Introduction
Lung cancer exhibits the highest mortality rate
among all types of cancer worldwide. Even though chemotherapy is
one of the standard therapies for lung cancer, <20% of patients
treated with chemotherapy live >5 years (1). This dismal number indicates that
advances in lung cancer treatment remain inadequate compared to
other types of cancer. The poor prognosis of patients with lung
cancer is mainly derived from the low response rate and resistance
to current chemotherapeutics (2).
As non-small cell lung cancer (NSCLC) is the most common type of
lung cancer (3), the development
of more effective and advanced therapeutic strategies for the
treatment of NSCLC is fundamental in order to improve the poor
prognosis of patients with lung cancer.
Epidermal growth factor receptor (EGFR) is a
transmembrane receptor tyrosine kinase protein which belongs to the
ErbB family. Upon the binding of a ligand, such as epidermal growth
factor (EGF), the intracellular domain of EGFR is phosphorylated
and activates downstream signal transduction pathways, including
RAS/RAF/MEK/mitogen-activated protein kinase (MAPK),
phosphoinositide 3-kinase (PI3K)/Akt, and signal transducer and
activator of transcription (STAT) signaling pathways. These signal
transductions finally result in cell proliferation and in the
inhibition of apoptosis (4). The
overexpression of EGFR has been implicated in the pathogenesis of
NSCLC (5,6). Studies have reported that EGFR
overexpression in NSCLC is associated with a reduced overall
survival, chemoresistance and frequent lymph node metastasis
(7-11). In addition, a quarter of NSCLCs
cases possess activating mutations in the tyrosine kinase domain of
EGFR (12). These mutations
sensitize NSCLCs to EGFR receptor tyrosine kinase inhibitors
(TKIs), such as gefitinib and erlotinib (13-15).
However, patients ultimately develop acquired resistance against
these drugs. The most common mechanism of resistance is a secondary
T790M mutation in EGFR exon 20 (16,17).
Thus, the identification of novel drugs to effectively suppress the
activity of EGFR, not only in naïve NSCLCs, but also in EGFR
TKI-resistant NSCLCs is imperative.
STAT3 is a transcription factor recognized as a key
oncogenic factor driving tumor development and progression. STAT3
is activated by phosphorylation at tyrosine 705 or serine 727 via
interleukin (IL)-6 receptor (IL-6R), growth factor receptors, and
non-receptor tyrosine kinase, such as Src (18). The activation of STAT3 mediates a
variety of cellular functions, including cell proliferation,
differentiation, angiogenesis, metastasis, and drug resistance
(19). Studies have reported that
STAT3 was activated in NSCLC and a high phosphorylation level of
STAT3 was a strong predictor of poor prognosis in NSCLC (19-21).
Specifically, STAT3 signaling has been related to the development
of resistance to EGFR TKIs (22-27).
Therefore, aberrant STAT3 phosphorylation appears to be a potential
therapeutic target for NSCLC.
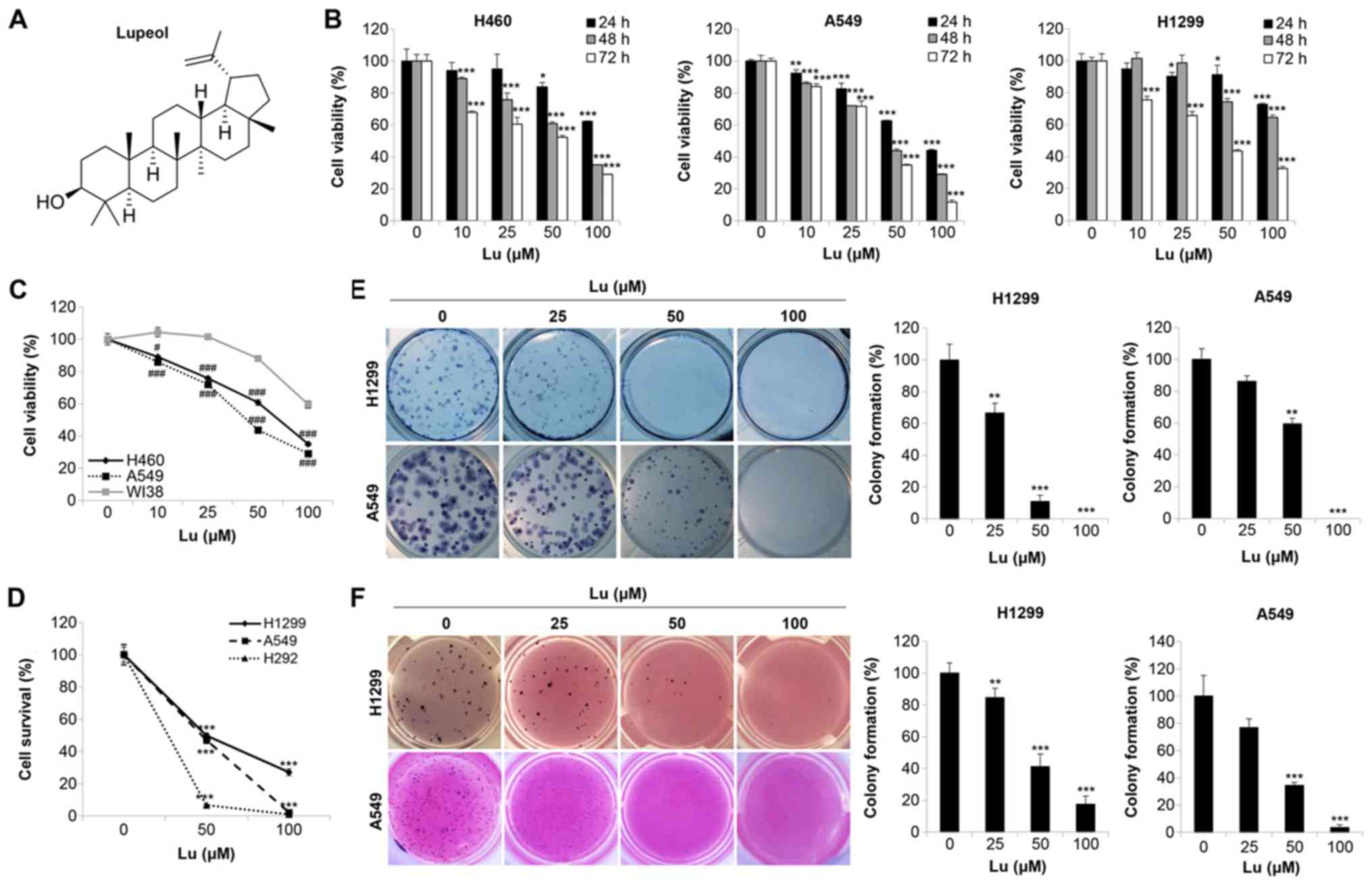
Lupeol (chemical structure shown in Fig. 1A) is a dietary triterpenoid present
in various types of fruits, vegetables and medicinal plants. Lupeol
has been reported to exhibit strong antioxidant, anti-inflammatory,
anti-microbial, anti-arthritic, anti-diabetic and anti-malarial
activities (28). Moreover, lupeol
has been shown to exert anticancer effects in various cancer cells.
The suppression of tumorigenesis, the induction of apoptosis, cell
cycle regulation, chemosensitization and the enhancement of the
cytotoxic function of natural killer cells have been reported as
the mechanisms of the anticancer effects of lupeol (7,29-37).
Notably, lupeol has been shown to suppress EGFR activity in oral
squamous cell carcinoma and gallbladder carcinoma (36,37).
It has also been shown to inhibit the STAT3 signaling cascade in
hepatocellular carcinoma cells (7). In NSCLC, lupeol has been reported to
downregulate COX2 and mTOR/PI3K/AKT pathways to induce apoptosis
(33,34). However, the regulatory effects of
lupeol on the EGFR/STAT3 signaling pathway in human NSCLC cells
have not yet been elucidated, at least to the best of our
knowledge. Thus, in the current study, we investigated the
mechanisms responsible for the anticancer activity of lupeol in
human NSCLC cells, focusing on the regulation of EGFR/STAT3
activity. We also aimed to verify whether lupeol exerts anticancer
effects on NSCLC cells that are resistant to EGFR TKIs.
Materials and methods
Cell lines and cell culture
The H1299, A549, H460, H292 human NSCLC cell lines
and WI38 human lung fibroblast were purchased from the American
Type Culture Collection (ATCC). The H1975 human NSCLC cell line was
kindly supplied by professor Ho-Young Lee (College of Pharmacy,
Seoul National University). The cells were grown in RPMI-1640
(WelGENE) supplemented with 10% fetal bovine serum (FBS, WelGENE)
and 1% antibiotics (WelGENE) at 37°C in a humidified incubator
under 5% CO2.
Reagents and antibodies
Lupeol was purchased from ChemFaces and was
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich). Trypan blue
was purchased from WelGENE, and MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] from
Duchefa. Hematoxylin, propidium iodide (PI), paraformaldehyde and
4,6-diamidino-2-phenylindole (DAPI) were obtained from
Sigma-Aldrich. Primary antibodies against phospho-EGFR (Y1068,
#2234S), EGFR (#4267S), phospho-STAT3 (Y705, #9145S), STAT3
(#9139S), phospho -A KT (S473, # 4 0 6 0S), phospho -E R K
(T202/Y204, #9106S), cleaved caspase-3 (#9661S) and cleaved
poly(ADP-ribose) polymerase (PARP, #5625S) were purchased from Cell
Signaling Technology. The other primary antibodies including AKT
(#sc-5298), ERK (#sc-514302), β-actin (#sc-47778), α-tubulin
(#sc-5286), Lamin B (#sc-374015), survivin (#sc-17779) and cyclin
D1 (#sc-450) were obtained from Santa Cruz Biotechnology. Goat
anti-mouse secondary antibody was purchased from Bethyl
Laboratories and goat anti-rabbit secondary antibody was purchased
from Enzo Life Sciences.
Cell viability assay
For the MTT assay, 3×103 cells were
seeded onto 96-well plates and treated with lupeol (10-100
µM) for various time periods (24-72 h) or treated with
erlotinib (LC Labs; 10-100 µM) for 72 h. MTT solution was
added to the media at a concentration of 0.4 mg/ml followed by
incubation for 4 h at 37°C. The media were then aspirated and 100
µl of DMSO were added to each well to dissolve the formazan.
The absorbance values at 540 nm were measured using a microplate
reader (SpectraMax M3; Molecular Devices). For the trypan blue
exclusion assay, 2×104 cells were seeded in 12-well
plates and treated with lupeol at 50 or 100 µM for 72 h. The
cells were then collected and stained with 0.4% trypan blue
solution at a final concentration of 0.1%. The number of viable
cells was evaluated by counting the unstained cells using a
hemocytometer under a microscope (Leica).
Anchorage-dependent and -independent
colony formation assay
For the anchorage-dependent 2D colony formation
assay, 3×102 cells were seeded in 12-well plates and
treated with lupeol for 2 weeks. The medium was changed every 3
days. The colonies were fixed with 100% methanol for 5 min and
stained with hematoxylin for 30 min at room temperature. Images of
the stained colonies were acquired using a digital camera (Canon)
and the number of colonies was counted using ImageJ software. For
the anchorage-independent colony formation assay (soft agar assay),
4% SeaPlaque agarose (Lonza) dissolved in PBS was melted and mixed
with warm media to yield 1% bottom agar. Bottom agar (1 ml) was
then added to 24-well plates and allowed to solidify at room
temperature. The cells (1×103) were suspended in 0.5 ml
of top agar (0.4%) and plated onto the bottom agar. The plate was
kept at room temperature until the top agar solidified. The cells
were then treated with lupeol at 25, 50 and 100 µM for 2
weeks and the medium was changed every 3 days. The colonies were
stained with MTT solution (final concentration, 0.5 mg/ml) for 2 h
at 37°C. Images of the stained colonies were acquired using a
digital camera (Canon) and the number of colonies was counted using
ImageJ software.
DAPI staining
The cells (1×105) were seeded in 6-well
plates and treated with lupeol at 100 µM for 72 h. The cells
were then harvested, fixed with 3.7% paraformaldehyde, and attached
to slide glasses using a cytospin (Shandon). After staining with
DAPI solution (2.5 µg/ml) for 20 min at room temperature in
the dark, the attached cells were washed with PBS and distilled
water and mounted with aqueous mounting medium (Crystal Mount). The
morphology of the nuclei was observed under a fluorescence
microscope (Carl Zeiss) at ×200 magnification.
Flow cytometric analysis
The cells (1×105) were seeded in 6-well
plates and treated with lupeol at 50 or 100 µM for 72 h. For
cell cycle analysis, the cells were collected, washed with cold
PBS, and fixed with cold 80% ethanol for 1 h at 4°C. Subsequently,
the cells were stained with 50 µg/ml of PI in the presence
of 30 µg/ml DNase-free RNase A (Sigma-Aldrich) for 30 min at
room temperature. The stained cell pellet was then resuspended in
500 µl of PBS. The relative DNA content in each phase of the
cell cycle was determined using a flow cytometer (FACSCalibeur, BD
Biosciences) and CellQuest Pro software (version 5.1). For the
Annexin V-PI double staining assay, the cells were harvested and
double-stained with annexin V-FITC and PI using the Annexin V-FITC
Apoptosis Detection kit I (BD Biosciences; PharMingen) according to
the manufacturer's instructions. Annexin V-positive cells were
determined using a flow cytometer and CellQuest software.
STAT3-luciferase reporter gene assay
The cells (3×104) were seeded in 24-well
plates and co-transfected with 100 ng of p-STAT3-TA-luc (Clontech)
and 5 ng of pRL-TK using Lipofectamine 2000 according to the
manufacturer's instructions (Invitrogen; Thermo Fisher Scientific).
At 24 h post-transfection, the cells were treated with lupeol for
an additional 24 to 48 h. The cells were then lysed and the STAT3
reporter gene activity was measured with the Dual-Luciferase
Reporter Assay System (Promega) as described in the manufacturer's
protocol.
EGFR stimulation by EGF treatment
The cells (5×105) were seeded in 6-well
plates and treated with lupeol at 50 or 100 µM for 24 h. EGF
(Lifeline Cell Technology) was then added at 20 ng/ml to the
culture media 1 h prior o harvesting to activate EGFR.
Nuclear/cytosol extraction
To extract cytosolic fractions, 1×107
cells were lysed with buffer A [10 mM HEPES (pH 7.9), 1.5 mM
MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM EDTA, 0.5% NP-40,
protease inhibitor cocktail (Thermo Fisher Scientific), and
phosphatase inhibitors (1 mM Na3VO4 and 100
mM NaF)] for 20 min on ice. The supernatant containing cytosolic
proteins was collected by centrifugation (900 × g, 10 min, 4°C) and
cleared again by high-speed centrifugation (16,000 × g, 10 min,
4°C). To extract the nuclear fraction, the pellet was washed with
buffer A for three times and lysed with buffer C [20 mM HEPES (pH
7.9), 1.5 mM MgCl2, 420 mM NaCl, 0.2 mM EDTA, 0.5 mM
DTT, 25% glycerol, protease inhibitor cocktail (Thermo Fisher
Scientific), and phosphatase inhibitors (1 mM
Na3VO4 and 100 mM NaF)] for 1 h on ice with
vigorous vortexing for 15 sec every 10 min. The supernatant
containing nuclear proteins was obtained by centrifugation (16,000
× g, 10 min, 4°C). To detect any cross-contamination between the
nuclear and cytosolic fractions, Lamin B (1:1,000 dilution) and
α-tubulin (1:1,000 dilution) were used as markers for the nuclear
and cytosolic fractions, respectively.
Constitutive activation of STAT3
The cells (5×105) were seeded in a 6-well
plate and transfected with 1 µg of pExpress1-stat3Y705D for
the constitutive activation of STAT3, or with 1 µg of
pExpress-1 as a control, using Lipofectamine 2000. pExpress-1 and
pExpress1-stat3Y705D were kindly provided by Professor Ho-Young Lee
(Seoul National University). At 48 h post-transfection, the cells
were trypsinized and seeded again in 6-well plates. The cells were
treated with lupeol at 50 µM for a further 72 h, and
subsequently examined by western blot analysis.
RT-PCR and semi-quantitative PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific) according to the
manufacturer's instructions. First-strand cDNA was synthesized with
the PrimeScript RT reagent kit (Takara) using 1 µg of total
RNA as described in the manufacturer's protocol. The primer
sequences used are as follows: Survivin forward, 5′-TCA AGG ACC ACC
GCA TCT CTA-3′ and reverse, 5′-TGA AGC AGA AGA AAC ACT GGG-3′;
cyclin D1 forward, 5′-CCT GTC CTA CTA CCG CCT CA-3′ and reverse,
5′-TCC TCC TCT TCC TCC TCC TC-3′; and actin forward, 5′-ACT ACC TCA
TGA AGA TC-3′ and reverse, 5′-GAT CCA CAT CTG CTG GAA-3′. cDNA was
amplified using a SimpliAmp Thermal Cycler (Applied Biosystems).
Cycle numbers corresponding to the exponential phase of the
reaction were determined to be 28 cycles at an annealing
temperature of 55°C for survivin and cyclin D1 and 20 cycles at an
annealing temperature of 55°C for actin. The PCR products were
resolved on a 1.5% agarose gel (Lonza), stained with nucleic acid
gel staining solution (RBC), and visualized by the Gel Imaging
System (Daihan Scientific).
Western blot analysis
The cells were lysed with cold
radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher
Scientific) supplemented with a protease inhibitor cocktail (Thermo
Fisher Scientific) and phosphatase inhibitors (1 mM
Na3VO4 and 100 mM NaF) and incubated for 1 h
on ice. The supernatants were collected by centrifugation at 16,000
× g at 4°C for 30 min. Protein concentrations were determined using
a bicinoconinic acid (BCA) protein assay kit (Pierce Biotechnology)
according to the manufacturer's instructions. The same amounts (20
µg) of protein were resolved by sodium dodecyl sulfate
(SDS)-polyacrylamide gels (8-12%) and transferred onto a polyvinyl
difluoride (PVDF) membrane. The membrane was then blocked with 3%
bovine serum albumin (BSA, GenDEPOT) in TBST [Tris-buffered saline
(TBS) containing 0.1% Tween-20] for 1 h at room temperature and
incubated overnight with primary antibodies (1:500 dilution for
p-STAT3 antibody and p-EGFR antibody; 1:1,000 dilution for the
other antibodies) at 4°C. Following several washes with TBST for 1
h, the membrane was incubated with secondary antibody solution
(1:10,000 dilution in blocking solution) for 1 h at room
temperature. Protein expression was detected by SuperSignal West
Pico Chemiluminescent Substrate (Thermo Fisher Scientific)
according to the manufacturer's instructions. The densitometric
analysis of the western blots was performed using ImageJ software
(ImageJ 1.38; National Institutes of Health).
Molecular docking
The SwissDock web server (http://www.swissdock.ch) was used for molecular
docking and prediction of the lowest free binding energy (38). The protein databank code (PDB) code
for EGFR (1M17) was obtained from the Protein Data Bank (39). UCSF Chimera 1.13 software was used
to explore the predicted binding modes. Among the clusters, the
conformation with the lowest binding free energy was selected.
Statistical analyses
Each result is expressed as the mean ± SD of data
obtained from triplicate experiments. Statistical analyses were
performed by Student's t-test or one-way ANOVA followed by a
Tukey's post hoc test to determine the significant differences
between groups. Differences with values of P<0.05 were
considered statistically significant. Statistical analysis was
performed using GraphPad Prism 5 software (GraphPad Prism Software
Inc.).
Results
Lupeol inhibits the growth and colony
formation of human NSCLC cells
To examine the effects of lupeol on the growth of
human NSCLC cell lines, the H1299, A549 and H460 cells were treated
with various concentrations of lupeol for different periods of
time. The results from MTT assay revealed that lupeol markedly
reduced cell viability in a concentration- and time-dependent
manner (Fig. 1B). We set 100
µg/ml as the maximum concentration of lupeol and 72 h as the
optimal incubation time for further experiments. In order to
examine the effects of lupeol on the viability of normal cells, we
performed an MTT assay using the WI38 human lung fibroblasts. As
shown in Fig. 1C, the viability of
the WI38 fibroblasts was higher than that of the NSCLC cells,
including the H460 and A549 cells, following 48 h of treatment with
lupeol, indicating that lupeol exhibited a higher sensitivity to
cancer cells than normal cells (Fig.
1C). To confirm the growth inhibitory effects of lupeol on
NSCLC cells, trypan blue exclusion assays were performed on the
H1299, A549 and H292 cells. The results revealed that the number of
surviving cells was reduced by lupeol in a dose-dependent manner
(Fig. 1D). These results clearly
demonstrated that lupeol inhibited the growth of human NSCLC
cells.
Subsequently, we examined the effects of lupeol on
colony formation, a critical step in tumorigenesis, in human NSCLC
cells. The results from the 2D colony formation assay indicated
that lupeol significantly reduced the number of colonies in a
dose-dependent manner in H1299 and A549 cells (Fig. 1E). To mimic the 3D tumorigenesis
environment, a soft agar assay was further conducted. As shown in
Fig. 1F, lupeol induced a
dose-dependent decrease in colony formation in these cell lines
(Fig. 1F). These results
collectively indicated that lupeol suppressed anchorage-dependent
and -independent colony formation in human NSCLC cells.
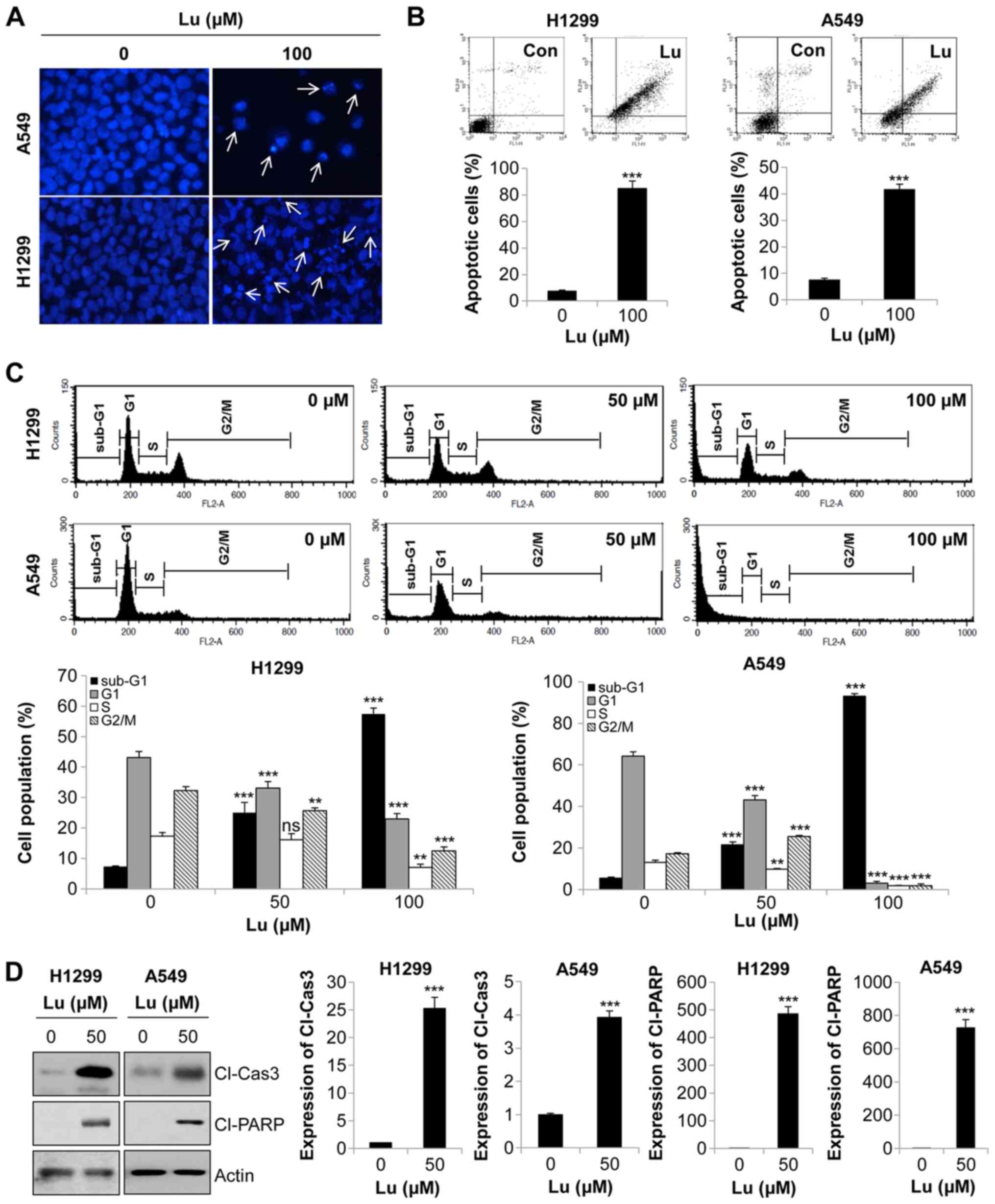
Lupeol induces the apoptosis of human
NSCLC cells
To gain insight into the mechanisms underlying the
anti-proliferative effects of lupeol on NSCLC cells, we performed
DAPI staining. The NSCLC cells treated with lupeol exhibited highly
condensed and fragmented nuclei, indicative of apoptotic cells
(Fig. 2A). To confirm this result,
we monitored apoptosis by flow cytometry. The results of Annexin
V-PI double staining assay revealed that 72 h of treatment with
lupeol markedly enhanced the Annexin V-positive cell population, an
apoptotic portion, in NSCLC cells (Fig. 2B). Similar results were obtained by
cell cycle analysis. The proportion of sub-G1 phase cells, i.e.,
apoptotic cells, was gradually increased in a dose-dependent manner
(Fig. 2C). Likewise, the
expression levels of cleaved PARP and cleaved caspase-3, apoptosis
marker proteins, were upregulated by lupeol treatment (Fig. 2D). Taken together, these results
demonstrated that lupeol triggered the apoptosis if human NSCLC
cells.
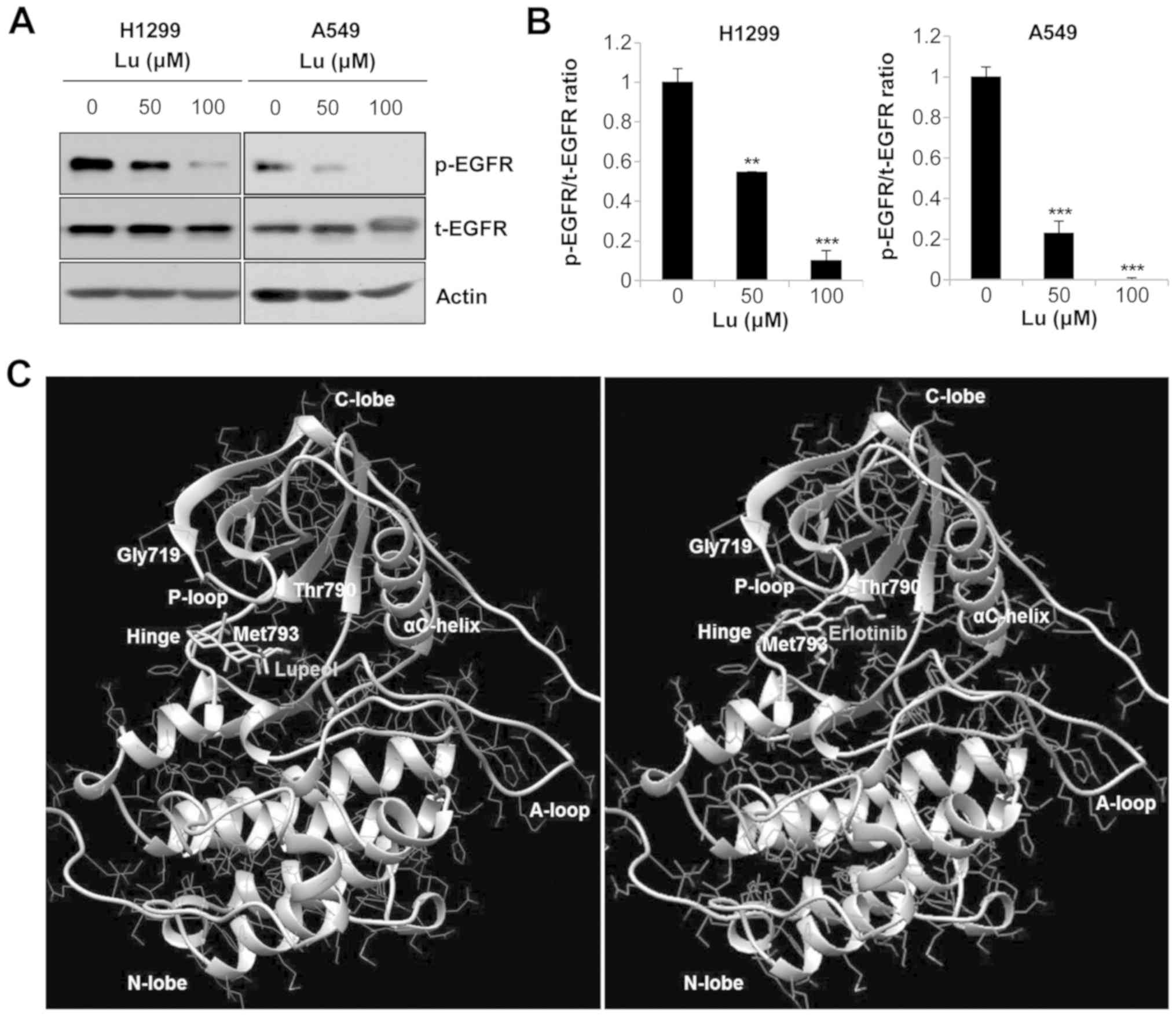
Lupeol inhibits EGFR activation by direct
binding to the EGFR TK domain in human NSCLC cells
As one of the pivotal oncogenic signaling pathways
involved in NSCLC is EGFR, we then examined the effects of lupeol
on the activity of EGFR. Western blot analysis indicated that the
phosphorylation of EGFR was decreased by lupeol in a dose-dependent
manner, while levels of the corresponding total proteins remained
unaltered (Fig. 3A and B). To
determine the mechanisms through which lupeol suppressed EGFR
phosphorylation, we performed a molecular docking analysis using
the SwissDock webservice (38).
The crystal structure for the TK domain of EGFR [PDB ID: 1M17] was
used for the analysis and erlotinib, a known EGFR TKI, was used as
a control (40,41). Five clusters with a total of 48
binding modes between lupeol and EGFR were generated and the
binding ∆G (−7.37 to −5.87 kcal/mol) was calculated. Among the
clusters, the conformation with the lowest binding ∆G was selected.
The 3D binding mode viewed by UCSF Chimera software clearly
revealed that lupeol bound to the hinge region of the TK domain
(Fig. 3C) with full fitness energy
below 2,145 kcal/mol, which was comparable to that of erlotinib
(Table I). These results indicate
that lupeol blocked EGFR activity by directly binding to the EGFR
TK domain and competing with adenosine trisphosphate (ATP).
 | Table IDocking of lupeol or erlotinib to the
TK domain of EGFR (PDB ID: 1M17). |
Table I
Docking of lupeol or erlotinib to the
TK domain of EGFR (PDB ID: 1M17).
| Compound | Binding site | Full fitness
(kcal/mol) | Estimated ∆G
(kcal/mol) |
|---|
| Erlotinib | Hinge region of TK
domain | −2,200.16 | −7.94 |
| Lupeol | Hinge region of TK
domain | −2,145.44 | −7.37 |
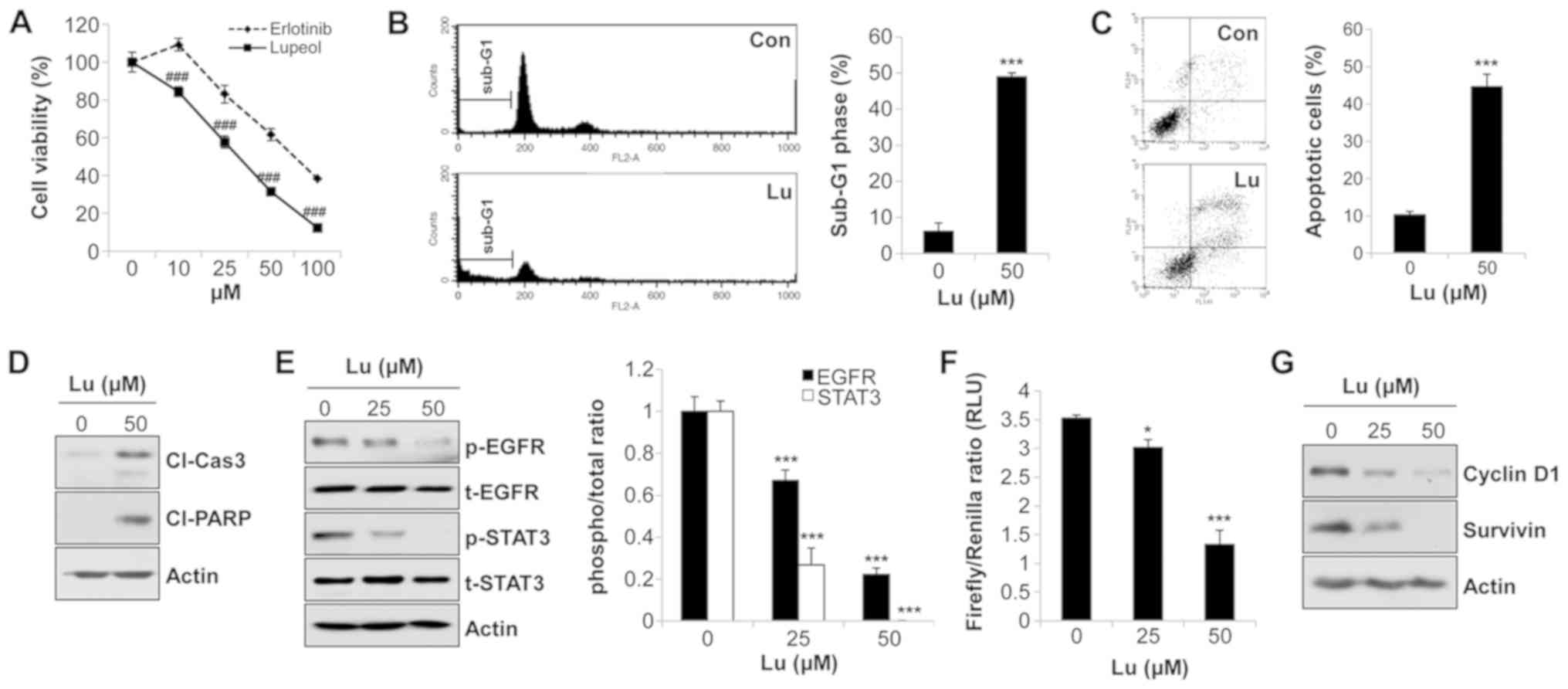
Inhibition of STAT3 activity by lupeol
induces the apoptosis of human NSCLC cells
We further examined the effects of lupeol on the
activity of downstream molecules of EGFR. As shown in Fig. S1, the phosphorylation levels of
ERK and AKT were not decreased by lupeol treatment (Fig. S1). On the contrary, lupeol
markedly suppressed the phosphorylation of STAT3 in NSCLC cells
(Fig. 4A and B). To verify whether
this event was dependent on EGFR regulation, we used EGF as a
stimulator of EGFR. The phosphorylation of EGFR and STAT3 was
enhanced by EGF treatment. However, the addition of lupeol reversed
the EGF-mediated phosphorylation of EGFR and STAT3 (Fig. 4C). The same expression pattern of
p-EGFR and p-STAT3 clearly indicated that lupeol suppressed STAT3
activity via EGFR regulation. In addition, the expression of
p-STAT3 in the nuclear extracts was significantly decreased by
lupeol in the H1299 cells, demonstrating that lupeol inhibited the
nuclear translocation of p-STAT3 (Fig.
4D). The results from the dual luciferase reporter assay also
revealed that the STAT3 reporter gene activity was reduced in a
dose-dependent manner in the H1299 cells (Fig. 4E). Consistently, the mRNA and
protein levels of STAT3 target genes, including survivin, an
apoptosis-inhibitory protein, and cyclin D1, a protein involved in
cell cycle progression, were decreased by lupeol in the H1299 cells
(Fig. 4F). Taken together, these
results indicated that lupeol blocked the transcriptional activity
of STAT3 in human NSCLC cells.
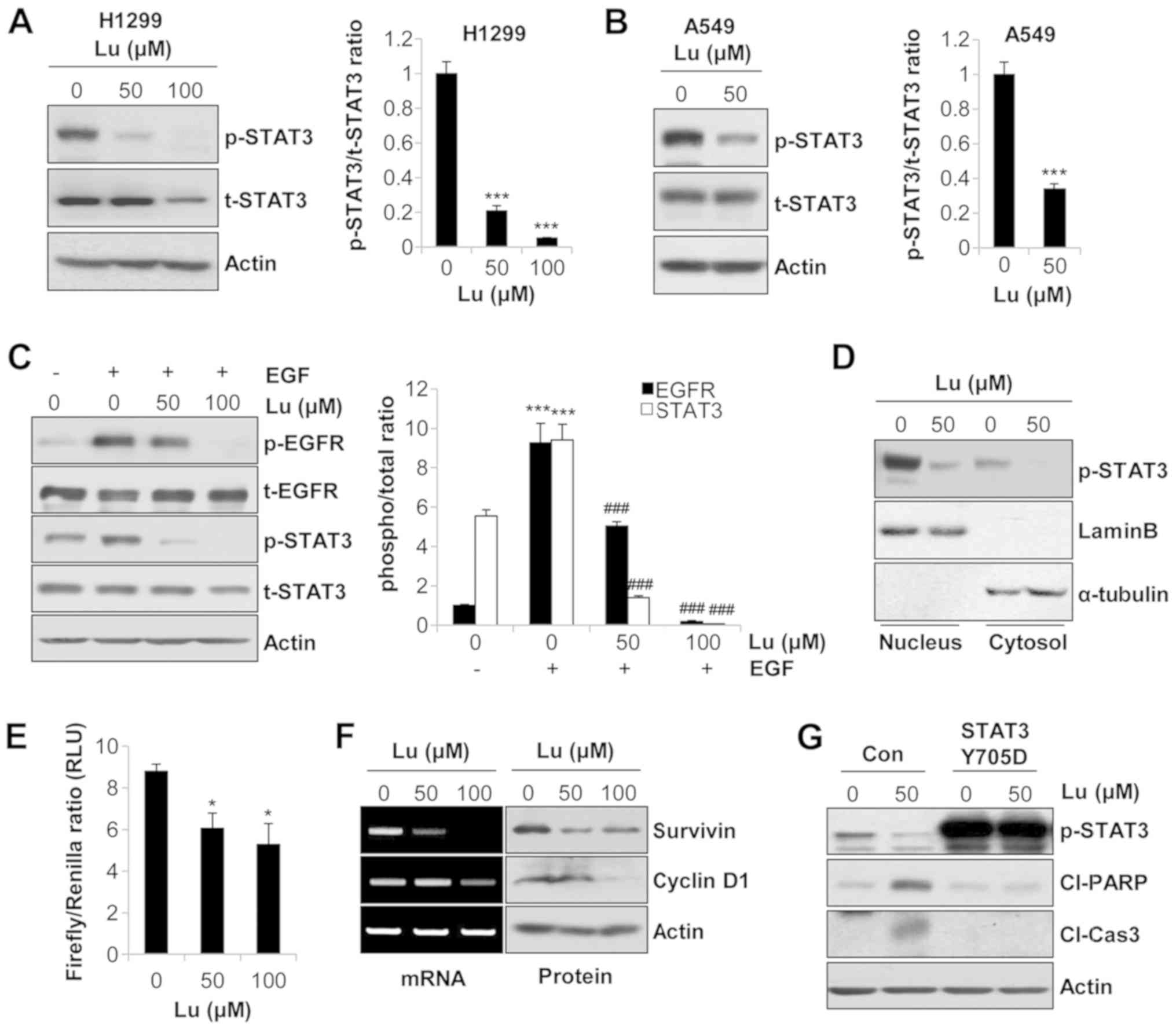 | Figure 4Suppression of the transcriptional
activity of STAT3 by lupeol induces the apoptosis of human NSCLC
cells. (A) H1299 and (B) A549 cells were treated with lupeol for 24
h. The expression levels of p-STAT3 and t-STAT3 were detected by
western blot analysis (left panels). The ratio of p-STAT3/t-STAT3
was calculated using ImageJ software (right panels). (C) A549 cells
were treated with lupeol for 24 h, and EGF (20 ng/ml) was then
added to the culture media 1 h before harvesting to activate EGFR.
The expression levels of the indicated proteins were evaluated by
western blot analysis (left panel). The ratio of phosphorylated
protein/total protein was analyzed with ImageJ software using actin
for normalization (right panel). (D) H1299 cells were treated with
lupeol for 24 h. The nuclear translocation of p-STAT3 was examined
by the nuclear/cytosol fractionation assay. Lamin B and α-tubulin
were used as markers of nuclear and cytosolic fractions,
respectively. (E) H1299 cells were transfected with a
STAT3-responsive Firefly luciferase construct (p-STAT3-TA-luc) and
a Renilla luciferase construct (pRL-TK). At 24 h
post-transfection, the cells were treated with lupeol for an
additional 24 h. The transcriptional activity of STAT3 was measured
by the Dual-Luciferase Reporter Assay System. (F) H1299 cells were
treated with lupeol for 72 h. mRNA levels (left panel) and protein
levels (right panel) of the STAT3 target genes were evaluated by
RT-PCR (and semi-quantitative PCR) and western blot analysis,
respectively. Actin was used as an internal control. (G) H1299
cells were transfected with control vector or STAT3 phospho-mimetic
mutant (Y705D). At 48 h post-transfection, the cells were treated
with the indicated concentrations of lupeol for an additional 72 h.
The expressions of p-STAT3, cleaved PARP and cleaved caspase-3 were
detected by western blot analysis. Actin was used as an internal
control. The data are expressed as the means ± SD of 3 independent
experiments. *P<0.05 and ***P<0.001 vs.
untreated controls; ###P<0.001 vs. EGF-treated cells.
NSCLC, non-small cell lung cancer; EGFR, epidermal growth factor
receptor; Con, control vector; Lu, lupeol; RLU, relative luciferase
unit; Cl-Cas3, cleaved caspase-3; Cl-PARP, cleaved PARP. |
We then examined whether the suppression of STAT3
activity by lupeol was sufficient to trigger the apoptosis of NSCLC
cells. As shown in Fig. 4G, the
H1299 cells transfected with pExpress1-stat3Y705D exhibited a
significantly upregulated p-STAT3 expression compared with that in
cells transfected with the control vector. In addition,
transfection with STAT3 Y705D downregulated the expression levels
of cleaved PARP and cleaved caspase-3 which had been increased by
lupeol (Fig. 4G). Thus, these data
suggested that the inhibition of STAT3 activity by lupeol
contributed to the induction of the apoptosis of NSCLC cells.
Lupeol exerts anticancer effects on EGFR
TKI-resistant NSCLC cells
To examine whether lupeol exhibits anticancer
activities in an EGFR TKI-resistant H1975 cell line with
L858R/T790M double mutations, we first evaluated the effects of
lupeol on the growth of H1975 cells. The results from MTT assay
revealed that the viability of the erlotinib-resistant H1975 cells
was markedly reduced by lupeol (Fig.
5A). Lupeol also increased the percentage of sub-G1 phase cells
and Annexin V-positive cells (Fig. 5B
and C) and upregulated the expression levels of cleaved PARP
and cleaved caspase-3 in H1975 cells (Fig. 5D). These results suggested that
lupeol suppressed the growth of H1975 cells by triggering
apoptosis. We then verified whether these events were derived from
the inhibition of the EGFR/STAT3 signaling pathway. Lupeol
downregulated the expression levels of p-EGFR and p-STAT3 in a
dose-dependent manner, while the corresponding total proteins
remained unaltered in the H1975 cells (Fig. 5E). To measure the transcriptional
activity of STAT3 following treatment with lupeol, the H1975 cells
were transfected with a STAT3-responsive reporter vector and
subsequently treated with lupeol for 48 h. As shown in Fig. 5F, the STAT3 reporter gene activity
was reduced in a dose-dependent manner (Fig. 5F). Consistently, the protein
expression levels of STAT3 target genes, including cyclin D1 and
survivin, were decreased by lupeol in the H1975 cells (Fig. 5G). Collectively, these results
clearly demonstrated that lupeol exerted anticancer effects on EGFR
TKI-resistant H1975 cells, as well as on EGFR wild-type NSCLC
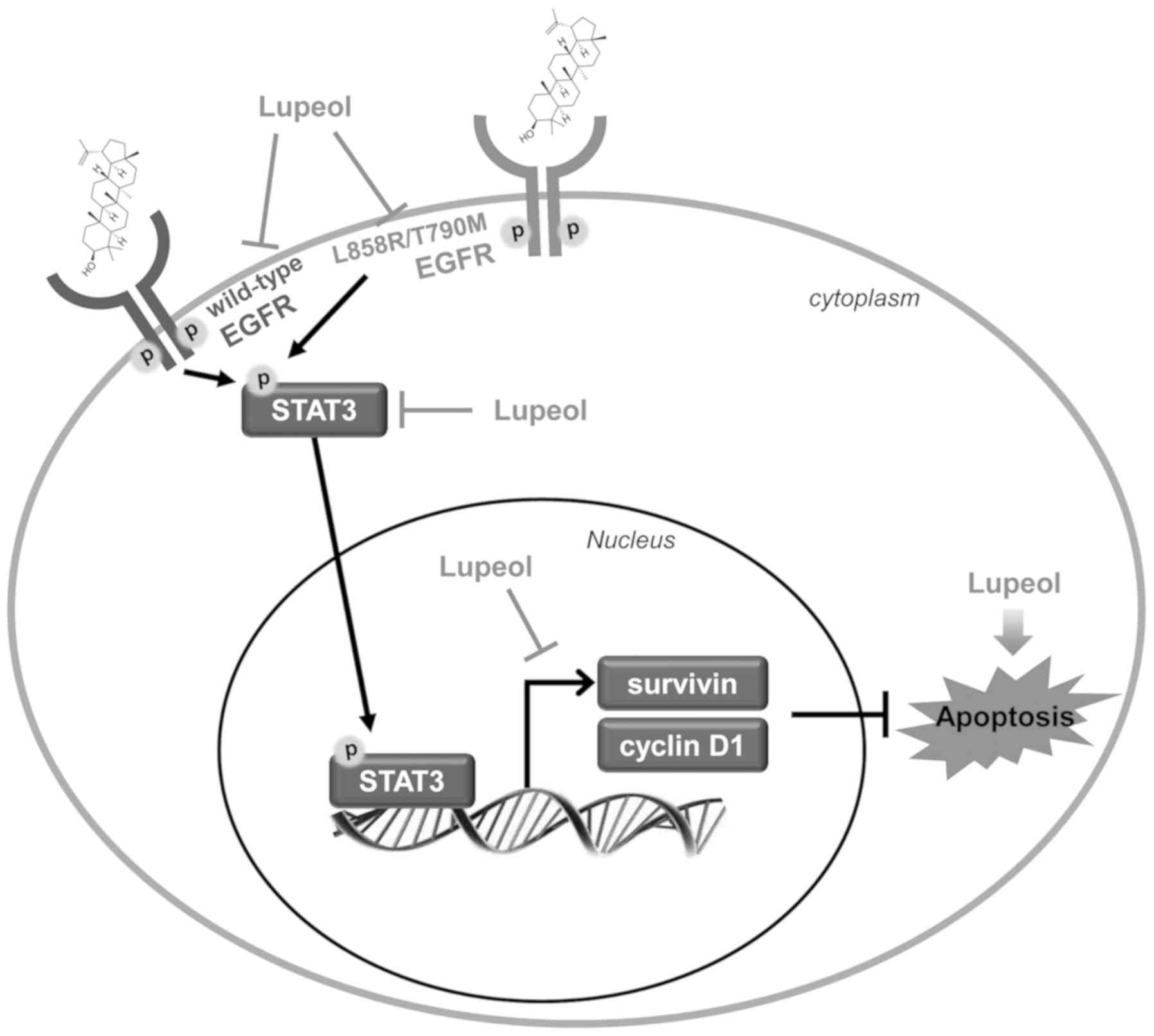
cells, through the suppression of EGFR/STAT3 activation (Fig. 6).
Discussion
The present study explored the potential anticancer
effects of lupeol on NSCLC cells, focusing specifically on the
regulation of the EGFR//STAT3 signaling pathway. Our results
demonstrated that lupeol triggered the apoptosis of and inhibited
the activity of EGFR and STAT3 in NSCLC cells, regardless of their
EGFR mutation status. The novelty of this study is as follows:
First, to the best of our knowledge, this is the first study to
demonstrate a contribution of the EGFR/STAT3 axis to the
lupeol-induced apoptosis of NSCLC cells. Although previous studies
have reported that lupeol suppresses EGFR activity in several
cancer cells, they usually identified AKT as a downstream target of
EGFR (36,37). In our case, the phosphorylation
levels of AKT, as well as the MAPK proteins, the main signaling
mediators activated by EGFR, were even slightly increased by lupeol
treatment (Fig. S1). Instead,
lupeol markedly suppressed the phosphorylation, nuclear
translocation, and transcriptional activity of STAT3, suggesting
that lupeol evoked anticancer effects in NSCLC cells by
deactivation of the EGFR/STAT3 signaling pathway. Second, we
reported the putative direct interaction between lupeol and EGFR.
The docking analysis implemented by SwissDock revealed that lupeol
effectively bound to the TK domain of EGFR with a low binding
energy. Finally, we demonstrated that erlotinib-resistant H1975
cells were highly sensitive to lupeol. Lupeol consistently
suppressed the activity of EGFR and STAT3 in H1975 cells. Given
that the T790M mutation in EGFR accounts for approximately half of
the acquired resistance to EGFR TKIs and lowers the response rate
and overall survival of NSCLC patients (16,17),
our results suggest that lupeol may be a putative therapeutic
option not only for EGFR TKI-naïve NSCLC patients, but also for
advanced NSCLC with acquired resistance to EGFR TKIs.
Notably, STAT3 signaling has been implicated in
primary and acquired resistance to EGFR TKIs. Various cell lines
and ex vivo-based resistant models and patient-derived tumor tissue
analyses have demonstrated that STAT3 activity may protect NSCLC
cancer cells from EGFR TKI (22-27).
These studies have suggested that STAT3 may be a desirable
molecular target for enhancing sensitivity to EGFR TKIs. Our data
also demonstrated that lupeol exerted potent anticancer effects in
erlotinib-resistant H1975 cells via the suppression of STAT3
activation. The sole study that reported the inhibitory effects of
lupeol on STAT3 activity was published by Siveen et al
(7). They revealed that lupeol
blocked the phosphorylation of STAT3 by enhancing the expression of
tyrosine phosphatase SHP-2. In our case, lupeol did not affect the
expression of SHP-2 and treatment with pervanadate, a phosphatase
inhibitor, did not abrogate lupeol-induced apoptosis in NSCLC cells
(data not shown). Instead, our results clearly demonstrated that
STAT3 activation was dependent on the activity of EGFR.
Despite the novelty of this study, further molecular
evidence should be provided to demonstrate the direct binding
between lupeol and EGFR. We also cannot exclude the possibility
that other upstream molecules of STAT3, such as IL6R and Src, are
regulated by lupeol. In addition, if lupeol does modulate various
upstream targets of STAT3, it would be of interest to examine the
effects of lupeol on the crosstalk between these targets to
strengthen its anticancer activity.
In conclusion, in this study, we propose a novel
molecular mechanism of lupeol which involves the induction of the
apoptosis of NSCLC cells. Our results revealed that lupeol can
directly bind to the TK domain of EGFR with a quite low binding
energy comparable to erlotinib, leading to the suppression of
EGFR/STAT3 activity. Lupeol exerted anticancer effects, not only in
EGFR wild-type NSCLC cells, but also on H1975 cells with a
secondary T790M mutation. These results collectively suggest that
lupeol may be an alternative therapeutic option for patients with
advanced NSCLC with acquired EGFR-TKI resistance, as well as for
patients with EGFR TKI-naïve NSCLC. Further preclinical and
clinical studies are required to evaluate the anticancer activity
of lupeol.
Supplementary Materials
Funding
This study was supported by grants from the National
Research Foundation of Korea (NRF), Republic of Korea (No.
NRF-2016R1C1B2015076).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article. The datasets used and/or
analyzed during the current study are available from the
corresponding author on reasonable request.
Authors' contributions
SHP conceived and designed the study. TRM and HJP
performed the majority of the in vitro experiments. SHP,
KTH, GYC and YHC analyzed the data and coordinated the project.
SHP, GYC and YHC wrote and reviewed the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Keith RL and Miller YE: Lung cancer
chemoprevention: Current status and future prospects. Nat Rev Clin
Oncol. 10:334–343. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Blackhall FH, Shepherd FA and Albain KS:
Improving survival and reducing toxicity with chemotherapy in
advanced non-small cell lung cancer: A realistic goal? Treat Respir
Med. 4:71–84. 2005. View Article : Google Scholar
|
|
3
|
Travis WD, Travis LB and Devesa SS: Lung
cancer. Cancer. 75(Suppl): 191–202. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mosesson Y and Yarden Y: Oncogenic growth
factor receptors: Implications for signal transduction therapy.
Semin Cancer Biol. 14:262–270. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohsaki Y, Tanno S, Fujita Y, Toyoshima E,
Fujiuchi S, Nishigaki Y, Ishida S, Nagase A, Miyokawa N, Hirata S,
et al: Epidermal growth factor receptor expression correlates with
poor prognosis in non-small cell lung cancer patients with p53
overexpression. Oncol Rep. 7:603–607. 2000.PubMed/NCBI
|
|
6
|
Inamura K, Ninomiya H, Ishikawa Y and
Matsubara O: Is the epidermal growth factor receptor status in lung
cancers reflected in clinicopathologic features? Arch Pathol Lab
Med. 134:66–72. 2010.PubMed/NCBI
|
|
7
|
Siveen KS, Nguyen AH, Lee JH, Li F, Singh
SS, Kumar AP, Low G, Jha S, Tergaonkar V, Ahn KS, et al: Negative
regulation of signal transducer and activator of transcription-3
signalling cascade by lupeol inhibits growth and induces apoptosis
in hepatocellular carcinoma cells. Br J Cancer. 111:1327–1337.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scagliotti GV, Selvaggi G, Novello S and
Hirsch FR: The biology of epidermal growth factor receptor in lung
cancer. Clin Cancer Res. 10:S4227–S4232. 2004. View Article : Google Scholar
|
|
9
|
Veale D, Kerr N, Gibson GJ, Kelly PJ,
Harris AL and Br J: The relationship of quantitative epidermal
growth factor receptor expression in non-small cell lung cancer to
long term survival. Br J Cancer. 68:162–165. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fontanini G, De Laurentiis M, Vignati S,
Chinè S, Lucchi M, Silvestri V, Mussi A, De Placido S, Tortora G,
Bianco AR, et al: Evaluation of epidermal growth factor-related
growth factors and receptors and of neoangiogenesis in completely
resected stage I-IIIA non-small-cell lung cancer: Amphiregulin and
microvessel count are independent prognostic indicators of
survival. Clin Cancer Res. 4:241–249. 1998.PubMed/NCBI
|
|
11
|
Ogawa J, Iwazaki M, Inoue H, Koide S and
Shohtsu A: Immunohistochemical study of glutathione-related enzymes
and proliferative antigens in lung cancer. Relation to cisplatin
sensitivity. Cancer. 71:2204–2209. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shigematsu H and Gazdar AF: Somatic
mutations of epidermal growth factor receptor signaling pathway in
lung cancers. Int J Cancer. 118:257–262. 2006. View Article : Google Scholar
|
|
13
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pao W, Miller V, Zakowski M, Doherty J,
Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al:
EGF receptor gene mutations are common in lung cancers from 'never
smokers' and are associated with sensitivity of tumors to gefitinib
and erlotinib. Proc Natl Acad Sci USA. 101:13306–13311. 2004.
View Article : Google Scholar
|
|
16
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Johnston PA and Grandis JR: STAT3
signaling: Anticancer strategies and challenges. Mol Interv.
11:18–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harada D, Takigawa N and Kiura K: The role
of STAT3 in non-small cell lung cancer. Cancers (Basel). 6:708–722.
2014. View Article : Google Scholar
|
|
20
|
Jiang R, Jin Z, Liu Z, Sun L, Wang L and
Li K: Correlation of activated STAT3 expression with
clinicopathologic features in lung adenocarcinoma and squamous cell
carcinoma. Mol Diagn Ther. 15:347–352. 2011. View Article : Google Scholar
|
|
21
|
Xu YH and Lu S: A meta-analysis of STAT3
and phospho-STAT3 expression and survival of patients with
non-small-cell lung cancer. Eur J Surg Oncol. 40:311–317. 2014.
View Article : Google Scholar
|
|
22
|
Gao SP, Chang Q, Mao N, Daly LA, Vogel R,
Chan T, Liu SH, Bournazou E, Schori E, Zhang H, et al: JAK2
inhibition sensitizes resistant EGFR-mutant lung adenocarcinoma to
tyrosine kinase inhibitors. Sci Signal. 9:ra332016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yao Z, Fenoglio S, Gao DC, Camiolo M,
Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V,
et al: TGF-beta IL-6 axis mediates selective and adaptive
mechanisms of resistance to molecular targeted therapy in lung
cancer. Proc Natl Acad Sci USA. 107:15535–15540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK,
Sica GL, Ramalingam SS, Curran WJ, Khuri FR and Deng X: Niclosamide
overcomes acquired resistance to erlotinib through suppression of
STAT3 in non-small cell lung cancer. Mol Cancer Ther. 12:2200–2212.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB,
Ko YG, Lee JS, Lee SJ, Lee JC and Park MJ: Upregulation of CXCR4 is
functionally crucial for maintenance of stemness in drug-resistant
non-small cell lung cancer cells. Oncogene. 32:209–221. 2013.
View Article : Google Scholar
|
|
26
|
Fan W, Tang Z, Yin L, Morrison B,
Hafez-Khayyata S, Fu P, Huang H, Bagai R, Jiang S, Kresak A, et al:
MET-independent lung cancer cells evading EGFR kinase inhibitors
are therapeutically susceptible to BH3 mimetic agents. Cancer Res.
71:4494–4505. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zulkifli AA, Tan FH, Putoczki TL, Stylli
SS and Luwor RB: STAT3 signaling mediates tumour resistance to EGFR
targeted therapeutics. Mol Cell Endocrinol. 451:15–23. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Siddique HR and Saleem M: Beneficial
health effects of lupeol triterpene: A review of preclinical
studies. Life Sci. 88:285–293. 2011. View Article : Google Scholar
|
|
29
|
Wu XT, Liu JQ, Lu XT, Chen FX, Zhou ZH,
Wang T, Zhu SP and Fei SJ: The enhanced effect of lupeol on the
destruction of gastric cancer cells by NK cells. Int
Immunopharmacol. 16:332–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu Y, Bi T, Dai W, Wang G, Qian L, Shen G
and Gao Q: Lupeol enhances inhibitory effect of 5-fluorouracil on
human gastric carcinoma cells. Naunyn Schmiedebergs Arch Pharmacol.
389:477–484. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tarapore RS, Siddiqui IA, Adhami VM,
Spiegelman VS and Mukhtar H: The dietary terpene lupeol targets
colorectal cancer cells with constitutively active Wnt/β-catenin
signaling. Mol Nutr Food Res. 57:1950–1958. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pitchai D, Roy A and Ignatius C: In vitro
evaluation of anticancer potentials of lupeol isolated from
Elephantopus scaber L. on MCF-7 cell line. J Adv Pharm Technol Res.
5:179–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sankaranarayanan S, Bama P, Sathyabama S
and Bhuvaneswari N: Anticancer compound isolated from the leaves of
Tridax procumbens against human lung cancer cell A-549. Asian J
Pharm Clin Res. 6:91–96. 2013.
|
|
34
|
He W, Li X and Xia S: Lupeol triterpene
exhibits potent antitumor effects in A427 human lung carcinoma
cells via mitochondrial mediated apoptosis, ROS generation, loss of
mitochondrial membrane potential and downregulation of
m-TOR/PI3Ksol;AKT signalling pathway. J BUON. 23:635–640.
2018.PubMed/NCBI
|
|
35
|
Lee TK, Castilho A, Cheung VC, Tang KH, Ma
S and Ng IO: Lupeol targets liver tumor-initiating cells through
phosphatase and tensin homolog modulation. Hepatology. 53:160–170.
2011. View Article : Google Scholar
|
|
36
|
Liu Y, Bi T, Shen G, Li Z, Wu G, Wang Z,
Qian L and Gao Q: Lupeol induces apoptosis and inhibits invasion in
gallbladder carcinoma GBC-SD cells by suppression of EGFR/MMP-9
signaling pathway. Cytotechnology. 68:123–133. 2016. View Article : Google Scholar :
|
|
37
|
Rauth S, Ray S, Bhattacharyya S, Mehrotra
DG, Alam N, Mondal G, Nath P, Roy A, Biswas J and Murmu N: Lupeol
evokes anticancer effects in oral squamous cell carcinoma by
inhibiting oncogenic EGFR pathway. Mol Cell Biochem. 417:97–110.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Grosdidier A, Zoete V and Michielin O:
SwissDock, a protein-small molecule docking web service based on
EADock DSS. Nucleic Acids Res. 39(Suppl): W270–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Berman HM, Westbrook J, Feng Z, Gilliland
G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE: The protein
data bank. Nucleic Acids Res. 28:235–242. 2000. View Article : Google Scholar
|
|
40
|
Choowongkomon K, Sawatdichaikul O,
Songtawee N and Limtrakul J: Receptor-based virtual screening of
EGFR kinase inhibitors from the NCI diversity database. Molecules.
15:4041–4054. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stamos J, Sliwkowski MX and Eigenbrot C:
Structure of the epidermal growth factor receptor kinase domain
alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol
Chem. 277:46265–46272. 2002. View Article : Google Scholar : PubMed/NCBI
|