Introduction
Hepatoblastoma (HB) is the most common type of liver
tumor in children worldwide and mainly affects infants and toddlers
between the ages of 6 months and 3 years (1). The characteristically early
manifestation of these embryonal tumors suggests that comparatively
few genetic alterations are necessary for the formation of a
malignant phenotype (2). In fact,
systematic analysis of HB by exome sequencing revealed mutations in
the β-catenin (CTNNB1) gene as the only recurrent
alteration, which occurs in approximately two-thirds of all
patients (3,4). The oncogenic mutation of
CTNNB1 is known to trigger the pathological activation of
the Wnt signaling pathway, which can also occur in
CTNNB1-wildtype HB via mutations in the genes, adenomatous
polyposis coli, axin 1 and axin 2 (5,6).
Notably, the activation of mutant Ctnnb1 in liver progenitor
cells has been shown to cause the development of HB in mice by 26
weeks of age (7). However,
although Wnt activation has been proven to drive the development of
pediatric liver tumors, identification of other molecular
mechanisms that are responsible for the manifestation of different
histopathological subtypes of HB and more importantly the varying
response to chemotherapy and thus the outcome is still warranted
(8).
The use of genome-wide expression analysis methods
has not only helped to improve our understanding of the molecular
mechanisms that drive tumorigenesis but, by identifying
subtype-specific changes, fuels the hope of achieving the
biomarker-based stratification of tumors, and thereby improve
prognosis estimation and risk-adapted therapies (9). A first step in this direction in the
field of HB has been the description of a unique gene signature
which allows for, based on an expression determination of only 16
genes, the classification of HB into 2 tumor subtypes (10). These subtypes not only reflect the
different phases of liver development, but also show a strong
prognostic divergence (10). Thus,
tumors of the so-called C1 subtype exhibit a higher degree of
differentiation with mostly fetal histology and a better prognosis,
while the C2 tumors usually have an embryonic differentiation with
a poorer prognosis and a higher tumor stage. One of the genes with
a significant differential expression between C1 and C2 tumor
subtypes was MYCN proto-oncogene basic-helix-loop-helix
transcription factor (MYCN), which maps to 2p24.1, a
chromosomal region known to be frequently duplicated in HB
(11). Upregulation of MYCN
expression serves a crucial role in several tumors, such as lung
and breast cancer as well as malignancies of neural origin
including glioblastoma, medulloblastoma and neuroblastoma (12). In neuroblastoma, transcriptional
upregulation is mainly caused by the amplification of the
MYCN locus (13).
MYCN encodes a nuclear phosphoprotein that dimerizes with
MYC interacting protein X to form a complex that binds to the
regulatory regions of MYCN-regulated genes (14). It is generally believed that
aberrant MYCN expression increases cellular proliferation by
inducing cell-cycle progression as well as inhibiting apoptosis
(15). Taken together, these
findings qualify MYCN as an interesting target for the treatment of
HB.
Materials and methods
Patients and tumor cell lines
A total of 61 liver tumor specimens [47 HB, 4
transitional liver cell tumors (TLCT), and 10 pediatric
hepatocellular carcinomas (HCC)] were obtained from 61 pediatric
patients (32 males, 29 females) undergoing surgical resection in
the Department of Pediatric Surgery (University Hospital, LMU
Munich, Munich, Germany). The inclusion criteria were that the
patients were aged <20 years and had a histologically proven
diagnosis of HB, TLCT or HCC. Patients with underlying liver
disease were excluded from the study. The clinicopathological
features of the patients are presented in Table I. Written informed consent was
obtained from each patient or the parents if younger than 12 years
and the study protocol was approved by the
Ludwig-Maximilians-University Ethics Committee (Munich, Germany;
no. 431-11). Furthermore, 3 human HB cell lines were used,
including HepT1 (provided by Dr. Torsten Pietsch, Institute of
Neuropathology, University of Bonn, Bonn, Germany), HUH6 (Japanese
Collection of Research Bioresources, Osaka, Japan) and HepG2
(American Type Culture Collection, Manassas, VA, USA) as well as
the hepatocellular carcinoma cell line HUH7 (kindly provided by Dr.
Enrico de Toni, Department of Medicine 2, University of Munich,
Munich, Germany). Cells were grown at 37°C in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% fetal calf serum (FCS; Gibco; Thermo Fisher
Scientific, Inc.), 1% antibiotics and glutamine supplement.
 | Table IClinicopathological features of
different types of pediatric liver cancers and their association
with MYCN and MYCNOS gene expression. |
Table I
Clinicopathological features of
different types of pediatric liver cancers and their association
with MYCN and MYCNOS gene expression.
| Category | Patients with
pediatric liver cancer, n
| P-value
|
|---|
| HB (n=47) | TLCT (n=4) | HCC (n=10)a | MYCN | MYCNOS |
|---|
| Age range
(months) | 0-56 | 11-128 | 92-199 | 0.8414 | 0.3881 |
| Metastasis | | | | 0.9195 | 0.5297 |
| No | 30 | 3 | 7 | | |
| Yes | 17 | 1 | 2 | | |
| Vascular
invasion | | | | 0.4371 | 0.5526 |
| No | 38 | 3 | 7 | | |
| Yes | 9 | 1 | 2 | | |
| Multifocal
growth | | | | 0.7421 | 0.8147 |
| No | 35 | 2 | 6 | | |
| Yes | 12 | 2 | 2 | | |
| Extrahepatic
growth | | | | 0.8020 | 0.8270 |
| No | 44 | 4 | 6 | | |
| Yes | 3 | 0 | 2 | | |
| PRETEXT stage | | | | 0.7402 | 0.5787 |
| I | 3 | 0 | 2 | | |
| II | 12 | 2 | 2 | | |
| III | 21 | 1 | 3 | | |
| IV | 11 | 1 | 0 | | |
| Embryonal
histology | | | | 0.0166 | 0.0130 |
| No | 34 | 4 | n.a. | | |
| Yes | 13 | 0 | n.a. | | |
| Outcome | | | | 0.2936 | 0.8646 |
| Alive | 40 | 3 | 8 | | |
| Succumbed | 7 | 1 | 2 | | |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA extraction and purification of tissues and cells
as well as cDNA synthesis were performed as described previously
(16). Quantification of gene
expression was conducted using TaqMan®Gene expression
assays (Applied Biosystems; Thermo Fisher Scientific, Inc.) for
MYCN (Hs00232074_m1), MYCNOS (Hs01040745_m1) and the
house-keeping gene TATA-box binding protein (TBP;
Hs00427620_m1) as well as the TaqMan universal MasterMix II
(Applied Biosystems; Thermo Fisher Scientific, Inc.) according to
the manufacturer’s instructions. Primer sequences are commercially
unavailable (Applied Biosystems; Thermo Fisher Scientific, Inc.).
RT-qPCR amplifications were conducted in doublets on a Mastercycler
Realplex2 instrument (Eppendorf, Hamburg, Germany) and
consisted of 2 min uracil-N-glycosylase incubation at 50°C, 10 min
DNA polymerase activation at 95°C, and 45 cycles of 15 sec
denaturation at 95°C, and primer annealing and extension for 1 min
at 60°C. The 2−∆∆Cq method was used to calculate the
relative mRNA expression levels from the means of
MYCN/MYNCOS and TBP (17).
Immunohistochemistry
Frozen tumor tissue sections were embedded in
Tissue-Tek Optimal Cutting Temperature compound (Sakura Finetek
USA, Inc., Torrance, CA, USA; 5 µm-thick), were fixed for 10
min in ice-cold acetone and endogenous peroxidase activity was
quenched using 0.3% hydrogen peroxide in bi-distilled water for 10
min. Following 1 h of blocking with 10% FCS at 37°C, the mouse
anti-human N-MYC antibody clone B8.4.B (cat. no. sc-53993; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) diluted to 1:200 in PBS
was applied to the sections, which were then incubated overnight at
4°C. Following three washes with PBS, the sections were then
covered with one drop of anti-mouse immunoglobulin G (IgG; ImmPRESS
HRP reagent kit; Vector Laboratories, Inc.; Maravai LifeSciences,
San Diego, CA, USA) and incubated for 30 min at room temperature.
Signal detection was conducted using the liquid
3,3′-diaminobenzidine substrate chromogen system (Dako; Agilent
Technologies, Inc., Santa Clara, CA, USA) for 30 min at 37°C.
Sections were then counterstained with hematoxylin for 5 min at
37°C and mounted with glycergel mounting medium (Dako; Agilent
Technologies, Inc.). Consecutive sections of the same tumor tissue
were then stained with hematoxylin and eosin without prior
immunohistochemical detection and used for histopathological
evaluation on an Axioplan light microscope (Zeiss GmbH, Jena,
Germany) at a magnification of ×100 and ×200.
Proliferation assays
Cell proliferation was assessed using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assays. Cells (of all cell lines) were seeded at a density of
5-10×103 cells per well into 96-well plates (NUNC,
Langenselbold, Germany) in 100 µl cell culture medium.
Following overnight attachment, cells were treated for 48 h at 37°C
with various concentrations (1, 10, 100 nM, 1 and 10 µM) of
MLN8237 (Alisertib; Axon Medchem, Groningen, Netherlands) and JQ1
(BIOMOL International; Enzo Life Sciences, Inc., Farmingdale, NY,
USA) or the solvent DMSO (Merck KGaA). Cell lines were
alternatively transfected with 200 nM small interfering (si)-RNA
Hs_MYCN_6 FlexiTube siRNA (SI03087518; 5′-CGTGCCGGAGTTGGTAAAGAA-3′;
Qiagen GmbH, Hilden, Germany) or 200 nM siGENOME non-targeting
siRNA #1 (GE Healthcare Dharmacon, Inc., Lafayette, CO, USA;
sequences are commercially unavailable) by electroporation and then
directly cultured at a density of 5-10×103 cells in
96-well plates overnight at 37°C prior to subsequent
experimentation.
To assess cell viability, 10 µl MTT
(Sigma-Aldrich; Dramstadt, Germany) labeling agent (5 mg/ml in PBS)
was added to each well prior to 4 h incubation at 37°C.
Media-containing wells without cells were used for background
estimation. For cell lysis, 100 µl of the SDS-HCl solution
(10% SDS/0.01M HCl) was added to each well. The plate was incubated
overnight at 37°C. Cell viability was detected by measuring the
optical density at a wavelength of 595 nm using the GENios multi
scanner microplate reader (Tecan Group, Ltd., Männedorf,
Switzerland). Each experiment was performed in duplicate.
Cell screening assay
Morphological changes were detected by means of
microscopy once the cells had been treated for 48 h at 37°C with
vehicle (DMSO), 10 µM MLN8237 (in HepG2 cells only) or 1.0
µM MLN8237 (in HepT1, HUH6 and HUH7 cells), or 10 µM
JQ1 (in HepT1 and HepG2 cells) or 0.5 µM JQ1 (in HUH6 and
HUH7 cells) using an inverted Axiovert 40 CFL microscope (Zeiss
GmbH) equipped with a Powershot G6 digital device (Canon, Inc.,
Tokyo, Japan).
Western blot analysis
Cells were seeded at a density of 1×106
per 10 cm cell culture dish. Following overnight attachment, cells
were treated for 48 h at 37°C with MLN8237, JQ1 (both at 10
µM for HepT1 and HepG2, and 0.5 µM for HUH6 and HUH7)
or DMSO. Once treated, non-adherent and adherent cells were pooled
together in ice-cold lysis buffer [0.5% Triton X-100, 1 mM sodium
orthovanadate and cOmplete Mini protease inhibitor (Roche
Diagnostics, Basel, Switzerland)]. Protein lysates were incubated
on ice for 30 min with occasional vortexing. Following
centrifugation for 30 min at 4°C and 13,000 × g, protein lysates
without cell debris were stored at 4°C until use. The protein
concentration was determined by a Bio-Rad Protein Assay (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Proteins (20 µg)
were loaded on a 4-12% BIS TRIS NuPage Gel (Novex; Thermo Fisher
Scientific, Inc.), separated under reducing conditions and then
transferred to a nitrocellulose blotting membrane (GE Healthcare
Life Sciences, Little Chalfont, UK). Thereafter, membranes were
blocked with PBS/0.1% Tween-20/5% non-fat dry milk overnight at
4°C. Next, antibodies including mouse anti-human N-MYC clone B8.4.B
(1:500; cat. no. sc-53993; Santa Cruz Biotechnology, Inc.), rabbit
anti-human poly(adenosine diphosphate-ribose) polymerase (PARP;
1:1,000; cat. no. 9542) or rabbit anti-human β-actin (1:2,500; cat.
no. 4967S; both Cell Signaling Technology, Inc., Danvers, MA, USA)
were added to the cells overnight at 4°C. For detection, membranes
were incubated for 1 h at room temperature with horseradish
peroxidase-conjugated polyclonal goat anti-rabbit immunoglobulin
secondary antibody (1:2,000; cat. no. PO448; Dako; Agilent
Technologies, Inc.). Signals were captured using the enhanced
western blotting reagent detection system and Hyperfilm high
performance autoradiography films (both GE Healthcare, Chicago, IL,
USA).
Apoptosis and cell cycle analysis
Cells (all cell lines) were seeded in 6-well plates
and following 24 h, exposed to DMSO, MLN8237 or JQ1 at various
concentrations (10 µM for HepG2, and 1 µM for HepT1,
HUH6 and HUH7) for 48 h at 37°C. Fixation and permeabilization of
cells were performed by the dropwise addition of 70% ethanol while
vortexing and incubating at −20°C for at least 2 h. Permeabilized
cells were washed with PBS and DNA was stained using 0.02 mg/ml
propidium iodide (Sigma-Aldrich; Merck KGaA) and 0.2 mg/ml RNaseA
(Qiagen, Inc., Valencia, CA, USA) in PBS/0.1% Triton X-100
(Sigma-Aldrich; Merck KGaA) for 30 min at room temperature in the
dark. The cell cycle was analyzed with a BD-LSRFORTESSA flow
cytometer (BD Biosciences; Becton, Dickinson and Company, Franklin
Lakes, NJ, USA) and Flowing software 2.5.1 (www.flowing-software.com/).
Immunofluorescence staining
A total of 7.5×104 cells (all cell lines)
were plated onto Lab-Tek II Chamber Slides (Thermo Fisher
Scientific, Inc.) with a diameter of 18 mm and then incubated in
12-well plates overnight at 37°C prior to treatment. Following 24 h
of treatment with MLN8237 or DMSO at the indicated concentrations
(10 µM for HepG2, and 1 µM for HepT1, HUH6 and HUH7),
cells were fixed in 4% formaldehyde-phosphate-buffered saline for
15 min at room temperature, permeabilized for 15 min with 0.15%
Triton X-100 in PBS at room temperature and blocked for 30 min with
1% bovine serum albumin (BSA; no. 8076.1; Carl Roth GmbH + Co., KG,
Karlsruhe, Germany) in PBS at room temperature. Cells were then
incubated with rat-anti-human α-tubulin (cat. no. CBL270; EMD
Millipore, Billerica, MA, USA) 1:100 diluted in 1% (v/v) BSA in PBS
overnight at 4°C in a wet chamber. Following several washing steps
with PBS, cells were permeabilized for 10 min with 0.15% Triton
X-100 in PBS at room temperature and blocked for 7 min at room
temperature in 1% (v/v) BSA in PBS. Cells were then incubated for
45 min in the dark with Alexa Fluor 555 Goat anti-rat IgG (H+L)
(Life technologies, Carlsbad, CA, USA) at a dilution of 1:200.
After three rinses with PBS, microscope slides were mounted with
Vectashield containing 4,6-diamidino-2-pheylindole (DAPI) (Vector
Laboratories Inc., Burlingame, CA, USA). Images were acquired using
the Olympus FluoView™ FV1000 confocal microscope.
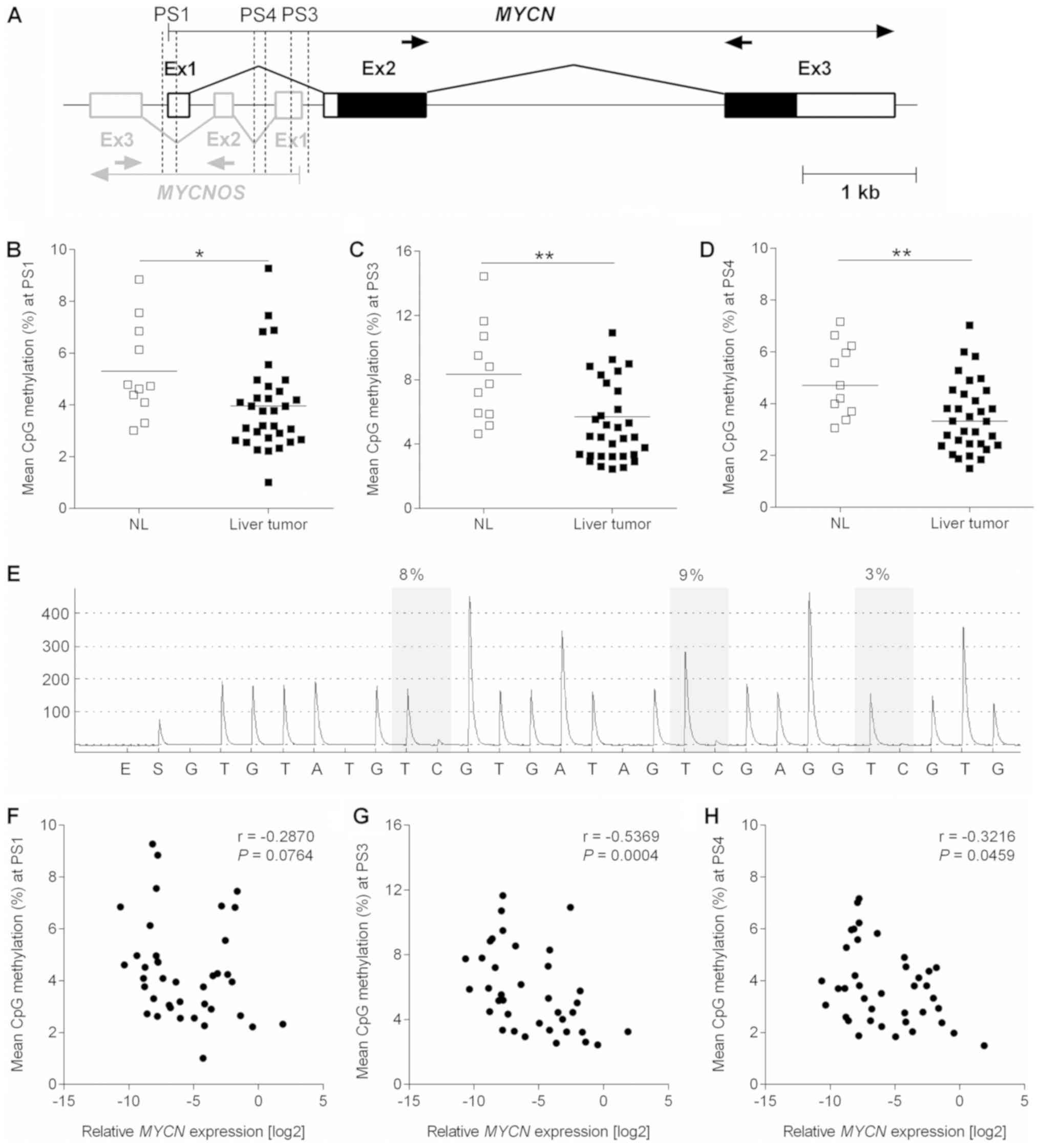
Bisulfite pyrosequencing
The methylation status of 3 CpG-rich regions at the
MYCN/MYCNOS locus was determined via bisulfite
pyrosequencing. Briefly, the genomic DNA of 30 HB and 11 normal
liver tissues was bisulfite modified using an Epitec Bisulfite kit
(Qiagen, Inc.) according to the manufacturer’s instructions.
Subsequent PCR amplification was performed using the following
forward (F) and reverse (R) pyrosequencing primers (PS) designed
with PyroMark Assay Design software (Qiagen, Inc.): PS1-F,
biotin-GGTGTGTTA-GATTTTTTAGTTAAT and PS1-R, ACAAACCCTACT CCTTACCT;
PS3-F, GAGAGTTAGAATTTTGTAGTTAGG AATAGT and PS3-R,
biotin-TCCCCCCCTCCTTTTATATACAAACTATCT; and PS4-F,
AGTTTTTAATTAAGTTATTGGTAGGAGTAT and PS4-R,
biotin-AAACACCCATATCCACAAATCC. Pyrosequencing was performed with
the sequencing primers PS1-S (ATATCCTCCAATAACTAC AATCTAT), PS3-S
(AATGGTAGTTTTTAAAGTT) and PS4-S (AGTTATTGGTAGGAGTATTTT), and data
analysis was performed with the PyroMark Q24 system (Qiagen, Inc.)
following the manufacturer’s instructions.
Statistical analyses
Data were expressed as the mean ± standard
deviation, and statistical analyses were performed with the
Student’s unpaired or paired t-test, the Spearman’s r correlation
test and the Mann-Whitney-U test. Grouped analyses were performed
by employing non-parametric one-way analysis of variance (ANOVA)
with a Dunn’s correction for multiple comparisons or two-way ANOVA
with a Bonferroni post hoc test. P<0.05 was considered to
indicate a statistically significant difference. GraphPad Prism 6
biostatistics software (GraphPad Software, Inc., La Jolla, CA, USA)
was used for all statistical analyses.
Results
MYCN is upregulated in pediatric liver
tumors due to the transcriptional activity of its antisense
transcript, MYCNOS
MYCN expression was initially measured via
RT-qPCR at the mRNA level in a comprehensive tumor collection
consisting of 47 primary HB, 4 TLCT, 10 pediatric HCC, 4 liver
tumor cell lines and 16 non-tumorous liver samples. The results
revealed a significant upregulation of MYCN expression in
all tumor samples and cell lines, when compared with normal liver
tissue (Fig. 1A). Notably, the
marked MYCN upregulation gradually increased across the
samples, from HCC (16.2-fold) to TLCT (57.3-fold) and finally to HB
(87.4-fold). In addition, histological (Fig. 1D) and immunohistochemical staining
for MYCN protein (Fig. 1E) in HB
tissues demonstrated that MYCN overexpression originated from tumor
cells. As pediatric liver cancers lack amplifications at the
MYCN locus (3), the present
study next investigated if the antisense transcript MYCNOS,
which maps to the same region on the opposite strand (Fig. 2A) and has been reported to
positively regulate MYCN transcription (18), is associated with MYCN
overexpression. The results revealed a significant overexpression
of MYCNOS in all tumor samples, again with highest
upregulation in HB tissues (Fig.
1B). Consistent with these results, a highly significant
correlation between MYCN and MYCNOS mRNA levels was
detected (Fig. 1C).
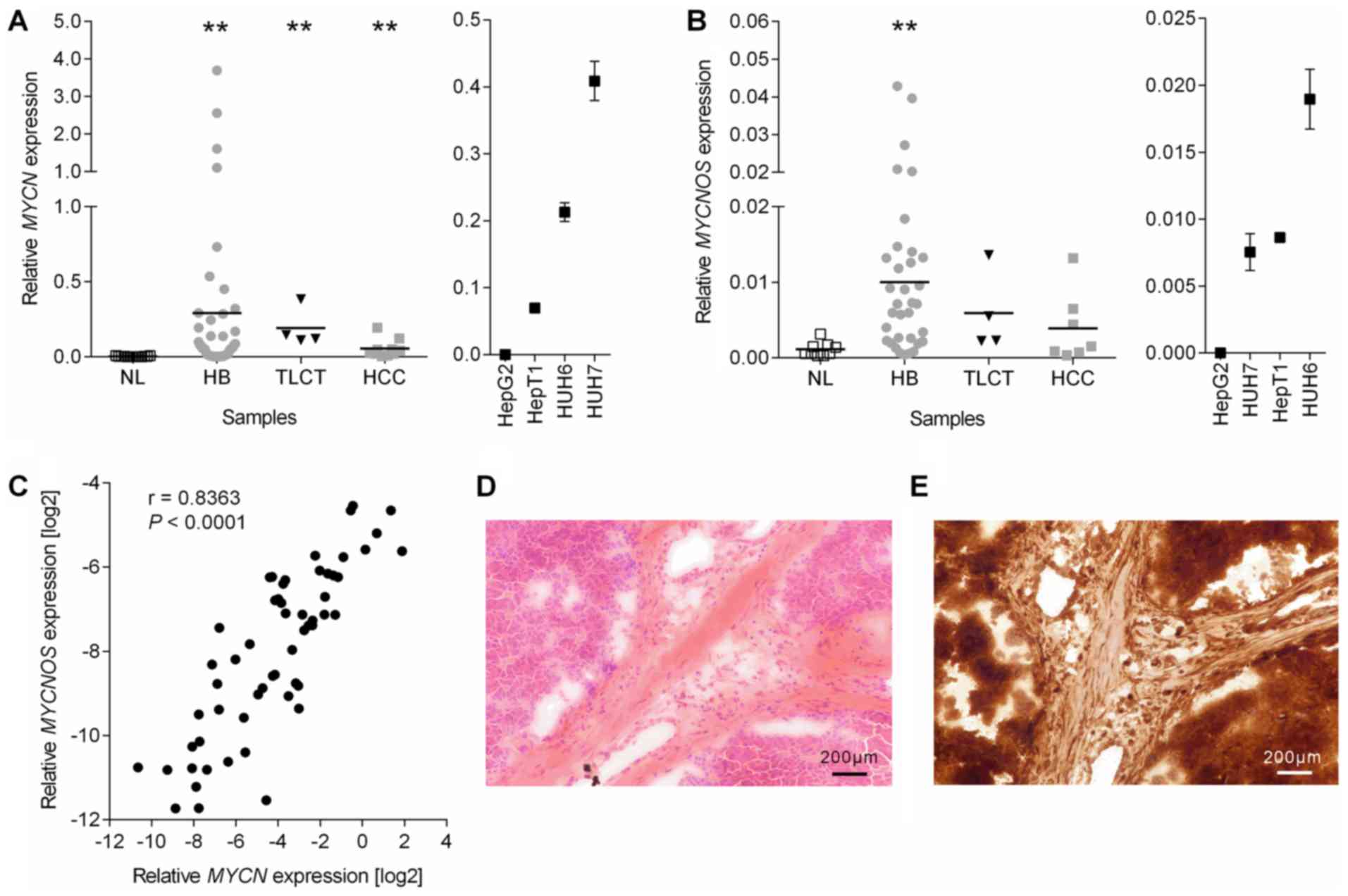 | Figure 1Expression analysis. Expression
levels of (A) MYCN and (B) MYCNOS in the NL, HB, TLCT
and HCC as well as the 4 liver tumor cell lines were measured by
reverse transcription-quantitative polymerase chain reaction and
normalized to the expression of the house-keeping gene TBP,
with the mean presented as a horizontal line across the data
points. Significances were determined by non-parametric one-way
analysis of variance with a Dunn’s correction for multiple
comparisons. **P<0.01 vs. NL. (C) The correlation between
log2-transformed MYCN and MYCNOS expression levels
was calculated using Spearman r correlation. (D and E)
Representative photographs (magnification, ×100; scale bars, 200
µm) of a (D) hematoxylin and eosin and (E) MYCN stained
hepatoblastoma. NL, normal liver; HB, hepatoblastoma; TLCT,
transitional liver cell tumor; HCC, hepatocellular carcinoma;
MYCN, MYCN proto-oncogene basic-helix-loop-helix
transcription; MYCNOS, MYCN opposite strand; TBP,
TATA-box binding protein. |
In order to validate the prognostic value of this
data, correlations between MYCN and MYCNOS expression
and clinicopathological characteristics were evaluated. By
comparing the expression levels between patients with or without
clinical high risk features such as metastasis, vascular invasion,
multifocal or extrahepatic growth, high PRETEXT stage, embryonal
histology, and age of onset >5 years, it was demonstrated that
embryonal histology was the only characteristic to be significantly
associated with high MYCN and MYCNOS expression
(Table I). There was no expression
difference in regard to outcome. Collectively, these results
suggest that concomitant overexpression of MYCN and
MYCNOS may be a general phenomenon in pediatric liver
tumors, especially in those with more undifferentiated
histology.
Hypomethylation of the MYCN/MYCNOS locus
is associated with high expression levels
DNA methylation has been described as an important
molecular mechanism to control the transcriptional activity of
genes (19). By examining 3
CpG-rich regions located at the interface of MYCN and
MYCNOS (Fig. 2A) by means
of pyrosequencing, the present study reported a low methylation
level of <15% in all of the investigated samples (Fig. 2B-E). Notably, the median
methylation level was significantly lower in the 30 pediatric liver
tumors when compared with the 11 normal liver samples.
Consequently, MYCN (Fig.
2F-H) and MYCNOS (data not shown) expression was
negatively correlated with DNA methylation levels.
Transient MYCN knockdown impedes the
growth of HB cells
Having revealed that MYCN was overexpressed
in the vast majority of pediatric liver tumors, the present study
then evulated whether liver tumor cell growth is dependent on MYCN.
siRNA-mediated knockdown of MYCN in HepT1, HUH6 and HUH7
cells (the cell lines that exhibited high MYCN expression
levels; Fig. 1A), was therefore
employed. Following transient transfection, the expression levels
of MYCN mRNA were significantly reduced after 24, 48 and 72
h when compared with the control transfected cells, being the most
efficient following 24 h (Fig.
3A). The levels of MYCNOS expression was unchanged (data
not shown). To assess whether the reduced mRNA levels of
MYCN had an impact on cell proliferation, the present study
generated cell growth curves over a time course of 0, 24, 48 and 72
h for MYCN depleted and control cells using MTT assays.
Transient knockdown of MYCN led to a slightly impeded growth
rate in HepT1, HUH6 and HUH7 cells when compared with control
transfected cells during the first 2 days (Fig. 3B). However, the cell growth rate of
HepT1 and HUH6 cells recovered 72 h post-transient siMYCN
transfection, which could be associated with the decline of
MYCN knockdown.
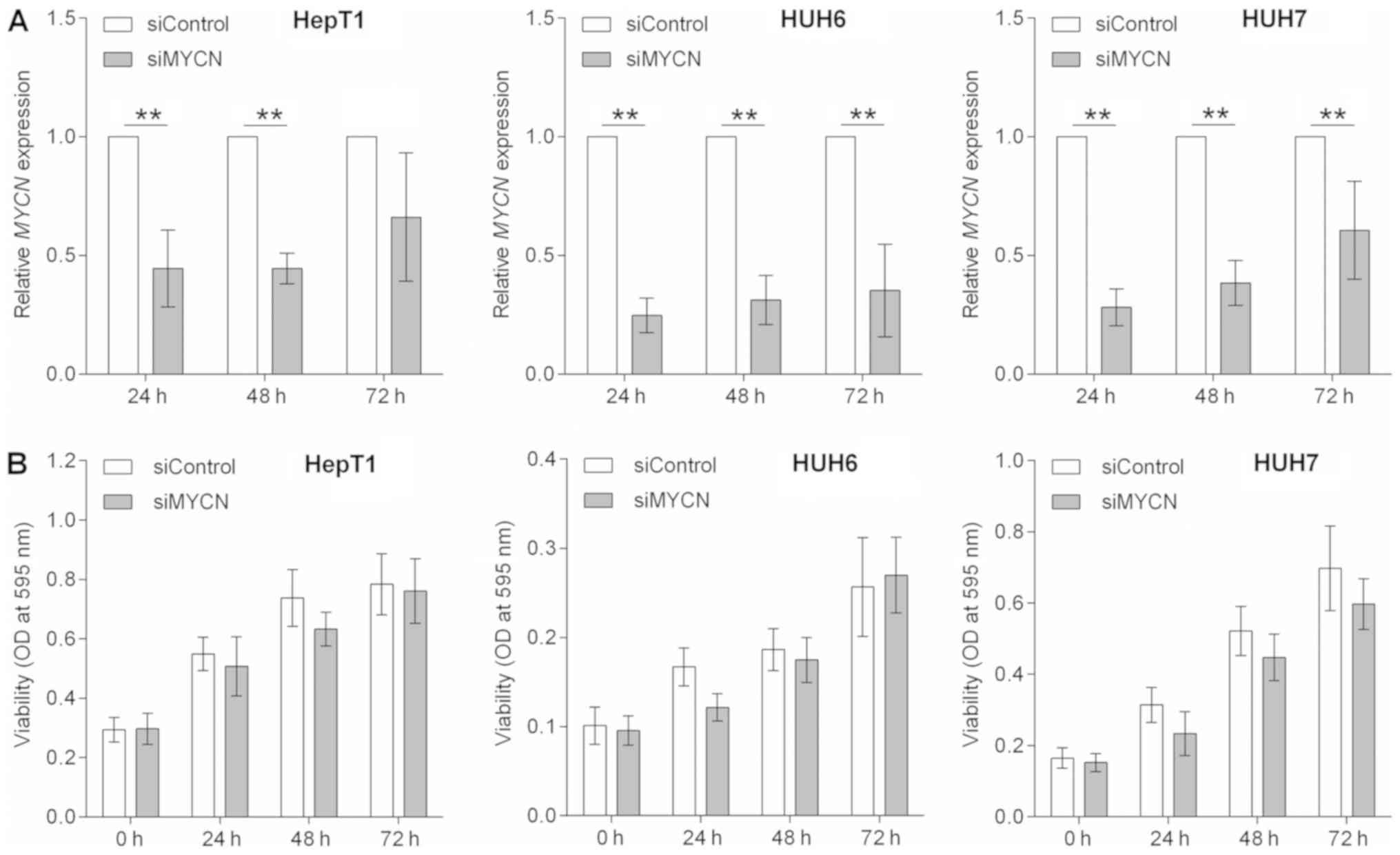 | Figure 3MYCN knockdown. (A) HepT1,
HUH6 and HUH7 cells were transiently transfected with either siRNA
against MYCN (siMYCN) or non-targeting control siRNA
(siControl), and the mRNA expression of MYCN was determined
by reverse transcription-quantitative polymerase chain reaction at
24, 48 and 72 h post-transfection and normalized to the
house-keeping gene TBP. Data are presented as the mean ±
standard deviation of 4 biological replicates and were standardized
to the siControl. Statistical significances were calculated using
the paired Student t-test. **P<0.01, as indicated.
(B) The cell viability of siMYCN or siControl transiently
transfected HepT1, HUH6 and HUH7 cells was assessed at the
indicated time points using MTT assays and OD measurements. The
values are presented as the mean of 6 biological replicates ±
standard deviation. MYCN, MYCN proto-oncogene
basic-helix-loop-helix transcription; MYCNOS, MYCN opposite
strand; siRNA/si-, small interfering RNA; TBP, TATA-box
binding protein; OD, optical density. |
MLN8237 and JQ1 impact MYCN either at the
protein or RNA level
In order to sustain the anti-proliferative effect
observed following siRNA-mediated MYCN inhibition, the
present study examined the effects of MLN8237 and JQ1, 2 known MYCN
inhibitors already pre-clinically tested in various types of
cancers (18,19), on the viability of liver cancer
cells. The small molecule MLN8237 is believed to disrupt the
Aurora-A/MYCN complex, thereby promoting the degradation of the
MYCN protein mediated by F-box and WD repeat domain containing 7
ubiquitin ligase (20). JQ1 is an
inhibitor of the bromodomain and extraterminal domain family of
bromodomains that displaces the bromodomain containing 4 domain
from the MYCN promoter and thus causes a potent depletion of
MYCN transcription (21).
As anticipated, MLN8237 treatment resulted in a marked reduction in
MYCN protein levels in the 3 tumor cell lines with high MYCN
expression, namely HepT1, HUH6 and HUH7 (Fig. 4A). Notably, a concomitant reduction
in MYCN transcripts was also observed in HepT1 and HUH7
cells, for which there is currently no explanation at the molecular
level. By contrast, the major effect of JQ1 treatment was the
downregulation of MYCN mRNA observed in all tumor cell
lines, which resulted in reduced protein levels in the three cell
lines that highly expressed MYCN (Fig. 4B). The low MYCN expressing
cell line, HepG2 (Fig. 1A),
exhibited unchanged protein levels upon treatment with JQ1 and
MLN8237 (Fig. 4A and B). In
conclusion, MLN8237 and JQ1 are potent MYCN inhibitors that
interfere with MYCN protein abundancy via different molecular
mechanisms.
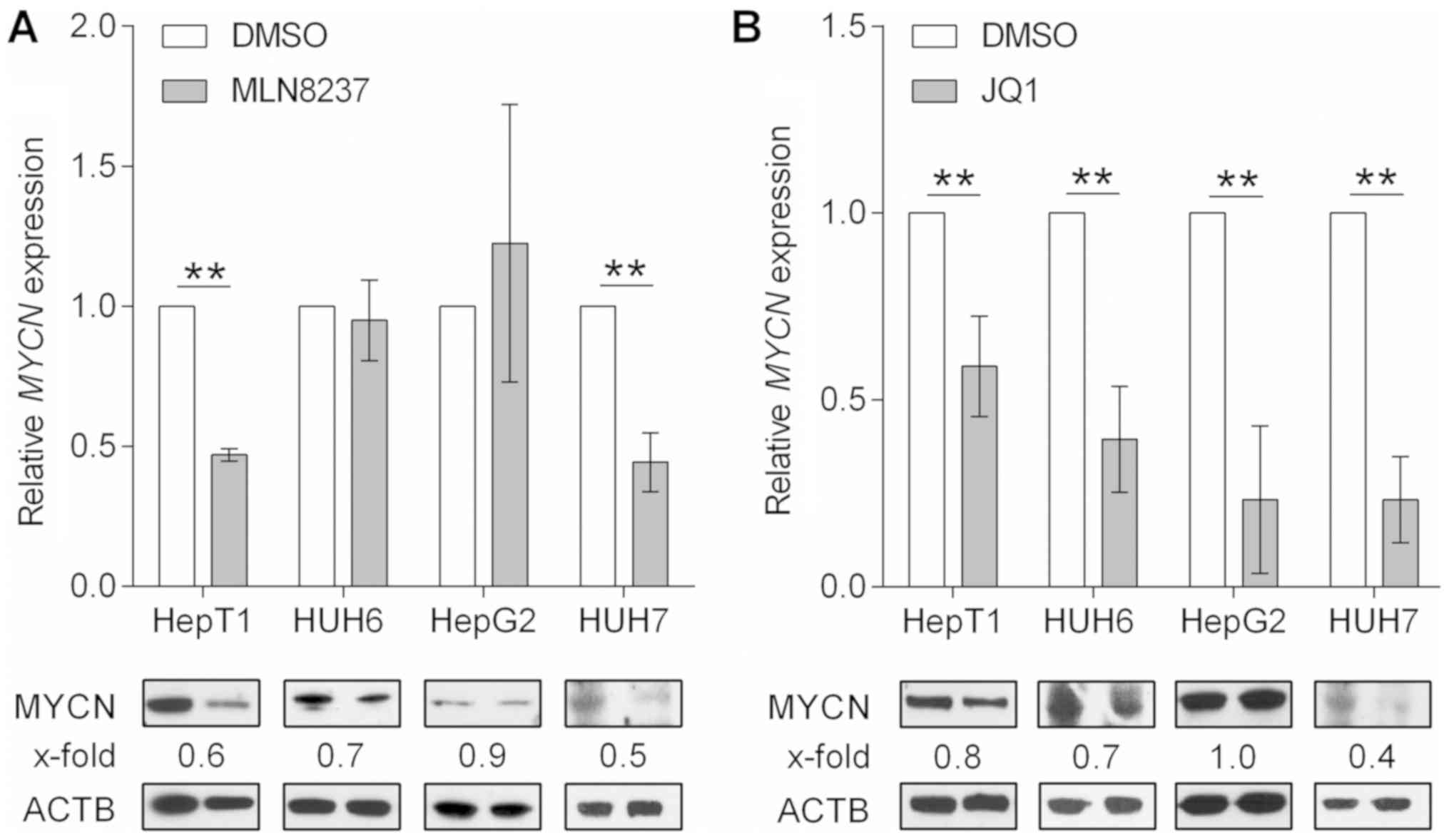 | Figure 4MYCN inhibition. Analysis of
MYCN/MYCN mRNA and protein levels at 48 h post-treatment
with vehicle (DMSO) and the MYCN inhibitors, (A) MLN8237 (10
µM for HepG2, and 1.0 µM for HepT1, HUH6, and HUH7)
and (B) JQ1 (10 µM for HepT1 and HepG2, and 0.5 µM
for HUH6 and HUH7) using reverse transcription-quantitative
polymerase chain reaction and western blotting, respectively. The
expression of MYCN was normalized to the mRNA levels of the
house-keeping gene TBP and is presented as fold changes to
the DMSO control. Data are presented as the mean ± standard
deviation of 3 biological replicates. **P<0.01, as
indicated. Quantification of the MYCN protein (67 kDa) was
performed via densitometry, with the fold reduction compared with
the house-keeping protein ACTB (45 kDa) displayed on top of each
band. The treated and untreated samples of each cell line were
always run on the same blot and simultaneously probed with MYCN and
ACTB antibodies. ACTB signals were detected with an exposure time
of 5 sec, MYCN of 6 min (HUH7, HepT1 and HUH6) and 15 min (HepG2)
due to their varying protein levels in the respective cell lines.
MYCN, MYCN protooncogene basic-helix-loop-helix
transcription; MYCNOS, MYCN opposite strand; ACTB, β-actin;
DMSO, dimethyl sulfoxide; TBP, TATA-box binding protein. |
MLN8237 and JQ1 induce dose-dependent
growth arrest by trapping cells in either the G1/G0 or G2
phase
To investigate the role of MYCN inhibition as a
possible treatment option in pediatric liver tumors, the present
study examined the effect of MLN8237 and JQ1 on the viability of 4
liver cancer cell lines using MTT assays. Treatment with MLN8237
resulted in a potent reduction of cell viability in a
dose-dependent manner in the 3 cell lines with high MYCN
expression, whereas the MYCN low expressing cell line HepG2
was unaffected by the treatment (Fig.
5A). Similarly, treatment with low doses of JQ1 led to
significant reductions in the viability of HUH6 and HUH7 cells
(Fig. 5B). Higher doses of 10
µM JQ1 were required to induce a response in HepT1 and HepG2
cells (Fig. 5B). In addition,
marked morphological changes were also noted following treatment
with MLN8237 and JQ1, as evidenced by enlarged, rounded and swollen
cells or detached and shrunken cells, respectively (Fig. 5C).
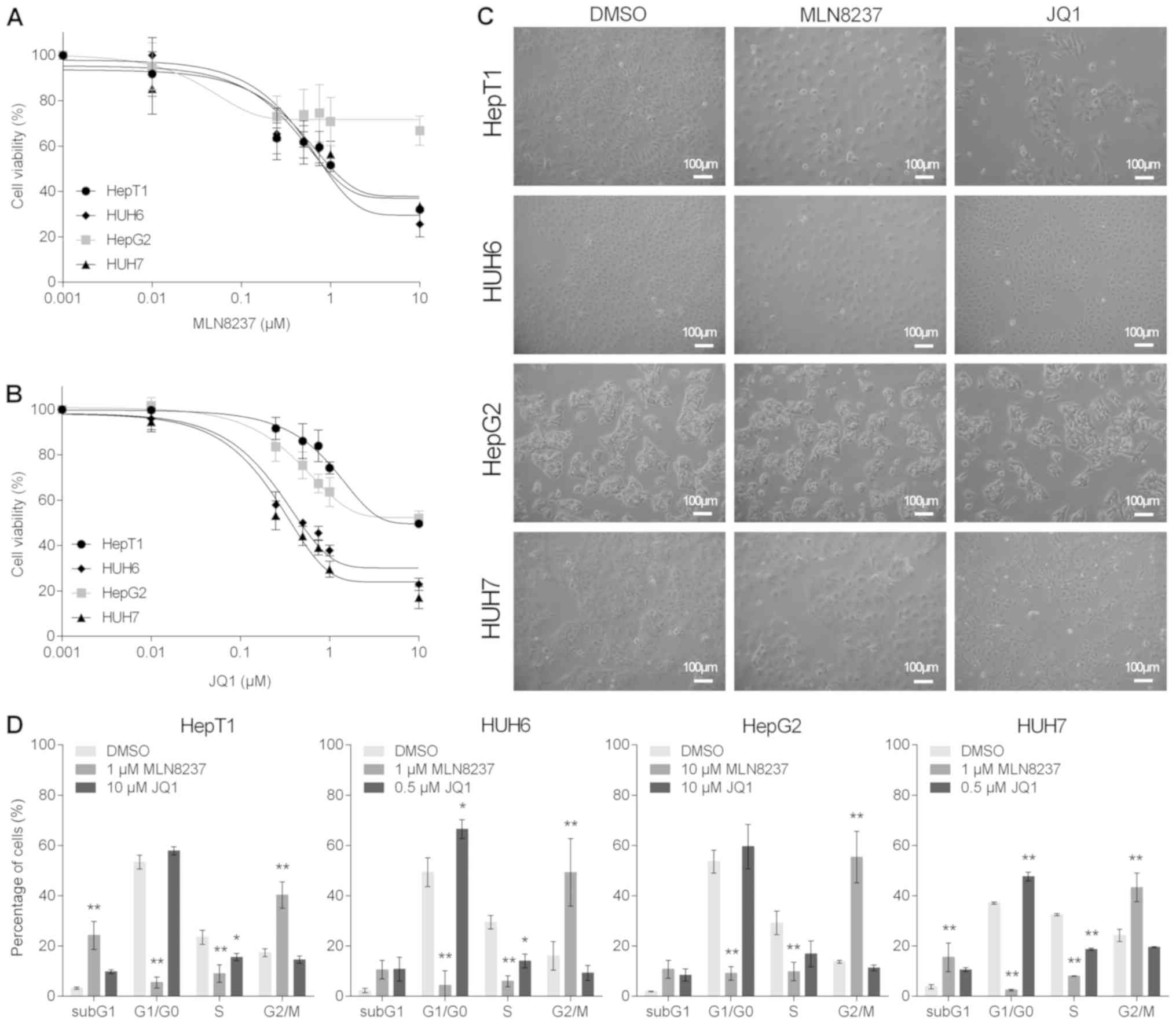 | Figure 5MYCN inhibition impairs cell
growth. The cell viability of the hepatoblastoma cell lines (HepT1,
HUH6, HepG2) and a hepatocellular carcinoma cell line (HUH7) was
evaluated by MTT assay following 48 h of treatment with different
concentrations (0.001, 0.01, 0.25, 0.5, 0.75, 1.0 and 10.0
µM) of (A) MLN8237 or (B) JQ1. Values are presented as the
mean ± standard deviation of 5 independent experiments performed in
duplicates. (C) Morphological changes in the cell lines following
48 h of treatment with vehicle (DMSO) and MLN8237 (10 µM for
HepG2, and 1.0 µM for HepT1, HUH6 and HUH7) or JQ1 (10
µM for HepT1 and HepG2, and 0.5 µM for HUH6 and HUH7)
was detected by microscopy (magnification, ×100; scale bars, 100
µm). (D) Flow cytometric analysis of cell cycle phases of
propidium iodide-stained liver tumor cells in response to 48 h of
treatment with vehicle (DMSO) and MLN8237 or JQ1 (concentrations as
aforementioned). Data are presented as the mean ± standard
deviation of 3 biological replicates. Statistical significances
from the two-way analysis of variance and the Bonferroni post hoc
test are presented. *P<0.05 and
**P<0.01 vs. DMSO. MYCN, MYCN protooncogene
basic-helix-loop-helix transcription; DMSO, dimethyl sulfoxide. |
In order to analyze the cause of these morphological
changes in more detail, the present study performed flow
cytometry-based cell cycle analyses. MLN8237 treatment resulted in
high levels of G2/M arrest and aneuploidy in all cell lines, while
the fraction of cells in the G1/G0 and S phases were significantly
reduced (Fig. 5D). Notably, all
liver tumor cell lines exhibited an increase in the subG1 peak,
which is indicative of apoptotic events. In JQ1-treated cells, a
strong induction of apoptosis was observed in all cell lines, as
indicated by the prominent subG1 peak. In addition, significant
G1/G0 arrest was detected in the HUH6 and HUH7 cells that were
highly sensitive to JQ1 treatment in the viability assays (Fig. 5D). In conclusion, the MYCN
inhibitors MLN8237 and JQ1 reduced cell viability and changed the
morphology of liver cancer cell lines by arresting cells either in
the G1/G0 or G2 phase.
MLN8237 and JQ1 cause spindle
disturbancies and/or apoptosis
As treatment with MLN8237, but not JQ1, led to
cellular swelling and marked G2/M arrest, and is known to induce
spindle pole abnormalities (22),
the present study wanted to analyze if this may be due to
disruption of the mitotic spindle apparatus. Using
immunofluorescent staining of α-tubulin and confocal microscopy,
discontinous spindles and missegregated chromosomes in
MLN8237-treated cells were observed, whereas control cells
displayed a proper spindle apparatus and nicely ordered chromosomes
in metaphase (Fig. 6A).
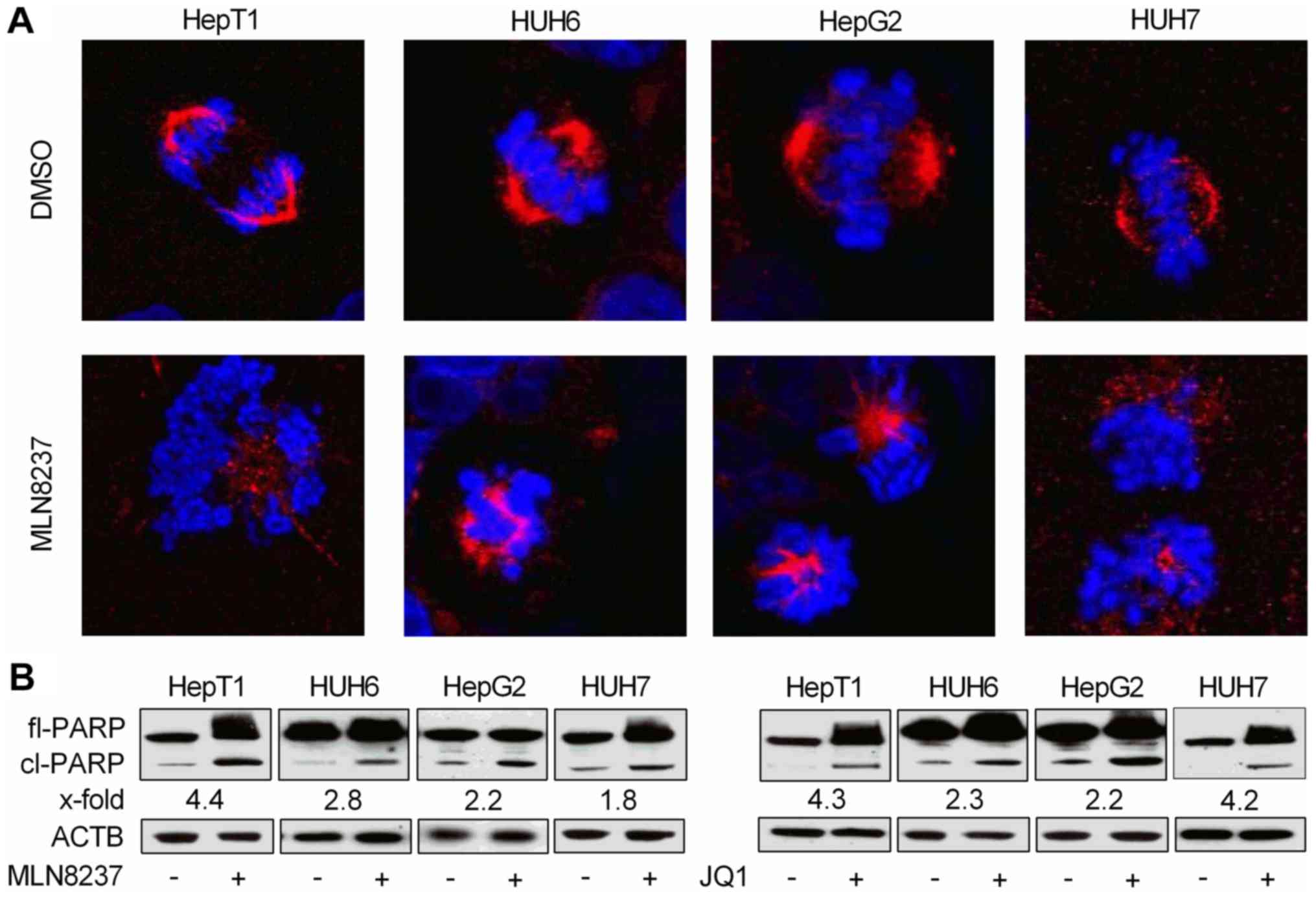 | Figure 6MYCN inhibition induces
apoptosis. (A) Immunofluorescent staining of the spindles of liver
tumor cells treated for 24 h with DMSO or MLN8237 (10 µM for
HepG2, and 1.0 µM for HepT1, HUH6 and HUH7) using an
antibody against α-tubulin (depicted in red). DNA was
counterstained with DAPI (depicted in blue). Magnification, ×400.
(B) Immunoblots presenting the levels of fl-PARP protein (116 kDa),
cl-PARP (89 kDa), and the house-keeping protein ACTB (45 kDa) at 48
h following the addition of DMSO, MLN8237 or JQ1 (10 µM for
HepT1 and HepG2, and 0.5 µM for HUH6 and HUH7). The numbers
below each of the PARP bands indicates the fold induction of
cl-PARP normalized to ACTB. Treated and untreated samples of each
cell line were always run on the same blot and simultaneously
probed with PARP and ACTB antibodies. ACTB signals were detected
with an exposure time of 5 sec, and for PARP 30 sec (HepT1 and
HUH7) and 5 min (HUH6 and HepG2) due to their varying protein
levels in the respective cell lines. MYCN, MYC
proto-oncogene basic-helix-loop-helix transcription; DMSO, dimethyl
sulfoxide; PARP, poly(adenosine diphosphate-ribose) polymerase;
fl-, full-length; cl-, cleaved; ACTB, β-actin. |
As flow cytometric analysis of MLN8237- and
JQ1-treated cells showed a marked induction of apoptosis as
evidenced by the fragmented DNA in the subG1 peak, the present
study wanted to further corroborate this finding via a qualitative
apoptosis assay. Western blot analysis revealed high levels of
proteolytically cleaved PARP, a known downstream event elicited by
caspase-induced apoptosis, following MYCN inhibition by MLN8237 and
JQ1. This result confirms the assumption that the two MYCN
inhibitors exert their anti-proliferative capabilities at least in
part through apoptosis (Fig.
6B).
Discussion
Molecular profiling of HB serves an important role
in stratifying patients into different risk groups (4). Due to the strikingly low mutation
rate, molecular stratification of HB is largely determined by gene
expression rather than genetic events (3). While the 16-gene HB classifier is
already able to divide patients into 2 distinct risk groups based
on gene expression signatures, additional biomarkers are required
to predict the efficacy of targeted therapeutics (10). The present study revealed that
MYCN and MYCNOS expression was significantly
upregulated in HB. Furthermore, MYCN expression appeared to
be a positive predictive marker for the response to MYCN
inhibition, since HB cells with high MYCN expression were
more susceptible to MLN8237 and JQ1 treatment. Therefore,
MYCN and MYCNOS might be useful biomarkers for
patients with HB in predicting their response to treatment with
MYCN inhibitors.
It is the patients in the high-risk group who would
particularly benefit from a more targeted therapy approach, as they
are normally treated with a combination of cisplatin and
doxorubicin (23). These two
chemotherapeutic agents cause common immediate side effects and
doxorubicin in particular has severe late effects including
cardiomyopathy, congestive heart failure and the development of
secondary malignancies that can arise years after the treatment has
been completed (24,25). Previous studies on other solid
types of cancer have shown promising synergies when combining a
cisplatin-backbone with either JQ1 or MLN8237 (26,27).
It seems reasonable that HB patients with MYCN
overexpressing tumors might also benefit from a combined regimen of
cisplatin and a MYCN inhibitor, thereby reducing overall
chemotherapy doses and preventing the subsequent effects from the
more toxic and untargeted agents such as doxorubicin.
While MYC is the most common deregulated
protooncogene in human cancers, MYCN deregulation is rare
and seems to serve a specific role in pediatric malignancies
(28,29). MYCN expression is strictly
controlled during embryonal development and the activation of MYCN
expression can be observed in a variety of peditatric tumors
including neuroblastoma, medulloblastoma, rhabdomyosarcoma and
gliomas (30). Activation of MYCN
in these tumors is generally caused by amplification or alterations
in other oncogenes that are able to enhance the expression of
MYCN or stabilize its protein. MYCNOS has been shown
to positively regulate MYCN expression in rhabdomyosarcoma and
neuroblastoma harbouring MYCN-amplifications (31). Although MYCN is not amplified in
HB, the present results suggest that MYCNOS may be an
important driver of MYCN overexpression in HB.
Notably, a recent study reported that MYCN was able
to directly bind the promoter region of Lin-28 homolog B
(LIN28B) and activate its transcription in neuroblastoma. In
fact, MYCN expression exhibited a strong postive correlation
to LIN28B expression in primary neuroblastoma (32). Another previous study demonstrated
that elevated LIN28B expression in neuroblastoma was in turn
capable of interfering with the let-7 mediated repression of
MYCN (33). LIN28B is able
to sequester let-7, leading to elevated MYCN expression and
neuroblastoma formation in mice (31). LIN28B is also a known driver
of HB as well as other liver-associated malignancies and its
overexpression was shown to be sufficient for HB initiation and
maintanence in mice (34).
Notably, the present study detected a strong correlation between
LIN28B and MYCN expression in primary HB tissues
(data not shown), which is suggestive of an additional mechanism to
trigger MYCN overexpression, similar to the mechanims
observed in neuroblastoma.
In conclusion, the results of the present study
suggest that MYCN overexpression may be a common feature of
pediatric liver malignancies, comprising HB, TLCT and HCC. While
there seem to be various mechanisms through which MYCN
overexpression arises, MYCN itself appears to be a promising
biomarker for HB. Targeting MYCN with small molecules such as
MLN8237 and JQ1 may help to improve outcomes and reduce the
long-term effects of conventional chemotherapy protocols in
patients with HB. However, preclinical testing of the therapeutic
efficacy of these inhibitors in patient-derived HB xenograft models
(35) is an absolute
necessity.
Funding
The present study was supported by the Bettina Bräu
Foundation (Munich, Germany) and the Gänseblümchen Foundation
(Voerde, Germany).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
CE designed experiments, acquired, analyzed and
interpreted the data, and wrote the manuscript. AB interpreted the
data and wrote the manuscript. CV analyzed the histopathology
results and provided the samples. KB and BH contributed and
analyzed the clinical data and gave scientific advice. DvS provided
the samples, contributed and analyzed clinical data and critically
reviewed the manuscript for important intellectual content. RK
conceived the study, obtained funding, designed the experiments,
analyzed and interpreted the data, wrote the manuscript, and
directed the overall research. All authors discussed the results
and implications, and reviewed and approved the final
manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from each
patient and the study protocol was approved by the Ethics Committee
of Ludwig-Maximilian-University (Munich, Germany; no. 431-11).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to acknowledge the assistance
of Ms. Fatemeh Promoli and Ms. Marion Bertow for their technical
support, Dr. Rebecca Maxwell for critically reviewing the
manuscript and Professor Torsten Pietsch (Institute of
Neuropathology, University of Bonn, Bonn, Germany) for supplying
the HepT1 cell line.
References
|
1
|
Mann JR, Kasthuri N, Raafat F, Pincott JR,
Parkes SE, Muir KR, Ingram LC and Cameron AH: Malignant hepatic
tumours in children: Incidence, clinical features and aetiology.
Paediatr Perinat Epidemiol. 4:276–289. 1990.
|
|
2
|
Zimmermann A: The emerging family of
hepatoblastoma tumours: From ontogenesis to oncogenesis. Eur J
Cancer. 41:1503–1514. 2005.
|
|
3
|
Eichenmüller M, Trippel F, Kreuder M, Beck
A, Schwarzmayr T, Häberle B, Cairo S, Leuschner I, von Schweinitz
D, Strom TM, et al: The genomic landscape of hepatoblastoma and
their progenies with HCC-like features. J Hepatol. 61:1312–1320.
2014.
|
|
4
|
Sumazin P, Chen Y, Treviño LR, Sarabia SF,
Hampton OA, Patel K, Mistretta TA, Zorman B, Thompson P, Heczey A,
et al: Genomic analysis of hepatoblastoma identifies distinct
molecular and prognostic subgroups. Hepatology. 65:104–121.
2017.
|
|
5
|
de La Coste A, Romagnolo B, Billuart P,
Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C,
Kahn A, et al: Somatic mutations of the beta-catenin gene are
frequent in mouse and human hepatocellular carcinomas. Proc Natl
Acad Sci USA. 95:8847–8851. 1998.
|
|
6
|
Taniguchi K, Roberts LR, Aderca IN, Dong
X, Qian C, Murphy LM, Nagorney DM, Burgart LJ, Roche PC, Smith DI,
et al: Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in
hepatocellular carcinomas and hepatoblastomas. Oncogene.
21:4863–4871. 2002.
|
|
7
|
Mokkapati S, Niopek K, Huang L, Cunniff
KJ, Ruteshouser EC, deCaestecker M, Finegold MJ and Huff V:
β-catenin activation in a novel liver progenitor cell type is
sufficient to cause hepatocellular carcinoma and hepatoblastoma.
Cancer Res. 74:4515–4525. 2014.
|
|
8
|
Buendia MA: Unravelling the genetics of
hepatoblastoma: Few mutations, what else? J Hepatol. 61:1202–1204.
2014.
|
|
9
|
Klijn C, Durinck S, Stawiski EW, Haverty
PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, et al: A
comprehensive transcriptional portrait of human cancer cell lines.
Nat Biotechnol. 33:306–312. 2015.
|
|
10
|
Cairo S, Armengol C, De Reyniès A, Wei Y,
Thomas E, Renard CA, Goga A, Balakrishnan A, Semeraro M, Gresh L,
et al: Hepatic stem-like phenotype and interplay of
Wnt/beta-catenin and Myc signaling in aggressive childhood liver
cancer. Cancer Cell. 14:471–484. 2008.
|
|
11
|
Weber RG, Pietsch T, von Schweinitz D and
Lichter P: Characterization of genomic alterations in
hepatoblastomas. A role for gains on chromosomes 8q and 20 as
predictors of poor outcome. Am J Pathol. 157:571–578. 2000.
|
|
12
|
Vita M and Henriksson M: The Myc
oncoprotein as a therapeutic target for human cancer. Semin Cancer
Biol. 16:318–330. 2006.
|
|
13
|
Schwab M, Westermann F, Hero B and
Berthold F: Neuroblastoma: Biology and molecular and chromosomal
pathology. Lancet Oncol. 4:472–480. 2003.
|
|
14
|
Kretzner L, Blackwood EM and Eisenman RN:
Transcriptional activities of the Myc and Max proteins in mammalian
cells. Curr Top Microbiol Immunol. 182:435–443. 1992.
|
|
15
|
Kress TR, Sabò A and Amati B: MYC:
Connecting selective transcriptional control to global RNA
production. Nat Rev Cancer. 15:593–607. 2015.
|
|
16
|
Eichenmüller M, Gruner I, Hagl B, Häberle
B, Müller-Höcker J, von Schweinitz D and Kappler R: Blocking the
hedgehog pathway inhibits hepatoblastoma growth. Hepatology.
49:482–490. 2009.
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
18
|
Vadie N, Saayman S, Lenox A, Ackley A,
Clemson M, Burdach J, Hart J, Vogt PK and Morris KV: MYCNOS
functions as an antisense RNA regulating MYCN. RNA Biol.
12:893–899. 2015.
|
|
19
|
Baylin SB and Ohm JE: Epigenetic gene
silencing in cancer - a mechanism for early oncogenic pathway
addiction? Nat Rev Cancer. 6:107–116. 2006.
|
|
20
|
Brockmann M, Poon E, Berry T, Carstensen
A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, et
al: Small molecule inhibitors of aurora-a induce proteasomal
degradation of N-myc in childhood neuroblastoma. Cancer Cell.
24:75–89. 2013.
|
|
21
|
Schnepp RW and Maris JM: Targeting MYCN: A
good BET for improving neuroblastoma therapy? Cancer Discov.
3:255–257. 2013.
|
|
22
|
Asteriti IA, Giubettini M, Lavia P and
Guarguaglini G: Aurora-A inactivation causes mitotic spindle pole
fragmentation by unbalancing microtubule-generated forces. Mol
Cancer. 10:1312011.
|
|
23
|
Hiyama E: Pediatric hepatoblastoma:
Diagnosis and treatment. Transl Pediatr. 3:293–299. 2014.
|
|
24
|
Sivaprakasam P, Gupta AA, Greenberg ML,
Capra M and Nathan PC: Survival and long-term outcomes in children
with hepatoblastoma treated with continuous infusion of cisplatin
and doxorubicin. J Pediatr Hematol Oncol. 33:e226–e230. 2011.
|
|
25
|
Lipshultz SE, Sambatakos P, Maguire M,
Karnik R, Ross SW, Franco VI and Miller TL: Cardiotoxicity and
cardioprotection in childhood cancer. Acta Haematol. 132:391–399.
2014.
|
|
26
|
Sehdev V, Peng D, Soutto M, Washington MK,
Revetta F, Ecsedy J, Zaika A, Rau TT, Schneider-Stock R, Belkhiri
A, et al: The aurora kinase A inhibitor MLN8237 enhances
cisplatin-induced cell death in esophageal adenocarcinoma cells.
Mol Cancer Ther. 11:763–774. 2012.
|
|
27
|
Zanellato I, Colangelo D and Osella D:
JQ1, a BET inhibitor, synergizes with cisplatin and induces
apoptosis in highly chemo-resistant malignant pleural mesothelioma
cells. Curr Cancer Drug Targets. 18:816–828. 2018.
|
|
28
|
Kalkat M, De Melo J, Hickman KA, Lourenco
C, Redel C, Resetca D, Tamachi A, Tu WB and Penn LZ: MYC
Deregulation in Primary Human Cancers. Genes (Basel). 8:82017.
|
|
29
|
Sala A: Editorial: Targeting MYCN in
Pediatric Cancers. Front Oncol. 4:3302015.
|
|
30
|
Rickman DS, Schulte JH and Eilers M: The
Expanding World of N-MYC-Driven Tumors. Cancer Discov. 8:150–163.
2018.
|
|
31
|
O’Brien EM, Selfe JL, Martins AS, Walters
ZS and Shipley JM: The long non-coding RNA MYCNOS-01 regulates MYCN
protein levels and affects growth of MYCN-amplified
rhabdo-myosarcoma and neuroblastoma cells. BMC Cancer.
18:2172018.
|
|
32
|
Beckers A, Van Peer G, Carter DR,
Gartlgruber M, Herrmann C, Agarwal S, Helsmoortel HH, Althoff K,
Molenaar JJ, Cheung BB, et al: MYCN-driven regulatory mechanisms
controlling LIN28B in neuroblastoma. Cancer Lett. 366:123–132.
2015.
|
|
33
|
Molenaar JJ, Domingo-Fernández R, Ebus ME,
Lindner S, Koster J, Drabek K, Mestdagh P, van Sluis P, Valentijn
LJ, van Nes J, et al: LIN28B induces neuroblastoma and enhances
MYCN levels via let-7 suppression. Nat Genet. 44:1199–1206.
2012.
|
|
34
|
Nguyen LH, Robinton DA, Seligson MT, Wu L,
Li L, Rakheja D, Comerford SA, Ramezani S, Sun X, Parikh MS, et al:
Lin28b is sufficient to drive liver cancer and necessary for its
maintenance in murine models. Cancer Cell. 26:248–261. 2014.
|
|
35
|
Nicolle D, Fabre M, Simon-Coma M, Gorse A,
Kappler R, Nonell L, Mallo M, Haidar H, Déas O, Mussini C, et al:
Patient-derived mouse xenografts from pediatric liver cancer
predict tumor recurrence and advise clinical management.
Hepatology. 64:1121–1135. 2016.
|