Introduction
Epidemiological data show that chronic stress in a
negative social and psychological state has an adverse effect on
cancer incidence and progression (1-3).
Additionally, patients with cancer have increased stress levels,
which further exacerbates cancer progression (4). Laboratorial studies have demonstrated
that catecholamines released from the
hypothalamic-pituitary-adrenal axis in response to stressors not
only affect cellular immunity, but also contribute to tumor
proliferation, metastasis and angiogenesis through various
signaling pathways (5-9). As an important target organ of stress
hormones, the stomach is frequently subjected to stress-related
injury, such as stress ulcers (10). However, few reports on the
association between stress and gastric cancer (GC) have been
published to date.
Autophagy is an evolutionarily conserved process in
eukaryotes. It is a process that degrades cytoplasmic components,
including long-lived, damaged proteins, and organelles (11). The by-products of autophagy are
recycled back to the cytosol as macromolecules for the synthesis of
new structures (12). The process
of autophagy can be divided into three stages: Sequestration of
cytosolic components by the autophagosome, fusion of autophagosomes
with lysosomes to form autolysosomes and degradation of the
components incorporated into autolysosomes. Dysregulation in
autophagy has been associated with several human disorders,
including metabolic diseases and cancer (13). Under different stress conditions,
autophagy primarily represents an essential adaptive response to
provide nutrients and energy (14). Increasing evidence has demonstrated
that autophagy is a key component of the stress response in cancer
cells (15-17). It is generally considered that
autophagy has two primary and opposing functions in tumor cells in
response to stress, the cytoprotective function and the cytotoxic
function (11). Autophagy can act
as a tumor suppressor by eliminating oncogenic proteins and
preventing oxidative stress and genome instability. By contrast,
autophagy can promote tumor development by providing substrates
that allow tumor cells to overcome nutrient limitation and hypoxia
(18). However, the effects of
stress hormones on GC cells and the role of stress hormone-induced
autophagy remain to be elucidated.
The present study is the first, to the best of our
knowledge, to provide direct preclinical evidence for the role of
chronic stress in the progression of GC. In addition, the present
study identified the positive role of autophagy on the
norepinephrine-induced progression of GC and revealed an important
regulatory mechanism of the activation of ADRB2 signaling in
autophagy.
Materials and methods
Cell lines and cell culture
The SGC-7901 and BGC-823 human GC cell lines were
purchased from the Shanghai Institutes for Biological Sciences,
Chinese Academy of Sciences (Shanghai China), and were incubated in
RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.).
All cell lines were grown in a humidified chamber supplemented with
5% CO2 at 37°C. Norepinephrine (10 µM; Sigma,
Shanghai, China), 5 µM terbutaline (Sigma), 1 µM
xamoterol (Tocris Chemicals, Shanghai, China), 10 µM
propranolol (Sigma), 50 nMH 89 2HCL (Selleck Chemicals, Shanghai,
China), 5 µM Chloroquine (Selleck Chemicals) in PBS were
used to treat the cells for 24 h prior to testing. In addition, 5
µg/ml E64d (Sigma) and 5 µg/ml pepstain A (Sigma)
were used to treat the cells following norepinephrine
treatment.
Patients and tissue microarray (TMA)
Paired tumor and adjacent non-tumor human gastric
tissues were obtained from 133 patients with GC who underwent
radical resection at the First Affiliated Hospital of Nanjing
Medical University (Nanjing, China) between 2008 and 2010. All
patients were diagnosed pathologically according to the criteria of
the American Joint Committee on Cancer (19) by two professional pathologists
independently. The clinicopathological details are provided in
Table I. Disease-free survival
(DFS) was the primary end-point. Formalin-fixed, paraffin-embedded
tissues were used to construct a TMA, as described previously
(20). The thickness of the tissue
section was 3 µm. All procedures performed in experiments
involving human participants were in accordance with the ethical
standards of the institutional and/or national research committee
and with the 1964 Helsinki declaration and its later amendments or
comparable ethical standards. The present study was approved by the
Institutional Ethical Board of the First Affiliated Hospital of
Nanjing Medical University. Informed consent was obtained from all
individual participants included in the study.
 | Table IClinical characteristics and
expression of ADRB2 in 133 patients with gastric cancer. |
Table I
Clinical characteristics and
expression of ADRB2 in 133 patients with gastric cancer.
| Clinical
characteristic | No. of patients
(n=133) | ADRB2 expression
| P-value |
|---|
| High (n=53) | Low (n=80) |
|---|
| Age (years) | | | | |
| ≤60 | 63 | 30 | 33 | 0.110 |
| >60 | 70 | 23 | 47 | |
| Sex | | | | |
| Male | 81 | 33 | 48 | 0.857 |
| Female | 52 | 20 | 32 | |
|
Differentiation | | | | |
| Well/moderate | 62 | 20 | 42 | 0.112 |
| Poor | 71 | 33 | 38 | |
| TNM stage | | | | |
| Stage I | 21 | 3 | 18 | 0.014 |
| Stage II/III | 112 | 50 | 62 | |
Immunohistochemistry and assessment
The tissue slide was heated at 60°C for 1 h,
followed by treatment with xylene, 100% ethanol and then decreasing
concentrations of ethanol. Following antigen retrieval, the tissues
were blocked with 5% bovine serum albumin (BSA) (Roche, Basel,
Switzerland) for 30 min at room temperature, and stained with
antibodies at 4°C overnight, followed by incubation with
biotinylated secondary antibody (1:200) for 30 min at 37°C and
visualizing using a standard avidin biotinylated peroxidase complex
method. The following antibodies were used: ADRB2 (cat. no.
ab61778, 1:100, Abcam, Cambridge, UK), SQSTM1 (cat. no. ab56416,
1:100, Abcam), Ki-67 (cat. no. ab15580, 1:100, Abcam) and
biotinylated goat anti-rabbit IgG (cat. no. ab64256, 1:200, Abcam).
Immunostaining was observed under a Zeiss fluorescence microscope
at a ×100 or ×200 magnification.The immunoreactivity was scored
independently by two pathologists using a semi-quantitative
immunoreactivity score (IRS) (21). The IRS was calculated by combining
the quantity score with the intensity score. Briefly, the quantity
score represented the percentage of immunoreactive cells as 1
(0-25%), 2 (26-50%), 3 (51-75%) and 4 (76-100%). The intensity
score represented the intensity of immunostaining as 0 (negative),
1 (weak), 2 (moderate) and 3 (strong). Multiplication of the
quantity score and intensity score resulted in an IRS ranging
between 0 and 12. The immunoreactivity was assessed as follows: −
(score 0); + (score 1-4); ++ (score 5-8); +++ (score 9-12). IRS ≤4
was divided into a low expression group and IRS >4 was divided
into a high expression group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the paired tumorous and
adjacent non-tumorous human gastric tissues with TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, and was reverse-transcribed into cDNA with
PrimeScript RT reagent (Takara Biotechnology Co., Ltd., Dalian,
China). qPCR was performed using a 7500 Real-time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) with a SYBR
Premix Ex Taq kit (Takara Biotechnology Co., Ltd.). The following
primers were used: ADRB2, forward 5′-AGAGCCT GCTGACCAAGAAT-3′ and
reverse 5′-TAGCAGTTGATGGC TTCCTG-3′; β-actin, forward
5′-AGAGCCTCGCCTTTGCCG ATCC-3′ and reverse
5′-CTGGGCCTCGTCGCCCACATA-3′. The cycling conditions were comprised
of 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and
60°C for 1 min. The 2−∆∆Cq method was used to calculate
relative expression (22). All
procedures were performed in triplicate.
Western blotting
Total proteins were extracted using lysis buffer [50
mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM
EDTA and protease inhibitor cocktail]. The concentrations were
determined using the Bradford assay. Proteins (30 µg/lane)
were loaded on concentration-appropriate gels. The cellular protein
was size-fractionated by 10% SDS-polyacrylamide gel electrophoresis
and transferred onto PVDF membranes (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Following blocking with PBS containing 5% BSA
for 2 h at room temperature, the membrane was incubated with the
appropriate primary antibodies at 4°C overnight, followed by
incubation with HRP-conjugated anti-mouse or anti-rabbit IgG (cat.
no. ab6728 and cat. no. ab6721, 1:1,000, Abcam) at room temperature
for 2 h. The protein bands were detected using an enhanced
chemiluminescence detection system following the manufacturer's
protocol. Anti-microtubule-associated protein 1 light chain 3 (LC3)
A/B (cat. no. 4108, 1:1,000), anti-p62 (cat. no. 88588, 1:1,000),
anti-adenosine 5′-monophosphate (AMP)-activated protein kinase
(AMPK) (cat. no. 5832, 1:1,000), anti-phosphorylated (p-)AMPK
(Thr172) (cat. no. 50081, 1:1,000), anti-unc-51 like autophagy
activating kinase 1 (ULK1) (cat. no. 6439, 1:1,000), anti-p-ULK1
(Ser555) (cat. no. 5869, 1:1,000), anti-p-CREB (Ser133) (cat. no.
9198, 1:1,000) and anti-CREB (cat. no. 9197, 1:1,000) were obtained
from Cell Signaling Technology, Inc. (Danvers, MA, USA). Anti-ADRB2
(cat. no. ab61778, 1:1,000), anti-Beclin 1 (cat. no. ab62557,
1:1,000), anti-caspase-3 (cat. no. ab13847, 1:1,000), anti-β-catin
(cat. no. ab8227, 1:1,000) antibodies were obtained from Abcam. All
procedures were performed in triplicate. Protein expression was
semi-quantified using ImageJ version 1.46 software (National
Institutes of Health, Bethesda, MD, USA).
Cell viability assay
The cells (2,000/well) were seeded into 96-well
plates and stained at the indicated time point using the Cell
Counting Kit-8 (Dojindo Molecular Laboratories, Inc., Kumamoto,
Japan), according to the manufacturer's protocol. The optical
density measured at 450 nm was used as an indicator of cell
viability. All procedures were performed in triplicate.
Colony formation assay
The GC cells were cultured in 6-well plates (500
cells/well). Following treatment, the cells were cultured for a
further 3 weeks. Colonies composed of ≥50 or more cells were
identified as positive colonies. The cells were stained with 0.2%
crystal violet (KeyGEN, Jiangsu, China) for 20 min at room
temperature. Colonies were observed under a Zeiss microscope at a
×100 magnification. All procedures were performed in
triplicate.
Cell apoptosis analysis
Flow cytometry and terminal-deoxynucleoitidyl
transferase-mediated nick end labeling (TUNEL) were used to analyze
cell apoptosis. Flow cytometry was used to analyze cell apoptosis.
The cells were treated with norepinephrine for 24 h prior to being
harvested. The staining of apoptotic cells was achieved by
incubating the cells with7-AAD and Annexin V-Alexa Fluor 647 (BD
Biosciences, Franklin Lakes, NJ, USA) in the dark for 15 min at
room temperature. The cells were then examined on a FACScan flow
cytometer (BD Biosciences). All procedures were performed in
triplicate. TUNEL assays were conducted using the TUNEL apoptotic
cell detection kit (Roche), according to the manufacturer's
instructions.
Electron microscopy
The cells were pre-treated with 10 µM
norepinephrine for 24 h. For electron microscopy, the cells were
trypsinized using 0.25% trypsin (Thermo Fisher Scientific,
Shanghai, China) for 1 min at 37°C and frozen at high pressure. The
frozen samples were rapidly transferred to a Leica EM AFS2 freeze
subsystem at −90°C for 96 h. The temperature was then gradually
increased to −60°C for 28 h and then to −20°C. The samples were
dehydrated with absolute ethanol and were infiltrated with LR gold
resin. The ultrathin sections (50-70 nm) were stained with 4%
uranyl acetate and lead citrate. Images were captured using a
Technai G2 Spirit TWIN transmission electron microscope (FEI;
Thermo Fisher Scientific, Inc.) equipped with a Gatan 1 k × 1 k CCD
camera. Quantification was performed by counting individual
autophagosomes and autolysosomes in 24 random cell sections of
100-µm2 cytoplasmic area.
Immunofluorescence
The cells were pre-treated with 10µM
norepinephrine for 24 h. The cells were then fixed with 4%
paraformaldehyde for 15 min, permeabilized and blocked with
blocking buffer (KeyGEN) containing 1% BSA, 0.3 M glycine, 0.1%
Tween-20 and 5% donkey serum in PBS (pH 8.0) for 1 h at room
temperature. Following incubation with antibodies (LC3A, cat. no.
4599, 1:200 and LC3B, cat. no. 2775, 1:200;Cell Signaling
Technology) overnight at 4°C, the cells were washed with PBS and
labeled with fluorescence-conjugated secondary antibodies (cat. no.
4412, 1:1,000, and cat. no. 8889, 1:1,000;Cell Signaling
Technology) for 2 h at room temperature and counterstained with
DAPI. Images were captured using a Zeiss LSM 780 confocal laser
scanning microscope.
GFP-mRFP-LC3 imaging
A lentivirus carrying GFP-mRFP-LC3 was purchased
from Shanghai GeneChem Co., Ltd. (Shanghai, China) and used to
infect GC cells following the manufacturer's protocol. The
transfected cells underwent puromycin selection for 2 months. The
cells were determined by confocal microscopy (Carl Zeiss,
Oberkochen, Germany) following Hoechst 33342 (2 µg/ml)
staining for 10 min at room temperature. Red puncta represent
autolysosomes and yellow' puncta represent autophagosomes. The
numbers of puncta were calculated per cell in five high-power
fields.
Animal models
Male BALB/c nude mice (5weeks old, weighing ~20 g)
were purchased from Vital River Laboratories, Co., Ltd. (Nanjing,
China) and were housed in the Laboratory Animal Centre of Nanjing
Medical University under the following conditions: Temperature,
22-25°C; humidity, 50-60%; 12 h light/dark cycle. Mice had free
access to water and food. Mice were randomly divided into 6 groups
(6 mice per group). The GC cells (106 cells/100
µl per mouse) were inoculated subcutaneously to form the
first-generation xenografts. The xenografts were serially
transplanted into the gastric wall by inoculating tumor fragments
(2×2×2 mm) through laparotomy. The kinetics of tumor formation were
assessed by measuring the tumor sizes every 4 days. The tumor
volume was calculated using the following formula: Volume =
(width2 × length)/2. For the chronic stress model, a
restraint-stress procedure was used for 21 days based on a previous
study (9). For β-blockade, Alzet
osmotic minipumps (Durect Corporation, Cupertino, CA, USA)
containing PBS or 2 mg/kg/d propranolol hydrochloride
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) were inserted on
the nape of the neck 7 days prior to the initiation of restraint
stress (9). The experimental
protocols were approved by the Animal Care and Use Committee of the
Laboratory Animal Center of Nantong University.
Plasma sampling and catecholamine
assay
The concentrations of stress hormones
(norepinephrine and corticosterone) were determined in plasma
samples collected immediately following the removal of the animals
from the stress situation. The mice were anesthetized and ~500
µl blood was withdrawn by cardiac puncture using a 1-ml
plastic syringe. Blood was centrifuged at 1,100 g for 10 min at
4°C. Plasma was collected and stored at -80°C until required. The
catecholamine concentration in plasma samples was measured by high
performance liquid chromatography with electrochemical detection,
as previously described (23).
RNA interference and plasmids
The synthesized DNA fragments encoding the short
hairpin RNA (shRNA) used for the knockdown of endogenous ADRB2 were
inserted into the pGPU6/GFP/Neo vector (GenePharma, Co. Ltd.,
Shanghai, China). The sequences of the shRNAs were as follows:
ADRB1-shRNA, 5′-CCGATAGCAGGTGAACTCGAACTC
GAGTTCGAGTTCACCTGCTATCGG-3′, ADRB2-shRNA,
5′-GCATCGTCATGTCTCTCATCGCTCGAGCGATGAGA GACATGACGATGC-3′, negative
control (NC)-shRNA, 5′-TTCACCGAACGTGAAACGTATCGAGACGTGACAAGTTCG
GAGAA-3′. All plasmids were verified by sequencing.
Statistical analysis
Statistical analysis was performed with SPSS
software (SPSS Standard version 13.0; SPSS, Chicago, IL, USA). Data
were analyzed using two-tailed Student's t-tests for two groups.
Datasets containing more than two groups were compared using
one-way analysis of variance and Dunnett's test. A χ2
test was used to determine the association between the clinical
characteristics of the patients and the expression of ADRB2.
Spearman's correlation test was used to evaluate the correlation
between ADRB2 and sequestosome-1 (SQSTM1). The probability of
differences in DFS was ascertained using the Kaplan-Meier method,
with a log-rank test probe for significance. P<0.05 was
considered to indicate a statistically significant difference.
Results
Activation of ADRB2 by stress hormones
promotes GC cell growth and survival
It is reported that catecholamines mediate the
effects of stress on cells via adrenergic receptor activation. The
present study first examined the effects of adrenergic receptor
agonists on tumor growth and survival in GC cells. Three adrenergic
receptor agonists, 10 µM norepinephrine (a non-specific
adrenergic receptor agonist), 5 µM terbutaline (a specific
ADRB2 agonist), 1 µM xamoterol (a specific ADRB1 agonist),
and 10 µM propranolol (a non-specific β-adrenergic receptor
antagonist) (24,25) were used to treat the SGC-7901 and
BGC-823 GC cell lines. The results showed that norepinephrine
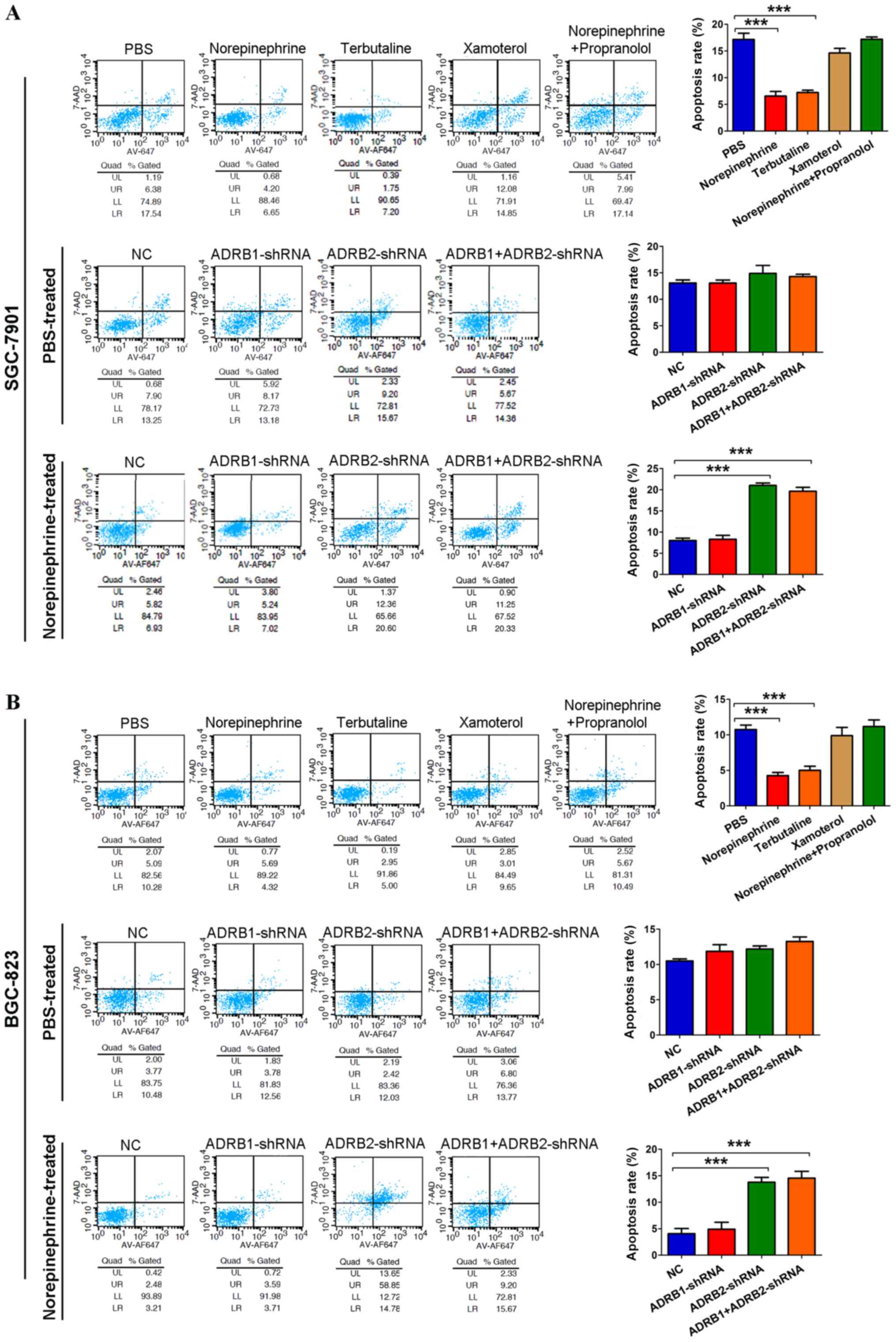
significantly promoted the proliferation of GC cells (Fig. 1A). A similar increase in cancer
cell proliferation was observed with the ADRB2 agonist terbutaline,
but not with theADRB1 agonist xamoterol. In addition, treating
cells with norepinephrine and propranolol simultaneously inhibited
the norepinephrine-induced increase in cancer cell proliferation
(Fig. 1A). This indicated that
β2-adrenergic signaling was essential for the enhanced cell
proliferation. To further identify the specific β-adrenergic
receptor subtype responsible for catecholamine effects, tumor cell
adrenoreceptor expression was inhibited using shRNA specific for
human ADRB1 or ADRB2. The RT-qPCR method was used to determine
which adrenergic receptor is present in GC cells and validate the
knockdown efficiency. The data showed that ADRB1 and ADRB2 were
expressed in SGC-7901 and BGC-823 cells. The RT-qPCR analysis
indicated ~80% downregulation at the mRNA level (Fig. 1A). In the PBS-treated GC cells,
neither ADRB1-shRNA nor ADRB2-shRNA affected cell growth. In the
norepinephrine-treated cells, ADRB2-shRNA significantly inhibited
norepinephrine-induced tumor cell growth, but NC and ADRB1-shRNA
failed to do so (Fig. 1A).
Anchorage-dependent and anchorage-independent colony formation
assays confirmed these observations (Fig. 1B and C). Taken together, these
results suggest a critical role of ADRB2 in catecholamine-mediated
GC cell proliferation.
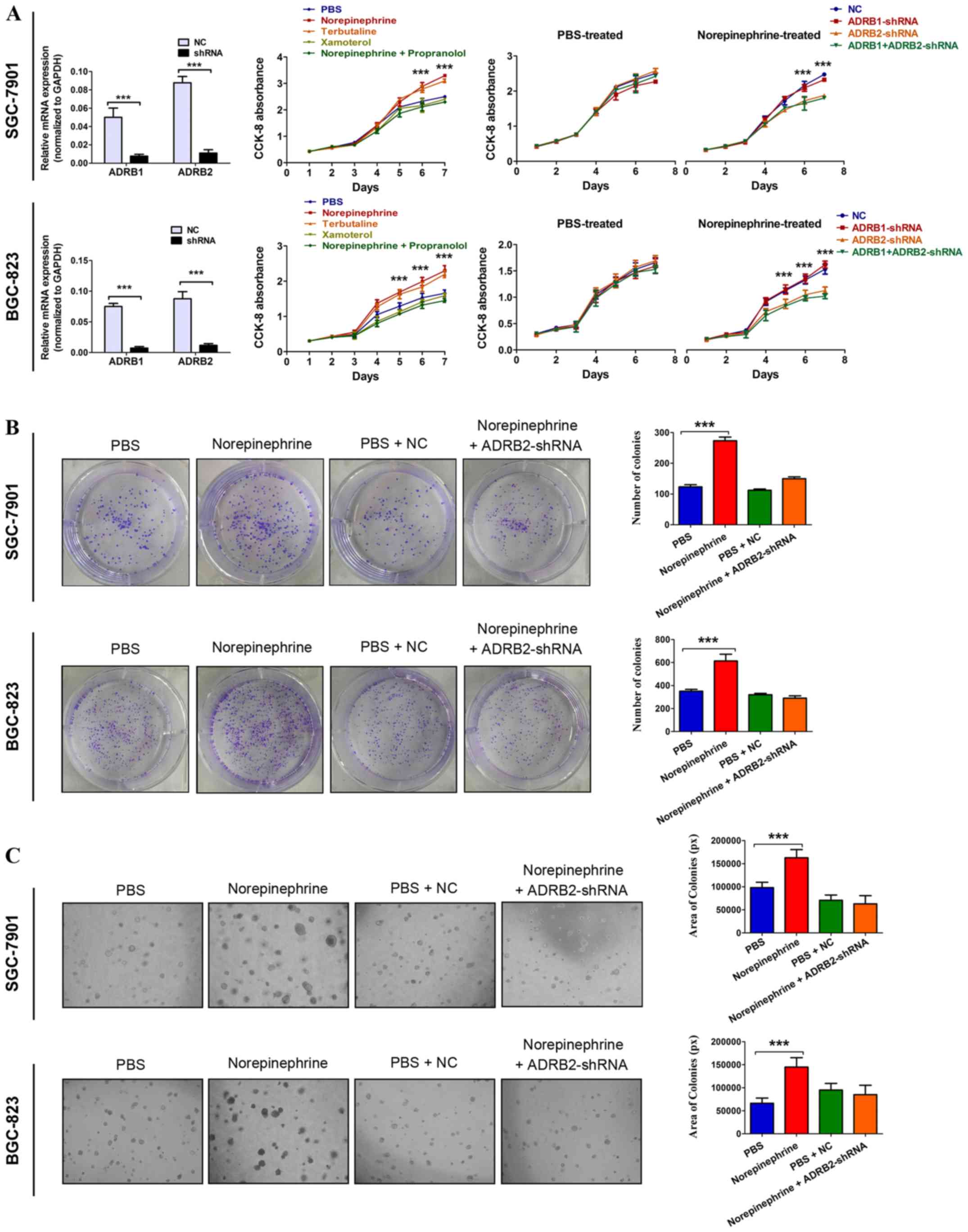 | Figure 1Activation of ADRB2 by stress hormone
promotes GC cell growth. (A) Cell viability was determined using
CCK-8 in GC cells treated with 10 µM norepinephrine (a
non-specific adrenergic receptor agonist), 5 µM terbutaline
(a specific ADRB2 agonist), 1 µM xamoterol (a specific ADRB1
ago-nist), and 10 µM propranolol (a non-specific
β-adrenergic receptor antagonist). The significant P-values were
derived from comparing the PBS group and norepinephrine/terbutaline
group (left panels). Cell viability was determined using CCK-8 in
ADRB1- or ADRB2-knockdown cells treated with 10 µM
norepinephrine. The significant P-values were derived from
comparing the NC group and ADRB2-shRNA/ADRB1+ADRB2-shRNA group
(right panels). Anchorage-dependent and anchorage-independent
colony formation assays were used to determine the effect of
norepinephrine and ADRB2 pathway on cell proliferation according to
(B) colony number (magnification, ×100) and (C) colony area
(magnification, ×200). Data represent the results from three
independent experiments. ***P<0.001. GC, gastric
cancer; ADRB2, β2-adrenergic receptor; ADRB1, β1-adrenergic
receptor; CCK-8, Cell Counting Kit-8; shRNA, short hairpin RNA; NC,
negative control. |
Tumor cells develop resistance to apoptosis,
allowing for their survival during stress. To evaluate the effect
of catecholamine on GC cell survival, tumor cells cultured in
serum-free medium were treated with adrenergic receptor agonists.
Norepinephrine significantly protected tumor cells from nutrient
deprivation (Fig. 2A and B). The
ADRB2 agonist terbutaline showed a similar effect on promoting
tumor cell survival, but not the ADRB1 agonist xamoterol. In
addition, propranolol inhibited the effects of norepinephrine.
Cells expressing ADRB1-shRNA and NC were protected from nutrient
deprivation by norepinephrine, but cells expressing ADRB2-shRNA
were not (Fig. 2A and B).
Collectively, these results indicated that β2-adrenergic signaling
is essential for tumor cell survival under stress.
Specific activation of ADRB2 leads to
autophagy in GC cells
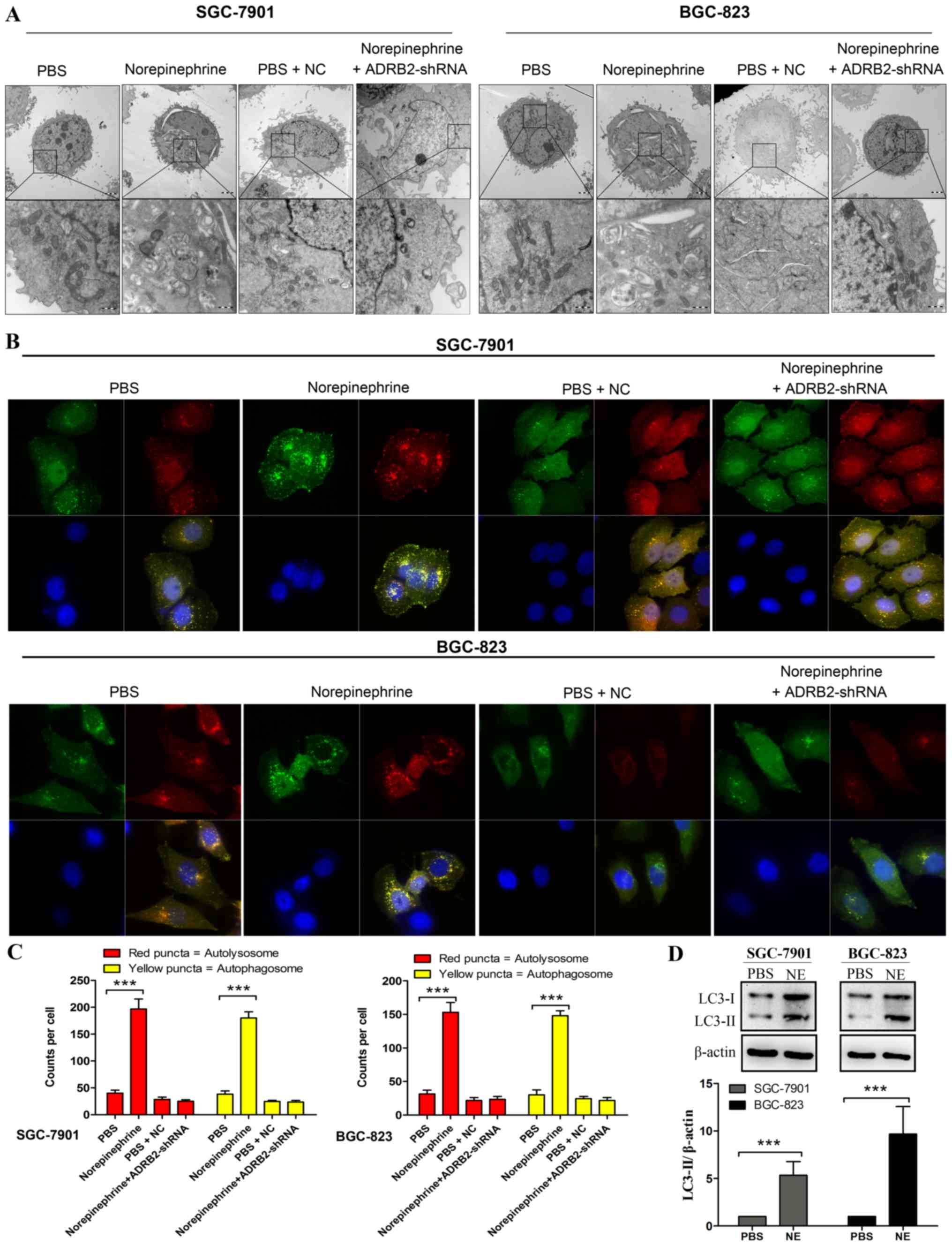
One of the most prominent morphological features of
cells treated with norepinephrine was the appearance of cytoplasmic
vesicles. Transmission electron microscopy (TEM) showed that these
vesicles were double-membrane vesicles containing cytoplasmic
components, which is a hallmark of autophagy (Fig. 3A). To further confirm that the
accumulation of autophagosome induced by norepinephrine was due to
the specific activation of ADRB2, the cells were transfected with
ADRB2-shRNA prior to norepinephrine treatment. The results revealed
that the emergence of autophagosomes was markedly inhibited
following transfection with ADRB2-shRNA (Fig. 3A). In addition to visually
detecting autophagosomes by TEM, LC3-II is widely used as a marker
of autophagy (26). Its lipidation
and specific recruitment to autophagosomal membranes provides a
shift from diffuse to punctate staining. In the present study,
autophagic flux was monitored by exogenously introducing
GFP-mRFP-LC3. As shown in Fig. 3B and
C, norepinephrine treatment led to a significant increase in
the number of GFP-LC3 puncta per cell, whereas shRNA-ADRB2 markedly
inhibited the autophagy phenotype induced by norepinephrine.
Consistently, norepinephrine treatment led to increased number of
red puncta (autolysosomes) and yellow puncta (autophagosomes).
These findings indicate that the accumulation of LC3-II induced by
norepinephrine was due to increased autophagosome formation rather
than the inhibition of autolysosomal degradation alone.
Given that the expression of exogenous LC3 can lead
to false positives of LC3-II aggregation, the processing of
endogenous LC3 in norepinephrine-treated cells was investigated by
western blotting. LC3 lipidation was determined by the LC3-II ratio
over β-actin instead of the LC3-II/LC3-I ratio as, during the
initial stages of autophagy, LC3-II increases and LC3-I is subject
to de novo synthesis, whereas the decrease of LC3-II at
later stages is due to lysosomal degradation (27). The GC cells treated with
norepinephrine exhibited a significant increase in the
LC3-II/β-actin ratio when compared with the control cells (Fig. 3D). To further confirm the effect of
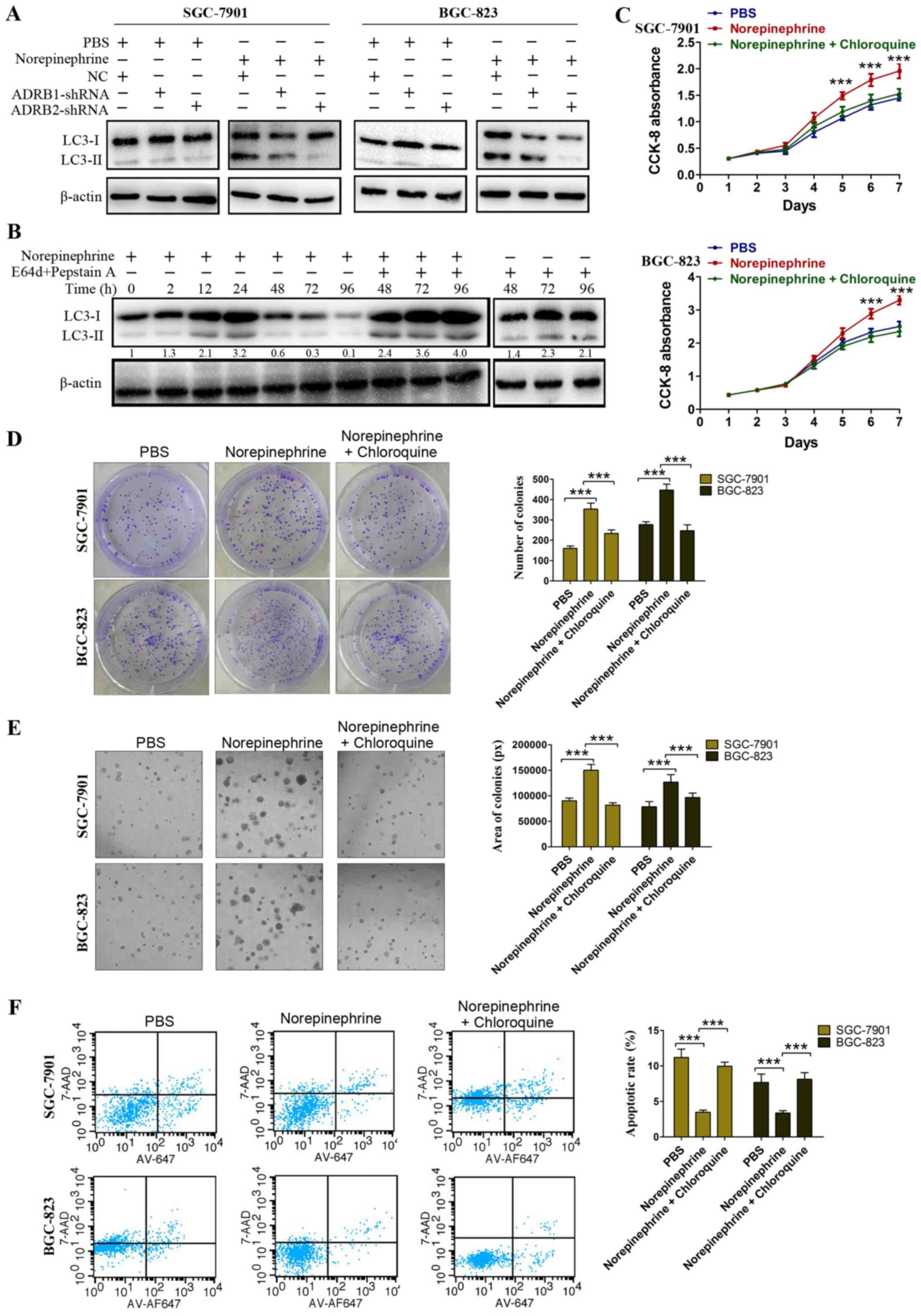
β-adrenergic receptor on LC3 lipidation, the LC3-II/β-actin ratio
was assessed following transfection with ADRB1-shRNA and
ADRB2-shRNA in the GC cells. As shown in Fig. 4A, in the PBS-treated cells, neither
ADRB1-shRNA nor ADRB2-shRNA affected the LC3-II/β-actin ratio.
However, in the norepinephrine treated cells, ADRB2-shRNA markedly
inhibited norepinephrine-induced LC3 lipidation, whereas the NC and
ADRB1-shRNA had no effect (Fig.
4A). In order to determine whether an increase in the number of
autophagosomes result from an induction of the autophagic process,
or from an arrest of lysosomal fusion/degradation, GC cells were
treated with norepinephrine in the presence of E64d plus pepstain
A, which is known to inhibit lysosomal acidic proteases and inhibit
the degradation of LC3-II. Western blotting demonstrated an
increased accumulation of LC3-II when lysosomal degradation was
inhibited by E64d plus pepstain A (Fig. 4B), and treatment with NE plus
lysosomal protease inhibitor increased LC3 accumulation compared
with that in cells treated with lysosomal protease inhibitor alone.
This is indicative of enhanced autophagosome formation induced by
norepinephrine, rather than an impairment of autophagosome
degradation. Taken together, these data suggest that ADRB2
signaling positively regulates autophagy in GC.
ADRB2-driven autophagy contributes to
catecholamine-enhanced GC cell proliferation and survival in
vitro
Autophagy has two opposing functions in tumor cells
in response to stress, the cytoprotective function and the
cytotoxic function. The activation of ADRB2 in GC cells has been
described as the regulation of autophagy. In order to evaluate the
physiological effects of ADRB2-driven autophagy, cell proliferation
and survival were monitored to discriminate whether these effects
were cytoprotective or cytotoxic. Chloroquine was used to inhibit
autophagy pharmacologically. The CCK-8 assay showed that cell
viability was further impaired by the inhibition of autophagy with
chloroquine (Fig. 4C).
Anchorage-dependent and anchorage-independent colony formation
assays confirmed these findings (Fig.
4D and E). In addition, chloroquine treatment significantly
attenuated the protective role of norepinephrine under nutrient
deprivation (Fig. 4F).
Collectively, the results indicate that ADRB2-driven autophagy
serves a vital role in GC cell proliferation and survival.
Immobilization stress accelerates GC
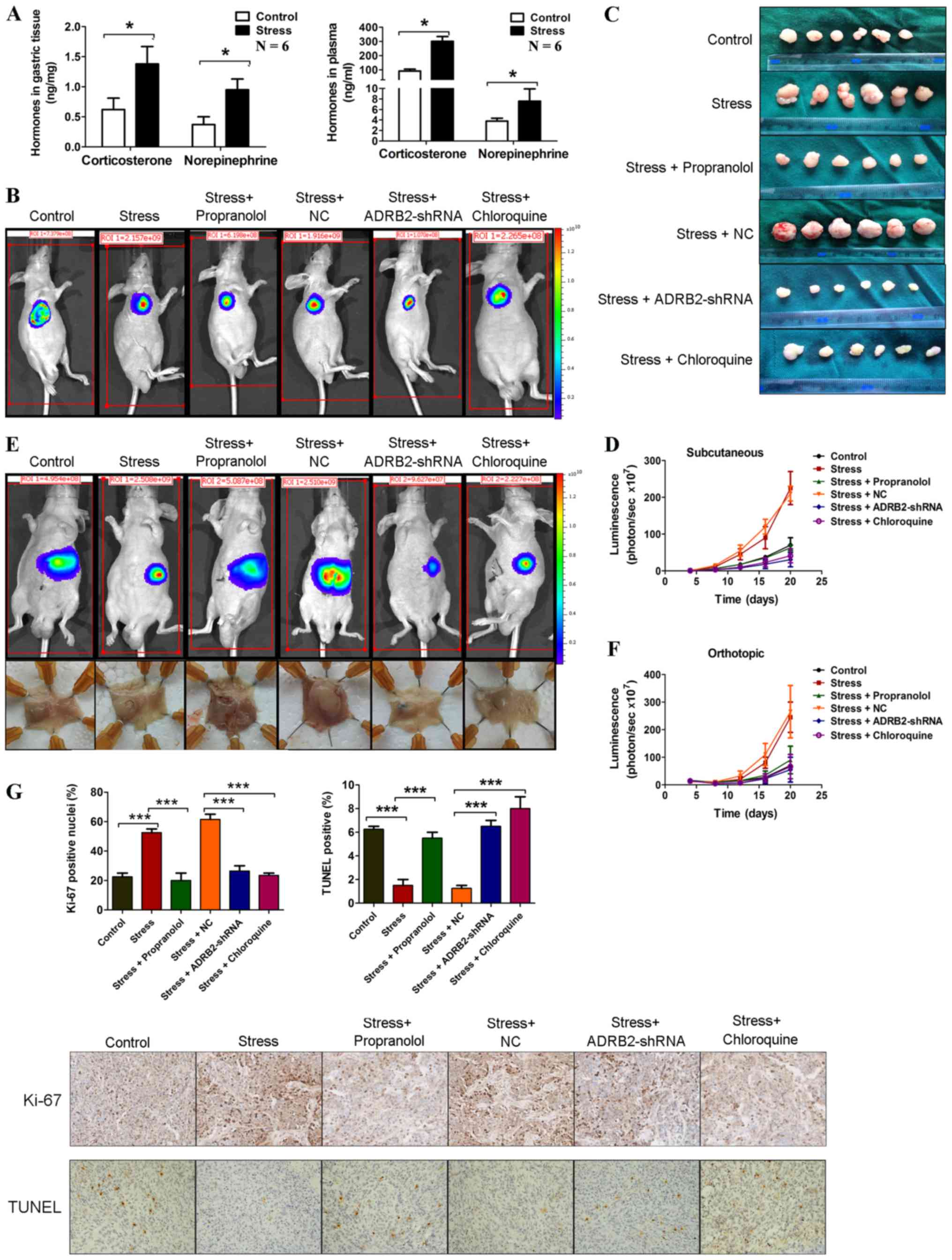
progression via ADRB2 signaling-induced autophagy in vivo
To investigate the effects of chronic stress on GC
in vivo, SGC-7901 cells stably expressing luciferase were
injected into BALB/c nude mice subcutaneously and orthotopically.
The mice were then subjected to immobilization stress. Sustained
chronic stress is known to elevate catecholamines and
corticosteroid cortisol levels (28). Stress hormone levels were measured
in the gastric tissues and plasma of these mice. As shown in
Fig. 5A, samples derived from mice
in the stress group had significantly higher stress hormone levels
compared with mice in the control group. In the subcutaneous
xenograft mouse model, after 20 days, the mice in the stress group
exhibited a marked increase in tumor size compared with mice in the
control group (Fig. 5B-D). Similar
results were observed in the orthotopic xenograft model (Fig. 5E and F). The mechanisms through
which chronic behavioral stress promotes GC progression were
investigated. ADRB2-driven autophagy was found to promote
catecholamine-enhanced GC cell proliferation and survival in
vitro. Subsequently, whether the activation of β-adrenergic
signaling similarly induces xenograft tumor growth of GC cells by
unregulating autophagy was investigated. Propranolol, a
β-adrenergic receptor antagonist, was used to inhibit β-adrenergic
signaling, and ADRB2-shRNA was used to knockdown the expression of
ADRB2. As shown in Fig. 5B-F,
propranolol treatment and ADRB2-shRNA transduction significantly
inhibited chronic stress-induced GC progression. Furthermore,
inhibition of autophagy using chloroquine attenuated chronic
stress-induced GC progression. Immunohistochemical staining of
Ki-67 and a terminal-deoxynucleoitidyl transferase mediated nick
end labeling (TUNEL) assay was used to determine cell proliferation
and survival in mice, respectively. Ki-67 staining was elevated in
tumors from mice in the stress group, however propranolol
treatment, ADRB2-shRNA transfection and chloroquine treatment
markedly attenuated this effect (Fig.
5G). The TUNEL assays showed that chronic stress reduced
apoptosis compared with that in the control group, whereas
propranolol treatment, ADRB2-shRNA transfection and chloroquine
treatment inhibited this effect (Fig.
5G). These results suggest that the activation of autophagy
induced by the β2-adrenergic signaling pathway promoted tumor
growth in GC xenograft mouse models.
AMPK-ULK1 pathway is required for
autophagy in response to ADRB2 signaling activation
The mechanisms underlying norepinephrine-mediated GC
cell autophagy and survival were subsequently investigated. CREB is
a key downstream effector of the β-adrenergic signaling pathway
(29). In line with this finding,
treatment with norepinephrine promoted CREB activation in GC cells
and this effect was inhibited by ADRB2 silencing (Fig. 6A). AMPK has been demonstrated to
induce autophagy by activating ULK1 (30). The AMPK-ULK1 pathway was activated
by norepinephrine treatment (Fig.
6A), and knockdown of ADRB2 significantly attenuated the
activation of AMPK-ULK1 induced by norepinephrine (Fig. 6A). Beclin1 serves an important role
in the initiation of autophagosome formation. The expression levels
of Beclin1 were analyzed by western blotting. As shown in Fig. 6A, norepinephrine treatment markedly
increased the protein levels of Beclin1, but had no effect in
ADRB2-depleted cells (Fig. 6A).
Additionally, norepinephrine treatment markedly decreased the
apoptosis marker, cleaved-caspase-3, whereas norepinephrine had no
effect on the levels of cleaved-caspase-3 in ADRB2-knockdown cells
(Fig. 6A). H89 2HCl, an inhibitor
of protein kinase A, abrogated the NE-induced phosphorylation of
AMPK and ULK1 (Fig. 6B).
Collectively these results suggest that autophagy induced by
norepinephrine was attributed to activation of the AMPK-ULK1
pathway.
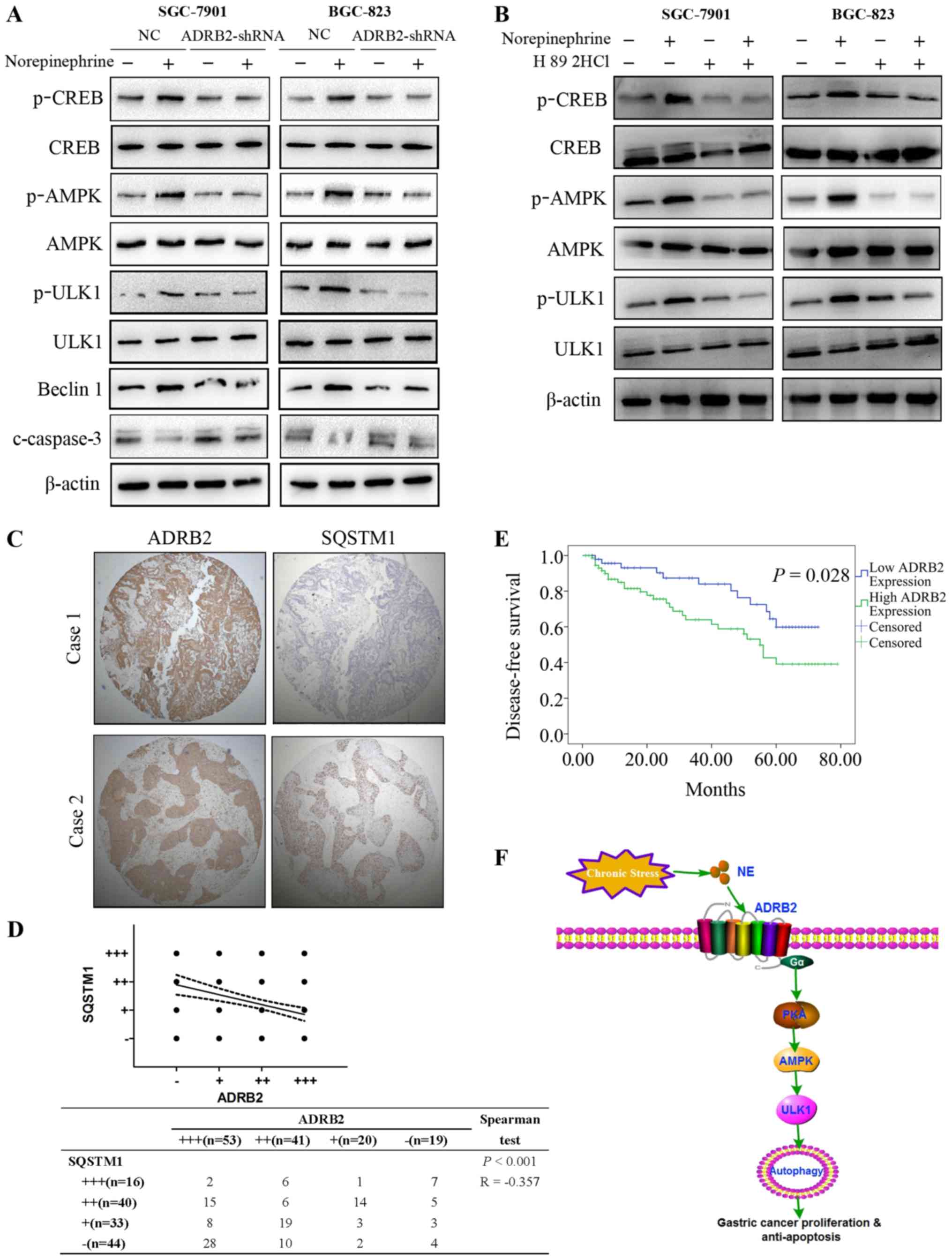 | Figure 6Activation of theAMPK-ULK1 pathway is
critical for autophagy induced by norepinephrine. (A) Expression
levels of AMPK-ULK1 pathway proteins were detected by western
blotting. (B) SGC-7901 and BGC-823 cells were treated with
norepinephrine with or without H89 2HCl (50 nM), and the expression
levels of AMPK-ULK1 pathway proteins were detected with western
blotting. (C) Representative images of the expression of ADRB2 and
SQSTM1 in the gastric cancer tissue microarray (magnification,
×100). (D) Correlation between the expression of ADRB2 and SQSTM1
was examined by Spearman's correlation test. (E) Survival analysis
of patients with gastric cancer with high ADRB2 expression and low
ADRB2 expression. (F) Schematic representation of the proposed
mechanism. Data represent the results from three independent
experiments. ADRB2, β2-adrenergic receptor; shRNA, short hairpin
RNA; NC, negative control AMPK, adenosine 5′-monophosphate
(AMP)-activated protein kinase; CREB, cAMP-response element binding
protein; p, phosphorylated; SQSTM1, sequestosome-1; ULK1, unc-51
like autophagy activating kinase 1; PKA, protein kinase A; NE,
norepinephrine. |
Association of ADRB2 with SQSTM1 in
clinical GC samples
To investigate the clinical relevance of the above
findings, immunohistochemical staining was performed to determine
the protein expression levels of ADRB2 in 133 GC tumor samples
(Table I). High protein levels of
ADRB2 were positively were associated with the TNM stage,
indicating that high protein levels of ADRB2 may serve as a
predictor for staging. High protein levels of ADRB2were also
associated with poor prognosis in patients with GC (Fig. 6E). The p62/SQSTM1 protein serves as
a link between LC3 and ubiquitinated substrates and is degraded in
autolysosomes, indicating that the protein levels of p62/SQSTM1
reflect the autophagic status (31). Therefore, the present study
investigated the correlation between ADRB2 and autophagic marker
p62/SQSTM1 in GC tumor samples. TMA showed that there was a
negative correlation between the expression levels of ADRB2 and
p62/SQSTM1(Fig. 6C and D). Taken
together, the results of the present study demonstrate the positive
effect of autophagy on norepinephrine-induced GC progression and
revealed an important regulatory mechanism of ADRB2 signaling
activation in autophagy (Fig.
6F).
Discussion
Emerging evidence has implicated that chronic
stress, specifically adrenergic signaling, is involved in the onset
and progression of various types of cancer (32,33).
The stomach is one of the most susceptible target organs of chronic
stress, and patients with GC experiencing severe psychological
distress at the time of diagnosis frequently experience prior
psychological stress (34).
However, limited reports of the association between stress and
gastric cancer have been published. Molecular insights into how
chronic stress affects gastric tumorigenesis may offer novel
treatment options. The results presented in the present study is
the first, to the best of our knowledge, to provide direct
preclinical evidence for the role of chronic stress in the
progression of GC.
The present study found that chronic stress
increased norepinephrine and corticosterone levels in plasma and
gastric tissues, and markedly enhanced GC development, as
demonstrated in subcutaneous and orthotopic xenograft mouse models.
Previous studies have demonstrated that increased levels of
circulating catecholamines, particularly norepinephrine, serve a
crucial role in cancer development. In the present study, it was
demonstrated that mice implanted with an osmotic pump that releases
physiologically relevant doses of norepinephrine enhanced the
progression of GC. The mechanism by which chronic stress promotes
gastric tumorigenesis remains to be fully elucidated. Previous
studies have shown that chronic stress induces cancer initiation
and development by impairing the immune system, which is essential
for immune surveillance. These immune dysfunctions include
cytotoxicity in natural killer cells, cytokine production and T
cell mitogenesis (35). However,
chronic stress enhances tumor development in immunodeficient nude
mice. This indicates that additional mechanisms are involved in
chronic stress induced tumor development. The present study
demonstrated that the norepinephrine-induced activation of
autophagy in gastric cancer cells was responsible for the
progression of GC.
Autophagy is an important cellular process that
serves a critical role in tumor initiation and progression.
However, there is a lack of information regarding the effects of
chronic stress on gastric cancer cell autophagy. In the present
study, it was demonstrated that stress hormone norepinephrine
triggers gastric cancer cell autophagy. The induction of autophagy
was a novel finding of β2-adrenergic activation in GC cells, as
demonstrated by the appearance of double-membrane vesicles, the
punctuate of GFP-RFP-LC3 distribution in the cytoplasm and the
corresponding increase in the LC3-II/β-actin ratios. Additionally,
autophagic flux was monitored by protein degradation assays.
Enhanced autophagosome formation was induced by norepinephrine,
rather than the impairment of autophagosome degradation alone. As
autophagy is involved in the degradation of proteins, the
correlation between ADRB2 and autophagic marker p62/SQSTM1 protein
was investigated in GC tumor samples to determine its clinical
relevance. The TMA showed a negative correlation between the
expression of ADRB2 and p62/SQSTM1.
However, there is contradictory information with
regards to whether autophagy is a tumor promoter or tumor
suppressor. Under conditions of severe stress, autophagy increases
cell death (36), whereas, in
certain instances, autophagy formation is necessary for maintaining
normal physiology (11). The
underlying question with regards to autophagy in gastric cancer is
whether its role is detrimental or protective. The present study is
the first, to the best of our knowledge, to demonstrate the
protective effects of autophagy in norepinephrine-induced GC
progression in vitro and in vivo; the results shed
light on the importance of autophagy regulation in the development
of GC.
The signaling pathway downstream of stress-activated
ADRB2 for regulating autophagy remains to be elucidated. The
present study demonstrated that silencing ADRB2 reduced the
autophagy-related gene Beclin1 and impaired cell survival via
inactivation of the AMPK-ULK1 pathway. It is established that AMPK
serves a major role in autophagy and is activated by the increase
of cellular AMP/ATP ratios. This in-turn favors the binding of
adenine nucleotides to the γ subunit of AMPK (37). Previously, AMPK was reported to be
activated by protein kinase A (38). The present study demonstrated that
the knockdown of ADRB2 significantly attenuated the activation of
AMPK-ULK1 pathway induced by norepinephrine. This indicates that
the knockdown of ADRB2 may reduce the expression of
autophagy-related genes, including Beclin1, and inactivate the
autophagic AMPK-ULK1 pathway, which in turn, attenuates autophagy
and inhibits growth of human GC cells. High protein levels of ADRB2
correlated positively with TNM stage and predicted an unfavorable
prognosis. A recent meta-analysis also demonstrated that
non-selective β-blockers have the potential to decrease tumor
incidence by targeting the β-adrenergic receptor. Therefore,
β-adrenergic receptors, specifically ADRB2, may be a promising
therapeutic target for cancer treatment and prevention.
In conclusion, based on the subcutaneous and
orthotopic xenograft mouse models, the present study is the first,
to the best of our knowledge, to provide preclinical evidence of
the role of chronic stress in the progression of GC. The novel
mechanism proposed outlines the chronic stress-mediated development
of GG cancer. Insights from the present study advance current
understanding of the mechanisms involved in chronic stress,
β-adrenergic signaling, autophagy regulation and cancer
progression. Due the efficacy of β2-adrenergic modulation on GC
inhibition, the present study supports the potential use of an
adrenoceptor antagonist, such as propranolol, for the treatment of
GC.
Abbreviations:
|
GC
|
gastric cancer
|
|
ADRB2
|
β2-adrenergic receptor
|
|
DFS
|
disease-free survival
|
|
TMA
|
tissue microarray
|
|
IRS
|
immunore-activity score
|
|
PVDF
|
polyvinylidene fluoride
|
|
ADRB1
|
β1-adrenergic receptor
|
|
NC
|
negative control
|
|
TEM
|
transmission electron microscopy
|
|
TUNEL
|
terminal-deoxynucleoitidyl transferase
mediated nick end labeling
|
|
AMPK
|
adenosine 5′-monophosphate-activated
protein kinase
|
|
CREB
|
cAMP-response element binding
protein
|
|
PKA
|
protein kinase A
|
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81702369),the China
Postdoctoral Science Foundation (grant nos. 2018T110534 and
2016M601868), the Postdoctoral Science Foundation of Jiangsu
Province (grant no. 1701033B), the National Natural Science
Foundation of China (grant no. 81572362), the National Natural
Science Foundation Project of International Cooperation (grant no.
81361120398), the Primary Research and Development Plan of Jiangsu
Province (grant no. BE2016786), the Program for Development of
Innovative Research Team at the First Affiliated Hospital of NJMU;
the Priority Academic Program Development of Jiangsu Higher
Education Institutions (grant no. JX10231801), the 333 Project of
Jiangsu Province (grant no. BRA2015474), Jiangsu Key Medical
Discipline (General Surgery) and Jiangsu Key Lab of Cancer
Biomarkers, Prevention and Treatment, Collaborative Innovation
Center for Cancer Personalized Medicine, Nanjing Medical
University.
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
XZ, BL, ZL and JZ performed the experiments,
acquired the data and drafted the manuscript. JY and LZ collected
the tissue samples. ZX analyzed and interpreted the data. All
authors have read and approved the final manuscript and agreed to
be accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All applicable international, national, and/or
institutional guidelines for the care and use of animals were
followed. Animal research was approved by the Animal Care and Use
Committee of the Laboratory Animal Center of Nantong University.
All procedures performed in experiments involving human
participants were in accordance with the ethical standards of the
institutional and/or national research committee and with the 1964
Helsinki declaration and its later amendments or comparable ethical
standards. The present study was approved by the Institutional
Ethical Board of the First Affiliated Hospital of Nanjing Medical
University. Informed consent was obtained from all individual
participants included in the study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Chida Y, Hamer M, Wardle J and Steptoe A:
Do stress-related psychosocial factors contribute to cancer
incidence and survival? Nat Clin Pract Oncol. 5:466–475. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ng BH and Tsang HW: Psychophysiological
outcomes of health qigong for chronic conditions: A systematic
review. Psychophysiology. 46:257–269. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bulli F, Miccinesi G, Maruelli A, Katz M
and Paci E: The measure of psychological distress in cancer
patients: the use of Distress Thermometer in the Oncological
Rehabilitation Center of Florence. Support Care Cancer. 17:771–779.
2009. View Article : Google Scholar
|
|
4
|
Moussas GI, Papadopoulou AG,
Christodoulaki AG and Karkanias AP: Psychological and psychiatric
problems in cancer patients: relationship to the localization of
the disease. Psychiatriki. 23:46–60. 2012.In Greek. PubMed/NCBI
|
|
5
|
Shan T, Cui X, Li W, Lin W, Li Y, Chen X
and Wu T: Novel regulatory program for norepinephrine-induced
epithelial-mesen-chymal transition in gastric adenocarcinoma cell
lines. Cancer Sci. 105:847–856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moretti S, Massi D, Farini V, Baroni G,
Parri M, Innocenti S, Cecchi R and Chiarugi P: β-adrenoceptors are
upregulated in human melanoma and their activation releases
pro-tumorigenic cytokines and metalloproteases in melanoma cell
lines. Lab Invest. 93:279–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hassan S, Karpova Y, Baiz D, Yancey D,
Pullikuth A, Flores A, Register T, Cline JM, D'Agostino R Jr,
Danial N, et al: Behavioral stress accelerates prostate cancer
development in mice. J Clin Invest. 123:874–886. 2013.PubMed/NCBI
|
|
8
|
Armaiz-Pena GN, Allen JK, Cruz A, Stone
RL, Nick AM, Lin YG, Han LY, Mangala LS, Villares GJ, Vivas-Mejia
P, et al: Src activation by β-adrenoreceptors is a key switch for
tumour metastasis. Nat Commun. 4:14032013. View Article : Google Scholar
|
|
9
|
Thaker PH, Han LY, Kamat AA, Arevalo JM,
Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori
M, et al: Chronic stress promotes tumor growth and angiogenesis in
a mouse model of ovarian carcinoma. Nat Med. 12:939–944. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Konturek SJ, Brzozowski T, Konturek PC,
Zwirska-Korczala K and Reiter RJ: Day/night differences in
stress-induced gastric lesions in rats with an intact pineal gland
or after pinealectomy. J Pineal Res. 44:408–415. 2008. View Article : Google Scholar
|
|
11
|
Gewirtz DA: The four faces of autophagy:
Implications for cancer therapy. Cancer Res. 74:647–651. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lozy F and Karantza V: Autophagy and
cancer cell metabolism. Semin Cell Dev Biol. 23:395–401. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine B and Kroemer G: Autophagy in
aging, disease and death: The true identity of a cell death
impostor. Cell Death Differ. 16:1–2. 2009. View Article : Google Scholar :
|
|
15
|
Farah BL, Sinha RA, Wu Y, Singh BK, Zhou
J, Bay BH and Yen PM: β-adrenergic agonist and antagonist
regulation of autophagy in HepG2 cells, primary mouse hepatocytes,
and mouse liver. PLoS One. 9:e981552014. View Article : Google Scholar
|
|
16
|
Lizaso A, Tan KT and Lee YH: β-adrenergic
receptor-stimulated lipolysis requires the RAB7-mediated
autolysosomal lipid degradation. Autophagy. 9:1228–1243. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Funakoshi T, Aki T, Unuma K and Uemura K:
Lysosome vacuolation disrupts the completion of autophagy during
norephedrine exposure in SH-SY5Y human neuroblastoma cells. Brain
Res. 1490:9–22. 2013. View Article : Google Scholar
|
|
18
|
Eskelinen EL: The dual role of autophagy
in cancer. Curr Opin Pharmacol. 11:294–300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dikken JL, van de Velde CJ, Gönen M,
Verheij M, Brennan MF and Coit DG: The New American Joint Committee
on Cancer/International Union Against Cancer staging system for
adenocarcinoma of the stomach: Increased complexity without clear
improvement in predictive accuracy. Ann Surg Oncol. 19:2443–2451.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Meng D, Chen Y, Zhao Y, Wang J, Yun D,
Yang S, Chen J, Chen H and Lu D: Expression and prognostic
significance of TCTN1 in human glioblastoma. J Transl Med.
12:2882014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang S, Wu X, Chen Y, Zhang J, Ding J,
Zhou Y, He S, Tan Y, Qiang F, Bai J, et al: Prognostic and
predictive role of JWA and XRCC1 expressions in gastric cancer.
Clin Cancer Res. 18:2987–2996. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Zamora-González EO, Santerre A,
Palomera-Avalos V and Morales-Villagrán A: A chronic combinatory
stress model that activates the HPA axis and avoids habituation in
BALB/C mice. J Neurosci Methods. 213:70–75. 2013. View Article : Google Scholar
|
|
24
|
Bernabé DG, Tamae AC, Biasoli ER and
Oliveira SH: Stress hormones increase cell proliferation and
regulates interleukin-6 secretion in human oral squamous cell
carcinoma cells. Brain Behav Immun. 25:574–583. 2011. View Article : Google Scholar
|
|
25
|
Nilsson MB, Armaiz-Pena G, Takahashi R,
Lin YG, Trevino J, Li Y, Jennings N, Arevalo J, Lutgendorf SK,
Gallick GE, et al: Stress hormones regulate interleukin-6
expression by human ovarian carcinoma cells through a Src-dependent
mechanism. J Biol Chem. 282:29919–29926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu FQ, Fang T, Yu LX, Lv GS, Lv HW, Liang
D, Li T, Wang CZ, Tan YX, Ding J, et al: ADRB2 signaling promotes
HCC progression and sorafenib resistance by inhibiting autophagic
degradation of HIF1α. J Hepatol. 65:314–324. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D
and Li W: Beta-adrenergic signaling promotes tumor angiogenesis and
prostate cancer progression through HDAC2-mediated suppression of
thrombospondin-1. Oncogene. 36:1525–1536. 2017. View Article : Google Scholar
|
|
29
|
Hayakawa Y, Sakitani K, Konishi M, Asfaha
S, Niikura R, Tomita H, Renz BW, Tailor Y, Macchini M, Middelhoff
M, et al: Nerve growth factor promotes gastric tumorigenesis
through aberrant cholinergic signaling. Cancer Cell. 31:21–34.
2017. View Article : Google Scholar :
|
|
30
|
Xie CM, Liu XY, Sham KW, Lai JM and Cheng
CH: Silencing of EEF2K (eukaryotic elongation factor-2 kinase)
reveals AMPK-ULK1-dependent autophagy in colon cancer cells.
Autophagy. 10:1495–1508. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Komatsu M, Wang QJ, Holstein GR, Friedrich
VL Jr, Iwata J, Kominami E, Chait BT, Tanaka K and Yue Z: Essential
role for autophagy protein Atg7 in the maintenance of axonal
homeo-stasis and the prevention of axonal degeneration. Proc Natl
Acad Sci USA. 104:14489–14494. 2007. View Article : Google Scholar
|
|
32
|
Le CP, Nowell CJ, Kim-Fuchs C, Botteri E,
Hiller JG, Ismail H, Pimentel MA, Chai MG, Karnezis T, Rotmensz N,
et al: Chronic stress in mice remodels lymph vasculature to promote
tumour cell dissemination. Nat Commun. 7:106342016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Krizanova O, Babula P and Pacak K: Stress,
catecholaminergic system and cancer. Stress. 19:419–428. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang B, Chen H, Deng Y, Yi T, Wang Y and
Jiang Y: Diagnosis, disease stage, and distress of Chinese cancer
patients. Ann Transl Med. 4:732016.PubMed/NCBI
|
|
35
|
Reiche EM, Nunes SO and Morimoto HK:
Stress, depression, the immune system, and cancer. Lancet Oncol.
5:617–625. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xie CM, Chan WY, Yu S, Zhao J and Cheng
CH: Bufalin induces autophagy-mediated cell death in human colon
cancer cells through reactive oxygen species generation and JNK
activation. Free Radic Biol Med. 51:1365–1375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hardie DG: AMP-activated protein kinase:
An energy sensor that regulates all aspects of cell function. Genes
Dev. 25:1895–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li X, Yuan Z, Liu R, Hassan HM, Yang H,
Sun R, Zhang L and Jiang Z: UDCA and CDCA alleviate
17α-ethinylestradiol-induced cholestasis through PKA-AMPK pathways
in rats. Toxicol Appl Pharmacol. 311:12–25. 2016. View Article : Google Scholar : PubMed/NCBI
|