Introduction
Androgen deprivation therapy (ADT) is considered a
milestone in the treatment of advanced prostate cancer. Although
the majority of cases of prostate cancer initially exhibit an
apparently good response to ADT, almost all treatments eventually
fail and the disease progresses to metastatic castration-resistant
prostate cancer (CRPC), which is the leading cause of mortality
(1-3). Recently, several novel agents, such
as the second-generation androgen receptor (AR) antagonist,
enzalutamide, have been administered to patients and are considered
to have better therapeutic effects than conventional AR
antagonists, such as bicalutamide and flutamide (4-8).
However, the majority of patients become resistant to all these
agents within a few years. To date, several molecular mechanisms
have been implicated in the progression of castration resistance,
the majority of which are associated with the AR signaling axis,
involving AR amplification, mutations, coregulators, activation,
aberrant post-translational modification and alternative splicing
(1,7,9,10).
However, it remains unclear as to whether other signaling pathways
contribute to resistance or can be targeted, and if so, which ones
and to what extent.
Caveolin-1 (Cav-1) is a scaffold protein of
caveolae, which are 50-100 nm Ω-shaped invaginations of the plasma
membrane in the majority of cell types (11). Previous studies have demonstrated
that Cav-1 is involved in the regulation of cholesterol homeostasis
and interacts with multiple signaling molecules, including small
GTPases, Src tyrosine kinases and endothelial nitric oxide synthase
(11,12). Numerous studies have also
demonstrated that Cav-1 is overexpressed in numerous human tumors
and is associated with poor clinical outcomes (13-15).
However, the tumorigenic effects of Cav-1 in various types of
cancer at different stages are controversial (16). Although Hehlgans and Cordes also
reported that radio- and chemoresistance were associated with
increased Cav-1 levels in certain types of cancer (17), the effects of Cav-1 on the
progression from primary prostate cancer (PPC) to CRPC remain
largely unknown.
Phosphoinositide-specific phospholipase Cε (PLCε),
the homolog of C. elegans PLC210, is a member of the human
phosphatidylinositol PLC family. PLCε is unique compared with other
phospholipase C isoforms in terms of its N-terminal GTP exchange
factor (GRF CDC25-like) domain and two C-terminal Ras-associating
(RA) domains, which reveals that it functions by activating Ras
family GTPases, and is also able to be regulated by Ras family
GTPases (18). Ras oncogenes are
involved in a high proportion of cancer types (19). However, the mechanisms through
which the Ras direct signaling effector, PLCε, contributes to tumor
development and is associated with Ras in this context, have not
yet been elucidated. It has been demonstrated that PLCε contributes
to carcinogenesis in different types of tumors (20). Consistent with these data, our
previous studies revealed that PLCε expression was significantly
higher in urologic neoplasms and promoted AR nuclear translocation
in prostate cancer (21,22). However, further studies are
required to determine whether the Ras/PLCε signaling pathway
regulates malignant progression and drug resistance in CRPC and to
elucidate the underlying mechanisms of action.
The aim of the present study was to examine whether
Cav-1 plays a key role in regulating metastasis and sensitization
to AR antagonists through the Ras/PLCε signaling cascade, resulting
in the development of CRPC, and whether the downregulation of the
expression of Cav-1 using cholesterol inhibitors can suppress this
lethal progression. For this purpose, a comprehensive analysis was
conducted using tissue and serum specimens from patients with PPC
and CRPC, as well as different cell lines in vitro to
determine treatment strategies using cellular models. Taken
together, the findings of this study may contribute to the
clarification of the critical biological pathways involved in
metastatic CRPC and may aid in the development of novel strategies
for the prevention and treatment of CRPC.
Materials and methods
Serum and tissue samples
A total of 70 PPC and 56 CRPC serum samples were
collected from patients prior to surgery between December, 2016 and
August, 2018, and 45 PPC and 36 CRPC tissue samples were collected
from patients between January, 2010 and August, 2018 at The First
Affiliated Hospital of Chongqing Medical University, Chongqing,
China. All the participants were informed of the aims and
procedures of the study. Patients with CRPC were selected according
to the European Association of Urology (EAU) guidelines (23). The present study retrospectively
collected clinical data, such as age, prostate-specific antigen
(PSA) levels and metastatic status from medical records. The study
was approved by the Ethics Committee of Chongqing Medical
University and was conducted according to the principles of the
Declaration of Helsinki.
Immunohistochemistry assay
All tissue samples were fixed in 10%
paraformaldehyde for 12 h at 25°C. They were then embedded in
paraffin and cut into 5-µm-thick sections.
Immunohistochemical staining was detected using a standard
immunoperoxidase staining procedure. The sections were incubated
overnight at 4°C with anti-Cav-1, (1:200, cat. no. 3267; Cell
Signaling Technology, Inc., Danvers, MA, USA) or anti-H-Ras (1:50,
cat. no. sc-53958), anti-K-Ras (1:50, cat. no. sc-521), anti-PLCε
(1:50, cat. no. sc-28402) (all from Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), followed by incubation with a secondary antibody
(1:100, SP9000 or PV9003; ZSGB-BIO, Beijing, China) at 37°C for 1
h, then sequentially incubated with avidinbiotin complex solution
(room temperature, 1 h). Protein expression was detected by
coloration with diaminobenzidine (DAB) (ZLI-9018; ZSGB-BIO) buffer
for 5 min, and the sections were counterstained with hematoxylin
for 5 min at room temperature. All images were captured using a
fluorescent microscope (Nikon Corp., Tokyo, Japan). The staining
intensity was scored as follows: 0 (no staining), 1 (light
staining), 2 (moderate staining) and 3 (strong staining). The
immunoreactivity ratio was scored as follows: 0 (0% immunoreactive
cells), 1 (<5% immunoreactive cells), 2 (5-50% immunoreactive
cells) and 3 (>50% immunoreactive cells). The final score was
defined as the sum of both parameters. Scores ≤2 denoted a negative
expression, while scores ≥3 denoted a positive expression.
Enzyme-linked immunosorbent assay
(ELISA)
Serum samples were centrifuged at 1,000 × g for 10
min (room temperature) and stored at -80°C until analysis. The
concentrations of Cav-1 were determined using a human Cav-1 ELISA
kit (detection range, 0.15-10 ng/ml; Signalway Antibody LLC,
College Park, MD, USA), according to the manufacturer’s
instructions. The absorbance was measured at a wavelength of 450±2
nm to detect the concentration of Cav-1 in serum using a microplate
reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Cells, cell culture and treatment
The human prostate cancer cell lines, LNCaP
(CRL-1740™), PC3 (CRL-1435™) and DU145 (HTB-81™), were obtained
from the American Type Culture Collection (ATCC, Manassas, VA,
USA). Castration-resistant derivatives of LNCaP cells
[specifically, bicalutamide-resistant cells (Bic-R) and
enzalutamide-resistant cells (En-R)] were constructed and
maintained as previously described (24). All cells were cultured in RPMI-1640
medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 1% penicillin and streptomycin antibiotics (Beyotime
Institute of Biotechnology, Haimen, China) and 10% fetal bovine
serum (Thermo Fisher Scientific, Inc.) at 37°C in a humidified
atmosphere of 5% CO2.
For transfection, 1×105 cells were seeded
in a 6-well plate and passaged every 2 days. When appropriate,
i.e., at 1 day after seeding or when the cell cultures were 40-60%
confluent, the cells were transfected with 4 µg Cav-1
knockdown plasmids or negative control (Shanghai GenePharma Co.,
Ltd., Shanghai, China) and 10 µl Lipofectamine 2000 (Thermo
Fisher Scientific, Inc.) in serum-free medium, according to the
manufacturer’s instructions. The transfection medium was replaced
with fresh culture medium after 6-8 h at 37°C. The transfection
process of the H-RasG12V or K-RasG12V plasmids (Shanghai GenePharma
Co., Ltd.) was similar to that of the Cav-1 knockdown plasmids.
However, for the transduction of PLCε knockdown lentiviral
particles, 1×105 cells were seeded in 6-well plates and
incubated with 3 ml complete medium until 40-60% confluency, and
subsequently 3 µl of PLCε knockdown lentivirus or negative
control lentivirus (Shanghai GenePharma Co., Ltd.) and 3 ml of
Polybrene (Thermo Fisher Scientific) were added to the culture
medium for 8 h. The original medium was then replaced with 6 ml
fresh medium containing 1 µg/ml puromycin. The cells were
harvested after 72 h and used for protein extraction, or were
cultured for 48 h and used for RNA extraction or in other
experiments.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted from all cell lines using TRIzol
reagent, and 1 µg RNA was reverse transcribed into
complementary DNA using the Prime Script™ RT reagent kit, according
to the manufacturer’s instructions (Takara Biotechnology Co., Ltd.,
Dalian, China). Messenger RNA (mRNA) levels were analyzed using the
SYBR PremixEx Taq™ II kit (Takara Biotechnology Co., Ltd.) and the
CFX96™ Real-Time PCR Detection System (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The thermocycling conditions were as previously
described (24), with the
exception of the annealing temperature. For Cav-1 analysis, the
annealing temperature was 52°C, while for H-RasG12V and K-RasG12V
analysis, the annealing temperature were 56°C and 60°C. The mRNA
expression levels were calculated using the comparative
2−ΔΔCq method (25) and
GAPDH was used as a calibrator. The primers used for the human
Cav-1, H-RasG12V, K-RasG12V and GAPDH genes were as follows: Cav-1
(107 bp), forward (F), 5′-CATCCCGATGGCACTCATCTG-3′ and reverse (R),
5′-TGCACTGAATCTCAATCAGGAAG-3′; H-RasG12V (129 bp), F,
5′-CTGAGGAGCGATGACGGAA-3′ and R, 5′-AGGCTCACCTCTATAGTGGG-3′;
K-RasG12V (165 bp), F, 5′-AAGGCCTGCTGAAAATGACTG-3′ and R,
5′-GGTCCTGCACCAGTAATATGCA-3′; and GAPDH (258 bp), F,
5′-AGAAGGCTGGGGCTCATTTG-3′, R, 5′-AGGGGCCAT CCACAGTCTTC-3′.
Reagents and treatment
Simvastatin (S1792; Selleck Chemicals, Houston, TX,
USA) was activated using absolute ethanol and adjusted to a final
concentration of 10 mM with PBS (pH 7.2). Methyl-β-cyclodextrin
(M-β-CD; C4555; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was
solubilized to 30 mM in PBS. Cholesterol (C3045; Sigma-Aldrich;
Merck KGaA) was solubilized to 10 mM in ethanol. All these
procedures were performed according to the manufacturer’s
instructions and the reagents were stored at −20°C.
Western blot analysis
Total protein extraction and subcellular
fractionations were performed as previously described (26,27),
although certain modifications were introduced to extract the
detergent-resistant fraction (DRF) from lipid-rich membranes and
the detergent-soluble fraction (DSF). Cell pellets were lysed in 1%
Triton X-100, 25 mM Tris (pH 8.0), 150 mM NaCl, 10 mM
Na-pyrophosphate, 10 mM NaF, 3 mM EDTA, 1 mM Phenylmethanesulfonyl
fluoride (PMSF), 10 µg/ml aprotinin, 1 mM benzamidine and 1
mM sodium orthovanadate for 10 min at 4°C. An ultrasonic homogenate
step was included to more finely disrupt cellular membranes, and
the homogenate was incubated for 30 min on ice and then centrifuged
at 100,000 × g for 30 min at 4°C. The insoluble pellets (containing
the DRF) and the supernatant (containing the DSF) were resuspended
by boiling in SDS-PAGE sample buffer.
Western blot analysis was performed as previously
described (24,28). The intensity analyses were
quantified using Image-Pro Plus 6.0 software. The membranes were
incubated with primary antibodies overnight at 4°C, then
sequentially incubated with secondary antibodies for 2 h at room
temperature. The primary and secondary antibodies used were as
follows: Anti-Cav-1 (1:1,000, cat. no. 3267), anti-matrix
metalloproteinase 2 (MMP2) (1:1,000, cat. no. 40994), anti-matrix
metalloproteinase 9 (MMP9) (1:1,000, cat. no. 13667), anti-Snail
(1:1,000, cat. no. 3879) and anti-GAPDH (1:1,000, cat. no. 5174)
antibodies were obtained from Cell Signaling Technology. Anti-H-Ras
(1:500, cat. no. sc-53958), anti-K-Ras (1:500, cat. no. sc-521),
anti-3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase
(HMGR) (1:500, cat. no. sc-271595) and anti-PLCε (1:500, cat. no.
sc-28402) antibodies were obtained from Santa Cruz Biotechnology.
Goat anti-mouse IgG (1:3,000, cat. no. SA00001-1), goat anti-rabbit
IgG (1:3,000, cat. no. SA00001-2) and rabbit anti-goat IgG
(1:3,000, cat. no. SA00001-4) were obtained from ProteinTech
(Rosemont, IL, USA).
Cell Counting kit-8 (CCK-8) assay
The cells (2,000 cells/well) examined for cell
viability using the CCK-8 assay were seeded in a 96-well plate and
incubated for 12 h at 37°C. Following 24-72 h of treatment with
various agents or concentrations, CCK-8 reagent solution (10
µl; Beyotime Institute of Biotechnology) was added to each
well followed by incubation for 2 h at 37°C. The absorbance was
measured at 450 nm using a microplate reader (Bio-Rad Laboratories,
Inc.). Cell viability data were calculated as percentage values for
each treatment condition compared with the control. The synergistic
effects of simvastatin and AR antagonists was calculated using
CalcuSyn® software version 2.0 (Biosoft, Cambridge, UK),
and the combination index (CI) quantitatively depicted additive
effects (CI=1), synergism (CI<1) or antagonism (CI>1).
Transwell and wound-healing assays
For the invasion assay, the cells (1×104
cells/well) incubated in serum-free medium were added to the upper
chamber of the insert with Matrigel (BD Biosciences, San Jose, CA,
USA). Following incubation at 37°C for 24 h, permeable cells were
fixed with 4% paraformaldehyde for 20 min and stained with 0.1%
crystal violet for 10 min at room temperature. The cells were
counted under an inverted microscope (Nikon Corp.) in 5 different
visual fields and averaged.
For the wound-healing assay, the cells
(5×104 cells/well) were seeded into 6-well plates and
incubated in serum-free medium for 24 h at 37°C. The cell monolayer
was scratched with a 200-µl pipette tip to form wound gaps.
The cells were then washed in PBS and then incubated for 24 h at
37°C continuously, and images were captured under an inverted
microscope (Nikon Corp.) at the indicated time-points.
Immunofluorescence staining
The cells (1×105 cells/well) plated into
6-well plates covered with glass were incubated overnight at 37°C.
Once the cells adhered to the walls, they were fixed with 4%
paraformaldehyde for 20 min, the experiment was then performed as
previously described (24). The
cells were then incubated with the primary antibody (anti-Cav-1;
1:200; cat. no. 3267; Cell Signaling Technology, Inc.) overnight at
4°C, and then with a secondary antibody (FITC conjugated goat
anti-rabbit IgG, 1:100, ZF0311; ZSGB-BIO) in a dark room at 37°C
for 30 min. Cell nuclei were stained with DAPI (ZLI-9557; ZSGB-BIO)
at 37°C for 5 min. Immunofluorescence images were obtained upon
incubation in 50% glycerol using a fluorescence microscope (Nikon
Corp.).
Statistical analysis
All experiments were repeated ≥3 times independently
with technical replicates and analyzed using SPSS software version
21.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism software
version 5 (GraphPad Software, Inc., La Jolla, CA, USA). Significant
differences among experimental groups were evaluated using the
Student’s t-test, one-way analysis of variance (ANOVA) and two-way
ANOVA, while the Bonferroni adjustment was employed for multiple
comparisons. Survival analyses were conducted using the
Kaplan-Meier method. The Cox proportional hazards model was used to
estimate hazard ratios (HRs). Other data were analyzed using
Spearman’s correlation analysis, the Chi-square test and
Mann-Whitney test. One-way ANOVA was used for multiple comparisons
followed by the Student-Newman-Keuls test as a post hoc test. A
value of P<0.05 was considered to indicate a statistically
significant difference.
Results
Cav-1 is overexpressed in CRPC and is
associated with genes in Ras signaling pathways
To determine the differential expression levels of
Cav-1, H-Ras, K-Ras and PLCε in the tissues of patients with PPC
and CRPC, and to explore the association between these proteins, 45
PPC and 36 CRPC tissue samples were collected from patients.
Immunohistochemical assays revealed that the expression of Cav-1,
H-Ras, K-Ras and PLCε was upregulated in the CRPC compared with the
PPC tissue samples (P=0.002, P=0.0272, P=0.0377 and P=0.0364,
respectively) (Fig. 1A and B).
Spearman’s correlation analysis revealed that there was a positive
correlation between the Cav-1 expression levels and the H-Ras
(r=0.4151, P=0.0118), K-Ras (r=0.3443, P=0.0398) and PLCε
expression levels (r=0.4338, P=0.0082) in CRPC (Fig. 1C). The demographic and clinical
characteristics of these patients and the association of these
characteristics with Cav-1 expression are summarized in Table I. The data revealed that 72.2%
(26/36) of the CRPC samples had a positive Cav-1 expression vs.
31.1% (14/45) of the PPC samples (Table I and Fig. 1B). Among the various clinical
parameters, only bone metastasis was positively associated with the
expression of Cav-1 in the PPC (P=0.021) and CRPC (P=0.006)
tissues, suggesting that Cav-1 overexpression may promote bone
metastasis (Table I).
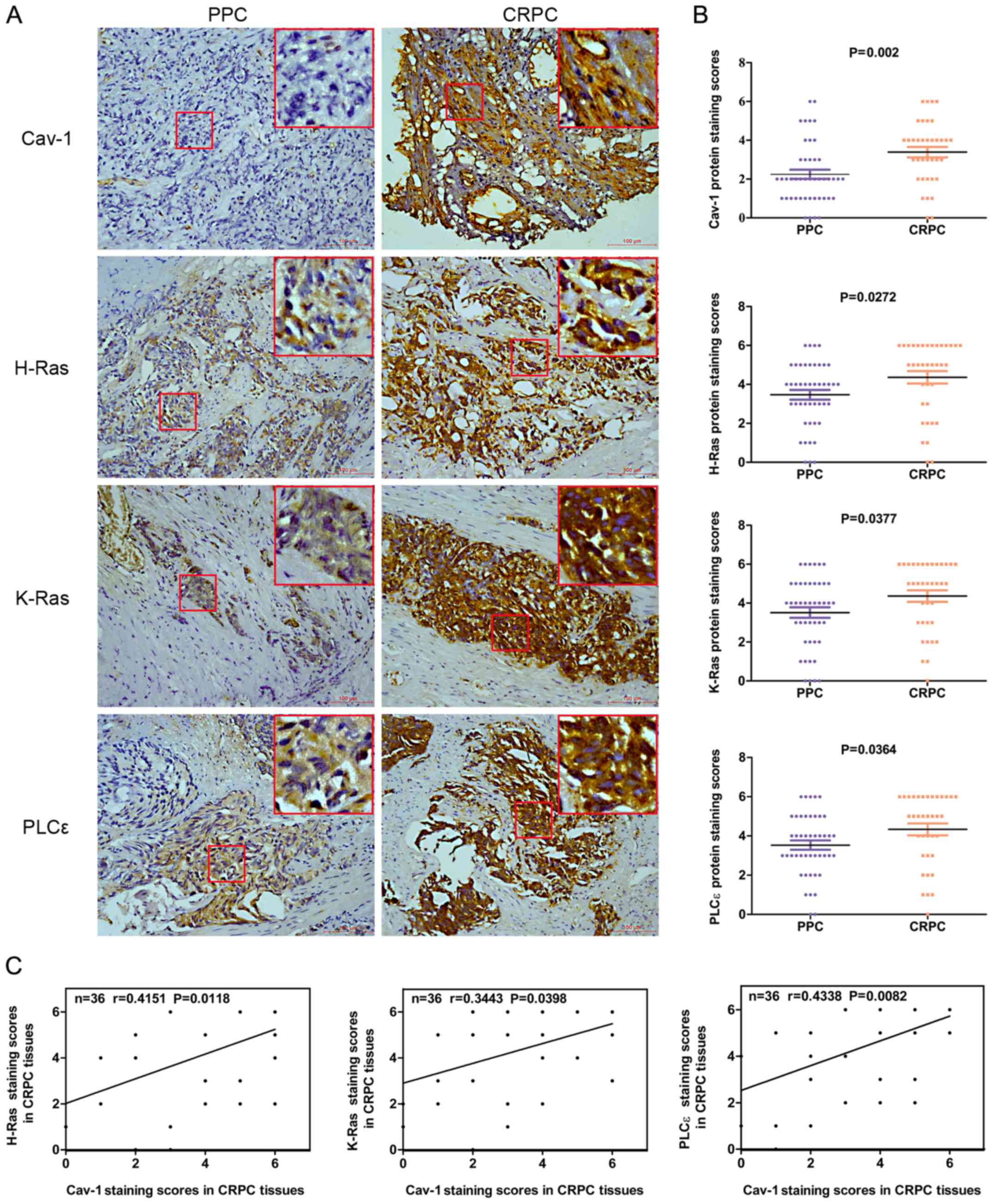 | Figure 1Expression levels of Cav-1, H-Ras,
K-Ras and PLCε in 45 PPC and 36 CRPC tumor samples. (A) Cav-1,
H-Ras, K-Ras and PLCε expression in PPC and CRPC was determined by
immunochemistry (magnification, ×200) (the big red boxes are an
enlargement of the area in the small red boxes, enlarged almost
7-fold). (B) Average staining scores for Cav-1, H-Ras, K-Ras and
PLCε in PPC and CRPC tissues. (C) Correlation curve analysis for
Cav-1 staining scores vs. H-Ras, K-Ras and PLCε staining scores in
CRPC tissues. PPC, primary prostate cancer; CRPC,
castration-resistant prostate cancer; Cav-1, caveolin-1; PLCε,
phosphoinositide-specific phospholipase Cε. |
 | Table IDemographic and clinical
characteristics of the patients and the correlation of these
characteristics with Cav-1 expression. |
Table I
Demographic and clinical
characteristics of the patients and the correlation of these
characteristics with Cav-1 expression.
|
Characteristics | Overall | Cav-1
| P-value |
|---|
| Negative (%) | Positive (%) |
|---|
| PPC | n=45 | 31 (68.9) | 14 (31.1) | |
| CRPC | n=36 | 10 (27.8) | 26 (72.2) | |
| Age of patients
with PPC (years) | | | | P=0.650a |
| Median | 66 | 66 | 67 | |
| Quartiles
(25-75) | 62-71 | 62-70 | 61-72 | |
| Age of patients
with CRPC (years) | | | | P=0.794a |
| Median | 72 | 71 | 72 | |
| Quartiles
(25-75) | 68-79 | 67-77 | 67-80 | |
| PSA of patients
with PPC (µg/l) | | | | P=0.418a |
| Median | 67.64 | 84.63 | 59.81 | |
| Quartiles
25-75 | 26.91-463.00 | 29.75-505.1 | 21.74-312.89 | |
| PSA of patients
with CRPC (µg/l) | | | | P=0.374a |
| Median | 23.64 | 13.115 | 28.25 | |
|
Quartiles(25-75) | 5.99-47.76 | 2.255-58.18 | 8.05-46.39 | |
| Metastases in
PPC | | | | |
| Bone | 17/45 (37.8) | 8/31 (25.8) | 9/14 (64.3) |
P=0.021b |
| Metastases in
CRPC | | | | |
| Bone | 27/36 (75) | 4/10 (40) | 23/26 (88.5) |
P=0.006b |
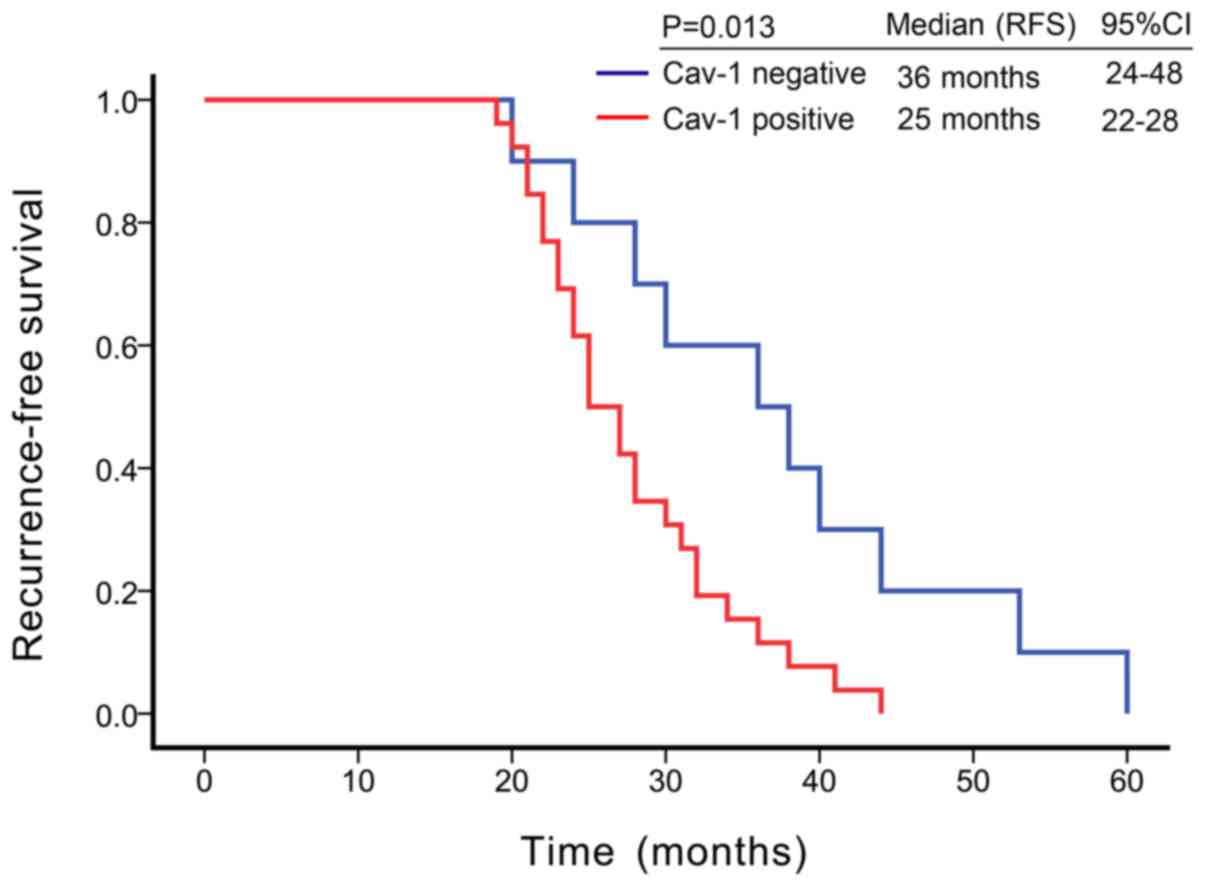
Kaplan-Meier survival analysis revealed that the
median recurrence-free survival (RFS) was 36 months [95% confidence
interval (CI) = 24-48 months] in patients with Cav-1-negative CRPC,
while the median RFS was 25 months (95% CI = 22-28 months) in
Cav-1-positive patients. Cav-1-positive tumor tissues were
associated with a shorter RFS in patients with CRPC (P=0.013, log
rank test) (Fig. 2). In univariate
Cox proportional hazards regression analyses, Cav-1 was found to be
significantly associated with recurrence following ADT (HR = 2.65,
95% CI = 1.162-6.029; P=0.02) (Table
II), suggesting that Cav-1 is an independent risk factor for
the occurrence of CRPC, and that the risk of CRPC occurring in
Cav-1-positive patients was 2.65-fold greater than in
Cav-1-negative patients.
| Table IIUnivariate Cox proportional hazards
regression analysis of Cav-1 for RSF in CRPC. |
Table II
Univariate Cox proportional hazards
regression analysis of Cav-1 for RSF in CRPC.
| Variable | Univariate analysis
|
|---|
| n | HR | 95% CI | P-value |
|---|
| Cav-1 | | | | |
| Negative | 10 | | | |
| Positive | 26 | 2.65 | 1.162-6.029 | 0.02a |
Expression of Cav-1 in serum may be
predictive of CRPC
To gain further insight into the functions of Cav-1
in CRPC, the Cav-1 levels in serum samples from 70 patients with
PPC and 56 patients with CRPC were determined by ELISA. Statistical
analysis revealed that the mean Cav-1 expression level in the serum
of patients with CRPC was higher than in the serum of patients with
PPC (CRPC, 1.57±0.83 ng/ml vs. PPC, 0.64±0.25 ng/ml; P<0.001)
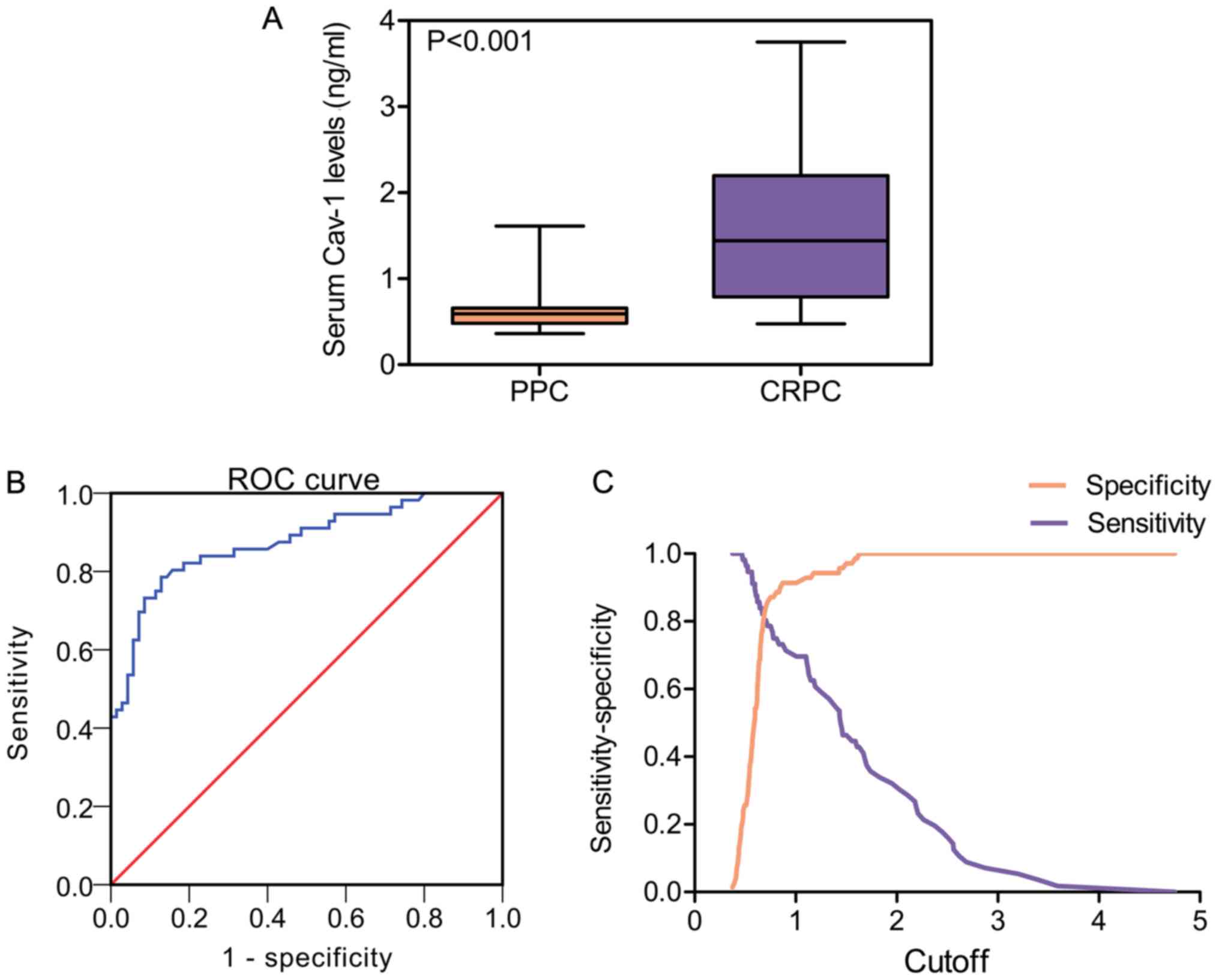
(Table III and Fig. 3A). Subsequently, the present study
explored whether Cav-1 can be used as a possible diagnostic factor
in CRPC by assessing receiver operating characteristic (ROC)
curves. As shown in Fig. 3B, the
area under the curve (AUC) was 0.876 (95% CI = 0.813-0.939),
suggesting that Cav-1 could be used for the accurate diagnosis of
CRPC. The diagnostic properties using different Cav-1 cut-off
values are shown in Fig. 3C.
| Table IIIStatistical comparison of serum Cav-1
in PPC and CRPC. |
Table III
Statistical comparison of serum Cav-1
in PPC and CRPC.
| Group | n | Mean ± SD
(ng/ml) | P-value (vs.
PPC) |
|---|
| PPC | 70 | 0.64±0.25 | |
| CRPC | 56 | 1.57±0.83 |
P<0.001 |
Since different cut-off values result in different
values of sensitivity and specificity, the cut-off was set at 0.75
ng/ml, as it maximized the Youden index, resulting in sensitivity
and specificity rates of 78.6 and 87.1%, respectively. However,
considering that our study population receiving ADT is known to be
at high risk of castration resistance, achieving a minimum of 80%
sensitivity and maximizing sensitivity over specificity were deemed
desirable. Using this criterion, the optimal cut-off value was set
at 0.68 ng/ml, which could improve sensitivity to 82.1% while
maintaining specificity at 80%. Consequently, Cav-1 levels were
statistically evaluated with certain clinical parameters in
patients with CRPC. The data revealed that an increased Cav-1
expression was positively associated with tumor metastasis
(P=0.003; Table IV). Taken
together, these data suggest that Cav-1 could be used as a
potential biomarker to predict metastasis in patients with
CRPC.
| Table IVAssociation of serum Cav-1 levels
with the clinico-pathological characteristics of patients with
CRPC. |
Table IV
Association of serum Cav-1 levels
with the clinico-pathological characteristics of patients with
CRPC.
|
Characteristics | No. | Cav-1
| P-value |
|---|
| Mean ± SD |
|---|
| Total | 56 | 1.57±0.83
ng/ml | |
| Age (years) | | | |
| <60 | 17 | 1.62±0.70
ng/ml | P=0.788a |
| ≥60 | 39 | 1.56±0.89
ng/ml | |
| PSA | | | |
| <4 | 12 | 1.46±0.61
ng/ml | P=0.776b |
| 4-10 | 11 | 1.67±0.81
ng/ml | |
| >10 | 33 | 1.59±0.93
ng/ml | |
| Metastasis | | | |
| Yes | 43 | 1.75±0.83
ng/ml |
P=0.003c |
| No | 13 | 0.99±0.55
ng/ml | |
Cav-1 knockdown suppresses CRPC
metastasis
Although we demonstrated shown that the Cav-1 levels
were higher in serum and tumor tissues from patients with CRPC than
those with PPC, there is little evidence to indicate whether this
increase regulates the progression or metastasis of CRPC. To
address this question, the present study examined the mRNA and
protein expression levels of Cav-1 by RT-qPCR and western blotting,
respectively, in an androgen-sensitive LNCaP cell line and in
castration-resistant Bic-R, En-R, PC3 and DU145 cell lines. The
protein level of Cav-1 was markedly low in the LNCaP cells, but was
considerably higher in the PC3, DU145, Bic-R and En-R cells. At the
mRNA level, similar results were obtained (Fig. 4A-C). This finding suggested that
Cav-1 expression was increased in the CRPC cells. Subsequently, to
determine the relevance of the increased Cav-1 expression on the
metastasis of castration-resistant tumors, a Cav-1 knockdown
plasmid was constructed and transfected into the PC3 and En-R cells
(Fig. 4D and E). The results of
western blot analysis revealed that Cav-1 knockdown downregulated
the expression levels of factors associated with invasion and
migration, such as Snail, MMP2 and MMP9 (Fig. 4F-I). Transwell and wound-healing
assays consistently demonstrated that the knockdown of Cav-1
suppressed the invasion and migration of CRPC cells (Fig. 4J-N).
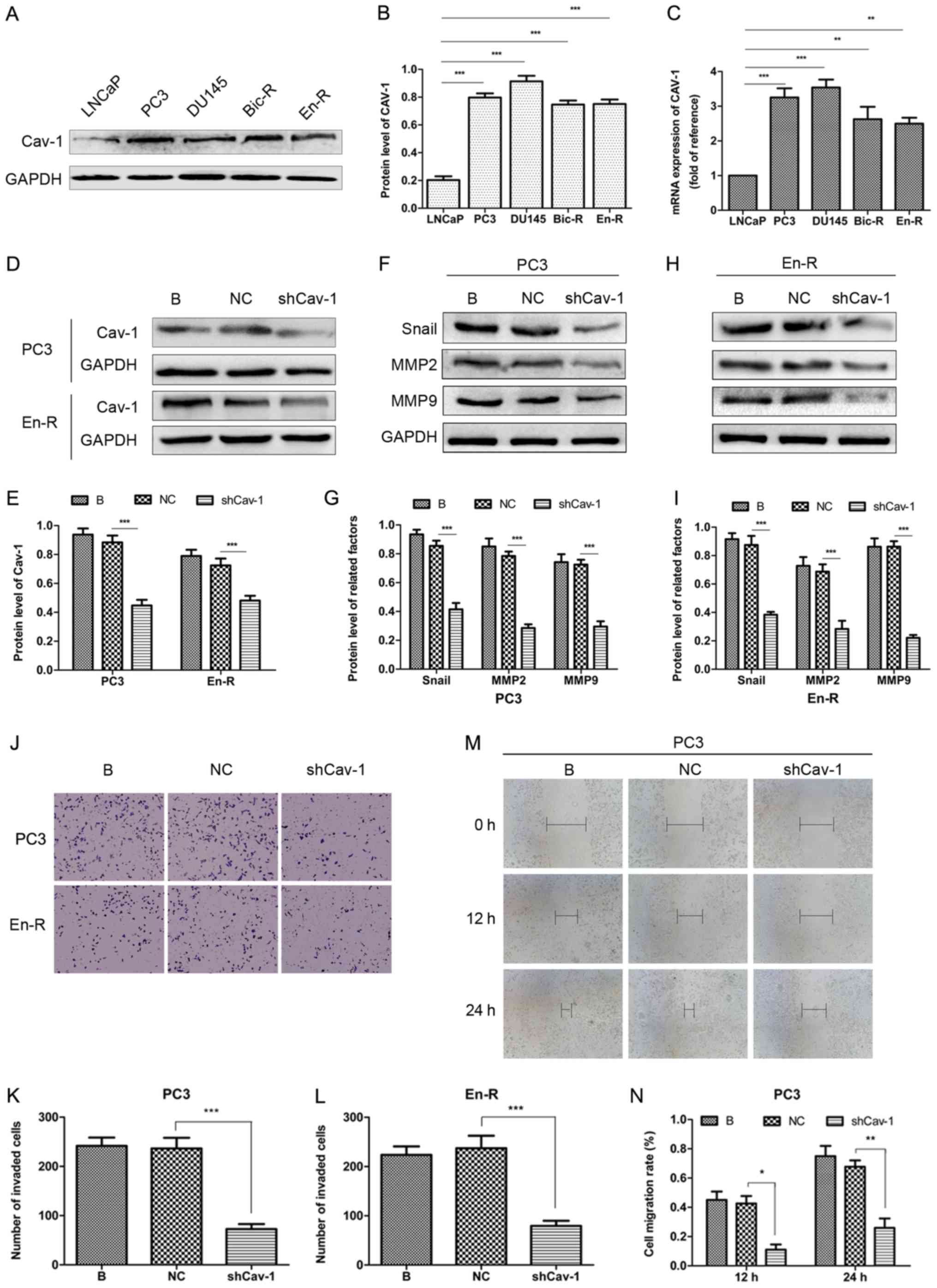 | Figure 4Knockdown of Cav-1 suppresses the
metastasis of CRPC. (A-C) The protein and mRNA expression of Cav-1
was upregulated in the PC3, DU145, Bic-R and En-R cell lines vs.
the LNCaP cells. (D and E) Knockdown of Cav-1 was performed by
transfection of the cells with plasmids containing shRNA targeting
Cav-1. (F-N) The B, NC and shCav-1 groups in PC3 and En-R cells
were compared. (F-I) The expression of Snail, MMP2 and MMP9 in PC3
and En-R cells was examined by western blot analysis. (J-N) The
cell invasive and migratory capacities were evaluated using
Transwell and wound-healing assays. Cells were transfected with a
negative control or shCav-1 for 72 h. GAPDH served as a loading
control. *P<0.05, **P<0.01 and
***P<0.001. Bic-R, bicalu-tamide-resistant LNCaP
cells; En-R, enzalutamide-resistant LNCaP cells; B, blank; NC,
negative control; sh, small hairpin; shCav-1, Cav-1 knockdown;
CRPC, castration-resistant prostate cancer; Cav-1, caveolin-1; MMP,
matrix metalloproteinase. |
Cav-1 knockdown attenuates the expression
of PLCε in DRFs through H-Ras
As stated above, the isoforms of Ras, H-Ras and
K-Ras and the downstream gene PLCε were overexpressed in CRPC and
exhibited a positive correlation with Cav-1 expression. Therefore,
we hypothesized that Cav-1 may regulate Ras signaling in cell
membranes and may be associated with CRPC metastasis.
To explore this concept, a series of Cav-1 knockdown
experiments were performed and the expression of H-Ras, K-Ras and
PLCε in DRFs was examined. H-Ras, K-Ras and PLCε were detected in
DRFs, which are composed of lipid rafts and caveolae, as confirmed
by the presence of Cav-1. The non-classical localization of PLCε
was specifically observed in caveolae and rafts by western blot
analysis. Additionally, the knockdown of Cav-1 downregulated the
expression of H-Ras and PLCε, but did not alter the expression of
K-Ras in DRFs (Fig. 5A-D).
 | Figure 5The Ras signaling pathway is
regulated by Cav-1. (A-D) Expression level of Cav-1, H-Ras, K-Ras
and PLCε in DRFs of PC3 and En-R cells in different groups. (E and
F) mRNA expression of verified the successful transfection of
H-RasG12V or K-RasG12V plasmids. (G-J) Expression of PLCε in PC3
and En-R cell DRFs by western blot analysis. Cells were treated
with activated Ras isoforms, H-RasG12V or K-RasG12V, and with
H-RasG12V or K-RasG12V combined with knockdown of Cav-1.
***P<0.001. NS, not significant. En-R,
enzalutamide-resistant LNCaP cells; DRF, detergent-resistant
fraction; Cav-1, caveolin-1; PLCε, phosphoinositide-specific
phospholipase Cε. B, blank; NC, negative control; sh, small
hairpin; shCav-1, Cav-1 knockdown; Vector, empty vector of
overexpression plasmid. |
To further elucidate the role of Cav-1 in regulating
PLCε through H-Ras, the cells were treated with H-RasG12V and
K-RasG12V (Fig. 5E and F), which
are the activated forms of H-Ras and K-Ras, respectively. The
results revealed that H-RasG12V upregulated the expression of PLCε
in DRFs in the different CRPC cells, and that this upregulation was
reversed by the knockdown of Cav-1 (Fig. 5G and I). K-RasG12V also increased
the expression of PLCε, although the knockdown of Cav-1 did not
reverse this process (Fig. 5H and
J). These results suggest that Cav-1 knockdown attenuates the
expression of PLCε in the plasma membrane, particularly in lipid
rafts, through H-Ras.
Cav-1 knockdown suppresses the metastasis
of CRPC through H-Ras/PLCε
Our previous study suggested that PLCε induced the
invasion and migration of bladder cancer cells (21); however, it remains unclear as to
whether PLCε, in particular PLCε in DRFs, is responsible for the
invasion and migration of prostate cancer. Therefore, in this
study, PLCε was knocked down using lentiviral transduction
particles. As shown in Fig. 6A-D,
the knockdown of PLCε in PCa cells decreased the expression of
factors involved in invasion and migration, such as Snail, MMP2 and
MMP9, suggesting that PLCε regulates invasion and migration in
CRPC.
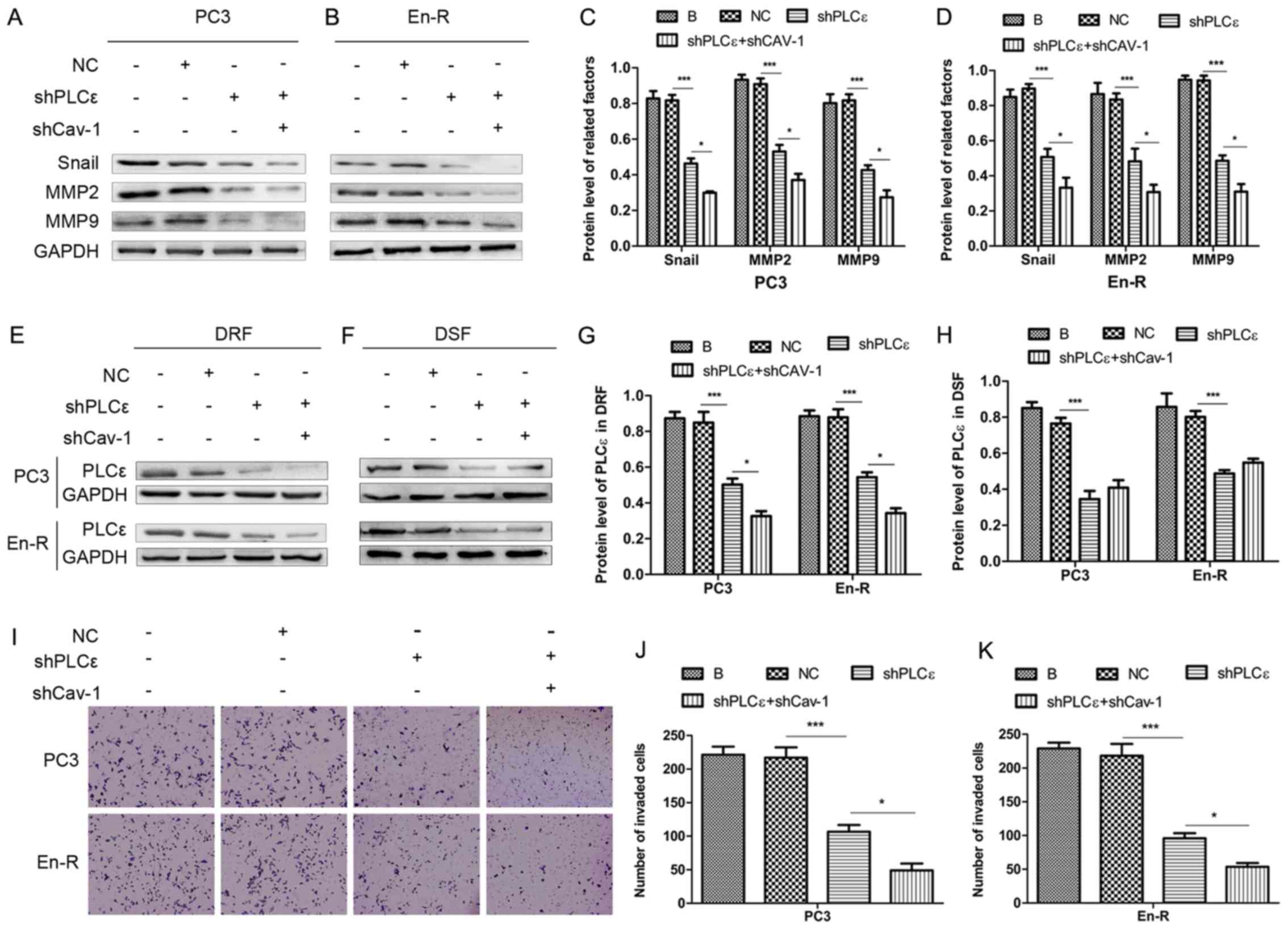
To determine whether the process of metastasis is
associated with the expression of PLCε in DRFs, PLCε was knocked
down in combination with the knockdown of Cav-1. The results
revealed that, compared with the knockdown of PLCε alone, the
concurrent knockdown of Cav-1 downregulated the expression of PLCε
only in DRFs and slightly upregulated the expression of PLCε in
DSFs (although the difference was not statistically significant)
(Fig. 6E-H). Consistently, the
knockdown of Cav-1 in combination with the knockdown of PLCε
downregulated more effectively the changes in downstream factors of
invasion and migration compared with knockdown of PLCε alone
(Fig. 6A-D). Using Transwell
assays, consistent results were obtained (Fig. 6I-K), suggesting that the knockdown
of Cav-1 suppressed invasion and migration in CRPC through the
attenuation of the expression of PLCε in plasma membrane
caveolae.
Simvastatin abrogates the expression of
Cav-1 associated with membrane cholesterol
Murata et al have previously reported that
Cav-1 can directly binds cholesterol, and that a threshold level of
membrane cholesterol is required for caveolae to form (29). To delineate whether cholesterol is
responsible for the expression of Cav-1, the cells were cultured in
serum-free conditions, which removed the effects of exogenous
cholesterol, and treated the cells with simvastatin, M-β-CD and
cholesterol. Simvastatin is an inhibitor of HMGR, which causes the
blockade of cholesterol biosynthesis at the rate-limiting step
(HMG-CoA conversion to mevalonate) (30). M-β-CD is the most effective agent
for stripping cholesterol from the cell membrane. Both drugs can
therefore deplete cholesterol through different pathways.
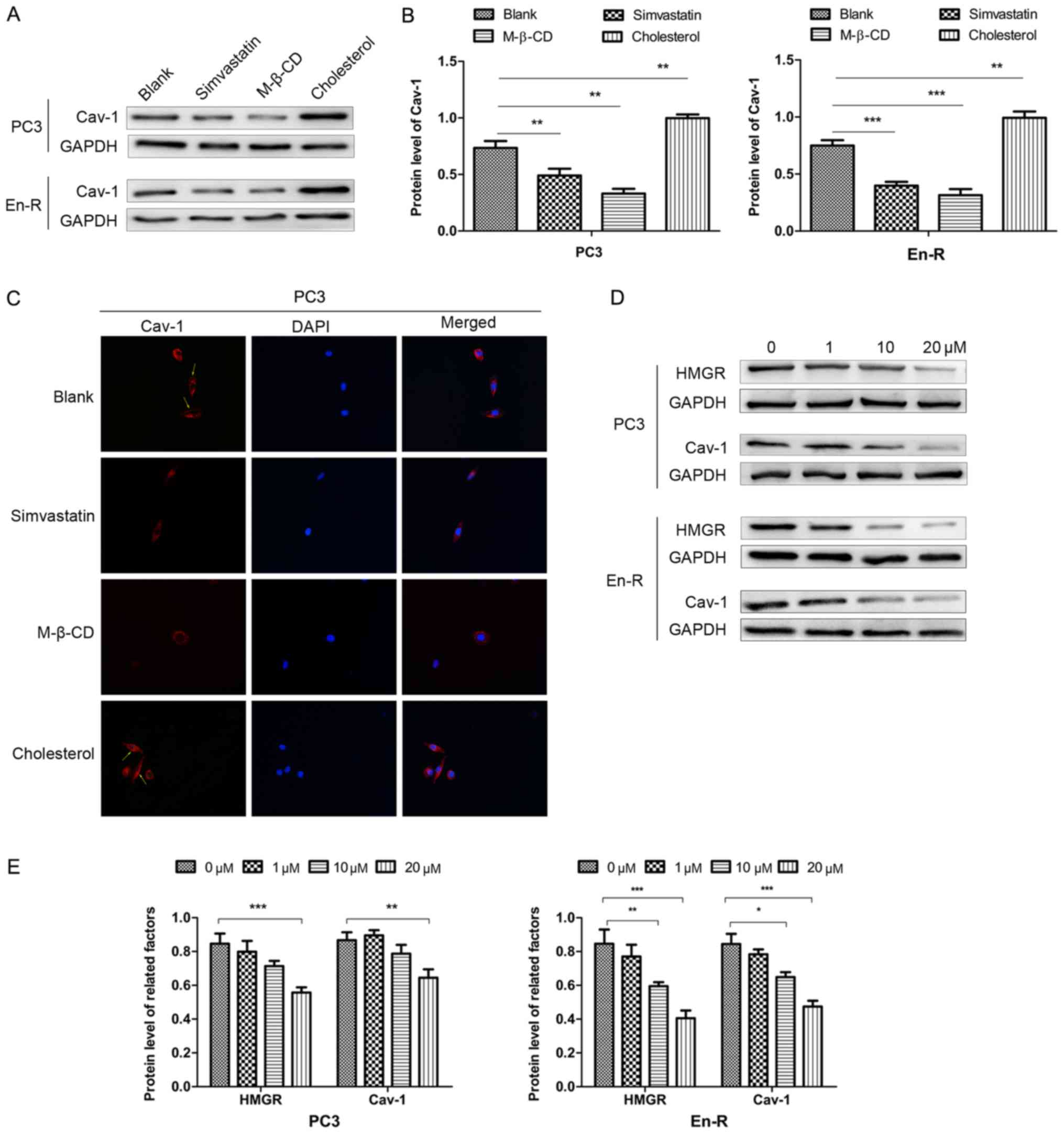
In this study, simvastatin decreased the expression
of Cav-1 in DRFs, although the level varied in different cells,
similar to what was observed with M-β-CD. Inversely, cholesterol
replenishment resulted in an increased membrane Cav-1 expression
compared with the untreated cells (Fig. 7A and B). The immunofluorescence
assay revealed similar results (Fig.
7C). Taken together, these results suggested that there was a
functional association between free membrane cholesterol and Cav-1
expression in CPRC cells.
To further delineate the association between
simvastatin and Cav-1, western blot analysis revealed that 10
µM simvastatin decreased the expression of HMGR and Cav-1 in
En-R cells, with an even greater suppression when 20 µM
simvastatin was used. A statistically significant decrease in the
expression of HMGR and Cav-1 also occurred when the PC3 cells were
cultured with 20 µM simvastatin, but not with 10 µM
simvastatin (Fig. 7D and E). Taken
together, these results indicate that simvastatin can markedly
suppress membrane Cav-1 expression through inhibition of de
novo cholesterol synthesis in CRPC cells.
Simvastatin enhances the effects of
anti-androgenic drugs through Cav-l
To further explore whether Cav-1 reverses the
sensitivity of the cells to AR antagonists, Cav-1 was knocked down
in Bic-R and En-R cells. Cell proliferation was not suppressed by
bicalutamide in the control cells, but was suppressed in the cells
in which Cav-1 was knocked down, and was further suppressed in the
cells in which Cav-1 was knocked down and treated with
bicalutamide. Replenishment with H-RasG12V modestly reversed the
suppression of cell proliferation caused by Cav-1 knockdown or by
Cav-1 knockdown combined with bicalutamide. The same results were
observed with enzalutamide treatment (Fig. 8A and B), suggesting that Cav-1 can
reverse the sensitivity of cells to AR antagonists partly through
H-RasG12V.
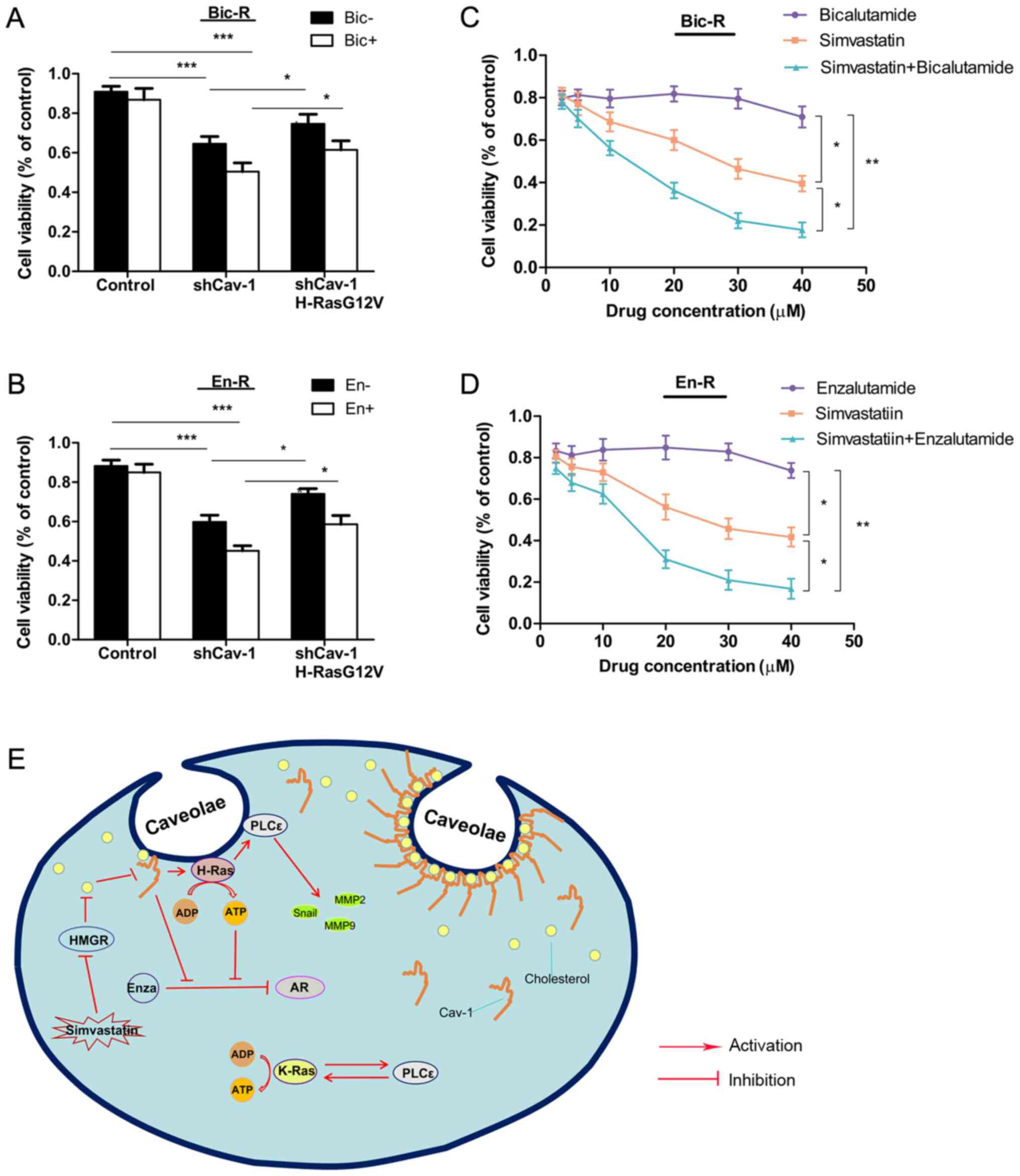 | Figure 8Effects of simvastatin and Cav-1 on
androgen receptor antagonist resistance. (A and B) Bic-R and En-R
cells were transfected with Cav-1 knockdown plasmids with or
without H-RasG12V. After 24 h, 10 µM of bicalutamide or
enzalutamide was applied and further incubated for 48 h, and then
cell survival rates were analyzed using CCK-8 analysis in
triplicate. (C and D) Various concentrations of bicalutamide or
enzalutamide and simvastatin were applied to Bic-R and En-R cells
in a 1:1 ratio. After 48 h, cell survival rates were analyzed using
CCK-8 in triplicate. P-values were calculated using one-way ANOVA,
followed by the Student-Newman-Keuls test as a post hoc test.
*P<0.05, **P<0.01 and
***P<0.001. (E) Schematic diagram summarizing our
findings: Cav-1 upregulates the expression of H-Ras/PLCε in
caveolae, which can promote the migration, invasion and drug
resistance in CRPC. Simvastatin, a 3-hydroxy-3-methylglutaryl
coenzyme A reductase inhibitor, can downregulate the expression of
Cav-1 in caveolae by blocking cholesterol biosynthesis, which in
turn delays the progression of CRPC. Bic-R, bicalutamide-resistant
LNCaP cells; En-R, enzalutamide-resistant LNCaP cells; shCav-1,
Cav-1 knockdown; sh, small hairpin; Cav-1, caveolin-1; CRPC,
castration-resistant prostate cancer. |
Finally, to explore whether simvastatin exerts
antitumor effects on CRPC, cell viability was evaluated by a CCK-8
assay in cells treated with various concentrations of simvastatin.
In contrast to enzalutamide or bicalutamide treatment, in these
drug-resistant cells, increasing concentrations of simvastatin
caused a gradual decrease in the viability of CPRC cells, and more
prominently when enzalutamide or bicalutamide was combined with
simvastatin (Fig. 8C and D).
Consistently, simvastatin enhanced the suppressive effect of
enzalutamide on the En-R or bicalutamide on Bic-R cells, as
indicated by the combination indexes (CI)<1. [En-R, CI = 0.35 at
effective concentration (EC) of 50, 0.19 at EC = 75 and 0.10 at EC
= 90; and Bic-R, CI = 0.38 at EC = 50, 0.21 at EC = 75 and 0.12 at
EC = 90] (Table V).
| Table VCombination index values for
drug-resistant cells treated with combination of simvastatin and
bicalutamide or enzalutamide. |
Table V
Combination index values for
drug-resistant cells treated with combination of simvastatin and
bicalutamide or enzalutamide.
| Cell line | Drug | CI value at
|
|---|
| EC50 | EC75 | EC90 |
|---|
| Bic-R | Sim + Bic | 0.38 | 0.21 | 0.12 |
| En-R | Sim + En | 0.35 | 0.19 | 0.10 |
Discussion
Determining the appropriate therapy for CRPC is
challenging due to castration-resistant progression and the
unavailability of measurable biomarkers or validated surrogate
markers. The present study demonstrated that Cav-1 was
overexpressed in tissue and serum specimens of patients with CRPC
compared to those with PPC, and this was also shown in different
PPC and CRPC cell lines. However, various studies have previously
observed that the expression of Cav-1 is downregulated in prostate
and breast cancer, and Cav-1 has been described as being a
tumor-suppressor gene (31,32).
Unlike these previous studies, which compared non-malignant with
malignant prostate cancer tissue or the expression of Cav-1 in the
tumor stroma, this study mainly explored the differential
expression of endothelial Cav-1 in CRPC and PPC. Similar to our
results, various previous studies have suggested that Cav-1 is
upregulated in cancers at the advanced stages, and that the
overexpression of Cav-1 is closely associated with aggressive
behavior and poor clinical outcomes in various types of cancer
(33-38). Burgermeister et al also
revealed that Cav-1 was ‘Janus-faced’ and tissue-specific in
cancers of different types and stages, and this conditional
functionality was also reflected in its potential tumor-suppressing
roles in early disease stages and oncogenic roles in later stages
(39). A similar observation has
also been documented for hepatocellular carcinoma and melanoma
(40,41). In addition, this upregulation of
the expression of Cav-1 either in tissue or serum specimens is
strongly associated with CRPC metastasis. This persists into the
late metastatic stage and may be identified as a signature
suggesting early aggressiveness, which provides a guidance for the
application of anti-Cav-1 therapy in early-stage disease. In this
study, a clear separation of RFS curves was observed in patients
with Cav-1-positive vs. Cav-1-negative tumors, suggesting that
Cav-1 may be associated with a poor prognosis of patients who
received ADT.
Furthermore, ROC analysis and AUC values underscored
the enhanced prognostic performance of Cav-1 in evaluating the risk
of castration resistance in patients with PPC who received ADT. The
determination of serum Cav-1 seems feasible as it is technically
simple and less invasive compared with conventional diagnostic
indexes that require a tissue biopsy. The limitations of this study
include the relatively small number of only Chinese subjects and
the fact that the analysis was retrospective. Further studies with
additional populations performed in a prospective manner are
required.
This study also suggests that, when castration
resistance occurs, not only Cav-1, but also H-Ras, K-Ras and PLCε
expression is upregulated. However, the findings suggest that Cav-1
may act only upstream of H-Ras and regulate the expression of H-Ras
and PLCε in the membrane caveolae. This is slightly less consistent
with our results from immunohistochemistry, which revealed that the
expression of Cav-1 was also positively associated with K-Ras in
patients with CRPC. Supporting our findings, Hancock reported that
each Ras isoform has a different lipid anchor and a different
hyper-variable flanking region that participates in membrane
interactions (42). Therefore, the
correlation between Cav-1 and K-Ras may involve other connections,
while K-Ras is not directly activated by Cav-1.
However, to date, at least to the best of our
knowledge, there are no reports that have elucidated the oncogenic
roles of membrane PLCε. Although various studies have shown that
Ras proteins are membrane-associated signal transducers and
participate in the malignant behavior of numerous human tumors
(43-46), studies on different types of tumors
have mainly implicated the Ras-mitogen-activated protein kinase
(MAPK)/ERK kinase signaling axis as important in cell proliferation
and resistance to apoptosis (19,45,46).
The findings of this study revealed that Cav-1 increased the
metastatic potential in CRPC through the relocation and activation
of the H-Ras/PLCε kinase cascade in membranes. Similarly, Smrcka
et al noted that Ras caused the translocation of PLCε from
the cytosol to the membrane where it would gain access to the
phosphatidylinositol-4,5-bisphosphate substrate, while the Rap
GTP-binding protein promotes the translocation of PLCε to
perinuclear regions in Cos-7 cells (18). Although membrane targeting may not
be the only factor driving activation, translocation from the
cytosol to the plasma membrane may, in part, underlie the mechanism
involved in the activation of PLCε, which then induces the invasion
and migration of CRPC cells.
In the present study, an appropriate strategy to
attenuate the progression of CRPC was demonstrated. The knockdown
of Cav-1 not only suppressed the metastasis of CRPC but also
decreased resistance to AR antagonists in CRPC cells. This finding
was supported by the findings of our previous study, which reported
that the blocking of PLCε reversed AR nuclear translocation
(22). However, activated H-Ras
mutants can only partly reverse sensitization to anti-androgenic
drugs that has been augmented by Cav-1 knockdown. Further studies
are warranted to determine whether other signaling cascades are
involved in this process.
Previous studies have demonstrated that numerous
cholesterol synthesis enzymes, including sterol-regulatory element
binding proteins (SREBPs) and its downstream targets, such as
HMG-CoA synthase, exhibit an increased expression at the mRNA and
protein level in CRPC disease following AR antagonist treatment
(47-50). Han et al also reported the
increased expression of AR-V7, which may be the result of more
intensive androgen-deprivation, such as abiraterone or enzalutamide
treatment, and is associated with therapeutic resistance,
potentially restored the lipid biosynthetic pathways and led to
intracellular cholesterol accumulation (51). Notably, the expression of Cav-1 in
the plasma membrane was regulated by cholesterol, whether
exogenously supplied or through endogenous de novo
biosynthesis. At the same time, as a significant factor associated
with cholesterol homeostasis (11), upregulated mRNA and protein levels
of Cav-1 may represent a metabolic feedback of this pathway and a
protective mechanism with which to limit cellular cholesterol
accumulation in these cells, although it is activated alongside
other carcinogenic mechanisms. In this context, the administration
of AR antagonists followed by simvastatin can enhance the
anticancer efficacy of enzalutamide or bicalutamide. Simvastatin is
used as first-line clinical treatment for hypercholesterolemia due
to its low price and few side-effects; however, its use in the
treatment of cancer has rarely been reported. The present study has
shed light onto the role of simvastatin in inhibiting resistance of
CRPC to AR antagonists. Therefore, our findings may provide a novel
treatment strategy for clinicians with which to delay the
progression of CRPC. Furthermore, advising patients to follow a
low-cholesterol diet may be an additional method to rationally
reduce the risk of CRPC.
In conclusion, and to the best of our knowledge, the
present study explored for the first time the effects of membrane
Cav-1/H-Ras/PLCε signaling on the metastasis and drug resistance of
CRPC. Cav-1 appears to be a promising predictive biomarker of CRPC
that may be used to identify patients requiring more intensive
treatment and as a putative candidate therapeutic target.
Simvastatin was identified as an inhibitor of the development of
castration resistance and a factor in delaying the progression of
resistance to AR antagonists (Fig.
8E). These findings provide data for the appropriate design of
other trials of drug resistance in CRPC, in particular
chemotherapeutic drug resistance, as well as longitudinal trials
in vivo that could validate this treatment modality for
clinical use.
Funding
The study was supported by a grant from the Natural
Science Foundation of China (no. 81802543).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors’ contributions
YG, CL and LL designed the experiments. YG, TL, LM,
ZD, ZQ, XW and MY collected the specimens and analyzed the clinical
data. YG, WS, HC, LN, carried out the experiments. YG wrote the
manuscript. ZQ, CL and XW provided technical support of this
research project and supervised the progress of the experiments.
YG, YF and JF analyzed the statistical data. YG, LL and TL
assembled and installed the figures. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Ethics Committee of
Chongqing Medical University. Informed consent was obtained from
the patients or their family members who agreed to the use of their
samples in this study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank The Department of
Urinary, The First Affiliated Hospitals of Chongqing Medical
University, China, for providing technical assistance and specimen
collection.
References
|
1
|
Watson PA, Arora VK and Sawyers CL:
Emerging mechanisms of resistance to androgen receptor inhibitors
in prostate cancer. Nat Rev Cancer. 15:701–711. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang C, Peng G, Huang H, Liu F, Kong DP,
Dong KQ, Dai LH, Zhou Z, Wang KJ, et al: Blocking the feedback loop
between neuroendocrine differentiation and macrophages improves the
therapeutic effects of enzalutamide (MDV3100) on prostate cancer.
Clin Cancer Res. 24:708–723. 2018. View Article : Google Scholar
|
|
3
|
Zhu G, Yan W, He HC, Bi XC, Han ZD, Dai
QS, Ye YK, Liang YX, Wang J and Zhong W: Inhibition of
proliferation, invasion, and migration of prostate cancer cells by
downregulating elongation factor-1alpha expression. Mol Med.
15:363–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shiota M, Fujimoto N, Imada K, Yokomizo A,
Itsumi M, Takeuchi A, Kuruma H, Inokuchi J, Tatsugami K, Uchiumi T,
et al: Potential role for YB-1 in castration-resistant prostate
cancer and resistance to enzalutamide through the androgen receptor
V7. J Natl Cancer Inst. Feb 8;1082016.Epub ahead of print.
View Article : Google Scholar
|
|
5
|
de Bono JS, Chowdhury S, Feyerabend S,
Elliott T, Grande E, Melhem-Bertrandt A, Baron B, Hirmand M,
Werbrouck P and Fizazi K: Antitumour activity and safety of
enzalutamide in patients with metastatic castration-resistant
prostate cancer previously treated with abiraterone acetate plus
prednisone for ≥24 weeks in Europe. Eur Urol. 74:37–45. 2018.
View Article : Google Scholar
|
|
6
|
Penson DF, Armstrong AJ, Concepcion R,
Agarwal N, Olsson C, Karsh L, Dunshee C, Wang F, Wu K, Krivoshik A,
et al: Enzalutamide versus bicalutamide in castration-resistant
prostate cancer: The STRIVE Trial. J Clin Oncol. 34:2098–2106.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Antonarakis ES, Lu C, Wang H, Luber B,
Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et
al: AR-V7 and resistance to enzalutamide and abiraterone in
prostate cancer. N Engl J Med. 371:1028–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Efstathiou E, Titus M, Wen S, Hoang A,
Karlou M, Ashe R, Tu SM, Aparicio A, Troncoso P, Mohler J, et al:
Molecular characterization of enzalutamide-treated bone metastatic
castration-resistant prostate cancer. Eur Urol. 67:53–60. 2015.
View Article : Google Scholar
|
|
9
|
Crona DJ, Milowsky MI and Whang YE:
Androgen receptor targeting drugs in castration-resistant prostate
cancer and mechanisms of resistance. Clin Pharmacol Ther.
98:582–589. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Coffey K and Robson CN: Regulation of the
androgen receptor by post-translational modifications. J
Endocrinol. 215:221–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Couet J, Belanger MM, Roussel E and Drolet
MC: Cell biology of caveolae and caveolin. Adv Drug Deliv Rev.
49:223–235. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sotgia F, Martinez-Outschoorn UE, Howell
A, Pestell RG, Pavlides S and Lisanti MP: Caveolin-1 and cancer
metabolism in the tumor microenvironment: Markers, models, and
mechanisms. Annu Rev Pathol. 7:423–467. 2012. View Article : Google Scholar
|
|
13
|
Chatterjee M, Ben-Josef E, Thomas DG,
Morgan MA, Zalupski MM, Khan G, Andrew Robinson C, Griffith KA,
Chen CS, Ludwig T, et al: Caveolin-1 is associated with tumor
progression and confers a multi-modality resistance phenotype in
pancreatic cancer. Sci Rep. 5:108672015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y, Roche O, Xu C, Moriyama EH, Heir
P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, et al: Hypoxia
promotes ligand-independent EGF receptor signaling via
hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc
Natl Acad Sci USA. 109:4892–4897. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nwosu ZC, Ebert MP, Dooley S and Meyer C:
Caveolin-1 in the regulation of cell metabolism: A cancer
perspective. Mol Cancer. 15:712016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Quest AF, Gutierrez-Pajares JL and Torres
VA: Caveolin-1: An ambiguous partner in cell signalling and cancer.
J Cell Mol Med. 12:1130–1150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hehlgans S and Cordes N: Caveolin-1: An
essential modulator of cancer cell radio-and chemoresistance. Am J
Cancer Res. 1:521–530. 2011.PubMed/NCBI
|
|
18
|
Smrcka AV, Brown JH and Holz GG: Role of
phospholipase Cε in physiological phosphoinositide signaling
networks. Cell Signal. 24:1333–1343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Samatar AA and Poulikakos PI: Targeting
RAS-ERK signalling in cancer: Promises and challenges. Nat Rev Drug
Discov. 13:928–942. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang RY, Du WQ, Zhang YC, Zheng JN and
Pei DS: PLCε signaling in cancer. J Cancer Res Clin Oncol.
142:715–722. 2016. View Article : Google Scholar
|
|
21
|
Du HF, Ou LP, Yang X, Song XD, Fan YR, Tan
B, Luo CL and Wu XH: A new PKCα/β/TBX3/E-cadherin pathway is
involved in PLCε-regulated invasion and migration in human bladder
cancer cells. Cell Signal. 26:580–593. 2014. View Article : Google Scholar
|
|
22
|
Wang Y, Wu X, Ou L, Yang X, Wang X, Tang
M, Chen E and Luo C: PLCε knockdown inhibits prostate cancer cell
proliferation via suppression of Notch signalling and nuclear
translocation of the androgen receptor. Cancer Lett. 362:61–69.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cornford P, Bellmunt J, Bolla M, Briers E,
De Santis M, Gross T, Henry AM, Joniau S, Lam TB, Mason MD, et al:
EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: Treatment of
relapsing, metastatic, and castration-resistant prostate cancer.
Eur Urol. 71:630–642. 2017. View Article : Google Scholar
|
|
24
|
Du Z, Li L, Sun W, Wang X, Zhang Y, Chen
Z, Yuan M, Quan Z, Liu N, Hao Y, et al: HepaCAM inhibits the
malignant behavior of castration-resistant prostate cancer cells by
downregulating Notch signaling and PF-3084014 (a γ-secretase
inhibitor) partly reverses the resistance of refractory prostate
cancer to docetaxel and enzalutamid in vitro. Int J Oncol.
53:99–112. 2018.PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Kortum RL, Fernandez MR, Costanzo-Garvey
DL, Johnson HJ, Fisher KW, Volle DJ and Lewis RE: Caveolin-1 is
required for kinase suppressor of Ras 1 (KSR1)-mediated
extracellular signal-regulated kinase 1/2 activation,
H-RasV12-induced senescence, and transformation. Mol Cell Biol.
34:3461–3472. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lingwood D and Simons K: Detergent
resistance as a tool in membrane research. Nat Protoc. 2:2159–2165.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Quan Z, He Y, Luo C, Xia Y, Zhao Y, Liu N
and Wu X: Interleukin 6 induces cell proliferation of clear cell
renal cell carcinoma by suppressing hepaCAM via the STAT3-dependent
up-regulation of DNMT1 or DNMT3b. Cell Signal. 32:48–58. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Murata M, Peränen J, Schreiner R, Wieland
F, Kurzchalia TV and Simons K: VIP21/caveolin is a
cholesterol-binding protein. Proc Natl Acad Sci USA.
92:10339–10343. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mullen PJ, Yu R, Longo J, Archer MC and
Penn LZ: The interplay between cell signalling and the mevalonate
pathway in cancer. Nat Rev Cancer. 16:718–731. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang B, Bhusari S, Kueck J, Weeratunga P,
Wagner J, Leverson G, Huang W and Jarrard DF: Methylation profiling
defines an extensive field defect in histologically normal prostate
tissues associated with prostate cancer. Neoplasia. 15:399–408.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sotgia F, Rui H, Bonuccelli G, Mercier I,
Pestell RG and Lisanti MP: Caveolin-1, mammary stem cells, and
estrogen-dependent breast cancers. Cancer Res. 66:10647–10651.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tahir SA, Yang G, Ebara S, Timme TL, Satoh
T, Li L, Goltsov A, Ittmann M, Morrisett JD and Thompson TC:
Secreted caveolin-1 stimulates cell survival/clonal growth and
contributes to metastasis in androgen-insensitive prostate cancer.
Cancer Res. 61:3882–3885. 2001.PubMed/NCBI
|
|
34
|
Timme TL, Goltsov A, Tahir S, Li L, Wang
J, Ren C, Johnston RN and Thompson TC: Caveolin-1 is regulated by
c-myc and suppresses c-myc-induced apoptosis. Oncogene.
19:3256–3265. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Senetta R, Stella G, Pozzi E, Sturli N,
Massi D and Cassoni P: Caveolin-1 as a promoter of tumour
spreading: When, how, where and why. J Cell Mol Med. 17:325–336.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mi L, Zhu F, Yang X, Lu J, Zheng Y, Zhao
Q, Wen X, Lu A, Wang M, Zheng M, et al: The metastatic suppressor
NDRG1 inhibits EMT, migration and invasion through interaction and
promotion of caveolin-1 ubiquitylation in human colorectal cancer
cells. Oncogene. 36:4323–4335. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z,
Nakamura Y and Fukami K: Lipid rafts and caveolin-1 are required
for invadopodia formation and extracellular matrix degradation by
human breast cancer cells. Cancer Res. 69:8594–8602. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Senetta R, Trevisan E, Rudà R, Maldi E,
Molinaro L, Lefranc F, Chiusa L, Lanotte M, Soffietti R and Cassoni
P: Caveolin 1 expression independently predicts shorter survival in
oligoden-drogliomas. J Neuropathol Exp Neurol. 68:425–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Burgermeister E, Liscovitch M, Röcken C,
Schmid RM and Ebert MP: Caveats of caveolin-1 in cancer
progression. Cancer Lett. 268:187–201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tse EY, Ko FC, Tung EK, Chan LK, Lee TK,
Ngan ES, Man K, Wong AS, Ng IO and Yam JW: Caveolin-1
overexpression is associated with hepatocellular carcinoma
tumourigenesis and metastasis. J Pathol. 226:645–653. 2012.
View Article : Google Scholar
|
|
41
|
Pandey V, Vijayakumar MV, Ajay AK, Malvi P
and Bhat MK: Diet-induced obesity increases melanoma progression:
Involvement of Cav-1 and FASN. Int J Cancer. 130:497–508. 2012.
View Article : Google Scholar
|
|
42
|
Hancock JF: Ras proteins: Different
signals from different locations. Nat Rev Mol Cell Biol. 4:373–384.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bivona TG and Philips MR: Ras pathway
signaling on endomembranes. Curr Opin Cell Biol. 15:136–142. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Castellano E, Molina-Arcas M, Krygowska
AA, East P, Warne P, Nicol A and Downward J: RAS signalling through
PI3-kinase controls cell migration via modulation of Reelin
expression. Nat Commun. 7:112452016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Okada T, Sinha S, Esposito I, Schiavon G,
López-Lago MA, Su W, Pratilas CA, Abele C, Hernandez JM, Ohara M,
et al: The Rho GTPase Rnd1 suppresses mammary tumorigenesis and EMT
by restraining Ras-MAPK signalling. Nat Cell Biol. 17:81–94. 2015.
View Article : Google Scholar
|
|
46
|
Mazur PK, Reynoird N, Khatri P, Jansen PW,
Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak
D, et al: SMYD3 links lysine methylation of MAP3K2 to Ras-driven
cancer. Nature. 510:283–287. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Leon CG, Locke JA, Adomat HH, Etinger SL,
Twiddy AL, Neumann RD, Nelson CC, Guns ES and Wasan KM: Alterations
in cholesterol regulation contribute to the production of
intra-tumoral androgens during progression to castration-resistant
prostate cancer in a mouse xenograft model. Prostate. 70:390–400.
2010.
|
|
48
|
Yuan X, Cai C, Chen S, Chen S, Yu Z and
Balk SP: Androgen receptor functions in castration-resistant
prostate cancer and mechanisms of resistance to new agents
targeting the androgen axis. Oncogene. 33:2815–2825. 2014.
View Article : Google Scholar
|
|
49
|
Holzbeierlein J, Lal P, LaTulippe E, Smith
A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, et
al: Gene expression analysis of human prostate carcinoma during
hormonal therapy identifies androgen-responsive genes and
mechanisms of therapy resistance. Am J Pathol. 164:217–227. 2004.
View Article : Google Scholar
|
|
50
|
Ettinger SL, Sobel R, Whitmore TG, Akbari
M, Bradley DR, Gleave ME and Nelson CC: Dysregulation of sterol
response element-binding proteins and downstream effectors in
prostate cancer during progression to androgen independence. Cancer
Res. 64:2212–2221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Han W, Gao S, Barrett D, Ahmed M, Han D,
Macoska JA, He HH and Cai C: Reactivation of androgen
receptor-regulated lipid biosynthesis drives the progression of
castration-resistant prostate cancer. Oncogene. 37:710–721. 2018.
View Article : Google Scholar :
|