Introduction
Hepatocellular carcinoma (HCC) is the second most
common cause of cancer mortality worldwide, and accounts for
500,000 to 1 million deaths annually worldwide (1). More than 80% of HCCs are discovered
at a late stage when surgery is not an option (2). The 5-year-survival rate for
resectable HCC ranges between 15 and 39% (3), largely due to tumor recurrence. Two
randomized controlled trials of sorafenib in patients with HCC
demonstrated an improvement in median overall survival of ~3
months, and established sorafenib as a standard of care in advanced
HCC (4,5). Although sorafenib improves overall
survival, the benefit is at best modest and confers a rather
transient clinical benefit (4,5).
Recently, FDA has approved lenvatinib and regorafenib as first line
and second line treatments, respectively, for HCC (6). Thus, effective novel therapies to
combat this deadly disease are urgently required.
Aberrant activation of the Wnt/β-catenin signaling
pathway is found in up to 67% of HCC cases, underlining its
importance in hepatocarcinogenesis (7). Tumors with nuclear and/or cellular
accumulation of β-catenin had, generally, poorly differentiated
morphology (8), high proliferative
activity (9), high vascular
invasion and a dismal prognosis (7-9).
Gene expression studies have revealed distinct molecular subclasses
in HCC, each associated with unique clinicopathological features.
CTNNB1 (β-catenin exon 3 mutated) class signature, which accounts
for 20-30% of all HCC cases, a subset of S3, is enriched in
hepatitis C virus (HCV)-related HCC and portends a better
prognosis. Notably, the S1 subclass, which is characterized by
non-mutated Wnt/β-catenin activation signaling, is associated with
non-HCV-related HCC, a larger tumor with moderately/poorly
differentiated histology, propensity for vascular invasion and the
development of satellite lesions, which is translated to early
recurrence compared with other subclasses (10,11).
Several non-mutational mechanisms, including mutations in Axin 1
and Axin 2, have been put forward to explain the increased
Wnt/β-catenin signaling (12).
Besides cooperation with transforming growth
factor-β, promoter methylation of secreted frizzled-related protein
members and the overexpression of frizzled (FZD) receptors have
been implicated (11). The
accumulation of Wnt/FZD-signaling dysregulation was also revealed
to be associated with increased activation of downstream effectors,
β-catenin, protein kinase C and c-Jun N-terminal kinase further
implicating the importance of Wnt/β-catenin signaling in
hepatocarcinogenesis (13). These
observations lend support to its functional relevance and
therapeutic value.
Selumetinib (AZD6244; AstraZeneca, Cambridge, UK) is
an allosteric adenosine triphosphate-uncompetitive inhibitor of
MEK1/2 (14). Refametinib (BAY
86-9766; Bayer AG, Leverkusen, Germany) is a nonadenosine
triphosphate competitive inhibitor targeting MEK 1/2 (15). Refametinib and selumetinib were
selected for the present study as both have similar mechanisms in
HCC. Although lack of activity was observed with selumetinib
(14) and refametinib (16) monotherapies, they exhibited
significant antitumor activity when combined with sorafenib in
clinical trials (16,17).
The aim of the present study was to investigate the
effects of sorafenib and MEK inhibitor on tumor growth and the
β-catenin signaling pathway in HCC.
Materials and methods
Reagents
Antibodies against disheveled segment polarity
protein (DVL)2 (#3224), DVL3 (#3218), protein kinase B (AKT;
#9272), β-catenin (#8480), Axin2 (#5863), Cyclin D1 (#2978), c-Myc
(#5605), c-Jun (#9165), low-density lipoprotein receptor-related
protein (LRP)6 (#2560), survivin (#2803), E-cadherin (#3195),
cleaved caspase 3 (#9661), cleaved poly (ADP-ribose) polymerase
(PARP; #5625), and phosphorylation-specific antibodies against AKT
Ser473 (#9271), glycogen synthase kinase (GSK)-3αβ (Ser21/9;
#9331), RanBP3 Ser58 (#9380), non-p (Active) β-catenin
Ser33/Ser37/Thr41 (#8814), phosphorylated (p)-histone 3 Ser10
(#9701), LRP6 Ser1490 (#2568), α-tubulin (#2144) and extracellular
signal-regulated kinase (ERK)1/2 Thr202/Tyr204 (#4370) were
obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA).
The antibodies against ERK1/2 (sc-94), glutamine synthetase
(SC-74430) were obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Anti-mouse cluster of differentiation (CD)31
antibody (#102502) was from BioLegend, Inc. (San Diego, CA, USA).
p-β-catenin Tyr142 (#CP1081) was purchased from ECM Biosciences LLC
(Versailles, KY, USA).
Conditioned medium
L-cells (ATCC CRL-2648) and L-Wnt-3A cells (ATCC
CRL-2647) cells were obtained from the American Type Culture
Collection (ATCC; Manassas, VA, USA). Control-conditioned medium
from L-cells and Wnt-3A-conditioned medium from L-Wnt-3A cells was
prepared according to the protocols provided by the ATCC. The
conditioned medium was concentrated 2X and sterile-filtered.
Cell culture
Between January 2004 and September 2018, 280 primary
HCCs were obtained during surgery at Department of General Surgery,
Singapore General Hospital (Singapore) and used for establishment
of HCC PDX models. Written informed consent was obtained from all
patients prior to tissue collection and the present study received
Ethics Committee approval from National Cancer Centre Singapore
(NCCS; Singapore) as well as Singapore General Hospital. HCC PDX
models were established as described previously (18). Primary HCC13-0109, HCC26-0808A, and
HCC06-0606 cells were freshly isolated from PDX tumors as follows:
HCC tumors were finely minced and washed 3 times with modified
Eagle's medium (MEM; Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany). The minced tissue was incubated with MEM containing 5%
fetal bovine serum (FBS; GE Healthcare Life Sciences, Logan, UT,
USA) and 5 mg/ml collagenase (Roche Diagnostics, Indianapolis, IN,
USA) at 37°C for 12 h. Cells were harvested by centrifuging at 4°C
at 800 × g for 10 min. The cell pellets were washed 3 times with
serum-free MEM, and primary HCC cells were plated at a density of
5.0×106 cells per well in MEM containing 10% FBS and 1%
penicillin-streptomycin (growth medium), and incubated at 37°C and
5% CO2 for 48 h. The primary cells were then used for
subsequent experiments.
Primary HCC06-0606 or HCC07-0409 cells were treated
with the vehicle, 0.5 µM refametinib, 2.5 µM
sorafenib or 2.5 µM sorafenib plus 0.5 µM refametinib
in growth medium at 37°C for 24 h then stimulated with concentrated
conditioned medium prepared either from the control L-cells or
L-Wnt-3A cells at 37°C for 2 h. The cells were harvested, and
changes in the levels of proteins of interest were determined by
western blotting.
Antisense β-catenin transfection
Phosphorothioate oligonucleotides (ODN) directed
against β-catenin antisense (5′-TAAGAGCTTAACCACAACTG-3′) and
scrambled control (5′-CAGTAATCGAATAGCTACCA-3′) were purchased from
TriLink Biotechnologies (San Diego, CA, USA). A further
5×106 HCC06-0606 cells were transfected with 150 nM
scrambled control or 150 nM ODN directed against β-catenin using
Plus™ Reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) at 37°C for 48 h. The cells were subsequently assayed for
β-catenin protein and its downstream targets by western
blotting.
Combined treatment with sorafenib plus
MEK inhibitor (refametinib or selumetinib)
The present study was approved by the Ethics Board
at NCCS (Singapore). Mice were maintained at NCCS according to the
'Guide for the Care and Use of Laboratory Animals' (19). All animal experiments were
performed at NCCS.
Sorafenib (Nexavar™; Bayer AG) and selumetinib
(14) (Selleck Chemicals, Houston,
TX, USA) were suspended in vehicle [30% captisol in water: 5%
glucose (50% v:v)] at 1.25 and 3.125 mg/ml, respectively.
Refametinib (Bayer AG) (15) was
diluted in dimethyl sulfoxide at a concentration of 100 mg/ml as
stock. Stock refametinib (0.150 ml) was further diluted in 3.925 ml
30% captisol in water followed by 3.925 ml 5% glucose to obtain an
appropriate concentration.
Establishment of HCC patient-derived
xenograft (PDX) model
Primary HCCs have previously been used to establish
HCC PDX xenograft lines (18), of
which the following 9 HCC lines (HCC01-0207; HCC25-0705A;
HCC10-0505; HCC09-0913; HCC01-0708; HCC29-0909A; HCC06-0606;
HCC26-0808A, and HCC07-0409), as well as one sorafenib-resistant
HCC26-0808ASora54, were used to establish tumors in 450 male C.B-17
SCID mice aged 9-10 weeks and weighed 23-25 g (InVivos Pte. Ltd.,
Singapore) as described previously (18,20).
Mice were provided with sterilized food and water ad
libitum, and housed in negative pressure isolators, which were
set at 23°C and 43% humidity, with 12-h light/dark cycles.
Development of a sorafenib-resistant HCC
model
C.B-17 SCID mice bearing HCC26-0808A tumors
(18) were treated once daily with
10 mg/kg sorafenib for 80 to 90 days. Following the initial
response to sorafenib, HCC26-0808A mice gradually acquired
resistance, leading to further tumor growth. Sorafenib-resistant
tumors were harvested for serial transplantation when they reached
the size of 1,500 mm3. The cycle was repeated until
sorafenib had minimal impact on the growth of treated tumors.
Mice bearing the indicated tumors (8-10 mice per
group) were orally administered either 200 µl vehicle, 10
mg/kg sorafenib, 15 mg/kg refametinib (or 25 mg/kg selumetinib) or
10 mg/kg sorafenib plus refametinib daily for 12-14 days. Treatment
commenced when the tumors reached 150-175 mm3 in size.
Bi-dimensional measurements were performed once every 2-3 days and
tumor volumes were calculated based on the following formula: Tumor
volume = [(length) x (width2) × (π/6)], and plotted as
the means ± standard error of the mean for each treatment group vs.
time, as previously described (18). Body and tumor weights were recorded
at the time of sacrifice. The tumors were harvested 2 h following
the final treatment and stored at -80°C for later biochemical
analysis. The efficacy of each treatment was determined using the
T/C ratio, where T and C are the median weight of drug-treated and
vehicle-treated tumors, respectively, at the end of treatment.
Western blot analysis
Tumors from vehicle- and drug-treated mice were
homogenized in buffer containing 50 mM Tris-HCl pH 7.4, 150 mM
NaCl, 0.5% NP-40, 1 mM EDTA, 25 mM NaF, supplemented with
proteinase inhibitors and 10 mM Na3VO4.
Protein concentration was determined by Bradford assay. In total,
80 µg protein/lane was resolved by either 8% or 14%
SDS-PAGE, and western blotting was performed as previously
described (21). All primary
antibodies were diluted in 1% low-fat skimmed milk powder dissolved
at a ratio of 1:1,000 to a final concentration of 1 µg/ml.
Blots were incubated with primary antibodies for 14-16 h at 4°C.
After 3 washes in TBST, blots were incubated with a secondary
antibody [horseradish peroxidase (HRP)-conjugated goat anti-rabbit
immunoglobulin (Ig)G (H+L; #31460; 1:5,000; Thermo Fisher
Scientific, Inc.] for 60 min at room temperature. All primary
antibodies were then visualized using WesternBight™ ECL
chemiluminescent detection reagents (Advansta, Inc., Menlo Park,
CA, USA). For quantification analysis, total density of the band
corresponding to protein blotting with the indicated antibody was
quantified using the GS-900 Calibrated Densitometer and Image Lab™
Software 6.0.1 (Bio-Rad Laboratories, Inc., Hercules, CA, USA),
normalized to both the tubulin loading control and the appropriate
phosphorylated/total protein where applicable, and expressed as the
fold-change.
Immunohistochemistry
Tumor tissues were fixed in PBS buffer containing 4%
formaldehyde (ICM Pharma, Pte. Ltd, Singapore) for 24 h at room
temperature and embedded in paraffin. Tissue sections (5-µm)
were blocked with 5% low-fat skimmed milk powder dissolved in TBS
containing 0.1% Tween (TBST) for 1 h at room temperature, followed
by 3 washes in TBS. Sections were then incubated with CD31 (1:100),
p-histone 3 Ser10 (1:300), cleaved PARP (1:100), and total
β-catenin (1:150) antibodies for 14-16 h at 4°C to assess
micro-vessel density, cell proliferation, and apoptosis,
cytoplasmic and nuclear β-catenin, respectively, as described
(20). Following 3 washes in TBST
(5 min each), sections were incubated with biotin-conjugated goat
anti-rabbit IgG secondary antibody (#31820; 1:150; Thermo Fisher
Scientific, Inc.) for 45 min at room temperature, followed by 3
washes in TBS and then incubated with streptavidin HRP-conjugated
antibody (#21126; 1:500; Thermo Fisher Scientific, Inc.). Slides
were counterstained with hematoxylin (Sigma Diagnostics, Inc.,
Livonia, MI, USA) for 10 sec at room temperature. Images were
captured on an Olympus BX60 light microscope (Olympus Corporation,
Tokyo, Japan). Five random 0.159-mm2 fields at ×100
magnification were captured for each tumor. The number of p-histone
3 Ser10 and cleaved PARP-positive cells among at least 500 cells
per field was counted and expressed as the number of positive cells
per 1,000 cells. For the quantification of mean microvessel density
in sections stained for CD31, 5 random fields at a magnification of
×100 were selected for each tumor.
Whole exome sequencing (WES)
The CTNNB1 (β-catenin) mutational status of 9 HCC
models was determined via WES as described previously (22).
Statistical analysis
Data were presented as the mean ± standard error of
the mean. Differences in the protein level, tumor weight at
sacrifice, p-histone 3 Ser10 index, mean micro-vessel density, and
the number of cleaved PARP-positive cells were compared. Student's
t-test was used for comparisons between two groups. One-way
analysis of variance followed by the Tukey-Kramer method post-hoc
test was used when comparing more than two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Sorafenib/refametinib treatment inhibits
tumor growth independent of β-catenin mutational status
WES analysis revealed that HCC29-0909A harbored a
T41A β-catenin mutation, Y126 p53 mutation and EZH2 intergenic
deletion; HCC10-0505 harbored a S45P β-catenin mutation, p53
deletion, and 2108G>T JAK1 mutation. HCC01-0708 harbored a T41A
β-catenin mutation, Y126 p53 mutation, and N-RAS amplification;
HCC07-0409 harbored a T41A β-catenin mutation, Y126 p53 mutation,
and CCND1 and N-RAS amplification; HCC26-0808A had a NF1 deletion,
H168R p53 mutation, 3G>T VHL mutation, and loss of CDKN2A and
CDKN2B; HCC06-0606 harbored a 407A>C p53 mutation and a MCL1
amplification; HCC01-0207 had a 341G>A RET mutation, R249S p53
mutation and LRP1B truncation; HCC25-0705A had a 341G>A RET
mutation and R249S p53 mutation; HCC09-0913 harbored a R249S p53
mutation. HCC01-0708, HCC07-0409 and HCC29-0909A harbored a T41A
β-catenin mutation, and HCC10-0505 had S45P β-catenin mutations.
These observations are consistent with those of previous studies,
indicating that mutations in the β-catenin gene account for 20-30%
of all HCC cases (10,11).
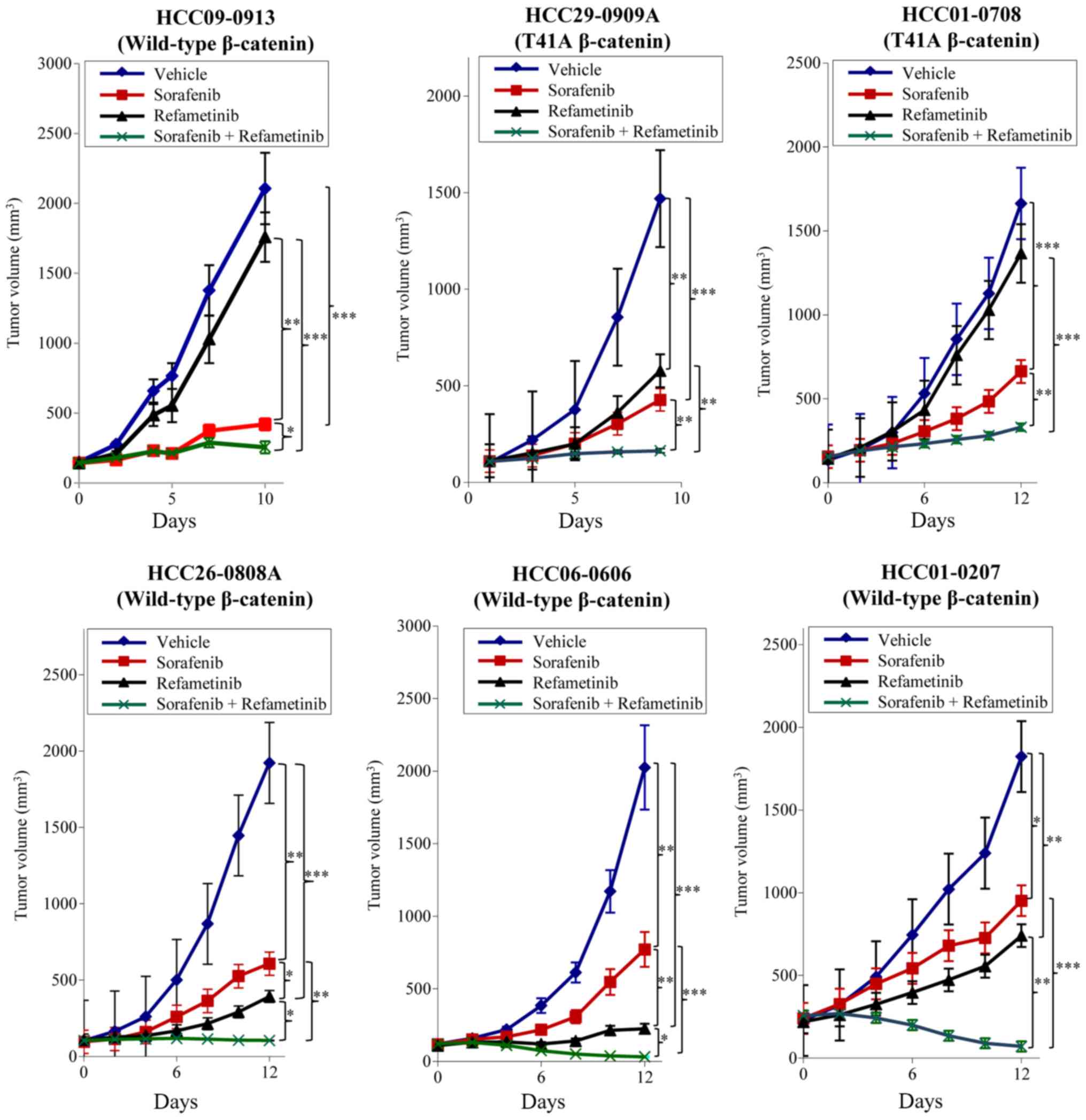
As treatment with sorafenib led to the upregulation
of p-ERK1/2 (21) and ERK kinase
activated Wnt/β-catenin signaling (23), the combined effects of
sorafenib/MEK inhibitor on tumor growth and the Wnt/β-catenin
signaling pathway were evaluated. Consistent with our previous
study (21), oral delivery of
sorafenib led to significant inhibition of tumor growth as compared
with the vehicle group (P<0.01). Addition of refametinib to
sorafenib resulted in significantly improved anti-tumor activity in
all HCC models tested when compared with either agent alone
(P<0.01; Fig. 1). As compared
with the vehicle group, refametinib significantly inhibited tumor
growth of HCC29-0909A, HCC26-0808A, HCC06-0606 and HCC01-0207
(P<0.01; Fig. 1) but not
HCC09-0913 and HCC01-0708. Tumor regression was observed in
wild-type β-catenin HCC26-0808A, HCC06-0606, HCC01-0207 and
HCC09-0913 models following sorafenib/refametinib treatment. The
antitumor activity of sorafenib/refametinib was independent of
β-catenin mutational status as both mutant and wild-type β-catenin
xenografts were inhibited by this combination (Fig. 1). Refametinib monotherapy was not
considered to be active in HCC01-0708 (mutant β-catenin) and
HCC09-0913 (wild-type β-catenin) models (Fig. 1), suggesting that factors other
than β-catenin were responsible for the responsiveness. As
predicted, sorafenib/refametinib inhibited cell proliferation and
caused increased apoptosis, as compared with sorafenib or
refametinib alone (P<0.05; Fig.
2). Similar results were obtained when selumetinib was combined
with sorafenib (data not shown).
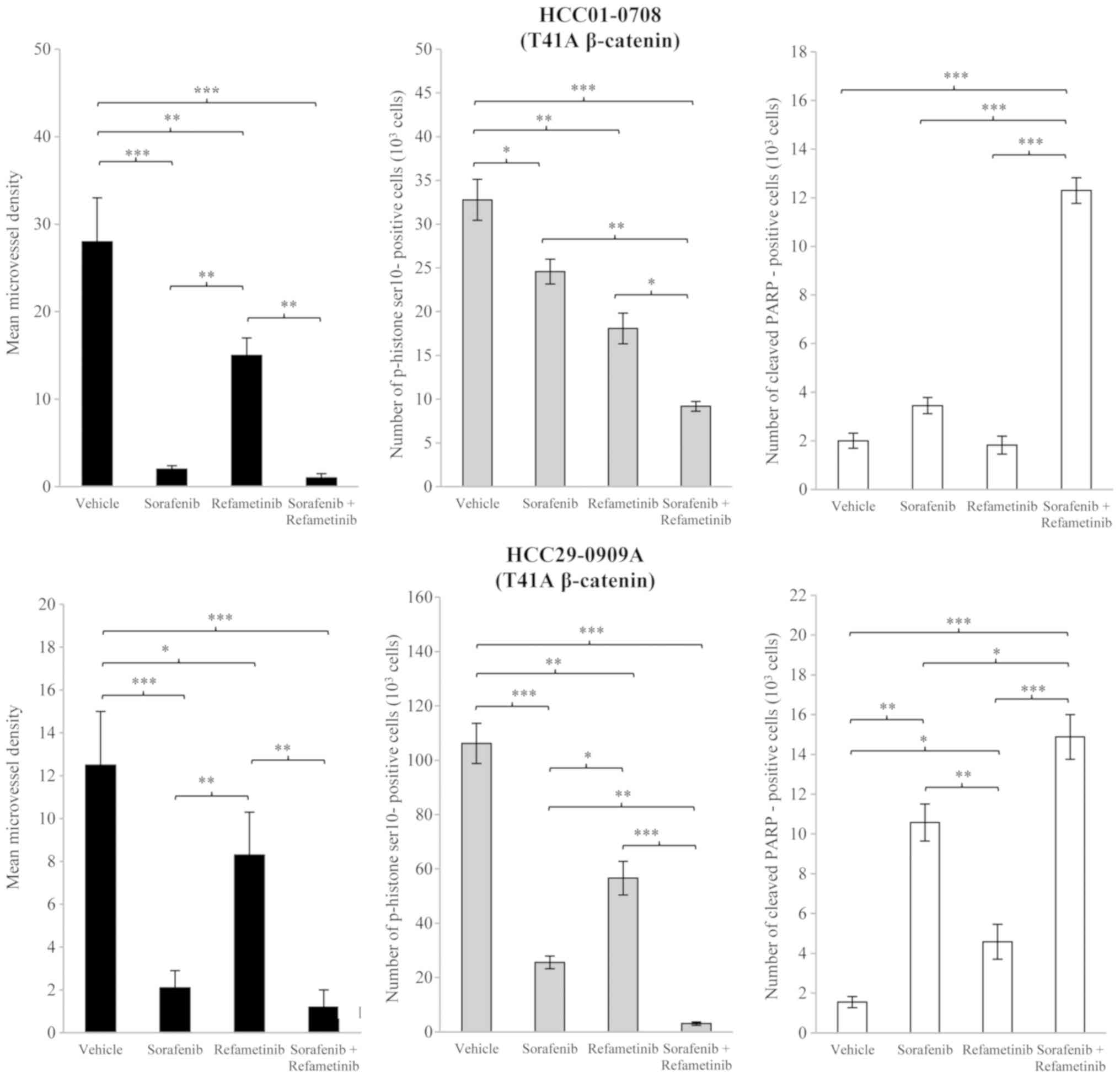 | Figure 2Effects of sorafenib/refametinib on
angiogenesis, cell proliferation and apoptosis in T41A mutant
β-catenin HCC01-0708 and HCC29-0909A models. Mice bearing the
indicated tumors were treated with 200 µl vehicle, 10 mg/kg
sorafenib, 15 mg/kg refametinib or sorafenib/refametinib for 10-15
days. Sorafenib and refametinib were given once daily. Tumor
tissues were collected 2 h following the final treatment. Each
treatment arm comprised 8 independent tumor-bearing mice. Sections
(5-µm) were immunostained with cluster of differentiation
31, p-histone 3 Ser10 and cleaved PARP antibodies to assess
microvessel density, cell proliferation and apoptosis,
respectively. The number of p-histone 3 Ser10 and cleaved
PARP-positive cells, among at least 500 cells counted per region,
was determined and plotted as the mean number of positive cells per
1,000 cells ± SE. For the quantification of the mean microvessel
density ± SE in each section, 5 random 0.159 mm2 fields
at a magnification of x 100 were observed for each tumor. Images
were captured via light microscopy. Differences in mean microvessel
density, p-histone 3 Ser10 and cleaved PARP-positive cells were
compared using one-way analysis of variance followed by the
Tukey-Kramer method post-hoc test. *P<0.05,
**P<0.01 and ***P<0.001. p,
phosphorylated; PARP, poly (ADP-ribose) polymerase; SE, standard
error of the mean. |
Sorafenib/refametinib combination
treatment suppresses the β-catenin signaling pathway independent of
its mutational status
The effects of sorafenib/refametinib combination on
β-catenin and its downstream targets in mutant β-catenin
HCC29-0909A and wild-type β-catenin HCC06-0606 xenografts were then
investigated. As presented in Fig.
3, sorafenib alone had minimal effect on p-LRP6 Ser1490 and
p-RanBP3 Ser58. Quantification of the immunoblots revealed that a
decrease in these proteins was observed in refametinib-treated
HCC06-0606 (Fig. 3A) and
HCC29-0909A xenografts (Fig. 3B).
Notably, sorafenib/refametinib resulted in a significant decrease
in p-LRP6 Ser1490 (P<0.05; Fig.
3). As presented in Fig. 3,
there was no significant difference in p-GSK-3αβ (Ser21/9) levels
between the treatment groups. Although total β-catenin was modestly
reduced, p-β-catenin Tyr142, p-RanBP-3 Ser58 and active β-catenin
in sorafenib/refametinib-treated tumors were significantly reduced
(P<0.05; Fig. 3). Significant
reduction of β-catenin downstream targets (Axin-2, survivin, c-Myc,
cyclin D1 and glutamine synthetase) in
sorafenib/refametinib-treated tumors was observed (P<0.05;
Fig. 3). Similar results were
observed when wild-type HCC09-0913 and mutant β-catenin HCC01-0708
were treated with sorafenib/selumetinib (data not shown).
 | Figure 3Effects of sorafenib/refametinib on
Wnt/β-catenin signaling in (A) wild-type β-catenin HCC06-0606 (B)
and T41A mutant β-catenin HCC29-0909A models. Mice bearing the
indicated tumors were treated with 200 µl vehicle, 10 mg/kg
sorafenib, 15 mg/kg refametinib and sorafenib/refametinib once
daily for 5 days. Each treatment arm comprised 3-4 independent
tumor-bearing mice. Tumors were collected 2 h following the final
dose for marker analysis. Tumor lysates were analyzed by western
blotting. Blot membranes were incubated with the indicated
antibodies. Representative blots are presented. Total density of
the band corresponding to protein blotting with the indicated
antibody was quantified, normalized to both the tubulin loading
control and the phosphory-lated/total protein (e.g., ERK and LRP6)
and expressed as the fold-change. *P<0.05,
**P<0.01 and ***P<0.001 vs. vehicle. p,
phosphorylated; ERK, extracellular signal-regulated kinase; LRP,
low-density lipoprotein receptor-related protein 6; GSK, glycogen
synthetase kinase; PARP, poly (ADP-ribose) polymerase. |
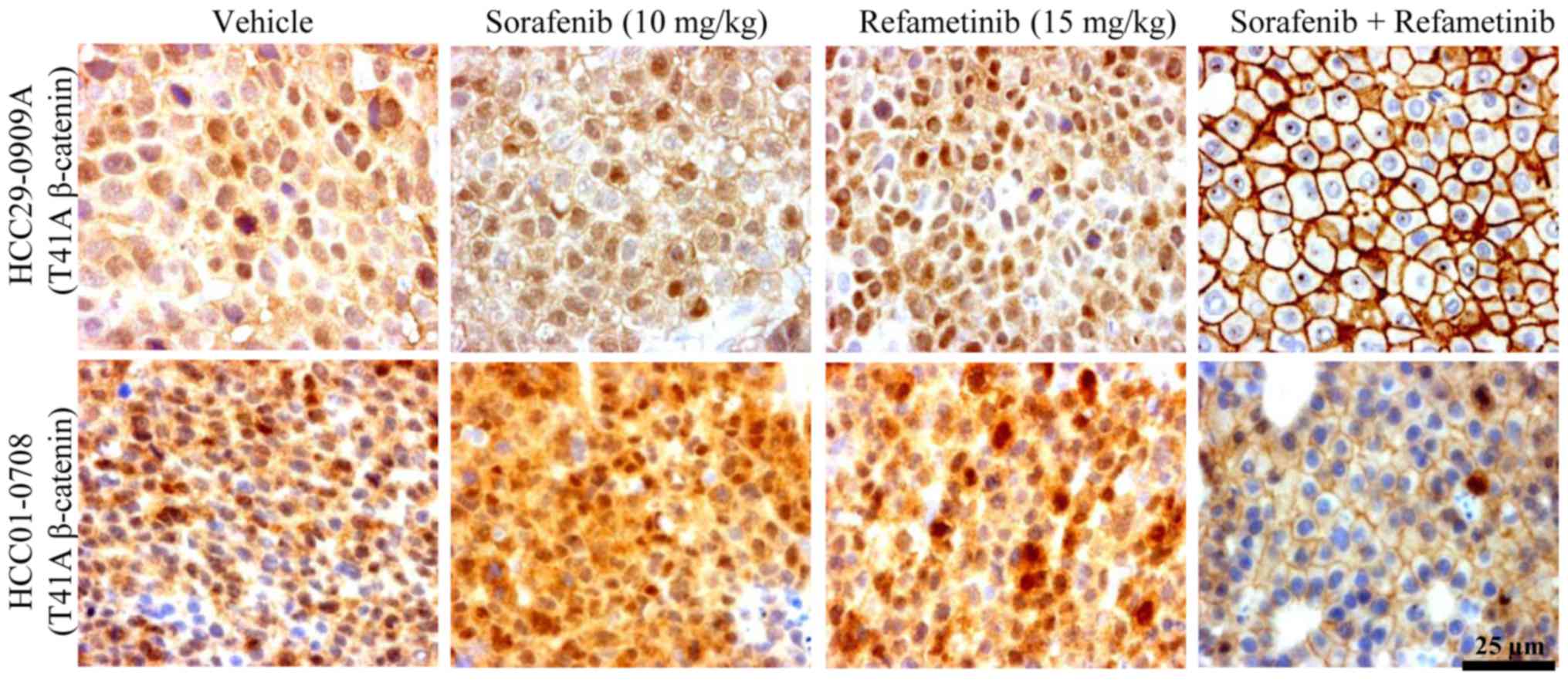
Sorafenib/refametinib combination
treatment causes export of β-catenin to the membrane
The tyrosine kinases have been demonstrated to
induce phosphorylation of β-catenin at tyrosine 142 (24,25).
As p-β-catenin Tyr142 (24) and
p-RanBP3 (26) have been reported
to be involved in β-catenin export, whether sorafenib/refametinib
treatment caused nuclear export was examined. Tumor sections among
different treatment groups were stained with total β-catenin
antibody. Intense nuclear and cytoplasmic β-catenin staining was
observed in vehicle-treated mutant β-catenin tumors (HCC29-0909A
and HCC01-0708) (Fig. 4). The
majority of β-catenin in sorafenib-treated tumors was located in
the cytoplasm and nucleus (Fig.
4). Refametinib markedly reduced nuclear and cytoplasmic
β-catenin concomitant with a marked increase in membranous
β-catenin (Fig. 4). There was no
clear difference in β-catenin relocalization between sorafenib- and
refametinib-treated tumors. Notably, nuclear β-catenin staining was
barely detectable in HCC29-0909A and HCC01-0708 tumors treated with
sorafenib/refametinib (Fig. 4).
The nuclear staining was less pronounced in the HCC10-0505 model
with the S45P mutant of β-catenin, suggesting that there may be
functional differences amongst the various mutations (data not
shown). In HCC29-0909A and HCC01-0708 models, a membranous signal
accumulated in areas of intercellular contact, likely representing
β-catenin associated with membranous cadherins (24,27).
These observations suggest that β-catenin export from the nucleus
and/or the cytoplasm to the membrane following
sorafenib/refametinib treatment likely occurs via the inhibition of
p-β-catenin Tyr142 (25) and
p-RanBP3 (26). β-catenin
relocalization also occurred when HCC09-0913 tumors were treated
with sorafenib/selumetinib (data not shown).
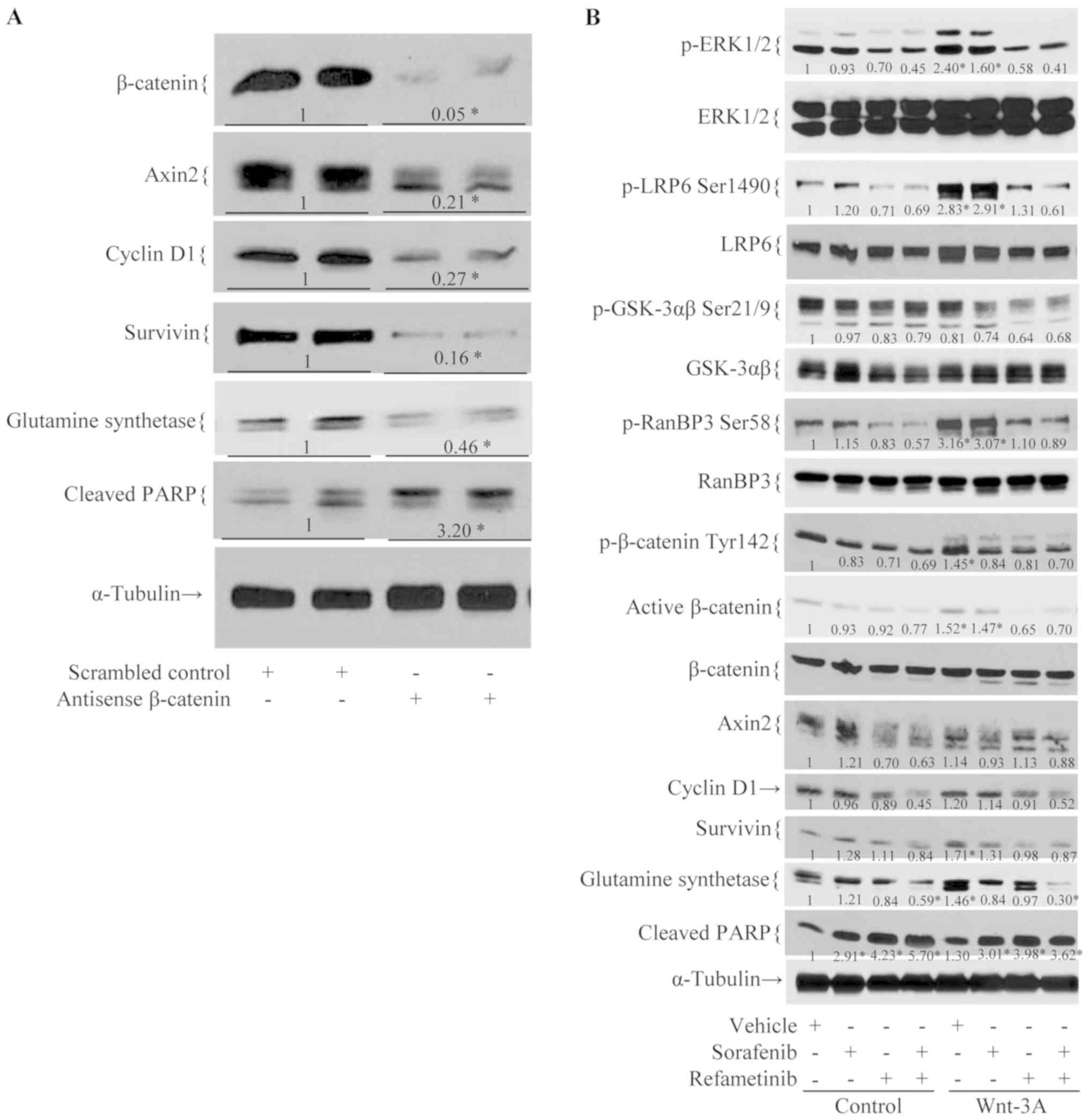
Sorafenib/refametinib inhibits
Wnt-3A-induced activation of the β-catenin signaling pathway
As presented in Fig.
5A, treatment with antisense, but not scrambled control,
β-catenin resulted in decreased cell viability, β-catenin levels
and those of its known downstream targets. Treatment of wild-type
β-catenin HCC06-0606 cells with Wnt-3A-conditioned medium led to
the significant upregulation of p-LRP6 Ser1490, p-ERK1/2
Thr202/Tyr204, p-RanBP3 Ser58 and p-β-catenin Tyr142 (P<0.01;
Fig. 5B) as compared with
control-conditioned medium. Marked increases in survivin, cyclin D1
and glutamine synthetase were also noted. Pre-treatment of
HCC06-0606 cells with refametinib or sorafenib/refametinib, but not
with sorafenib, for 24 h abolished Wnt-3A-induced upregulation of
p-LRP6 Ser1490, p-β-catenin Tyr142, p-RanBP3 Ser58 and p-ERK1/2
Thr202/Tyr204 (P<0.05; Fig.
5B). These findings suggest that sorafenib/refametinib
profoundly suppressed β-catenin activity, resulting in decreased
proliferation and elevated levels of apoptosis. Similar
observations were obtained when mutant β-catenin HCC07-0409 cells
were treated with Wnt-3A-conditioned medium (data not shown).
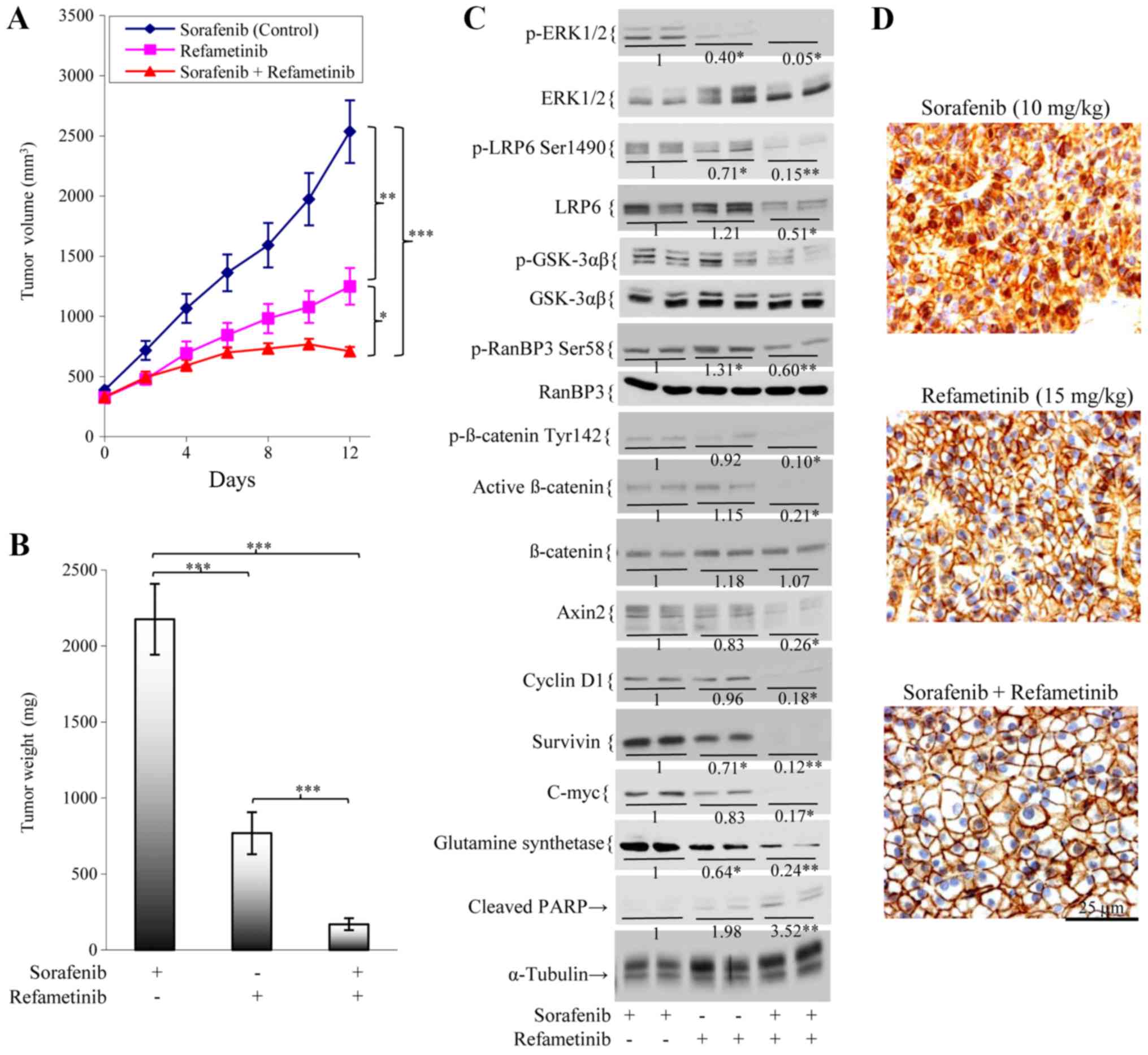
Sorafenib/refametinib treatment has
anti-tumor effects in a sorafenib-resistant HCC model
While growth of the parental HCC26-0808A line was
significantly reduced by sorafenib, growth of the HCC26-0808ASora54
line was unaffected (data not shown). This indicates that
HCC26-0808ASora54 is resistant to sorafenib treatment. As presented
in Fig. 6A, sorafenib-resistant
HCC26-0808ASora54 tumors that continued to grow in the presence of
sorafenib experienced a significant reduction in tumor growth when
ERK activity was blocked by refametinib (P<0.001). The addition
of refametinib to sorafenib abolished tumor growth (Fig. 6A) and the final tumor weight of
sorafenib/refametinib group was significantly smaller than that of
the refametinib or sorafenib groups (P<0.001; Fig. 6B). This was associated with
inhibition of the β-catenin signaling pathway, as evidenced by a
significant reduction in p-LRP6 Ser1490, p-β-catenin Tyr142,
p-RanBP-3 Ser58, and active non-phospho-β-catenin, Axin2, survivin,
cyclin D1, c-Myc, and glutamine synthetase levels (P<0.05;
Fig. 6C). Nuclear exportation of
β-catenin was also observed when HCC26-0808ASora54 was treated with
sorafenib/refametinib (Fig.
6D).
Discussion
HCC is the second leading cause of cancer mortality
worldwide, often presenting in the advanced stage when cure is no
longer possible. Although sorafenib improves the overall survival
of patients with HCC, the benefit is modest and treatment confers
only a transient clinical benefit (4,5). As
such, there is an urgent need for more efficacious systemic
therapies. Mutations affecting the Wnt/β-catenin pathway have been
found in 26-40% of HCC cases (28-30).
Activation of the Wnt/β-catenin pathway leads to upregulation of
downstream target genes that regulate cell proliferation and tumor
progression (31,32). As such, agents targeting the
Wnt/β-catenin pathway may be an alternative therapy for patients
with β-catenin-dependent tumors. Targeting Wnt/β-catenin has been
comprehensively discussed (33,34).
Wnt/β-catenin pathway inhibitor ICG001, FH535 and other small
inhibitors have been tested in HCC models (35-37).
Combinations of sorafenib/ICG001 or sorafenib/FH535 had better
treatment outcomes in their experimental models (38,39).
These studies have demonstrated that the promoted downregulation or
degradation of wild-type β-catenin is the mechanism of β-catenin
pathway inhibition in HCC.
In the present study, combinations of
sorafenib/refametinib or sorafenib/selumetinib inhibited cell
proliferation and induced apoptosis, both of which were associated
with the downregulation of p-ERK1/2, p-RanBP3 Ser58, p-LRP6
Ser1490, p-β-catenin Tyr142, and β-catenin target genes. In
addition, the sorafenib/refametinib combination suppressed
Wnt-3A-induced activation of the Wnt/β-catenin signaling pathway in
HCC cells. Although the overall levels of β-catenin were not
significantly changed, those of non-p- (active) β-catenin in
sorafenib/refametinib-treated tumors were significantly reduced.
Whereas vehicle-treated and sorafenib-treated mutant β-catenin
tumors exhibited strong cytoplasmic and nuclear β-catenin
localization, the majority of the β-catenin in
sorafenib/refametinib-treated tumors was accumulated at the
membrane. The precise mechanisms by which sorafenib/refametinib or
sorafenib/selumetinib inactivates the Wnt/β-catenin pathway remain
unclear. It is possible that sorafenib/refametinib or
sorafenib/selumetinib inactivates the Wnt/β-catenin pathways by
inhibiting p-β-catenin Tyr142 (40) and p-RanBP3 Ser58 (26). This leads to a reduction in nuclear
β-catenin and β-catenin-dependent transcription (40-43).
As phosphorylation of β-catenin at tyrosine 142 is also essential
for binding to E-cadherin (41,43)
and α-catenin (27,40,42,44),
respectively, inhibition of p-β-catenin Tyr142 by sorafenib/MEK
inhibitor may facilitate the interaction of E-cadherin and
α-catenin with β-catenin at the membrane, leading to a decrease of
the cytosolic pool without reducing total β-catenin levels
(45). In the present study,
suppressing β-catenin by sorafenib/refametinib reduces p-histone 3
Ser10, cyclin B1, cyclin D1, c-Myc, and survivin protein
expression, providing the link between β-catenin signaling pathway
and cell cycle. In addition, sorafenib/refametinib also reduced
glutamine synthetase, which in turn may deplete cellular glutamine.
As activation of Wnt/β-catenin is implicated in maintenance of
tumor initiating cells, drug resistance, tumor progression and
metastasis (31), it remains to be
determined if sorafenib/refametinib inhibits the proliferation of
liver cancer stem cells.
Sorafenib/refametinib or sorafenib/selumetinib
targets wild-type as well as T41A- and S45P-mutated β-catenin,
which have been identified in 2-18.8% of analyzed HCC cases
(7,46-50).
These mutations were revealed to activate the β-catenin pathway by
preventing GSK3-mediated phosphorylation at Ser33/37, further
avoiding β-TrCP recognition (51-53).
In the present study, it was observed that the levels of p-GSK-3αβ
(Ser21/9) and total β-catenin were not significantly altered by
sorafenib/refametinib or sorafenib/selumetinib treatment. It is
unlikely that downregulation or degradation of β-catenin by GSK3 is
the mechanism by which sorafenib/refametinib or
sorafenib/selumetinib inhibits the β-catenin pathway. Enhanced
nuclear β-catenin export due to the inhibition of p-β-catenin
Tyr142 (40,42) and p-RanBP3 Ser58 (26) may have a key role in sorafenib/MEK
inhibitor mediated inactivation of β-catenin signaling pathway.
Treatment with sorafenib, although associated with
inhibition of tumor growth and angiogenesis in in vivo
studies, led to the upregulation of p-ERK1/2 (21), which is one of the most critical
cellular signaling pathways supporting hepatocarcinogenesis. The
addition of a MEK inhibitor (selumetinib or refametinib) to
sorafenib resulted in the attenuation of p-ERK1/2 activity and
greater antitumor effects (17,21).
In the present study, 'adaptive' tumor growth in the presence of
sorafenib experienced a marked reduction when MEK/ERK activity was
blocked. These findings suggest that resistance to sorafenib may be
overcome by adding refametinib (or selumetinib) to sorafenib.
We recently reported that a phase Ib study of
selumetinib (AZD6244) in combination with sorafenib in advanced HCC
demonstrated encouraging anti-tumor activity with acceptable
adverse events (17). In the SHARP
trial, sorafenib monotherapy was associated with an median
progression-free survival (mPFS) and median overall survival (mOS)
of 5.5 and 10.7 months, respectively (5). A phase III study of sorafenib in
patients in the Asia-Pacific region with advanced hepatocellular
carcinoma reported an mPFS and mOS of 2.8 and 6.5 months,
respectively (4). mPFS and mOS of
5.6 and 14.4 months, respectively, achieved with combination
sorafenib and selumetinib is encouraging. Lack of activity observed
with selumetinib monotherapy in a previous study suggests
synergistic anti-tumor effect are possible with this combination
(17). The robust anti-tumor
activity of sorafenib/selumetinib presented in HCC PDX models and
in phase 1b clinical trial warrants the development of phase II
clinical trials. In a phase 1b study of selumetinib in combination
with sorafenib in HCC, a high incidence of grade 3/4 diarrhea (44%)
and the finding that 66% of SAEs were GI-related
(vomiting/diarrhea) are testament of overlapping toxicities between
sorafenib and selumetinib (17).
In the present study, dose reductions were required for sorafenib.
Patient education and prompt aggressive management strategies may
potentially mitigate some of the toxicities. A phase II study of
refametinib and sorafenib also demonstrated these agents to be
clinically active in patients with HCC and mutant KRAS tumors
(16). These results also suggest
that a therapy regimen involving the combination of sorafenib and a
MEK inhibitor could possibly reverse sorafenib-resistance in
patients with HCC.
Funding
The present study was supported by grants
(NMRC/MOHIAFCAT2/006/2016, NMRC/MOHIAFCat1/0042/2016,
NMRC/MOHIAFCat1/0040/2016, NMRC/MOHIAFCat1/0022/2015,
NMRC/MOHIAFCat1/0003/2014, and NMRC/MOHIAFCat1/0004/2014) from
National Medical Research Council. This project was supported in
part by Bayer AG (Leverkusen, Germany).
Availability of data and materials
All data generated or analyzed during this study are
available from the corresponding author on reasonable request.
Authors' contributions
HH, LL and RO were responsible for collection and
assembly of data. FP, AS, OP, DM, KZ and HH were responsible for
financial support, provision of study materials and study design.
HH and KG were responsible for data analysis, interpretation and
writing the manuscript. All authors provided final approval of the
article.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients prior to tissue collection. The present study was approved
by the Ethics Board at National Cancer Centre Singapore (Singapore)
and at Singapore General Hospital (Singapore). All mice were
maintained according to the 'Guide for the Care and Use of
Laboratory Animals'.
Patient consent for publication
Not applicable.
Competing interests
FP, AS, OP, DM and KZ are employees of Bayer AG
(Leverkusen, Germany).
Acknowledgments
The authors would like to thank Dr Dieter Zopf
(Bayer AG, Leverkusen, Germany) for his critical reading of this
manuscript.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nagasue N, Kohno H, Chang YC, Taniura H,
Yamanoi A, Uchida M, Kimoto T, Takemoto Y, Nakamura T and Yukaya H:
Liver resection for hepatocellular carcinoma. Results of 229
consecutive patients during 11 years. Ann Surg. 217:375–384. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Takenaka K, Kawahara N, Yamamoto K,
Kajiyama K, Maeda T, Itasaka H, Shirabe K, Nishizaki T, Yanaga K
and Sugimachi K: Results of 280 liver resections for hepatocellular
carcinoma. Arch Surg. 131:71–76. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomised, double-blind,
placebo-controlled trial. Lancet Oncol. 10:25–34. 2009. View Article : Google Scholar
|
|
5
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dawkins J and Webster RM: The
hepatocellular carcinoma market. Nat Rev Drug Discov. 18:13–14.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wong CM, Fan ST and Ng IO: beta-Catenin
mutation and over-expression in hepatocellular carcinoma:
Clinicopathologic and prognostic significance. Cancer. 92:136–145.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Endo K, Ueda T, Ueyama J, Ohta T and
Terada T: Immunoreactive E-cadherin, alpha-catenin, beta-catenin,
and gamma-catenin proteins in hepatocellular carcinoma:
Relationships with tumor grade, clinicopathologic parameters, and
patients' survival. Hum Pathol. 31:558–565. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Inagawa S, Itabashi M, Adachi S, Kawamoto
T, Hori M, Shimazaki J, Yoshimi F and Fukao K: Expression and
prognostic roles of beta-catenin in hepatocellular carcinoma:
Correlation with tumor progression and postoperative survival. Clin
Cancer Res. 8:450–456. 2002.PubMed/NCBI
|
|
10
|
Hoshida Y, Nijman SM, Kobayashi M, Chan
JA, Brunet JP, Chiang DY, Villanueva A, Newell P, Ikeda K,
Hashimoto M, et al: Integrative transcriptome analysis reveals
common molecular subclasses of human hepatocellular carcinoma.
Cancer Res. 69:7385–7392. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lachenmayer A, Alsinet C, Savic R,
Cabellos L, Toffanin S, Hoshida Y, Villanueva A, Minguez B, Newell
P, Tsai HW, et al: Wnt-pathway activation in two molecular classes
of hepatocellular carcinoma and experimental modulation by
sorafenib. Clin Cancer Res. 18:4997–5007. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Taniguchi K, Roberts LR, Aderca IN, Dong
X, Qian C, Murphy LM, Nagorney DM, Burgart LJ, Roche PC, Smith DI,
et al: Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in
hepatocellular carcinomas and hepatoblastomas. Oncogene.
21:4863–4871. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bengochea A, de Souza MM, Lefrançois L, Le
Roux E, Galy O, Chemin I, Kim M, Wands JR, Trepo C, Hainaut P, et
al: Common dysregulation of Wnt/Frizzled receptor elements in human
hepatocellular carcinoma. Br J Cancer. 99:143–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
O'Neil BH, Goff LW, Kauh JS, Strosberg JR,
Bekaii-Saab TS, Lee RM, Kazi A, Moore DT, Learoyd M, Lush RM, et
al: Phase II study of the mitogen-activated protein kinase 1/2
inhibitor selumetinib in patients with advanced hepatocellular
carcinoma. J Clin Oncol. 29:2350–2356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iverson C, Larson G, Lai C, Yeh LT, Dadson
C, Weingarten P, Appleby T, Vo T, Maderna A, Vernier JM, et al:
RDEA119/BAY 869766: A potent, selective, allosteric inhibitor of
MEK1/2 for the treatment of cancer. Cancer Res. 69:6839–6847. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lim HY, Heo J, Choi HJ, Lin CY, Yoon JH,
Hsu C, Rau KM, Poon RT, Yeo W, Park JW, et al: A phase II study of
the efficacy and safety of the combination therapy of the MEK
inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian
patients with unresectable hepatocellular carcinoma. Clin Cancer
Res. 20:5976–5985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tai WM, Yong WP, Lim C, Low LS, Tham CK,
Koh TS, Ng QS, Wang WW, Wang LZ, Hartano S, et al: A phase Ib study
of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib
in advanced hepatocellular carcinoma (HCC). Ann Oncol.
27:2210–2215. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huynh H, Soo KC, Chow PK, Panasci L and
Tran E: Xenografts of human hepatocellular carcinoma: A useful
model for testing drugs. Clin Cancer Res. 12:4306–4314. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; US, Washington, DC: 2011
|
|
20
|
Huynh H, Chow PK, Palanisamy N,
Salto-Tellez M, Goh BC, Lee CK, Somani A, Lee HS, Kalpana R, Yu K,
et al: Bevacizumab and rapamycin induce growth suppression in mouse
models of hepatocellular carcinoma. J Hepatol. 49:52–60. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Huynh H, Ngo VC, Koong HN, Poon D, Choo
SP, Toh HC, Thng CH, Chow P, Ong HS, Chung A, et al: AZD6244
enhances the anti-tumor activity of sorafenib in ectopic and
orthotopic models of human hepatocellular carcinoma (HCC). J
Hepatol. 52:79–87. 2010. View Article : Google Scholar
|
|
22
|
Huynh H, Lee LY, Goh KY, Ong R, Hao H-X,
Huang A, Wang Y, Graus Porta D, Chow P and Chung A: Infigratinib
mediates vascular normalization, impairs metastasis and improves
chemotherapy in hepatocellular carcinoma. Hepatology. Dec 21–2018,
Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Červenka I, Wolf J, Mašek J, Krejci P,
Wilcox WR, Kozubík A, Schulte G, Gutkind JS and Bryja V:
Mitogen-activated protein kinases promote WNT/beta-catenin
signaling via phosphorylation of LRP6. Mol Cell Biol. 31:179–189.
2011. View Article : Google Scholar
|
|
24
|
Brembeck FH, Rosário M and Birchmeier W:
Balancing cell adhesion and Wnt signaling, the key role of
beta-catenin. Curr Opin Genet Dev. 16:51–59. 2006. View Article : Google Scholar
|
|
25
|
Krejci P, Aklian A, Kaucka M, Sevcikova E,
Prochazkova J, Masek JK, Mikolka P, Pospisilova T, Spoustova T,
Weis M, et al: Receptor tyrosine kinases activate canonical
WNT/β-catenin signaling via MAP kinase/LRP6 pathway and direct
β-catenin phosphorylation. PLoS One. 7:e358262012. View Article : Google Scholar
|
|
26
|
Hendriksen J, Fagotto F, van der Velde H,
van Schie M, Noordermeer J and Fornerod M: RanBP3 enhances nuclear
export of active (beta)-catenin independently of CRM1. J Cell Biol.
171:785–797. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pokutta S and Weis WI: Structure of the
dimerization and beta-catenin-binding region of alpha-catenin. Mol
Cell. 5:533–543. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
de La Coste A, Romagnolo B, Billuart P,
Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C,
Kahn A, et al: Somatic mutations of the beta-catenin gene are
frequent in mouse and human hepatocellular carcinomas. Proc Natl
Acad Sci USA. 95:8847–8851. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miyoshi Y, Iwao K, Nagasawa Y, Aihara T,
Sasaki Y, Imaoka S, Murata M, Shimano T and Nakamura Y: Activation
of the beta-catenin gene in primary hepatocellular carcinomas by
somatic alterations involving exon 3. Cancer Res. 58:2524–2527.
1998.PubMed/NCBI
|
|
30
|
Laurent-Puig P and Zucman-Rossi J:
Genetics of hepatocellular tumors. Oncogene. 25:3778–3786. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Katoh M: Canonical and non-canonical WNT
signaling in cancer stem cells and their niches: Cellular
heterogeneity, omics reprogramming, targeted therapy and tumor
plasticity (Review). Int J Oncol. 51:1357–1369. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pez F, Lopez A, Kim M, Wands JR, Caron de
Fromentel C and Merle P: Wnt signaling and hepatocarcinogenesis:
molecular targets for the development of innovative anticancer
drugs. J Hepatol. 59:1107–1117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vilchez V, Turcios L, Marti F and Gedaly
R: Targeting Wnt/β-catenin pathway in hepatocellular carcinoma
treatment. World J Gastroenterol. 22:823–832. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Delgado ER, Yang J, So J, Leimgruber S,
Kahn M, Ishitani T, Shin D, Mustata Wilson G and Monga SP:
Identification and characterization of a novel small-molecule
inhibitor of β-catenin signaling. Am J Pathol. 184:2111–2122. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gedaly R, Galuppo R, Daily MF, Shah M,
Maynard E, Chen C, Zhang X, Esser KA, Cohen DA, Evers BM, et al:
Targeting the Wnt/β-catenin signaling pathway in liver cancer stem
cells and hepatocellular carcinoma cell lines with FH535. PLoS One.
9:e992722014. View Article : Google Scholar
|
|
37
|
Handeli S and Simon JA: A small-molecule
inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma
and PPARdelta activities. Mol Cancer Ther. 7:521–529. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Galuppo R, Maynard E, Shah M, Daily MF,
Chen C, Spear BT and Gedaly R: Synergistic inhibition of HCC and
liver cancer stem cell proliferation by targeting RAS/RAF/MAPK and
WNT/β-catenin pathways. Anticancer Res. 34:1709–1713.
2014.PubMed/NCBI
|
|
39
|
Lin HH, Feng WC, Lu LC, Shao YY, Hsu CH
and Cheng AL: Inhibition of the Wnt/β-catenin signaling pathway
improves the anti-tumor effects of sorafenib against hepatocellular
carcinoma. Cancer Lett. 381:58–66. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Aberle H, Schwartz H, Hoschuetzky H and
Kemler R: Single amino acid substitutions in proteins of the
armadillo gene family abolish their binding to alpha-catenin. J
Biol Chem. 271:1520–1526. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roura S, Miravet S, Piedra J, García de
Herreros A and Duñach M: Regulation of E-cadherin/Catenin
association by tyrosine phosphorylation. J Biol Chem.
274:36734–36740. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Piedra J, Miravet S, Castaño J, Pálmer HG,
Heisterkamp N, García de Herreros A and Duñach M: p120
Catenin-associated Fer and Fyn tyrosine kinases regulate
beta-catenin Tyr-142 phosphorylation and beta-catenin-alpha-catenin
Interaction. Mol Cell Biol. 23:2287–2297. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Piedra J, Martinez D, Castano J, Miravet
S, Dunach M and de Herreros AG: Regulation of beta-catenin
structure and activity by tyrosine phosphorylation. J Biol Chem.
276:20436–20443. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brembeck FH, Schwarz-Romond T, Bakkers J,
Wilhelm S, Hammerschmidt M and Birchmeier W: Essential role of
BCL9-2 in the switch between beta-catenin's adhesive and
transcriptional functions. Genes Dev. 18:2225–2230. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li Y, Lu W, He X, Schwartz AL and Bu G:
LRP6 expression promotes cancer cell proliferation and
tumorigenesis by altering beta-catenin subcellular distribution.
Oncogene. 23:9129–9135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Austinat M, Dunsch R, Wittekind C,
Tannapfel A, Gebhardt R and Gaunitz F: Correlation between
beta-catenin mutations and expression of Wnt-signaling target genes
in hepatocellular carcinoma. Mol Cancer. 7:212008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Boyault S, Rickman DS, de Reyniès A,
Balabaud C, Rebouissou S, Jeannot E, Hérault A, Saric J, Belghiti
J, Franco D, et al: Transcriptome classification of HCC is related
to gene alterations and to new therapeutic targets. Hepatology.
45:42–52. 2007. View Article : Google Scholar
|
|
48
|
Cleary SP, Jeck WR, Zhao X, Chen K,
Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, et al:
Identification of driver genes in hepatocellular carcinoma by exome
sequencing. Hepatology. 58:1693–1702. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Guichard C, Amaddeo G, Imbeaud S, Ladeiro
Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M,
Degos F, et al: Integrated analysis of somatic mutations and focal
copy-number changes identifies key genes and pathways in
hepatocellular carcinoma. Nat Genet. 44:694–698. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kan Z, Zheng H, Liu X, Li S, Barber TD,
Gong Z, Gao H, Hao K, Willard MD, Xu J, et al: Whole-genome
sequencing identifies recurrent mutations in hepatocellular
carcinoma. Genome Res. 23:1422–1433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: beta-catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Orford K, Crockett C, Jensen JP, Weissman
AM and Byers SW: Serine phosphorylation-regulated ubiquitination
and degradation of beta-catenin. J Biol Chem. 272:24735–24738.
1997. View Article : Google Scholar : PubMed/NCBI
|