Introduction
Mitotane (also termed o,p′-DDD) is the only
drug approved for the treatment of metastatic adrenocortical
carcinoma (ACC) (1); however, its
molecular mechanisms of action remain to be fully elucidated. The
recommended therapeutic window of plasma mitotane levels in
patients is between 14 and 20 mg/l, corresponding approximately to
50 µM (1). We previously
reported that mitotane induces mitochondrial dysfunction in
NCI-H295R human adrenocortical cells, including respiratory chain
inhibition and mitochondrial fragmentation (2). Moreover, a mitochondrial uptake of
mitotane leading to cell apoptosis has been shown (3). We have also previously demonstrated
that mitotane disrupts the integrity of mitochondrial-associated
membranes (MAMs) using metabolomic, lipidomic and imaging
approaches (4). Indeed, MAMs
constitute pivotal intracellular structures controlling key
cellular processes, such as apoptosis, calcium homeostasis,
phospholipid metabolism, mitochondrial function, cholesterol
metabolism and steroid synthesis, notably in adrenocortical cells.
Recently, sterol-O-acyl transferase 1 (SOAT1), the enzyme
that metabolizes free cholesterol to cholesterol esters, was also
proposed as a new potential target of mitotane (5). Accordingly, Sbiera et al
(5) hypothesized that mitotane
could induce endoplasmic reticulum (ER) stress, through SOAT1
inhibition, leading to increased intracellular free cholesterol
concentrations followed by apoptosis. In steroidogenic cells, such
as adrenocortical cells, cholesterol metabolism plays a major role
since cholesterol is the main precursor for steroid biosynthesis.
There are at least 4 sources of free cholesterol in the
adrenocortical cell: An exogenous source of cholesteryl esters
(CEs) originating from: i) low-density lipoprotein (LDL) through
low-density lipoprotein receptor (LDL-R); and ii) high-density
lipoprotein (HDL) through scavenger receptor B (SrB1); iii) lipid
droplets; and iv) de novo cholesterol synthesis through
3-hydroxy-3-methyl-glutaryl-coenzyme A (HMGCoA) reductase (HMGCR)
activity, also known as the mevalonate pathway. Free cholesterol
may then be transported by steroidogenic acute regulatory protein
(StAR) to MAMs to be converted in pregnenolone by cytochrome P450,
family 11, subfamily A, polypeptide 1 (CYP11A1) whereas an efflux
of free cholesterol may also occur through ATP-binding cassette
transporter (ABCA1). Mitotane is a lipophilic molecule that
circulates either free or is bound to lipoproteins. Furthermore,
mitotane induces dyslipidemia with increased LDL, HDL and
triglycerides concentrations (6).
This dyslipidemia strikingly reduces mitotane efficacy in
vitro as demonstrated by the higher anti-proliferative and
pro-apoptotic effects of mitotane when NCI-H295R cells are cultured
in lipoprotein-free medium (3).
Moreover, this dyslipidemia leads to an overestimation of plasma
mitotane levels in patients (7),
and is generally treated by statins. Lastly, in a retrospective
study of 26 patients with ACC (3),
the combination of mitotane and statins was shown to be
significantly associated with a better tumor control according to
Response Evaluation Criteria In Solid Tumors criteria (RECIST)
(8). Thus, the mechanisms through
which statins may potentiate the effects of mitotane are therefore
considered of relevance for investigation.
Statins inhibit HMGCR and exert an
anti-proliferative effect in vitro on several cancer cell
lines, such as lung, prostate, breast, ovary, leukemia and myeloma
cells (9). These effects could be
linked to an inhibition of the mevalonate pathway (10); however, they have never been
investigated in adrenocortical cells to date, at least to the best
of our knowledge.
The aim of the present study was to evaluate the
effects of mitotane alone or in association with statins in
NCI-H295R human ACC cells.
Materials and methods
Human adrenocortical cell culture
For in vitro experiments, NCI-H295R (hereon
referred to as H295R) human ACC cells (from passage 7 to 12)
obtained from Gustave Roussy, Universite Paris Sud, Villejuif,
France and used in our previous studies (2-4,11),
were cultured as previously described (3). The H295R cells were cultured in
DMEM/HAM’S F-12 (PAA, Les Mureaux, France) supplemented with 20 mM
HEPES (Invitrogen, Life Technologies/Thermo Fisher Scientific,
Waltham, MA, USA), antibiotics (penicillin 100 IU/ml and
streptomycin 100 µg/ml) and 2 mM glutamine. The medium for
H295R cell culture was enriched with 10% fetal bovine serum and a
mixture of insulin/transferrin/selenium. The cells were cultured at
37°C in a humidified incubator with 5% CO2.
o,p′-DDD (Sigma-Aldrich, St. Louis, MO, USA), and
rosuvastatin (Sigma-Aldrich) were solubilized in dimethyl sulfoxide
(DMSO; Sigma-Aldrich) and used at the indicated concentrations
ranging from 0 to 100 µM. In all experiments, the percentage
of DMSO in the culture medium never exceeded 0.1% v/v. Given that
o,p′-DDD induces hepatic CYP3A4 activity (12), we selected rosuvastatin for use in
our experiments, a statin not metabolized by CYP3A4.
Cell viability and apoptosis
analysis
Cell viability assays were performed using WST1
assay (Roche, Basel, Switzerland) and apoptosis tests were
performed using the Caspase-Glo 3/7 assay (Promega, Madison, WI,
USA) according to the manufacturer’s recommendations. The cells
were cultured in 96-well plates and treated with 0-100 µM
mitotane alone or with rosuvastatin for various periods of time (0
to 72 h). The number of cells per well was 3 to 10×103.
Optical densities were measured 4 h after the addition of WST1
solution (10 µl per well) by spectrophotometry at 450 nm
(Viktor multilabel plate reader; PerkinElmer, Waltham, MA, USA).
The results were validated by cell counting with the cell counter
method (TC20 automated cell counter; Bio-Rad Laboratories,
Hercules, CA, USA). Luminescence was measured 1 h after the
addition of Caspase-Glo 3/7 solution (equal volume) by luminometry
(Viktor multilabel plate reader; PerkinElmer).
Western blot analysis
Total protein extracts were prepared and western
blot analyses were performed as previously described (11). Total protein extracts were prepared
from cells lysed in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM
NaCl, 5 mM EDTA, 30 mM Na pyrophosphate, 50 mM Na fluoride and 1%
Triton X-100) and 1X protease inhibitor (Sigma-Aldrich), 40
µg proteins were loaded by lane. Following protein blotting
on an Odyssey nitrocellulose membrane (LI-COR, Lincoln, NE, USA),
the blots were incubated for 1 h at room temperature in a blocking
buffer [5% fat-free milk in phosphate-buffered saline (PBS) with
0.1% Tween-20] before an overnight incubation at 4°C. Primary
antibodies were a rabbit polyclonal anti-poly(ADP-ribose)
polymerase (PARP) antibody (dilution, 1:200; #9542; Cell Signaling
Technology, Danvers, MA, USA). The antibody detected total PARP
(116 kDa) and cleaved PARP (89 kDa), the ratio of which reflects
the pro-apoptotic status. The normalizing antibody was the
anti-α-tubulin (dilution 1:1,000; AB_10013740; Sigma-Aldrich).
Secondary antibodies were goat anti-mouse IgG (H+L) cross adsorbed
secondary antibody (DyLight 680 conjugated, AB_614942) and goat
anti-rabbit IgG (H+L) DyLight 800 conjugated (dilution 1:10,000,
AB_614947) (both from Thermo Fisher Scientific).
Antibodies were diluted in PBS 0.1% Tween-20 buffer
5% non-fat milk and added to the membranes for 1 h at room
temperature or overnight at 4°C, followed by incubation with the
indicated secondary antibody for 1 h at room temperature. Target
proteins were detected using Odyssey Fc, Dual-Mode Western Imaging
(LI-COR) by fluorescence (680 nm wavelength for anti-mouse antibody
and 800 nm for anti-rabbit antibody) and quantified using the
Odyssey Fc Dual-mode Western Imaging apparatus from LI-COR as
indicated.
Intracellular free cholesterol
measurement
The concentrations of free intracellular cholesterol
were measured using the Cholesterol Quantification kit
(Sigma-Aldrich) according to the manufacturer’s recommendations.
The cells were cultured in F12 plates (4 wells per condition)
treated with mitotane and/or rosuvastatin for 24 h. In these
experiments, we used serum-free culture media to exclude exogenous
cholesterol uptake. The absorbance was read at 570 nm 1 h after the
addition of 2 µl of cholesterol probe and 2 µl of
cholesterol enzyme mixture per well. Cholesterol concentrations
were calculated according to the established standard curve and
normalized to the initial cell count.
Measurement of HMGCR activity
The measurement of HMGCR was carried out using
HMGCoA Reductase Assay (Sigma-Aldrich), according to the
manufacturer’s recommendations. Incubation medium included HMGCoA
(enzyme substrate), NADPH (reduced nicotinamide adenine
dinucleotide phosphate), buffer solution and HMG-CoA reductase
(provided in the kit). Specific absorbance at 340 nm was compared
in the presence or in the absence of pravastatin, a potent HMGCR
activity inhibitor. HMGCR activity was determined by the difference
in absorbance slope between these two conditions.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the H295R cells with
the RNeasy kit (Qiagen) according to the manufacturer’s
recommendations. A total of 1 µg total RNAwere subjected to
DNase I treatment (Invitrogen/Thermo Fisher Scientific) and
reverse-transcribed with 200 units of reverse transcriptase
(Superscript II, Invitrogen/Thermo Fisher Scientific). PCR was
performed with 100 ng cDNA in the presence of qPCR™ Mastermix Plus
for Sybr™-Green I (Eurogentec, Seraing, Belgium) containing 300 nM
of specific primers (Table SI).
qPCR was carried on an ABI Step One Plus (Applied Biosystems,
Foster City, CA, USA) whose parameters were as follows: A pre-cycle
at 95°C for 20 sec then 40 cycles at 95°C for 1 sec followed by 40
cycles at 60°C for 20 sec. The amount of cytochrome c
oxidase subunit II COX2 transcript in the samples was determined by
comparison with the standard range and related to the amount of the
18S gene of nuclear origin. For standards preparation, amplicons
were subcloned into pGEMT-easy plasmid (Promega) and sequenced to
confirm the identity of each sequence. Standard curves were
generated using serial dilutions of linearized standard plasmids.
Samples were amplified in duplicate or triplicate. Ribosomal 18S
was used as an internal control for data normalization. qPCR was
performed using the Fast SYBR Green Master Mix (Life
Technologies/Thermo Fisher Scientific) and carried out on a
QuantStudio 6 Flex (Life Technologies/Thermo Fisher Scientific).
The relative expression of each gene was expressed as the ratio of
attomoles of specific gene to attomoles of 36B4 mRNA or
femtomoles of 18S rRNA.
Steroidogenesis
Steroid measurements (progesterone, 17OHP) were
assayed in the cell supernatants under various conditions after
48-h treatment, by means of liquid chromatography (LC)-mass
spectrometry (MS)/MS analysis. LC-MS/MS was performed using a
Waters Xevo TQS triple-quadrupole mass spectrometer connected to a
Waters Acquity UPLC H-class (Waters SAS, Saint Quentin Yvelines,
France). Chromatographic separation was performed on a BEH C18
column (1.7 µm, 100×2.1) at a flow rate of 0.3 ml/min at
40°C. The mobile phase consisting of methanol and 5 mmol/l ammonium
formate in water was delivered according to the following gradient:
35% methanol from 0 to 1.5 min, linear increase to 45% methanol
(1.5-3 min), then to 63% methanol (3-7 min) followed by 100%
methanol (7-9 min). Following column washing with 100% methanol
(9-11.5 min), the gradient was reversed to reach initial conditions
at 14 min. The injection volume was 10 µl and the sample
manager was maintained at 10°C. Detection was performed on a Xevo
TQS tandem mass spectrometer (Waters, Paris, France). Instrument
optimization for the analytes was conducted by infusing standard
solution (100 pg/ml) of the analytes by the built-in syringe pump
at a flow rate of 10 µl/min. The following optimized
operating conditions were used for the multiple reaction monitoring
mode: Capillary voltage, 3.5 kV; cone voltage, 4-60 V; collision
energy, 15-34 eV; dwell time, 0.03-0.1 sec, depending on the
steroid. The mass spectrometer parameters were configured as
follows: Desolvation temperature, 500°C; desolvation nitrogen flow,
790 l/h; source temperature, 150°C; cone nitrogen flow, 145 l/h.
Argon was used as collision gas with a flow rate of 0.14 ml/min.
Two mass transitions were monitored for each steroid. System
control and data acquisition were achieved with the MassLynx 4.0
software (Waters). Cells were cultured in F6 plates (3 wells per
condition) treated with 25 µM mitotane (M25) and/or 50
µM rosuvastatin (R50) for 48 h.
Statistical analysis
Results are expressed as the means ± SEM of n
independent replicates performed in the same experiment or from
separate experiments (n). The non-parametric Mann-Whitney U test
was used when appropriate and differences between groups were
analyzed using non-parametric Kruskall-Wallis multiple comparison
tests followed by a post hoc Dunn’s test (Prism software, GraphPad,
CA, USA). A P-value of 0.05 was considered to indicate a
statistically significant difference.
Results
Effect of mitotane and rosuvastatin on
ACC cell viability and apoptosis
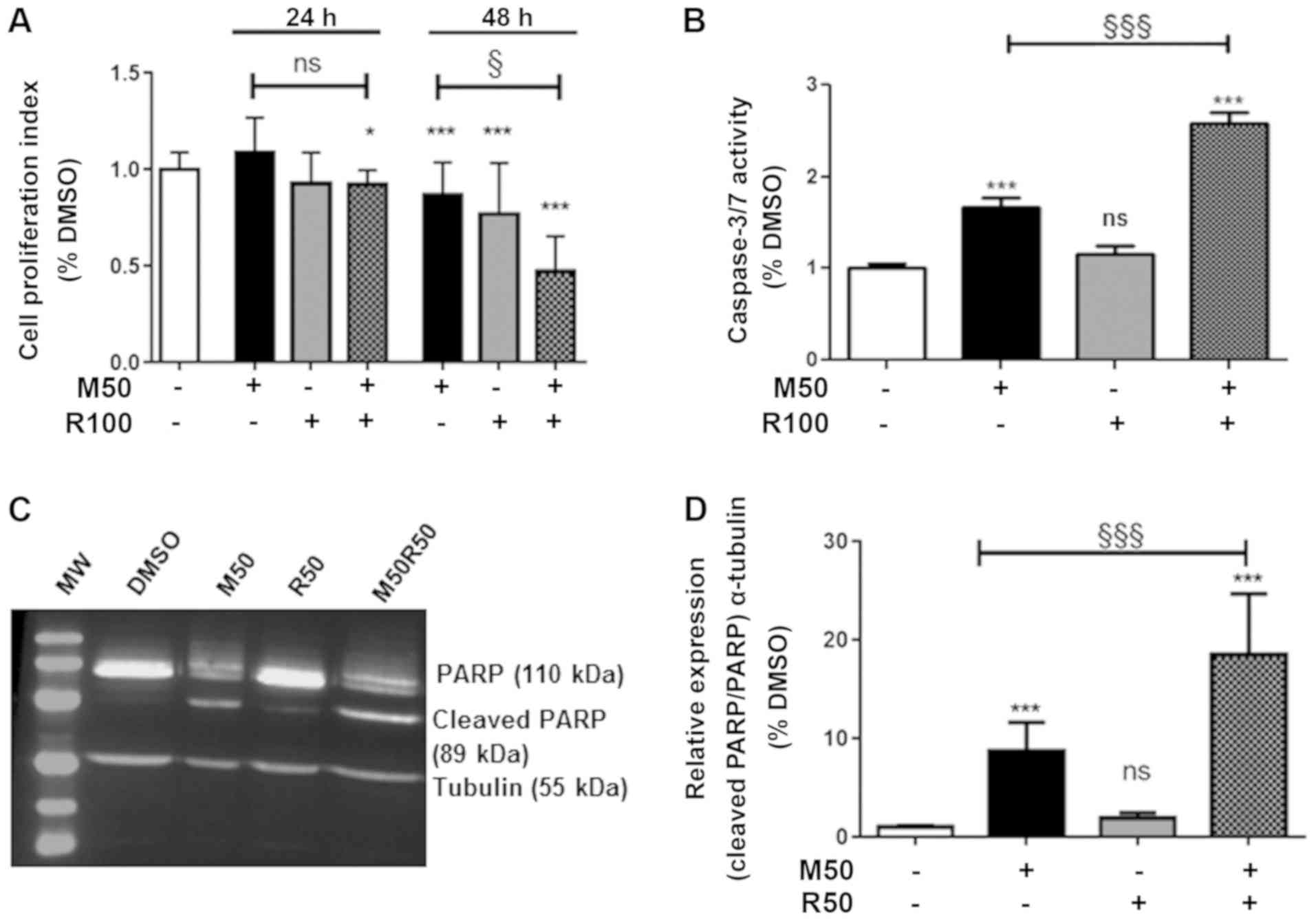
First, we examined the effects of mitotane or
rosuvastatin alone on cell viability at the concentration to 100
µM and at different time periods (up to 48 h). Mitotane (50
µM; M50) (Fig. 1A) reduced
the absorbance in a time-dependent manner, confirming its
anti-proliferative effect. Rosuvastatin alone also induced a
time-dependent inhibition of cell viability, providing support for
a specific effect of rosuvastatin. This effect, however, was only
observed at high concentrations starting from 50 µM (R50)
(Fig. S1). The combination of
mitotane (50 µM; M50) and rosuvastatin (100 µM; R100)
potentiated the inhibition of cell viability at 48 h (Fig. 1A), while rosuvastatin (100
µM) had no additive effect on the anti-proliferative effects
of mitotane at 72 h (Fig.
S2).
Using similar experimental conditions, we then
analyzed the index of H295R cell apoptosis using caspase-3/7
activity (Fig. 1B) and cleaved
PARP expression (Fig. 1C and D).
As expected, mitotane (50 µM; M50) alone induced a
significant increase in caspase-3/7 activity, while rosuvastatin
(100 µM; R100) alone had no effect. However, the combination
of mitotane and rosuvastatin led to an over-induction of
caspase-3/7 activity. Moreover, similar potentiation effects
between mitotane and rosuvastatin were observed when examining the
expression of cleaved PARP (Fig. 1C
and D), confirming the induction of H295R cell apoptosis when
both molecules were used in combination (M50 and R50). As the
effects of rosuvastatin were observed at 50 µM, this
concentration was considered as the most relevant for use in our
experiments.
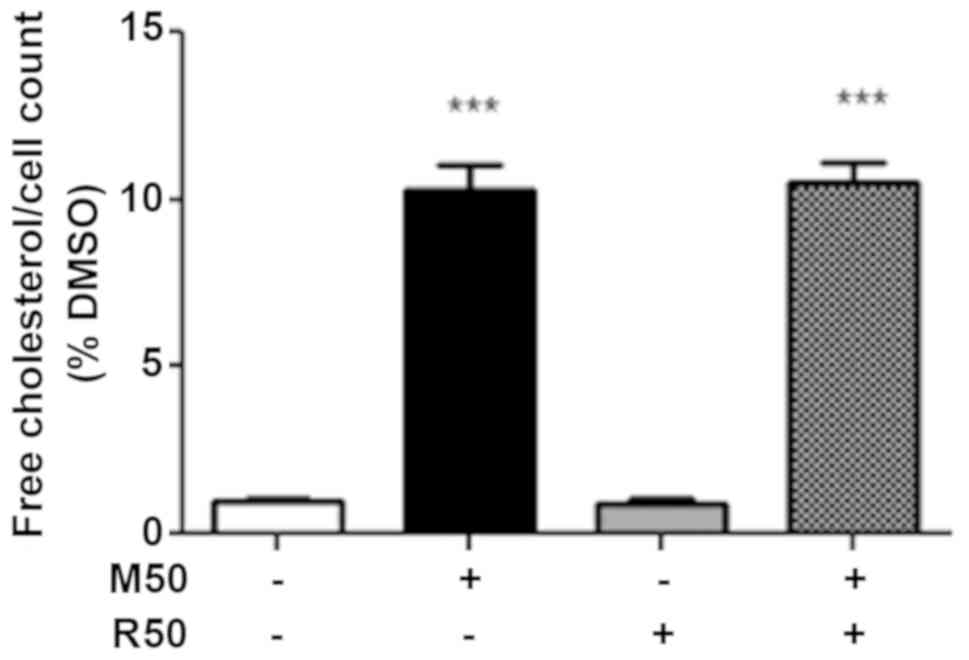
Effect of mitotane and rosuvastatin on
the intracellular free cholesterol concentration
Since statins and mitotane alter intracellular lipid
metabolism (9,13), in this study, we examined the
effects of mitotane, rosuvastatin and their association on the
level of intracellular free cholesterol, the precursor of
steroidogenesis. As shown in Fig.
2, we confirmed that mitotane (50 µM; M50) for 24 h
significantly increased the concentration of intracellular free
cholesterol, whereas rosuvastatin (50 µM; R50) alone had no
effect. However, no potentiating effect on the free intracellular
cholesterol concentration was observed when the cells were exposed
to both mitotane and rosuvastatin.
Mitotane does not alter HMGCR
activity
Since mitotane increases the intracellular free
cholesterol concentrations, we then sought to examine the
hypothesis that mitotane may directly increase the activity of
HMGCR and may thereby stimulate the mevalonate pathway.
HMGCR activity was measured at 1,815 U/mg protein
under control conditions and at 1,876 U/mg following the addition
of up to 100 µM mitotane, indicating that mitotane exerts no
direct effect on the activity of HMGCR at least in vitro.
The lack of a direct effect of mitotane in vitro associated
with the difficulties to accurately evaluate HMGCR activity (data
not shown) in the cells did not prompt us to examine the activity
in H295R cells under various experimental settings.
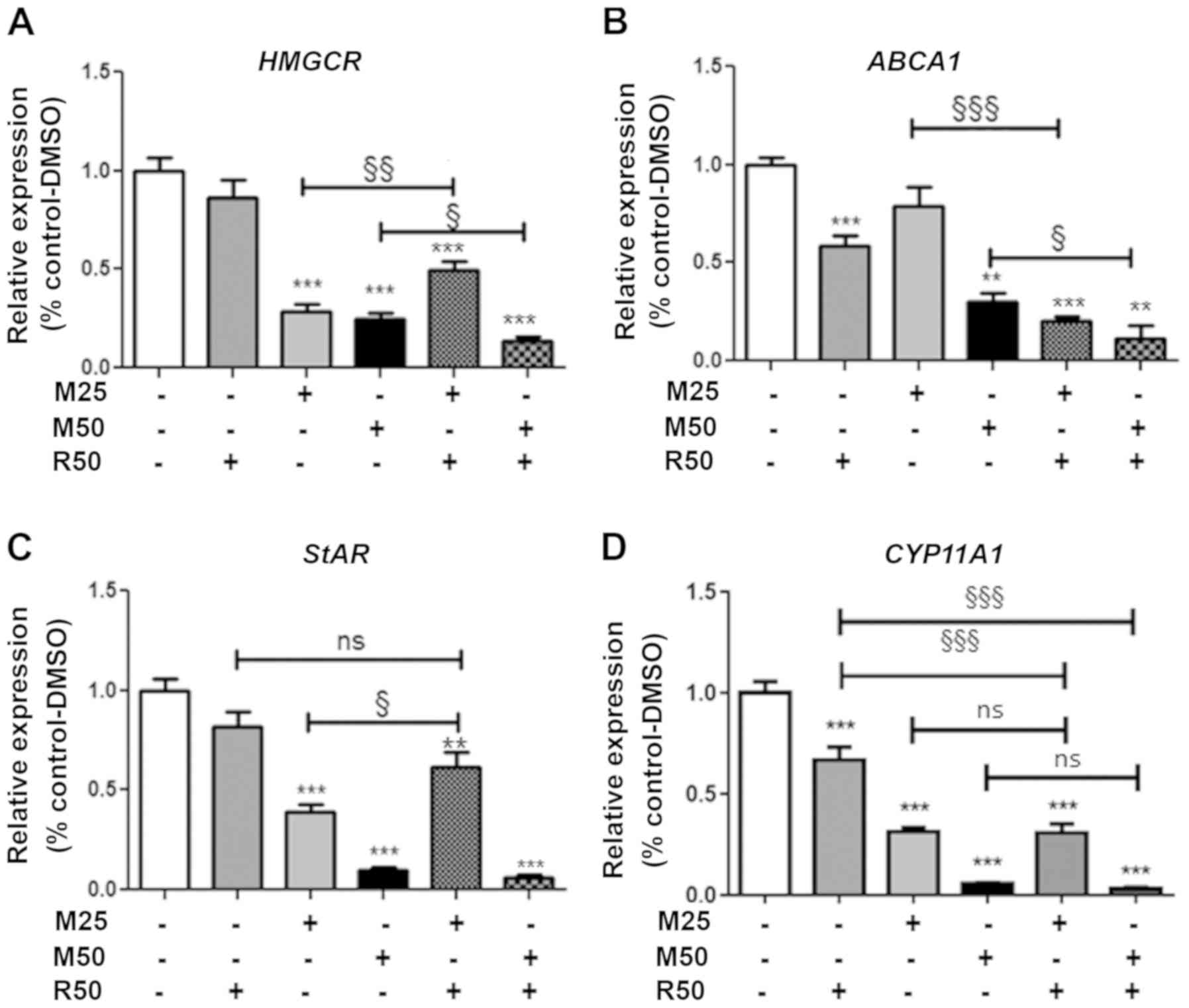
Effect of mitotane and rosuvastatin on
cholesterol metabolism-related gene expression
We then examined the expression of several genes
involved in cholesterol metabolism, including the HMGCR
gene, encoding a key player in the intracellular free cholesterol
balance, and the ABCA1 gene, encoding a protein that allows
cholesterol efflux from the cell.
Rosuvastatin (50 µM; R50) alone did not exert
any effect, but acted in combination with mitotane at 50 µM
to significantly reduce HMGCR expression (Fig. 3A). Rosuvastatin (50 µM; R50)
significantly reduced ABCA1 gene expression, alone or in
combination with mitotane 25 and 50 µM (Fig. 3B). However, rosuvastatin (50
µM) did not alter LDLR or SrB1 gene
expression, regardless of the duration of treatment, while mitotane
inhibited the expression of these genes (data not shown).
Effect of mitotane and rosuvastatin on
steroidogenesis
We then examined the expression of genes involved in
steroidogenesis, including StAR, encoding the cholesterol
transporter facilitating transfer to the mitochondria, and
CYP11A1, encoding the first limiting step of steroid
synthesis catalyzing cholesterol to pregnenolone (Fig. 3C and D). The expression of
StAR was significantly reduced by mitotane in a
concentration- (25 and 50 µM; M25 and M50). Rosuvastatin (50
µM; R50) did not exert any significant effect when used
alone, but slightly prevented the mitotane-induced reduction in
StAR expression. With respect to CYP11A1 expression,
mitotane alone significantly reduced its expression (25 and 50
µM; M25 and M50) (Fig. 3D).
Rosuvastatin (50 µM; R50) alone significantly reduced the
expression of CYP11A1.
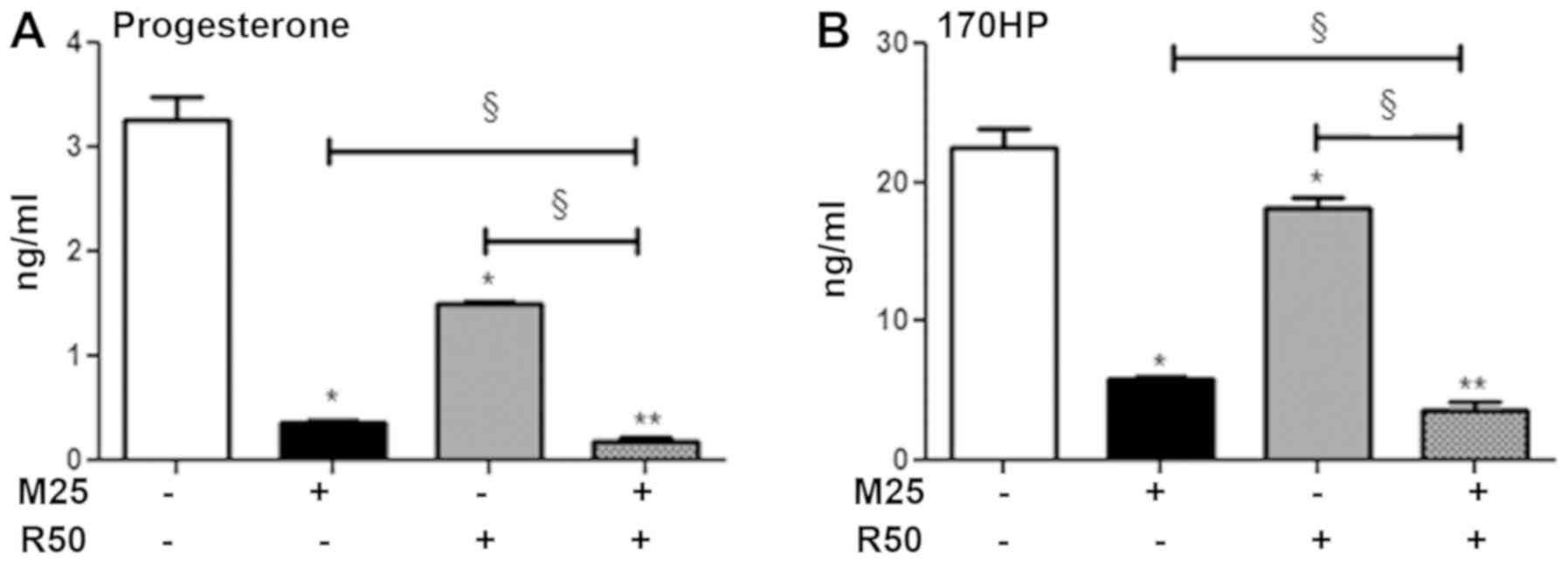
We then evaluated the steroid-secreting capacities
of the H295R cells (Fig. 4A). As
previously demonstrated (2,11),
mitotane (25 µM; M25) alone decreased the concentration of
cortisol and corticosterone in the supernatants of H295R cells
following 48 h of treatment (Fig.
S3). Rosuvastatin (50 µM; R50) alone decreased the
progesterone and 17OHP concentrations. The combination of mitotane
25 µM and rosu-vastatin 50 µM exhibited a significant
potentiation effect in inhibiting steroidogenesis, with
progesterone secretion reduced by 47% (Fig. 4A) and that of 17OHP reduced by 37%
(Fig. 4B).
A visual summary of the mechanisms of action of
mitotane and rosuvastatin in the H295R cells described in this
study is presented in Fig. 5.
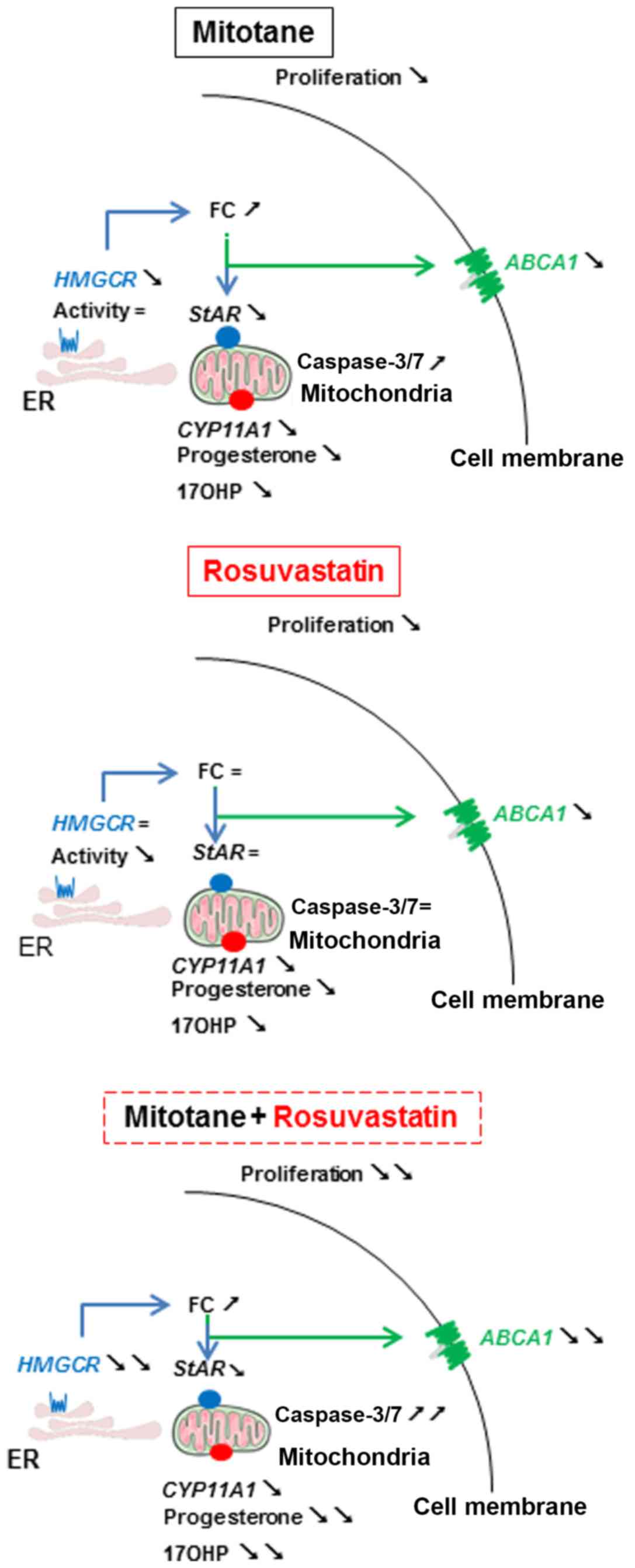 | Figure 5Mechanisms of action of mitotane and
rosuvastatin in H295R cells. Arrows facing to the bottom right
indicate decreased expression, concentration or activity compared
to DMSO. Arrows facing to the top and right indicate an increased
expression, concentration or activity compared to DMSO. The ‘=’
symbol indicates the same expression, concentration or activity
compared to DMSO. FC, free cholesterol; LDL-R, LDL receptor; SrB1,
scavenger receptor B1; ABCA1, ATP-binding cassette transporter; ER,
endoplasmic reticulum; HMGCR or HMGCoA reductase:
3-hydroxy-3-methyl-glu-taryl-coenzyme A reductase; StAR,
steroidogenic acute regulatory protein; CYP11A1, cytochrome P450,
family 11, subfamily A, polypeptide 1; 17OHP, 17
hydroxyprogesterone. |
Discussion
The objective of this study was to better understand
the effects of mitotane and rosuvastatin in H295R human
adrenocortical carcinoma cells. The main results are summarized in
Fig. 5. In this study, we
confirmed that mitotane induced apoptosis, reduced cell viability
and inhibited steroidogenesis (13,14)
We also confirmed that mitotane induced an increase in the
intracellular free cholesterol concentration, as recently described
by Sbiera et al (5). To
gain further insight into the mechanisms involved, we examined the
effect of mitotane on the expression of genes involved in
cholesterol metabolism. Mitotane significantly decreased the
expression of genes involved in the cellular intake of exogenous
cholesterol, such as LDLR and SrB1 (data not shown),
but also in de novo cholesterol synthesis (HMGCR). On
the other hand, mitotane reduces the expression of ABCA1,
which is involved in the cellular efflux of cholesterol (14) and inhibits SOAT1 (5), which esterifies free cholesterol. One
limitation of this study was that no western blot analysis was
carried out to confirm/complete our findings. The observed increase
in the intracellular free cholesterol concentration shows the
predominance of the effects of the latter over those leading to
decrease the cholesterol concentration. These observations are
reminiscent of the mechanisms of action of ATR-101, a potent
inhibitor of SOAT1 (15) and ABCA1
(14) and currently under clinical
development (phase II) for the treatment of adrenocortical
carcinoma (Atterocor Inc., Ann Arbor, MI, USA) (16). We hypothesized that mitotane
induces MAM dysfunction (4),
together with a decrease in steroidogenesis via CYP11A1
inhibition, an increase in free cholesterol inducing ER stress via
TSPO, as well as SOAT1 inhibition and an increase in
intramitochondrial calcium responsible for apoptosis (2,13).
Statins might play a relevant role in oncology as
they induce antiproliferative effects in vivo (9). For the first time in this study, at
least to the best of our knowledge, we examined the effect of
rosuvastatin in ACC cells. Rosuvastatin alone reduced cell
viability at high concentrations, without inducing apoptosis, nor
altering intracellular free cholesterol. Other cell lines in
vitro (breast and glioblastoma cells) have been used to
demonstrate the anti-proliferative properties of rosuvastatin at
similar concentrations (IC50 between 18 and 75 µM
rosuvastatin) (17). The lack of
an effect of statins on intracellular free cholesterol has already
been shown in H295R cells with simvastatin (18), whereas a decrease of HMGCoA
reductase activity would be expected. Thus, we may assume that the
role of the HMGCoA reductase pathway is negligible in
adrenocortical cells.
In this study, we demonstrated that in vitro,
rosuvastatin potentiated the effects of mitotane by increasing
apoptosis and decreasing cell viability. However, the underlying
mechanisms remain unknown. Alternate mechanisms, such as autophagy
or necroptosis could be involved and thus further investigations
are required into this issue. Partial potentiation also occurred
for the inhibition of the expression of HMGCR and
ABCA1, but no effect was observed on LDLR and
SrB1 expression (data not shown) nor on intracellular free
cholesterol. Taken together, these observations confirm a
potentiation effect of rosuvastatin on mitotane action at the
cellular level explained by the inhibition of genes involved in
cholesterol metabolism, while no argument supports an influence of
rosuvastatin on either mitotane capture or efflux. We have
previously demonstrated that a mitochondrial uptake of mitotane
significantly increased when cells are cultured with BLT1, an SrB1
receptor inhibitor suggesting an involvement of SrB1 in mitotane
efflux (3). However, in the
present study, no effect on mitochondrial mitotane concentrations
was observed when the cells were exposed to both mitotane and
rosuvastatin.
Statins are already prescribed in clinical practice
for the treatment of mitotane-induced dyslipidemia (6). Such dyslipidemia reduces the efficacy
of mitotane (3) and overestimates
plasma mitotane level measurements (7). This study demonstrated that
rosuvastatin also had a direct effect at the cellular level.
Statins could therefore be used for their dual action on mitotane
transport and bioavailability by reducing lipoprotein
concentrations, thus facilitating mitotane efficacy in vivo
(3) as well as potentiating
cellular action of mitotane. However, not all statins are suitable
for such a combined treatment, given that mitotane activates
hepatic cytochrome CYP3A4, statins that are not metabolized by this
cytochrome seem to be more appropriate for combined therapy
(12). Based on the findings of
this study, rosuvastatin seems to be a good candidate given its
potentiating action with mitotane in vitro. Further
prospective studies are warranted to explore the potential benefits
of combining mitotane and statins in patients with ACC treated with
mitotane.
To conclude, this study demonstrates a potentiating
action of mitotane and rosuvastatin in H295R cells. The clinical
benefit of this combination remains to be validated in patients
and, if confirmed, should lead to a better management of patients
with ACC.
Supplementary Materials
Funding
This study was supported in part by grants from
Institut National de la Santé et de la Recherche Médicale (Inserm)
and Université Paris-Sud.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
GB performed and analyzed cell viability, apoptosis
analysis, intracellular free cholesterol measurements, RT-qPCR
experiments. LA and AN performed and analyzed PARP expression by
western blotting. AS and AP performed and analyzed mitotane
measurements. AL performed and analyzed HMGCoA reductase activity.
EP performed the LC-MS/MS experiments. EB, SH and ML designed the
study. GB, ML and SH interpreted the data, and were major
contributors to the writing the manuscript. EB contributed to the
drafting of the manuscript. All authors have read and approved the
version to be published and approved its submission.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Acknowledgments
GB was a recipient of an HRA PHARMA/SFE (Société
Française d’Endocrinologie) fellowship.
References
|
1
|
Berruti A, Baudin E, Gelderblom H, Haak
HR, Porpiglia F and Fassnacht M: Adrenal cancer: ESMO Clinical
Practice Guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 23(Suppl 7): vii131–vii138. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hescot S, Slama A, Lombès A, Paci A, Remy
H, Leboulleux S, Chadarevian R, Trabado S, Amazit L, Young J, et
al: Mitotane alters mitochondrial respiratory chain activity by
inducing cytochrome c oxidase defect in human adrenocortical cells.
Endocr Relat Cancer. 20:371–381. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hescot S, Seck A, Guerin M, Cockenpot F,
Huby T, Broutin S, Young J, Paci A, Baudin E and Lombès M:
Lipoprotein-free mitotane exerts high cytotoxic activity in
adrenocortical carcinoma. J Clin Endocrinol Metab. 100:2890–2898.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hescot S, Amazit L, Lhomme M, Travers S,
DuBow A, Battini S, Boulate G, Namer IJ, Lombes A, Kontush A, et
al: Identifying mitotane-induced mitochondria-associated membranes
dysfunctions: Metabolomic and lipidomic approaches. Oncotarget.
8:109924–109940. 2017. View Article : Google Scholar
|
|
5
|
Sbiera S, Leich E, Liebisch G, Sbiera I,
Schirbel A, Wiemer L, Matysik S, Eckhardt C, Gardill F, Gehl A, et
al: Mitotane inhibits sterol-O-acyl transferase 1 triggering
lipid-mediated endoplasmic reticulum stress and apoptosis in
adrenocortical carcinoma cells. Endocrinology. 156:3895–3908. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shawa H, Deniz F, Bazerbashi H, Hernandez
M, Vassilopoulou-Sellin R, Jimenez C and Habra MA: Mitotane-induced
hyper-lipidemia: A retrospective cohort study. Int J Endocrinol.
2013:6249622013. View Article : Google Scholar
|
|
7
|
Paci A, Hescot S, Seck A, Jublanc C,
Mercier L, Vezzosi D, Drui D, Quinkler M, Fassnacht M, Bruckert E,
et al: Dyslipidemia causes overestimation of plasma mitotane
measurements. Endocrinol Diabetes Metab Case Rep.
2016:1501352016.PubMed/NCBI
|
|
8
|
Eisenhauer EA, Therasse P, Bogaerts J,
Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S,
Mooney M, et al: New response evaluation criteria in solid tumours:
Revised RECIST guideline (version 1.1). Eur J Cancer. 45:228–247.
2009. View Article : Google Scholar
|
|
9
|
Pisanti S, Picardi P, Ciaglia E,
D’Alessandro A and Bifulco M: Novel prospects of statins as
therapeutic agents in cancer. Pharmacol Res. 88:84–98. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clendening JW, Pandyra A, Boutros PC, El
Ghamrasni S, Khosravi F, Trentin GA, Martirosyan A, Hakem A, Hakem
R, Jurisica I, et al: Dysregulation of the mevalonate pathway
promotes transformation. Proc Natl Acad Sci USA. 107:15051–15056.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hescot S, Paci A, Seck A, Slama A,
Viengchareun S, Trabado S, Brailly-Tabard S, Al Ghuzlan A, Young J,
Baudin E, et al: The lack of antitumor effects of o,p′DDA excludes
its role as an active metabolite of mitotane for adrenocortical
carcinoma treatment. Horm Cancer. 5:312–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takeshita A, Igarashi-Migitaka J, Koibuchi
N and Takeuchi Y: Mitotane induces CYP3A4 expression via activation
of the steroid and xenobiotic receptor. J Endocrinol. 216:297–305.
2013. View Article : Google Scholar
|
|
13
|
Touitou Y, Moolenaar AJ, Bogdan A, Auzéby
A and Luton JP: o,p′-DDD (mitotane) treatment for Cushing’s
syndrome: Adrenal drug concentration and inhibition in vitro of
steroid synthesis. Eur J Clin Pharmacol. 29:483–487. 1985.
View Article : Google Scholar
|
|
14
|
Burns VE and Kerppola TK: ATR-101 inhibits
cholesterol efflux and cortisol secretion by ATP-binding cassette
transporters, causing cytotoxic cholesterol accumulation in
adrenocortical carcinoma cells. Br J Pharmacol. 174:3315–3332.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
LaPensee CR, Mann JE, Rainey WE, Crudo V,
Hunt SW III and Hammer GD: ATR-101, a selective and potent
inhibitor of Acyl-CoA acyltransferase 1, induces apoptosis in H295R
adre-nocortical cells and in the adrenal cortex of dogs.
Endocrinology. 157:1775–1788. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
U.S National Library of Medicine: A Study
of ATR-101 for the Treatment of Endogenous Cushing’s Syndrome.
(Identification No. NCT03053271). https://clinicaltrials.gov/ct2/show/NCT03053271
Accessed February 15, 2019.
|
|
17
|
Jiang P, Mukthavaram R, Chao Y, Nomura N,
Bharati IS, Fogal V, Pastorino S, Teng D, Cong X, Pingle SC, et al:
In vitro and in vivo anticancer effects of mevalonate pathway
modulation on human cancer cells. Br J Cancer. 111:1562–1571. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guldvang A, Hansen CH, Weisser JJ,
Halling-Sørensen B and Styrishave B: Simvastatin decreases steroid
production in the H295R cell line and decreases steroids and FSH in
female rats. Reprod Toxicol. 58:174–183. 2015. View Article : Google Scholar : PubMed/NCBI
|