Introduction
Epidermal growth factor (EGF) receptor (EGFR) is a
receptor tyrosine kinase (RTK) and a member of the ErbB family.
EGFR agonists, such as EGF, transforming growth factor-α and
epiregulin, bind to the extracellular domain of EGFR and cause the
homodimerization or heterodimerization of Erb family receptors,
which leads to the phosphorylation of intracellular EGFR sites,
such as the Tyr1173 and Tyr1068 residues. The phosphorylation of
these sites activates signalling pathways, such as the
RAS/mitogen-activated protein kinase (MAPK),
phosphoinositide-3-kinase (PI3K)/serine/threonine-specific protein
kinase (Akt) and Janus kinase/signal transducer and activator of
transcription (Jak/Stat) pathways (1-5). The
activation of these pathways mediates cellular proliferation and
transformation, which may cause cancer occurrence, growth, invasion
and metastasis.
Triple-negative breast cancer (TNBC) does not
express human epidermal growth factor receptor type (HER)2,
progesterone receptor (PR) or oestrogen receptor (ER), but does
express EGFR (6,7). There is evidence that EGFR
overexpression is associated with an aggressive phenotype and a
poor clinical outcome in breast cancers, including TNBC (8-12).
Therefore, blocking the EGFR-mediated signalling
pathway is a therapeutic target for treating EGFR-expressing cancer
types, such as TNBC. Cetuximab (CTX) was developed to bind to the
extracellular domain of EGFR, which blocks the binding of EGF and
inhibits the subsequent phosphorylation and activation of EGFR and
downstream signalling pathways, such as RAS/MAPK, PI3K/Akt and
Jak/Stat (13-15). CTX has been used in the treatment
of metastatic colorectal cancer and head and neck squamous cell
carcinoma, both as monotherapy and in combination with chemotherapy
and radiotherapy (14,16).
However, certain types of cancer, such as TNBC and
lung and colon cancer are resistant to CTX treatment (4,16,17).
Primary and acquired resistance to anti-EGFR treatment has been
described (4), with several
studies reporting that genetic alterations in key regulators, such
as KRAS, BRAF, phosphatase and tensin homologue (PTEN) and
phosphoinositide-3-kinase, catalytic subunit α (PIK3CA), in the
signalling pathways of RTKs are an important mechanism underlying
resistance to anti-EGFR treatment (4,6,17-20).
These alterations lead to the activation of RAS/MAPK and PI3K/Akt,
independently of the activation status of growth factor receptors.
As a result, the blocking of EGFR by antibodies or tyrosine kinase
inhibitors becomes ineffective in shutting down these signalling
pathways. TNBC cells, such as MDA-MB-231, harbour mutations in KRAS
and BRAF, whereas MDA-MB-468 cells do not have the Akt signalling
pathway-inhibiting gene, PTEN, as a tumour suppressor (21). Hence, these cell lines often
exhibit resistance to anti-EGFR treatment. However, the mutations
in KRAS, BRAF, PTEN and PIK3CA cannot fully explain this
resistance, since it is also observed in wild-type breast and colon
cancer cells, which do not harbour these mutations (20,22).
The ligands of RTKs possess partial agonist
properties, which cause homo- or heterodimer formation and,
subsequently, receptor phosphorylation and activation (23). Some antibodies directed against
RTKs to block the binding of growth factors may also act as partial
agonists (24-26). One study evaluating the targeting
strategies of RTKs emphasised that the binding of an antibody with
partial agonist properties to the receptor may cause
phosphorylation, which triggers the ubiquitination and degradation
of the receptor, resulting in irreversible antagonism of the RTK
(26), the inhibition of the
malignant response and cell proliferation, which may lead to a
significant anticancer response in cancer cells.
On the other hand, the partial agonistic action of
an antibody on the RTK may induce the phosphorylation of receptor
tyrosine residues, activating PI3K/Akt, RAS/MAPK and PLCγ/PKC,
similar to EGF or a full agonist. Therefore, a partial agonist
antibody may lead to RTK activation, which may cause resistance to
this antibody in cancer cells. For example, trastuzumab binds to
ErbB2, induces phosphorylation and exerts an agonistic effect on
this receptor (27-29). Yoshida et al (30) demonstrated that matuzumab and CTX
activated EGFR by inducing the phosphorylation of the Tyr845,
Tyr1068 and Tyr1173 residues in the non-small-cell lung cancer cell
lines, H292 and H460. Similarly, Raben et al (31) observed that CTX induced the
phosphorylation of the Tyr845, Tyr992 and Tyr1068 residues of EGFR
in H322 and H292 cells. Furthermore, CTX treatment has been shown
to enhance the phosphorylation of EGFR Tyr845 residues in
MDA-MB-231 cells (32) and Tyr1068
in MDA-MB-468 cells (33).
The aim of the present study was to examine the
partial agonistic effect of CTX on EGFR in the TNBC cell lines,
MDA-MB-231 and MDA-MB-468. For this purpose, by treating TNBC cells
with EGF or CTX, we measured the phosphorylation levels of EGFR
(Tyr1173), PI3K, Akt, extracellular signal-regulated kinase
(ERK)1/2 and Src kinase, the cellular level of EGFR and cellular
morphology, using impedance measurement as an indication of RTK
activity. Since the agonistic action on EGFR may lead to the
transactivation or cross-activation of insulin-like growth factor
receptor (IGF-1R) and vascular endothelial growth factor receptor
(VEGFR)-2 (34,35), the phosphorylation status of these
receptors with CTX or EGF treatment of TNBC cells was also
determined. Finally, the anti-proliferative response of the cells
to CTX was evaluated in the presence or absence of the Src kinase
inhibitor, PP2, which inhibits the partial agonist action of CTX on
EGFR.
Materials and methods
Reagents and antibodies
The EGFR kinase inhibitor, AG1478, the PI3K
inhibitor, LY294002
[2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride],
the Src kinase inhibitor, PP2 [4-amino-3-(4-chlo
rophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d]pyrimidine], EGF and CTX
were obtained from Sigma-Aldrich, Merck KGaA (Darmstadt, Germany).
The antibodies used were as follows (dilution, catalogue number,
host and clonality used for western blot analysis):
p-VEGFR-2-Tyr1175 (1:1,000, 3770, rabbit monoclonal), p-Src-Tyr416
(1:1,000, 2101, rabbit polyclonal) from Cell Signalling Technology,
Inc., Danvers, MA, USA; p-EGFR-Tyr1173 (1:1,000, sc-12351-R, rabbit
polyclonal), EGFR (1:1,000, sc-53274, mouse monoclonal),
p-IGF-1R-Tyr1161 (1:1,000, sc-101703, rabbit polyclonal), p-Erk1/2
(1:5,000, sc-136521, mouse monoclonal), ERK1/2 (1:1,000, sc-514302,
mouse monoclonal), p-Akt-Ser473 (1:1,000, sc-7985, rabbit
polyclonal), Akt (1:1,000, sc-8312, rabbit polyclonal), p-PI3K
(1:1,000, sc-12929, goat polyclonal), GAPDH (1:5,000, sc-47724,
mouse monoclonal), mouse anti-rabbit (1:10,000, sc-2357), rabbit
anti-goat IgG-HRP (1:10,000, sc-2768) and donkey anti-mouse IgG-HRP
(1:10,000, sc-2314) all from Santa Cruz Biotechnology Inc. (Santa
Cruz, CA, USA). In all control experiments, the cells were
incubated with the corresponding dilution of solvent used for the
ligand.
Cell culture, treatments, protein
isolation and western blot analysis
The MDA MB 231 and MDA MB 468 TNBC cell lines, which
are negative for ER, PR and HER2, but positive for EGFR, were
employed in this study. The cell lines were obtained from the
American Type Culture Collection (Manassas, VA, USA). The
MDA-MB-231 and MDA-MB-468 cells were grown in 75-cm2
non-treated cell culture flasks (Corning, Tewksbury, MA, USA) in
Dulbecco's modified Eagle's medium and Eagle's minimal essential
medium, respectively, enriched with 10% foetal bovine serum and 1%
penicillin/streptomycin at 5% CO2, 37°C, with 90-95%
humidity. For each experiment, the cells (2.5×105
cells/well) were plated in 6-well plates and treated with CTX
(0.01, 0.1, 1, 10 or 100 nM) or EGF (1 nM) on the third day
following overnight serum starvation. The concentration of ligands
and the duration of cell treatment are specified in the results
section. In some experiments, prior to CTX or EGF stimulation, the
cells were pre-treated with one of the following inhibitors: AG1478
(10 µM), LY294002 (10 µM) or PP2 (10 µM) for
30 min. Following stimulation, the cells were immediately placed on
ice and washed with ice-cold phosphate-buffered saline and
homogenised in 100 µl lysis buffer (Roche Molecular
Diagnostics, Mannheim, Germany) containing 1% Nonidet P40, 0.02 M
sodium orthovanadate and protease inhibitors. Following
homogenization, the cells were incubated for 15 min and centrifuged
at 5,000 × g for 5 min at 4°C. After collecting the supernatant,
the protein concentration was determined using the Bradford protein
assay and the samples were stored at −80°C. Electrophoresis and
western blot analysis were performed as previously described
(36). Band intensities were
corrected against t-ERK1/2, t-AKT or GAPDH expression. As a control
of the loading amount of protein, GAPDH bands were used and the
band intensities of p-EGFR, p-IGF-1R or p-VEGFR-2 were corrected
against the GAPDH bands. The band intensities of EGFR, IGF-1R and
VEGFR-2 were not used for the corrections, due to internalization
and/or degradation of these receptors following ligand
stimulation.
Real-time cellular morphology and cell
proliferation assay by impedance measurements
These assays were performed as previously described
(37-39), using the xCelligence Real-Time Cell
Analyzer (RTCA) DP system (ACEA Biosciences, Inc., San Diego, CA,
USA). Briefly, the MDA-MB-231 or MDA-MB-468 cells (6,500
cells/well; E-plate) were seeded into each well. Subsequently, the
impedance of each well of the E-plate was measured continuously for
20-24 h at 37°C with 5% CO2. For the cell proliferation
assay, after the first 20-24 h, CTX or EGF was added to the wells
and the impedance measurement was recorded for 72-96 h. For
cellular morphology measurement, after the first 20-24 h, the
medium in each well was replaced with serum-free medium and
incubated for 4 h at 37°C with 5% CO2. CTX or EGF was
then added to the wells and impedance was recorded for 24 h. The
results were analysed using RTCA data analysis software 1.0 (ACEA
Biosciences, Inc., San Diego, CA, USA).
Cell viability was also assessed by the WST-1
proliferation assay according to the manufacturer's instructions
(Roche Applied Science, Mannheim, Germany).
Statistical analysis
Data are reported as the means ± standard error of
the mean, while ‘n’ represents the number of independent
experiments for each indicated condition. The results were obtained
from 4-5 independent experiments. Statistical analysis was
performed using SPSS 17.0 software for Windows (SPSS, Inc.,
Chicago, IL, USA). Comparisons between multiple groups were
performed with one-way analysis of variance followed by Tukey's
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
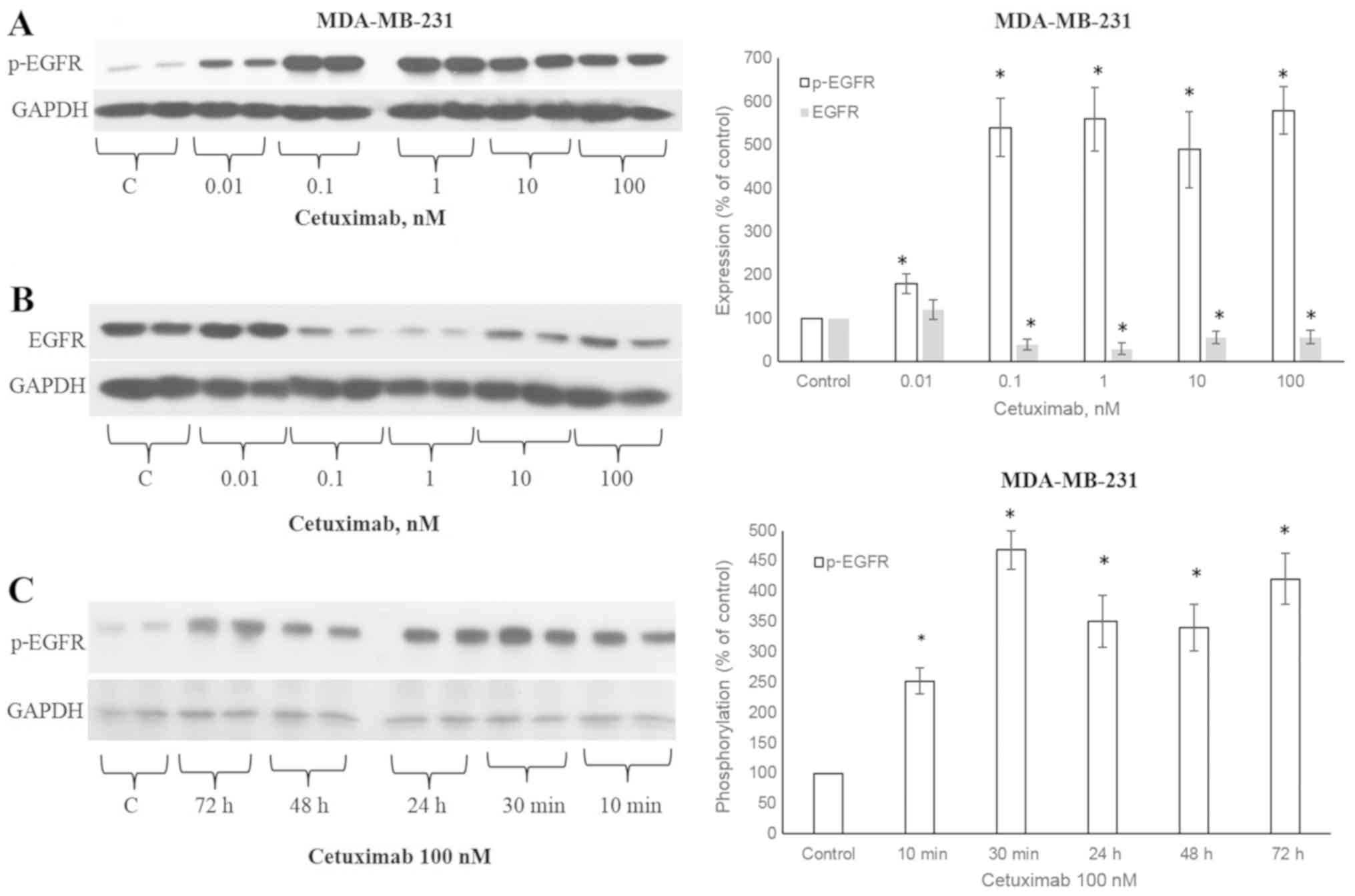
CTX induces the phosphorylation of
EGFR
The ligands that activate EGFR lead to the
phosphorylation of the 1173 tyrosine residue of the EGFR receptor
(5). CTX at concentrations of
0.01, 0.1, 1, 10 and 100 nM significantly increased the
phosphorylation of the 1173 tyrosine residue of the EGFR receptor
(Fig. 1A), similar to EGF
(Fig. 2A). The CTX-stimulated
phosphorylation was observable after 10 min of incubation at 100 nM
and persisted for 72 h in the MDA-MB-231 cells (Fig. 1C). CTX also induced EGFR
phosphorylation in the MDA-MB-468 TNBC cells (Fig. 2B).
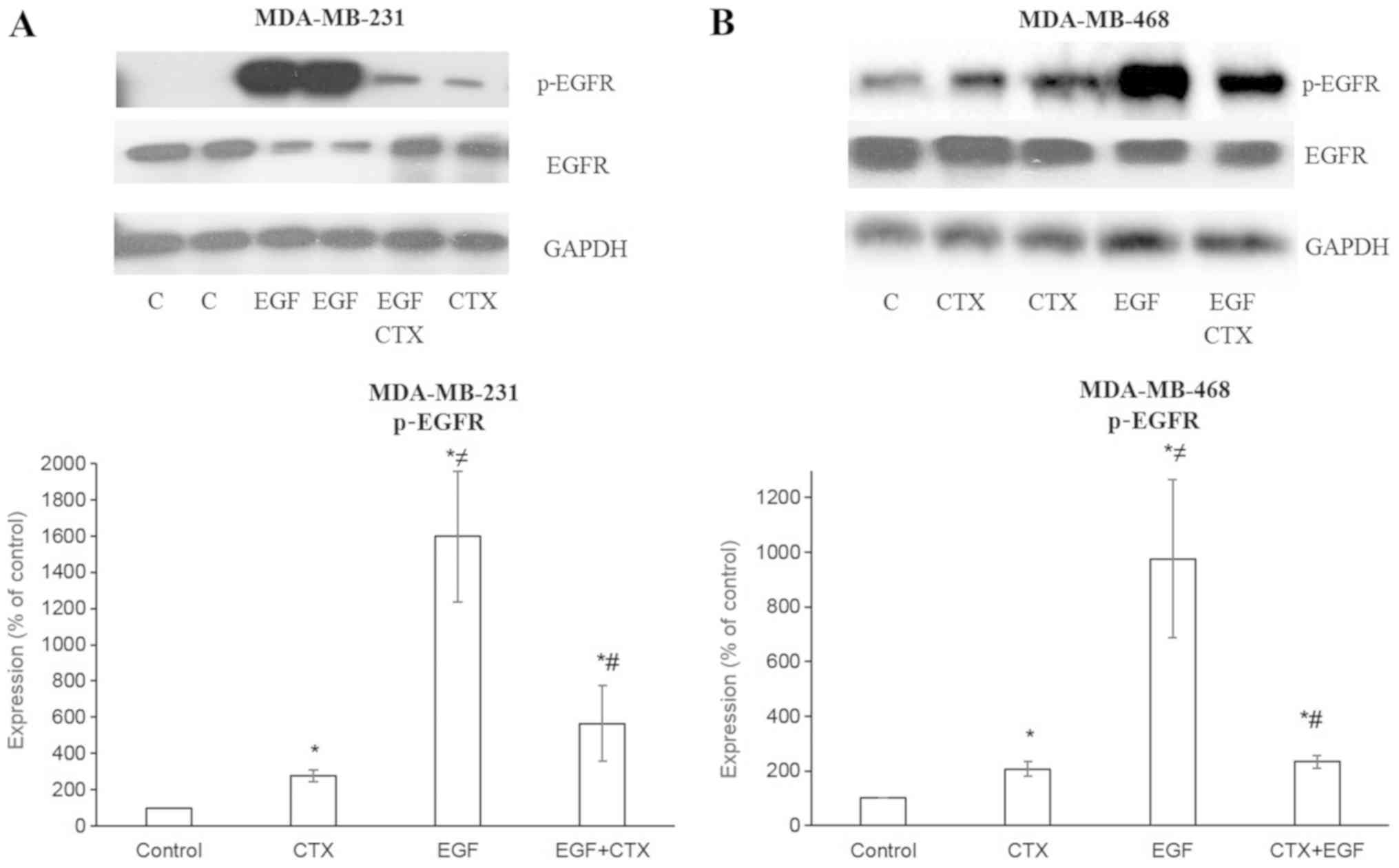 | Figure 2Cetuximab partially antagonizes the
EGF-stimulated phosphorylation of EGFR. Cetuximab (100 nM, 30 min)
produced significantly less EGFR phosphorylation than EGF (1 nM, 30
min). Pre-treatment of the cells with CTX (100 nM, 30 min)
significantly, but not completely inhibited the EGF-induced
phosphorylation (1 nM, 30 min) of EGFR in (A) MDA-MB-231 and (B)
MDA-MB-468. Representative western blot bands for p-EGFR and EGFR
are presented along with two replicate bands for the control
(indicated by ‘C’), for the EGF treatment groups in (A), and for
CTX in (B). Band intensities were normalized to GAPDH and presented
as a percentage of phosphate buffered saline-treated control cells.
Data are presented as the means ± standard error of the mean, n=4-5
experiments. *P<0.05 vs. the control cells;
≠P<0.05 vs. CTX; #P<0.05 vs. EGF. CTX,
cetuximab; EGF, epidermal growth factor; EGFR, epidermal growth
factor receptor. |
CTX induces less prominent EGFR
phosphorylation compared with EGF
CTX at a concentration of 100 nM [used as the
maximum concentration, which is >200-fold its Kd (dissociation
constant) of 0.39 nM and sufficient to saturate EGFR] produced
significantly less EGFR phosphorylation compared with 1 nM EGF, a
full agonist, in both the MDA-MB-231 (Fig. 2A) and MDA-MB-468 (Fig. 2B) cells.
CTX partially inhibits EGF-mediated EGFR
phosphorylation
It is well known that a partial agonist causes the
incomplete inhibition of the response induced by a full agonist.
Accordingly, CTX (100 nM) incompletely inhibited EGF-induced EGFR
phosphorylation at the 1 nM EGF concentration in both the
MDA-MB-231 (Fig. 2A) and
MDA-MB-468 (Fig. 2B) cells.
CTX causes the degradation of EGFR and
partially inhibits the EGF-mediated degradation of EGFR
When measuring the EGFR levels following treatment
of the cells with CTX (100 nM) or EGF (1 nM), a decreased level of
EGFR was observed in the MDA-MB-231 (Figs. 1B and 3A) and MDA-MB-468 (Fig. 3B) cells. These results indicate
that CTX functions like a partial agonist in the degradation of
EGFR and, as expected, CTX reduces the EGFR levels to a lesser
extent compared with EGF (Fig. 3).
Furthermore, CTX only partially inhibited the EGF-mediated decrease
in EGFR levels (Fig. 3).
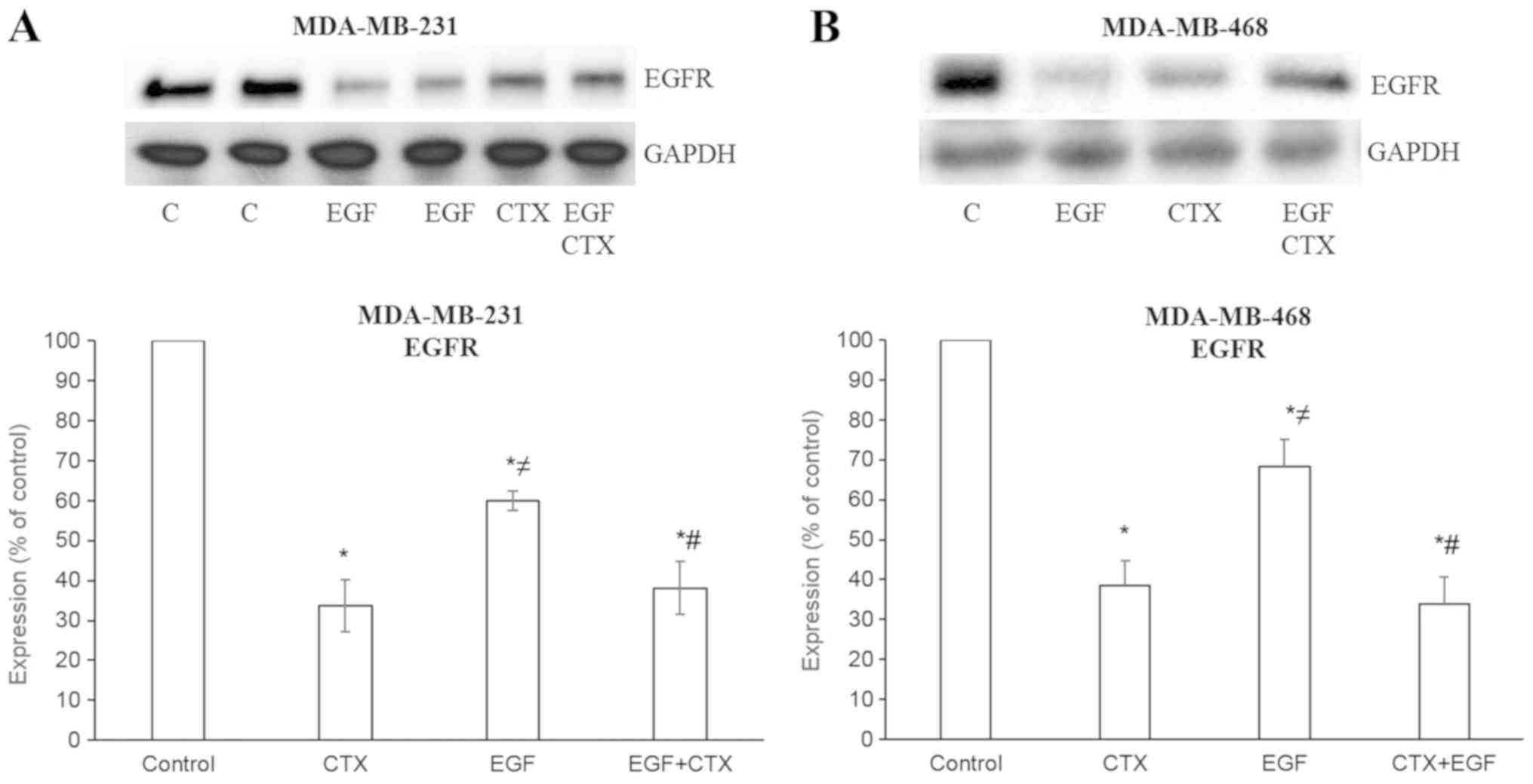 | Figure 3Cetuximab causes the degradation of
EGFR. EGF (1 nM, 2 h) and CTX (100 nM, 2 h) both reduced the EGFR
level, but CTX has a significantly less prominent effect on the
EGFR level than EGF. Pre-treatment of the cells with CTX (100 nM,
30 min) significantly, but not completely inhibited the EGF-induced
degradation (1 nM, 2 h) of EGFR in the (A) MDA-MB-231 and (B)
MDA-MB-468 cells. Representative western blot bands for EGFR are
presented. Two replicate bands are presented for the control
(indicated as ‘C’) and for the EGF treatment groups in (A). Band
intensities are normalized to GAPDH and presented as a percentage
of phosphate-buffered saline-treated control cells. Data are
presented as the mean ± standard error of the mean, n=4-5
experiments. *P<0.05 vs. the control cells;
≠P<0.05 vs. CTX; #P<0.05 vs. EGF. CTX,
cetuximab; EGF, epidermal growth factor; EGFR, epidermal growth
factor receptor. |
CTX triggers the EGFR downstream
pathway
After observing Tyr1173 phosphorylation of EGFR with
CTX, the downstream signalling of EGFR was investigated. In the
MDA-MB-231 cells, CTX treatment significantly enhanced the
phosphorylation of Src kinase, PI3K and Akt, but not ERK1/2
(Fig. 4A and B). The
phosphorylation of Akt was antagonized by the Src kinase inhibitor,
PP2, and the PI3K inhibitor, LY294002 (Fig. 4B). It was therefore deduced that
the stimulation of EGFR by CTX activates Src kinase and PI3K, and
causes the phosphorylation of Akt. The CTX-mediated phosphorylation
of PI3K and Akt was less prominent compared with that induced by
EGF, and it also partially inhibited the EGF-stimulated
phosphorylation of PI3K and Akt in the MDA-MB-231 cells (Fig. 4C).
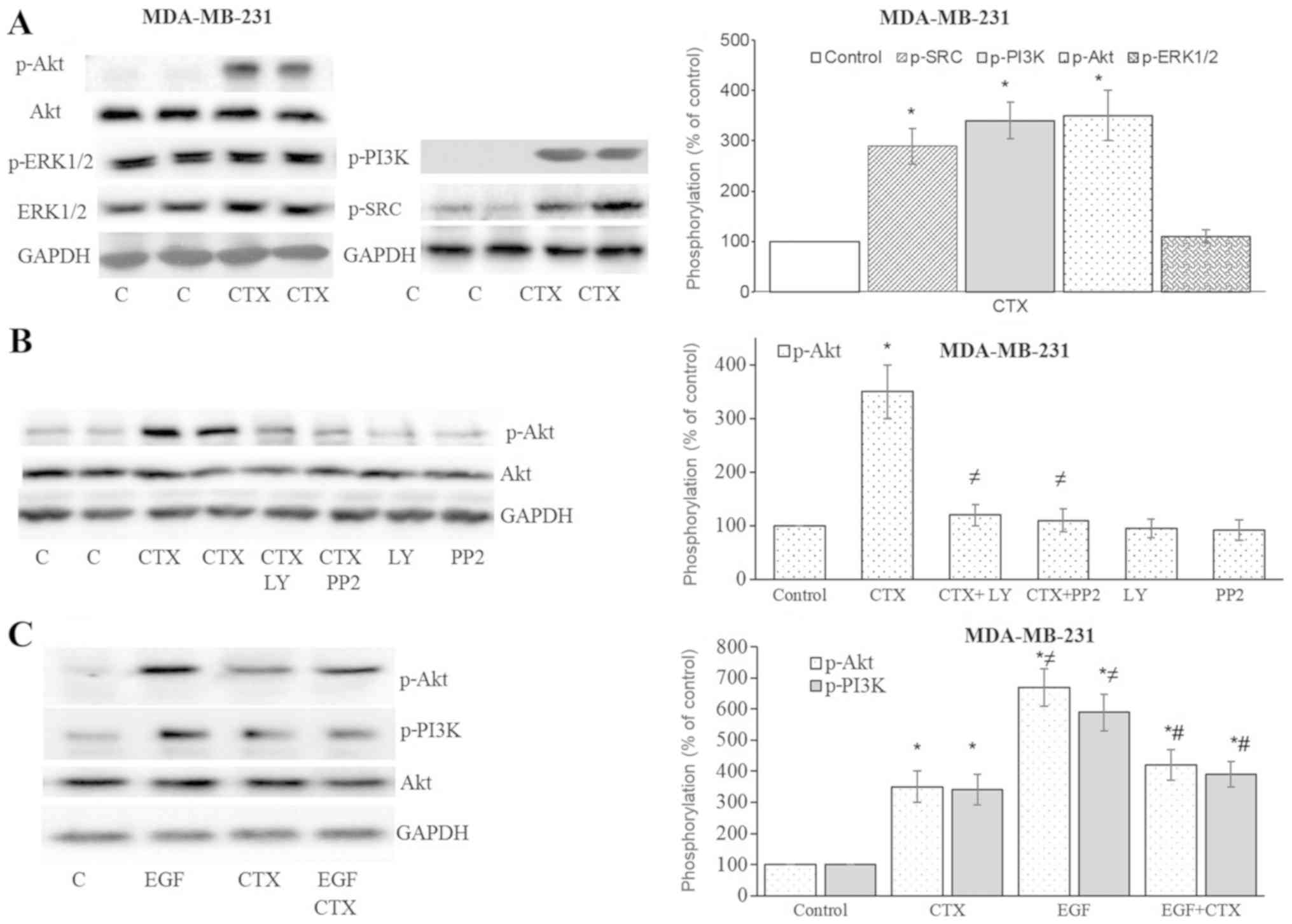 | Figure 4Cetuximab triggers the downstream
signalling of EGFR in MDA-MB-231 cells. (A and B) Cetuximab (100
nM, 30 min) induced the phosphorylation of Src kinase, PI3K and
Akt, but not that of ERK1/2 in the MDA-MB-231 cells. (B)
Pre-treatment of cells with the PI3K inhibitor, LY294002 (10
µM, 30 min), or the Src kinase inhibitor, PP2 (10 µM,
30 min), significantly inhibited CTX-induced Akt phosphorylation.
(C) CTX produced significantly less Akt or PI3K phosphorylation
than EGF did, while pre-treatment of the cells with CTX (100 nM, 30
min) significantly, but not completely inhibited the EGF-induced
phosphorylation (1 nM, 30 min) of PI3K and Akt in the MDA-MB-231
cells. Representative western blot bands for p-SRC, p-PI3K, p-Akt,
p-ERK1/2, Akt and GADPH are presented. Two replicate bands are
presented for the control or for the CTX treatment groups in (A and
B). The p-Akt and p-ERK1/2 band intensities are normalized to Akt
and ERK1/2, respectively, and the p-SRC and p-PI3K band intensities
are normalized to GADPH and presented as a percentage of
phosphate-buffered saline-treated control cells. Data are presented
as the means ± standard error of the mean, n=4-5 experiments.
*P<0.05 vs. control cells; ≠P<0.05 vs.
CTX; #P<0.05 vs. EGF. CTX, cetuximab; EGF, epidermal
growth factor; EGFR, epidermal growth factor receptor. |
In the MDA-MB-468 cells, CTX also increased the
phosphorylation of Src kinase and PI3K, but not that of Akt and
ERK1/2 (Fig. 5A). The CTX-mediated
phosphorylation of PI3K was less prominent compared with that
induced by EGF, and it also partially inhibited the EGF-stimulated
phos-phorylation of PI3K in the MDA-MB-468 cells (Fig. 5B). These results indicate that the
partial agonistic activity of CTX is observable in the EGFR
downstream signalling pathway.
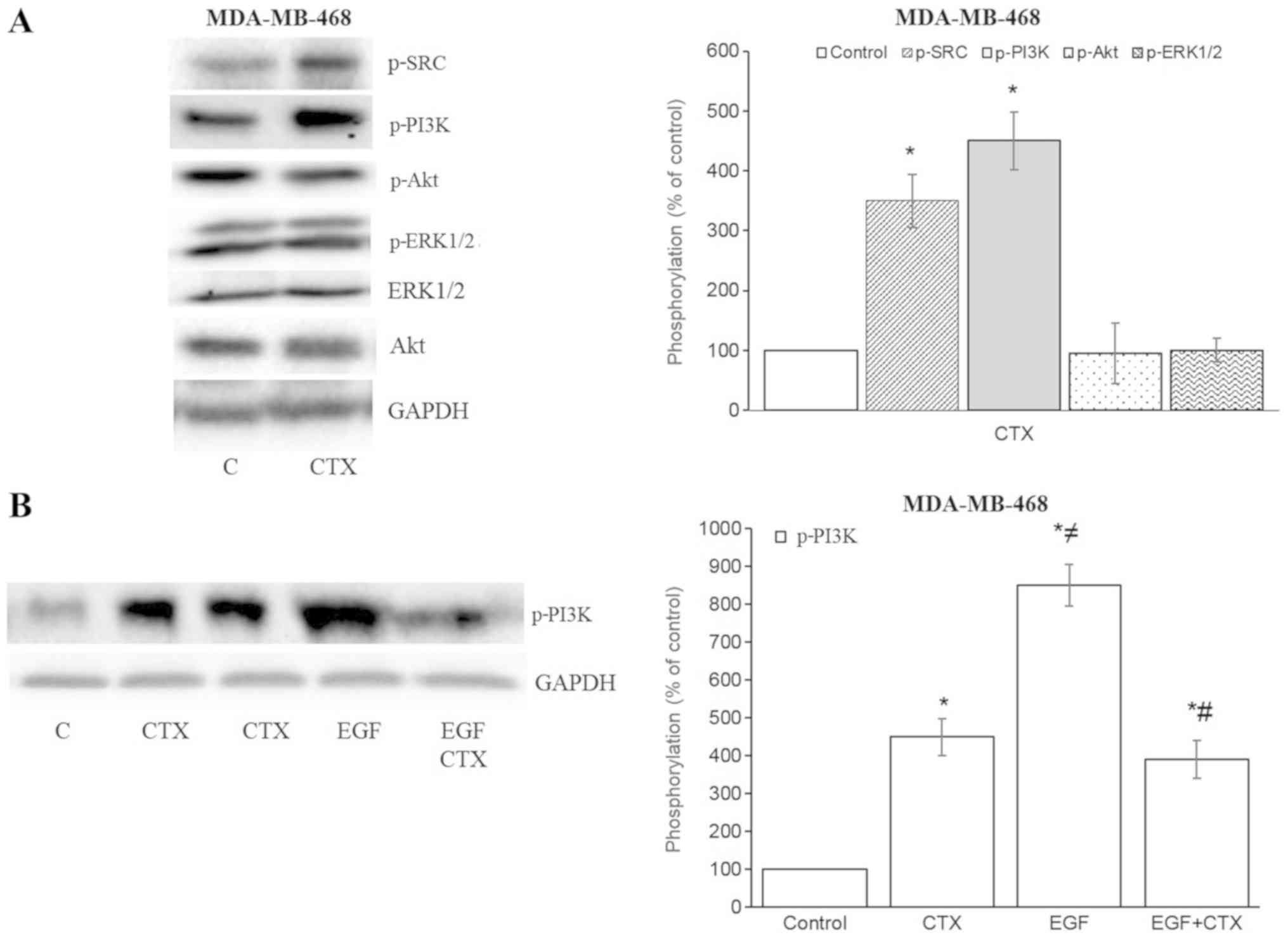 | Figure 5Cetuximab triggers the downstream
signalling of EGFR in MDA-MB-468 cells. (A and B) Cetuximab (100
nM, 30 min) induced the phosphorylation of Src kinase and PI3K, but
not that of Akt or ERK1/2 in MDA-MB-468 cells. (B) CTX produced
significantly less PI3K-phosphorylation than EGF did, while
pre-treatment of the cells with CTX (100 nM, 30 min) significantly,
but not completely inhibited the EGF-induced phosphorylation (1 nM,
30 min) of PI3K in MDA-MB-468 cells. Representative western blot
bands for pSrc, pPI3K, pAkt, pERK1/2, Akt and GADPH are presented.
Two replicate bands are presented for CTX in (B). The p-Akt and
p-ERK1/2 band intensities are normalized to Akt and ERK1/2,
respectively, and the p-Src, p-PI3K band intensities are normalized
to GADPH and presented as a percentage of phosphate-buffered
saline-treated control cells. Data are presented as the means ±
standard error of the mean, n=4-5 experiments.
*P<0.05 vs. the control cells; ≠P<0.05
vs. CTX; #P<0.05 vs. EGF. CTX, cetuximab; EGF,
epidermal growth factor; EGFR, epidermal growth factor
receptor. |
The possible reasons as to why CTX did not increase
the basal phosphorylation of ERK1/2 in the MDA-MB231 cells or the
phosphorylation of ERK1/2 and Akt in the MDA-MB-468 cells are
addressed below in the Discussion.
CTX induces IGF-1R and VEGFR-2
phosphorylation
In the present study, the effects of CTX or EGF on
the phosphorylation status of IGF-1R and VEGFR-2 were also
evaluated. CTX and EGF induced the phosphorylation of both IGF-1R
and VEGFR-2 in the MDA-MB231 (Fig.
6) and MDA-MB-468 (Fig. 7)
cells. This induced phosphorylation was inhibited by the EGFR
tyrosine kinase inhibitor, AG1478, and the Src kinase inhibitor,
PP2, in both the MDA-MB-231 (Fig. 6A
and B) and MDA-MB-468 (Fig. 7A and
B) cells. These results suggest that the CTX-mediated
activation of EGFR and Src kinase trigger the phosphorylation of
IGF-1R and VEGFR-2.
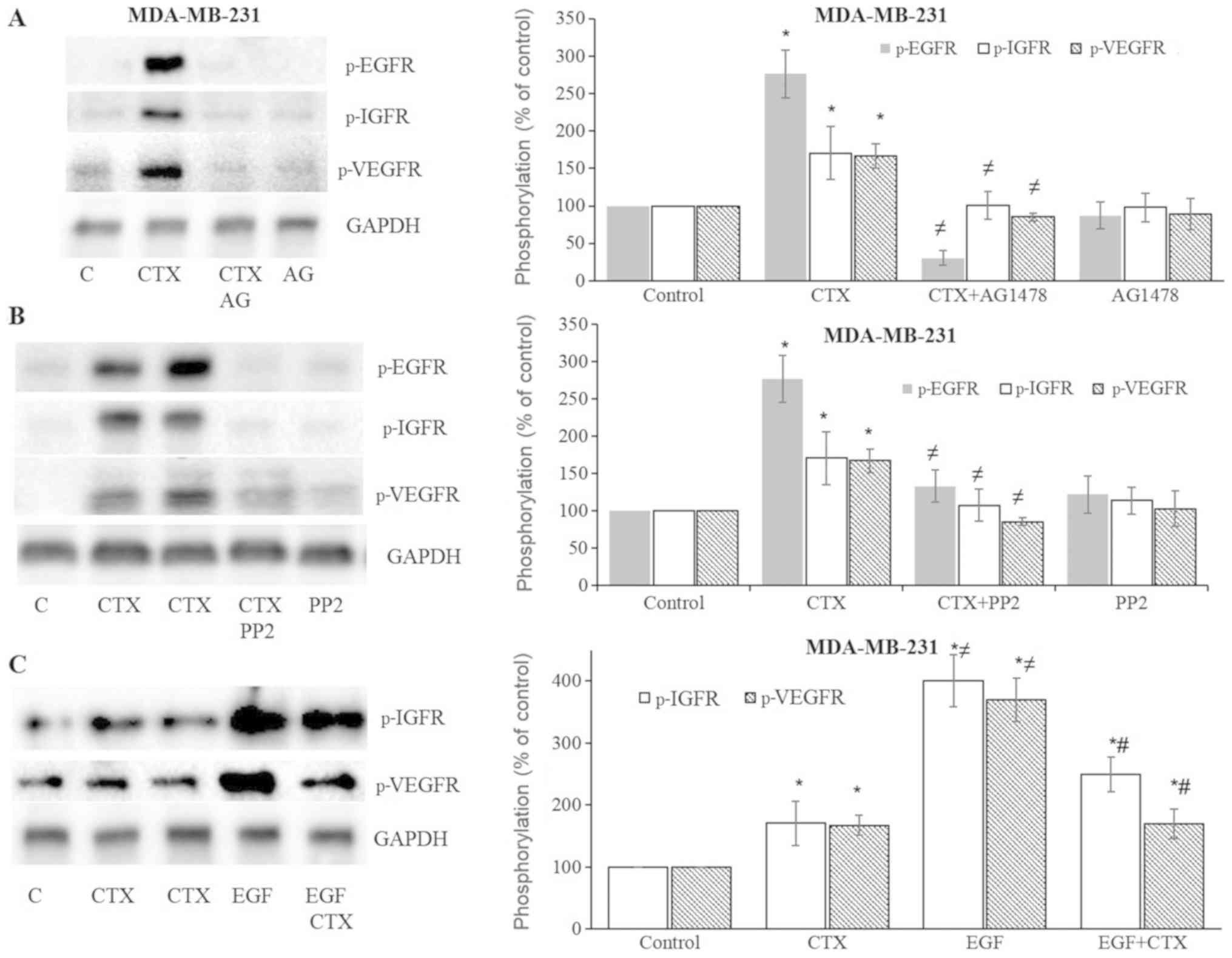 | Figure 6Cetuximab leads to the
phosphorylation of IGF-1R and VEGFR-2 in MDA-MB-231 cells. (A and
B) Cetuximab (100 nM, 30 min) induced the phosphorylation of EGFR,
IGF-1R and VEGFR-2, while pre-treatment of the cells with the EGFR
tyrosine kinase inhibitor, AG1478 (10 µM, 30 min), or the
Src kinase inhibitor, PP2 (10 µM, 30 min), significantly
inhibited EGFR, IGF-1R and VEGFR-2 phosphorylation in MDA-MB-231
cells. (C) CTX produced significantly less IGF-1R and
VEGFR-2-phosphorylation than EGF did, while pre-treatment of the
cells with CTX (100 nM, 30 min) significantly, but not completely
inhibited EGF-induced phosphorylation (1 nM, 30 min) of IGF-1R and
VEGFR-2 in MDA-MB-231cells. Representative western blot bands for
p-EGFR, p-IGF-1R, p-VEGFR-2 and GADPH are presented. Two replicate
bands are presented for CTX in (B and C). The band intensities are
normalized to GADPH and presented as a percentage of
phosphate-buffered saline-treated control cells. Data are presented
as the means ± standard error of the mean, n=4-5 experiments.
*P<0.05 vs. control cells; ≠P<0.05 vs.
CTX; #P<0.05 vs. EGF. CTX, cetuximab; EGF, epidermal
growth factor; EGFR, epidermal growth factor receptor. |
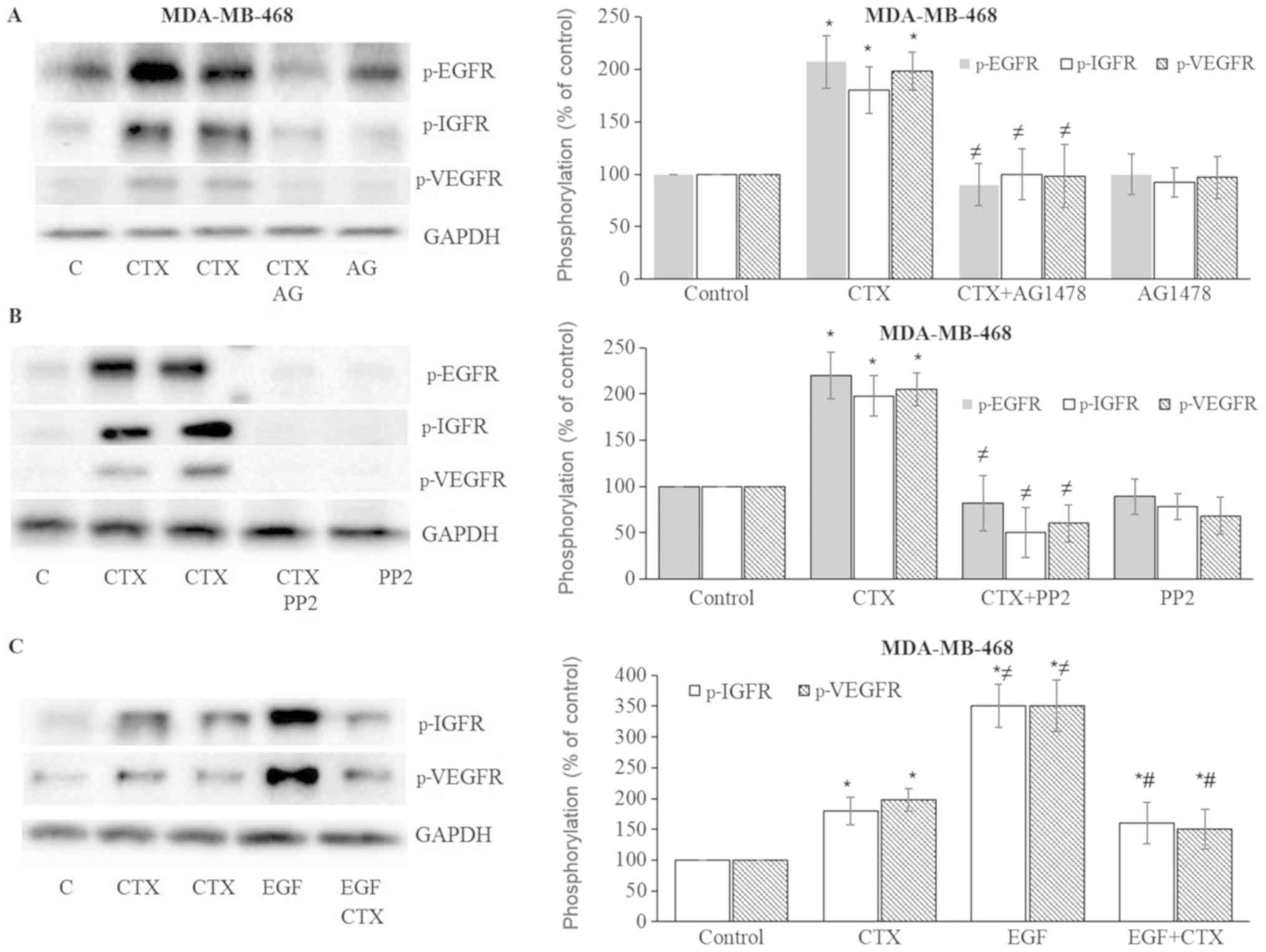 | Figure 7Cetuximab leads to the
phosphorylation of IGF-1R and VEGFR-2 in MDA-MB-468 cells. (A and
B) Cetuximab (100 nM, 30 min) induced the phosphorylation of EGFR,
IGF-1R, VEGFR-2, while pre-treatment of the cells with the EGFR
tyrosine kinase inhibitor, AG1478 (AG) (10 µM, 30 min), or
the Src inhibitor, PP2 (10 µM, 30 min), significantly
inhibited EGFR, IGF-1R and VEGFR-2 phosphorylation in MDA-MB-468
cells. (C) CTX produced signifi-cantly less IGF-1R and VEGFR-2
phosphorylation than EGF did, while pre-treatment of the cells with
CTX (100 nM, 30 min) significantly but not completely inhibited the
EGF-induced phosphorylation (1 nM, 30 min) of IGF-1R and VEGFR-2 in
MDA-MB-468 cells. Representative western blot bands for p-EGFR,
p-IGF-1R, p-VEGFR-2 and GADPH are presented. Two replicate bands
are presented for CTX. The band intensities are normalized to GADPH
and presented as a percentage of phosphate-buffered saline-treated
control cells. Data are presented as the means ± standard error of
the mean, n=4-5 experiments. *P<0.05 vs. control
cells; ≠P<0.05 vs. CTX; #P<0.05 vs.
EGF. CTX, cetuximab; EGF, epidermal growth factor; EGFR, epidermal
growth factor receptor. |
CTX partially inhibits EGF-mediated
IGF-1R and VEGFR-2 phosphorylation
As with EGFR phosphorylation, CTX treatment at 100
nM produced significantly less IGF-1R and VEGFR-2 phosphorylation
compared with the full agonist, EGF at 1 nM (Figs. 6C and 7C). CTX also functioned as a partial
agonist, partially inhibiting the IGF-1R and VEGFR-2
phosphorylation induced by EGF (1 nM) in the MDA-MB-231 (Fig. 6C) and MDA-MB-468 cells (Fig. 7C).
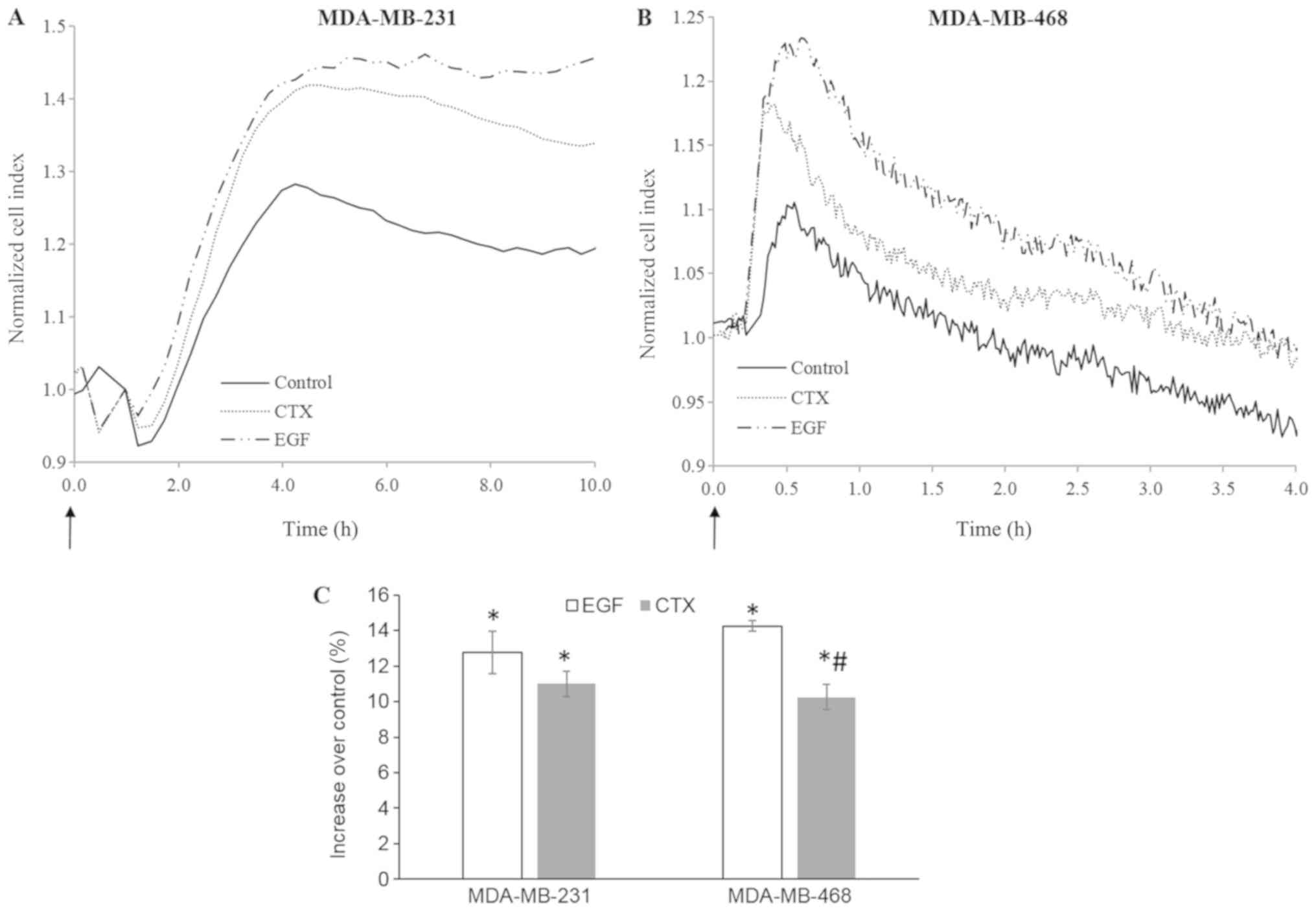
CTX alters cellular morphology in a
manner similar to EGF
Immediate morphological changes induced by growth
factors can be detected using real-time cell-based impedance
measurement (38-40). In this study, cell-based impedance
technology was also used to evaluate the rapid morphological
changes induced by RTK activity triggered by EGF or CTX in the
MDA-MB-231 and MDA-MB-468 cells. Both CTX (100 nM) and EGF (1 nM)
induced significant morphological changes during 4-10 h of cell
stimulation (Fig. 8). These
results indicate that CTX induces RTK activity similar to EGF in
terms of cellular morphology in both the MDA-MB-231 and MDA-MB-468
cells.
Src kinase inhibitor enhances the
CTX-mediated anti-prolife- rative effect
The effect of CTX on cellular proliferation was
examined in the presence of the Src kinase inhibitor, PP2, which
antagonizes the CTX-induced phosphorylation of Src kinase, PI3K,
Akt, IGF-1R and VEGFR-2. CTX alone did not affect the proliferation
of the MDA-MB-231 cells, but induced a 15% inhibition of the
proliferation of the MDA-MB-468 cells (Fig. 9). The Src kinase inhibitor, PP2, at
10 µM induced a 20 and 27% inhibition of MDA-MB-231 and
MDA-MB-468 cell proliferation, respectively. In combination with
CTX, however, this response increased to 38% in the MDA-MB-231
cells and to 45% in the MDA-MB-468 cells (Fig. 9). The results obtained by assessing
cell viability by WST-1 assay (data not shown) and the RTCA system
were similar.
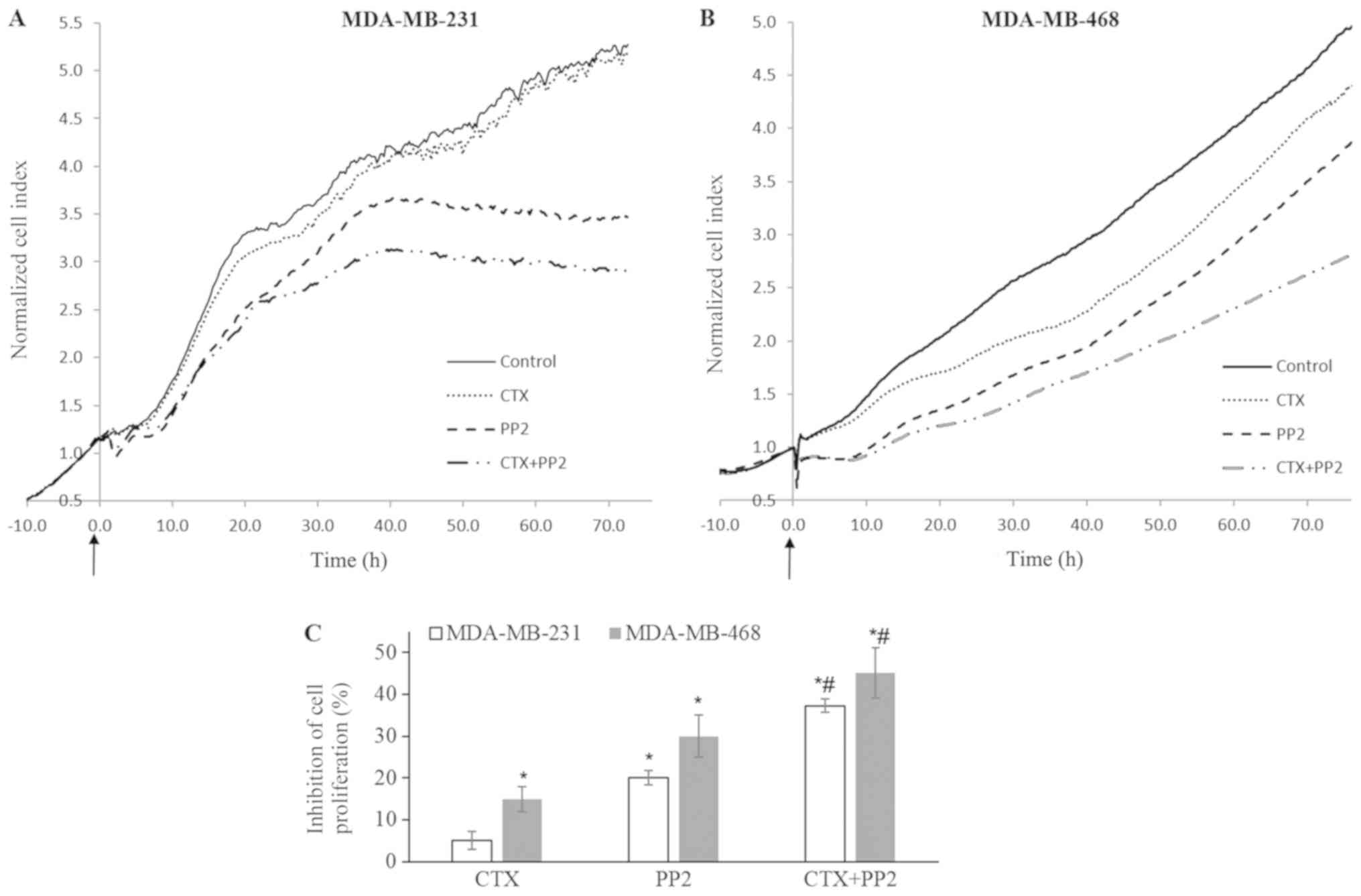 | Figure 9Src kinase inhibition enhances the
cetuximab-mediated anti-proliferative response. (A-C) Impedance
measurement of cell proliferation with the real-time cell
electronic sensing system with cetuximab (100 nM), PP2 (10
µM) and CTX (100 nM) + PP2 (10 µM) treatment of
MDA-MB-231 and MDA-MB-468 cells in medium with FBS (10%). CTX alone
did not alter the proliferation of MDA-MB-231 cells, but it induced
a 15% inhibition of the proliferation of MDA-MB-468 cells. The Src
kinase inhibitor, PP2, at 10 µM induced 20% and 27%
inhibition of cellular proliferation. This response significantly
increased to reach 38% and 45% when combined with CTX in MDA-MB-231
and MDA-MB-468 cells, respectively. The cell index recorded just
before CTX, PP2 or CTX+PP2 treatment was defined as the baseline
and fold change of the baseline was determined for further
measurements and shown as cell index fold change in (A and B). The
arrows indicate when the treatments were started. The cell index
fold change at 72 h for each group and the percentage of
phosphate-buffered saline-treated control cells was determined and
percent inhibition of the cell index obtained from phosphate
buffered saline-treated control cells was presented as a bar graph
in (C). Data are presented as the means ± standard error of the
mean, n=4-5 experiments. *P<0.05 vs. the control
cells; #P<0.05 vs. EGF. CTX, cetuximab; EGF,
epidermal growth factor; EGFR, epidermal growth factor
receptor. |
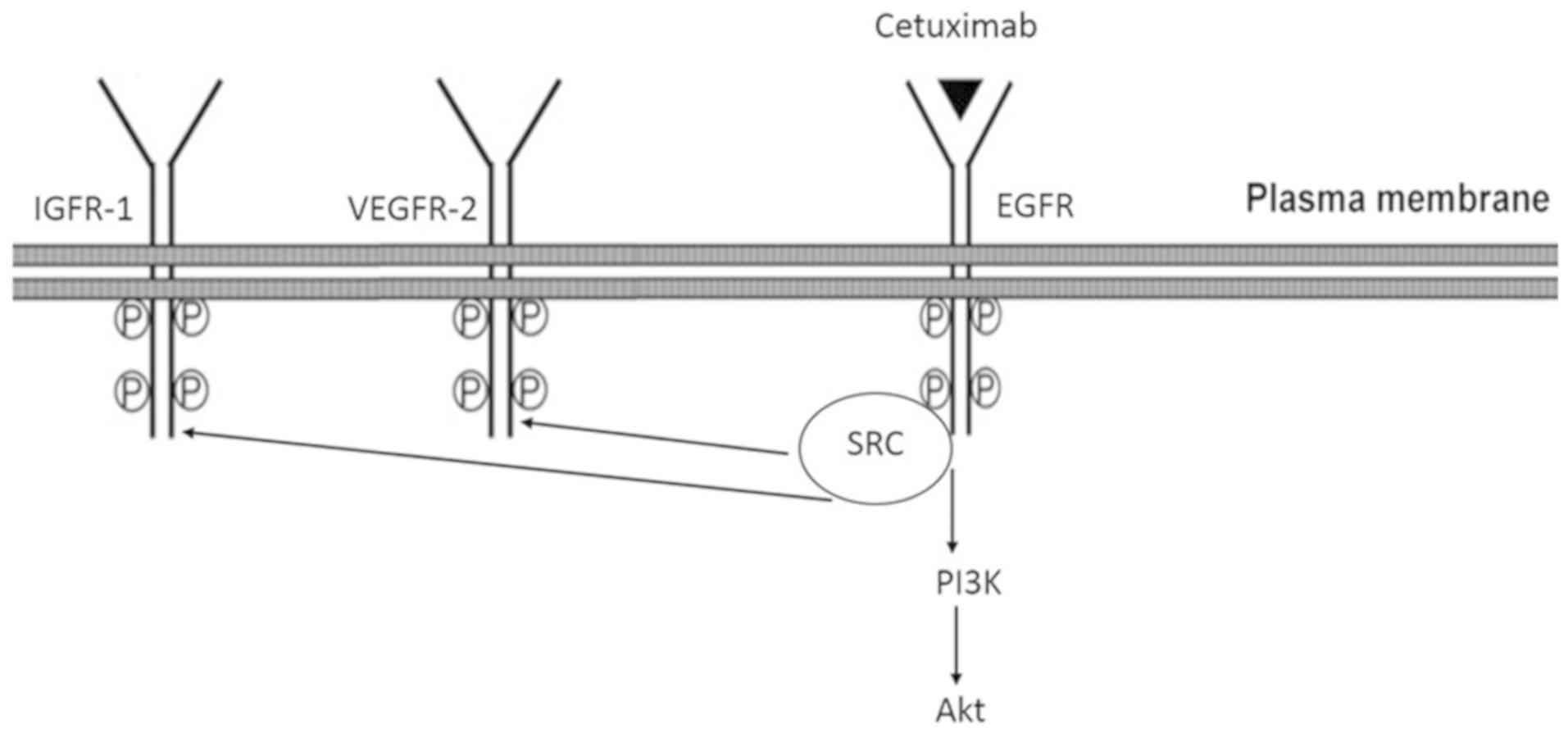
Model of CTX-mediated partial agonistic
action
Based on the observations that the CTX-mediated
phosphorylation of EGFR, IGF-1R and VEGFR-2, PI3K and Akt were
inhibited by the EGFR tyrosine kinase inhibitor, AG1478, and the
Src kinase inhibitor, PP2, it was suggested that the partial
agonistic action of CTX leads to the phosphorylation of EGFR and
Src kinase, which mediates the phosphorylation of IGF-1R, VEGFR-2,
PI3K and Akt (Fig. 10).
Discussion
The blocking and/or silencing of the EGFR signalling
pathway is one of the fundamental treatment strategies for TNBC,
since EGFR overexpression is associated with an aggressive
phenotype and a poor clinical outcome (8-12).
Several antibodies have been developed to shut down EGFR signaling
by blocking the binding of growth factors. However, these
antibodies may not only antagonize the binding of growth factors or
completely shut down EGFR signalling, but may also exert
agonist-like effects or act like partial agonists (24-26).
One of the unavoidable results of this action may be triggering
malignant signalling in cancer cells, similar to growth factors,
which may explain the lack of an anticancer response pattern in
TNBC cells. TNBCs are resistant to treatment with the EGFR
antibody, CTX. The primary focus of the present study was to
determine whether CTX possesses partial agonist-like properties and
triggers the EGFR signalling pathway; we then investigated whether
these properties are partly responsible for TNBC cell resistance to
CTX.
To define an antibody as a partial agonist, it must
exert an agonistic effect upon binding to a receptor. To elucidate
this, we initially determined whether CTX causes the
auto-phosphorylation of EGFR and triggers its downstream signalling
pathway similar to an EGFR agonist. It was observed that the
treatment of MDA-MB-231 and MDA-MB-468 cells with CTX significantly
increased the Tyr1173 phosphorylation of EGFR, which was the first
indication that CTX may function as a partial agonist of EGFR. To
determine whether the CTX-mediated Tyr1173 phosphorylation of EGFR
triggers downstream signalling, the activity status of Src kinase,
PI3K, Akt and ERK1/2 in the CTX-treated cells was then evaluated.
CTX treatment was found to markedly enhance the phosphorylation of
Src kinase, PI3K and Akt in the MDA-MB-231 cells, and the
phosphorylation of Src kinase and PI3K in the MDA-M468 cells.
However, we observed no increase in the phosphorylation of ERK1/2
with CTX treatment in either the MDA-MB-231 or MDA-MB-468 cells.
This may be explained by the high level of ERK1/2 expression in
these cells, together with the high basal phosphorylated ERK1/2
level (41), which may also be
linked to KRAS and BRAF mutations in MDA-MB-231 cells (21). Therefore, CTX cannot further
promote the phosphorylation of ERK1/2 in these cell lines.
Similarly, CTX did not enhance Akt phosphorylation in MDA-MB-468
cells, which is also most likely associated with the high basal
level of phosphorylated Akt in MDA-MB-468 cells, which lack the
functional tumour suppressor Akt-inhibiting gene, PTEN (21,42).
By contrast, EGF, as a full agonist, did enhance ERK1/2 and Akt
phosphorylation in these cell lines (data not shown). These results
indicate that the signal strength must be at the level induced by
the full agonist (EGF) on EGFR in order to observe Akt or ERK1/2
phosphorylation, whereas the weak signal induced by the partial
agonist (CTX), is not sufficient to achieve phosphorylation of
ERK1/2 and Akt in the MDA-MB-468 and ERK1/2 in MDA-MB-231
cells.
Growth factors cause the activation of both their
own specific receptors and different types of growth factor
receptors (34,35). The EGF stimulation of EGFR leads to
the phosphorylation of IGF-1R in an Src kinase-dependent manner
(34). There is also a synergistic
interaction between EGFR and VEGFR-2, while EGF stimulation
increases both the expression and phosphorylation of VEGFR-2
(35), and activation of Src
kinase also mediates transactivation of VEGFR-2 (43). After observing the CTX-mediated
phosphorylation of EGFR and Src kinase, the effect of CTX on IGF-1R
and VEGFR-2 phosphorylation was examined, and it was observed that
CTX enhanced the phosphorylation of both, and that this induced
phosphorylation was inhibited by the EGFR tyrosine kinase
inhibitor, AG1478, and the Src kinase inhibitor, PP2. In our
experiments, although CTX treatment led to an increase in the level
of phosphorylated IGF-1R and VEGFR-2, it caused some decline or no
change in the level of IGR-1 and VEGF-2. After observing this
variability in the total level of IGF-1R or VEGF-2 we did not
measure their level in each experiment. The stimulation or
phosphorylation of tyrosine kinase residues of EGFR, VEGFR-2 and
IGF-1R leads to their internalization and degradation but does not
increase their cellular level (44-47).
We also observed that CTX stimulation significantly decreased the
level of EGFR (Figs. 1Figure 2-3). Taken together, our data suggest that
the CTX-mediated phosphorylation of EGFR, VEGFR-2 and IGF-1R is not
related to an increase in their expression level. Based on these
findings, it is suggested that the CTX-mediated activation of EGFR
tyrosine kinase leads to the phosphorylation of EGFR and triggers
Src kinase activation, which mediates the phosphorylation of IGF-1R
and VEGFR-2 (Fig. 10).
As mentioned above, the stimulation of EGFR by
agonists, such as EGF, causes the rapid internalization and
degradation of the receptor (44).
Based on these and our initial observations indicating the decline
in the level of EGFR with both CTX and EGF treatments, we measured
the EGFR levels following treatment with CTX to further evaluate
the agonist-like action of CTX. It was observed that CTX treatment
led to a decline in EGFR levels in both the MDA-MB-231 and
MDA-MB468 cells. In this response pattern, CTX acted as an agonist,
but did not reduce the EGFR levels to a greater extent than EGF.
Riese (26) proposed that partial
agonist/antagonist antibodies trigger the phosphorylation of the
Tyr residues of RTKs, leading to the degradation and irreversible
inhibition of the receptor. This partial agonistic property of
monoclonal antibodies may be an advantage for EGFR-targeted
anticancer effectiveness. Ferraro et al (48) also suggested an alternative
strategy for inhibiting tumour growth by enhancing the
internalization of EGFR, increasing its degradation and inhibiting
its recycling through the use of EGFR-targeted monoclonal
antibodies. From our results, it may be concluded that although CTX
causes the degradation of EGFR, this is not sufficient to exert a
pronounced anti-proliferative effect on TNBC cells.
Several studies have demonstrated that growth
factors and RTK agonists induce immediate morphological or
behavioural changes that may be detected using real-time cell-based
impedance measurement (38-40).
Thus, in this study, we also measured the effect of CTX on cellular
behaviour or morphology as another approach to evaluating its
partial agonist-like activity. CTX was found to induce significant
morphological changes in the cells, similar to EGF, indicating that
CTX has a partial agonistic action in terms of cellular morphology.
It would also be better to visualize the morphological changes
induced with CTX in these cells by using microscopic techniques in
a future study.
The second important indication to define an
antibody as a partial agonist is that it has a notably lower signal
strength compared with a full agonist. We therefore compared CTX-
and EGF-mediated responses. All the responses induced by CTX,
namely the phosphorylation of EGFR, IGF-1R, VEGFR-2, Akt and PI3K,
and reduced EGFR levels, were significantly less pronounced
compared with those induced by EGF as a full agonist. These results
clearly indicate that CTX can partially, but not fully, activate
EGFR. A third typical indication of a partial agonist is that it
partially inhibits the response induced by a full agonist. Our
results clearly demonstrated that CTX partially, but not fully,
inhibited the EGF-mediated responses.
An important aspect of the results of the present
study is that the partial agonistic action of CTX causes EGFR
phosphorylation on the Tyr1173 residue, leading to the
phosphorylation of Src kinase, PI3K, Akt, IGF-1R and VEGFR-2. All
these signalling molecules are coupled to a malignant phenotype.
Previous research, as well as the present study, have demonstrated
that MDA-MB-231 and MDA-MB-468 cells are resistant to CTX treatment
(33,49,50).
This partial agonistic property may therefore be implicated in the
mechanism underlying the resistance of these cell lines to CTX.
Based on the observation that the inhibition of Src kinase
significantly inhibited the CTX-mediated phosphorylation of PI3K,
Akt, IGF-1R and VEGFR-2, we investigated whether the activation of
Src kinase could partly explain the resistance of these cells to
CTX. For this purpose, the effect of CTX on cellular proliferation
was examined in the presence of the Src kinase inhibitor, PP2, in
the MDA-MB-231 and MDA-MB-468 cells. Indeed, PP2 significantly
enhanced the CTX-mediated anti-proliferative response in these cell
lines. As expected, however, the partial agonistic action of CTX
could not fully explain the resistance observed in these cell
lines, since these cells harbour mutations in KRAS/BRAF and PTEN
that cause EGFR-independent activation of malignant signalling.
Various results reported in the literature also
support our findings that CTX can cause the phosphorylation of
EGFR, IGF-1R and Src kinase in several different cancer cell lines,
which are primarily refractory or have acquired resistance
(30-32,51,52).
In particular, CTX-sensitive cancer cells may become resistant
following CTX treatment (18,53-55).
There is evidence to indicate that EGFR, HER2, HER3, c-Met and
VEGFR-2 are overexpressed, and also that Src kinase and PI3K/Akt
activity increase in acquired CTX resistance (18-20,54,56-58).
CTX can phosphorylate and activate EGFR in non-small cell lung
cancer cells or in head and neck squamous cancer cells (30,31,54).
Yoshida et al (30)
reported that CTX led to the dimerization of EGFR and caused the
phosphorylation of the Tyr845, Tyr1068 and Tyr1173 residues in the
non-small cell lung cancer cells lines, H292 and H460. Accordingly,
it is very important to examine and elucidate a possible partial
agonist action of CTX and its role in acquired resistance of cancer
cells. The CTX-mediated phosphorylation or activation of growth
factor receptors have not been specifically addressed to date as a
mechanism underlying resistance, and this phenomenon has not been
defined as partial agonism. The results of the present study are
novel, as they clearly demonstrated the partial agonist action of
CTX and, therefore, have drawn attention to the possibility that
the partial agonistic action of monoclonal antibodies may be key to
the resistance to EGFR-targeted therapies.
To further elucidate the effectiveness of
EGFR-targeted antibodies in cancer cells exhibiting high EGFR
signalling, it would be valuable to compare the anticancer
effectiveness of antibodies with different properties or
efficacies; one may act like a neutral antagonist on EGFR, thereby
preventing any type of phosphorylation or activation of EGFR or its
downstream signalling molecules, whereas another may act like a
biased agonist that selectively triggers a specific EGFR signalling
pathway. The binding of this type of antibody to EGFR may lead to
the phosphorylation of the Tyr residues that trigger its
ubiquitination and facilitate its degradation, but without the
phosphorylation of the Tyr residues that trigger malignant
signalling. Such an antibody could completely shut down EGFR
signalling. Therefore, it is crucial to understand the therapeutic
effectiveness of EGFR-targeted antibodies with different
properties, such as partial agonists, neutral antagonists and
biased agonists, in the treatment of cancers that retain high EGFR
signalling.
In conclusion, the results of the present study
indicate that CTX exerts a partial agonistic effect on EGFR, which
leads to the phosphorylation of EGFR, Src kinase, PI3K, Akt, IGF-1R
and VEGFR-2, whereas the inhibition of this induced phosphorylation
by the Src kinase inhibitor, PP2, enhances its anti-proliferative
effect. To the best of our knowledge, this study is the first to
emphasize the partial agonist properties of CTX, which are likely
implicated in the mechanisms underlying the resistance of MDA-MB231
and MDA-MB468 cells to CTX. The anticancer effectiveness of CTX
should thus be examined further by blocking its partial agonist
action through Src kinase inhibition in preclinical and clinical
studies.
Funding
The present study was supported by a research grant
from the Scientific and Technological Research Council of Turkey
(TÜBITAK; grant no. SBAG-113S396).
Availability of data and materials
All data generated or analysed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
HG, MMT and SYB participated in the design of the
study. MMT, SYB and BD performed the experiments and generated the
data. HG, MMT, SYB and BD analysed and reviewed the Results and
Discussion. HG and SYB prepared the manuscript and revised it. All
authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Bala Gur Dedeoğlu
(Biotechnology Institute, University of Ankara, Ankara, Turkey) for
her critical reading of the manuscript.
Abbreviations:
|
Akt
|
serine/threonine-specific protein
kinase
|
|
EGFR
|
epidermal growth factor receptor
|
|
ERK1/2
|
extracellular signal-regulated kinase
1/2
|
|
IGF-1R
|
insulin-like growth factor
receptor
|
|
GAPDH
|
glyceraldehyde 3-phosphate
dehydrogenase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
SDS-PAGE
|
sodium dodecyl sulphate-polyacrylamide
gel electrophoresis
|
|
PI3K
|
phosphoinositide-3-kinase
|
|
TNBC
|
triple-negative breast cancer
|
|
VEGFR
|
vascular endothelial growth factor
receptor
|
References
|
1
|
Lewis TS, Shapiro PS and Ahn NG: Signal
transduction through MAP kinase cascades. Adv Cancer Res.
74:49–139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Klapper LN, Kirschbaum MH, Sela M and
Yarden Y: Biochemical and clinical implications of the ErbB/HER
signaling network of growth factor receptors. Adv Cancer Res.
77:25–79. 2000. View Article : Google Scholar
|
|
3
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Downward J, Parker P and Waterfield MD:
Autophosphorylation sites on the epidermal growth factor receptor.
Nature. 311:483–485. 1984. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brenton JD, Carey LA, Ahmed AA and Caldas
C: Molecular classification and molecular forecasting of breast
cancer: Ready for clinical application? J Clin Oncol. 23:7350–7360.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hurvitz S and Mead M: Triple-negative
breast cancer: advancements in characterization and treatment
approach. Curr Opin Obstet Gynecol. 28:59–69. 2016.
|
|
8
|
DiGiovanna MP, Stern DF, Edgerton SM,
Whalen SG, Moore D II and Thor AD: Relationship of epidermal growth
factor receptor expression to ErbB-2 signaling activity and
prognosis in breast cancer patients. J Clin Oncol. 23:1152–1160.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bhargava R, Gerald WL, Li AR, Pan Q, Lal
P, Ladanyi M and Chen B: EGFR gene amplification in breast cancer:
Correlation with epidermal growth factor receptor mRNA and protein
expression and HER-2 status and absence of EGFR-activating
mutations. Mod Pathol. 18:1027–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rimawi MF, Shetty PB, Weiss HL, Schiff R,
Osborne CK, Chamness GC and Elledge RM: Epidermal growth factor
receptor expression in breast cancer association with biologic
phenotype and clinical outcomes. Cancer. 116:1234–1242. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Burness ML, Grushko TA and Olopade OI:
Epidermal growth factor receptor in triple-negative and basal-like
breast cancer: Promising clinical target or only a marker? Cancer
J. 16:23–32. 2010. View Article : Google Scholar
|
|
12
|
Nielsen TO, Hsu FD, Jensen K, Cheang M,
Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler
L, et al: Immunohistochemical and clinical characterization of the
basal-like subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sunada H, Magun BE, Mendelsohn J and
MacLeod CL: Monoclonal antibody against epidermal growth factor
receptor is internalized without stimulating receptor
phosphorylation. Proc Natl Acad Sci USA. 83:3825–3829. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vincenzi B, Zoccoli A, Pantano F, Venditti
O and Galluzzo S: Cetuximab: From bench to bedside. Curr Cancer
Drug Targets. 10:80–95. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kawamoto T, Sato JD, Le A, Polikoff J,
Sato GH and Mendelsohn J: Growth stimulation of A431 cells by
epidermal growth factor: Identification of high-affinity receptors
for epidermal growth factor by an anti-receptor monoclonal
antibody. Proc Natl Acad Sci USA. 80:1337–1341. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ciardiello F and Tortora G: EGFR
antagonists in cancer treatment. N Engl J Med. 358:1160–1174. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carey LA, Rugo HS, Marcom PK, Mayer EL,
Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A,
et al: TBCRC 001: Randomized phase II study of cetuximab in
combination with carboplatin in stage IV triple-negative breast
cancer. J Clin Oncol. 30:2615–2623. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brand TM, Iida M and Wheeler DL: Molecular
mechanisms of resistance to the EGFR monoclonal antibody cetuximab.
Cancer Biol Ther. 11:777–792. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bardelli A and Siena S: Molecular
mechanisms of resistance to cetuximab and panitumumab in colorectal
cancer. J Clin Oncol. 28:1254–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sforza V, Martinelli E, Ciardiello F,
Gambardella V, Napolitano S, Martini G, Della Corte C, Cardone C,
Ferrara ML, Reginelli A, et al: Mechanisms of resistance to
anti-epidermal growth factor receptor inhibitors in metastatic
colorectal cancer. World J Gastroenterol. 22:6345–6361. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hollestelle A, Elstrodt F, Nagel JHA,
Kallemeijn WW and Schutte M: Phosphatidylinositol-3-OH kinase or
RAS pathway mutations in human breast cancer cell lines. Mol Cancer
Res. 5:195–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hsu HC, Thiam TK, Lu YJ, Yeh CY, Tsai WS,
You JF, Hung HY, Tsai CN, Hsu A, Chen HC, et al: Mutations of
KRAS/NRAS/BRAF predict cetuximab resistance in metastatic
colorectal cancer patients. Oncotarget. 7:22257–22270. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Macdonald-Obermann JL and Pike LJ:
Different epidermal growth factor (EGF) receptor ligands show
distinct kinetics and biased or partial agonism for homodimer and
heterodimer formation. J Biol Chem. 289:26178–26188. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Prat M, Oltolina F and Basilico C:
Monoclonal Antibodies against the MET/HGF Receptor and Its Ligand:
Multitask Tools with Applications from Basic Research to Therapy.
Biomedicines. 2:359–383. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deb TB, Zuo AH, Barndt RJ, Sengupta S,
Jankovic R and Johnson MD: Pnck overexpression in HER-2
gene-amplified breast cancer causes Trastuzumab resistance through
a paradoxical PTEN-mediated process. Breast Cancer Res Treat.
150:347–361. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Riese DJ II: Ligand-based receptor
tyrosine kinase partial agonists: New paradigm for cancer drug
discovery? Expert Opin Drug Discov. 6:185–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Scott GK, Dodson JM, Montgomery PA,
Johnson RM, Sarup JC, Wong WL, Ullrich A, Shepard HM and Benz CC:
p185HER2 signal transduction in breast cancer cells. J Biol Chem.
266:14300–14305. 1991.PubMed/NCBI
|
|
28
|
Nagata Y, Lan K-H, Zhou X, Tan M, Esteva
FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al: PTEN
activation contributes to tumor inhibition by trastuzumab, and loss
of PTEN predicts trastuzumab resistance in patients. Cancer Cell.
6:117–127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Esteva FJ, Yu D, Hung M-C and Hortobagyi
GN: Molecular predictors of response to trastuzumab and lapatinib
in breast cancer. Nat Rev Clin Oncol. 7:98–107. 2010. View Article : Google Scholar
|
|
30
|
Yoshida T, Okamoto I, Okabe T, Iwasa T,
Satoh T, Nishio K, Fukuoka M and Nakagawa K: Matuzumab and
cetuximab activate the epidermal growth factor receptor but fail to
trigger downstream signaling by Akt or Erk. Int J Cancer.
122:1530–1538. 2008. View Article : Google Scholar
|
|
31
|
Raben D, Helfrich B, Chan DC, Ciardiello
F, Zhao L, Franklin W, Barón AE, Zeng C, Johnson TK and Bunn PA Jr:
The effects of cetuximab alone and in combination with radiation
and/or chemotherapy in lung cancer. Clin Cancer Res. 11:795–805.
2005.PubMed/NCBI
|
|
32
|
Molli PR, Adam L and Kumar R: Therapeutic
IMC-C225 antibody inhibits breast cancer cell invasiveness via
Vav2-dependent activation of RhoA GTPase. Clin Cancer Res.
14:6161–6170. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
El Guerrab A, Bamdad M, Bignon YJ,
Penault-Llorca F and Aubel C: Anti-EGFR monoclonal antibodies
enhance sensitivity to DNA-damaging agents in BRCA1-mutated and
PTEN-wild-type triple-negative breast cancer cells. Mol Carcinog.
56:1383–1394. 2017. View Article : Google Scholar
|
|
34
|
Hallak H, Moehren G, Tang J, Kaou M, Addas
M, Hoek JB and Rubin R: Epidermal growth factor-induced activation
of the insulin-like growth factor I receptor in rat hepatocytes.
Hepatology. 36:1509–1518. 2002.PubMed/NCBI
|
|
35
|
Saryeddine L, Zibara K, Kassem N, Badran B
and El-Zein N: EGF-Induced VEGF Exerts a PI3K-Dependent Positive
Feedback on ERK and AKT through VEGFR2 in Hematological In Vitro
Models. PLoS One. 11:e01658762016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tuglu MM, Bostanabad SY, Ozyon G, Dalkiliç
B and Gurdal H: The role of dual specificity phosphatase 1 and
protein phosphatase 1 in β2 adrenergic receptor mediated
inhibition of extracellular signal regulated kinase 1/2 in triple
negative breast cancer cell lines. Mol Med Rep. 17:2033–2043.
2018.
|
|
37
|
Solly K, Wang X, Xu X, Strulovici B and
Zheng W: Application of real-time cell electronic sensing (RT-CES)
technology to cell-based assays. Assay Drug Dev Technol. 2:363–372.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Atienza JM, Yu N, Wang X, Xu X and Abassi
Y: Label-free and real-time cell-based kinase assay for screening
selective and potent receptor tyrosine kinase inhibitors using
microelectronic sensor array. J Biomol Screen. 11:634–643. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dynamic Monitoring of Receptor Tyrosine
Kinase Activation in Living Cells Dynamic Monitoring of Receptor
Tyrosine Kinase Activation in Living Cells. ACEA Biosciences, Inc.;
San Diego, CA: 2013
|
|
40
|
Chinkers M, McKanna JA and Cohen S: Rapid
induction of morphological changes in human carcinoma cells A-431
by epidermal growth factors. J Cell Biol. 83:260–265. 1979.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bartholomeusz C, Gonzalez-Angulo AM, Liu
P, Hayashi N, Lluch A, Ferrer-Lozano J and Hortobágyi GN: High ERK
protein expression levels correlate with shorter survival in
triple-negative breast cancer patients. Oncologist. 17:766–774.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Stemke-hale K, Gonzalez-Angulo AM, Lluch
A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, et
al: An integrative genomic and proteomic analysis of PIK3CA, PTEN,
and AKT mutations in breast cancer. Cancer Res. 68:6084–6092. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Petreaca ML, Yao M, Liu Y, Defea K and
Martins-Green M: Transactivation of vascular endothelial growth
factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for
IL-8/CXCL8-induced endothelial permeability. Mol Biol Cell.
18:5014–5023. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vieira AV, Lamaze C and Schmid SL: Control
of EGF receptor signaling by clathrin-mediated endocytosis.
Science. 274:2086–2089. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Carpentier JL: Insulin receptor
internalization: Molecular mechanisms and physiopathological
implications. Diabetologia. 37(Suppl 2): S117–S124. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bruns AF, Herbert SP, Odell AF, Jopling
HM, Hooper NM, Zachary IC, Walker JH and Ponnambalam S:
Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated
trafficking and proteolysis. Traffic. 11:161–174. 2010. View Article : Google Scholar
|
|
47
|
Chow JC, Condorelli G and Smith RJ:
Insulin-like growth factor-I receptor internalization regulates
signaling via the Shc/mitogen-activated protein kinase pathway, but
not the insulin receptor substrate-1 pathway. J Biol Chem.
273:4672–4680. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ferraro DA, Gaborit N, Maron R,
Cohen-Dvashi H, Porat Z, Pareja F, Lavi S, Lindzen M, Ben-Chetrit
N, Sela M, et al: Inhibition of triple-negative breast cancer
models by combinations of antibodies to EGFR. Proc Natl Acad Sci
USA. 110:1815–1820. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
El Guerrab A, Bamdad M, Kwiatkowski F,
Bignon YJ, Penault-Llorca F and Aubel C: Anti-EGFR monoclonal
antibodies and EGFR tyrosine kinase inhibitors as combination
therapy for triple-negative breast cancer. Oncotarget.
7:73618–73637. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sohn J, Liu S, Parinyanitikul N, Lee J,
Hortobagyi GN, Mills GB, Ueno NT and Gonzalez-Angulo AM: cMET
activation and EGFR-directed therapy resistance in triple-negative
breast cancer. J Cancer. 5:745–753. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li X, Xu L, Li H, Zhao L, Luo Y, Zhu Z,
Liu Y and Qu X: Cetuximab-induced insulin-like growth factor
receptor I activation mediates cetuximab resistance in gastric
cancer cells. Mol Med Rep. 11:4547–4554. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Rebucci M, Peixoto P, Dewitte A, Wattez N,
De Nuncques MA, Rezvoy N, Vautravers-Dewas C, Buisine MP, Guerin E,
Peyrat JP, et al: Mechanisms underlying resistance to cetuximab in
the HNSCC cell line: Role of AKT inhibition in bypassing this
resistance. Int J Oncol. 38:189–200. 2011.
|
|
53
|
Troiani T, Napolitano S, Vitagliano D,
Morgillo F, Capasso A, Sforza V, Nappi A, Ciardiello D, Ciardiello
F and Martinelli E: Primary and acquired resistance of colorectal
cancer cells to anti-EGFR antibodies converge on MEK/ERK pathway
activation and can be overcome by combined MEK/EGFR inhibition.
Clin Cancer Res. 20:3775–3786. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bianco R, Rosa R, Damiano V, Daniele G,
Gelardi T, Garofalo S, Tarallo V, De Falco S, Melisi D, Benelli R,
et al: Vascular endothelial growth factor receptor-1 contributes to
resistance to anti-epidermal growth factor receptor drugs in human
cancer cells. Clin Cancer Res. 14:5069–5080. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Leto SM and Trusolino L: Primary and
acquired resistance to EGFR-targeted therapies in colorectal
cancer: Impact on future treatment strategies. J Mol Med (Berl).
92:709–722. 2014. View Article : Google Scholar
|
|
56
|
Wheeler DL, Huang S, Kruser TJ,
Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT and
Harari PM: Mechanisms of acquired resistance to cetuximab: Role of
HER (ErbB) family members. Oncogene. 27:3944–3956. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Iida M, Brand TM, Campbell DA, Starr MM,
Luthar N, Traynor AM and Wheeler DL: Targeting AKT with the
allosteric AKT inhibitor MK-2206 in non-small cell lung cancer
cells with acquired resistance to cetuximab. Cancer Biol Ther.
14:481–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kim SM, Kim JS, Kim JH, Yun CO, Kim EM,
Kim HK, Solca F, Choi SY and Cho BC: Acquired resistance to
cetuximab is mediated by increased PTEN instability and leads
cross-resistance to gefitinib in HCC827 NSCLC cells. Cancer Lett.
296:150–159. 2010. View Article : Google Scholar : PubMed/NCBI
|