Introduction
The initiation and progression of colorectal cancer
are associated with a loss of cellular response to the growth
inhibition exerted by tumor suppressor proteins, including
transforming growth factor (TGF)-β1. The TGF-β1 family of growth
factors serve fundamental roles in cell regulation, including cell
growth, differentiation, apoptosis and migration (1-3).
TGF-β1 members transduce signals from the plasma membrane to the
nucleus through type I and type II receptors (TβR-I and TβR-II) and
the Smad family of proteins (4).
The heterodimerization of TβR-II and TβR-I provokes the
phosphorylation of Smad2 and Smad3, and activated Smad2/3
subsequently combines with Smad4 and migrates to the nucleus to
regulate transcription (5). In
addition, TGF-β1 also signals through a number of non-canonical
pathways, including the PI3K/AKT/m-TOR, RhoA, Ras, p38
mitogen-activated protein kinase (MAPK), PP2A/p70s6K and c-Jun
N-terminal kinase (JNK) pathways (6).
TGF-β1 inhibits cell proliferation and promotes
apoptosis and differentiation in colon epithelial cells (5,7). In
a high percentage of colorectal tumors, the growth-inhibiting role
of TGF-β1 is disrupted by mutations in its receptors and downstream
effectors (3,8,9).
However, TGF-β1 levels are elevated in the plasma of patients with
cancers, including colorectal cancer (10,11).
TGF-β1 is overexpressed by malignant tumor cells, and it increases
the tumorigenicity of several types of tumor cells, indicating the
oncogenic switching of TGF-β1 function during malignant tumor
progression (12). TGF-β1 thus has
biphasic functions in tumorigenesis, with a growth inhibitory
effect in the early stages and exacerbation of the malignant
properties of tumors in later stages (13,14).
It has been shown that TGF-β1 contributes to malignant progression
via activation of the extracellular signal-activated kinase (Erk)
signaling pathway. Crosstalk between TGF-β1 signaling and the Erk,
JNK and MAPK pathways is important in the specificity of various
tumor-promoting effects of TGF-β1, including immune inhibition, the
stimulation of angiogenesis and improved cell mobility (15). Additionally, TGF-β1 has
anti-apoptotic functions and increases cell survival (16,17).
However, the signaling pathways underlying the TGF-β1-mediated
inhibition of apoptosis remain poorly characterized.
Apoptotic cell death serves an important role in the
elimination of defective or potentially dangerous cells, and
inhibits malignant transformation (18). Impairment of apoptosis disrupts the
process of physiological cell death, leading to tumor initiation,
progression and metastasis, by allowing the continued growth of
potentially dangerous cells and the accumulation of gene mutations.
Resistance to anticancer therapies is also caused by defects in
apoptotic mechanisms (19).
Apoptotic processes are controlled by several pro- and
anti-apoptotic families of genes (20,21).
The human inhibitor of apoptosis (IAP) family of proteins,
including c-IAP1, c-IAP2 and X-linked inhibitor of apoptosis
protein (XIAP), inhibits specific members of the caspase family
(22,23). The caspase-inhibitory effects of
IAPs are antagonized by apoptosis-promoting proteins. Two
mitochondrial proteins, Smac/DIABLO and HtrA2, promote caspase
activation by antagonizing the caspase-inhibitory activity of XIAP
(24,25). XIAP-associated factor 1 (XAF1) was
first isolated based on its ability to bind to and antagonize XIAP.
XAF1 consists of eight exons and is located on chromosome 17p13.2,
a region just telomeric to the p53 gene (26). XAF1 increases the apoptotic
response of tumor cells to chemotherapeutic agents, such as
etoposide and 5-fluorouracil (5-FU) (26). XAF1 mRNA is expressed in all
normal adult tissues, but is absent or present at very low levels
in various cancer cell lines, including colorectal tumor cells
(27). Some regulatory factors of
XAF1 expression have previously been revealed, but the underlying
signaling pathways are not well understood (28,29).
In the present study, whether the anti-apoptotic
function of TGF-β1 is associated with XAF1 expression was
investigated. The specific aim of the study was to determine
whether TGF-β1 evokes anti-apoptotic effects in human colon cancer
cells via the regulation of XAF1, and to elucidate the underlying
mechanism.
Materials and methods
Human cancer cell lines and reagents
Human colorectal cancer cell lines Colo205, RKO and
HT29 were purchased from the American Type Culture Collection
(Rockville, MD, USA) and Caco2 cells were obtained from Korea Cell
Line Bank (Seoul, Korea). The cells were maintained in RPMI-1640
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C in a humidified
atmosphere with 5% CO2. RKO sub-lines with short hairpin
(sh) RNA-mediated knockdown of XAF1 were established by
transfection with a shXAF1 construct (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) and Zeocin selection. The shRNA plasmid for
XAF1 was constructed by Genolution Pharmaceuticals, Inc. (Seoul,
Korea). The mitogen-activated protein kinase kinase (MEK) inhibitor
U0126 was purchased from New England Biolabs, Inc. (Ipswich, MA,
USA). Porcine TGF-β1 (1010-B1001) and anti-TGF-β1 neutralizing
antibodies (nAb; MAB240-100) were obtained from R&D Systems,
Inc. (Minneapolis, MN, USA).
Drug treatment
To evoke apoptotic stress, cells were exposed to
5-FU (20 µM), etoposide (50 µM),
H2O2 (50 µM), γ-IR (6 Gy) or hypoxic
conditions (1% O2). γ-irradiation and hypoxic stress
assays were carried out using an irradiator (IBL-437-C; Syncor
Intl. Corp., Sylmar, CA, USA) and hypoxic chamber (2000 Hypoxia
Workstation; Ruskinn Technology Ltd., Leeds, UK), respectively.
After 24 or 48 h exposure, the cells were harvested for further
molecular analysis. TGF-β1 (2 ng/ml) was added to the cells 2 h
prior to drug exposure, and TGF-β1 nAb (2 ng/ml) was added 2 h
before TGF-β1 treatment.
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total cellular RNA was extracted from cells by a
single-step method (30). One
microgram total cellular RNA was converted to complementary DNA
(cDNA) by RT using random hexamer primers and M-MLV reverse
transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.). PCR was
initially performed over a range of cycles (24-40 cycles), and 2
µl 1:4 diluted cDNA (12.5 ng/50 µl PCR) subjected to
30-36 cycles was observed to be within the logarithmic phase of
amplification with primers used for XAF1 (sense,
5′-CAGAAGTCCTCGCTGGAGTTTC-3′ and antisense,
5′-TGAAATTCTTTCCCCTTTCC-3′), PAI-1 (sense,
5′-CTGCCTAGTCCTGGGCCTGGCC-3′ and antisense,
5′-ATGAGCTCCTTGTACAGATGCC-3′) and the endogenous expression
standard gene glyceraldehyde-3-phosphate dehydrogenase
(GAPDH) (sense, 5′-CATGTGGGCCATGAGGTCCAC CAC-3′ and
antisense, 5′-AACCATGAGAAGTATGACAAC AGC-3′). PCR conditions
comprised 32-38 cycles at 95°C for 1 min, 58-62°C for 0.5 min, and
72°C for 1 min in 1.5 mM MgCl2-containing reaction
buffer (PCR buffer; Takara Bio, Inc., Shiga, Japan). A total of
10-15 µl PCR product was resolved by 2% agarose gel
electrophoresis. Quantitation was achieved by densitometric
scanning of the ethidium bromide-stained gels. Integration and
analysis were performed using Quantity One software (version 4.6.9;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). RT-PCR was repeated
at least three times for each specimen and the mean value was
determined.
Immunoblot (IB) assay
Cells were lysed with buffer containing 20 mM Tris
(pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1%
sodium dodecyl sulfate (SDS), 50 mM NaF, 2 mM sodium pyrophosphate,
1 mM sodium orthovanadate, protease inhibitor cocktail and 1 mM
phenylmethylsulfonyl fluoride. The cell lysate was clarified by
centrifugation (20,000 × g, 30 min) and the protein concentration
was determined using a BCA Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Total proteins (20-40 µg) were
supplemented with Laemmli buffer and loaded onto a 10%
SDS-polyacrylamide gel for electrophoresis. The proteins were
transferred to the nitrocellulose blotting membrane (10600001; GE
Healthcare Life Sciences, Little Chalfont, UK). The membranes were
incubated with specific antibodies for 12-18 h at 4°C. Antibodies
specific for XAF1 (1:1,000; sc-19194) and PAI-1 (1:2,500; sc-5297)
were obtained from Santa Cruz Biotechnology, Inc. and antibodies
specific for cleaved poly (ADP-ribose) polymerase (1:2,000; CST no.
9541), cleaved caspase-3 (1:2,000; CST no. 9661S) and Erk (1:2,500;
CST no. 910) were purchased from Cell Signaling Technology
(Danvers, MA, USA). Antibody for the loading control β-tubulin
(1:10,000; T0198) was obtained from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). Antibody binding was detected by enhanced
chemiluminescence using 1 h incubation at 24°C with a secondary
antibody conjugated to horseradish peroxidase (1:5,000;
PI-1000/2000; Vector Laboratories, Inc., Burlingame, CA, USA).
Expression plasmids, small interfering
RNA (siRNA) and transfection
Expression vector for green fluorescent protein
(GFP)-tagged Ras was constructed using pEGFP-N3 vector (Clontech,
Mannheim, Germany) and the Expand™ High Fidelity PCR System (Roche
Molecular Diagnostics, Pleasanton, CA, USA). Approximately
3.3×105 cells were plated per 60-mm plate in medium
containing 10% FBS. When 50-60% confluence was reached, the
transfection of constructs was performed using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) or Turbofect™ in
vitro Transfection Reagent (Pierce; Thermo Fisher Scientific,
Inc.) according to the manufacturer’s protocol. Each transfection
experiment was carried out in triplicate. The transfection
efficiency was monitored by fluorescence microscopy for GFP
detection. The siRNA duplexes against XAF1
(5′-AUGUUGUCCAGACUCAGAG-3′), Erk1 (5′-CCC
UGGAAGCCAUGAGAGAUGUCUA-3′), Erk2 (5′-CACCAU
UCAAGUUCGACAU-3′) and plasminogen activator inhibitor-1
(PAI-1; 5′-AAGCACAACUCCCUUAAGGUC-3′), and control siRNA
duplexes used as a negative control were synthesized by Dharmacon
(D-001210-0X; Lafayette, CO, USA) or Bioneer, Inc. (SN-1001-CFG;
Daejeon, Korea). The transfection of siRNAs was performed using the
Neon™ Transfection System (Invitrogen; Thermo Fisher Scientific,
Inc.). Transfected cells were stabilized in serum-supplemented
media for 24 h before further experiments.
Reporter constructs and luciferase
assay
The XAF1 promoter region (nucleotides
-221/+1) was cloned into the pGL3-basic vector (Promega
Corporation, Madison, WI, USA). RKO cells were cotransfected with
0.5 µg XAF1 promoter-containing luciferase plasmid
(Pro221-Luc) and 0.5 µg pPGL3-basic plasmid DNA using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.).
The culture medium was changed following 18 h of transfection, and
the cells were maintained for another 48 h prior to lysis with 200
µl lysis buffer (E3971; Promega Corporation). Following the
normalization of each extract for protein content, luciferase
activity was measured using the Luciferase Assay System (Promega
Corporation).
Apoptosis assay using flow cytometry
A total of 5×104 cells were seeded and
transfected with expression vector or siRNA. For the sub-G1
fraction analysis, cells were fixed with 70% ethanol and
resuspended in 1 ml phosphate-buffered saline containing 100 mg/ml
RNase and 50 mg/ml propidium iodide. (P4170; Sigma-Aldrich; Merck
KGaA). The assay was performed using a FACSCalibur flow cytometer
(BDBiosciences, San Jose, CA, USA) and the sub-G1 fraction was
analyzed using MultiCycle software (version 3.21; Phoenix Flow
Systems, San Diego, CA, USA).
Statistical analysis
Flow cytometry was performed in triplicate and data
are expressed as mean ± standard deviation. Statistical analysis
was performed using Student’s t-test when comparing two groups or
analysis of variance with the Bonferroni correction as a post hoc
test when making multiple comparisons. Statistical analyses were
carried out using GraphPad Prism 6.0 software (GraphPad Software,
Inc., La Jolla, CA, USA). P≤0.05 was considered to indicate a
statistically significant difference.
Results
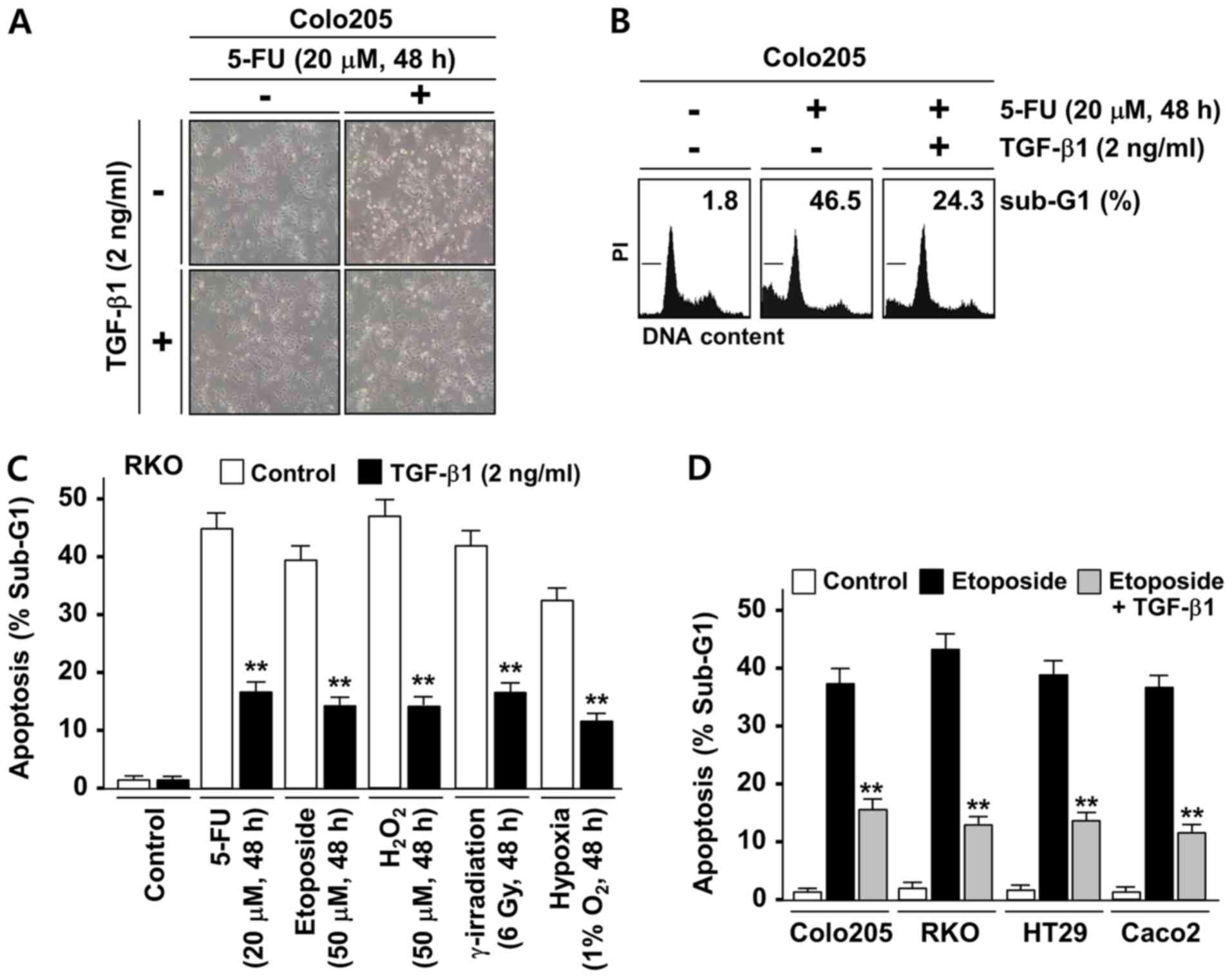
TGF-β1 protects colorectal tumor cells
from stress-induced apoptosis
TGF-β1 is known to promote colonic tumorigenesis
partially through its protective role against apoptotic stresses.
To investigate whether the anti-apoptotic function of TGF-β1 is
associated with its regulation of XAF1 expression, an initial
examination of the effect of TGF-β1 on the apoptosis induced by the
chemotherapeutic drug 5-FU was conducted using the Colo205 cell
line, which is known to have no genetic alterations of TGF-β
receptors (30,31). The Colo205 cells exhibited a
sensitive cytotoxic response to 5-FU (20 µM, 48 h), but this
response was markedly attenuated when cells were co-treated with
TGF-β1 (Fig. 1A). A flow
cytometric analysis of the apoptotic sub-G1 fraction also indicated
that TGF-β1 exerts a strong inhibitory effect on 5-FU-induced
apoptosis (Fig. 1B). To further
evaluate the cytoprotective role of TGF-β1 against various types of
apoptotic stress, RKO cells, which are widely used for the study of
stress-induced cell death, were utilized. RKO cells are known to
express low level of TGF-β receptor II due to a premature
termination mutation in one allele of the gene but have a
functional TGF-β1/Smad signaling pathway (9,31,32).
As shown in Fig. 1C, TGF-β1
treatment significantly attenuated the apoptotic response of RKO
cells to genotoxic (5-FU, etoposide and γ-irradiation), oxidative
(H2O2) and hypoxic (1% O2)
stresses. To further evaluate these findings, the inhibitory
effects of TGF-β1 on etoposide-induced apoptosis in Colo205, RKO
and two other cancer cell lines (HT29 and Caco2) that have no
alterations of TGF-β receptors were compared (30,31).
In all the cell lines tested, TGF-β1 evoked a significant
inhibitory effect on etoposide-induced apoptosis (Fig. 1D). These results indicate that
TGF-β1 evokes a strong cytoprotective effect on human colon cancer
cells under various apoptotic stress conditions.
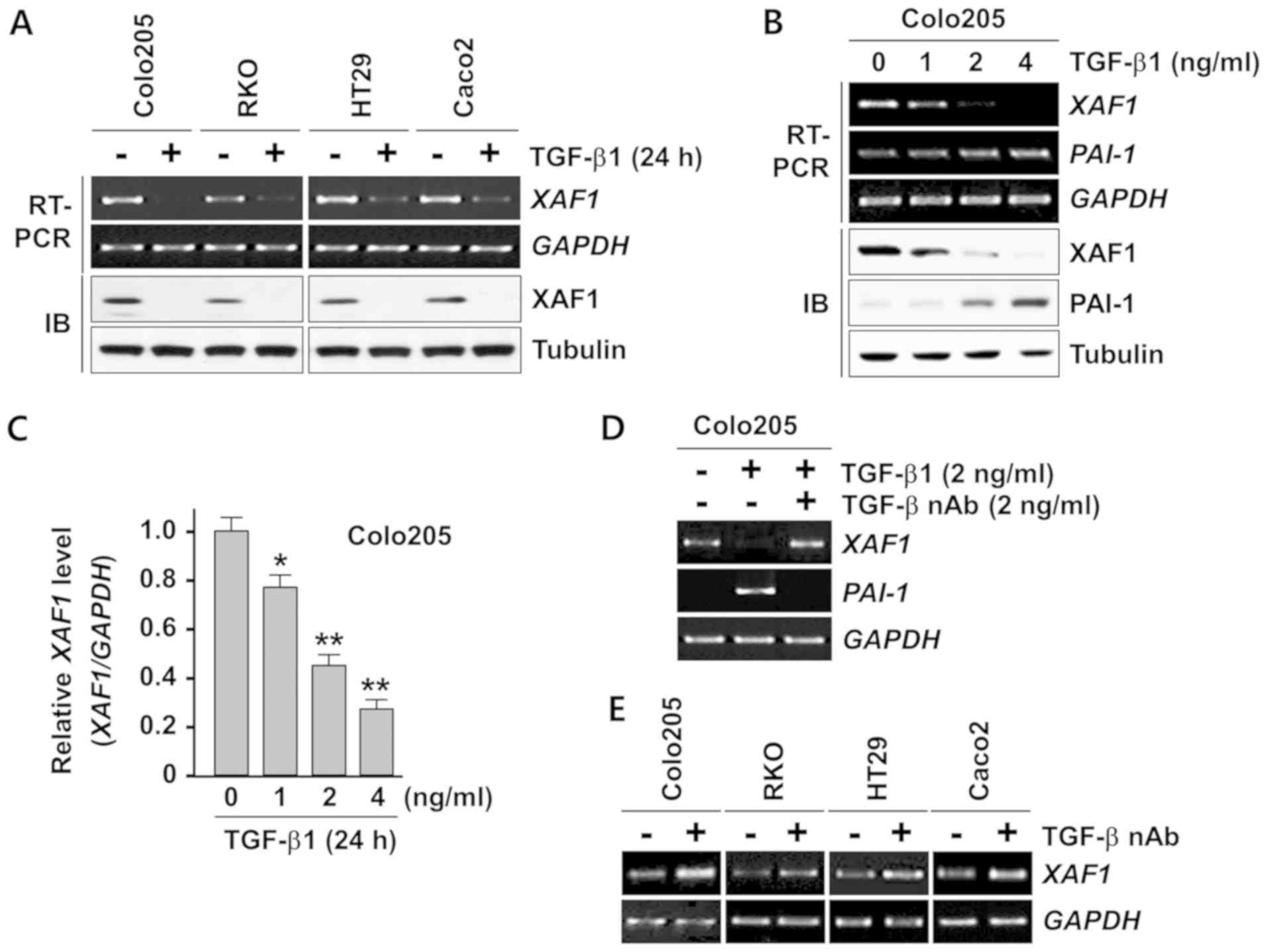
TGF-β1 suppresses XAF1 mRNA
expression
Next, the effect of TGF-β1 on XAF1 expression was
investigated. Semi-quantitative RT-PCR analysis demonstrated that
TGF-β1 treatment resulted in a strong reduction of XAF1 mRNA
levels in all four cell lines that were tested (Fig. 2A). An IB assay revealed that the
TGF-β1-induced downregulation of XAF1 mRNA expression was reflected
in a clear reduction in the levels of its protein product in these
cells. Furthermore, the TGF-β1-induced inhibition of XAF1
mRNA expression appeared to be concentration-dependent (Fig. 2B). In this assay, the activation of
PAI-1, a transcription target of TGF-β1, as utilized as an
indicator of the functionality of TGF-β1 signaling. Quantitative
analysis of the RT-PCR products revealed that 4 ng/ml TGF-β1
significantly repressed ~75% of XAF1 mRNA expression in
Colo205 cells (Fig. 2C). The
inhibitory effect of TGF-β1 on XAF1 mRNA expression was
abrogated when the cells were pretreated with TGF-β1 nAb (Fig. 2D). Next, whether TGF-β1 produced by
the tumor cells exerted an inhibitory effect on XAF1
expression was examined. Notably, the basal expression level of
XAF1 mRNA was elevated in all four cancer cell lines exposed
to TGF-β1 nAb, supporting the hypothesis that tumor cell-produced
TGF-β1 acts as a negative regulator of XAF1 expression
(Fig. 2E). Together, these results
indicate that XAF1 expression is repressed at the mRNA level by
TGF-β1 in human colon cancer cells.
TGF-β1 represses basal and stress-induced
XAF1 gene transcription levels
To elucidate the inhibitory effect of TGF-β1 on the
pro-apoptotic function of XAF1, the role of XAF1 in stress-induced
apoptosis was evaluated using the siRNA-mediated knockdown of XAF1
expression (Fig. 3A). RT-PCR
analysis revealed that XAF1 mRNA expression was strongly
activated in Colo205 cells exposed to 5-FU, and IB assays of
cleaved PARP and caspase-3 indicated that 5-FU-induced apoptosis
was attenuated by the siRNA-mediated blockade of XAF1 induction,
and the attenuation appeared to be XAF1 concentration-dependent
(Fig. 3B). Likewise, the
etoposide-induced apoptosis of RKO cells was markedly suppressed by
siXAF1 transfection in an apparently concentration-associated
manner (Fig. 3C). These results
indicate that XAF1 induction serves a key role in the apoptotic
response of colon cancer cells to genotoxic chemotherapeutic drugs.
The regulation of chemotherapeutic drug-mediated XAF1
induction by TGF-β1 was then evaluated. As shown in Fig. 3D, TGF-β1 exerted a strong
inhibitory effect on the induction of XAF1 mRNA by 5-FU,
etoposide and Adriamycin. To further elucidate the mechanistic
basis for the TGF-β1-induced suppression of XAF1 mRNA
expression, a promoter luciferase assay was performed using the
XAF1-Pro221-Luc reporter, which includes the proximal region of the
XAF1 promoter (nucleotides −221/+1 relative to ATG; Fig. 3E). The reporter was activated by
5-FU and this responsiveness was substantially attenuated by TGF-β1
in a dose-dependent manner, indicating that the TGF-β1-mediated
inhibition of XAF1 expression occurs through transcriptional
repression of the gene (Fig.
3F).
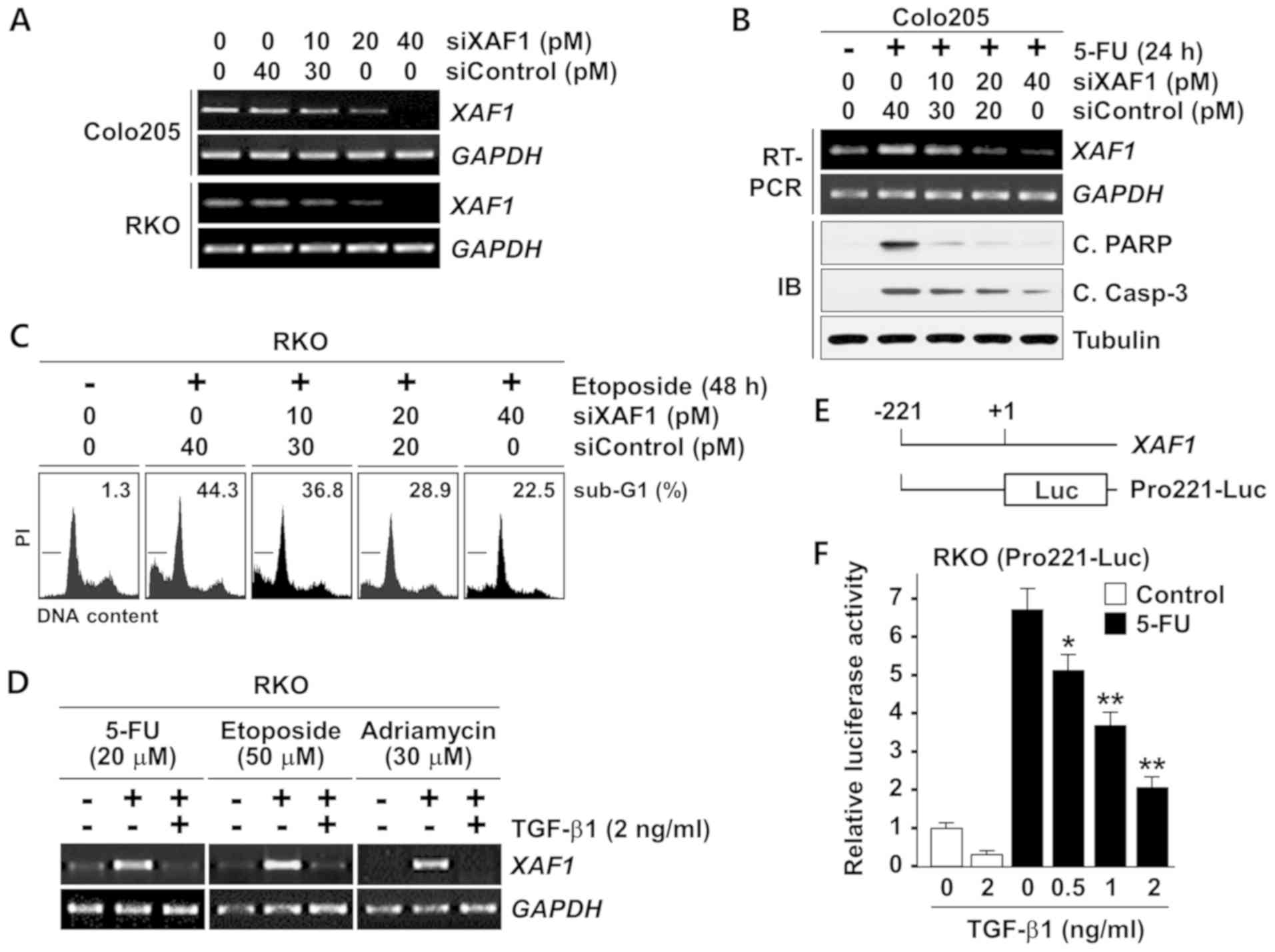 | Figure 3TGF-β1 blocks the stress-mediated
activation of XAF1 gene transcription. (A) XAF1
mRNA-depleting effect of siXAF1 duplexes. Colo205 and RKO cells
were transfected with increasing concentrations of siXAF1 as
indicated. Following 24 h transfection, XAF1 mRNA levels
were determined by semi-quantitative RT-PCR assay. (B) Effect of
XAF1 knockdown on 5-FU-induced apoptosis. Colo205 cells were
transfected with siXAF1 as indicated. Following 24 h transfection,
the cells were exposed to 5-FU (20 µM) for 24 h. (C) Flow
cytometric analysis of the sub-G1 fraction showing the effect of
XAF1 knockdown on etoposide-induced apoptosis. RKO cells
transfected with siXAF1 were exposed to etoposide (50 µM)
for 48 h. (D) Semi-quantitative RT-PCR assay showing the inhibitory
effect of TGF-β1 on therapeutic drug-induced XAF1 mRNA
expression. (E) Construction of the XAF1 reporter for
luciferase assay. (F) Attenuation of the Pro221-Luc responsiveness
to 5-FU by TGF-β1. RKO cells were treated with 5-FU (20 µM)
for 12 h in the absence or presence of TGF-β1 (2 ng/ml). Data
represent means ± SD of triplicate assays. *P<0.05,
**P<0.01 vs. the 0 ng/ml TGF-β1 + 5-FU group
(analysis of variance with the Bonferroni post hoc test). TGF-β1,
transforming growth factor-β1; XAF1, X-linked inhibitor of
apoptosis protein-associated factor 1; siXAF1, XAF1 small
interfering RNA; siControl, control small interfering RNA; RT-PCR,
reverse transcription-quantitative polymerase chain reaction; IB,
immunoblot; 5-FU, 5-fluorouracil; C. PARP, cleaved poly
(ADP-ribose) polymerase; C. Casp-3, cleaved caspase-3; SD, standard
deviation. |
TGF-β1 protects tumor cells from
drug-induced apoptosis via XAF1 repression
Next, whether the anti-apoptotic effect of TGF-β1 is
linked to its ability to repress XAF1 was assessed. Western
blot analysis revealed that the cleavage of PARP and caspase-3
triggered by the apoptosis-inducing chemotherapeutic drugs 5-FU,
etoposide and Adriamycin was strongly suppressed by pretreatment
with TGF-β1, and this effect was accompanied by the inhibition of
XAF1 induction (Fig. 4A). A
crucial link between the anti-apoptotic function of TGF-β1 and its
XAF1 repressive activity was further characterized using a stable
XAF1 knockdown sub-line of RKO cells constructed using a
shRNA-mediated knockdown system (Fig.
4B). Compared with the shControl sub-line, the shXAF1 sub-line
displayed a markedly attenuated apoptotic response to 5-FU
(Fig. 4C). The anti-apoptotic
effect of TGF-β1 was negligible in shXAF1 cells, further suggesting
that the TGF-β1-induced inhibition of stress-induced apoptosis is
highly dependent on its XAF1-repressing activity. Additionally,
based on the results indicating that tumor cell-produced TGF-β1 is
a negative regulator of XAF1 basal expression, whether tumor
cell sensitivity to apoptotic stresses is increased by the blockade
of TGF-β1 production by tumor cells was tested. As shown in
Fig. 4D, RKO cells pretreated with
TGF-β nAb displayed an increased apoptotic response to various
apoptotic stresses compared with untreated control cells. Together
these results indicate that TGF-β1 protects colon cancer cells by
blocking XAF1 induction under various apoptotic stress
conditions.
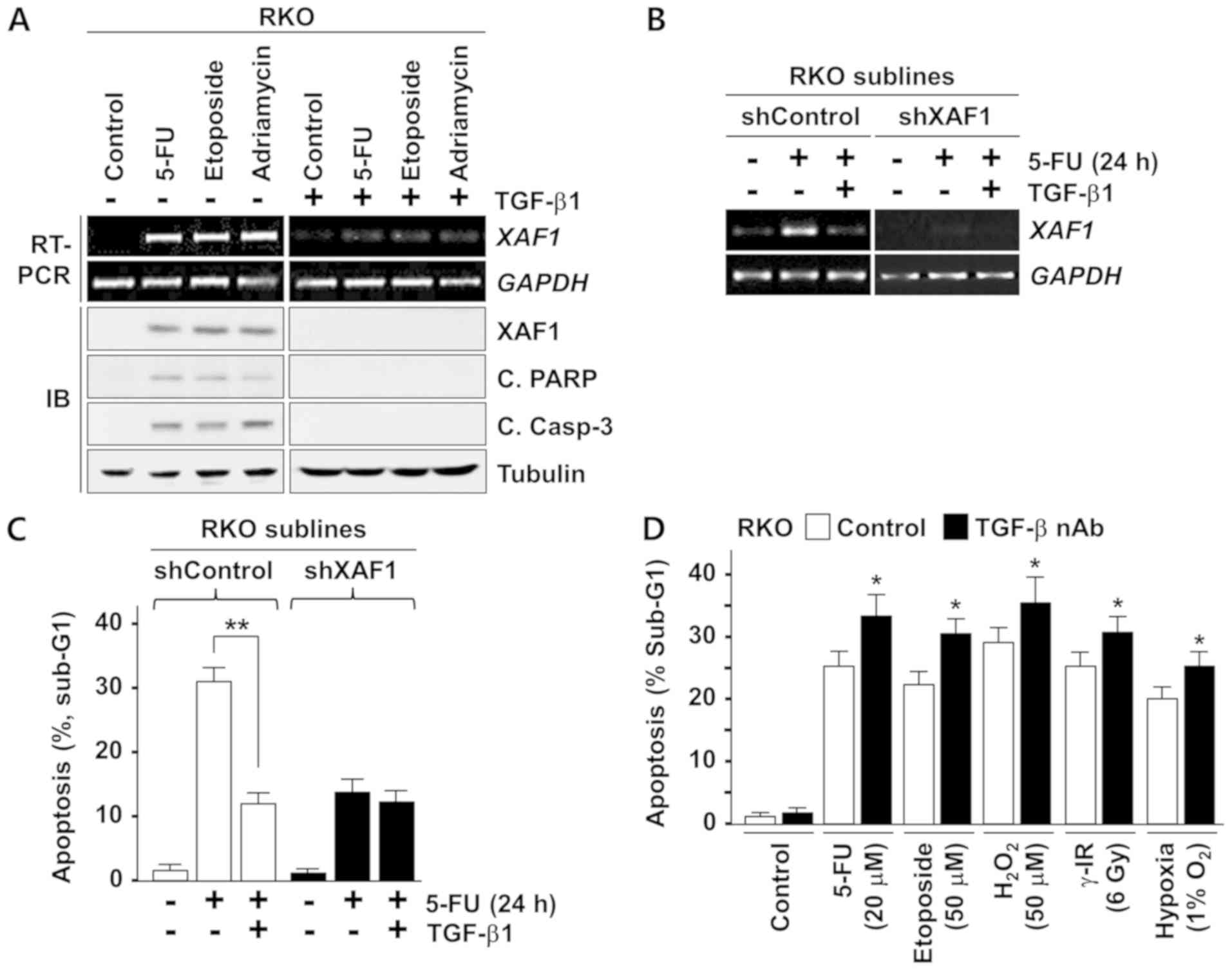 | Figure 4TGF-β1 suppresses stress-induced
apoptosis by blocking XAF1 induction. (A) Effect of TGF-β1
on chemotherapeutic drug-induced XAF1 and apoptosis. RKO cells were
treated with 5-FU (20 µM), etoposide (50 µM) or
Adriamycin (30 µM) for 24 h. TGF-β1 (2 ng/ml) was added to
the cells 2 h prior to drug treatment. (B) Effect of TGF-β1 on
5-FU-mediated XAF1 mRNA induction. RKO subline cells
(shControl and shXAF1) were treated with 5-FU (20 µM) and/or
TGF-β1 (2 ng/ml) for 24 h and XAF1 mRNA levels were
determined by semi-quantitative RT-PCR assay. (C) The inhibitory
effect of TGF-β on apoptosis is XAF1-dependent. shControl and
shXAF1 RKO subline cells were treated with 5-FU (20 µM)
and/or TGF-β1 (2 ng/ml). Apoptosis induction was measured by flow
cytometric analysis of the sub-G1 fraction. Data represent means ±
SD of triplicate assays. **P<0.01 (analysis of
variance with the Bonferroni post hoc test). (D) Effect of TGF-β
nAb pretreatment on stress-induced apoptosis. RKO cells were
exposed to various stresses for 24 h in the absence or presence of
TGF-β nAb (2 ng/ml). Apoptosis induction was measured by flow
cytometric analysis of the sub-G1 fraction. Data represent means ±
SD of triplicate assays. *P<0.05 vs. control
(Student’s t-test). TGF-β1, transforming growth factor-β1; XAF1,
X-linked inhibitor of apoptosis protein-associated factor 1; 5-FU,
5-fluorouracil; shXAF1, XAF1 short hairpin RNA; shControl, control
short hairpin RNA; RT-PCR, reverse transcription-quantitative
polymerase chain reaction; IB, immunoblot; C. PARP, cleaved poly
(ADP-ribose) polymerase; C. Casp-3, cleaved caspase-3; SD, standard
deviation. |
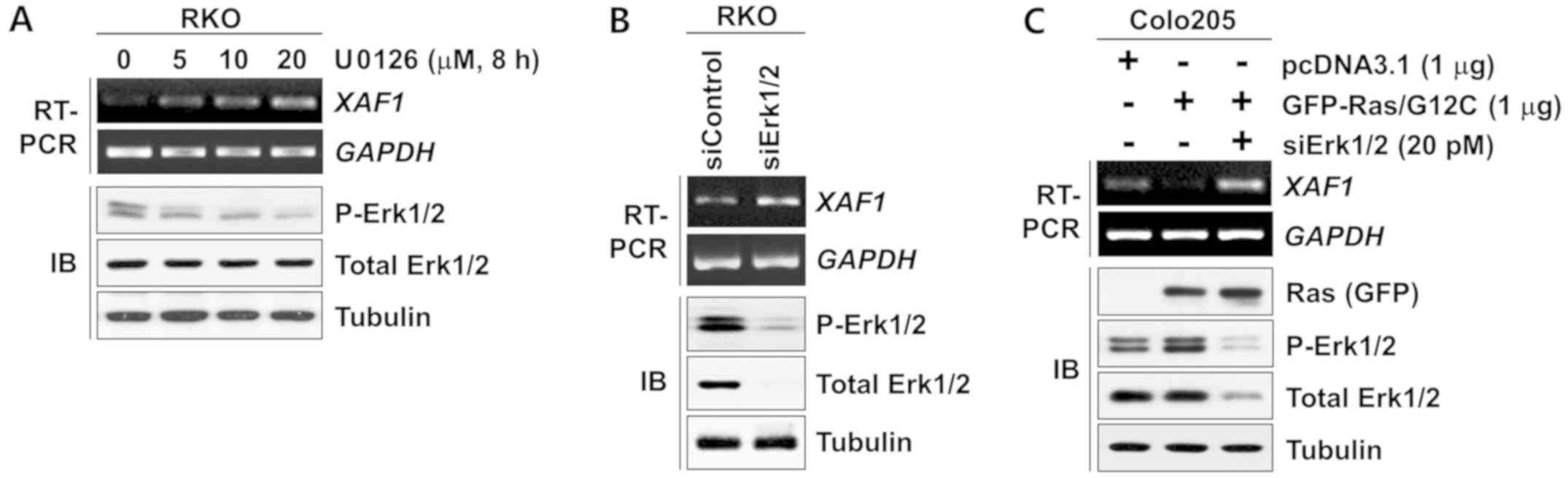
XAF1 expression is suppressed by Ras-Erk
activation
Activation of the Ras-Erk signaling pathway is
common in colorectal tumorigenesis and serves a key role in the
proliferation, survival and malignant progression of tumor cells.
Studies have shown that TGF-β1 activates Ras-Erk signaling to
promote tumor progression (15,33).
These findings suggest that the Ras-Erk signaling pathway is
involved in the TGF-β1-mediated repression of XAF1. Notably, in the
present study it was observed that the expression level of
XAF1 mRNA increased substantially following treatment with
U0126, an inhibitor of MEK, which is upstream of Erk (Fig. 5A). Consistent with this result,
XAF1 was upregulated by the siRNA-mediated knockdown of
Erk1/2 expression (Fig. 5B).
Furthermore, XAF1 levels were markedly decreased by the
ectopic expression of the activated form of K-Ras (Ras/G12C;
Fig. 5C). Together, these results
indicate that XAF1 expression is suppressed at the transcription
level by activation of the Ras-Erk signaling pathway.
TGF-β1 suppresses XAF1 expression by the
activation of Ras-Erk signaling
On the basis of the findings that XAF1 mRNA
expression is repressed by Ras-Erk signaling and that TGF-β1
activates the Ras-Erk pathway, the role of Ras-Erk signaling in the
TGF-β1-induced repression of XAF1 by TGF-β1 was examined. The
induction of XAF1 mRNA expression by 5-FU was strongly
inhibited by TGF-β1, and the effect appear to be dependent on
TGF-β1 concentration (Fig. 6A).
Intriguingly, the inhibitory effect of TGF-β1 on 5-FU-mediated
induction and apoptosis was markedly attenuated when Erk1/2
expression was depleted by siErk1/2 transfection (Fig. 6B and C). Consistently, the
inhibitory effect of TGF-β1 on 5-FU-induced apoptosis was greatly
abrogated by pretreatment with U0126 (Fig. 6D). This observation suggests TGF-β1
represses XAF1 mRNA expression via the Ras-Erk signaling
pathway. However, the depletion of PAI-1, which has been reported
to exert apoptosis-modulating activity (34,35),
did not affect the TGF-β1-mediated inhibition of apoptosis and XAF1
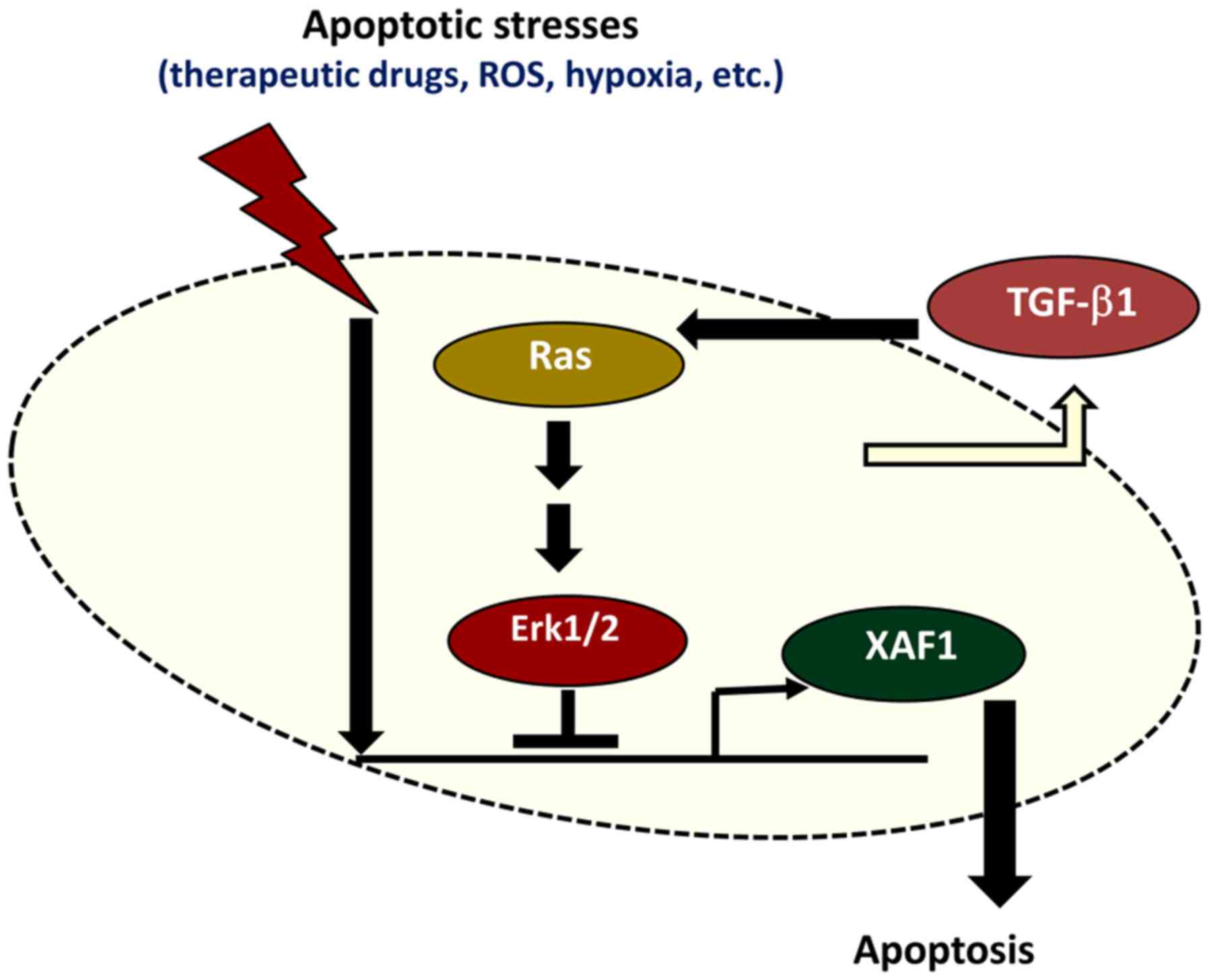
expression. Collectively, these results demonstrate that TGF-β1
protects colorectal tumor cells from various apoptotic stresses by
blocking XAF1 mRNA induction via activation of the Ras-Erk
signaling pathway (Fig. 7).
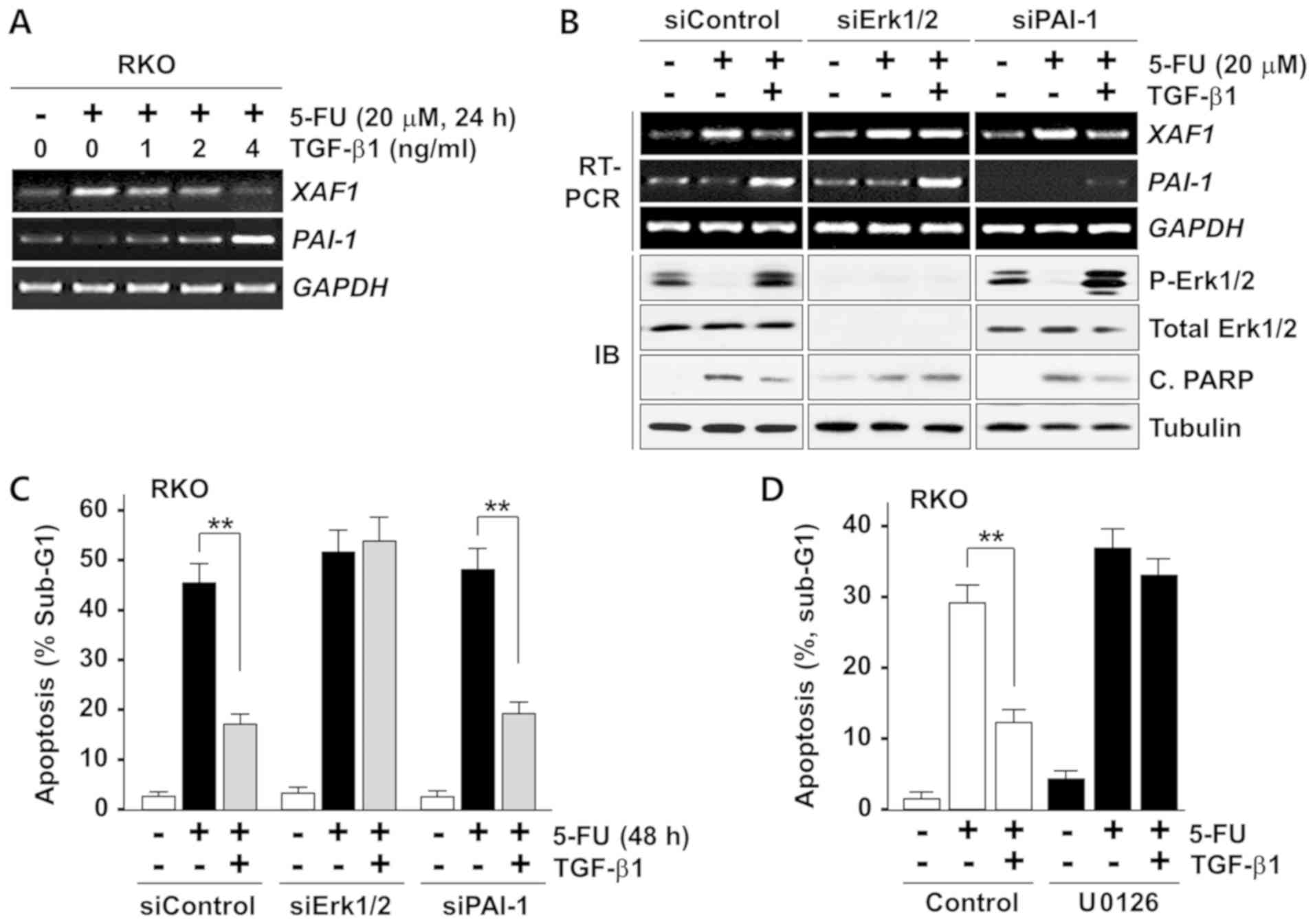 | Figure 6TGF-β1 represses XAF1
expression in a Ras-Erk signaling-dependent manner. (A)
Semi-quantitative RT-PCR analysis showing a dose-dependent
inhibitory effect of TGF-β1 on 5-FU-mediated XAF1 mRNA
induction in RKO cells. TGF-β1 was added to the cells 2 h prior to
5-FU treatment. (B) Failure of TGF-β1 to inhibit 5-FU-mediated
induction of XAF1 and apoptosis in Erk1/2-depleted cells.
RKO cells were transfected with siRNAs (20 pM) as indicated. The
transfected cells were treated with 5-FU (24 h). TGF-β1 (2 ng/ml)
was added to the cells 2 h prior to 5-FU treatment. PAI-1 depletion
was included for comparison. (C) Flow cytometric analysis of the
sub-G1 fraction showing TGF-β1 inhibition of apoptosis in an
Erk-dependent manner. RKO cells transfected with siRNAs (20 pM)
were incubated with TGF-β1 (2 ng/ml) 2 h prior to 5-FU (20
µM) treatment. Data represent means ± SD of triplicate
assays. **P<0.01 (analysis of variance with the
Bonferroni post hoc test). (D) Effect of U0126 treatment on the
TGF-β1-induced inhibition of 5-FU-induced apoptosis. RKO cells were
treated with 5-FU (20 µM) for 24 h. U0126 (20 µM) and
TGF-β1 (2 ng/ml) were added to the cells 2 h prior to 5-FU
treatment. Data represent means ± SD of triplicate assays.
**P<0.01 (analysis of variance with the Bonferroni
post hoc test). TGF-β1, transforming growth factor-β1; XAF1,
X-linked inhibitor of apoptosis protein-associated factor 1; Erk,
extracellular signal-activated kinase; P-Erk, phosphorylated Erk;
RT-PCR, reverse transcription-quantitative polymerase chain
reaction; IB, immunoblot; 5-FU, 5-fluorouracil; PAI-1, plasminogen
activator inhibitor-1; siErk, Erk small interfering RNA; siPAI-1,
PAI-1 small interfering RNA; siControl, control small interfering
RNA; C. PARP, cleaved poly (ADP-ribose) polymerase; SD, standard
deviation. |
Discussion
XAF1 was originally described as an antagonist of
XIAP-mediated anti-caspase activity (26,36).
XAF1 enhances the apoptotic sensitivity of tumor cells to various
genotoxic stresses, including γ-irradiation, 5-FU, etoposide and
H2O2, as well as non-genotoxic stresses,
including tumor necrosis factor-α and starvation (29). XAF1 is downregulated in various
human cancers, including colorectal cancer, by promoter
hypermethylation and a reduction in XAF1 expression is correlated
with advanced stage and high tumor grade (28,29).
The low-level transcription of XAF1 confers a survival advantage to
tumor cells by desensitizing the apoptotic response to various
stress conditions (29). However,
the signaling pathways and transcription factors involved in the
regulation of XAF1 gene expression remain largely undefined.
The present research team reported that XAF1 is activated at
the transcription level by various apoptotic stresses, including
chemotherapeutic drugs such as 5-FU, etoposide and cisplatin, and
that p53 and interferon-regulatory factor-1 serve key roles in
activating the XAF1 promoter in response to these stresses
(37,38). One of these studies also
demonstrated that numerous tumor-promoting growth factors
negatively regulate XAF1 mRNA expression (38). Therefore, the present study aimed
to determine whether XAF1 expression is influenced by TGF-β1.
TGF-β1 is a multifunctional cytokine that controls various aspects
of cellular functions including cell proliferation, differentiation
and death (3,39). TGF-β1 contributes to the malignant
progression of human colorectal tumors via inhibitory effects on
stress-induced tumor cell death (16,17).
However, the downstream mechanisms underlying the TGF-β1-mediated
protection of tumor cells from apoptotic stresses remain
unclear.
Based on expression analyses using four human colon
cancer cell lines, the present study aimed to determine whether
TGF-β1 regulates XAF1 expression to evoke its anti-apoptotic
effect. The results indicated that TGF-β1 repressed basal and
stress-mediated XAF1 gene transcription levels, and these
effects were tightly associated with its tumor cell-protective role
under various apoptotic conditions. Previous studies have provided
evidence that TGF-β1 activates Ras-Erk signaling to promote the
malignant transformation of colorectal epithelial cells in part by
attenuating the induction of apoptosis (40). Specifically, the activity and
crosstalk between TGF-β1 and Ras-Erk signaling pathways are
associated with the acquisition of invasion and metastatic
potential by epithelial tumor cells (2).
Notably, a previous study demonstrated that XAF1
mediates apoptosis through Erk in colon cancer (41). In the present study, it was
identified that activation of the Ras-Erk pathway is crucial for
the TGF-β1-mediated repression of XAF1 expression. In a promoter
luciferase assay, TGF-β1 abrogated the stress-mediated activation
of the XAF1 promoter via the Ras-Erk pathway. This was further
supported by the finding that siRNA-mediated Erk1/2 depletion or
treatment with the MEK inhibitor U0126 eradicated the inhibitory
effect of TGF-β1 on XAF1 mRNA expression. Together, these results
strongly suggest that TGF-β1 blocks XAF1 induction through the
activation of Ras-Erk signaling to protect human colorectal cancer
cells from a variety of apoptotic stresses. Previous literature has
reported that TGF-β1 is overexpressed in colorectal cancer and that
high serum or plasma levels of TGF-β1 in cancer patients are
associated with a poor prognosis (10,42).
Indeed, cancer recurrence following treatment has been shown to be
increased in individuals with high TGF-β1 expression (43). TGF-β1 has also been demonstrated to
induce a variety of pro-metastatic activities that range from the
induction of the epithelial-to-mesenchymal transition to the
expression of genes that allow metastatic colonization (44-46).
In the current study, a novel function of tumor-produced TGF-β1 was
identified, namely its ability to increase the resistance of cancer
cells to chemotherapeutic drug-induced apoptosis by blocking XAF1
induction. This finding also implicates XAF1 in the development of
drug resistance and disease progression. It is thus conceivable
that the restoration of XAF1 expression through the blockade of
Ras-Erk signaling could be a useful therapeutic strategy to improve
the efficiency of chemotherapeutic treatment and prevent the
progression of colorectal cancer.
In conclusion, the present study demonstrated that
TGF-β1 repressed XAF1 mRNA induction in human colon cancer cells
under various stressful conditions and increased the resistance of
tumor cells to therapeutic drug-induced apoptosis. The
TGF-β1-mediated suppression of XAF1 mRNA induction occurs through
the activation of Ras-Erk signaling. Restoration of XAF1 function
by blocking TGF-β1 or Ras-Erk signaling may increase tumor cell
sensitivity to apoptotic stimuli and may therefore be an effective
strategy for the treatment of drug-resistant colorectal tumors.
Funding
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Science and ICT (grant no.
NRF-2017R1A5A2014768).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
JRM analyzed and interpreted the data, and prepared
the first manuscript and revised it. HJK and SGC participated in
the conception and design of the study. SGC performed experiments
and statistical analysis. SJO and CKL reviewed the results and
participated in the discussion of the data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Eastham JA, Truong LD, Rogers E, Kattan M,
Flanders KC, Scardino PT and Thompson TC: Transforming growth
factor-beta 1: Comparative immunohistochemical localization in
human primary and metastatic prostate cancer. Lab Invest.
73:628–635. 1995.PubMed/NCBI
|
|
2
|
Park BJ, Park JI, Byun DS, Park JH and Chi
SG: Mitogenic conversion of transforming growth factor-beta1 effect
by oncogenic Ha-Ras-induced activation of the mitogen-activated
protein kinase signaling pathway in human prostate cancer. Cancer
Res. 60:3031–3038. 2000.PubMed/NCBI
|
|
3
|
Massagué J, Cheifetz S, Boyd FT and Andres
JL: TGF-beta receptors and TGF-beta binding proteoglycans: Recent
progress in identifying their functional properties. Ann NY Acad
Sci. 593(1 Transforming): 59–72. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Massagué J, Seoane J and Wotton D: Smad
transcription factors. Genes Dev. 19:2783–2810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mu Y, Gudey SK and Landström M: Non-Smad
signaling pathways. Cell Tissue Res. 347:11–20. 2012. View Article : Google Scholar
|
|
7
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massagué J: A very private TGF-beta
receptor embrace. Mol Cell. 29:149–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Markowitz S, Wang J, Myeroff L, Parsons R,
Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein
B, et al: Inactivation of the type II TGF-beta receptor in colon
cancer cells with microsatellite instability. Science.
268:1336–1338. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsushima H, Kawata S, Tamura S, Ito N,
Shirai Y, Kiso S, Imai Y, Shimomukai H, Nomura Y, Matsuda Y, et al:
High levels of transforming growth factor beta 1 in patients with
colorectal cancer: Association with disease progression.
Gastroenterology. 110:375–382. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Friedman E, Gold LI, Klimstra D, Zeng ZS,
Winawer S and Cohen A: High levels of transforming growth factor
beta 1 correlate with disease progression in human colon cancer.
Cancer Epidemiol Biomarkers Prev. 4:549–554. 1995.PubMed/NCBI
|
|
12
|
Park JI, Lee MG, Cho K, Park BJ, Chae KS,
Byun DS, Ryu BK, Park YK and Chi SG: Transforming growth
factor-beta1 activates interleukin-6 expression in prostate cancer
cells through the synergistic collaboration of the Smad2,
p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene.
22:4314–4332. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Principe DR, Doll JA, Bauer J, Jung B,
Munshi HG, Bartholin L, Pasche B, Lee C and Grippo PJ: TGF-β:
Duality of function between tumor prevention and carcinogenesis. J
Natl Cancer Inst. 106:djt3692014. View Article : Google Scholar
|
|
14
|
Yan Z, Winawer S and Friedman E: Two
different signal transduction pathways can be activated by
transforming growth factor beta 1 in epithelial cells. J Biol Chem.
269:13231–13237. 1994.PubMed/NCBI
|
|
15
|
Javelaud D and Mauviel A: Crosstalk
mechanisms between the mitogen-activated protein kinase pathways
and Smad signaling downstream of TGF-beta: Implications for
carcinogenesis. Oncogene. 24:5742–5750. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chin BY, Petrache I, Choi AM and Choi ME:
Transforming growth factor beta1 rescues serum deprivation-induced
apoptosis via the mitogen-activated protein kinase (MAPK) pathway
in macrophages. J Biol Chem. 274:11362–11368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang Y, Hutter D, Liu Y, Wang X, Sheikh
MS, Chan AM and Holbrook NJ: Transforming growth factor-beta 1
suppresses serum deprivation-induced death of A549 cells through
differential effects on c-Jun and JNK activities. J Biol Chem.
275:18234–18242. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Thompson CB: Apoptosis in the pathogenesis
and treatment of disease. Science. 267:1456–1462. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haq R and Zanke B: Inhibition of apoptotic
signaling pathways in cancer cells as a mechanism of chemotherapy
resistance. Cancer Metastasis Rev. 17:233–239. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reed JC: Dysregulation of apoptosis in
cancer. J Clin Oncol. 17:2941–2953. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kaufmann SH and Vaux DL: Alterations in
the apoptotic machinery and their potential role in anticancer drug
resistance. Oncogene. 22:7414–7430. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fraser M, Leung BM, Yan X, Dan HC, Cheng
JQ and Tsang BK: p53 is a determinant of X-linked inhibitor of
apoptosis protein/Akt-mediated chemoresistance in human ovarian
cancer cells. Cancer Res. 63:7081–7088. 2003.PubMed/NCBI
|
|
23
|
Salvesen GS and Duckett CS: IAP proteins:
Blocking the road to death’s door. Nat Rev Mol Cell Biol.
3:401–410. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Du C, Fang M, Li Y, Li L and Wang X: Smac,
a mitochondrial protein that promotes cytochrome c-dependent
caspase activation by eliminating IAP inhibition. Cell. 102:33–42.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Suzuki Y, Imai Y, Nakayama H, Takahashi K,
Takio K and Takahashi R: A serine protease, HtrA2, is released from
the mitochondria and interacts with XIAP, inducing cell death. Mol
Cell. 8:613–621. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liston P, Fong WG, Kelly NL, Toji S,
Miyazaki T, Conte D, Tamai K, Craig CG, McBurney MW and Korneluk
RG: Identification of XAF1 as an antagonist of XIAP anti-Caspase
activity. Nat Cell Biol. 3:128–133. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ma TL, Ni PH, Zhong J, Tan JH, Qiao MM and
Jiang SH: Low expression of XIAP-associated factor 1 in human
colorectal cancers. Chin J Dig Dis. 6:10–14. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee MG, Huh JS, Chung SK, Lee JH, Byun DS,
Ryu BK, Kang MJ, Chae KS, Lee SJ, Lee CH, et al: Promoter CpG
hyper-methylation and downregulation of XAF1 expression in human
urogenital malignancies: Implication for attenuated p53 response to
apoptotic stresses. Oncogene. 25:5807–5822. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chung SK, Lee MG, Ryu BK, Lee JH, Han J,
Byun DS, Chae KS, Lee KY, Jang JY, Kim HJ, et al: Frequent
alteration of XAF1 in human colorectal cancers: Implication for
tumor cell resistance to apoptotic stresses. Gastroenterology.
132:2459–2477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chomczynski P and Sacchi N: Single-step
method of RNA isolation by acid guanidinium
thiocyanate-phenol-chloroform extraction. Anal Biochem.
162:156–159. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ilyas M, Efstathiou JA, Straub J, Kim HC
and Bodmer WF: Transforming growth factor beta stimulation of
colorectal cancer cell lines: Type II receptor bypass and changes
in adhesion molecule expression. Proc Natl Acad Sci USA.
96:3087–3091. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Liu C, Wang J, Fan Y, Wang Z and
Wang Y: Oxymatrine inhibits the migration of human colorectal
carcinoma RKO cells via inhibition of PAI-1 and the TGF-β1/Smad
signaling pathway. Oncol Rep. 37:747–753. 2017. View Article : Google Scholar
|
|
33
|
Janda E, Lehmann K, Killisch I, Jechlinger
M, Herzig M, Downward J, Beug H and Grünert S: Ras and TGFβ
cooperatively regulate epithelial cell plasticity and metastasis:
Dissection of Ras signaling pathways. J Cell Biol. 156:299–313.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Higgins SP, Samarakoon R, Higgins CE,
Freytag J, Wilkins-Port CE and Higgins PJ: TGF-β1-induced
expression of the anti-apoptotic PAI-1 protein requires EGFR
signaling. Cell Commun Insights. 2:1–11. 2009. View Article : Google Scholar
|
|
35
|
Chen SC, Henry DO, Reczek PR and Wong MK:
Plasminogen activator inhibitor-1 inhibits prostate tumor growth
through endothelial apoptosis. Mol Cancer Ther. 7:1227–1236. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Byun DS, Cho K, Ryu BK, Lee MG, Kang MJ,
Kim HR and Chi SG: Hypermethylation of XIAP-associated factor 1, a
putative tumor suppressor gene from the 17p13.2 locus, in human
gastric adenocarcinomas. Cancer Res. 63:7068–7075. 2003.PubMed/NCBI
|
|
37
|
Lee MG, Han J, Jeong SI, Her NG, Lee JH,
Ha TK, Kang MJ, Ryu BK and Chi SG: XAF1 directs apoptotic switch of
p53 signaling through activation of HIPK2 and ZNF313. Proc Natl
Acad Sci USA. 111:15532–15537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jeong SI, Kim JW, Ko KP, Ryu BK, Lee MG,
Kim HJ and Chi SG: XAF1 forms a positive feedback loop with IRF-1
to drive apoptotic stress response and suppress tumorigenesis. Cell
Death Dis. 9:8062018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Massagué J: How cells read TGF-beta
signals. Nat Rev Mol Cell Biol. 1:169–178. 2000. View Article : Google Scholar
|
|
40
|
McCubrey JA, Steelman LS, Chappell WH,
Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M,
Tafuri A, et al: Roles of the Raf/MEK/ERK pathway in cell growth,
malignant transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar
|
|
41
|
Yu LF, Wang J, Zou B, Lin MC, Wu YL, Xia
HH, Sun YW, Gu Q, He H, Lam SK, et al: XAF1 mediates apoptosis
through an extracellular signal-regulated kinase pathway in colon
cancer. Cancer. 109:1996–2003. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Levy L and Hill CS: Alterations in
components of the TGF-beta superfamily signaling pathways in human
cancer. Cytokine Growth Factor Rev. 17:41–58. 2006. View Article : Google Scholar
|
|
43
|
Calon A, Espinet E, Palomo-Ponce S,
Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung
P, Zhang XH, et al: Dependency of colorectal cancer on a
TGF-β-driven program in stromal cells for metastasis initiation.
Cancer Cell. 22:571–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Massagué J: TGFbeta in cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jung B, Staudacher JJ and Beauchamp D:
Transforming growth factor β superfamily signaling in development
of colorectal cancer. Gastroenterology. 152:36–52. 2017. View Article : Google Scholar
|
|
46
|
Mao L, Li Y, Zhao J, Li Q, Yang B, Wang Y,
Zhu Z, Sun H and Zhai Z: Transforming growth factor-β1 contributes
to oxaliplatin resistance in colorectal cancer via epithelial to
mesenchymal transition. Oncol Lett. 14:647–654. 2017. View Article : Google Scholar : PubMed/NCBI
|