sobala GM, O'Connor HJ, Dewar EP, King RF,
Axon AT and Dixon MF: Bile reflux and intestinal metaplasia in
gastric mucosa. J Clin Pathol. 46:235–240. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dixon MF, Mapstone NP, Neville PM,
Moayyedi P and Axon AT: Bile reflux gastritis and intestinal
metaplasia at the cardia. Gut. 51:351–355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tatsugami M, Ito M, Tanaka S, Yoshihara M,
Matsui H, Haruma K and Chayama K: Bile acid promotes intestinal
metaplasia and gastric carcinogenesis. Cancer Epidemiol Biomarkers
Prev. 21:2101–2107. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang JX, Liu Q, Zhao B, Zhang HH, Sang
HM, Djaleel SM, Zhang GX and Xu SF: Risk factors for intestinal
metaplasia in a southeastern Chinese population: An analysis of
28,745 cases. J Cancer Res Clin Oncol. 143:409–418. 2017.
View Article : Google Scholar
|
|
20
|
Tamagawa Y, Ishimura N, Uno G, Aimi M,
Oshima N, Yuki T, Sato S, Ishihara S and Kinoshita Y: Bile acids
induce Delta-like 1 expression via Cdx2-dependent pathway in the
development of Barrett's esophagus. Lab Invest. 96:325–337. 2016.
View Article : Google Scholar
|
|
21
|
Xu Y, Watanabe T, Tanigawa T, Machida H,
Okazaki H, Yamagami H, Watanabe K, Tominaga K, Fujiwara Y, Oshitani
N, et al: Bile acids induce cdx2 expression through the farnesoid x
receptor in gastric epithelial cells. J Clin Biochem Nutr.
46:81–86. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Qin R, Wang NN, Chu J and Wang X:
Expression and significance of homeodomain protein Cdx2 in gastric
carcinoma and precancerous lesions. World J Gastroenterol.
18:3296–3302. 2012.PubMed/NCBI
|
|
24
|
Chen Q, Zheng PS and Yang WT:
EZH2-mediated repression of GSK-3β and TP53 promotes Wnt/β-catenin
signaling-dependent cell expansion in cervical carcinoma.
Oncotarget. 7:36115–36129. 2016.PubMed/NCBI
|
|
25
|
Kauer WK, Peters JH, DeMeester TR,
Feussner H, Ireland AP, Stein HJ and Siewert RJ: Composition and
concentration of bile acid reflux into the esophagus of patients
with gastroesophageal reflux disease. Surgery. 122:874–881. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu T, Zhang X, So CK, Wang S, Wang P, Yan
L, Myers R, Chen Z, Patterson AP, Yang CS, et al: Regulation of
Cdx2 expression by promoter methylation, and effects of Cdx2
transfection on morphology and gene expression of human esophageal
epithelial cells. Carcinogenesis. 28:488–496. 2007. View Article : Google Scholar
|
|
27
|
Huo X, Zhang HY, Zhang XI, Lynch JP,
Strauch ED, Wang JY, Melton SD, Genta RM, Wang DH, Spechler SJ, et
al: Acid and bile salt-induced CDX2 expression differs in
esophageal squamous cells from patients with and without Barrett's
esophagus. Gastroenterology. 139:194–203.e1. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Merga YJ, O'Hara A, Burkitt MD, Duckworth
CA, Probert CS, Campbell BJ and Pritchard DM: Importance of the
alternative NF-κB activation pathway in inflammation-associated
gastrointestinal carcinogenesis. Am J Physiol Gastrointest Liver
Physiol. 310:G1081–G1090. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gambhir S, Vyas D, Hollis M, Aekka A and
Vyas A: Nuclear factor kappa B role in inflammation associated
gastrointestinal malignancies. World J Gastroenterol. 21:3174–3183.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang YD, Chen WD, Wang M, Yu D, Forman BM
and Huang W: Farnesoid X receptor antagonizes nuclear factor kappaB
in hepatic inflammatory response. Hepatology. 48:1632–1643. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim S, Domon-Dell C, Wang Q, Chung DH, Di
Cristofano A, Pandolfi PP, Freund JN and Evers BM: PTEN and
TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene
through a PI3K, PKB/Akt, and NF-appaB-dependent pathway.
Gastroenterology. 123:1163–1178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen BJ, Zeng S, Xie R, Hu CJ, Wang SM, Wu
YY, Xiao YF and Yang SM: hTERT promotes gastric intestinal
metaplasia by upregulating CDX2 via NF-κB signaling pathway.
Oncotarget. 8:26969–26978. 2017.PubMed/NCBI
|
|
33
|
Vageli DP, Prasad ML and Sasaki CT:
Gastro-duodenal fluid induced nuclear factor-kappaB activation and
early pre-malignant alterations in murine hypopharyngeal mucosa.
Oncotarget. 7:5892–5908. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Spence AD, Cardwell CR, McMenamin UC,
Hicks BM, Johnston BT, Murray LJ and Coleman HG: Adenocarcinoma
risk in gastric atrophy and intestinal metaplasia: A systematic
review. BMC Gastroenterol. 17:1572017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsuhisa T, Arakawa T, Watanabe T,
Tokutomi T, Sakurai K, Okamura S, Chono S, Kamada T, Sugiyama A,
Fujimura Y, et al: Relation between bile acid reflux into the
stomach and the risk of atrophic gastritis and intestinal
metaplasia: a multicenter study of 2283 cases. Dig Endosc.
25:519–525. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Silberg DG, Sullivan J, Kang E, Swain GP,
Moffett J, Sund NJ, Sackett SD and Kaestner KH: Cdx2 ectopic
expression induces gastric intestinal metaplasia in transgenic
mice. Gastroenterology. 122:689–696. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Boudreau F, Rings EH, van Wering HM, Kim
RK, Swain GP, Krasinski SD, Moffett J, Grand RJ, Suh ER and Traber
PG: Hepatocyte nuclear factor-1 alpha, GATA-4, and caudal related
homeodomain protein Cdx2 interact functionally to modulate
intestinal gene transcription. Implication for the developmental
regulation of the sucrase-isomaltase gene. J Biol Chem.
277:31909–31917. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Debruyne PR, Witek M, Gong L, Birbe R,
Chervoneva I, Jin T, Domon-Cell C, Palazzo JP, Freund JN, Li P, et
al: Bile acids induce ectopic expression of intestinal guanylyl
cyclase C Through nuclear factor-kappaB and Cdx2 in human
esophageal cells. Gastroenterology. 130:1191–1206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu J, Gong J, Geng J and Song Y:
Deoxycholic acid induces the overexpression of intestinal mucin,
MUC2, via NF-kB signaling pathway in human esophageal
adenocarcinoma cells. BMC Cancer. 8:3332008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee HY, Crawley S, Hokari R, Kwon S and
Kim YS: Bile acid regulates MUC2 transcription in colon cancer
cells via positive EGFR/PKC/Ras/ERK/CREB,
PI3K/Akt/IkappaB/NF-kappaB and p38/MSK1/CREB pathways and negative
JNK/c-Jun/AP-1 pathway. Int J Oncol. 36:941–953. 2010.PubMed/NCBI
|
|
41
|
Yuan ZQ and Li KW: Role of farnesoid X
receptor in cholestasis. J Dig Dis. 17:501–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong B, Young M, Liu X, Singh AB and Liu
J: Regulation of lipid metabolism by obeticholic acid in
hyperlipidemic hamsters. J Lipid Res. 58:350–363. 2017. View Article : Google Scholar :
|
|
43
|
Lax S, Schauer G, Prein K, Kapitan M,
Silbert D, Berghold A, Berger A and Trauner M: Expression of the
nuclear bile acid receptor/farnesoid X receptor is reduced in human
colon carcinoma compared to nonneoplastic mucosa independent from
site and may be associated with adverse prognosis. Int J Cancer.
130:2232–2239. 2012. View Article : Google Scholar
|
|
44
|
Liu J, Tong SJ, Wang X and Qu LX:
Farnesoid X receptor inhibits LNcaP cell proliferation via the
upregulation of PTEN. Exp Ther Med. 8:1209–1212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Giordano C, Barone I, Vircillo V, Panza S,
Malivindi R, Gelsomino L, Pellegrino M, Rago V, Mauro L, Lanzino M,
et al: Activated FXR Inhibits Leptin Signaling and Counteracts
Tumor-promoting Activities of Cancer-Associated Fibroblasts in
Breast Malignancy. Sci Rep. 6:217822016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
De Gottardi A: Dumonceau JM, Bruttin F,
Vonlaufen A, Morard I, Spahr L, Rubbia-Brandt L, Frossard JL,
Dinjens WN, Rabinovitch PS, et al Expression of the bile acid
receptor FXR in Barrett's esophagus and enhancement of apoptosis by
guggulsterone in vitro. Mol Cancer. 5:482006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li S, Chen X, Zhou L and Wang BM:
Farnesoid X receptor signal is involved in deoxycholic acid-induced
intestinal metaplasia of normal human gastric epithelial cells.
Oncol Rep. 34:2674–2682. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yamada T, Osawa S, Hamaya Y, Furuta T,
Hishida A, Kajimura M and Ikuma M: Guggulsterone suppresses bile
acid-induced and constitutive caudal-related homeobox 2 expression
in gut-derived adenocarcinoma cells. Anticancer Res. 30:1953–1960.
2010.PubMed/NCBI
|
|
49
|
Lian F, Xing X, Yuan G, Schäfer C, Rauser
S, Walch A, Röcken C, Ebeling M, Wright MB, Schmid RM, et al:
Farnesoid X receptor protects human and murine gastric epithelial
cells against inflammation-induced damage. Biochem J. 438:315–323.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zollner G, Wagner M, Moustafa T, Fickert
P, Silbert D, Gumhold J, Fuchsbichler A, Halilbasic E, Denk H,
Marschall HU, et al: Coordinated induction of bile acid
detoxification and alternative elimination in mice: Role of
FXR-regulated organic solute transporter-alpha/beta in the adaptive
response to bile acids. Am J Physiol Gastrointest Liver Physiol.
290:G923–G932. 2006. View Article : Google Scholar
|
|
51
|
Sarkar FH, Li Y, Wang Z and Kong D:
NF-kappaB signaling pathway and its therapeutic implications in
human diseases. Int Rev Immunol. 27:293–319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Abdel-Latif MM, Inoue H, Kelleher D and
Reynolds JV: Factors regulating nuclear factor-kappa B activation
in esophageal cancer cells: Role of bile acids and acid. J Cancer
Res Ther. 12:364–373. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lee JY, Lee KT, Lee JK, Lee KH, Jang KT,
Heo JS, Choi SH, Kim Y and Rhee JC: Farnesoid X receptor,
overexpressed in pancreatic cancer with lymph node metastasis
promotes cell migration and invasion. Br J Cancer. 104:1027–1037.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gadaleta RM, Oldenburg B, Willemsen EC,
Spit M, Murzilli S, Salvatore L, Klomp LW, Siersema PD, van Erpecum
KJ and van Mil SW: Activation of bile salt nuclear receptor FXR is
repressed by pro-inflammatory cytokines activating NF-κB signaling
in the intestine. Biochim Biophys Acta. 1812:851–858. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kazumori H, Ishihara S, Rumi MA, Kadowaki
Y and Kinoshita Y: Bile acids directly augment caudal related
homeobox gene Cdx2 expression in oesophageal keratinocytes in
Barrett's epithelium. Gut. 55:16–25. 2006. View Article : Google Scholar
|