Introduction
Glioblastoma (GB) is the most common and aggressive
malignant tumor of the central nervous system, characterized by a
high degree of proliferation, angiogenesis, necrosis and
invasiveness (1). According to the
most recent World Health Organization guidelines, GB is classified
as a grade IV diffuse astrocytic tumor (2), which can develop either de
novo (primary GB) or through the malignant progression of
lower-grade astrocytomas (secondary GB).
The current standard treatment for GB consists of
surgical resection, followed by radiotherapy with concomitant and
adjuvant temozolomide (TMZ) chemotherapy (3,4).
However, despite this intensive approach, almost all patients
experience recurrence (1) and the
prognosis of patients with this malignancy remains extremely poor,
with the median survival ranging between 12 and 15 months from
diagnosis (5). Therefore, more
effective therapies are urgently required.
The inactivation of the p53 tumor suppressor is one
of the most common molecular alterations occurring in GB. Indeed,
the p53 pathway has been found to be altered in 87% of GB cases
(6), by either mutations/deletions
of the TP53 gene itself (6,7) or
defects affecting other members of the pathway, such as the
amplification of the two main p53 negative regulators, murine
double minute (MDM)2 and MDM4, and mutations/deletions of the
cyclin-dependent kinase inhibitor 2A (CDKN2A) locus,
encoding the MDM2 inhibitor, p14ARF (6). Similar high frequencies of p53
mutations have been observed among lower-grade astrocytomas and
secondary GBs, suggesting an important role of p53 alterations in
the early stages of GB development (8). Individuals carrying p53 germline
mutations are predisposed to the development of astrocytomas,
further supporting a crucial role of p53 mutations in driving
gliomagenesis (9). Consistently,
different mouse models analyzing the role of p53 mutations in GB
development, either alone or in combination with other molecular
alterations, have confirmed a central role of p53 defects in the
early stages of gliomagenesis, although additional alterations,
such as phosphatase and tensin homolog (PTEN) inactivation, are
required to drive p53-mediated GB development (10-13).
Of note, p53 can negatively regulate the expression
of O(6)-methylguanine-DNA
methyltransferase (MGMT) (14,15),
a DNA repair enzyme, whose high level of activity underlies GB
resistance to TMZ (16). This
suggests that the re-activation of p53 may be an effective strategy
with which to overcome both the growth advantage and the tumor
resistance to TMZ treatment conferred by p53 inactivation in GB. In
addition to p53 inactivation, TP53 gain-of-function (GOF)
mutations are associated with a poor prognosis and response to TMZ
in GB (17,18). Thus, restoring wild-type functions
in p53 mutants could represent a promising therapeutic approach
against GB.
Owing to its crucial role in apoptosis and cell
growth control, and considering that its pathway is altered in the
majority of human cancers (19),
p53 represents one of the most appealing targets for cancer therapy
and therefore, to date, several strategies targeting p53 and its
pathway have been developed (20-22).
Approaches aimed at restoring the oncosuppressive function of p53,
through either wild-type TP53 gene transfer systems
(12,23,24)
or small molecule/peptide-based methods (25-39),
have proven promising for GB treatment, owing not only to their
pro-apoptotic and anti-proliferative effects, but also to their
ability to sensitize GB cells to TMZ (23,24,28,30,33,37).
The majority of the above-mentioned p53
re-activation strategies are designed to prevent MDM2 from
targeting p53 for proteasomal degradation. For instance, the
well-known p53 re-activating molecule, nutlin-3, functions by
binding MDM2, and thus inducing the release and accumulation of
p53. In GB cells, nutlin-3 has been observed to principally cause
growth arrest, rather than apoptosis, and to be effective
exclusively in p53 wild-type cells (27).
Another molecule, RITA (re-activation of p53 and
induction of tumor cell apoptosis), was initially identified as a
compound preventing the p53-MDM2 interaction and inducing
p53-dependent apoptosis in various tumor cell lines (40). Unlike nutlin-3, RITA was found to
bind the p53 N terminus (40) and
was suggested to induce a conformational change of p53, thus
preventing its binding to MDM2 (41). However, some subsequent studies
questioned the inhibitory effects of RITA on the p53-MDM2
interaction (42,43) and it has emerged that the molecular
mechanisms underlying the cellular effects of RITA are much more
complex than just p53 stabilization. In particular, RITA can
function not only through p53-dependent mechanisms, but also
through other yet to be fully elucidated p53-independent
mechanisms. Indeed, accumulating data have indicated that RITA is
also effective on p53 null cells or on cells experimentally
depleted of p53 (44-49). Thus, despite the progress in
understanding the molecular mechanisms through which RITA
functions, mainly involving reactive oxygen species (ROS) induction
(48,50,51),
c-Jun N-terminal kinase (JNK) signaling activation (48,51,52),
DNA damage response (DDR) induction (49,51,53-57)
and the suppression of anti-apoptotic and pro-survival factors
(48,51,58),
the role of p53 in these RITA-triggered events remains
controversial. Indeed, RITA has been suggested to induce these
processes both dependently (50-52,54-56,58)
and independently of p53 (48,49,57).
Although the exact mode of action of RITA warrants
further investigation, this small molecule has exhibited several
appealing properties, including: i) Its pro-apoptotic rather than
growth-arresting effects, which can be crucial for efficiently
eliminating cancer cells, while avoiding possible interference with
the effects of chemotherapeutic drugs in the clinical setting
(59,60); ii) its preferential cytotoxicity to
malignant cells (40,48,51,57,58,60-62),
which supports the possible use of RITA for a safe anti-cancer
treatment; iii) its potential ability to restore wild-type
functions in p53 mutants, possibly by stabilizing a wild-type-like
conformation (60,63-67),
which renders RITA an attractive anti-cancer drug for use against
tumors characterized by a high frequency of p53 mutations, such as
GB.
Recently, RITA has been shown to reduce the
viability of a p53 wild-type GB cell line and to sensitize it to
TMZ treatment (68), thus
suggesting that RITA may represent, indeed, a novel and feasible
approach against GB. In the present study, we evaluated the effects
of RITA on different GB cell lines, expressing either wild-type or
mutant p53.
Materials and methods
Cell lines and culture conditions
The human GB cell line, PRT-HU2 (69), was kindly provided by Professor
Sergio Comincini (University of Pavia, Pavia, Italy), whereas the
GB cell line of unknown origin, U-87MG, and the GB cells T98G [Cat.
no. ATCC HTB-14 and ATCC CRL-1690, respectively; American Type
Culture Collection (ATCC), Manassas, VA, USA] were a kind gift from
Professor Annamaria Cimini (University of L’Aquila, L’Aquila,
Italy). Normal human astrocytes (NHAs) were purchased from Tebu-Bio
(Magenta, Italy; Cat. no. 882-05) and 293FT cells, used to generate
lentiviral particles, were purchased from ATCC (Cat. no. ATCC
PTA-5077). The GB and 293FT cells were grown in DMEM containing 10%
fetal bovine serum, 0.5% penicillin-streptomycin and 1% glutamine
at 37°C in a humidified atmosphere containing 5% CO2.
All cell culture reagents were purchased from Sigma-Aldrich (Milan,
Italy). The NHAs were maintained in astrocyte growth medium (Cat.
no. 821-500; Sigma-Aldrich,) in Petri dishes coated with 15
µg/ml poly-L-lysine (Cat. no. P4707; Sigma-Aldrich). All
cells were maintained at low passage numbers and periodically
tested for the presence of mycoplasma with the PlasmoTest™
Mycoplasma Detection kit (Cat. no. rep-pt1; Invivogen, San Diego,
CA, USA).
TP53 sequencing
Total RNA was extracted using TRIzol reagent (Cat.
no. 15596026; Thermo Fisher Scientific, Waltham, MA, USA) and 1
µg of RNA was then retrotranscribed using SuperScript
reverse transcriptase III (Cat. no. 18080085; Thermo Fisher
Scientific). Full-length TP53 was amplified by PCR using the
Pfu DNA polymerase (Cat. no. 600380; Agilent Technologies,
Santa Clara, CA, USA) and the following primers:
5′-CGTCCAGGGAGCAGGTAG-3′ (forward) and 5′-CAAGCAAGGGTTCAAAGAC-3′
(reverse). The reaction mixture was denatured at 94°C for 2 min and
subjected to 40 amplification cycles consisting of 20 sec at 94°C,
20 sec at 61°C, 40 sec at 72°C each, followed by a 3-min extension
at 72°C. PCR products were purified using the QIAquick gel
extraction kit (Cat. no. 28704; Qiagen S.r.l., Hilden, Germany).
Sequencing reactions were performed by PRIMM S.r.l., and analyzed
using Sequencher software 4.10.1 (Gene Codes Corp., Ann Arbor, MI,
USA). The human TP53 wild-type sequence used as a reference
was NM_000546.4 (National Center for Biotechnology Information,
Bethesda, MD, USA).
Cell treatment with RITA and nutlin-3,
MTS and clonogenic assay
RITA (Cat. no. CAY-10006426-5; Vincibiochem S.r.l,
Florence, Italy) and nutlin-3 (Cat. no. 675576; Sigma-Aldrich) were
dissolved in DMSO as a stock and then diluted in culture medium.
The cells were seeded in 96-well plates 24 h prior to treatment
with increasing concentrations (0.01-10 µM) of RITA or
increasing concentrations (0.62-20 µM) of nutlin-3. As a
control, cells were treated with the maximum amount of DMSO used to
deliver the compounds. At 72 h after treatment, cell viability was
evaluated by MTS assay (CellTiter 96® AQueous One
Solution Cell Proliferation Assay; Cat. no. G3582; Promega, Milan,
Italy), following the manufacturer’s instructions. The half maximal
inhibitory concentration (IC50) values were calculated
using GraphPad Prism Software, version 5.01 for Windows. DMSO
exhibited no toxic effect on any of the cell lines (data not
shown).
For clonogenic assays, 1.5×102 cells were
seeded in each well of 6-well plates and, 24 h after seeding, they
were treated with RITA for 24 h at its IC50 values or
DMSO, as a control. The medium containing RITA was then replaced,
according to a previously published protocol (63), to avoid excessive cell death in the
very sparse cell cultures. After 2 weeks, colonies were fixed with
methanol and stained at room temperature for 30 min with crystal
violet (Cat. no. HT90132; Sigma-Aldrich).
Cytoflourimetric analyses of cell cycle
profile and apoptosis
GB cells were plated in 100-mm diameter Petri dishes
and, at 24 h after seeding, were treated with RITA at its
IC50 values or DMSO. At 48/72 h after treatment, the
cells were collected, washed with PBS and then fixed in 70%
ice-cold ethanol. The cells were then incubated at 37°C for 1 h
with 50 µg/ml propidium iodide (PI; Cat. no. P4170;
Sigma-Aldrich) and 20 µg/ml RNase (Cat. no. 9001-99-4;
Sigma-Aldrich) and then analyzed with a BD FACSCalibur flow
cytometer (BD Biosciences, Milan, Italy).
Apoptosis was evaluated through FACS analysis
following cell staining with Annexin V-FITC and PI (Annexin V-FITC
kit; Cat. no. 130-092-052; Miltenyi Biotec Inc., Bologna, Italy)
according to the manufacturer’s instructions. Pifithrin-α (PFTα;
Cat. no. 6320882-82-2; Sigma-Aldrich) was dissolved in DMSO and
diluted to 25 µM in culture medium.
Silencing of p53 in T98G, U-87MG and
PRT-HU2 cells
To silence TP53, 2×106 293FT cells
were transfected with 2.25 µg of PAX2 packaging plasmid
(Cat. no. 12260; Addgene, Cambridge, MA, USA), 0.75 µg of
PMD2G envelope plasmid (Cat. no. 12259; Addgene) and 3 µg of
pLKO.1 hairpin vector, utilizing 30 µl of Attractene (Cat.
no. 301005; Qiagen S.r.l). The following pLKO.1 vectors were used:
Scrambled short hairpin RNA (shRNA; pLKO.1 shSCR, gift from
Professor S. Stewart, Washington University School of Medicine, St.
Louis, MO, USA; Cat. no. 17920; Addgene) and p53 shRNA (shp53
pLKO.1 puro, gift from Professor Bob Weinberg, Whitehead Institute
for Biomedical Research, Cambridge, MA, USA; Cat. no. 19119;
Addgene). Beginning from 24 h following transfection, supernatants
were collected at 24-h intervals for 3 days, filtered and used for
the transduction of the T98G, U-87MG and PRT-HU2 cell lines in the
presence of 1 µg/ml polybrene (Cat. no. 107689;
Sigma-Aldrich). At 3 days post-infection, the cells were selected
with 2.5 µg/ml puromycin (Cat. no. P7255;
Sigma-Aldrich).
Protein extraction and western blot
analysis
The cells were lysed on ice for 30 min in a buffer
consisting of 1 mM EDTA, 150 mM NaCl, 1% NP-40, 50 mM Tris-HCl pH
7.5, and 10 mg/ml each of aprotinin, leupeptin and pepstatin.
Proteins were quantified by the Bradford assay (Cat. no. 5000201;
Bio-Rad, Segrate, Italy). Equal amounts of proteins (50 µg)
per sample were electrophoresed onto 12.5% SDS-polyacrylamide gels
and blotted onto nitrocellulose membranes (Cat. no. 1620115;
Bio-Rad), which were then blocked in 5% non-fat dry milk and
incubated at 4°C overnight with monoclonal antibodies against p53
(Cat. no. sc-126), survivin (Cat. no. sc-17779), MGMT (Cat. no.
sc-56432) and GAPDH (Cat. no. sc-32233) (all from Santa Cruz
Biotechnology, Santa Cruz, CA, USA), and a polyclonal antibody
against phosphor-histone H2AX (γ-H2AX) (Cat. no. ab11174; Abcam,
Branford, CT, USA). The antibodies were diluted according to
manufacturers’ recommendations. Following incubation at room
temperature for 1 h with horseradish peroxidase-conjugated
secondary antibodies (goat anti-mouse IgG, Cat. no. sc-2005, and
goat anti-rabbit IgG, Cat. no. sc-2004, both from Santa Cruz
Biotechnology), signals were detected through ECL (Cat. no.
RPN2232; Amersham Biosciences, Little Chalfont, UK).
Drug combination studies
For drug combination studies, we first determined
the 72-h IC50 values of TMZ (Cat. no. 85622-93-1; Santa
Cruz Biotechnology) through MTS assay in the GB cells, as described
above for RITA. Subsequently, based on the RITA and TMZ
IC50 values, we challenged the GB cells for 72 h with
the two drugs, both alone and in combination at various
concentrations in a constant ratio (2-fold serial dilutions above
and below the RITA and TMZ IC50 values), and assessed
cell viability through MTS assay. Synergism, additivity or
antagonism were determined calculating the combination index (CI)
according to the Chou-Talalay equation, using CalcuSyn software
1.1.1 (BioSoft, Cambridge, UK). CI <1 indicates synergism, CI =
1 additive effect, and CI >1 antagonism. The r value represents
the linear correlation coefficient of the median-effect plot, which
indicates the conformity of the data to the mass-action law.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism Software, version 5.01 for Windows. Statistically significant
differences were evaluated by one-way repeated measures ANOVA with
Dunnett’s post hoc test, to compare all data vs. the controls.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Anti-proliferative effects of RITA on GB
cell lines
We examined the effects of RITA on 3 GB cell lines
(T98G, U-87MG and PRT-HU2), expressing either wild-type or mutant
p53 (Fig. 1A). In particular, the
TP53 mutational status was evaluated in previous studies in
both T98G cells, in which TP53 was found to be mutated
(Sanger Institute, Catalogue of Somatic Mutations in Cancer,
http://cancer.sanger.ac.uk/cancergenome/projects/cell_lines/)
(70) and in U-87MG cells, in
which it was found to be wild-type (70). Since the data on TP53
mutational status were, conversely, not available for the PRT-HU2
cells, we performed TP53 cDNA sequencing in these cells,
which were found to express wild-type TP53 (data not
shown).
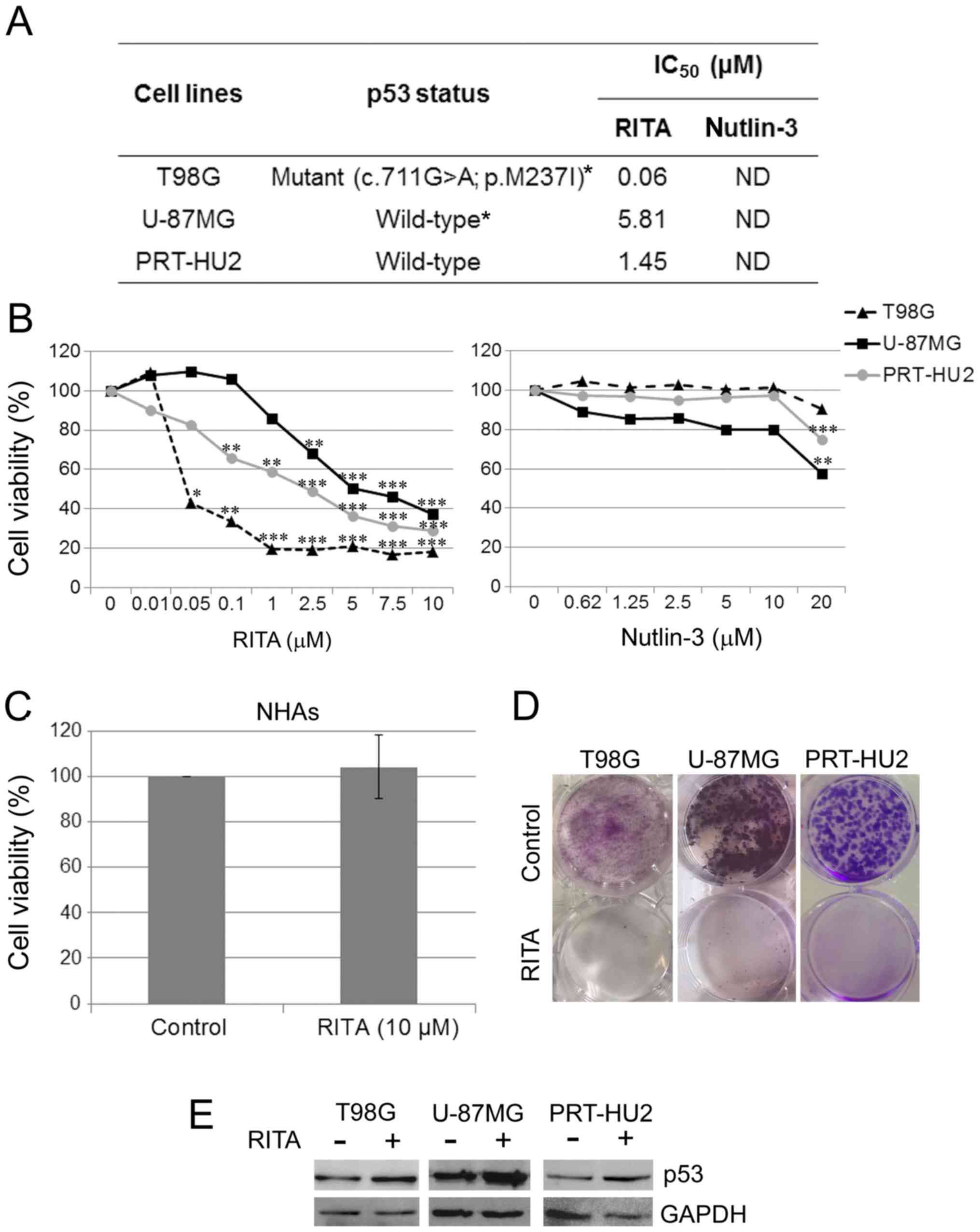 | Figure 1Effect of RITA and nutlin-3 on
glioblastoma (GB) cell viability. (A) Table reporting the p53
mutational status in T98G, U-87MG and PRT-HU2 cell lines, and RITA
IC50 values, as determined through MTS assay in these
cell lines at 72 h after treatment. Nutlin-3 IC50 values
were not determinable (ND) at the range of concentrations used. The
asterisks indicate TP53 mutational status, which was
previously reported (Sanger Institute, Catalogue of Somatic
Mutations in Cancer, http://cancer.sanger.ac.uk/cancergenome/projects/cell_lines/)
(70). (B) Dose-response curves
obtained through MTS assay in T98G, U-87MG and PRT-HU2 cell lines
at 72 h after treatment with either RITA or nutlin-3 at the
indicated concentrations. Results are reported as the means of at
least 2 independent experiments, each conducted in triplicate, and
expressed as percentages of cell viability calculated with respect
to the control cells treated with DMSO alone. The absorbance values
of the treated and control samples were subjected to one-way ANOVA
with Dunnett’s post hoc test. Statistically significant differences
between the treated and control cells are indicated as follows:
*P<0.05, significant; **P<0.01, very
significant; and ***P<0.001, extremely significant.
(C) Histogram showing that 72 h of treatment with 10 µM RITA
had no toxic effect on normal human astrocytes (NHAs), as
determined by MTS assay. Results are reported as the mean of 4
independent experiments and expressed as percentage of cell
viability calculated with respect to control cells treated with
DMSO alone. (D) Representative dishes, out of 2 independent
clonogenic assays, showing the long-term effect of RITA treatment
on T98G, U-87MG, and PRT-HU2 cell lines. Control cells were treated
with DMSO alone. (E) Western blot analysis of p53 in T98G, U-87MG
and PRT-HU2 cell lines treated for 24 h with RITA or DMSO, as a
control. An anti-GAPDH antibody was used for a loading control. A
representative experiment, out of at least 2 independent ones, is
shown. |
We treated the 3 GB cell lines with RITA at
concentrations ranging from 0.01 to 10 µM. After 72 h, we
evaluated cell viability by MTS assay and observed that RITA
exerted significant cytotoxic effects on all GB cell lines
(Fig. 1B). Therefore, as expected,
RITA was effective not only on p53 wild-type cells, but also on p53
mutant cells. We also compared the anti-proliferative effects of
RITA with those of the more well-studied p53 re-activating
molecule, nutlin-3, and observed that nutlin-3 was much less
effective than RITA in exerting anti-proliferative effects on the
GB cell lines, only affecting the viability of the p53 wild-type
cell lines (U-87MG and PRT-HU2) at the highest concentration used
(Fig. 1B). These data are
consistent with those of a previous study reporting that nutlin-3
was ineffective on the T98G cells, whereas it affected U-87MG cell
viability (27). We calculated the
RITA IC50 values for the 3 GB cell lines at 72 h after
treatment, whereas the nutlin-3 IC50 values were not
determinable under our experimental conditions (Fig. 1A).
To rule out the possible cytotoxic effects of RITA
on non-neoplastic brain cells, we treated NHAs with RITA at a
concentration of 10 µM, which corresponds to the maximum
amount of RITA used in the MTS assays on the GB cells. After 72 h,
we evaluated cell viability by MTS assay and observed no toxic
effect on the NHAs (Fig. 1C).
To evaluate whether RITA was able to exert a
long-term inhibitory effect on GB cell growth, we performed
clonogenic assays in the T98G, U-87MG and PRT-HU2 cells and
observed that treatment with RITA markedly inhibited colony
formation in all the cell lines (Fig.
1D).
We also verified the effects of RITA on p53 protein
levels through western blot analysis of total protein extracts from
GB cells treated with RITA for 24 h at its IC50 values.
We observed that treatment with RITA increased the p53 levels in
both wild-type and mutant GB cells (Fig. 1E).
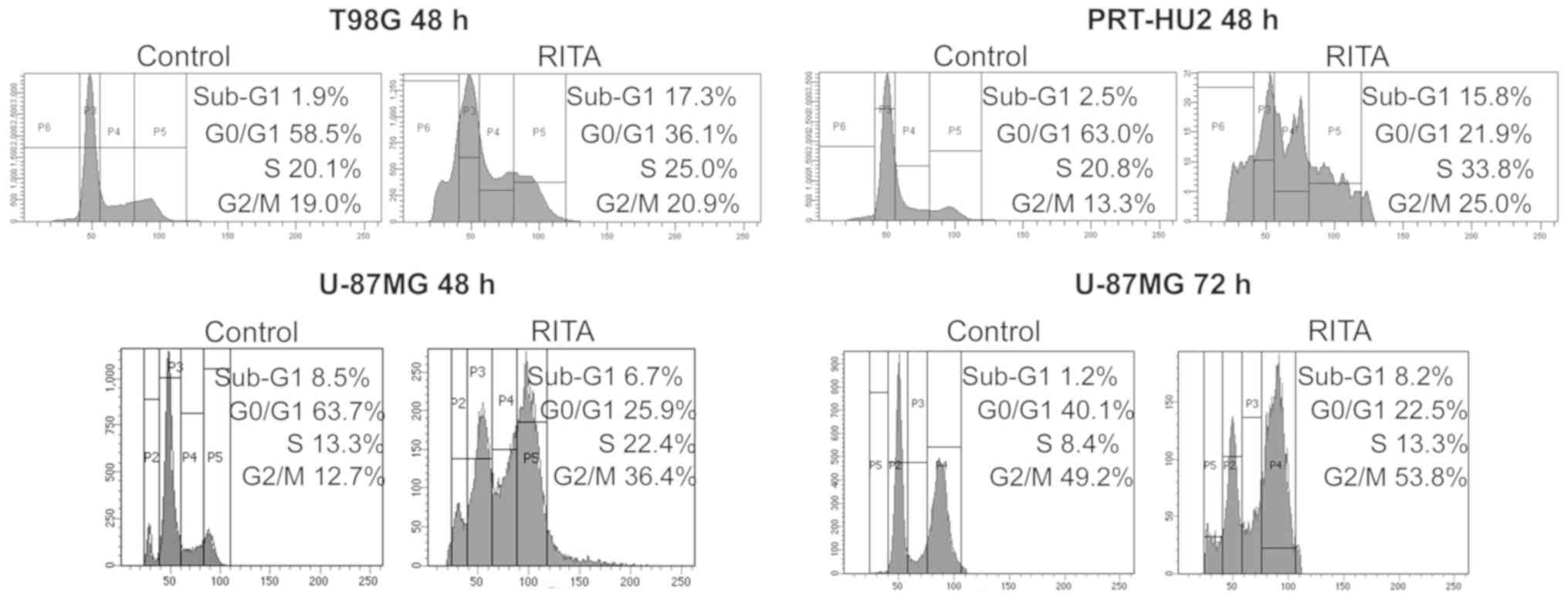
RITA affects cell cycle progression and
induces p53-dependent apoptosis of GB cell lines
To assess the effects of RITA on GB cell cycle
progression, we analyzed by FACS the cell cycle profiles of the
T98G, U-87MG and PRT-HU2 cell lines treated with RITA at the
IC50 values reported in Fig. 1A. We observed an increase in the
sub-G1 peak, which could be indicative of apoptosis, at 48 h
following treatment with RITA in the T98G and PRT-HU2 cells, and at
72 h following treatment in the less RITA-responsive U-87MG cells
(Fig. 2). Moreover, we observed
cell accumulation in the S and/or G2/M phases in all cell lines,
consistent with the findings of previous studies showing that RITA
can stall replication fork elongation (55,56)
and induce G2 arrest (56).
To verify the ability of RITA to induce apoptosis of
GB cells, we analyzed cell staining with Annexin V-FITC and PI by
FACS analysis at 72 h following treatment with RITA at its
IC50 values. These analyses revealed that RITA induced a
massive apoptosis of all GB cell lines (Fig. 3A). Moreover, to assess whether p53
has a role in the observed RITA-induced apoptosis of GB cells, we
treated these cells with PFTα, an agent reported to prevent p53
transcription-dependent apoptosis (71). PFTα markedly decreased the
RITA-induced apoptosis of all GB cell lines (Fig. 3A), thus suggesting that p53
transcriptional activity could be involved in the apoptotic program
triggered by RITA. To confirm the role of p53 in RITA-induced
apoptosis, we used this compound (at its 72-h IC50
values) to treat the T98G, U-87MG and PRT-HU2 cells in which p53
expression was stably silenced through transduction with lentiviral
vectors expressing TP53-specific shRNAs. We performed
Annexin V assays at shorter treatment times (24/48 h) as the
transduced cells underwent apoptosis earlier than the
non-transduced parental cells upon RITA treatment and also in order
to analyze early apoptotic events, which, compared with later
apoptotic events, are better distinguishable from possible necrotic
processes. We observed that in all the p53-silenced cells, the
percentage of apoptosis upon RITA treatment was markedly decreased
compared with that observed in the control p53-expressing cells,
stably transduced with non-targeting shRNAs (Fig. 3B). In addition, the p53 levels in
the GB cells expressing either non-targeting or
TP53-specific shRNAs were assessed by western blot analysis
in parallel experiments, which revealed that p53 was efficiently
knocked down by the shRNAs (Fig.
3C).
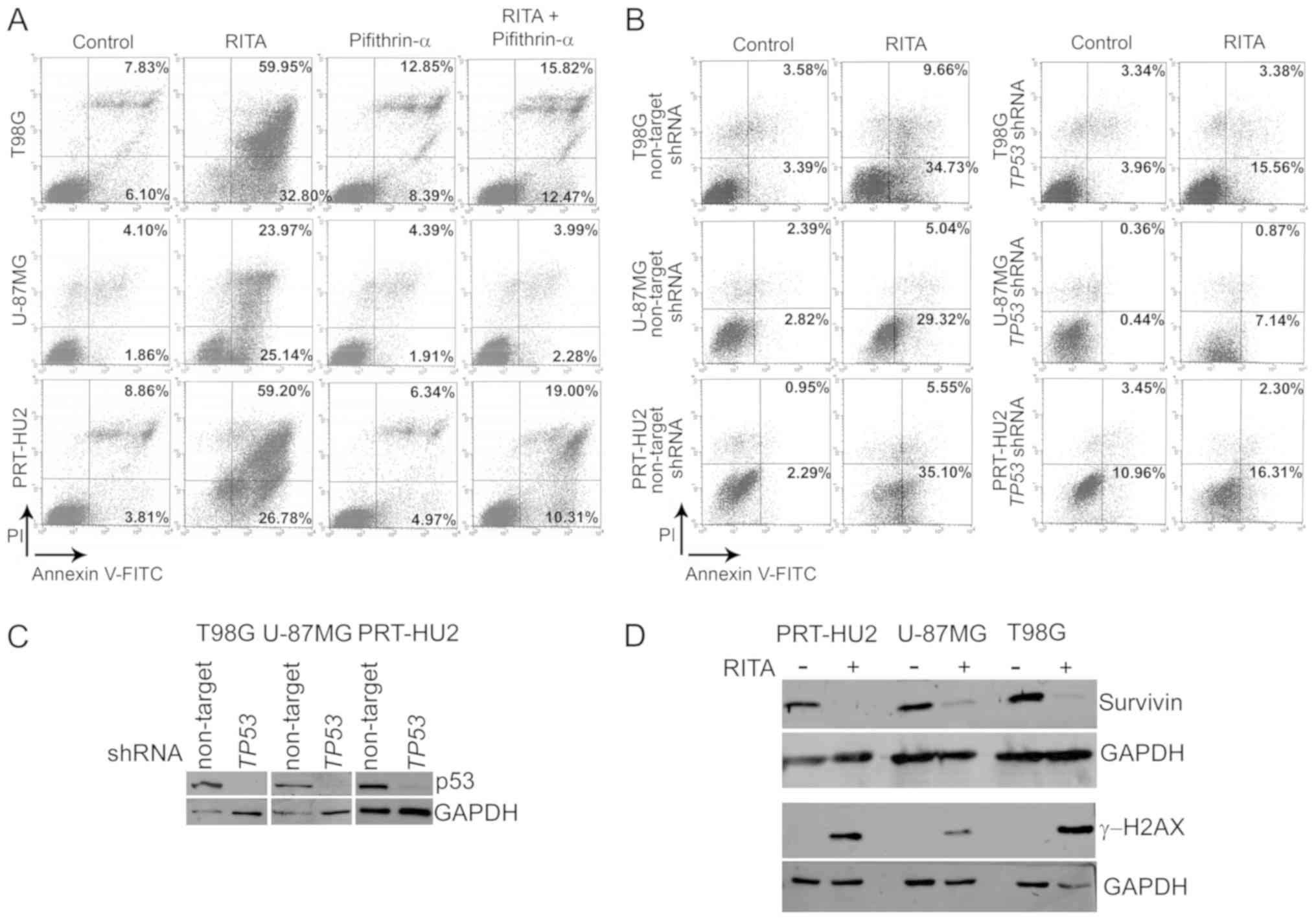 | Figure 3Induction of apoptosis of the
RITA-treated glioblastoma (GB) cell lines. (A) FACS analysis to
investigate apoptosis by cell staining with Annexin V-FITC and
propidium iodide (PI) in the T98G, U-87MG and PRT-HU2 cells at 72 h
after treatment with RITA and/or pifithrin-α or DMSO, as a control.
The graphs show the percentages of early apoptosis (Annexin
V-positive and PI-negative) and late apoptosis/necrosis (Annexin
V-positive and PI-positive). A representative experiment, out of
two independent ones, is shown. (B) Representative Annexin-V assays
(out of 2 independent experiments) in T98G, U-87MG and PRT-HU2
cells expressing either non-targeting or TP53-specific
shRNAs and treated for 48 h (T98G) and 24 h (U-87MG and PRT-HU2)
with RITA or DMSO, as a control. The graphs show the percentages of
early apoptosis and late apoptosis/necrosis. (C) Western blot
analysis of p53 in T98G, U-87MG and PRT-HU2 cells expressing either
non-targeting or TP53-specific shRNAs. An anti-GAPDH
antibody was used for a loading control. A representative
experiment, out of 2 independent ones, is shown. (D) Western blot
analysis of survivin and γ-H2AX expression in PRT-HU2, U-87MG and
T98G cell lines after 24 h of RITA treatment. Control cells were
treated with DMSO alone. An anti-GAPDH antibody was used for a
loading control. A representative experiment, out of 2 independent
ones, is shown. |
Both wild-type p53 (72,73)
and RITA/nutlin-3-activated p53 (27,58)
have previously been shown to inhibit the expression of the
anti-apoptotic protein, survivin, which has been implicated in GB
cell growth (74), prognosis
(75,76) and resistance to treatment (77-79),
and also seems a promising target for immunotherapeutic approaches
against gliomas (80). Thus, in
this study, we examined survivin expression in the GB cell lines
treated with RITA at its 72-h IC50 values. We observed,
through western blot analysis, that the survivin levels were
sharply decreased after 24 h of treatment with RITA in all GB cell
lines (Fig. 3D), thus suggesting
that the decrease in survivin expression may be involved in
RITA-triggered apoptosis.
Accumulating data have also suggested that RITA
activity largely depends on DDR induction (49,51,53-57).
Therefore, in this study, we examined the effect of RITA on γ-H2AX,
a marker of DNA damage, and found, indeed, that RITA increased the
γ-H2AX levels in all GB cell lines (Fig. 3D). This observation is consistent
with the S-phase cell accumulation described above, which can
indeed depend on RITA ability to activate an S-phase DNA damage
checkpoint, as previously described (55,56).
RITA synergizes with TMZ in GB cell
lines
We then examined whether RITA synergizes with TMZ, a
chemotherapeutic agent currently used in GB therapy. We first
determined the 72-h IC50 values of TMZ on the T98G,
U-87MG, PRT-HU2 cell lines by MTS assay (Fig. 4A). In line with what has been
previously reported (81), we
observed that the T98G cells were resistant to TMZ, as revealed by
its high IC50 value (987.4 µM). Subsequently,
based on these IC50 values and those previously
calculated for RITA (Fig. 1A), we
challenged the 3 GB cell lines for 72 h with the two drugs, both
alone and in combination at various concentrations in a constant
ratio (Fig. 4B). MTS data analysis
through the Chou-Talalay method (82) revealed CI values <1 for all cell
lines (Fig. 4C), which indicate
synergism between RITA and TMZ, although high concentrations of TMZ
were still required to markedly reduce the viability of resistant
cells (T98G).
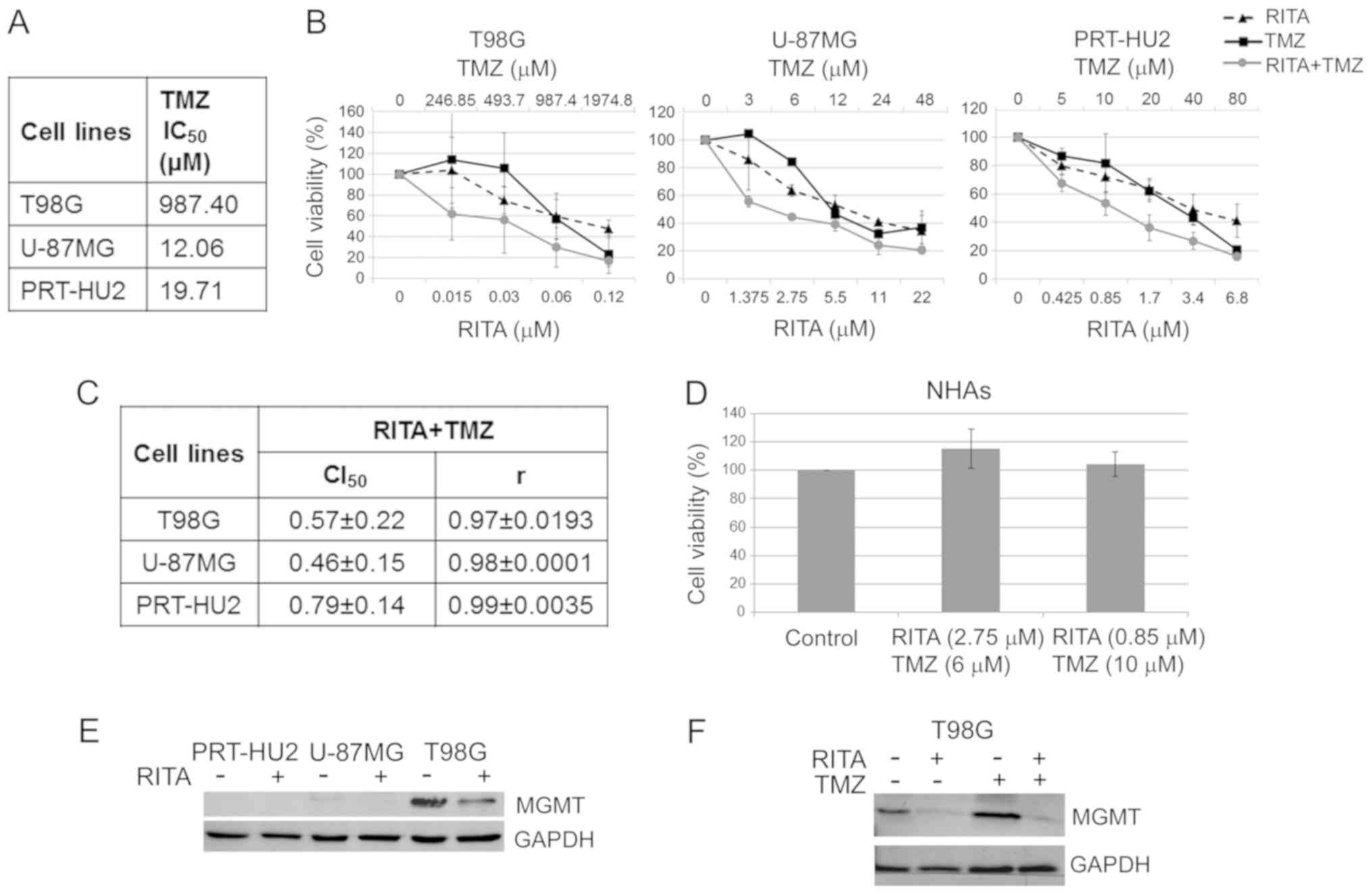 | Figure 4Synergistic effects of RITA-TMZ
combination on glioblastoma (GB) cell lines. (A) The table reports
the TMZ IC50 values on GB cell lines. These values were
calculated from cell viability data obtained through MTS after 72 h
of treatment with TMZ. (B) Dose-response curves for RITA alone, TMZ
alone and RITA-TMZ combinations in T98G, U-87MG and PRT-HU2 cell
lines at 72 h after treatment. Results represent the means of 2
independent experiments, each conducted in triplicate, and are
expressed as percentages of cell viability calculated with respect
to control cells treated with DMSO alone. (C) Table reporting the
means ± standard deviations of combination index (CI) and r values
of RITA-TMZ combination at 50% of cell killing (CI50)
following 72 h of treatment, calculated by the CalcuSyn software
for each of the 2 independent experiments. CI values <1 indicate
synergism. (D) Histogram showing that 72 h of treatment with
RITA-TMZ at the indicated combination doses had no toxic effect on
normal human astrocytes (NHAs), as determined through MTS assay.
Results are reported as the means of 2 independent experiments and
expressed as percentages of cell viability calculated with respect
to control cells treated with DMSO alone. (E) Western blot analysis
of MGMT expression in PRT-HU2, U-87MG and T98G cell lines at 24 h
after RITA treatment. Control cells were treated with DMSO alone.
An anti-GAPDH antibody was used for a loading control. A
representative experiment, out of 3 independent ones, is shown. (F)
Western blot analysis of MGMT expression in T98G cells at 24 h
after treatment with RITA and TMZ, both alone and in combination.
Control cells were treated with DMSO alone. An anti-GAPDH antibody
was used for a loading control. A representative experiment, out of
3 independent ones, is shown. |
To rule out possible cytotoxic effects of this drug
combination on non-neoplastic cells, we treated the NHAs with two
different RITA-TMZ combination doses, which corresponded to the
two-drug concentrations leading to a ~50% reduction in the
viability of the two TMZ-sensitive GB cell lines (U-87MG and
PRT-HU2), respectively (as shown by the MTS assays in Fig. 4B). Through MTS assay, we observed
no toxic effect of these drug combinations on NHAs 72 h following
treatment (Fig. 4D).
Considering the crucial role of MGMT in conferring
resistance to TMZ in GB cells (16), we examined, through western blot
analysis, the MGMT expression levels in GB cell lines, both
untreated and treated for 24 h with RITA at its 72-h
IC50 values. Consistent with what has previously been
reported for T98G and U-87MG cells (83), in this study, we observed that MGMT
was expressed only in the TMZ-resistant T98G cells, whereas it was
not expressed at detectable levels in the TMZ-sensitive U-87MG and
PRT-HU2 cell lines (Fig. 4E).
Importantly, RITA treatment decreased MGMT expression in T98G
cells. Moreover, we observed that RITA was able to decrease the
MGMT levels in the T98G cells also when combined with TMZ
(following 24 h of treatment with both drugs used at their
IC50 values), inhibiting the increase in MGMT
expression, which was observed upon treatment with TMZ alone
(Fig. 4F). Thus, these data
further suggest the potential of RITA to overcome TMZ resistance in
GB cells.
Discussion
Despite current intensive treatment regimens,
consisting of surgical resection followed by radiotherapy with
concomitant and adjuvant TMZ chemotherapy (3,4), GB
remains universally fatal, with a median survival ranging between
12 and 15 months from diagnosis (5).
Several advances have been achieved in the
understanding of the molecular mechanisms leading to GB development
and resistance to therapy, which are crucial for identifying
specific therapeutic targets. An appealing target for GB therapy is
the tumor suppressor p53, since the inactivation of its pathway
plays a key role in both GB development (6,8,12)
and resistance to TMZ treatment (14-16).
Moreover, TP53 GOF mutations have been shown to be
associated with a poor prognosis and response to TMZ in GB
(17,18). Therefore, the re-activation of p53
wild-type functions holds the potential to be an effective
therapeutic approach for GB.
In the present study, we tested RITA, a molecule
originally identified as a p53 re-activator preventing the p53-MDM2
interaction (40), on 3 GB cell
lines, expressing either wild-type or mutant p53. In particular,
the T98G cells have previously been reported to carry a
homozygously mutated form of TP53 (Sanger Institute,
Catalogue of Somatic Mutations in Cancer, http://cancer.sanger.ac.uk/cancergenome/projects/cell_lines/)
(70). Conversely, both the U-87MG
and PRT-HU2 cells bear wild-type TP53, as previously
reported for U-87MG cells (70)
and found in the present study for PRT-HU2 cells.
In this study, we observed that RITA increased the
p53 levels and reduced the viability and clonogenic potential of
all GB cell lines, irrespectively of the TP53 mutational
status. This observation is consistent with the findings of
previous studies by both our group and others, showing that RITA
was effective not only on p53 wild-type cells, but also on p53
mutant cells (57,60,63-67,84).
To date, different other compounds targeting the
MDM2-p53 interaction have been designed, which have proven to be
promising also for GB treatment (27-33,35-39).
In this study, we compared the anti-proliferative effects of RITA
with those of the well-known p53 re-activating molecule, nutlin-3,
and observed that nutlin-3 was much less effective on GB cell lines
under our experimental conditions, significantly reducing only the
viability of the p53 wild-type cell lines (U-87MG and PRT-HU2) at
the highest concentration. These observations are consistent with
those of a previous study showing that nutlin-3 was ineffective on
T98G cells, whereas it affected U-87MG cell viability, although
with a seemingly higher efficacy with respect to that observed in
the present study, owing to the longer treatment time used in the
previous study (27).
Importantly, in this study, we observed that RITA,
at the maximum concentration used on GB cells, had no toxic effect
on NHAs, consistent with the findings of previous studies showing
that RITA was preferentially cytotoxic to malignant cells (40,48,51,57,58,60-62).
Conversely, nutlin-3 has previously been shown to reduce the
viability of both NHAs (27) and
other non-cancer cells (60),
suggesting that RITA may be more tumor-selective with respect to
nutlin-3.
We then investigated whether the cytotoxic effects
of RITA on GB cells were due to cell cycle arrest or to cell death
and, in line with what has previously been reported (55,56),
we found that RITA induced cell accumulation in the S and G2/M
phases, but not in the G1 phase. Moreover, RITA induced massive
apoptosis of all GB cell lines. This is noteworthy, considering the
intrinsic resistance to apoptosis of GB cells, which is a key
mechanism whereby these cells evade death induced by anticancer
treatments (85).
Although RITA was initially identified as a
compound preventing the p53-MDM2 interaction and inducing a
p53-dependent apoptosis (40),
subsequent studies failed to demonstrate RITA inhibitory effect on
the p53-MDM2 interaction (42,43)
and indicated that RITA can act also through p53-independent
mechanisms (44-49,57,84).
Thus, in this study, to investigate the role of p53 in RITA-induced
apoptosis in GB cells, we treated these cells with RITA in
combination with the p53 inhibitor, PFTα, and observed that this
inhibitor markedly decreased the RITA-induced apoptosis of all GB
cells. Although this observation suggests that p53 can contribute
to the apoptotic program triggered by RITA, PFTα has also been
shown to be able to inhibit p53-independent apoptotic processes
induced by DNA damage (86).
Therefore, the ability of PFTα to inhibit apoptosis induced by RITA
may also be due to a PFTα action against p53-independent RITA
pro-apoptotic effects, as previously observed (48). In this study, to confirm the role
of p53 in RITA-induced apoptosis, we used this compound to treat GB
cells in which p53 was stably silenced and observed a markedly
decreased apoptosis of all p53-silenced cell lines. Thus,
RITA-induced apoptosis was at least in part dependent on p53 in
both p53 wild-type and mutant GB cells.
To further dissect the molecular mechanisms
underlying the pro-apoptotic effects of RITA on GB cells, we
analyzed the expression of the anti-apoptotic protein, survivin, in
RITA-treated GB cell lines and observed a sharp decrease in its
levels in all cell lines. This observation is consistent with
previous data showing the ability of both wild-type p53 (72,73)
and RITA/nutlin-3-activated p53 (27,58)
to inhibit the expression of survivin. In particular, a similar
decrease in survivin levels was previously observed in
nutlin-3-treated GB cells, although the decrease in survivin
expression was only found in p53 wild-type cells (27). The ability of RITA to decrease the
survivin levels is noteworthy, considering the involvement of this
protein in GB cell growth (74),
prognosis (75,76) and resistance to treatment (77-79).
In line with the S-phase cell accumulation observed
in this study, it has previously been reported that RITA is able to
activate an S-phase DNA damage checkpoint, stalling replication
fork elongation (55,56). DDR induction has been suggested to
be the primary RITA mechanism of action, leading to an indirect p53
stabilization or operating also independently of p53 (49,57).
Therefore, in this study, we analyzed the effect of RITA on the DNA
damage marker γ-H2AX and found, indeed, that RITA increased its
levels in all GB cell lines. Thus, although further studies are
required to elucidate the mechanisms of action of RITA in GB cells,
the effects of this molecule could, at least in part, depend on the
induction of DDR also in these cells.
Given that resistance to TMZ is a major cause of GB
treatment failure (16), we then
assessed whether RITA could increase GB cell sensitivity to TMZ. We
observed that RITA synergized with TMZ in TMZ-sensitive cells
(U-87MG and PRT-HU2) and enhanced the sensitivity of TMZ-resistant
cells (T98G) to this agent, although high TMZ concentrations were
still required to markedly reduce the viability of resistant cells.
Moreover, we also observed that the two-drug combination had no
toxic effects on NHAs, at least at the same concentrations causing
approximately a 50% reduction in the viability of the two
TMZ-sensitive GB cell lines.
Considering the crucial role of MGMT in the TMZ
resistance of GB (16), we
analyzed the expression of this DNA repair enzyme in GB cell lines
and observed that it was expressed only in T98G cells, consistent
with what has been previously reported (83). Notably, RITA was able to reduce
MGMT expression in T98G cells, not only when used as a single
agent, but also when combined with TMZ. Thus, RITA proved to be
able to overcome the increase in MGMT expression observed upon TMZ
treatment, which was previously described as a response to the
TMZ-induced DNA damage (87).
These data further support the possible use of RITA to sensitize GB
cells to TMZ treatment. Of note, it was previously suggested that
GOF activities of mutant p53 could contribute to TMZ resistance in
T98G cells (70). Indeed, the
knockdown of mutant p53 in these cells sensitized them to TMZ and
reduced their MGMT expression (70). Since we observed that RITA was
similarly able to sensitize T98G cells to TMZ and reduce MGMT
expression, despite the RITA-induced increase in mutant p53 levels
in these cells, we can speculate that RITA stabilized a
wild-type-like conformation of mutant p53 and restored its
wild-type functions in T98G cells. Analogously, another study
demonstrated that in pancreatic adenocarcinoma cells, in which a
gemcitabine (GEM)-induced adverse stabilization of mutant p53 was
shown to produce chemoresistance, either the knockdown of mutant
p53 or treatment with p53 re-activating molecules, including RITA,
sensitized these cancer cells to GEM (66). Such data suggest that the used
compounds stabilized a wild-type-like conformation of mutant p53,
which enabled cells to respond to GEM treatment (66). However, the mechanisms through
which RITA could help overcome resistance to chemotherapy warrant
further investigation.
In conclusion, in this study, we observed that RITA
induced massive p53-dependent apoptosis of both p53 wild-type and
mutant GB cell lines and synergized with TMZ, without affecting
non-neoplastic brain cells. Therefore, although additional studies
are required to fully clarify the mechanisms of action of RITA, our
data suggest a potential application of this approach in GB
therapy.
Funding
This study was supported by the Sbarro Health
Research Organization (http://www.shro.org), the Commonwealth of
Pennsylvania, and the AIRC-Associazione Italiana per la Ricerca sul
Cancro, under Grant IG 2014-15690.
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding authors on
reasonable request.
Authors’ contributions
IMF performed most of the experiments, contributing
to the experimental design and data analysis; PI defined the
experimental design, analyzed the data and wrote the manuscript; FP
conceived and supervised the study; AG supervised the whole work
and critically contributed to the study development; CAI, DC, DDM,
DB and FC each performed some of the experiments. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors report no conflict of interest.
Abbreviations:
|
CDKN2A
|
cyclin-dependent kinase inhibitor
2A
|
|
CI
|
combination index
|
|
DDR
|
DNA damage response
|
|
γ-H2AX
|
phospho-histone H2AX
|
|
GB
|
glioblastoma
|
|
GEM
|
gemcitabine
|
|
GOF
|
gain-of-function
|
|
IC50
|
half maximal inhibitory
concentration
|
|
JNK
|
c-Jun N-terminal kinase
|
|
MGMT
|
O(6)-methylguanine-DNA
methyltransferase
|
|
NHAs
|
normal human astrocytes
|
|
PFTα
|
pifithrin-α
|
|
PI
|
propidium iodide
|
|
PTEN
|
phosphatase and tensin homolog
|
|
RITA
|
reactivation of p53 and induction of
tumor cell apoptosis
|
|
ROS
|
reactive oxygen species
|
|
shRNA
|
short hairpin RNA
|
|
TMZ
|
temozolomide
|
|
MDM
|
murine double minute
|
Acknowledgments
The authors wish to thank Professor Sergio
Comincini (University of Pavia, Pavia, Italy) and Professor
Annamaria Cimini (University of L’Aquila, L’Aquila, Italy) for
providing the glioblastoma cell lines. AG is also Director of the
Cell Cycle and Cancer Research Line at CROM, Istituto Nazionale
Tumori - IRCCS - Fondazione G. Pascale, Naples. FP is also Adjunct
Associate Professor at Temple University, Department of Biology,
Philadelphia, PA, USA. PI is also a Research Fellow at the
Institute for High Performance Computing and Networking, ICAR-CNR,
Naples. Donatella Cirillo and Domenico Di Marzo were research
fellows at Oncology Research Center of Mercogliano (CROM), Istituto
Nazionale Tumori - IRCCS - Fondazione G. Pascale, I-80131 Napoli,
Italy until 31/01/2016 and 24/11/2015, respectively.
References
|
1
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
Classification of Tumors of the Central Nervous System: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al European Organisation for Research and Treatment of Cancer
Brain Tumor and Radiotherapy Groups; National Cancer Institute of
Canada Clinical Trials Group: Radiotherapy plus concomitant and
adjuvant temozolomide for glioblastoma. N Engl J Med. 352:987–996.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Perry JR, Laperriere N, O’Callaghan CJ,
Brandes AA, Menten J, Phillips C, Fay M, Nishikawa R, Cairncross
JG, Roa W, et al Trial Investigators: Short-course radiation plus
temozolomide in elderly patients with glioblastoma. N Engl J Med.
376:1027–1037. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shajani-Yi Z, de Abreu FB, Peterson JD and
Tsongalis GJ: Frequency of somatic TP53 mutations in combination
with known pathogenic mutations in colon adenocarcinoma, non-small
cell lung carcinoma, and gliomas as identified by mext-generation
sequencing. Neoplasia. 20:256–262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ohgaki H, Dessen P, Jourde B, Horstmann S,
Nishikawa T, Di Patre PL, Burkhard C, Schüler D, Probst-Hensch NM,
Maiorka PC, et al: Genetic pathways to glioblastoma: A
population-based study. Cancer Res. 64:6892–6899. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kleihues P, Schäuble B, zur Hausen A,
Estève J and Ohgaki H: Tumors associated with p53 germline
mutations: A synopsis of 91 families. Am J Pathol. 150:1–13.
1997.PubMed/NCBI
|
|
10
|
Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang
P, McKeever PE, Lee EY and Zhu Y: Expression of mutant p53 proteins
implicates a lineage relationship between neural stem cells and
malignant astrocytic glioma in a murine model. Cancer Cell.
15:514–526. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chow LM, Endersby R, Zhu X, Rankin S, Qu
C, Zhang J, Broniscer A, Ellison DW and Baker SJ: Cooperativity
within and among Pten, p53, and Rb pathways induces high-grade
astro-cytoma in adult brain. Cancer Cell. 19:305–316. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
England B, Huang T and Karsy M: Current
understanding of the role and targeting of tumor suppressor p53 in
glioblastoma multiforme. Tumour Biol. 34:2063–2074. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kang YJ, Balter B, Csizmadia E, Haas B,
Sharma H, Bronson R and Yan CT: Contribution of classical
end-joining to PTEN inactivation in p53-mediated glioblastoma
formation and drug-resistant survival. Nat Commun. 8:140132017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Srivenugopal KS, Shou J, Mullapudi SR,
Lang FF Jr, Rao JS and Ali-Osman F: Enforced expression of
wild-type p53 curtails the transcription of the
O(6)-methylguanine-DNA methyltransferase gene in human tumor cells
and enhances their sensitivity to alkylating agents. Clin Cancer
Res. 7:1398–1409. 2001.PubMed/NCBI
|
|
15
|
Sato A, Sunayama J, Matsuda K, Seino S,
Suzuki K, Watanabe E, Tachibana K, Tomiyama A, Kayama T and
Kitanaka C: MEK-ERK signaling dictates DNA-repair gene MGMT
expression and temozolomide resistance of stem-like glioblastoma
cells via the MDM2-p53 axis. Stem Cells. 29:1942–1951. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Messaoudi K, Clavreul A and Lagarce F:
Toward an effective strategy in glioblastoma treatment Part I:
Resistance mechanisms and strategies to overcome resistance of
glioblastoma to temozolomide. Drug Discov Today. 20:899–905. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang X, Chen JX, Liu JP, You C, Liu YH and
Mao Q: Gain of function of mutant TP53 in glioblastoma: Prognosis
and response to temozolomide. Ann Surg Oncol. 21:1337–1344. 2014.
View Article : Google Scholar
|
|
18
|
Ham SW, Jeon HY, Jin X, Kim EJ, Kim JK,
Shin YJ, Lee Y, Kim SH, Lee SY, Seo S, et al: TP53 gain-of-function
mutation promotes inflammation in glioblastoma. Cell Death Differ.
May 21–2018.Epub ahead of print. View Article : Google Scholar
|
|
19
|
Pentimalli F: Updates from the TP53
universe. Cell Death Differ. 25:10–12. 2018. View Article : Google Scholar
|
|
20
|
Stegh AH: Targeting the p53 signaling
pathway in cancer therapy - the promises, challenges and perils.
Expert Opin Ther Targets. 16:67–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pflaum J, Schlosser S and Müller M: p53
Family and cellular stress responses in cancer. Front Oncol.
4:2852014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Merkel O, Taylor N, Prutsch N, Staber PB,
Moriggl R, Turner SD and Kenner L: When the guardian sleeps:
Reactivation of the p53 pathway in cancer. Mutat Res. 773:1–13.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim SS, Rait A, Kim E, Pirollo KF, Nishida
M, Farkas N, Dagata JA and Chang EH: A nanoparticle carrying the
p53 gene targets tumors including cancer stem cells, sensitizes
glio-blastoma to chemotherapy and improves survival. ACS Nano.
8:5494–5514. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim SS, Rait A, Kim E, Pirollo KF and
Chang EH: A tumor-targeting p53 nanodelivery system limits
chemore-sistance to temozolomide prolonging survival in a mouse
model of glioblastoma multiforme. Nanomedicine (Lond). 11:301–311.
2015. View Article : Google Scholar
|
|
25
|
Wischhusen J, Naumann U, Ohgaki H,
Rastinejad F and Weller M: CP-31398, a novel p53-stabilizing agent,
induces p53-dependent and p53-independent glioma cell death.
Oncogene. 22:8233–8245. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weinmann L, Wischhusen J, Demma MJ,
Naumann U, Roth P, Dasmahapatra B and Weller M: A novel p53 rescue
compound induces p53-dependent growth arrest and sensitises glioma
cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ.
15:718–729. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Villalonga-Planells R, Coll-Mulet L,
Martínez-Soler F, Castaño E, Acebes JJ, Giménez-Bonafé P, Gil J and
Tortosa A: Activation of p53 by nutlin-3a induces apoptosis and
cellular senescence in human glioblastoma multiforme. PLoS One.
6:e185882011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Costa B, Bendinelli S, Gabelloni P, Da
Pozzo E, Daniele S, Scatena F, Vanacore R, Campiglia P, Bertamino
A, Gomez-Monterrey I, et al: Human glioblastoma multiforme: p53
reactivation by a novel MDM2 inhibitor. PLoS One. 8:e722812013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Daniele S, Taliani S, Da Pozzo E,
Giacomelli C, Costa B, Trincavelli ML, Rossi L, La Pietra V,
Barresi E, Carotenuto A, et al: Apoptosis therapy in cancer: The
first single-molecule co-activating p53 and the translocator
protein in glioblastoma. Sci Rep. 4:47492014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen X, Tai L, Gao J, Qian J, Zhang M, Li
B, Xie C, Lu L and Lu W and Lu W: A stapled peptide antagonist of
MDM2 carried by polymeric micelles sensitizes glioblastoma to
temozolomide treatment through p53 activation. J Control Release.
218:29–35. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Daniele S, Barresi E, Zappelli E,
Marinelli L, Novellino E, Da Settimo F, Taliani S, Trincavelli ML
and Martini C: Long lasting MDM2/Translocator protein modulator: A
new strategy for irreversible apoptosis of human glioblastoma
cells. Oncotarget. 7:7866–7884. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Daniele S, Costa B, Zappelli E, Da Pozzo
E, Sestito S, Nesi G, Campiglia P, Marinelli L, Novellino E,
Rapposelli S, et al: Combined inhibition of AKT/mTOR and MDM2
enhances Glioblastoma Multiforme cell apoptosis and differentiation
of cancer stem cells. Sci Rep. 5:99562015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Daniele S, La Pietra V, Barresi E, Di Maro
S, Da Pozzo E, Robello M, La Motta C, Cosconati S, Taliani S,
Marinelli L, et al: Lead optimization of
2-phenylindolylglyoxylyldipeptide murine double minute
(MDM)2/translocator protein (TSPO) dual inhibitors for the
treatment of gliomas. J Med Chem. 59:4526–4538. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Patyka M, Sharifi Z, Petrecca K, Mansure
J, Jean-Claude B and Sabri S: Sensitivity to PRIMA-1MET is
associated with decreased MGMT in human glioblastoma cells and
glioblastoma stem cells irrespective of p53 status. Oncotarget.
7:60245–60269. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Verreault M, Schmitt C, Goldwirt L, Pelton
K, Haidar S, Levasseur C, Guehennec J, Knoff D, Labussière M, Marie
Y, et al: Preclinical efficacy of the MDM2 inhibitor RG7112 in
MDM2-amplified and TP53 wild-type glio-blastomas. Clin Cancer Res.
22:1185–1196. 2016. View Article : Google Scholar
|
|
36
|
Renner G, Janouskova H, Noulet F, Koenig
V, Guerin E, Bär S, Nuesch J, Rechenmacher F, Neubauer S, Kessler
H, et al: Integrin α5β1 and p53 convergent pathways in the control
of anti-apoptotic proteins PEA-15 and survivin in high-grade
glioma. Cell Death Differ. 23:640–653. 2016. View Article : Google Scholar
|
|
37
|
Wang H, Cai S, Bailey BJ, Reza Saadatzadeh
M, Ding J, Tonsing-Carter E, Georgiadis TM, Zachary Gunter T, Long
EC, Minto RE, et al: Combination therapy in a xenograft model of
glio-blastoma: Enhancement of the antitumor activity of
temozolomide by an MDM2 antagonist. J Neurosurg. 126:446–459. 2017.
View Article : Google Scholar
|
|
38
|
Mai WX, Gosa L, Daniels VW, Ta L, Tsang
JE, Higgins B, Gilmore WB, Bayley NA, Harati MD, Lee JT, et al:
Cytoplasmic p53 couples oncogene-driven glucose metabolism to
apoptosis and is a therapeutic target in glioblastoma. Nat Med.
23:1342–1351. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Merlino F, Daniele S, La Pietra V, Di Maro
S, Di Leva FS, Brancaccio D, Tomassi S, Giuntini S, Cerofolini L,
Fragai M, et al: Simultaneous targeting of RGD-integrins and dual
murine double minute proteins in glioblastoma multiforme. J Med
Chem. 61:4791–4809. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Issaeva N, Bozko P, Enge M, Protopopova M,
Verhoef LG, Masucci M, Pramanik A and Selivanova G: Small molecule
RITA binds to p53, blocks p53-HDM-2 interaction and activates p53
function in tumors. Nat Med. 10:1321–1328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dickinson ER, Jurneczko E, Nicholson J,
Hupp TR, Zawacka-Pankau J, Selivanova G and Barran PE: The use of
ion mobility mass spectrometry to probe modulation of the structure
of p53 and of MDM2 by small molecule inhibitors. Front Mol Biosci.
2:392015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Krajewski M, Ozdowy P, D’Silva L,
Rothweiler U and Holak TA: NMR indicates that the small molecule
RITA does not block p53-MDM2 binding in vitro. Nat Med.
11:1135–1136; author reply 1136–1137. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Du Z, Yu J, Li F, Deng L, Wu F, Huang X,
Bergstrand J, Widengren J, Dong C and Ren J: In situ monitoring of
p53 protein and MDM2 protein interaction in single living cells
using single-molecule fluorescence spectroscopy. Anal Chem.
90:6144–6151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Di Conza G, Buttarelli M, Monti O,
Pellegrino M, Mancini F, Pontecorvi A, Scotlandi K and Moretti F:
IGF-1R/MDM2 relationship confers enhanced sensitivity to RITA in
Ewing sarcoma cells. Mol Cancer Ther. 11:1247–1256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Henze J, Mühlenberg T, Simon S, Grabellus
F, Rubin B, Taeger G, Schuler M, Treckmann J, Debiec-Rychter M,
Taguchi T, et al: p53 modulation as a therapeutic strategy in
gastrointestinal stromal tumors. PLoS One. 7:e377762012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chuang HC, Yang LP, Fitzgerald AL, Osman
A, Woo SH, Myers JN and Skinner HD: The p53-reactivating small
molecule RITA induces senescence in head and neck cancer cells.
PLoS One. 9:e1048212014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Surget S, Descamps G, Brosseau C, Normant
V, Maïga S, Gomez-Bougie P, Gouy-Colin N, Godon C, Béné MC, Moreau
P, et al: RITA (Reactivating p53 and Inducing Tumor Apoptosis) is
efficient against TP53abnormal myeloma cells
independently of the p53 pathway. BMC Cancer. 14:4372014.
View Article : Google Scholar
|
|
48
|
Weilbacher A, Gutekunst M, Oren M,
Aulitzky WE and van der Kuip H: RITA can induce cell death in
p53-defective cells independently of p53 function via activation of
JNK/SAPK and p38. Cell Death Dis. 5:e13182014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wanzel M, Vischedyk JB, Gittler MP, Gremke
N, Seiz JR, Hefter M, Noack M, Savai R, Mernberger M, Charles JP,
et al: CRISPR-Cas9-based target validation for p53-reactivating
model compounds. Nat Chem Biol. 12:22–28. 2016. View Article : Google Scholar :
|
|
50
|
Hedström E, Eriksson S, Zawacka-Pankau J,
Arnér ES and Selivanova G: p53-dependent inhibition of TrxR1
contributes to the tumor-specific induction of apoptosis by RITA.
Cell Cycle. 8:3584–3591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shi Y, Nikulenkov F, Zawacka-Pankau J, Li
H, Gabdoulline R, Xu J, Eriksson S, Hedström E, Issaeva N, Kel A,
et al: ROS-dependent activation of JNK converts p53 into an
efficient inhibitor of oncogenes leading to robust apoptosis. Cell
Death Differ. 21:612–623. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Saha MN, Jiang H, Yang Y, Zhu X, Wang X,
Schimmer AD, Qiu L and Chang H: Targeting p53 via JNK pathway: A
novel role of RITA for apoptotic signaling in multiple myeloma.
PLoS One. 7:e302152012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nieves-Neira W, Rivera MI, Kohlhagen G,
Hursey ML, Pourquier P, Sausville EA and Pommier Y: DNA protein
cross-links produced by NSC 652287, a novel thiophene derivative
active against human renal cancer cells. Mol Pharmacol. 56:478–484.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang J, Ahmed A, Poon E, Perusinghe N, de
Haven Brandon A, Box G, Valenti M, Eccles S, Rouschop K, Wouters B,
et al: Small-molecule activation of p53 blocks hypoxia-inducible
factor 1alpha and vascular endothelial growth factor expression in
vivo and leads to tumor cell apoptosis in normoxia and hypoxia. Mol
Cell Biol. 29:2243–2253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ahmed A, Yang J, Maya-Mendoza A, Jackson
DA and Ashcroft M: Pharmacological activation of a novel
p53-dependent S-phase checkpoint involving CHK-1. Cell Death Dis.
2:e1602011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
de Lange J, Verlaan-de Vries M, Teunisse
AF and Jochemsen AG: Chk2 mediates RITA-induced apoptosis. Cell
Death Differ. 19:980–989. 2012. View Article : Google Scholar :
|
|
57
|
Wiegering A, Matthes N, Mühling B, Koospal
M, Quenzer A, Peter S, Germer CT, Linnebacher M and Otto C:
Reactivating p53 and Inducing Tumor Apoptosis (RITA) Enhances the
Response of RITA-Sensitive Colorectal Cancer Cells to
Chemotherapeutic Agents 5-Fluorouracil and Oxaliplatin. Neoplasia.
19:301–309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Grinkevich VV, Nikulenkov F, Shi Y, Enge
M, Bao W, Maljukova A, Gluch A, Kel A, Sangfelt O and Selivanova G:
Ablation of key oncogenic pathways by RITA-reactivated p53 is
required for efficient apoptosis. Cancer Cell. 15:441–453. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Enge M, Bao W, Hedström E, Jackson SP,
Moumen A and Selivanova G: MDM2-dependent downregulation of p21 and
hnRNP K provides a switch between apoptosis and growth arrest
induced by pharmacologically activated p53. Cancer Cell.
15:171–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Di Marzo D, Forte IM, Indovina P, Di
Gennaro E, Rizzo V, Giorgi F, Mattioli E, Iannuzzi CA, Budillon A,
Giordano A, et al: Pharmacological targeting of p53 through RITA is
an effective antitumoral strategy for malignant pleural
mesothelioma. Cell Cycle. 13:652–665. 2014. View Article : Google Scholar
|
|
61
|
Saha MN, Jiang H, Mukai A and Chang H:
RITA inhibits multiple myeloma cell growth through induction of
p53-mediated caspase-dependent apoptosis and synergistically
enhances nutlin-induced cytotoxic responses. Mol Cancer Ther.
9:3041–3051. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Shin D, Kim EH, Lee J and Roh JL: RITA
plus 3-MA overcomes chemoresistance of head and neck cancer cells
via dual inhibition of autophagy and antioxidant systems. Redox
Biol. 13:219–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhao CY, Grinkevich VV, Nikulenkov F, Bao
W and Selivanova G: Rescue of the apoptotic-inducing function of
mutant p53 by small molecule RITA. Cell Cycle. 9:1847–1855. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jones RJ, Bjorklund CC, Baladandayuthapani
V, Kuhn DJ and Orlowski RZ: Drug resistance to inhibitors of the
human double minute-2 E3 ligase is mediated by point mutations of
p53, but can be overcome with the p53 targeting agent RITA. Mol
Cancer Ther. 11:2243–2253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Burmakin M, Shi Y, Hedström E, Kogner P
and Selivanova G: Dual targeting of wild-type and mutant p53 by
small molecule RITA results in the inhibition of N-Myc and key
survival oncogenes and kills neuroblastoma cells in vivo and in
vitro. Clin Cancer Res. 19:5092–5103. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Fiorini C, Cordani M, Padroni C, Blandino
G, Di Agostino S and Donadelli M: Mutant p53 stimulates
chemoresistance of pancreatic adenocarcinoma cells to gemcitabine.
Biochim Biophys Acta. 1853:89–100. 2015. View Article : Google Scholar
|
|
67
|
Menendez D, Lowe JM, Snipe J and Resnick
MA: Ligand dependent restoration of human TLR3 signaling and death
in p53 mutant cells. Oncotarget. 7:61630–61642. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
He XY, Feng XL, Song XP, Zeng HC, Cao ZX,
Xiao WW, Zhang B and Wu QH: RITA combined with temozolomide
inhibits the proliferation of human glioblastoma U87 cells. Nan
Fang Yi Ke Da Xue Xue Bao. 36:1423–1428. 2016.In Chinese.
PubMed/NCBI
|
|
69
|
Bacciocchi G, Gibelli N, Zibera C,
Pedrazzoli P, Bergamaschi G, De Piceis Polver P, Danova M, Mazzini
G, Palomba L, Tupler R, et al: Establishment and characterization
of two cell lines derived from human glioblastoma multiforme.
Anticancer Res. 12:853–861. 1992.PubMed/NCBI
|
|
70
|
Wang X, Chen JX, Liu YH, You C and Mao Q:
Mutant TP53 enhances the resistance of glioblastoma cells to
temozolomide by up-regulating O(6)-methylguanine
DNA-methyltransferase. Neurol Sci. 34:1421–1428. 2013. View Article : Google Scholar
|
|
71
|
Komarov PG, Komarova EA, Kondratov RV,
Christov-Tselkov K, Coon JS, Chernov MV and Gudkov AV: A chemical
inhibitor of p53 that protects mice from the side effects of cancer
therapy. Science. 285:1733–1737. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mirza A, McGuirk M, Hockenberry TN, Wu Q,
Ashar H, Black S, Wen SF, Wang L, Kirschmeier P, Bishop WR, et al:
Human survivin is negatively regulated by wild-type p53 and
participates in p53-dependent apoptotic pathway. Oncogene.
21:2613–2622. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hoffman WH, Biade S, Zilfou JT, Chen J and
Murphy M: Transcriptional repression of the anti-apoptotic survivin
gene by wild type p53. J Biol Chem. 277:3247–3257. 2002. View Article : Google Scholar
|
|
74
|
Uchida H, Tanaka T, Sasaki K, Kato K,
Dehari H, Ito Y, Kobune M, Miyagishi M, Taira K, Tahara H, et al:
Adenovirus-mediated transfer of siRNA against survivin induced
apoptosis and attenuated tumor cell growth in vitro and in vivo.
Mol Ther. 10:162–171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Saito T, Sugiyama K, Takeshima Y, Amatya
VJ, Yamasaki F, Takayasu T, Nosaka R, Muragaki Y, Kawamata T and
Kurisu K: Prognostic implications of the subcellular localization
of survivin in glioblastomas treated with radiotherapy plus
concomitant and adjuvant temozolomide. J Neurosurg. 128:679–684.
2018. View Article : Google Scholar
|
|
76
|
Faccion RS, Bernardo PS, de Lopes GPF,
Bastos LS, Teixeira CL, de Oliveira JA, Fernandes PV, Dubois LG,
Chimelli L and Maia RC: p53 expression and subcellular survivin
localization improve the diagnosis and prognosis of patients with
diffuse astrocytic tumors. Cell Oncol (Dordr). 41:141–157. 2018.
View Article : Google Scholar
|
|
77
|
Chakravarti A, Zhai GG, Zhang M, Malhotra
R, Latham DE, Delaney MA, Robe P, Nestler U, Song Q and Loeffler J:
Survivin enhances radiation resistance in primary human
glioblastoma cells via caspase-independent mechanisms. Oncogene.
23:7494–7506. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Guvenc H, Pavlyukov MS, Joshi K, Kurt H,
Banasavadi-Siddegowda YK, Mao P, Hong C, Yamada R, Kwon CH, Bhasin
D, et al: Impairment of glioma stem cell survival and growth by a
novel inhibitor for Survivin-Ran protein complex. Clin Cancer Res.
19:631–642. 2013. View Article : Google Scholar
|
|
79
|
Dahan P, Martinez Gala J, Delmas C,
Monferran S, Malric L, Zentkowski D, Lubrano V, Toulas C,
Cohen-Jonathan Moyal E and Lemarie A: Ionizing radiations sustain
glioblastoma cell dedifferentiation to a stem-like phenotype
through survivin: Possible involvement in radioresistance. Cell
Death Dis. 5:e15432014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fenstermaker RA, Figel SA, Qiu J, Barone
TA, Dharma SS, Winograd EK, Galbo PM, Wiltsie LM and Ciesielski MJ:
Survivin monoclonal antibodies detect survivin cell surface
expression and inhibit tumor growth in vivo. Clin Cancer Res.
24:2642–2652. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lee SY: Temozolomide resistance in
glioblastoma multiforme. Genes Dis. 3:198–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hermisson M, Klumpp A, Wick W, Wischhusen
J, Nagel G, Roos W, Kaina B and Weller M: O6-methylguanine DNA
meth-yltransferase and p53 status predict temozolomide sensitivity
in human malignant glioma cells. J Neurochem. 96:766–776. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gottlieb A, Althoff K, Grunewald L, Thor
T, Odersky A, Schulte M, Deubzer HE, Heukamp L, Eggert A, Schramm
A, et al: RITA displays anti-tumor activity in medulloblastomas
independent of TP53 status. Oncotarget. 8:27882–27891. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ziegler DS, Kung AL and Kieran MW:
Anti-apoptosis mechanisms in malignant gliomas. J Clin Oncol.
26:493–500. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sohn D, Graupner V, Neise D, Essmann F,
Schulze-Osthoff K and Jänicke RU: Pifithrin-alpha protects against
DNA damage-induced apoptosis downstream of mitochondria independent
of p53. Cell Death Differ. 16:869–878. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kitange GJ, Carlson BL, Schroeder MA,
Grogan PT, Lamont JD, Decker PA, Wu W, James CD and Sarkaria JN:
Induction of MGMT expression is associated with temozolomide
resistance in glioblastoma xenografts. Neuro-oncol. 11:281–291.
2009. View Article : Google Scholar :
|