Introduction
In 2018, kidney cancer is estimated to be diagnosed
in nearly 403,200 people worldwide and to lead to almost 175,000
cancer-related deaths according to the latest data released by the
International Agency for Research on Cancer (1). The course of kidney cancer is
commonly palliative and uneventful without initial distinct
clinical symptoms and signs (2).
Hence, patients fail to be diagnosed at the early stage of cancer.
For localized tumours, surgical excision by partial nephrectomy or
radical nephrectomy is now recognized as the preferred choice of
treatment, while for the patients who present with metastatic or
unresectable tumours, systemic treatment is needed (3). Unfortunately, renal cell carcinoma
(RCC), especially clear cell (cc)RCC, which accounts for up to 90%
of all kidney cancers, originates from the proximal convoluted
tubule, which highly expresses multidrug resistant protein-1,
resulting in the high resistance of RCC to chemotherapy (4). Although the development of drugs
targeting neovascularization or mammalian target of rapamycin
(mTOR) improves the survival of RCC patients, drug resistance
inevitably develops (5). Recently,
checkpoint immunotherapy has shown promise; however, the complete
response rate still remains at a low level (6). Therefore, it is imperative that
insight into the mechanisms underlying the development of RCC is
gained and that novel therapeutic targets to improve the prognosis
of RCC are identified.
The Cancer Genome Atlas (TCGA) was launched in 2005
with the aim of categorizing all genetic alterations contributing
to cancer formation and development to explore new clinical
therapies as well as diagnostic and preventive strategies. By 2015,
over 30 human tumour types had been incorporated into the database,
including RCC (7). By analysing
the RCC gene expression data in TCGA database and then performing a
high-throughput cell viability screening, the authors found that
pleckstrin homology domain-containing family O member 1 (PLEKHO1)
was aberrantly expressed in tumour tissue and probably contributed
to tumour growth. PLEKHO1 [also known as casein kinase
(CK)2-interacting protein 1] was originally identified as a novel
CK2-binding protein and has been shown to mediate the specific
function of CK2 by sequestering or recruiting CK2 to the plasma
membrane (8-10). The PLEKHO1 protein possesses
multiple active sites, including a pleckstrin homology domain at
the N-terminus, a putative leucine zipper (LZ) motif at the
C-terminus and five proline-rich motifs throughout the protein,
which mediate diverse protein-protein interactions (11).
On the basis of its structural characteristics,
PLEKHO1 was implicated in different signalling pathways involved in
diverse biological processes, such as cell proliferation (9,12),
cell apoptosis (13), cell
differentiation (9), cell
morphology (14,15), macrophage migration/proliferation
(16,17) and protein metabolism (18,19).
Recently, it was found that PLEKHO2, which belongs to the same
superfamily as PLEKHO1, is a key factor for macrophage survival
(20). Lu et al (18) reported that the expression of
PLEKHO1 negatively regulated bone formation, and after that study,
the 6-liposome system (AspSerSer) was developed to guide PLEKHO1
small interfering (si) RNAs to bone formation surfaces to treat
osteoporosis (21). In addition,
other studies have demonstrated that PLEKHO1 plays roles in many
diseases in humans and animal models, such as cancer (22-24),
diabetic nephropathy (25), fatty
liver disease (26) and chronic
heart failure (27). All these
findings suggest the potential significance of PLEKHO1 in both
biological and pathological processes of the human body.
Nevertheless, the functional roles and detailed mechanisms of
PLEKHO1 in diseases, especially in neoplasms, remain to be
elucidated. In the current study, the authors investigated the
function of PLEKHO1 in RCC and explored the mechanism in which
PLEKHO1 functions.
Materials and methods
Bioinformatic analysis
The raw expression data and clinical data, such as
tumour stage and survival status of RCC patients, were downloaded
from TCGA, which was searched with the following term:
'KIRC_RNA-seq_HTSeq-Counts' (https://cancergenome.nih.gov/; Tables SI and SII). In the present study, the raw
RNA-sequencing (high throughput sequencing-counts) data were
arranged and exported using R-project (R version 3.5.0; https://cran.r-project.org/src/base/R-3/), in which
'Edge R', 'gplots' and 'survminer' were used for differential,
clustering and survival analyses, respectively. Additionally,
co-expression analysis was performed based on the data obtained
from cBioPortal for Cancer Genomics using the search term
'Kidney_Kidney Renal Clear Cell Carcinoma (TCGA, Nature 2013)'
(http://www.cbioportal.org/) (28,29).
Packages were freely accessible from the following sources:
'edgeR': http://www.bioconductor.org/packages/release/bioc/html/edgeR.html;
'gplots': https://cran.r-project.org/web/packages/gplots/;
'survminer': https://cran.rstudio.com/web/packages/survminer/index.html.
Cell lines and cell culture
Human RCC cell lines (Caki-1, 786-O, ACHN, 769-P and
OS-RC-2) were obtained from the Cell Type Culture Collection in the
Institute of Biochemistry and Cell Biology of the Chinese Academy
of Sciences (Shanghai, China). Caki-1 cells were cultured in
McCoy's 5A medium (HyClone; GE Healthcare Life Sciences), while
786-O, 769-P and OS-RC-2 cells were cultured in RPMI-1640 medium
(HyClone; GE Healthcare Life Sciences), and ACHN cells was cultured
in minimum essential media (HyClone; GE Healthcare Life Sciences)
at 37°C with 5% CO2. All the culture media were
supplemented with 10% foetal bovine serum (HyClone; GE Healthcare
Life Sciences). 786-O and Caki-1 cell lines were authenticated, and
no mycoplasma contamination was identified.
Tissue samples
Paired tumour and para-tumour normal tissues were
acquired from 30 patients (19 males and 11 females aging from 35 to
71 years old) diagnosed with RCC who had undergone surgical
treatment at the Department of Urology, Cancer Hospital of China
Medical University and the Department of Urology, The First
Hospital of China Medical University (Shenyang, China) from
September 2014 to July 2016. The excised tissue samples were
snap-frozen in liquid nitrogen and saved at -80°C until their use.
The current study was approved by the Research Ethics Committee of
China Medical University and all patients provided signed written
informed consent.
High-throughput cell viability
screening
A high-throughput Celigo cytometry system was used
to evaluate cell viability as previously described (30,31).
In the present study, 786-O cells were chosen as the cell model of
RCC and transfected with a short hairpin (sh)RNA to a specific gene
or scrambled shRNA (sh-Ctrl; both GeneChem) in the presence of 6
µg/ml polybrene (Sigma-Aldrich; Merck KGaA). To guarantee the shRNA
silencing efficiency, three shRNAs [20 µg hU6-MCS-CMV-EGFP
(GeneChem)] targeting different sites on each of the 16 candidate
genes and the positive control (PC) gene (RNA-binding protein NOB1)
were pooled in equal proportions in the packaging viruses. These 16
candidate genes were chosen based on their relevance to the
development of RCC after analysing the TCGA dataset: DNA
dC->dU-editing enzyme APOBEC-3H, enkurin, α-(1,3)-fucosyltransferase 11, Golgi-associated
plant pathogenesis-related protein 1, mixed lineage kinase
domain-like protein, nucleoredoxin-like protein 2, opa interacting
protein 5, oncoprotein-induced transcript 3 protein, PLAC8-like
protein 1, PLEKHO1, protein prune homolog 2, Ras association
domain-containing protein 6, spindle and kinetochore-associated
protein 3, pachytene checkpoint protein 2 homolog,
ubiquitin-conjugating enzyme E2 T and zinc finger protein 320. Two
days after transfection, the transfected cells were subjected to
cell viability screening, in which a total of 2,000 cells were
seeded into each well of 96-well plates and scanned every day using
a Celigo Imaging Cytometer equipped with integrated Celigo software
(both Nexcelom Bioscience) at a magnification of ×100 for 5 days.
The fluorescence signal, which was proportional to the live cell
number in each well, was quantified automatically and recorded as
the cell viability in real time.
Lentivirus construction, and shRNA and
siRNA transfection
Lentiviruses carrying shRNAs targeting PLEKHO1
(sh-PLEK; GeneChem) or scrambled shRNA (sh-Ctrl) were constructed
using hU6-MCS-CMV-EGFP. The RCC cells were transfected with sh-PLEK
or sh-Ctrl lentivirus (1×108 TU/ml) at a multiplicity of
infection (MOI) of ~5 for 786-O cells or ~10 for Caki-1 cells in
the presence of 6 μg/ml polybrene (Sigma-Aldrich; Merck
KGaA) according to the manufacturer's protocol. Lentivirus volume
used was calculated as the following formula: V = (MOI x
N)/1×108 (V = lentivirus volume, N = cell number).
Additionally, siRNAs targeting PLEKHO1 (si-PLEK 1# and si-PLEK 2#)
or non-targeting control siRNA (si-Ctrl) were purchased from JTS
Scientific. Lipofectamine® 3000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for siRNA
transfection according to the manufacturer's protocol. The
efficiency of gene knockdown was identified by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting 48 h post-transfection. The targeting sequence of
sh-PLEK was 5′-GCTGAGAGACCTGTACAGA-3′ and for sh-Ctrl, the sequence
was 5′-TTCTCCGAACGTGTCACGT-3′. The sense strand sequence was
5′-AGCUCUACAUCUCUGAGAATT-3′ for si-PLEK 1#,
5′-GGACAGCUAUCUUGCCCAUTT-3′ for si-PLEK 2# and
5′-UUCUUCGAACGUGUCACGUTT-3′ for si-Ctrl.
RNA extraction and RT-qPCR
Total RNA was extracted from cultured cells (Caki-1,
786-O, ACHN, 769-P and OS-RC-2) or tissue samples (RCC tumour and
para-tumour tissues) using RNAiso Plus reagent (Takara
Biotechnology Co., Ltd.) following the manufacturer's protocol and
quantified using a BioDrop Duo UV/VIS spectrophotometer (BioDrop
Ltd.). For RT-qPCR analysis, total RNA was converted into cDNA
using PrimeScript™ RT Master Mix (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. A total of 1 μg
cDNA was added to SYBR Premix EX Taq™ (Takara Biotechnology Co.,
Ltd.). The data were collected on a LightCycler™ 480 II system
(Roche Diagnostics) as follows: 50°C for 2 min, 95°C for 10 min,
and 45 cycles at 95°C for 10 sec, 60°C for 30 sec and 72°C for 45
sec. A melting curve was obtained by increasing the temperature
from 40°C to 97°C at a rate of 0.4°C/sec. The results were
standardized with the expression level of GAPDH. Relative
quantitative analysis was performed with the 2−ΔΔCt
method (32). The primer sequences
were as follows: PLEKHO1, forward 5′-GAATCGTGGATCAATGCCCTC-3′ and
reverse 5′-GCGGGAGTGCTGGATTTTTG-3′; and GAPDH, forward
5′-TGACTTCAACAGCGACACCCA-3′ and reverse
5′-CACCCTGTTGCTGTAGCCAAA-3′.
Western blot analysis and antibodies
Proteins were extracted from 786-O and Caki-1 cells
transfected with shRNAs or siRNAs using 10X (wt/vol)
radioimmunoprecipitation assay lysis buffer containing 1 mM
phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology). The lysate was collected and centrifuged (12,000 ×
g, 30 min, 4°C). The supernatants containing proteins were divided
and quantified and then denatured at 100°C for 10 min. A BCA kit
(Beyotime Institute of Biotechnology) was used for protein
quantification. Equal amounts of protein (30 μg/lane) were
separated by SDS-PAGE on 10% gels and subsequently transferred to
polyvinylidene fluoride membranes (EMD Millipore). The membranes
were blocked with 5% skim milk in Tris-buffered saline with 0.1%
Tween (TBST) for 1 h at room temperature and then incubated with
primary antibodies at 4°C overnight. The next day, membranes were
washed with TBST three times for 5 min each and then incubated in
secondary antibodies at room temperature for 1.5 h. After another
three washes with TBST, immunoblot detection was performed using
enhanced chemiluminescent reagents (Thermo Fisher Scientific,
Inc.). GAPDH was used as an internal reference for total protein
detection. The antibodies used were listed as follows: Rabbit
anti-PLEKHO1 (cat. no. SAB1401681; 1:200; Sigma-Aldrich, Merck
KGaA), mouse anti-GAPDH (cat. no. sc-32233), horseradish
peroxidase-conjugated goat anti-mouse IgG (cat. no. sc-2005)
horseradish peroxidase-conjugated and goat anti-rabbit IgG (cat.
no. sc-2004; all 1:2,000; Santa Cruz Biotechnology, Inc.).
Cell proliferation assay
786-O and Caki-1 cells were transfected with si-Ctrl
and siRNAs (si-PLEK 1# and si-PLEK 2#). Twenty-four hours later,
cell proliferation was explored using the Cell Counting Kit 8
(CCK-8) assay (Dojindo Molecular Technologies, Inc.). Briefly,
cells were collected 24 h after the siRNA transfection, seeded at a
density of 2,000 cells/well into a 96-well plate (150
μl/well) and incubated at 37°C. At 0, 24, 48, 72 and 96 h,
10 μl of CCK-8 working solution was added into each well and
incubated for an additional 1.5 h. The absorbance (optical density)
of each well was detected using a microplate reader (Model 680;
Bio-Rad Laboratories, Inc.) at a wavelength of 450 nm. For all
proliferation assays, three separate experiments were
performed.
Cell apoptosis assay
786-O and Caki-1 cells were cultured in six-well
plates and transfected with sh-PLEK and sh-Ctrl as described
previously. After 5 days of transfection, cells were collected,
washed with D-Hanks (pH 7.2-7.4) pre-cooled at 4°C and then washed
with 1X binding buffer. The centrifuged cells (500 × g for 5 min at
room temperature) were resuspended using 200 μl 1X binding
buffer with an additional 10 μl Annexin V from the Annexin V
Apoptosis Detection kit APC (cat. no. 88-8007; eBioscience; Thermo
Fisher Scientific, Inc.). Then, the cells were incubated in the
dark for 15 min at room temperature, after which 300 μl 1X
binding buffer was added to dilute the solution. Flow cytometry
analysis was performed on a Guava® easyCyte HT equipped
with InCyte™ 3.1 (both EMD Millipore). Three separate experiments
were performed.
Xenografted tumour in nude mice
A total of 20 4-week-old female BALB/c nude mice
were purchased from LingChang Science and Technology Ltd. and
randomized into two groups: The sh-PLEK (KD) group and the sh-Ctrl
(NC) group. 1×107 786-O cells transfected with sh-PLEK
and sh-Ctrl as described above. Two days after transfection, cells
were observed under an inverted fluorescence microscope using the
light and fluorescence modes at a magnification of ×100. 786-O
cells without any shRNA transfection (NULL) was used as normal
control for cell morphology. Then the shRNA-transfected cells were
selected with 3 μg/ml puromycin (Sigma-Aldrich, Merck KGaA).
After a stable cell line was established, 1×107 786-O
cells mixed with Matrigel (BD Biosciences) at a ratio of 1:1 were
subcutaneously inoculated into the right armpit of nude mice. The
body weight of each mouse and the tumour diameter were measured
every week from day 43 after the inoculations of stably transfected
cells. The tumour volume (V) was calculated using the following
formula: V = 3.14/6 × L × W × W (L, length; W, width). All mice
were euthanized with the carbon dioxide method on day 87 with a
flow rate set at 25% according to the recommendation of AVMA
Guidelines (33). The current
study was carried out in strict accordance with the recommendations
of the Guide for the Care and Use of Laboratory Animals of the
National Institutes of Health (34). The protocol was approved by the
Committee on the Ethics of Animal Experiments of China Medical
University.
Expression microarrays and Ingenuity
Pathway Analysis (IPA)
For expression profile analysis after PLEKHO1
knockdown, GeneChip primeview human (cat. no. 901838; Affymetrix;
Thermo Fisher Scientific, Inc.) was used. The raw microarray data
were arranged with Expression Console™ software (version 1.4;
Affymetrix; Thermo Fisher Scientific, Inc.), and differential
analysis was performed with the GeneSpring (version 11.5; Agilent
Technologies, Inc.). The microarray data were submitted to the NCBI
Gene Expression Omnibus public database with the accession code
GSE126305 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126305)
and the data analysis was based on IPA (version 43605602; Qiagen,
Inc.; www.qiagen.com/ingenuity) (35,36).
Additionally, co-expression analysis was conducted
in TCGA dataset to check if PLEKHO1 expression correlated with
another protein. In order to diminish the interference raised from
the maximum and minimum values, the data of patients whose PLEKHO1
expression levels were in the top 1% when all the patients from
TCGA dataset were ranked according to PLEKHO1 mRNA expression were
excluded.
Statistical analysis
Statistical analyses were performed with SPSS 21.0
statistical software (IBM Corp.). In cell proliferation, apoptosis
and xenograft tumour formation assays, the data with normal
distribution are presented as mean ± standard deviation, and the
2-tailed Student's t-test was used to evaluate the significance of
differences between two groups. To detect the efficiency of
silencing, one-way analysis of variance followed by Dunnett's
multiple comparisons tests was used to compare differences among
multiple groups. The expression data downloaded from TCGA database
with non-normal distribution were presented as the median with the
interquartile range and the Mann-Whitney U test was performed to
compare groups. The Kaplan-Meier assay and Log-rank test was used
for survival analysis. Non-parametric Spearman correlation was used
for co-expression analysis. A value of P<0.05 was considered
statistically significant.
Results
Identifying PLEKHO1 as a potential
modulator that promotes cell proliferation in RCC
To explore genes that are potentially relevant to
the development of RCC, TCGA database was searched, and the gene
expression data derived from 72 RCC and paired para-tumour samples
from the same patient were analysed. The gene expression profiles
in 69 of the 72 paired samples showed a clear distinction between
the cancer and normal control tissues according to the clustering
analysis (Fig. 1A). The data of
these 69 paired samples were further analysed, and 16 genes that
were potentially related to RCC development were screened. These
genes were selected on the basis of their high expression levels in
tumour samples as well as their research significance based on the
literature (Fig. 1A; Table SIII). Next, to screen the
potential biological relevance of the 16 candidate genes in RCC,
each gene was knocked down in 786-O cells using lentiviruses
carrying specific shRNAs, separately. As shown in Fig. 1B and C, only treatment with
sh-OIP5, sh-PLEKHO1 or sh-PC, the positive control, markedly
inhibited cell proliferation. Although other genes, such as FUT11,
were similarly highly expressed in the tumour samples compared with
para-tumour samples in TCGA data, treatment with sh-FUT11 did not
impact the cell viability of 786-O cells. OIP5 has been shown to be
associated with the prognosis of RCC, and reduction of its mRNA
expression inhibits cell proliferation in 786-O and Caki-2 cells
(37). On the other hand, there
have been no reports on the relevance of the PLEKHO1 gene in RCC.
Thus, the authors chose to further investigate the functional role
of the PLEKHO1 gene in RCC.
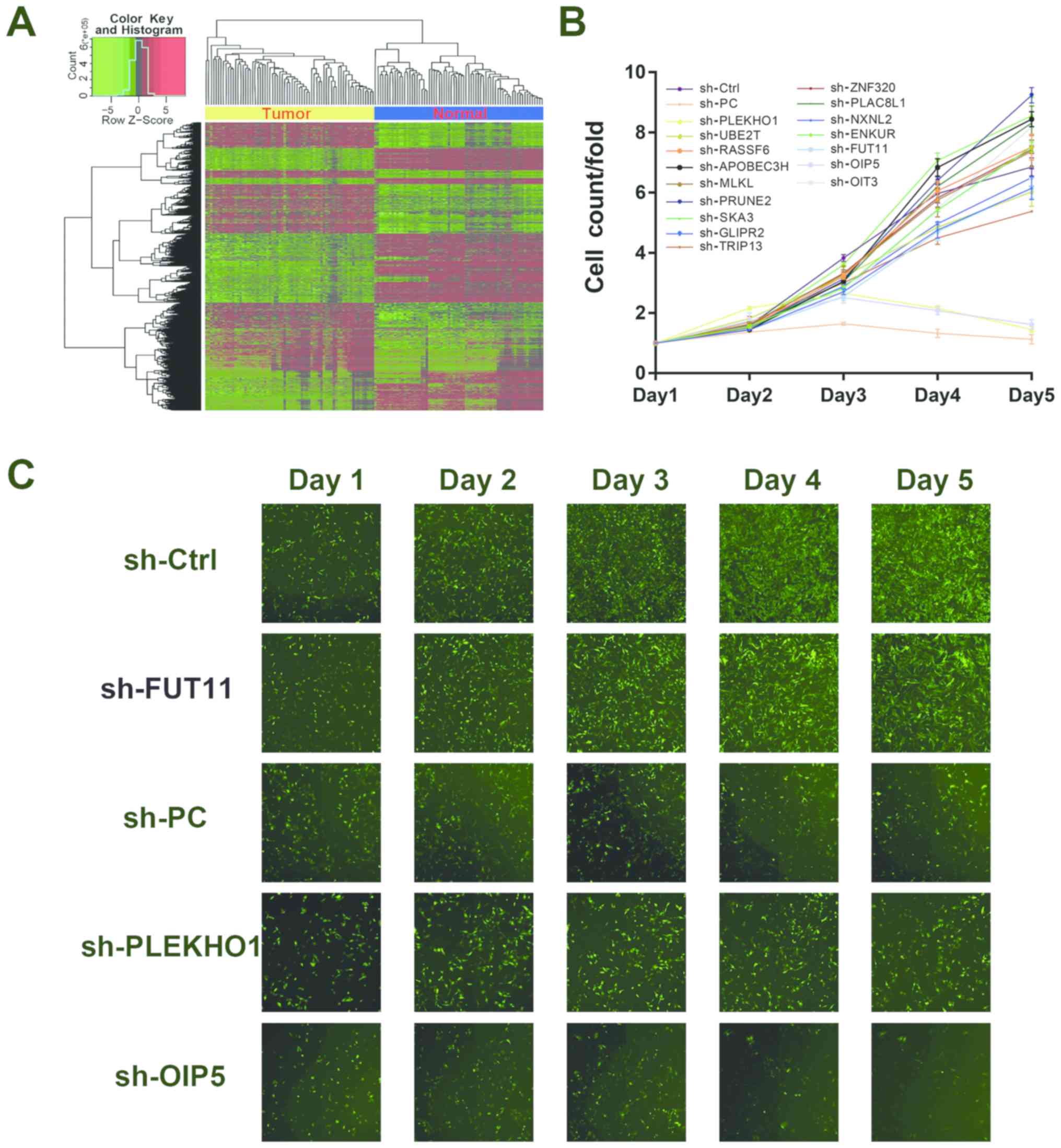 | Figure 1The Cancer Genome Atlas data analysis
and high-throughput screening identified PLEKHHO1 as a potential
modulator that promotes RCC cell proliferation. (A) Heatmap showing
the differentially expressed gene set between paired tumour and
para-tumour samples. Each row indicates a gene and each column
indicates a patient sample. (B) A total of 16 genes were selected
for validation of viability by high-throughput screening. (C)
Representative fluorescence images of high-throughput shRNA
screening. PLEKHO1, pleckstrin homology domain containing O1; RCC,
renal cell carcinoma; sh, short hairpin; sh-Ctrl, negative control
shRNA; sh-PC, positive control shRNA targeting NOB1; NOB1,
RNA-binding protein NOB1; FUT11, α-(1,3)-fucosyltransferase 11; OIP5, opa
interacting protein 5; UBE2T, ubiquitin-conjugating enzyme E2 T;
RASSF6, Ras association domain-containing protein 6; APOBEC3H, DNA
dC->dU-editing enzyme APOBEC-3H; MLKL, mixed lineage kinase
domain-like protein; PRUNE2, protein prune homolog 2; SKA3, spindle
and kinetochore-associated protein 3; GLIPR2, Golgi-associated
plant pathogenesis-related protein 1; TRIP13, pachytene checkpoint
protein 2 homolog; ZNF320, zinc finger protein 320; PLAC8L1,
PLAC8-like protein 1; NXNL2, nucleoredoxin-like protein 2; ENKUR,
enkurin; OIT3, oncoprotein-induced transcript 3 protein. |
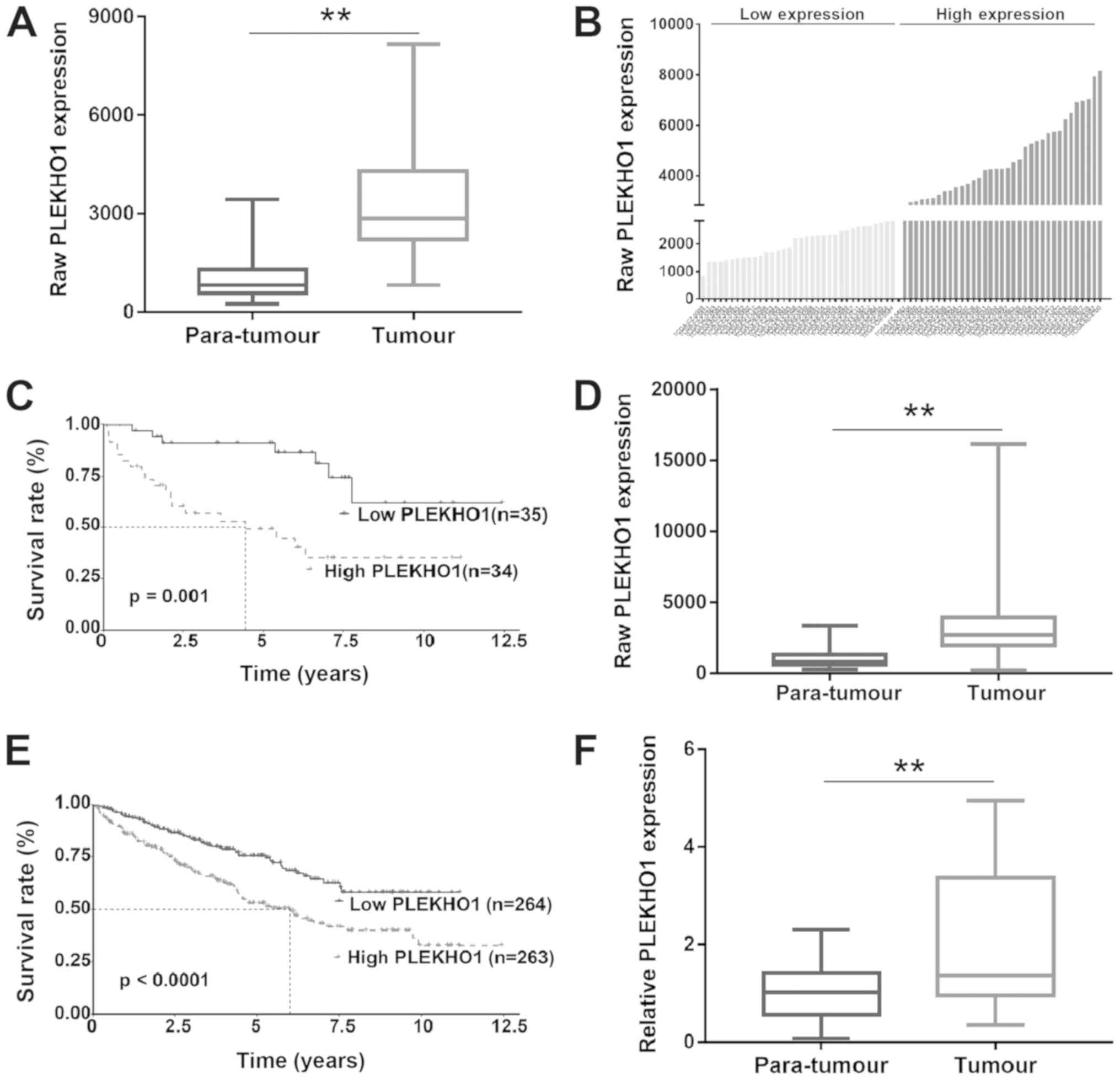
Upregulation of PLEKHO1 expression in RCC
samples is associated with poor prognosis
It has been reported that PLEKHO1 expression is
downregulated in some cancers, such as colon cancer (23), breast cancer (24) and gastric cancer (38). In contrast, in the present study,
by researching TCGA data, the authors found that PLEKHO1 expression
was upregulated in RCC tissue compared with normal para-tumour
tissue (Fig. 2A). To further
assess the correlation between PLEKHO1 mRNA expression and
clinicopathological characteristics, the 69 patients with primary
RCC were divided into two groups by using the median value (raw
expression, 3,005) of PLEKHO1 mRNA expression as the cut-off
(Fig. 2B). As shown in Fig. 2C, the overall survival rate of
patients in the low-PLEKHO1 group was significantly higher than
that of patients in the high-PLEKHO1 group. To test this
hypothesis, the RNA-seq data of an additional 458 unpaired RCC
samples from TCGA database were downloaded. The survival analysis
of this dataset containing 527 RCC patients showed a similar
result, in which PLEKHO1 expression was increased in tumour tissues
compared with 69 normal para-tumour tissues and patients with high
PLEKHO1 expression exhibited a worse prognosis compared with those
with low PLEKHO1 expression (Fig. 2D
and E). Next, the mRNA expression level of PLEKHO1 was detected
by RT-qPCR in 30 paired localized RCC and para-tumour normal
tissues. As shown in Fig. 2F,
PLEKHO1 expression was indeed significantly upregulated in the RCC
tissue samples compared with the para-tumour normal tissue samples.
These results suggest that the aberrant expression of PLEKHO1 may
contribute to the development of RCC.
Reduction of PLEKHO1 mRNA expression
inhibits cell proliferation in RCC
To explore the functional role of PLEKHO1 in RCC,
several cell models were tested. As shown in Fig. 3A, the mRNA level of PLEKHO1 was
high in all five tested cell lines, including 786-O and Caki-1
cells. Then, one shRNA and two siRNAs that specifically target
PLEKHO1 were transfected into 786-O and Caki-1 cells either
permanently or transiently. At 48 h after transfection, both the
mRNA and protein levels of PLEKHO1 were effectively reduced by
transfection of either the shRNA or siRNAs (Fig. 3B and Fig. S1).
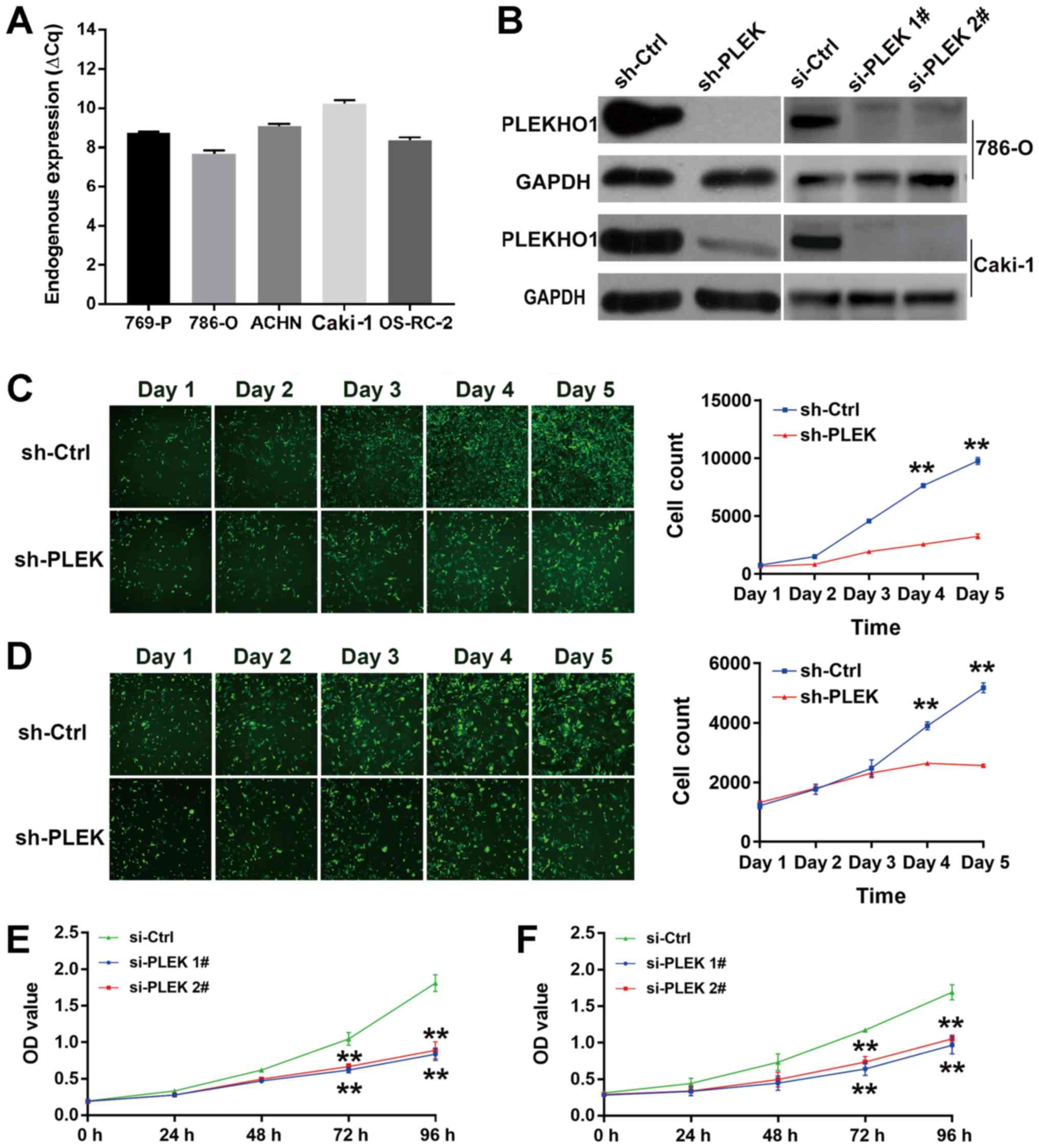 | Figure 3PLEKHO1 downregulation inhibits RCC
cell viability. (A) Endogenous PLEKHO1 expression in five RCC cell
lines (769-P, 786-O, ACHN, Caki-1, OS-RC-2) was detected by
RT-qPCR. (B) The efficacy of shRNA and siRNA in 786-O or Caki-1
cells was detected by western blotting. Fluorescence analysis was
used to determine the viability of sh-PLEK-transfected and
sh-Ctrl-transfected cancer cells: Representative fluorescence
images (left) and the cell growth curve (right) of sh-PLEK and
sh-Ctrl (C) 786-O and (D) Caki-1 cells. The cell count is presented
as mean ± standard deviation; N=3. **P<0.01 vs.
sh-Ctrl. Cell Counting Kit-8 assay showed that the viability of (E)
786-O and (F) Caki-1 cells after the transfection of si-PLEK1#,
si-PLEK 2# and si-Ctrl. The OD value is presented as mean ±
standard deviation; N=3. **P<0.01 vs. si-Ctrl. RCC,
renal cell carcinoma; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; sh, short hairpin; si, small
interfering; Ctrl, negative control; PLEKHO1 and PLEK, pleckstrin
homology domain containing O1; OD, optical density. |
Cells with stable knockdown of PLEKHO1 expression by
the shRNA were subjected to a Celigo cell viability analysis. As
expected, 786-O and Caki-1 cell proliferation was significantly
suppressed by sh-PLEK transfection compared with sh-Ctrl
transfection on days 4 and 5 (Figs.
3C and 2D). To further
validate the results, a CCK-8 assay was performed with 786-O and
Caki-1 cells after treatment with si-Ctrl or two different siRNAs
targeting PLEKHO1 (si-PLEK 1# and si-PLEK 2#). Again, treatment
with both siRNAs (si-PLEK 1# and si-PLEK 2#) significantly
inhibited cell viability in the two cell lines in comparison with
si-Ctrl treatment at 72 and 96 h (Fig.
3E and F). Together, these results suggest that PLEKHO1 may
play a role in RCC cell viability.
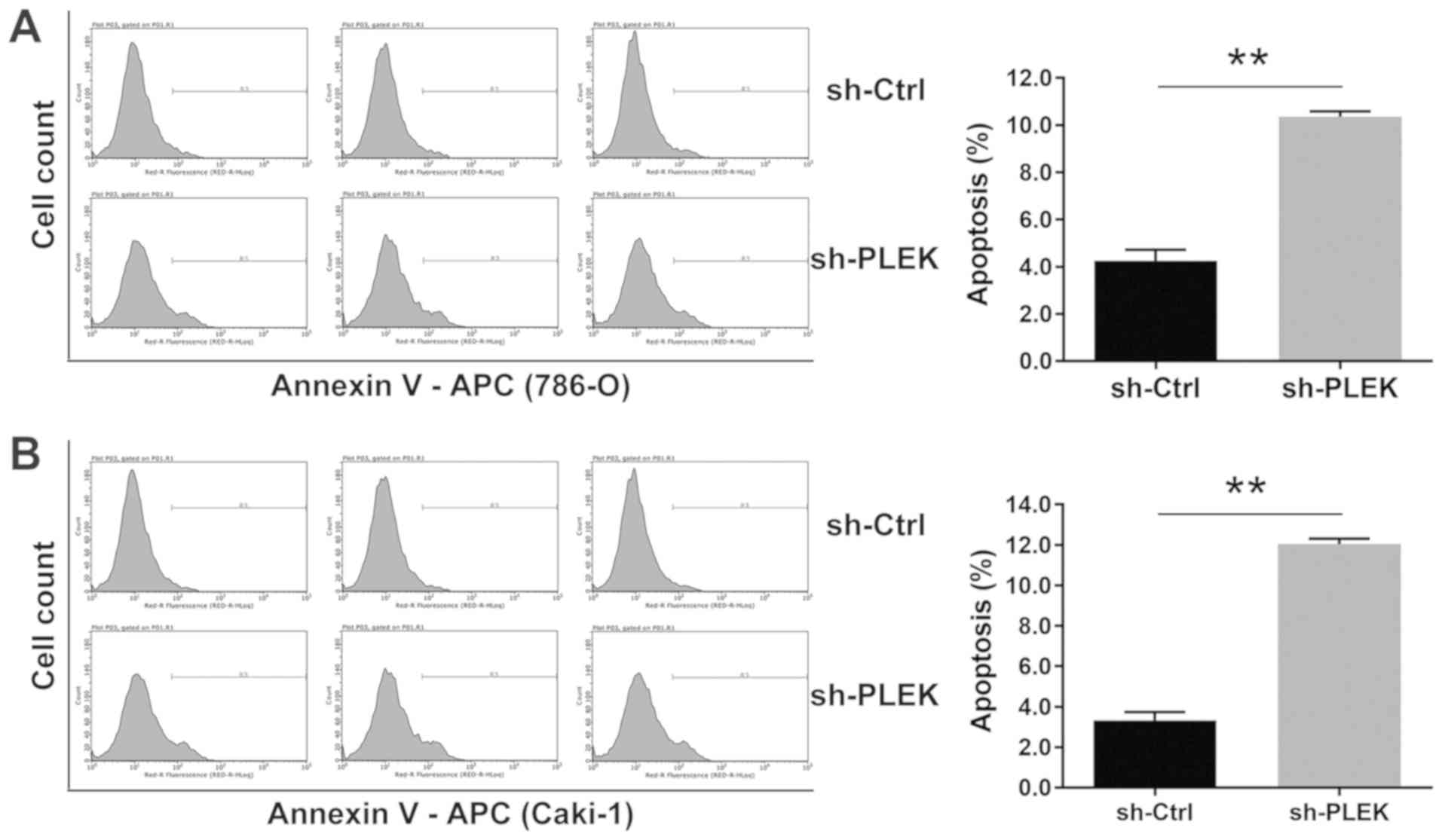
Downregulation of PLEKHO1 expression
induces cell apoptosis
As decreasing the expression level of PLEKHO1
greatly inhibited cell viability, it was hypothesised that PLEKHO1
may affect cell apoptosis in RCC cell lines. Indeed, a
flow-cytometric analysis with Annexin V-APC staining showed that
the percentage of apoptotic cells in cells transfected with sh-PLEK
or sh-Ctrl was 10.36 and 4.25%, respectively, in 786-O cells
(Fig. 4A), and 12.05 and 3.33%,
respectively, in Caki-1 cells (Fig.
4B). The apoptosis rate was significantly higher in cells
transfected with sh-PLEK compared with those transfected with
sh-Ctrl. Thus, these results suggest that PLEKHO1 affects the
viability of RCC cells at least in part by regulating cell
apoptosis.
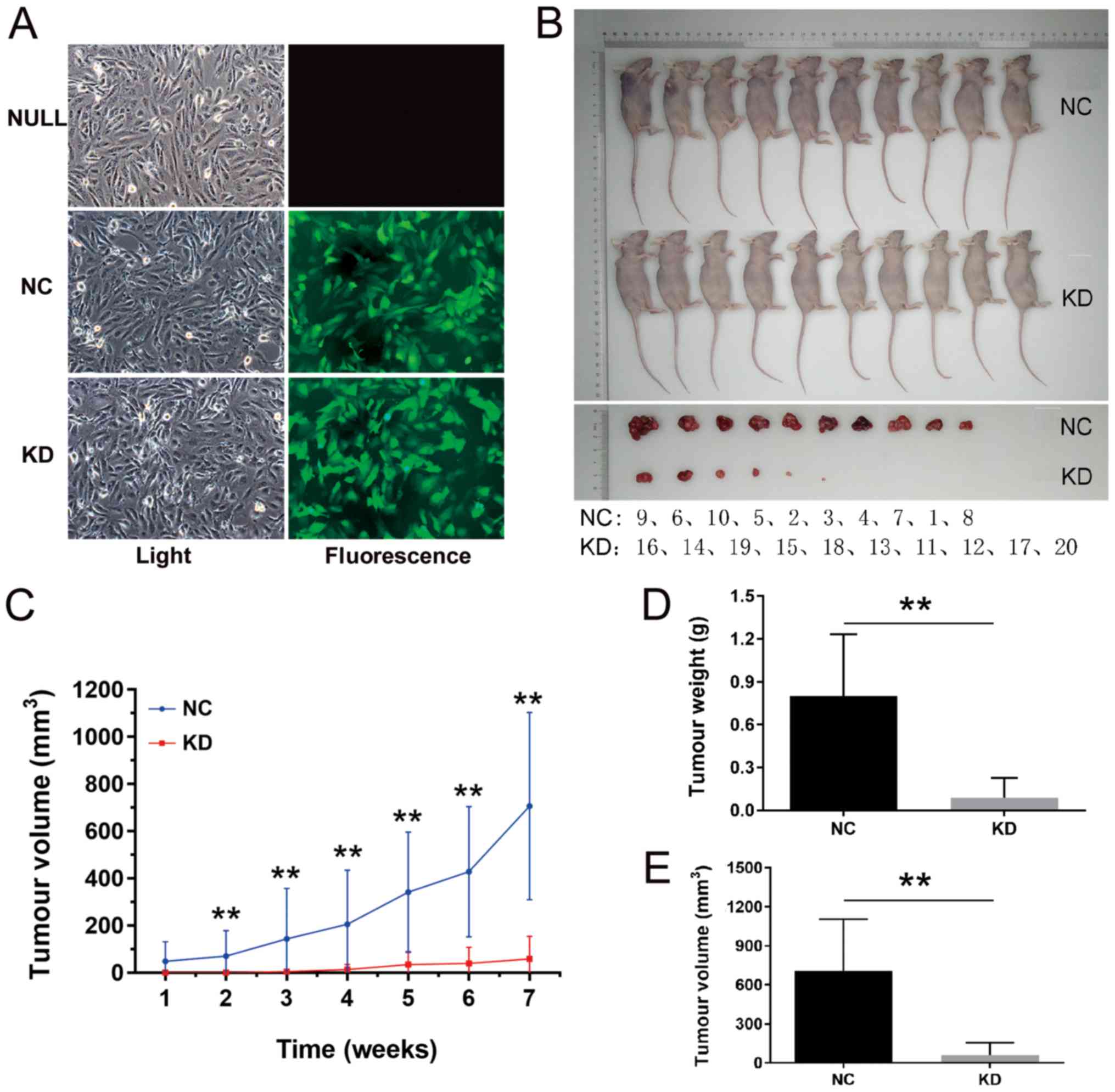
Reducing the expression level of PLEKHO1
attenuates RCC tumour growth in vivo
To further investigate whether PLEKHO1 affects RCC
tumour growth in vivo, a xenograft mouse model was used.
Compared with the NULL group, cell morphology in the NC and KD
groups did not markedly change (Fig.
5A). At the same time, a high transfection efficiency was
confirmed with observation under a fluorescence microscope. As
shown in Fig. 5B, at the end of
the experiment, small tumours formed in only a few mice in the KD
group. While in the NC group, xenograft tumours developed in every
mouse in which the longest length of these tumours was 19.04 mm
(Fig. 5B). Additionally, during
the experiment, the tumour growth in the KD group was significantly
slower compared with that in the NC group from week 2 to week 7
(Fig. 5C). Accordingly, the tumour
weight and tumour volume were both significantly lower in the KD
group compared with the NC group (Fig.
5D and E). These results support the conclusion that PLEKHO1
expression is significantly associated with the proliferative
capacity of 786-O cells in vivo.
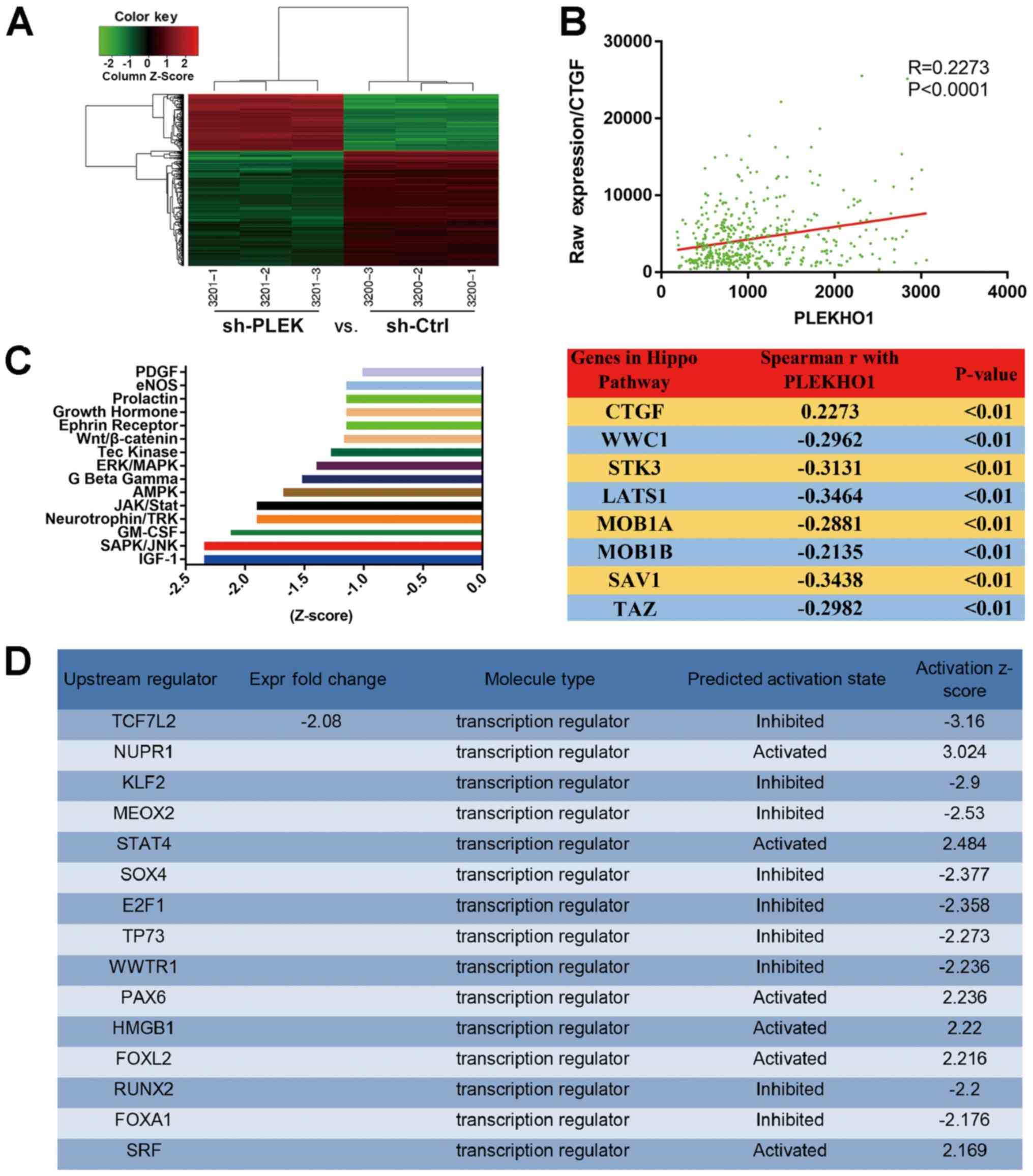
PLEKHO1 may be involved in regulating the
serine/threonine-protein kinase hippo and JNK signalling
pathways
To further explore the mechanisms underlying the
impact of PLEKHO1 on RCC cell viability, total RNA was extracted
from 786-O cells with or without knockdown of PLEKHO1 expression
and the RNA was subjected to DNA microarray analysis. Upon reducing
the expression of PLEKHO1, 196 genes were upregulated, while 403
genes were downregulated (Fig. 6A
and Table SIV). Subsequently,
upstream IPA showed that PLEKHO1 downregulation impacted the
expression of several transcription regulators, including WW domain
containing transcription regulator 1 (WWTR1, also known as TAZ;
Fig. 6D and Table SV). Additionally, through
co-expression analysis on TCGA dataset, it was found that
connective tissue growth factor (CTGF), a downstream molecule of
TAZ in the Hippo signalling axis, was positively correlated with
the expression of PLEKHO1, but negatively correlated with most of
the key factors in the Hippo signalling pathway (28,29)
(Fig. 6B). Therefore, the authors
postulated that PLEKHO1 may function by participating in the Hippo
signalling pathway.
In addition to the upstream analysis based on the
microarray data, IPA of canonical pathway was performed. Knockdown
of PLEKHO1 expression greatly repressed the canonical insulin-like
growth factor 1, stress-activated protein kinase (SAPK)/JNK and
granulocyte-macrophage colony-stimulating factor signalling
pathways (Fig. 6C and Table SVI). As shown in Fig. 6C, the top 15 pathways were highly
inclined to be inactivated and JNK was the most canonical and
obvious pathway with one of the most negative Z-scores. These
results suggest that PLEKHO1 potentially functions through the JNK
and Hippo signalling pathways, although further studies are clearly
needed.
Discussion
Since PLEKHO1 was first reported, its structural and
localization characteristics, and essential functional roles in
biological processes have been explored over the past decade
(8,9,12-14,17,18).
It has been shown that PLEKHO1 expression is downregulated in some
cancers and that PLEKHO1 is involved in some key signalling
pathways in cancer cells (12,13,23,24,38).
For example, Tokuda et al (12) reported that PLEKHO1 interacted with
Akt through its LZ motif and inhibited PI3K/Akt signalling. Another
study found that PLEKHO1 disturbed the endogenous protein level of
E3 ubiquitin-protein ligase SMURF1 (Smurf1) by regulating
PI3K/Akt/mTOR signalling and therefore promoted the
auto-degradation of Smurf1 in colon cancer (23). PLEKHO1 clearly functions as a
tumour suppressor gene in these cancers. However, in the present
study, aberrant overexpression of PLEKHO1 in RCC tissue samples was
observed compared with para-tumour normal kidney tissue samples and
found that the downregulation of PLEKHO1 gene expression markedly
compromised RCC cell viability. Additionally, reducing PLEKHO1
expression significantly impeded the growth of xenograft tumours in
mouse models. Thus, the present results suggest that, at least in
RCC, PLEKHO1 promotes cancer development.
It has been reported that PLEKHO1 protein is cleaved
into fragments by caspase-3 and that these fragments promote
apoptosis through inhibiting the anti-apoptotic activity of c-Jun
(13). In contrast, the present
study found that PLEKHO1 may protect RCC cells from apoptosis
because downregulating the expression of PLEKHO1 induces apoptosis.
Thus, it is speculated that PLEKHO1 may function in a
context-dependent manner in different cancers through diverse
mechanisms.
Furthermore, a gene expression array was performed
and it was found that the downregulation of PLEKHO1 impacted the
expression of numerous transcription factors, including TAZ. It is
known that dephosphorylated TAZ can translocate into the nucleus
and function as a co-activator along with its partner AP-1-like
transcription factor YAP1 to regulate the expression of CTGF, which
is negatively modulated by the Hippo signalling axis (39-41).
In mammals, the Hippo signalling pathway plays important roles in
organ and cancer development (42). As the main effector of the Hippo
pathway, TAZ is destabilized and restricted to the cytoplasm, and
therefore loses its transcriptional function when it is
phosphorylated by the activated Hippo signalling cascade (43-45).
The present co-expression analysis based on TCGA data showed that
high expression levels of PLEKHO1 may suppress Hippo signalling and
increase endogenous CTGF expression, which is consistent with the
result of the microarray analysis. In this context, the authors
hypothesized that aberrant PLEKHO1 expression may inhibit the Hippo
signalling pathway and activate the activity of TAZ.
Additionally, IPA of canonical pathways showed that
PLEKHO1 may interfere with the SAPK/JNK signalling pathway. With
sh-PLEK downregulating the level of PLEKHO1 expression, it was
found that SAPK/JNK was probably inactivated due to the low
Z-score. As a member of the mitogen-activated protein kinase
signalling network, the JNK signalling pathway has been proposed to
play pivotal roles in cell proliferation, differentiation, death
and survival (46). Deregulation
of this signalling pathway has been reported in various disease
conditions, such as malignancies, diabetes, inflammation and
neurodegenerative diseases (46).
However, JNK signalling appears to play opposing roles in different
cancers. For example, JNK signalling is hyperactivated and promotes
cancer development in hepatocellular carcinoma (47,48),
lung cancer (49,50), myeloma (51) and skin carcinoma (52). On the other hand, JNK signalling
functions as a tumour suppressor in melanoma, lymphoma and chronic
myeloid leukaemia (53,54). In the present study, PLEKHO1
knockdown distinctly inhibited RCC cell viability. Simultaneously,
microarray analysis showed that PLEKHO1 knockdown potentially
restrained the JNK signalling pathway due to the low Z-score.
Therefore, the authors of the current study preliminarily
hypothesised that JNK functions as a tumour promoter, which
mediated PLEKHO1 function and that PLEKHO1 impacts cell viability
in RCC partly via regulating the JNK signalling pathway. However,
further studies are warranted.
In conclusion, the present study sheds light on the
promotive effect of PLEKHO1 on cell viability in RCC, although this
protein was speculated to be a tumour suppressor in some other
cancers. Thus, further studies are needed to explore the potential
of PLEKHO1 as a cancer biomarker as well as a therapeutic target
specific for RCC.
Supplementary Materials
Funding
This study is partially supported by the National
Natural Science Foundation of China (grant nos. 81372766 and
81572532), and the Liaoning 'Climbing' scholarship.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ZY and QL conducted the experiments. GZ, CL, QD, and
CF participated in data collection and analysis. CK and YZ
participated in the design of the study. ZY and YZ participated in
the writing of the manuscript and data interpretation. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Experiments using tissue samples from human subjects
were approved by the Ethics Committee of China Medical University
(Shenyang, China). All participants provided written informed
consent for the whole study. Experiments on animals were performed
following approval from the Animal Ethical and Welfare Committee of
China Medical University.
Patient consent for publication
All participants provided written informed consent
for the whole study.
Competing interests
No competing interests.
Acknowledgments
Not applicable
References
|
1
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: Cancer Incidence and Mortality World wide
GLOBOCAN2012.v10.2013. https://publications.iarc.fr/Databases/Iarc-Cancerbases/GLOBOCAN-2012-Estimated-Cancer-Incidence-Mortality-And-Prevalence-Worldwide-In-2012-V1.0-2012.
|
|
2
|
Ljungberg B, Cowan NC, Hanbury DC, Hora M,
Kuczyk MA, Merseburger AS, Patard JJ, Mulders PF and Sinescu IC;
European Association of Urology Guideline Group: EAU guidelines on
renal cell carcinoma: The 2010 update. Eur Urol. 58:398–406. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rini BI, Campbell SC and Escudier B: Renal
cell carcinoma. Lancet. 373:1119–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walsh N, Larkin A, Kennedy S, Connolly L,
Ballot J, Ooi W, Gullo G, Crown J, Clynes M and O'Driscoll L:
Expression of multidrug resistance markers ABCB1 (MDR-1/P-gp) and
ABCC1 (MRP-1) in renal cell carcinoma. BMC Urol. 9:62009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Poletto V, Rosti V, Biggiogera M, Guerra
G, Moccia F and Porta C: The role of endothelial colony forming
cells in kidney cancer's pathogenesis, and in resistance to
anti-VEGFR agents and mTOR inhibitors: A speculative review. Crit
Rev Oncol Hematol. 132:89–99. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bedke J, Stühler V, Stenzl A and Brehmer
B: Immunotherapy for kidney cancer: Status quo and the future. Curr
Opin Urol. 28:8–14. 2018.
|
|
7
|
Tomczak K, Czerwińska P and Wiznerowicz M:
The Cancer Genome Atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19A:A68–A77. 2015.
|
|
8
|
Bosc DG, Graham KC, Saulnier RB, Zhang C,
Prober D, Gietz RD and Litchfield DW: Identification and
characterization of CKIP-1, a novel pleckstrin homology
domain-containing protein that interacts with protein kinase CK2. J
Biol Chem. 275:14295–14306. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Safi A, Vandromme M, Caussanel S, Valdacci
L, Baas D, Vidal M, Brun G, Schaeffer L and Goillot E: Role for the
pleckstrin homology domain-containing protein CKIP-1 in
phosphatidylinositol 3-kinase-regulated muscle differentiation. Mol
Cell Biol. 24:1245–1255. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Olsten ME, Canton DA, Zhang C, Walton PA
and Litchfield DW: The Pleckstrin homology domain of CK2
interacting protein-1 is required for interactions and recruitment
of protein kinase CK2 to the plasma membrane. J Biol Chem.
279:42114–42127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nie J, Liu L, He F, Fu X, Han W and Zhang
L: CKIP-1: A scaffold protein and potential therapeutic target
integrating multiple signaling pathways and physiological
functions. Ageing Res Rev. 12:276–281. 2013. View Article : Google Scholar
|
|
12
|
Tokuda E, Fujita N, Oh-hara T, Sato S,
Kurata A, Katayama R, Itoh T, Takenawa T, Miyazono K and Tsuruo T:
Casein kinase 2-interacting protein-1, a novel Akt pleckstrin
homology domain-interacting protein, down-regulates PI3K/Akt
signaling and suppresses tumor growth in vivo. Cancer Res.
67:9666–9676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang L, Xing G, Tie Y, Tang Y, Tian C, Li
L, Sun L, Wei H, Zhu Y and He F: Role for the pleckstrin homology
domain-containing protein CKIP-1 in AP-1 regulation and apoptosis.
EMBO J. 24:766–778. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Canton DA, Olsten ME, Kim K, Doherty-Kirby
A, Lajoie G, Cooper JA and Litchfield DW: The pleckstrin homology
domain-containing protein CKIP-1 is involved in regulation of cell
morphology and the actin cytoskeleton and interaction with actin
capping protein. Mol Cell Biol. 25:3519–3534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Canton DA, Olsten ME, Niederstrasser H,
Cooper JA and Litchfield DW: The role of CKIP-1 in cell morphology
depends on its interaction with actin-capping protein. J Biol Chem.
281:36347–36359. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang L, Xia X, Zhang M, Wang Y, Xing G,
Yin X, Song L, He F and Zhang L: Integrated analysis of genomics
and proteomics reveals that CKIP-1 is a novel macrophage migration
regulator. Biochem Biophys Res Commun. 436:382–387. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Wang Y, Xiao F, Wang S, Xing G,
Li Y, Yin X, Lu K, Wei R, Fan J, et al: CKIP-1 regulates macrophage
proliferation by inhibiting TRAF6-mediated Akt activation. Cell
Res. 24:742–761. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu K, Yin X, Weng T, Xi S, Li L, Xing G,
Cheng X, Yang X, Zhang L and He F: Targeting WW domains linker of
HECT-type ubiquitin ligase Smurf1 for activation by CKIP-1. Nat
Cell Biol. 10:994–1002. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Y, Nie J, Wang Y and Zhang L, Lu K,
Xing G, Xie P, He F and Zhang L: CKIP-1 couples Smurf1 ubiquitin
ligase with Rpt6 subunit of proteasome to promote substrate
degradation. EMBO Rep. 13:1004–1011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang P, Zhou C, Lu C, Li W, Li W, Jing B,
Chen W, Zha Y, Zhang P, Bai C, et al: PLEKHO2 is essential for
M-CSF-dependent macrophage survival. Cell Signal. 37:115–122. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang G, Guo B, Wu H, Tang T, Zhang BT,
Zheng L, He Y, Yang Z, Pan X, Chow H, et al: A delivery system
targeting bone formation surfaces to facilitate RNAi-based anabolic
therapy. Nat Med. 18:307–314. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koskimaki JE, Rosca EV, Rivera CG, Lee E,
Chen W, Pandey NB and Popel AS: Serpin-derived peptides are
antiangiogenic and suppress breast tumor xenograft growth. Transl
Oncol. 5:92–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nie J, Liu L, Xing G, Zhang M, Wei R, Guo
M, Li X, Xie P, Li L, He F, et al: CKIP-1 acts as a colonic tumor
suppressor by repressing oncogenic Smurf1 synthesis and promoting
Smurf1 autodegradation. Oncogene. 33:3677–3687. 2014. View Article : Google Scholar
|
|
24
|
Alvarez C, Aravena A, Tapia T, Rozenblum
E, Solís L, Corvalán A, Camus M, Alvarez M, Munroe D, Maass A, et
al: Different Array CGH profiles within hereditary breast cancer
tumors associated to BRCA1 expression and overall survival. BMC
Cancer. 16:2192016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gong W, Li J, Chen Z, Huang J, Chen Q, Cai
W, Liu P and Huang H: Polydatin promotes Nrf2-ARE anti-oxidative
pathway through activating CKIP-1 to resist HG-induced
up-regulation of FN and ICAM-1 in GMCs and diabetic mice kidneys.
Free Radic Biol Med. 106:393–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhan Y, Xie P, Li D, Li L, Chen J, An W,
Zhang L and Zhang C: Deficiency of CKIP-1 aggravates high-fat
diet-induced fatty liver in mice. Exp Cell Res. 355:40–46. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burgess MR, Hwang E, Mroue R, Bielski CM,
Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L,
et al: KRAS allelic imbalance enhances fitness and modulates MAP
kinase dependence in cancer. Cell. 168:817–829. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBio-Portal. Sci Signal. 6:112013. View Article : Google Scholar
|
|
30
|
Vinci M, Gowan S, Boxall F, Patterson L,
Zimmermann M, Court W, Lomas C, Mendiola M, Hardisson D and Eccles
SA: Advances in establishment and analysis of three-dimensional
tumor spheroid-based functional assays for target validation and
drug evaluation. BMC Biol. 10:292012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng L, Sun X, Csizmadia E, Han L, Bian S,
Murakami T, Wang X, Robson SC and Wu Y: Vascular CD39/ENTPD1
directly promotes tumor cell growth by scavenging extracellular
adenosine triphosphate. Neoplasia. 13:206–216. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Leary S: AVMA Guidelines for the
Euthanasia of Animals: 2013 Edition. 2013, https://www.avma.org/KB/Policies/Documents/euthanasia.pdf.
|
|
34
|
National Research Council: Guide for the
Care and Use of Laboratory Animals: Eighth Edition. The National
Academies Press; Washington, DC: 2011, https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf.
Ilar Journal 56 NP-NP, 2015 https://doi.org/10.1093/ilar/ilv024.
|
|
35
|
Patel SJ, Sanjana NE, Kishton RJ,
Eidizadeh A, Vodnala SK, Cam M, Gartner JJ, Jia L, Steinberg SM,
Yamamoto TN, et al: Identification of essential genes for cancer
immunotherapy. Nature. 548:537–542. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J,
Liang X, Chen JR, Shah S, Shang X, et al: Synthetic essentiality of
chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature.
542:484–488. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gong M, Xu Y, Dong W, Guo G, Ni W, Wang Y,
Wang Y and An R: Expression of Opa interacting protein 5 (OIP5) is
associated with tumor stage and prognosis of clear cell renal cell
carcinoma. Acta Histochem. 115:810–815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ma HW, Xie M, Sun M, Chen TY, Jin RR, Ma
TS, Chen QN, Zhang EB, He XZ, De W, et al: The pseudogene derived
long noncoding RNA DUXAP8 promotes gastric cancer cell
proliferation and migration via epigenetically silencing PLEKHO1
expression. Oncotarget. 8:52211–52224. 2016.
|
|
39
|
Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu
J, Lin JD, Wang CY, Chinnaiyan AM, et al: TEAD mediates
YAP-dependent gene induction and growth control. Genes Dev.
22:1962–1971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hao Y, Chun A, Cheung K, Rashidi B and
Yang X: Tumor suppressor LATS1 is a negative regulator of oncogene
YAP. J Biol Chem. 283:5496–5509. 2008. View Article : Google Scholar
|
|
41
|
Zhao B, Li L, Tumaneng K, Wang CY and Guan
KL: A coordinated phosphorylation by Lats and CK1 regulates YAP
stability through SCF(beta-TRCP). Genes Dev. 24:72–85. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maugeri-Saccà M and De Maria R: The Hippo
pathway in normal development and cancer. Pharmacol Ther.
186:60–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ando T, Charindra D, Shrestha M, Umehara
H, Ogawa I, Miyauchi M and Takata T: Tissue inhibitor of
metalloproteinase-1 promotes cell proliferation through YAP/TAZ
activation in cancer. Oncogene. 37:263–270. 2018. View Article : Google Scholar
|
|
44
|
Lei Q-Y, Zhang H, Zhao B, Zha ZY, Bai F,
Pei XH, Zhao S, Xiong Y and Guan KL: TAZ promotes cell
proliferation and epithelial-mesenchymal transition and is
inhibited by the hippo pathway. Mol Cell Biol. 28:2426–2436. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu C-Y, Zha Z-Y, Zhou X, Zhang H, Huang
W, Zhao D, Li T, Chan SW, Lim CJ, Hong W, et al: The hippo tumor
pathway promotes TAZ degradation by phosphorylating a phosphodegron
and recruiting the SCF{beta}-TrCP E3 ligase. J Biol Chem.
285:37159–37169. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bubici C and Papa S: JNK signalling in
cancer: In need of new, smarter therapeutic targets. Br J
Pharmacol. 171:24–37. 2014. View Article : Google Scholar :
|
|
47
|
Chang Q, Zhang Y, Beezhold KJ, Bhatia D,
Zhao H, Chen J, Castranova V, Shi X and Chen F: Sustained JNK1
activation is associated with altered histone H3 methylations in
human liver cancer. J Hepatol. 50:323–333. 2009. View Article : Google Scholar
|
|
48
|
Barbarulo A, Iansante V, Chaidos A, Naresh
K, Rahemtulla A, Franzoso G, Karadimitris A, Haskard DO, Papa S and
Bubici C: Poly(ADP-ribose) polymerase family member 14 (PARP14) is
a novel effector of the JNK2-dependent pro-survival signal in
multiple myeloma. Oncogene. 32:4231–4242. 2013. View Article : Google Scholar
|
|
49
|
Takahashi H, Ogata H, Nishigaki R, Broide
DH and Karin M: Tobacco smoke promotes lung tumorigenesis by
triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell.
17:89–97. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Luan L, Zhao Y, Xu Z, Jiang G, Zhang X,
Fan C, Liu D, Zhao H, Xu K, Wang M, et al: Diversin increases the
proliferation and invasion ability of non-small-cell lung cancer
cells via JNK pathway. Cancer Lett. 344:232–238. 2014. View Article : Google Scholar
|
|
51
|
Liu Q, Tao B, Liu G, Chen G, Zhu Q, Yu Y,
Yu Y and Xiong H: Thromboxane A2 receptor inhibition suppresses
multiple myeloma cell proliferation by inducing p38/c-Jun
N-terminal kinase (JNK) mitogen-activated protein kinase
(MAPK)-mediated G2/M progression delay and cell apoptosis. J Biol
Chem. 291:4779–4792. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Alameda JP, Fernández-Aceñero MJ,
Moreno-Maldonado R, Navarro M, Quintana R, Page A, Ramírez A, Bravo
A and Casanova ML: CYLD regulates keratinocyte differentiation and
skin cancer progression in humans. Cell Death Dis. 2:e2082011.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gao Y, Tao J, Li MO, Zhang D, Chi H,
Henegariu O, Kaech SM, Davis RJ, Flavell RA and Yin Z: JNK1 is
essential for CD8+ T cell-mediated tumor immune
surveillance. J Immunol. 175:5783–5789. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Xiao X, Jiang K, Xu Y, Peng H, Wang Z, Liu
S and Zhang G: (-)-Epigallocatechin-3-gallate induces cell
apoptosis in chronic myeloid leukaemia by regulating
Bcr/Abl-mediated p38-MAPK/JNK and JAK2/STAT3/AKT signalling
pathways. Clin Exp Pharmacol Physiol. 46:126–136. 2019. View Article : Google Scholar
|