Introduction
Multiple myeloma (MM) is the second most frequent
hematological malignancy and represents 1% of all types of types of
cancer. MM is a malignant hematological disorder characterized by
the accumulation of abnormal plasma cells in the bone marrow, the
secretion of monoclonal immunoglobulin (Ig) into the serum and
urine, and the absence of IgM expression. Symptomatic MM exhibits
characteristics, such as hypercalcemia, renal failure, osteopenic
or osteolytic bone disease and anemia; however, patients with MM
may be diagnosed at an asymptomatic stage by chance (1). The pathogenesis of MM is
characterized by the progressive acquisition of genetic lesions,
such as translocations, deletions and mutations in the regulating
genes of plasma cells, promoting, in the early stage, binding with
bone marrow stromal cells. Nuclear factor (NF)-kB is activated by the
mutations that occur during the progression of MM. It upregulates
cell surface adhesion molecules and increases growth,
anti-apoptotic and angiogenic cytokine production, including
interleukin (IL)-6, tumor necrosis factor-α, insulin-like growth
factor-1 and vascular endothelial growth factor (VEGF), in addition
to the secretion of IL-10 and transforming growth factor-β
secretion by tumor plasma cells, leading to the dysregulation of B-
and T-cells immune surveillance. This whole process supports the
survival, proliferation and chemoresistance of tumor plasma cells
(2,3).
Despite recent improvements in treatment leading to
significantly increased overall survival rates, the majority of
patients with MM have a differential level of residual disease,
leading to cyclic relapse and finally, to refractory disease
(4). Following disease relapse,
the main therapies are predominantly old classes of drugs,
including corticosteroids [e.g., prednisone and dexamethasone
(Dex)] used individually, and novel active classes of drugs,
including proteasome inhibitors [bortezomib (BTZ) and carfilzomib]
and immunomodulatory drugs (IMiDs), in combination with each other
or in combination with other less active agents (5,6). The
IMiD class of drugs, including lenalidomide (Len) and pomalidomide
(Pom), exhibit potent anti-myeloma properties in various MM models,
and have shown significant clinical activity in patients with MM.
Len is a 4-aminoglutarimide derivative of thalidomide, which
possesses markedly fewer neurologic toxic effects associated with
thalidomide (7). It has
pro-apoptotic and antiproliferative effects, boosts antitumor
immunity via T-cell proliferation, IL-2 and interferon-γ production
and the activation of cell adhesion molecules, and promotes
antiangiogenic activity by reducing VEGF and fibroblast growth
factor (FGF) production by the endothelium and bone marrow stroma
(7-11). Recent studies have demonstrated
that thalidomide and other IMiDs directly bind cereblon (CRBN), an
E3 ubiquitin ligase that controls the ubiquitination and
degradation of IKAROS family zinc finger (IKZF)1 (known as Ikaros)
and IKZF3 (known as Aiolos) (12-14).
The reduced expression of IKZF1/IKZF3 leads to decreased levels of
NF-κB, c-Myc and interferon regulatory factor 4 (IRF4), which play
a pivotal role in the pathogenesis of MM, and the increased
expression of p21, resulting in anti-MM activity in a CRBN- and
p21-dependent, but p53-indendent manner (15). Similar to thalidomide and Len, Pom
exerts its antitumor activity by direct antiproliferative and
pro-apoptotic effects on plasma cells, by immunomodulation, and by
the modulation of the bone marrow microenvironment. Importantly,
Pom has been shown to be more active than the two previously
mentioned IMiDs (16).
HDAC inhibitors (HDACis) have emerged as promising
agents for the treatment of MM and other tumor types (17-19).
HDACis kill cells through multiple mechanisms (20). Two pan-HDACis, suberoylanilide
hydroxamic acid (SAHA or vorinostat) and romidepsin, have been
approved by the US Food and Drug Administration for the treatment
of cutaneous T cell lymphoma (21-23).
Previous studies have shown the potential clinical activity of a
combination of the non-selective HDACi, SAHA (24), or LBH589 (panobinostat) (25) and Dex with Len or BTZ (26) in patients with MM. Recently,
panobinostat was approved as a combination drug regimen (BTZ + Dex)
for the treatment of MM; however, it exhibits serious toxicities
that limits its clinical utility (27). Therefore, more tolerable HDAC
inhibitors are required. As MM cells are sensitive to HDAC6
inhibition, HDAC6-selective inhibitors are attractive candidates
for MM treatment. HDAC6-selective inhibitors target the aggresome
and proteasome protein degradation pathways without substantially
altering gene expression and may thus have an improved safety
profile compared with pan-HDACis. Several HDAC6-selective
inhibitors have been previously reported (28-32).
ACY-1215 (ricolinostat) is the only first-in-class clinically
relevant HDAC6i with minimal effects on class I HDACs (31,33).
Combination treatment with ricolinostat and Dex with Len (34) or BTZ (35) is under clinical assessment in
relapsed/refractory (R/R) MM. Therefore, there is a need for the
further development of HDAC6-selective inhibitors that do not
produce side-effects, such as diarrhea, fatigue, nausea, vomiting,
neutropenia and thrombocytopenia, due to non-selective class I HDAC
inhibition.
In our recent study, we developed an HDAC6-selective
inhibitor A452 (36), which also
exerts significant cell growth inhibition and decreases cell
viability in various cancer cells (37) and in vivo murine xenograft
colon cancer model (36).
Therefore, the present study aimed to examine whether the novel
HDAC6-selective HDACi, A452, together with IMiDs induces
synergistic anti-MM activity, in order to provide the rationale for
combination clinical trials. The present study also investigated
whether A452 in combination with IMiD overcomes drug resistance in
MM.
Materials and methods
Cells and cell culture
The Dex-sensitive, MM.1S (CRL-2974), and
Dex-resistant, H929 (CRL-9068), MM cell lines and bone marrow
derived mesenchymal stromal cells (BM-MSCs; PCS-500-012) were
purchased from the American Type Culture Collection (ATCC). The
cells were cultured in medium (HyClone; GE Healthcare Life
Sciences) containing 10% fetal bovine serum (HyClone; GE Healthcare
Life Sciences), 100 U/ml penicillin and 100 μg/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere of 5% CO2 and 95% air at 37°C.
Reagents
SAHA and Dex were purchased from Sigma-Aldrich
(Merck KGaA). Bortezomib, Len, Pom, and ACY-1215 were purchased
from Selleck Chemicals. A452 (purity 99%) is a γ-lactam based HDAC6
inhibitor (36) and was kindly
provided by Dr Gyoonhee Han (Yonsei University).
Cell growth and viability assay
Each cell culture of MM cells was performed in
triplicate, and cell growth and viability were determined using
Cell Counting kit (CCK)-8 kit assays (Dojindo Molecular
Technologies, Inc.) according to the manufacturer's protocol, as
described previously (38).
Inhibitory assays
Drug concentrations that inhibited 50% of cell
growth (GI50) and 50% of cell viability
(IC50) were determined using a CCK-8 assay. All cell
lines were treated for 72 h on day 2, unless otherwise stated.
gi50 and ic50 were determined using
Prism Version 6.0 software (GraphPad Software, Inc.).
Short hairpin RNA (shRNA) infection and
generation of stable knockdown cell lines
The lentiviral shRNA sets for HDAC6 were purchased
from Sigma-Aldrich; Merck KGaA. The following sequences within
human HDAC6 were targeted: CCGGCATCCCATCCTGAATATCCTTCTCGAGAAGGAT
ATTCAGGATGGGATGTTTTTG(#1,NM_006044.2-3840s1c1 TRCN0000314976) and
CCGGCCTCACTGATCAGGCC ATATTCTCGAGAATATGGCCTGATCAGTGAGGTTTTT (#2,
NM_006044.2-2049s1c1, TRCN0000004843). The non-targeted shRNA
sequence was CCGGGCGCGATAGCG CTAATAATTTCTCGAGAAATTATTAGCGCTATCGCGC
TTTTT (SHC016; Sigma-Aldrich). To generate respective lentivirus,
293T cells (CRL-11268; ATCC) were co-transfected with the shRNA
vector and necessary packaging plasmids. Supernatants containing
lentivirus were collected 48 and 72 h after transfection and passed
through a 0.45-μm filter. The cells were then infected 3
times (every 12 h) with the lentivirus in the presence of
hexadimethrine bromide. Subsequently, cells were selected for 2
days in 2 _μg/ml puromycin, as the pLKO.1 vector encodes the
respective antibiotic resistance gene.
Western blot analysis
The treated cells were collected and lysed with
NP-40 lysis buffer [0.5% NP-40, 50 mM Tris-HCl (pH 7.4), 120 mM
NaCl, 25 mM NaF, 25 mM glycerol phosphate, 1 mM EDTA, 5 mM EGTA, 1
mM PMSF and 1 mM bezamidine]. The protein concentration was
measured using a bicinchoninic acid kit (Pierce; Thermo Fisher
Scientific, Inc.). Cell lysates containing 50-80 μg total
protein were subjected to 8-15% by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Western blot
analysis was performed as described previously (38). The nictrocellulose membranes were
blocked for 1 h in PBS containing 0.1% Tween-20 and 10% (v/v) skim
milk (Bio-Rad Laboratories, Inc.) and incubated overnight at 4°C
with primary antibodies (1:500-1:2,000). The following primary
antibodies were used. Acetylated-α-tubulin (DM1A, T6793-2ML;
1:2,000) was purchased from Sigma-Aldrich (Merck KGaA). Protein
kinase B (AKT; H-136, sc-8312; 1:1,000), phosphorylated (p)-AKT
(Ser473, sc-7985; 1:500), B-cell lymphoma (Bcl)-2 homologous
antagonist/killer (Bak; G-23, sc-832; 1:1,000), extracellular
signal-regulated kinase (ERK; K-23, sc-94; 1:1,000), HDAC6 (H-300,
sc-11420; 1:1,000), IRF4 (M-17, sc-6059, 1:2000), c-Myc (9E10,
sc-40, 1:500), p38 (C-20, sc-535, 1:1,000), and a-tubulin
(sc-32293; 1:2,000) were obtained from Santa Cruz Biotechnology,
Inc. Anti-HIF-1a (610958, 1:500), poly (ADP-ribose) polymerase
(PARP; 551024; 1:1,000), and XIAP (10716, 1:1000) antibodies was
purchased from BD Biosciences. Bax (2772, 1:500), Bcl-2 (2870,
1:500), B-cell lymphoma-extra large protein (Bcl-xL; 2762; 1:500),
caspase-3 (9662S; 1:1,000), caspase-8 (9746, 1:500), caspase-9
(9508, 1:500), p-ERK (Thr202/Tyr204; 4376; 1:1,000), GAPDH (14C10,
2118, 1:1,000), IKZF1 (5443, 1:500), IKZF3 (12720, 1:500), signal
transducer and activator of transcription 3 (STAT3; 12640;
1:1,000), p-STAT3 (Y705, 9138; 1:500), and phospho-p38
(Thr180/Tyr182; 9211, 1:500) were obtained from Cell Signaling
Technology, Inc. Acetylated-histone H3 (06-599, 1:1,000) and
histone H3 (06-755; 1:500) antibodies were obtained from EMD
Millipore. Anti-CRBN antibody (ARP56882-P050, 1:1,000) was
purchased from Aviva Systems Biology. Anti-programmed death-ligand
1 (PD-L1) antibody (PA5-28115; 1:1,000) was purchased from
Invitrogen; Thermo Fisher Scientific, Inc. The membranes were then
washed with 0.1% Tween-20/PBS and incubated at room temperature for
1 h with horseradish peroxidase-conjugated anti-rabbit
(111-035-003; 1:5,000) and anti-mouse (115-035-003; 1:10,000)
secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.).
The bound antibodies were detected using an enhanced
chemiluminescence western blotting analysis system (NCI4080KR,
Thermo Fisher Scientific, Inc.) and the blots were semi-quantified
using FusionCapt software version 16.08a (Viber Lourmat Sté,
Collégien).
Acid extraction of histones
The acid extraction of histones from the MM cells
was performed as previously described (38).
Apoptosis assay
Apoptosis was assessed using Annexin V/propidium
iodide (PI) double staining, according to the manufacturer's
protocol (BD Biosciences). Following treatments, the cells were
stained with 0.5 mg/ml Annexin V in binding buffer (10 mM HEPES
free acid, 0.14 M NaCl, and 2.5 mM CaCl2) for 30 min at
room temperature. Subsequently, PI (5 mg/ml final concentration)
was added, and the cells were incubated for a further 15 min at
room temperature. The cells were then analyzed using a flow
cytometer and BD FACSDiva software version 7 (both from BD
Biosciences).
Drug combination analysis
For combined drug analysis, a constant ratio
combination of A452 and other compounds was evaluated. Drug
dilutions and combinations were prepared in medium immediately
before use. Cells (2×104/well) in 96-well plates were
incubated with the drugs for 72 h at 37°C. A CCK-8 assay was
performed to determine cell viability. Drug interactions were
determined according to the combination index (CI) method described
by Chou (39); CI >1 implies
antagonism, CI = 1 is additive and CI <1 implies synergism. CIs
for the combination treatment groups were generated using CalcuSyn
software version 2.11 (Biosoft). The fraction affected
(FA) was calculated from the percent viability, as
follows: fa = (100 - percentage
viability)/100.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software version 5.01 (Version 5.0, Graphpad Software Inc.).
All data are presented as the means ± standard deviation from 3
independent experiments. Statistical significance was determined by
one-way analysis of variance with post hoc analysis using Tukey's
multiple comparison test. P<0.05 was considered to indicate a
statistically significant difference.
Results
A452 selectively inhibits HDAC6 in
MM
We, as well as others have previously reported that
A452 is a specific HDAC6 inhibitor in solid tumors (36,37).
To confirm the specific HDAC6 inhibitory effect of A452 in MM, its
effects on the acetylation of a-tubulin and histone H3 were
examined. The MM.1S and H929 cells were treated with increasing
concentrations of A452 for 24 h. A dose-dependent increase in
acetylated α-tubulin was observed following treatment with low
concentrations (0.05 μM) of A452, without affecting histone
acetylation (<0.5 μM), in the MM.1S cells, confirming its
more selective inhibitory effect on HDAC6 activity (Fig. 1A). To determine the selectivity of
A452 for HDAC6 over class I HDACs, the concentration required to
increase acetylated levels of α-tubulin was compared with that
required to increase acetylated levels of histone H3. The levels of
acetylated-α-tubulin and acetylated-H3 were semi-quantified
relative to α-tubulin and histone H3, respectively, and a
>2-fold increase in the acetylated levels was considered to
indicate a significant inhibitory effect. Based on the results of
western blot analysis, A452 was ~10-fold (HDAC6 inhibitory
concentration, 0.05 μM; vs. class I HDAC inhibitory
concentration, 0.5 μM) and 500-fold (0.001 vs. 0.5
μM) less active against class I HDACs in the MM.1S and H929
cells, respectively (Figs. 1A and
B and S1). The pan-HDAC
inhibitor, SAHA, was used as a positive control for HDAC inhibition
(40). These results indicated
that A452 may be an HDAC6-selective inhibitor with minimal class I
HDAC activity in MM cells.
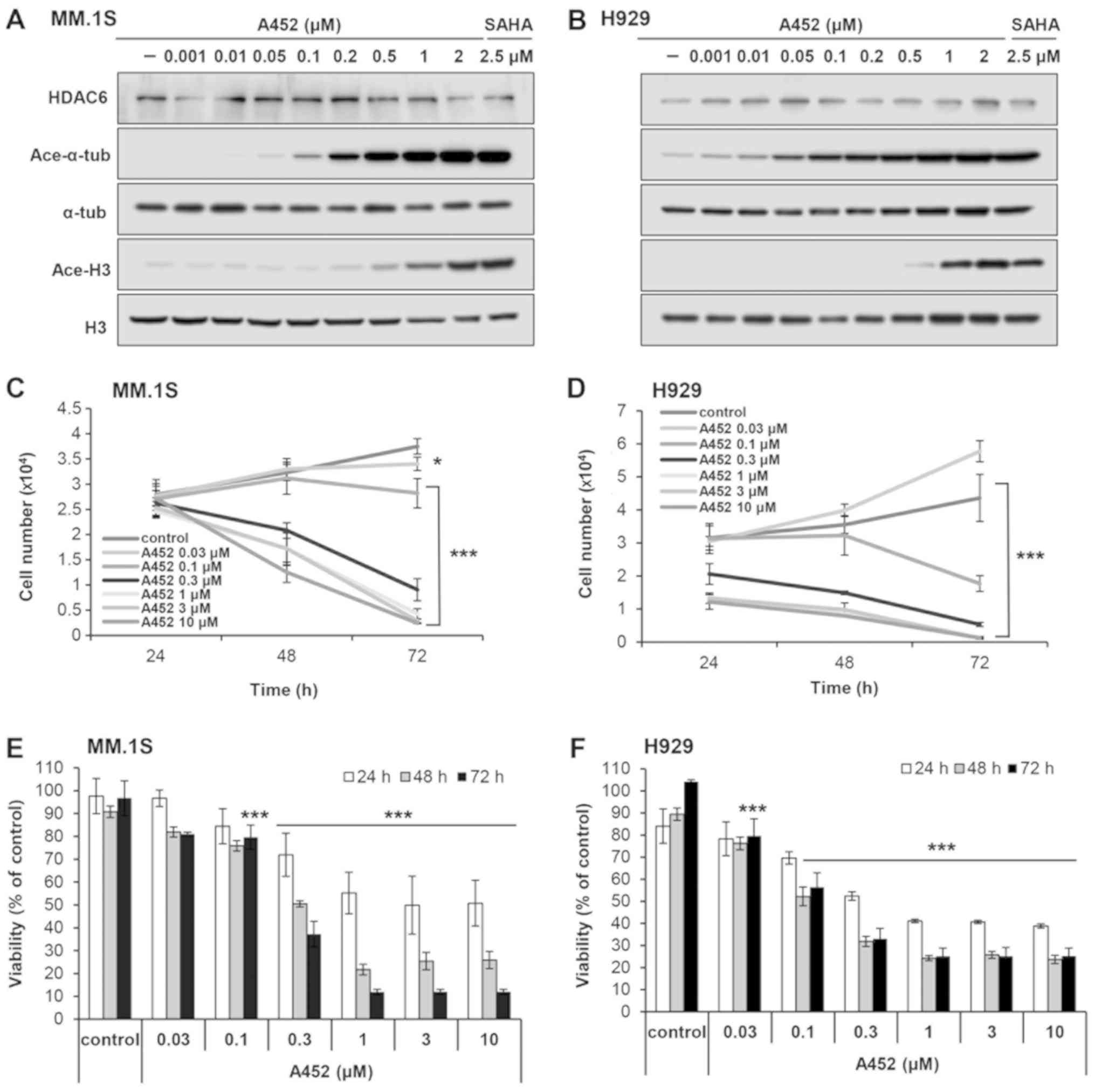 | Figure 1A452, an HDAC6-selective inhibitor,
suppresses the growth and viability of MM cells. (A) MM.1S and (B)
H929 cells were treated with A452 at the indicated concentrations
(0.001, 0.01, 0.05, 0.1, 0.2, 0.5, 1 and 2 μM) for 24 h and
western blot analysis was performed with antibodies against
acetylated a-tubulin (Ace-a-tub), acetylated histone H3 (Ace-H3)
and a-tubulin (a-tub) and total histone H3 (H3). a-tubulin and
histone H3 are shown as equal loading controls. The pan-HDAC
inhibitor, SAHA (2.5 μM), was used as a positive control for
HDAC inhibition. Growth and viability of (C and E) MM.1S and (D and
F) H929 cells cultured with 0.1% DMSO (control) or A452 at
indicated concentrations (0.03, 0.1, 0.3, 1, 3 and 10 μM)
for 72 h. Viable cell numbers and viability were measured using
CCK-8 assays. Cell counts were estimated indirectly from a standard
curve generated using solutions of known cell counts. Absorbance
was normalized to that of the negative control at each time
interval. Data are expressed as the means ± standard deviation from
3 independent experiments. *P<0.05 and *"P<0.001, vs. the
DMSO control (analysis of variance). HDAC, histone deacetylase. |
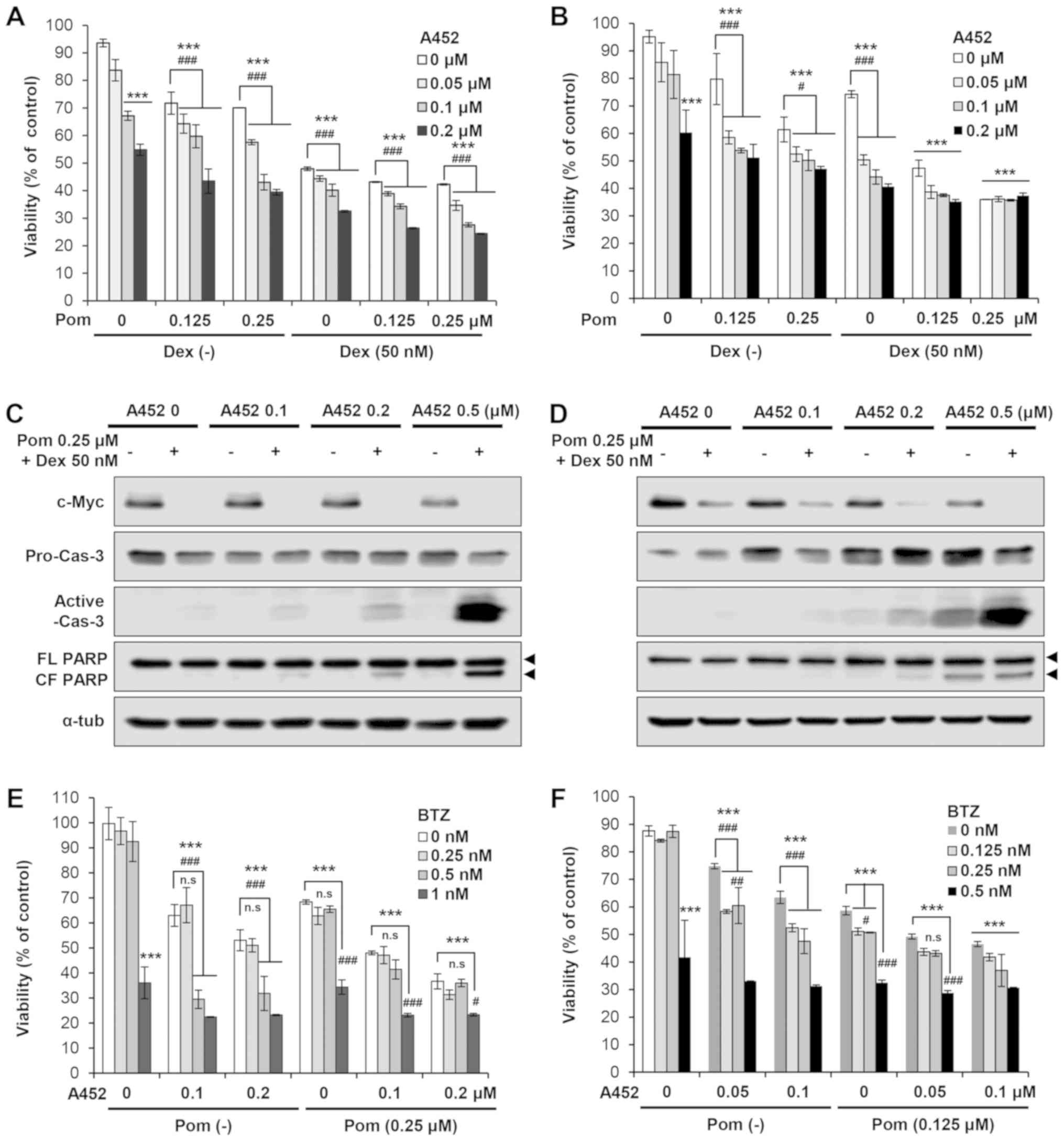
A452 induces time- and does-dependent
cytotoxicity in MM cells
The effects of A452 on cell growth and viability
were examined in the MM.1S and H929 MM cells. The cells were
treated with A452 for up to 72 h, and cell growth and viability
were measured using CCK-8 assays. In addition, to determine whether
A452 can overcome resistance to Dex, Dex-resistant H929 cells and
Dex-sensitive MM.1S cells were used. Compared with the control
cells, A452 (except at the dose of 0.03 μM) resulted in a
time- and dose-dependent decrease in the growth and viability of
both MM cells, with GI50 and IC50 values of 0.24-0.69
μM and 0.17-0.44 μM, respectively (Fig. 1C-F). In addition, the inhibitory
effect of A452 on the growth (1.3-1.5-fold) and viability
(2.1-2.6-fold) of the MM cells was greater than that of ACY-1215
(Table I). By contrast, at the
concentration of ≤1 μM, A452 and ACY-1215 did not alter the
total number of viable BM-MSCs, whereas only higher concentrations
(~2 μM) inhibited cell viability (data not shown). In
particular, A452 and ACY-1215 exhibited significant
anti-proliferative activity in the Dex-resistant H929 MM cell line,
suggesting the ability of A452 and ACY-1215 to overcome Dex
resistance. Taken together, these findings suggest that the
selective inhibition of HDAC6 by A452 induces cell death and
overcomes drug resistance in MM.
 | Table IGrowth and viability inhibitory
effects of A452 and ACY-1215 in MM cell lines. |
Table I
Growth and viability inhibitory
effects of A452 and ACY-1215 in MM cell lines.
| Cell line | Time (h) | A452 (μM)
| ACY-1215
(μM)
|
|---|
|
GI50 |
IC50 |
GI50 |
IC50 |
|---|
| MM.1S | 48 | 0.688 | 0.435 | 0.875 | 1.137 |
| 72 | 0.139 | 0.159 | 0.817 | 1.148 |
| H929 | 48 | 0.243 | 0.174 | 0.371 | 0.369 |
| 72 | 0.047 | 0.196 | 0.424 | 0.435 |
A452 in combination with IMiDs exerts
synergistic cytotoxic effects
The combined effect of IMiDs and an HDAC6-selective
inhibitor on MM cytotoxicity was then assessed. The MM.1S and H929
MM cells were treated with either A452 alone, or in combination
with Len and Pom, and a CCK-8 assay was performed to measure cell
growth and viability for 72 h. This combined treatment resulted in
synergistic growth inhibitory effect on both cell lines (Fig. 2A and D). A substantial decrease in
viability was observed following combined treatment compared with a
single agent. Synergism was evaluated by using the Chou-Talalay
method (39). The combination of
A452 and Len or Pom exhibited synergistic anti-MM activity with a
combination index of <1.0 (Fig. 2B,
C, E and F, and Table II).
Similarly, the combination of ACY-1215 and IMiDs exerted
synergistic cytotoxic effects (Table
II) These data confirmed the robust anti-proliferative effect
when A452 and IMiD were used in combination in Dex-sensitive and
Dex-resistant MM cells.
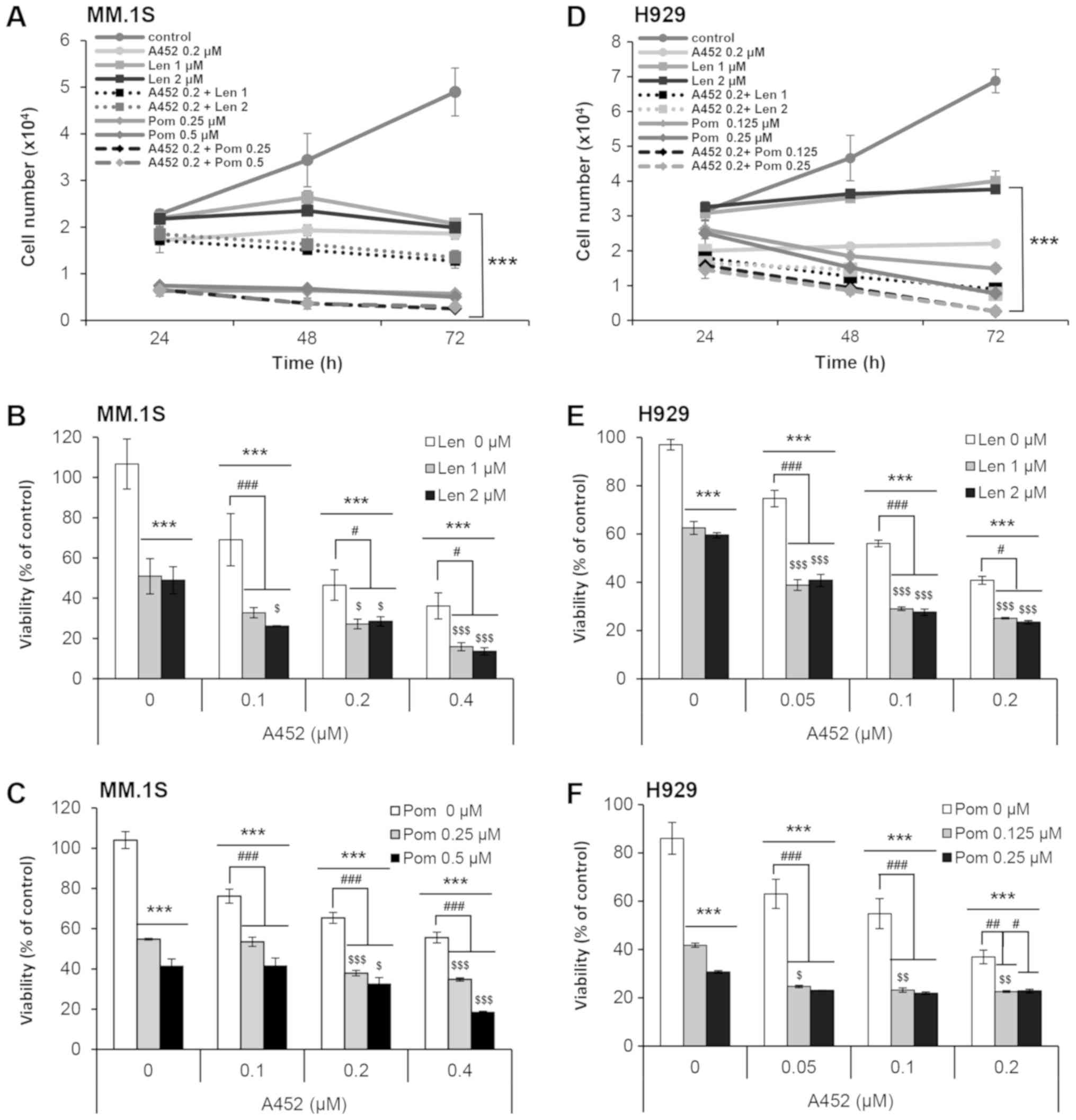 | Figure 2Co-treatment with IMiD and A452
triggers synergistic cytotoxicity. (A-C) MM.1S and (D-F) H929 cells
were treated with 0.1% DMSO (control), A452 (0.05-0.4 μM),
Len (1, 2 μM) or Pom (0.125-0.5 μM) or in combination
with these compounds as indicated for 72 h. Cell growth and
viability were measured using CCK-8 assays. Combination treatments
were then performed in (A-C) MM. 1S and (D-F) H929 cells
maintaining a constant ratio between the dose of the A452 and
IMiDs. Cell growth (24-72 h) and viability (72 h) were measured
using CCK-8 assays. Cell counts were indirectly estimated from a
standard curve generated using solutions of known cell counts.
Absorbance was normalized to that of the negative control at each
time interval. Data are expressed as the means ± standard deviation
from 3 independent experiments. ***P<0.001, vs. the
DMSO control; #P<0.05, ##P<0.01 and
###P<0.001, vs. the A452-treated group;
$P<0.05, $$P<0.01 and
$$$P<0.001, vs. the Len or Pom-treated group
(analysis of variance). IMiDs, immunomodulatory drugs; Len,
lenalidomide; Pom, pomalidomide. |
| Table IIMedian CI for cells exposed to
various drug combinations. |
Table II
Median CI for cells exposed to
various drug combinations.
| Cell line | Drug | Mean CI value | CI range | Synergy |
|---|
| MM.1S | A452 + Len | 0.301 | 0.168-0.383 | Synergism |
| A452 + Pom | 0.614 | 0.303-0.855 | Synergism |
| ACY + Len | 0.582 | 0.367-0.901 | Synergism |
| ACY + Pom | 0.634 | 0.423-0.805 | Synergism |
| H929 | A452 + Len | 0.405 | 0.211-0.581 | Synergism |
| A452 + Pom | 0.645 | 0.434-0.939 | Synergism |
| ACY + Len | 0.138 | 0.072-0.197 | Synergism |
| ACY + Pom | 0.620 | 0.516-0.743 | Synergism |
A452 in combination IMiDs synergistically
induces apoptosis
To investigate the mechanisms of the synergistic
cytotoxicity induced by the combination treatment, the activation
of apoptotic pathways was evaluated by Annexin V/PI staining. The
population of Annexin V-positive cells following treatment with
A452, Len or Pom was 18.9, 23.6 and 34.3% in the MM.1S cells and
18.4, 14.4 and 14.8% in the H929 cells, respectively, which
increased to 39 and 49.7% in the MM.1S, and 31.9 and 41.6% in the
H929 cells, respectively, following combination treatment (Figs. 3A and B, and S2). To examine the molecular mechanisms
of apoptosis, western blot analysis was performed. Combination
treatment markedly downregulated the levels of the Bcl-xL and XIAP
anti-apoptotic proteins without altering the levels of Bcl-2
anti-apoptotic protein, and Bax and Bak pro-apoptotic proteins.
Combination treatment triggered the synergistic cleavage of
caspase-3 and PARP (Figs. 3C and
D, and S3). However,
combination treatment caused the cleavage of caspase-8 and
caspase-9 in the H929 cells, but not the MM.1S cells. In addition,
combination treatment with ACY-1215 and IMiDs triggered apoptosis
(data not shown). However, ACY-1215 was less effective than A452.
Overall, these results indicated that A452 and Len or Pom induced
apoptosis by activating caspases and downregulating anti-apoptotic
factors.
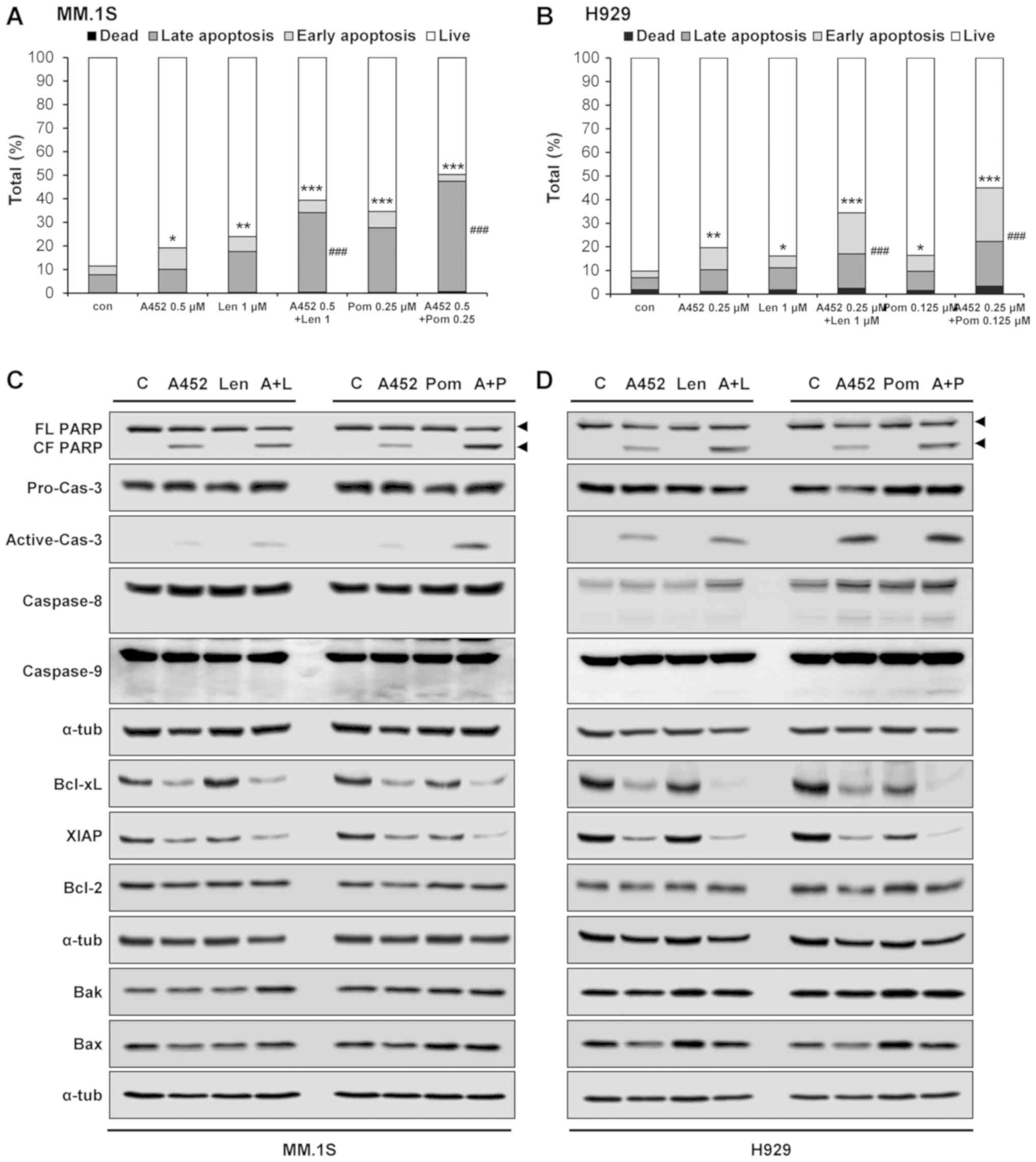 | Figure 3Co-treatment with IMiD and A452 leads
to synergistic apoptosis induction. (A) MM.1S and (B) H929 cells
were treated with 0.1% DMSO (control), A452 (0.25 and 0.5
μM), Len (1 μM) or Pom (0.125 and 0.25 μM) or
in combination with these compounds, as indicated, for 5 days and
stained with Annexin V and propidium iodide for 45 min. Apoptosis
induced by these compounds was assessed by flow cytometry (n=3).
Data are expressed as the means ± standard deviation from 3
independent experiments. *P<0.05,
**P<0.01 and ***P<0.001, vs. apoptotic
cells in the DMSO control; ###P<0.001, vs. apoptotic
cells in A452-treated group (analysis of variance). (C) MM.1S and
(D) H929 cells were treated with 0.1% DMSO (control), A452 (0.5
μM), Len (1 μM) or Pom (0.25 μM) or in
combination with these compounds, as indicated, for 24 h.
Whole-cell lysates were subjected to immunoblotting with indicated
antibodies. α-tubulin was used as a loading control. IMiDs,
immunomodulatory drugs; Len, lenalidomide; Pom, pomalidomide. |
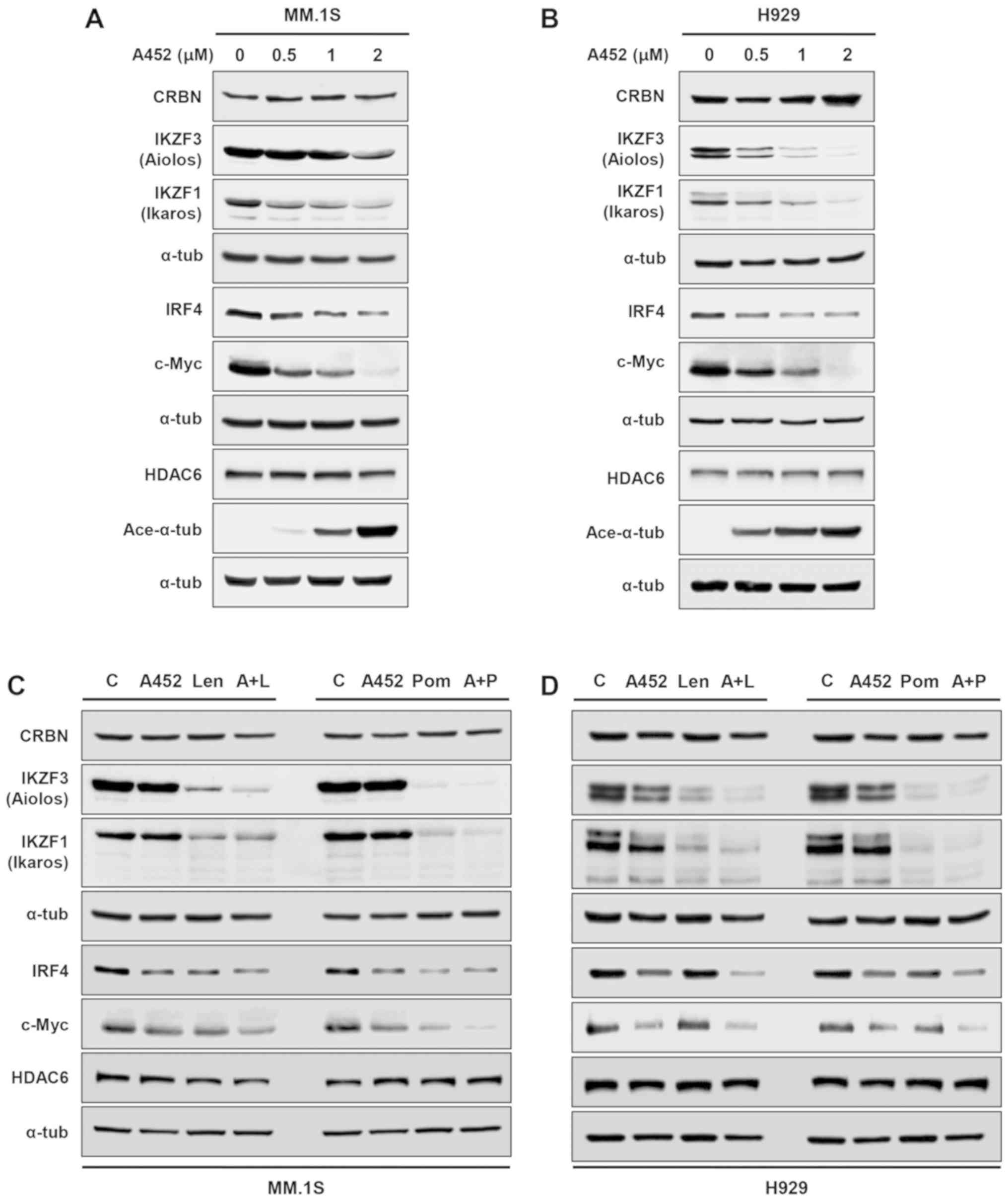
A452 in combination with IMiDs
synergistically downregulates the expression of c-Myc, IRF4 and
IKZF1/3
c-Myc and IRF4 play a pivotal role in the
progression of MM, and previous studies have shown that IMiDs
(41) and ACY-1215, an HDAC6
inhibitor with minimal effects on class I HDACs (33), downregulate the expression of c-Myc
and IRF4 in MM cells. Therefore, in this study, the inhibitory
effects of A452 on c-Myc and IRF4 expression levels were examined.
A452 downregulated the expression of c-Myc and IRF4 in the MM. 1S
and H929 cells, in a dose-dependent manner (Fig. 4A and B). It has previously been
reported that IMiDs bind to CRBN, which subsequently causes the
degradation of IKZF1 and IKZF3 (42,43).
Furthermore, downregulation of CRBN leads to resistance to IMiD
treatment (13,44). Therefore, the present study
examined whether A452 modulates the expression and/or function of
CRBN. A452 did not alter the expression of CRBN, but downregulated
the expression of IKZF1 and IKZF3 in both cell lines.
As ACY-1215 with Len synergistically downregulate
c-Myc, IRF4 and IKZF1/3 *expression in MM (33), the present study, we examined
whether combination treatment with A452 and IMiDs would cause this
effect. As shown in Fig. 4C and D,
combined treatment with A452 and Len or Pom synergistically
decreased the expression of c-Myc, IRF4 and IKZF1/3 without
reducing the expression of CRBN in the MM cells compared to the
control, even A452 slightly increased its level. (Figs. S4 and S5). Overall, these results
indicate that combined treatment with A452 and IMiDs enhances the
downregulation of c-Myc, IRF4 and IKZF1/3 in MM.
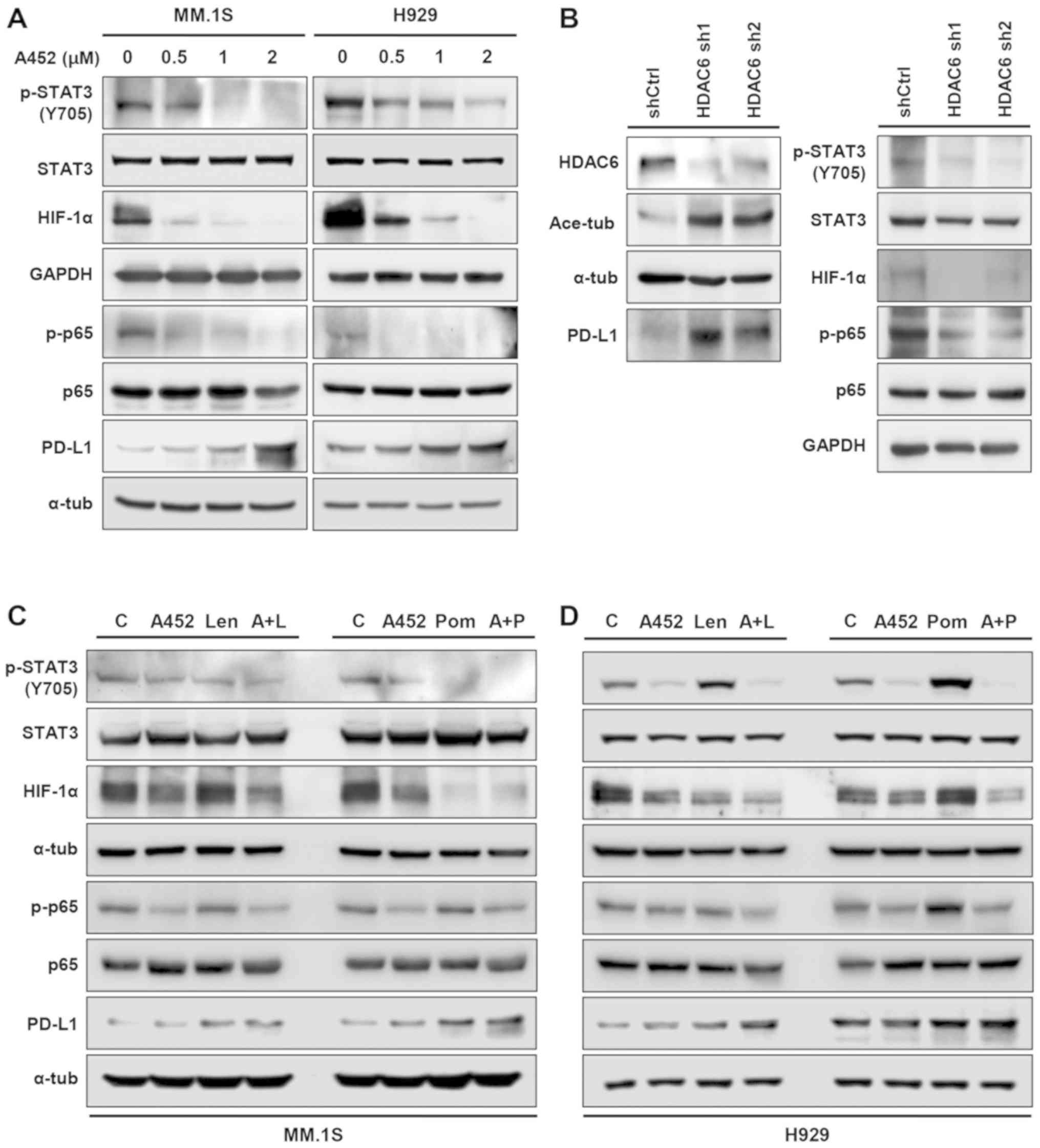
A452 controls the upregulation of PD-L1
in MM cells
A recent study demonstrated that pan-HDACis (LBH589
and PDX101) and class I HDACis (MS275 and MGCD0103) upregulated
PD-L1 via the STAT3 pathway in melanoma, whereas HDAC6i ACY-1215
and Nexturastat and HDAC8i PCI34051 did not alter the expression of
PD-L1 (45). In addition,
transcription factors, such as STAT3, HIF-1a and nf-kb have been found to regulate
PD-L1 transcriptionally (46).
Therefore, the present study examined the effect of HDAC6i on
expression of PD-L1 via the modulation of transcription factors in
MM cells. A452 treatment resulted in increased levels of PD-L1 in
the MM cells, whereas the phosphorylation levels of STAT3 and p65,
and the level of HIF-1a decreased in the MM.1S and H929 cells
(Figs. 5A and S6). Similar results were observed in
other U266 MM cells (data not shown). Similar to A452 treatment,
HDAC6 knockdown by shRNA (Fig. 5B)
led to similar effects, suggesting that this effect was due to the
inhibition of HDAC6. In addition, combination treatment with A452
and IMiDs slightly enhanced the induction of PD-L1 in both MM cell
lines (Figs. 5C and D and S6). Similar results were also observed
in other U266 MM cells (data not shown). However, ACY-1215 did not
upregulate PD-L1 expression (Fig.
S7). Thus, these results indicate that HDAC6 regulates the
expression level of PD-L1 irrespective of its known transcription
factors.
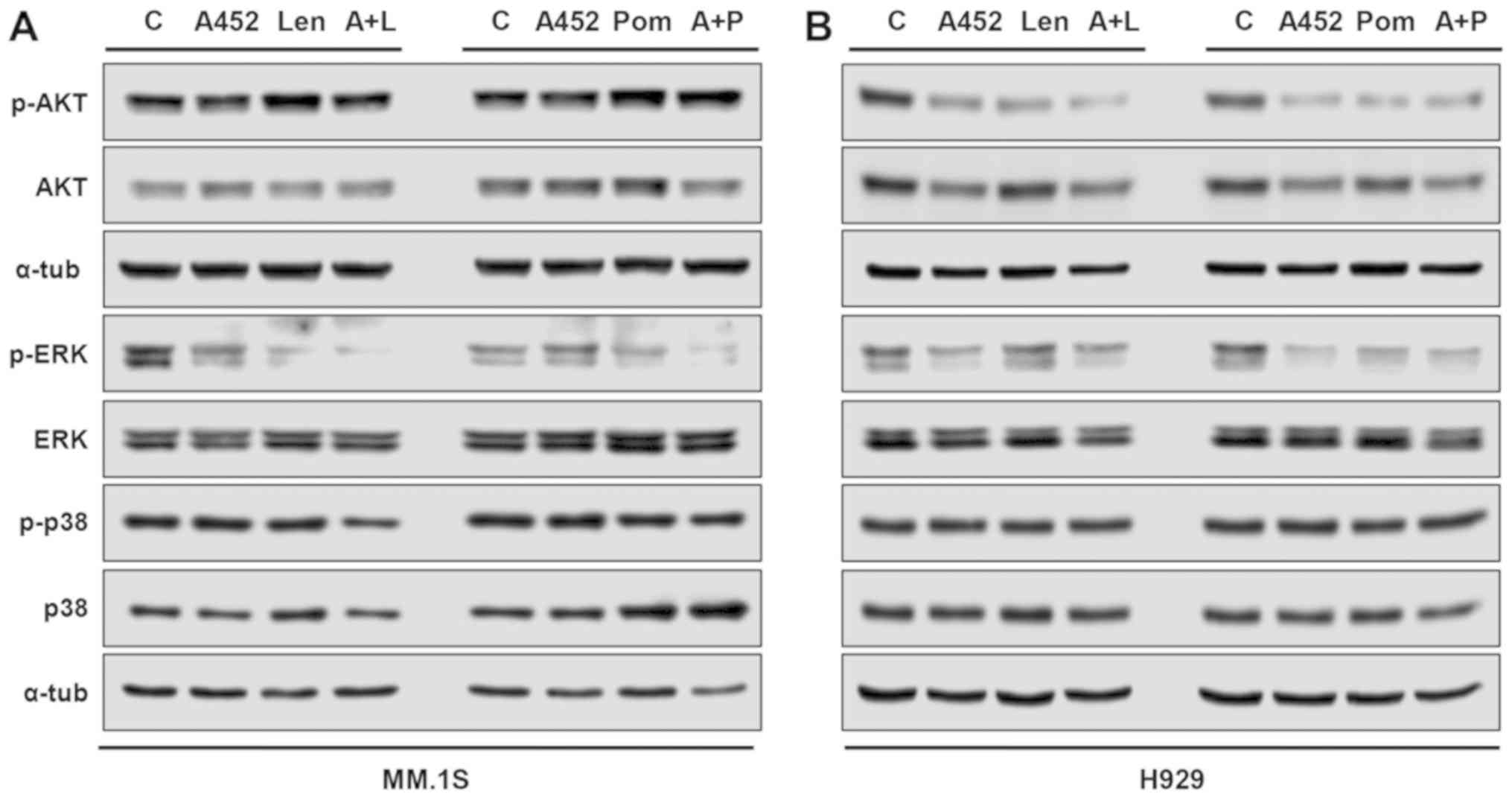
ERK and AKT inactivation plays functional
roles in the synergistic effects of A452 and IMiDs
To further address the synergism at the molecular
level, several signaling pathways were evaluated in the MM.1S and
H929 MM cells. As shown in Fig. 6,
within 24 h, both HDAC6i and IMiDs modulated either AKT or ERK.
A452 induced a decrease in ERK-MAPK phosphorylation and AKT
activation in both MM cells compared with the untreated cells. The
combination of A452 with Len or Pom synergistically decreased the
phosphorylation of ERK in both cells and the phosphorylation of AKT
in the H929 cells, compared with each compound alone (Fig. S8). By contrast, p38-MAPK
activation remained relatively unaltered (although some significant
differences were observed) in both MM cells. Although less evident,
similar results were observed in the MM.1S cells. Therefore, these
results indicate that combined treatment with A452 and IMiDs leads
to the suppression of cytoprotective ERK and AKT.
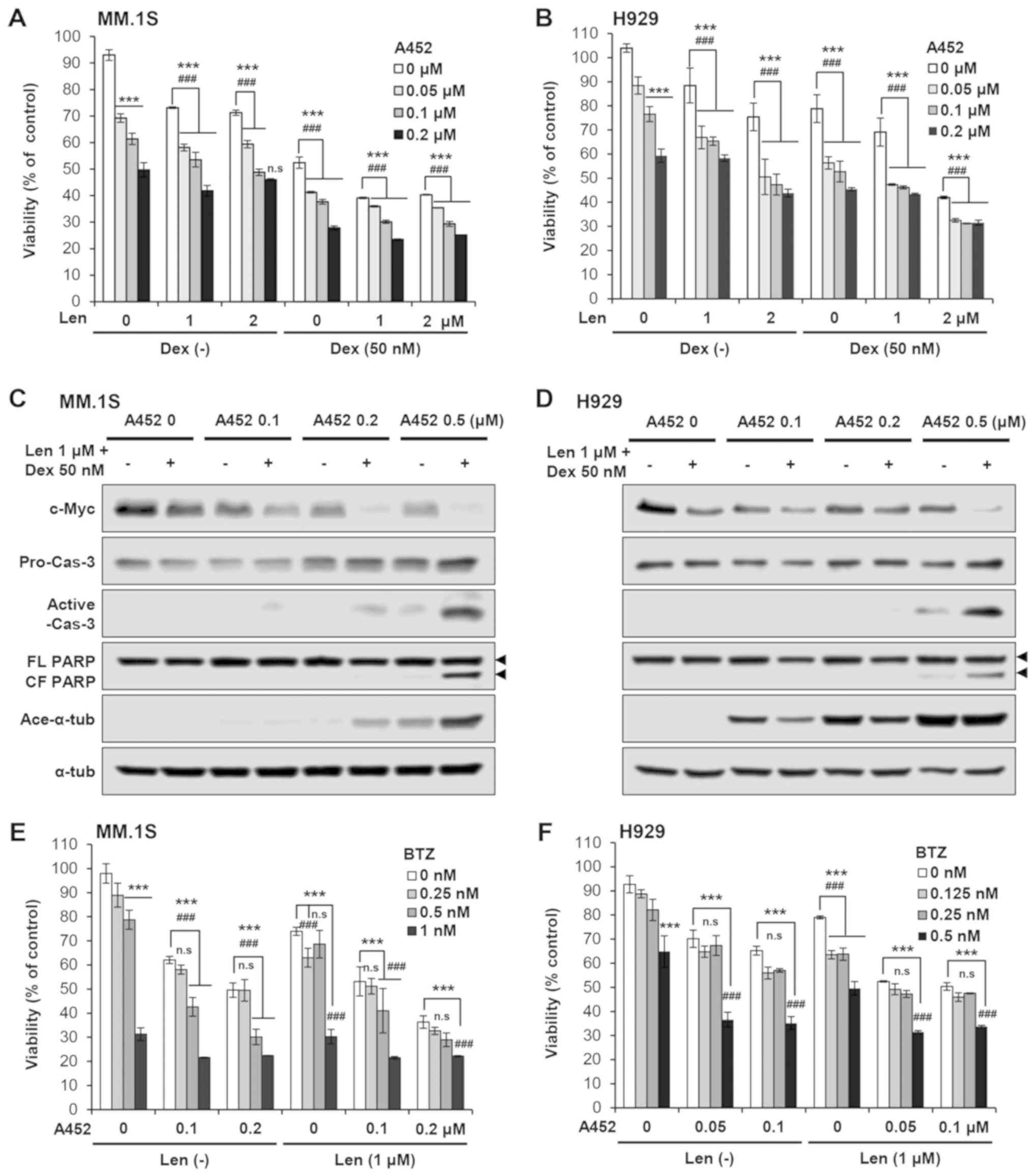
A452 enhances the cytotoxicity induced by
IMiDs with bortezomib or dexamethasone
As Len together with Dex is a standard treatment
regimen for MM, in the present study, we then examined whether A452
enhances the cytotoxicity triggered by this drug treatment. A452
enhanced the decrease in MM cell viability induced by Len or Pom
with Dex in both the Dex-sensitive MM. 1S and Dex-resistant H929
cells (Fig. 7A and B). The results
of western blot analysis clearly revealed that A452 downregulated
c-Myc expression, and upregulated the cleavage of caspase-3 and
PARP, in a dose-dependent manner (Figs. 7C and D, S9 and S10). The combination of Len and
BTZ is another standard treatment option for MM. Other studies have
shown that treatment with the HDAC6 inhibitor, ACY-E2E5, or tubacin
with BTZ or carfilzomib exerts synergistic MM cytotoxicity,
associated with endoplasmic reticulum stress due to the
accumulation of polyubiquitinated proteins (31,47,48).
Therefore, the present study also examined whether A452 enhances
the cytotoxicity induced by Len or Pom with BTZ. A452 enhanced the
inhibition of viability induced by Len with BTZ in a dose-dependent
manner (Fig. 7E and F). Similar
results were obtained in the MM.1S and H929 cells treated with A452
and BTZ with Pom (Fig. 8). Taken
together, these results indicate that A452 enhances the MM
cytotoxicity triggered by standard MM treatment options that
include Len/Pom and/or BTZ or Dex, and abrogates the resistance
conferred by Dex-acquired resistance.
Discussion
Despite progress due to the development of IMiDs and
proteasome inhibitors which have significantly extended patient
overall survival rates, MM remains incurable and eventually leads
to death (1). Therefore, novel
drugs and/or combination treatment strategies are required to
further improve MM patient survival rates. The results of the
present study indicated that the novel HDAC6-selective inhibitor,
A452, interacted synergistically with the IMIDs, Len or Pom, to
induce cell death in MM, including that of Dex-resistant cells.
These studies were prompted by several considerations. First, the
HDAC6-selective inhibitor may limit adverse effects associated with
pan-HDACi and improve efficacy. Second, the HDAC6-selective
inhibitor may enhance the antitumor activity of IMiDs in MM. Third,
the HDAC6-selective inhibitor may overcome resistance to other
drugs.
Synergistic interactions between HDAC6i ricolinostat
and IMiDs in MM cells have been reported (33). The mechanisms underlying these
interactions are multifactorial; for example, the inhibition of
both proteasomal and aggresomal protein degradation and
downregulation of IKZF1/3, c-Myc, and IRF4 by IMiDs, which may
function together with HDAC inhibition to increase the expression
of differentiation and cell death related genes. Consistent with
previous studies, A452 significantly downregulated the expression
of IKZF1/3, c-Myc and IRF4 in MM cells. Furthermore, A452
synergistically enhanced the cytotoxicity of Len or Pom in MM by
downregulating the expression of IKZF1/3, c-Myc and IRF4, and by
inhibiting the cytoprotective AKT and ERK pathways. It was further
demonstrated that A452 with Len or Pom exerted synergistic
cytotoxicity associated with the induction of caspase-9 and
caspase-8 cleavage, activating the extrinsic and intrinsic
apoptotic pathways. XIAP plays a pivotal role in MM cell survival
by inhibiting apoptosis (49). In
the present study, Len or Pom with A452 markedly decreased the
expression of Bcl-xL and XIAP. These findings indicate that
combination treatment enhances MM cytotoxicity by activating
apoptotic signaling and inhibiting anti-apoptotic protein
expression.
The results of the present study demonstrated that
IMiDs with HDAC6i induced synergistic cytotoxicity in MM without
reducing the level of CRBN compared to the control. The E3 ligase
protein CRBN has been identified as a direct molecular target for
the teratogenecity of thalidomide (12). Thalidomide and its analogs, Len and
Pom, block the autoubiquitination of CRBN (13). The CRBN-dependent ubiquitination
and proteasomal degradation of IKZF1 and IKZF3 sequentially leads
to the downregulation of c-Myc followed by IRF4, thereby resulting
in growth inhibition and apoptosis induction (43,44,50).
Therefore, high CRBN concentrations in MM are associated with an
increased responsiveness to IMiDs (51,52).
However, pan-HDACi is able to downregulate CRBN and to antagonize
Len (33). By contrast, more
HDAC6-selective HDACis with weak class I HDAC inhibitory activity
of ACY-1215 or A452 do not downregulate CRBN, thereby resulting in
synergistic MM cytotoxicity. Importantly, A452 synergistically
enhances the reduced expression of IKZF1/3, c-Myc and IRF4 induced
by Len or Pom. Therefore, the selection of HDAC6i and treatment
schedules should be optimized to augment cytotoxicity without
reducing the expression of CRBN. Although the underlying mechanisms
are unknown, it is possible that HDAC6 may be associated with CRBN
and modulates its acetylation and ubiquitination, leading to
prevention of its autoubiqutination and degradation. Therefore,
further investigations are required to determine the molecular
mechanisms through which HDAC6 regulates CRBN.
Triple combination therapy for MM is a common
treatment regimen. For instance, RVD [(Revlimid (Len) + Velcade
(BTZ) + Dex)] is a one of the most effective combination treatment
strategies for R/R MM (53), newly
diagnosed MM (54), and
maintenance therapy in MM (55).
Therefore, the present study further investigated the combination
treatment of Dex and Len or Pom with or without A452, and observed
that A452 further enhanced cytotoxicity and apoptosis. Ricolinostat
has been examined as single agent or in combination with BTZ and
Dex in R/R MM in a phase I/II clinical trial (35,56).
Ricolinostat is also currently being investigated in a phase Ib
trial in combination with Len and Dex (34). As expected, BTZ significantly
enhanced the cytotoxicity triggered by Len- or Pom-Dex-A452
combination treatment. Taken together, the results of the present
study indicated that A452 may enhance the anti-MM activities of Len
or Pom in combination BTZ or Dex, providing a preclinical rationale
for RVD + A452 clinical trials to further improve patient outcomes.
Further studies are warranted to confirm our findings in patients
with MM.
In conclusion, the results of the present study
demonstrate that IMiDs with HDAC6-selective inhibitor induce
synergistic cytotoxicity in MM, associated with the downregulation
of IKZF1/3, c-Myc, and IRF4 without the reducing CRBN expression
compared to the control. By contrast, as previously demonstrated
A452 at a concentration of up to 2 μM does not inhibit
growth and decrease the cell viability of normal fetal colon
epithelial (FHC), foreskin fibroblast (BJ) and human dermal
fibroblasts (HDF) cells (37). In
addition, in a previous study, ACY-1215 and tubastatin A added at
day 0 of culture at a concentration of up to 1 μM did not
alter the total number of viable megakaryocytes, which are highly
specialized bone marrow cells that give rise to anucleate blood
cells known as platelets. At ≤1 μM, neither HDAC6 inhibitors
induced cell death by apoptosis. Only higher doses (2-5 μM)
inhibited cell viability (57).
Consistent with previous results, in this study, at ≤1 μM,
A452 and ACY-1215 did not alter the total number of viable BM-MSCs,
whereas only higher doses (~2 μM) inhibited cell viability
(data not shown). Although the present study did not investigate
the cytotoxicity of HDAC6-selective inhibitors on normal plasma
cells, these findings suggest that combination treatment with
HDAC6i and IMiDs induce cell death in MM but not normal cells such
as BM-MSCs. The data show that enhanced cell death is associated
with the inactivation of AKT and ERK1/2. Furthermore, the combined
treatment of HDAC6i and IMiD controlled the expression level of
PD-L1 in MM cells irrespective of its known transcription factors,
suggesting that HDAC6 may serve an important role in immune-related
pathways. A recent clinical study of anti-PD-L1 antibodies revealed
a better response rate in patients with MM with higher PD-L1 levels
and progression free survival rates was weakly correlated with PD-1
levels on lymphocytes (58).
Therefore, these findings support that the ability of A452 to
increase the expression of PD-L1 and suggest that it may be of
therapeutic benefit in immunotherapy using PD-L1 antibodies.
Finally, the data obtained in the present study reveal that
combined treatment of HDAC6i and IMiD is effective in Dex-resistant
MM cells. Taken together, the results of this study suggest that
rational combinations of IMiD with a targeted inhibitor for HDAC6
may provide beneficial therapeutic opportunities for patients with
MM exhibiting resistance to conventional drugs
Supplementary Data
Abbreviations:
|
BTZ
|
bortezomib
|
|
Bcl-xL
|
B-cell lymphoma-extra large
protein
|
|
CRBN
|
cereblon
|
|
CI
|
combination index
|
|
DEX
|
dexamethasone
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FA
|
Fraction affected
|
|
HDAC
|
histone deacetylase
|
|
HDACi
|
histone deacetylase inhibitor
|
|
γH2AX
|
phosphorylated H2AX
|
|
IMiD
|
immunomodulatory drugs
|
|
IRF4
|
interferon regulatory factor 4
|
|
Len
|
lenalidomide
|
|
MM
|
multiple myeloma
|
|
PARP
|
poly(ADP ribose) polymerase
|
|
Pom
|
pomalidomide
|
|
SAHA
|
suberoylanilide hydroxamic acid
|
Acknowledgments
The authors would like to thank Dr Gyoonhee Han
(Yonsei University) for providing A452.
Funding
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(2016R1D1A1A02937071 and 2018R1A6A1A03023718).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HRW, DHL and SKY designed and performed experiments,
as well as analyzed the data. HWR and GWK performed the experiments
and analyzed the data. SHK conceived the general design of the
study, participated in the development of the approaches, wrote the
initial draft of the manuscript, extensively edited the manuscript
and supervised the work. All authors have read and approved the
final manuscript
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Palumbo A and Anderson K: Multiple
myeloma. N Engl J Med. 364:1046–1060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morgan GJ, Walker BA and Davies FE: The
genetic architecture of multiple myeloma. Nat Rev Cancer.
12:335–348. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Offidani M, Corvatta L, Caraffa P, Leoni
P, Pautasso C, Larocca A and Palumbo A: Pomalidomide for the
treatment of relapsed-refractory multiple myeloma: A review of
biological and clinical data. Expert Rev Anticancer Ther.
14:499–510. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Moehler T and Goldschmidt H: Therapy of
relapsed and refractory multiple myeloma. Recent Results Cancer
Res. 183:239–271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laubach JP, Mitsiades CS, Mahindra A,
Luskin MR, Rosenblatt J, Ghobrial IM, Schlossman RL, Avigan D, Raje
N, Munshi NC, et al: Management of relapsed and relapsed/refractory
multiple myeloma. J Natl Compr Canc Netw. 9:1209–1216. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lonial S, Mitsiades CS and Richardson PG:
Treatment options for relapsed and refractory multiple myeloma.
Clin Cancer Res. 17:1264–1277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dredge K, Horsfall R, Robinson SP, Zhang
LH, Lu L, Tang Y, Shirley MA, Muller G, Schafer P, Stirling D, et
al: Orally administered lenalidomide (CC-5013) is anti-angiogenic
in vivo and inhibits endothelial cell migration and Akt
phosphorylation in vitro. Microvasc Res. 69:56–63. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davies FE, Raje N, Hideshima T, Lentzsch
S, Young G, Tai YT, Lin B, Podar K, Gupta D, Chauhan D, et al:
Thalidomide and immunomodulatory derivatives augment natural killer
cell cytotoxicity in multiple myeloma. Blood. 98:210–216. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dredge K, Marriott JB, Macdonald CD, Man
HW, Chen R, Muller GW, Stirling D and Dalgleish AG: Novel
thalidomide analogues display anti-angiogenic activity
independently of immunomodulatory effects. Br J Cancer.
87:1166–1172. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reddy N, Hernandez-Ilizaliturri FJ, Deeb
G, Roth M, Vaughn M, Knight J, Wallace P and Czuczman MS:
Immunomodulatory drugs stimulate natural killer-cell function,
alter cytokine production by dendritic cells, and inhibit
angiogenesis enhancing the anti-tumour activity of rituximab in
vivo. Br J Haematol. 140:36–45. 2008.
|
|
11
|
Verhelle D, Corral LG, Wong K, Mueller JH,
Moutouh-de Parseval L, Jensen-Pergakes K, Schafer PH, Chen R,
Glezer E, Ferguson GD, et al: Lenalidomide and CC-4047 inhibit the
proliferation of malignant B cells while expanding normal
CD34+ progenitor cells. Cancer Res. 67:746–755. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ito T, Ando H, Suzuki T, Ogura T, Hotta K,
Imamura Y, Yamaguchi Y and Handa H: Identification of a primary
target of thalidomide teratogenicity. Science. 327:1345–1350. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lopez-Girona A, Mendy D, Ito T, Miller K,
Gandhi AK, Kang J, Karasawa S, Carmel G, Jackson P, Abbasian M, et
al: Cereblon is a direct protein target for immunomodulatory and
antiproliferative activities of lenalidomide and pomalidomide.
Leukemia. 26:2326–2335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aldana-Masangkay GI, Rodriguez-Gonzalez A,
Lin T, Ikeda AK, Hsieh YT, Kim YM, Lomenick B, Okemoto K, Landaw
EM, Wang D, et al: Tubacin suppresses proliferation and induces
apoptosis of acute lymphoblastic leukemia cells. Leuk Lymphoma.
52:1544–1555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fecteau JF, Corral LG, Ghia EM, Gaidarova
S, Futalan D, Bharati IS, Cathers B, Schwaederle M, Cui B,
Lopez-Girona A, et al: Lenalidomide inhibits the proliferation of
CLL cells via a cereblon/p21(WAF1/Cip1)-dependent mechanism
independent of functional p53. Blood. 124:1637–1644. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schey S and Ramasamy K: Pomalidomide
therapy for myeloma. Expert Opin Investig Drugs. 20:691–700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mehnert JM and Kelly WK: Histone
deacetylase inhibitors: Biology and mechanism of action. Cancer J.
13:23–29. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Richon VM, Garcia-Vargas J and Hardwick
JS: Development of vorinostat: Current applications and future
perspectives for cancer therapy. Cancer Lett. 280:201–210. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ropero S and Esteller M: The role of
histone deacetylases (hdacs) in human cancer. Mol Oncol. 1:19–25.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu WS, Parmigiani RB and Marks PA: Histone
deacetylase inhibitors: Molecular mechanisms of action. Oncogene.
26:5541–5552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Campas-Moya C: Romidepsin for the
treatment of cutaneous T-cell lymphoma. Drugs Today (Barc).
45:787–795. 2009. View Article : Google Scholar
|
|
22
|
Duvic M, Olsen EA, Breneman D, Pacheco TR,
Parker S, Vonderheid EC, Abuav R, Ricker JL, Rizvi S, Chen C, et
al: Evaluation of the long-term tolerability and clinical benefit
of vorinostat in patients with advanced cutaneous T-cell lymphoma.
Clin Lymphoma Myeloma. 9:412–416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marks PA and Breslow R: Dimethyl sulfoxide
to vorinostat: Development of this histone deacetylase inhibitor as
an anticancer drug. Nat Biotechnol. 25:84–90. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Siegel DS, Richardson P, Dimopoulos M,
Moreau P, Mitsiades C, Weber D, Houp J, Gause C, Vuocolo S, Eid J,
et al: Vorinostat in combination with lenalidomide and
dexamethasone in patients with relapsed or refractory multiple
myeloma. Blood Cancer J. 4:e2022014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ocio EM, Vilanova D, Atadja P, Maiso P,
Crusoe E, Fernández-Lázaro D, Garayoa M, San-Segundo L,
Hernández-Iglesias T, de Alava E, et al: In vitro and in vivo
rationale for the triple combination of pano- binostat (LBH589) and
dexamethasone with either bortezomib or lenalidomide in multiple
myeloma. Haematologica. 95:794–803. 2010. View Article : Google Scholar
|
|
26
|
San-Miguel JF, Richardson PG, Günther A,
Sezer O, Siegel D, Bladé J, LeBlanc R, Sutherland H, Sopala M,
Mishra KK, et al: Phase Ib study of panobinostat and bortezomib in
relapsed or relapsed and refractory multiple myeloma. J Clin Oncol.
31:3696–3703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sivaraj D, Green MM and Gasparetto C:
Panobinostat for the management of multiple myeloma. Future Oncol.
13:477–488. 2017. View Article : Google Scholar
|
|
28
|
Bergman JA, Woan K, Perez-Villarroel P,
Villagra A, Sotomayor EM and Kozikowski AP: Selective histone
deacetylase 6 inhibitors bearing substituted urea linkers inhibit
melanoma cell growth. J Med Chem. 55:9891–9899. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Butler LM, Agus DB, Scher HI, Higgins B,
Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA and Richon
VM: Suberoylanilide hydroxamic acid, an inhibitor of histone
deacetylase, suppresses the growth of prostate cancer cells in
vitro and in vivo. Cancer Res. 60:5165–5170. 2000.PubMed/NCBI
|
|
30
|
Inks ES, Josey BJ, Jesinkey SR and Chou
CJ: A novel class of small molecule inhibitors of HDAC6. ACS Chem
Biol. 7:331–339. 2012. View Article : Google Scholar :
|
|
31
|
Santo L, Hideshima T, Kung AL, Tseng JC,
Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et
al: Preclinical activity, pharmacodynamic, and pharmacokinetic
properties of a selective HDAC6 inhibitor, ACY-1215, in combination
with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smil DV, Manku S, Chantigny YA, Leit S,
Wahhab A, Yan TP, Fournel M, Maroun C, Li Z, Lemieux AM, et al:
Novel HDAC6 isoform selective chiral small molecule histone
deacetylase inhibitors. Bioorg Med Chem Lett. 19:688–692. 2009.
View Article : Google Scholar
|
|
33
|
Hideshima T, Cottini F, Ohguchi H,
Jakubikova J, Gorgun G, Mimura N, Tai YT, Munshi NC, Richardson PG
and Anderson KC: Rational combination treatment with histone
deacetylase inhibitors and immunomodulatory drugs in multiple
myeloma. Blood Cancer J. 5:e3122015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yee AJ, Bensinger WI, Supko JG, Voorhees
PM, Berdeja JG, Richardson PG, Libby EN, Wallace EE, Birrer NE,
Burke JN, et al: Ricolinostat plus lenalidomide, and dexamethasone
in relapsed or refractory multiple myeloma: A multicentre phase 1b
trial. Lancet Oncol. 17:1569–1578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Raje N, Vogl DT, Hari PN, et al: ACY-1215,
a selective histone deacetylase (HDAC) 6 inhibitor: Interim results
of combination therapy with bortezomib in patients with multiple
myeloma (MM). Blood. 122:7592013.
|
|
36
|
Choi E, Lee C, Park JE, Seo JJ, Cho M,
Kang JS, Kim HM, Park SK, Lee K and Han G: Structure and property
based design, synthesis and biological evaluation of γ-lactam based
HDAC inhibitors. Bioorg Med Chem Lett. 21:1218–1221. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ryu HW, Shin DH, Lee DH, Won HR and Kwon
SH: A potent hydroxamic acid-based, small-molecule inhibitor A452
preferentially inhibits HDAC6 activity and induces cytotoxicity
toward cancer cells irrespective of p53 status. Carcinogenesis.
39:72–83. 2018. View Article : Google Scholar
|
|
38
|
Lee DH, Won HR, Ryu HW, Han JM and Kwon
SH: The HDAC6 inhibitor ACY-1215 enhances the anticancer activity
of oxali- platin in colorectal cancer cells. Int J Oncol.
53:844–854. 2018.PubMed/NCBI
|
|
39
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Witter DJ, Harrington P, Wilson KJ,
Chenard M, Fleming JC, Haines B, Kral AM, Secrist JP and Miller TA:
Optimization of biaryl Selective hdac1&2 Inhibitors (SHI-1:2).
Bioorg Med Chem Lett. 18:726–731. 2008. View Article : Google Scholar
|
|
41
|
López R, Cuca LE and Delgado G:
Antileishmanial and immunomodulatory activity of Xylopia discreta.
Parasite Immunol. 31:623–630. 2009. View Article : Google Scholar
|
|
42
|
Kronke J, Udeshi ND, Narla A, Grauman P,
Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al:
Lenalidomide causes selective degradation of IKZF1 and IKZF3 in
multiple myeloma cells. Science. 343:301–305. 2014. View Article : Google Scholar :
|
|
43
|
Lu G, Middleton RE, Sun H, Naniong M, Ott
CJ, Mitsiades CS, Wong KK, Bradner JE and Kaelin WG Jr: The myeloma
drug lenalidomide promotes the cereblon-dependent destruction of
Ikaros proteins. Science. 343:305–309. 2014. View Article : Google Scholar :
|
|
44
|
Zhu YX, Braggio E, Shi CX, Bruins LA,
Schmidt JE, Van Wier S, Chang XB, Bjorklund CC, Fonseca R,
Bergsagel PL, et al: Cereblon expression is required for the
antimyeloma activity of lenalidomide and pomalidomide. Blood.
118:4771–4779. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
M L, P PV, T K, M P, E S, J P, K v W, C L,
F C, S D, et al: Essential role of HDAC6 in the regulation of PD-L1
in melanoma. Mol Oncol. 10:735–750. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen J, Jiang CC, Jin L and Zhang XD:
Regulation of PD-L1: A novel role of pro-survival signalling in
cancer. Ann Oncol. 27:409–416. 2016. View Article : Google Scholar
|
|
47
|
Hideshima T, Bradner JE, Wong J, Chauhan
D, Richardson P, Schreiber SL and Anderson KC: Small-molecule
inhibition of proteasome and aggresome function induces synergistic
antitumor activity in multiple myeloma. Proc Natl Acad Sci USA.
102:8567–8572. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hideshima T, Mazitschek R, Santo L, Mimura
N, Gorgun G, Richardson PG, Raje N and Anderson KC: Induction of
differential apoptotic pathways in multiple myeloma cells by
class-selective histone deacetylase inhibitors. Leukemia.
28:457–460. 2014. View Article : Google Scholar
|
|
49
|
Desplanques G, Giuliani N, Delsignore R,
Rizzoli V, Bataille R and Barillé-Nion S: Impact of XIAP protein
levels on the survival of myeloma cells. Haematologica. 94:87–93.
2009. View Article : Google Scholar :
|
|
50
|
Bjorklund CC, Lu L, Kang J, Hagner PR,
Havens CG, Amatangelo M, Wang M, Ren Y, Couto S, Breider M, et al:
Rate of CRL4(CRBN) substrate Ikaros and Aiolos degradation
underlies differential activity of lenalidomide and pomalidomide in
multiple myeloma cells by regulation of c-Myc and IRF4. Blood
Cancer J. 5:e3542015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Broyl A, Kuiper R, van Duin M, van der
Holt B, el Jarari L, Bertsch U, Zweegman S, Buijs A, Hose D,
Lokhorst HM, et al: Dutch-Belgian HOVON group; German gmmg Group.
High cereblon expression is associated with better survival in
patients with newly diagnosed multiple myeloma treated with
thalidomide maintenance. Blood. 121:624–627. 2013. View Article : Google Scholar
|
|
52
|
Heintel D, Rocci A, Ludwig H, Bolomsky A,
Caltagirone S, Schreder M, Pfeifer S, Gisslinger H, Zojer N, Jäger
U, et al: High expression of cereblon (CRBN) is associated with
improved clinical response in patients with multiple myeloma
treated with lenalidomide and dexamethasone. Br J Haematol.
161:695–700. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Richardson PG, Xie W, Jagannath S,
Jakubowiak A, Lonial S, Raje NS, Alsina M, Ghobrial IM, Schlossman
RL, Munshi NC, et al: A phase 2 trial of lenalidomide, bortezomib,
and dexamethasone in patients with relapsed and relapsed/refractory
myeloma. Blood. 123:1461–1469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Richardson PG, Weller E, Lonial S,
Jakubowiak AJ, Jagannath S, Raje NS, Avigan DE, Xie W, Ghobrial IM,
Schlossman RL, et al: Lenalidomide, bortezomib, and dexamethasone
combination therapy in patients with newly diagnosed multiple
myeloma. Blood. 116:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nooka AK, Kaufman JL, Muppidi S, Langston
A, Heffner LT, Gleason C, Casbourne D, Saxe D, Boise LH and Lonial
S: Consolidation and maintenance therapy with lenalidomide,
bortezomib and dexamethasone (RVD) in high-risk myeloma patients.
Leukemia. 28:690–693. 2014. View Article : Google Scholar
|
|
56
|
Ashjian E and Redic K: Multiple myeloma.
Updates for pharmacists in the treatment of relapsed and refractory
disease. J Oncol Pharm Pract. 22:289–302. 2016. View Article : Google Scholar
|
|
57
|
Messaoudi K, Ali A, Ishaq R, Palazzo A,
Sliwa D, Bluteau O, Souquére S, Muller D, Diop KM, Rameau P, et al:
Critical role of the HDAC6-cortactin axis in human megakaryocyte
maturation leading to a proplatelet-formation defect. Nat Commun.
8:17862017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Badros A, Hyjek E, Ma N, Lesokhin A, Dogan
A, Rapoport AP, Kocoglu M, Lederer E, Philip S, Milliron T, et al:
Pembrolizumab, pomalidomide, and low-dose dexamethasone for
relapsed/refractory multiple myeloma. Blood. 130:1189–1197. 2017.
View Article : Google Scholar : PubMed/NCBI
|