Introduction
Anaplastic thyroid cancer (ATC) is a rare orphan
disease, accounting for 1-2% of all thyroid cancer cases (1,2). Due
to its highly malignant potential, patients with ATC often succumb
within 6 months of diagnosis despite intensive multimodal
therapies, including surgery, chemotherapy and/or radiation therapy
(1,2). At present, no effective therapeutic
method has been established; thus, development of novel therapeutic
strategies for ATC, including molecular-targeted therapy, is highly
anticipated. Our previous studies demonstrated the possible effects
of molecular therapies targeting peroxisome proliferator activated
receptor-γ (3), epithelial growth
factor receptor (4),
B-RAF/mitogen-activated protein kinase kinase (MEK) (5), as well as the effects of an mTOR
inhibitor (6). However, the
efficacy of these single molecule-targeted agents were limited and
depended on the characteristics of specific genetic alteration in
the cancer cells. These observations indicated the importance of
developing novel therapies targeting multiple molecules or
epigenetic mechanisms (7,8).
Sorafenib is a multi-kinase inhibitor targeting RAF,
vascular endothelial growth factor (VEGF) receptor (VEGFR) and
platelet-derived growth factor receptor (PDGFR) (9), and has been demonstrated to have
significant anticancer effects in renal cell carcinoma and
hepatocellular carcinoma by prolonging progression-free survival
(PFS) and/or overall survival in patients (10,11).
In addition, the DECISION trial showed that the mean PFS of
patients with radioiodine-refractory differentiated thyroid cancer
(RR-DTC) could be extended from 5.8 to 10.8 months following
sorafenib therapy compared with that of patients receiving the
placebo, leading to approval of sorafenib for clinical treatment of
RR-DTC in several countries (12).
Phase II trials have also been conducted for the effects of
sorafenib in ATC. Although no clinically relevant response was
demonstrated, disease stabilization was confirmed in certain cases
(13,14). Currently, lenvatinib is the only
drug approved for clinical use in Japan for patients with
unresectable ATC (15,16). Lenvatinib is also a multi-kinase
inhibitor targeting similar molecules as sorafenib, including VEGFR
and PDGFR, but not the RAF signaling pathway (17). Mutated BRAF is widely known
as an important driver gene that promotes the aberrant
proliferation of cancer cells (18-20).
A BRAF inhibitor has already been applied as
a clinically important therapeutic agent in several types of
cancers (21,22). Recent observations indicated that
BRAF mutations were more frequent in ATC tumors (~40%)
(23) than previously considered
(24). As previously proposed
(5), inhibition of BRAF may
be a promising strategy to control cases of ATC involving
BRAF mutations. Additionally, ATC cells have been shown to
secrete VEGF (25); thus,
VEGF-mediated tumor neovascularization is hypothesized to be a
strong contributor to the aggressive progression of ATC.
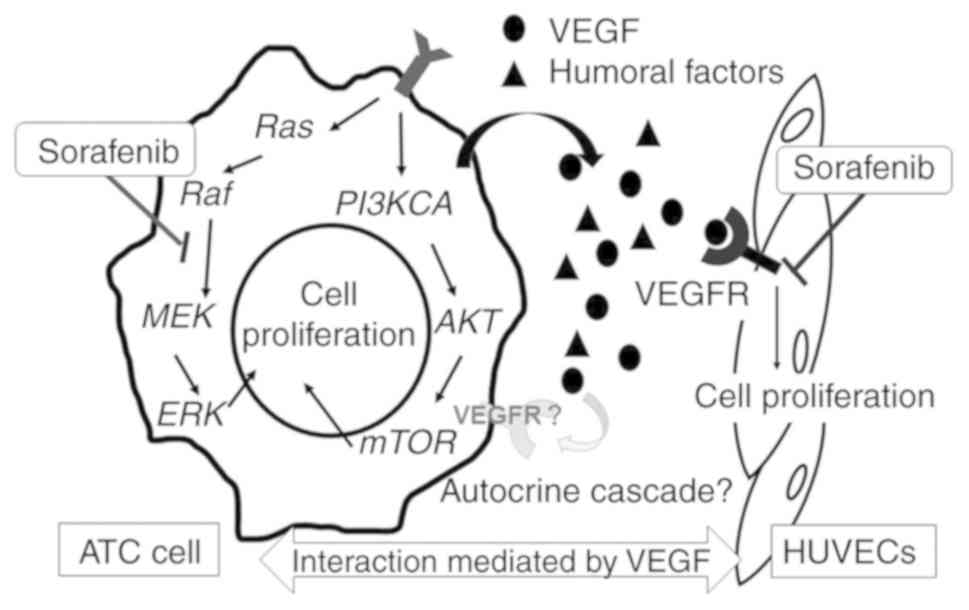
Based on this background, in the present study, the
mechanisms underlying the antitumor effects of sorafenib as a
multi-molecular targeted therapy agent were investigated using
authenticated human ATC cell lines. Additionally, the effects of
sorafenib on the impairment of cancer cell-secreted VEGF-mediated
tumor neovasculature were evaluated, as well as the inhibition of
signal transduction mediated by the RAS/RAF/MEK pathway (Fig. 1).
Materials and methods
Cell lines and culture conditions
Four authenticated human ATC cell lines were used in
the present study, including three cell lines (OCUT-2, OCUT-4 and
OCUT-6) established and characterized in our laboratory (4-6,25).
These three cell lines were authenticated via STR profiling. The
BRAF V600E mutation was found in OCUT-2 and OCUT-4 cells.
OCUT-2 cells harbor a mutation of PI3KCA in addition to the
BRAF mutation, and a NRAS mutation was detected in
the OCUT-6 and ACT-1 cell lines (Table
I). ACT-1 cells were kindly provided by Dr Seiji Ohata
(Tokushima University) (4). Each
cell line was cultured in DMEM (Wako Pure Chemical Industries,
Ltd.) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich;
Merck KGaA), 100 IU/ml penicillin and 100 µg/ml streptomycin
at 37˚C with 5% CO2 in humidified conditions.
 | Table ICharacteristics of the anaplastic
thyroid cancer cell lines. |
Table I
Characteristics of the anaplastic
thyroid cancer cell lines.
| Cell line | Gene mutation
| VEGF
(pg/ml)a | IC50
(nM)
|
|---|
| NRAS | BRAF | AKT1 | PI3KCA | P53 | hTERT | Sorafenib | Paclitaxel |
|---|
| ACT-1 | Q61K | wt | wt | wt | C242S | C250T Hetero | 409 | 680±50b | 4.45±1.26 |
| OCUT-2 | wt | V600E | wt | H1047R | wt | C250T Homo | 849 | 700±75b | 8.70±0.05 |
| OCUT-4 | wt | V600E | wt | wt | wt | C228T Homo | 64.5 | 200±30 | 3.35±0.76 |
| OCUT-6 | Q61R | wt | wt | wt | wt | C228T Hetero | 142 | 550±50b | 3.67±0.31 |
Viability assay
The inhibitory effects of sorafenib on the viability
of the ATC cells were measured by an MTT assay (4). Cells (1×103) were seeded
in each well of a 96-well plastic culture plate and incubated
overnight under the aforementioned culture conditions. They were
then treated with the indicated dose of sorafenib (50, 100, 250,
500 and 1,000 nM; Santa Cruz Biotechnology, Inc.) for 72 h.
Subsequently, MTT reagent (Dojindo Molecular Technologies, Inc.)
was added to each well at a final concentration of 0.5 mg/ml, and
the cells were incubated for a further 2 h under the same
conditions. The culture plate was centrifuged at 200 × g for 5 min
at 25˚C and the supernatant was removed. Dimethyl sulfoxide was
added to dissolve the formazan crystals, and the absorbance at 570
nm was measured using a microplate reader (Model 550; Bio-Rad
Laboratories, Inc.) and calculated using the supplied software
(LS-PLATE Manager 2004; Wako Pure Chemical Industries, Ltd.). The
experiments were conducted three times independently, in triplicate
each time, and the average values of the three independent
experiments were calculated. The efficacy of paclitaxel alone or in
combination with sorafenib on cell viability was also measured in
the same manner to investigate the synergistic effect of these two
drugs in all four cell lines. Cells were exposed to 1-100 nM of
paclitaxel for 72 h with or without 100 nM of sorafenib (equivalent
to the estimated IC80 value for each cell line) concomitantly.
Western blotting
BRAF signaling was measured in OCUT-6 cells. The
expression and phosphorylation of MEK, a protein downstream of
RAF (5), were measured via
western blot analysis. OCUT-4 and OCUT-6 cells (5×106)
were incubated for 24 h with sorafenib (50 nM). After treatment,
the cells were lysed in 400 µl of 1% Triton in PBS and
gently agitated for 20 min. Protein was then extracted via
centrifugation at 8,050 × g for 5 min at 4˚C. The concentration of
the protein was measured by BCA method (5). Total protein (60 µg) was
electrophoresed on a 10% polyacrylamide gel and transferred to a
polyvinylidene fluoride membrane (Trans-Blot Turbo Transfer Pack;
Bio-Rad Laboratories, Inc.). The membrane was blocked with 5% skim
milk for 2 h at room temperature and incubated with a 1:1,000
dilution of anti-human MEK1/2 (cat. no. 8727S), phosphorylated
(p)-MEK1/2 (S217/221; cat. no. 9154S; both Cell Signaling
Technology, Inc.) and β-actin (cat. no. A5441; Sigma-Aldrich; Merck
KGaA) for 12 h at 4˚C. After washing three times (10 min each) with
0.1% Tween 20 in PBS at room temperature, the membrane was
incubated for 1 h at room temperature with a 1:5,000 dilution of
peroxidase-conjugated secondary antibody (cat. no. NA934; GE
Healthcare Life Sciences), and washed three more times with PBS
under the same conditions. The peroxidase activity of the secondary
antibody was detected with an enhanced ECL reagent (Immuno Star
Zeta; Wako Pure Chemical Industries, Ltd.) and chemiluminescence
detection system (ImageQuant LAS 4000mini; GE Healthcare Life
Sciences). Densitometry was performed using software (Image Quant
TL version 7.0; GE Healthcare Life Sciences). Expression of VEGFR2
on the ATC cells was examined with the same protocol. Anti-VEGFR2
antibody (cat. no. 9698; Cell Signaling Technology, Inc.) was used
as the primary antibody, and cultured human umbilical vein
endothelial cells (HUVECs; Kurabo Industries Ltd.) were used as a
positive control for VEGFR expression.
Cell cycle analysis by flow
cytometry
Flow cytometry was used to assess the cell cycle
distribution of OCUT-4 cells treated with sorafenib. OCUT-4 cells
(5×106/ml) treated with 200 nM sorafenib for 1 h were
collected after brief trypsinization, washed with PBS, and fixed
with 70% cold ethanol at 4°C for 2 h. The samples were then treated
with ribonuclease at 37°C for 15 min (cat. no. R4875;
Sigma-Aldrich; Merck KGaA) and 10 mg/l propidium iodide at 4°C for
30 min (Sigma-Aldrich; Merck KGaA), and analyzed using a cell
sorter (FACScan LSR II; BD Biosciences). Cell cycle distributions
were quantified using ModFit LT version 5.0 software (Verity
Software House).
Measurement of VEGF in the culture
medium
Cells of all four cell lines (1×105) were
seeded on a 10-mm plastic culture plate in 5 ml of fresh medium,
and cultured for 48 h. The supernatant was sampled and filtered
(MILLEX-GV Filter Unit; Merck KGaA) to prepare conditioned medium.
The concentration of VEGF in the conditioned medium was measured
via ELISA (cat. no. DVE00; LSI Medience Corporation) according to
the manufacturer's protocols. The concentration of VEGF in the
fresh medium was confirmed to be below the detectable level of the
assay.
Effects of ATC conditioned medium and
sorafenib on HUVECs
To investigate the effects of sorafenib on the
impairment of cancer-secreted VEGF-mediated tumor neovasculature,
the proliferation of HUVECs was measured following stimulation by
conditioned medium from the supernatant of all four ATC cells, in
the presence or absence of sorafenib. Cryopreserved HUVECs were
purchased and prepared according to the manufacturer's protocols
before use for the experiment. HUVECs were thawed and incubated at
37°C with 5% CO2 in low-serum primary culture medium
(HuMedia-EB2 plus 2% fetal bovine serum, 10 ng/ml human epidermal
growth factor, 1.34 µg/ml hydrocortisone, 50 µg/ml
gentamicin, 50 µg/ml amphotericin B, 5 ng/ml human
fibroblast growth factor-B and 10 µg/ml heparin; all Kurabo
Industries, Ltd.) to promote constant growth. Primary culture
medium was completely removed prior to each experiment. Prepared
HUVECs (3×103) were seeded in each well of a 96-well
plastic culture plate and left overnight in a total of 100
µl culture medium. Subsequently, 100 µl of either
fresh culture medium or conditioned medium was added to each well
along with 1 nM VEGF (positive control; rhVEGF-A165; Wako Pure
Chemical Industries, Ltd.), 1 nM VEGF-blocking antibody (block
control; bevacizumab; Chugai Pharmaceutical Co., Ltd.) and/or
sorafenib (1 or 10 nM). After culturing for a further 48 h, the
supernatant was discarded, and the HUVECs were stained with Mayer's
hematoxylin solution (Wako Pure Chemical Industries, Ltd.) for 30
min at room temperature. Each well was washed with tap water and
dried. The number of cells in the middle of the well was counted
under three separate high-power fields (magnification, ×40) of a
light microscope. The average counts of three separate experiments
were calculated.
Statistics
Statistical analysis was performed using SPSS
version 22 (IBM Corp.). The differences of variables were examined
using ANOVA (parametric) or Kruskal-Wallis (non-parametric) tests
to analyze differences between multiple groups. Tukey (parametric)
or Games-Howell (non-parametric) tests were used as post hoc tests.
P<0.05 was considered statistically significant.
Results
Effects of sorafenib on cell
viability
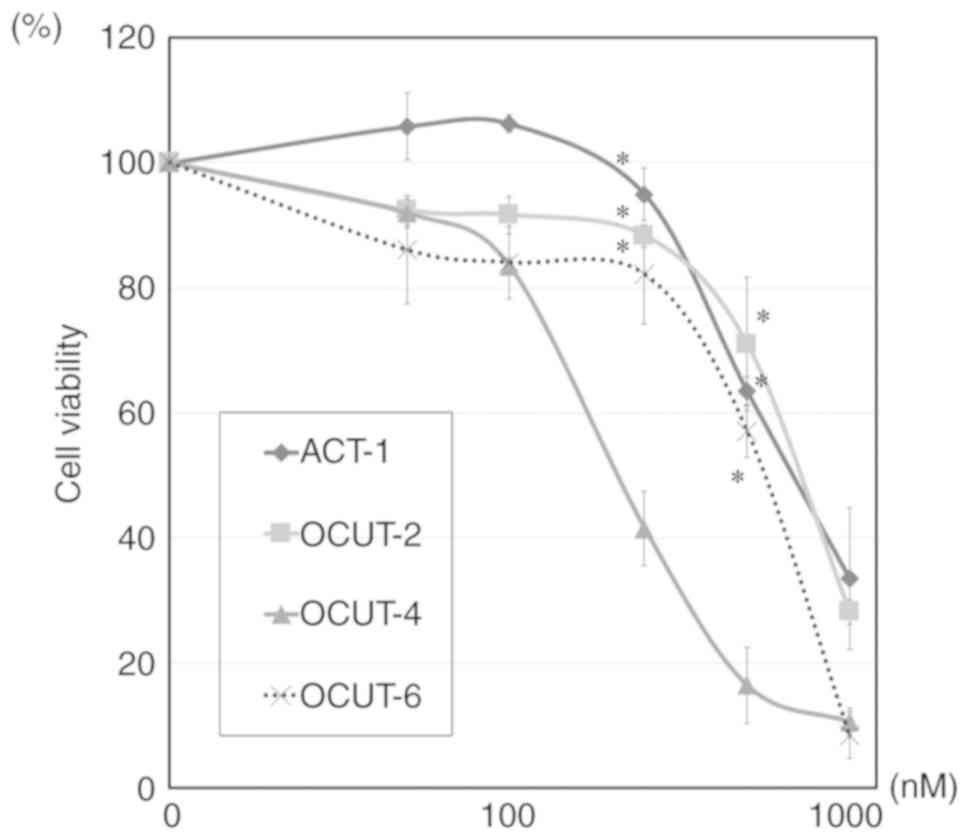
The inhibitory effects of sorafenib on ATC cells
were most pronounced in OCUT-4 cells, which possess BRAF
mutations only, when treated at moderately high concentrations
(>100 nM). Higher concentrations (>500 nM) of sorafenib were
required to observe equivalent inhibitory effects on cell viability
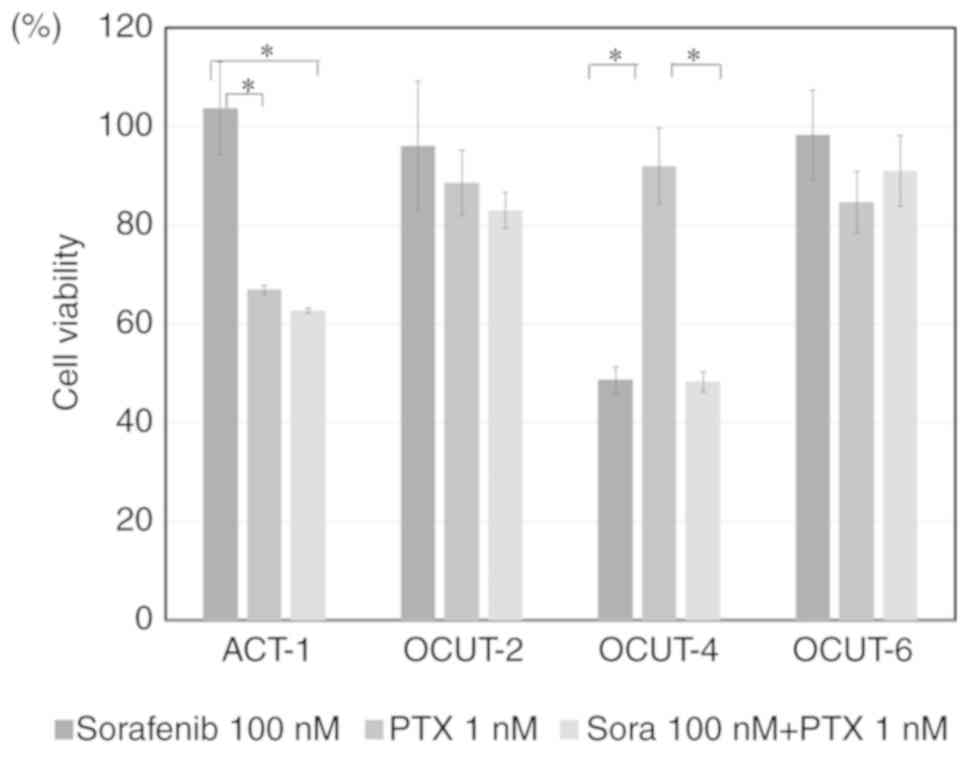
in the OCUT-2, OCUT-6 and ACT-1 cell lines (Fig. 2). Paclitaxel showed strong
inhibitory effects regardless of gene mutation status or
sensitivity to sorafenib (Table I;
only IC50 values are shown). With combined treatment of
sorafenib + paclitaxel, only additive effects were observed; no
synergistic effects were reported in any cell line investigated
(Fig. 3).
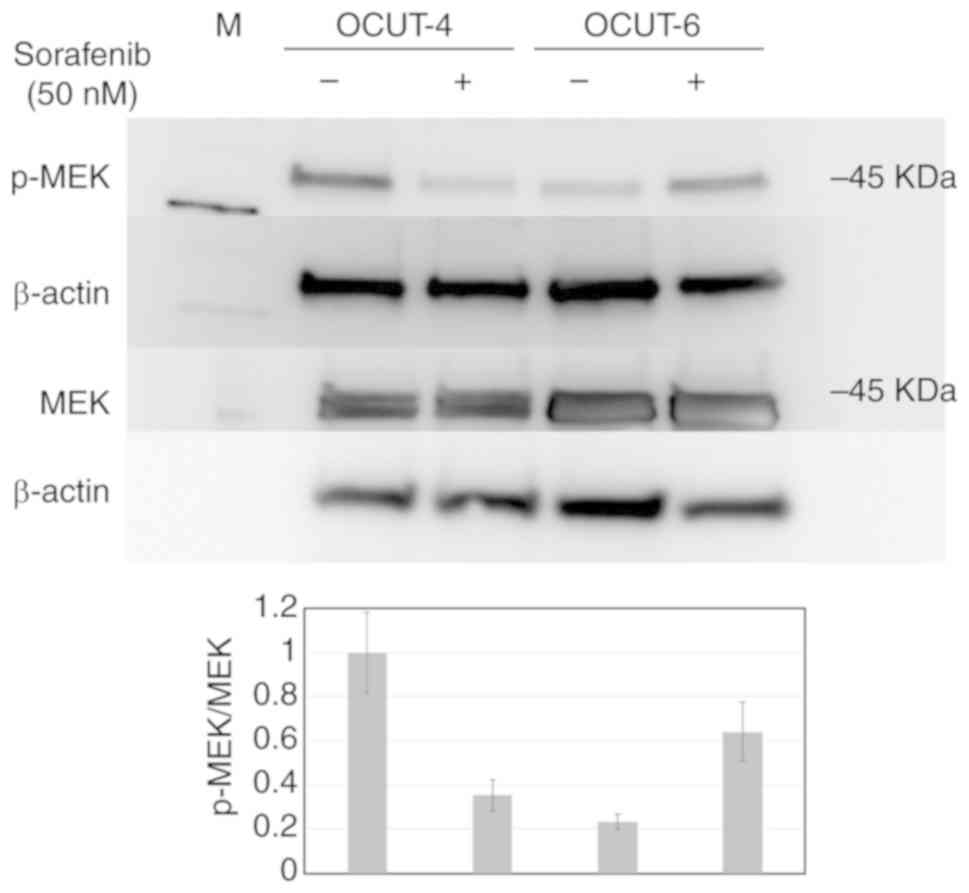
Effects of sorafenib on RAF/MEK
signaling
The mechanism via which sorafenib caused cellular
damage was investigated by evaluating alterations in downstream
signal transduction from the RAF gene and cell cycle
analysis. The phosphorylation of MEK, a direct downstream kinase of
RAF, was clearly down-regulated by sorafenib treatment in OCUT-4
cells. In contrast, p-MEK was upregulated by sorafenib in OCUT-6
cells, a sorafenib-insensitive cell line with a RAS mutation
(Fig. 4). Moreover, there was a
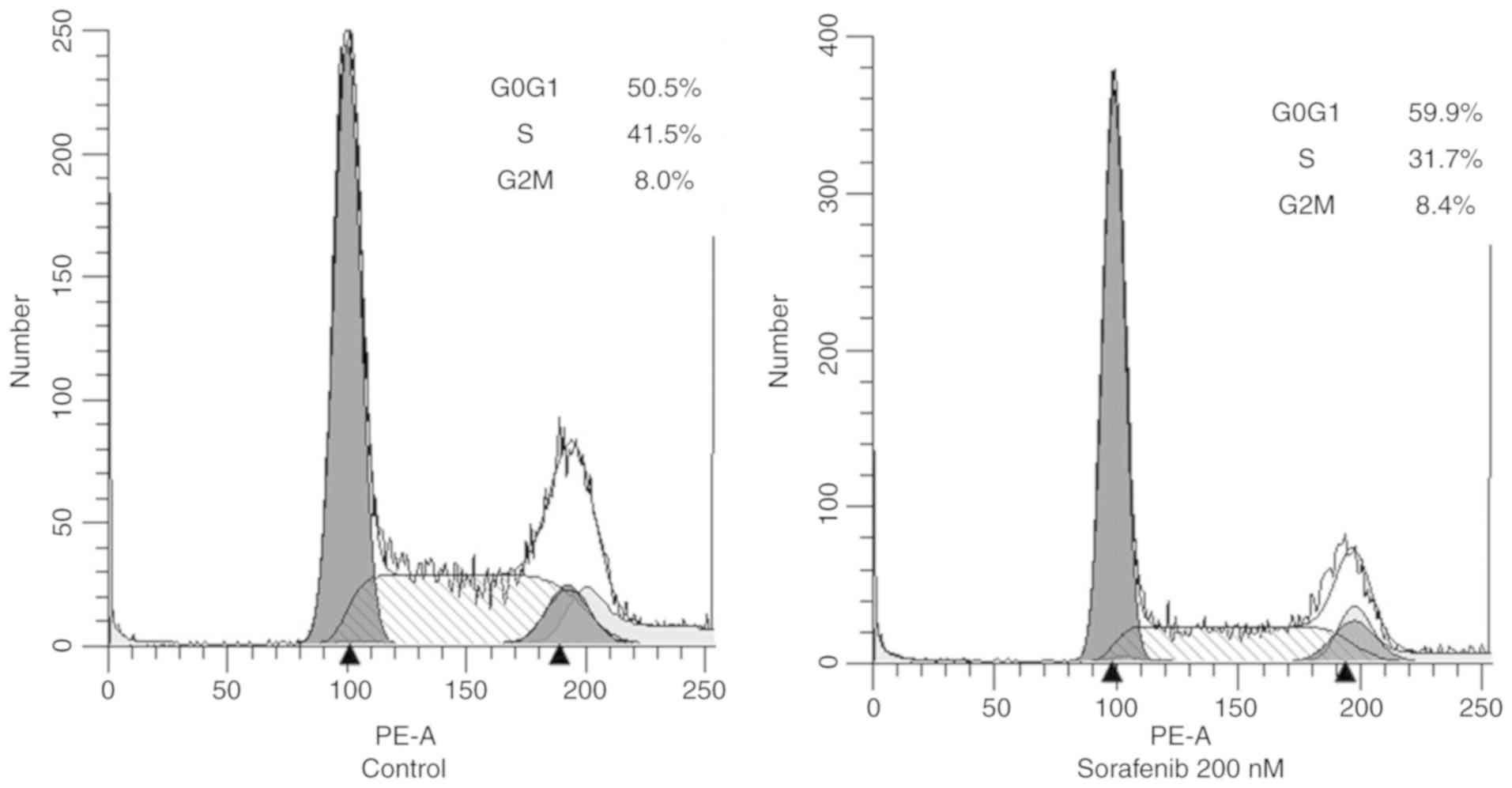
clear increase of the G0/G1-phase cell fraction (50.5 to 59.9%) and
a simultaneous decrease in the S-phase fraction (41.5 to 31.7%)
after treatment with sorafenib in OCUT-4 cells. No sub-G0 cell
fraction, indicative of apoptotic cell death, was identified after
sorafenib treatment (Fig. 5).
VEGF secretion by ATC cells
The secretion of VEGF was confirmed in every ATC
cell line examined. The concentration of VEGF varied among cell
lines without a clear association with the efficacy of sorafenib
(Table I). OCUT-4 cells secreted
the lowest concentration of VEGF in the culture medium but showed
the highest sensitivity to sorafenib. Moreover, the expression of
VEGFR could not be confirmed in any of the ATC cell lines
investigated (data not shown). Therefore, the existence of an
autocrine cell growth stimulation cascade mediated by VEGF and its
receptor was not identified in the experimental ATC cells.
Effect of VEGF and ATC cells on HUVEC
proliferation
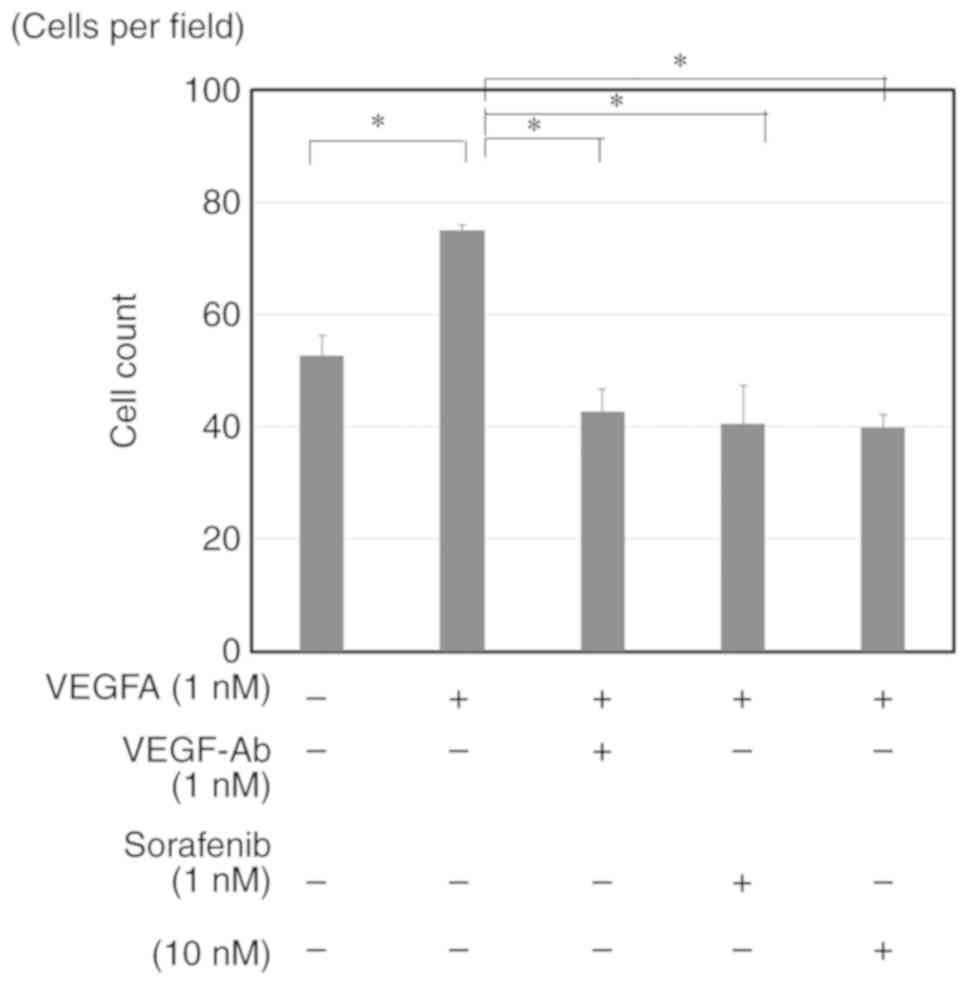
VEGF clearly stimulated the proliferation of HUVECs,
and this effect was completely blocked by sorafenib to a similar
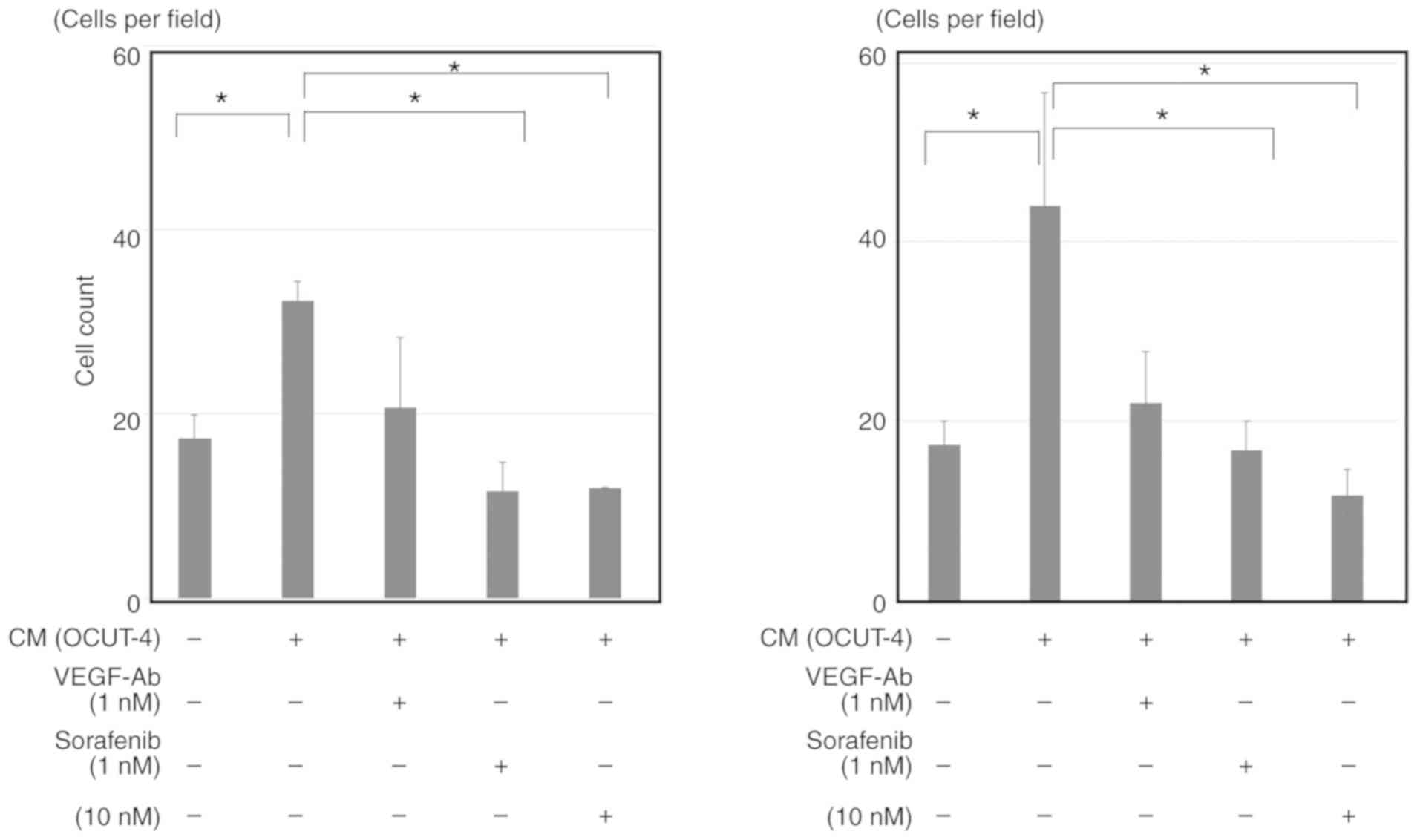
extent as observed with anti-VEGF antibody (Fig. 6). Moreover, the conditioned medium
of all ATC cell lines could also significantly stimulate the
proliferation of HUVECs (Fig. 7;
data of OCUT-2 and ACT-1 not shown). Although this stimulation was
not completely suppressed by VEGF blockade, sorafenib significantly
inhibited the conditioned medium-induced proliferation of HUVECs.
The inhibitory effect of sorafenib on VEGF/ATC cell-stimulated
HUVEC proliferation was evident within a much lower concentration
range (1-10 nM) than that required to reduce cancer cell viability
(>100 nM). Similar results were observed for all ATC cell lines
investigated, despite variation between cell lines in the quantity
of VEGF secreted in the conditioned medium.
Discussion
Sorafenib, a multi-kinase inhibitor, impairs the
signal transduction generated by RAF-family genes. Sorafenib
also inhibits the tyrosine kinase activities of VEGFR-1, -2, -3,
PDGFR-β, RET, c-Kit and Fms-like tyrosine kinase 3, and blocks
initiating downstream signals (14). Theoretically, and based on
experiments using various cancer cell lines, these mechanisms may
underlie aberrant cancer cell proliferation (26). ATC tumors commonly harbor driver
gene alterations in the RAS/RAF/MEK signaling cascade that
contribute to the aggressive proliferation of cancer cells
(23), and ATC cells often secrete
several growth factors or cytokines, such as VEGF, to establish
suitable microenvironments for cancer progression (27). Therefore, sorafenib may be a viable
agent for the treatment of ATC. Nevertheless, its detailed
mechanism of action in ATC warrants further investigation.
It was demonstrated that sorafenib was more
effective at regulating cellular proliferation in ATC cells
harboring BRAF mutations than cell lines possessing a
RAS mutation, or simultaneous BRAF and PI3KCA
mutations. The relationship between genetic abnormalities and
effects on cellular viability has been demonstrated using breast,
colon or pancreatic cancer cell lines (26), as well as ATC cell lines (28). Sorafenib has been shown to inhibit
cell lines possessing wild-type BRAF, or mutated BRAF
or CRAF. However, in all these previous reports, a high dose
of sorafenib was required to suppress the abundant growth signals
generated by mutated RAS, a gene upstream of RAF
(26). Consistent with these
reports, it was observed that >500 nM of sorafenib was required
to impair the viability of ATC cells with RAS mutations. Kim
et al (28) examined both
in vitro and in vivo effects of sorafenib in their
study using 5 ATC cell lines of known BRAF mutation status.
Although they reported the apoptotic cell death of ATC cells
irrespective of BRAF mutation status after treatment with
sorafenib, they used very high concentrations (>5,000 nM). Here,
the induction of cell cycle arrest in sorafenib-sensitive,
BRAF-mutated OCUT-4 cells by low concentrations of sorafenib
(50-200 nM) was demonstrated, potentially due to inhibition of MEK
phosphorylation. Thus, the main mechanism of cancer cell damage by
sorafenib appears to be due to cell cycle arrest via targeting RAF
as opposed to apoptosis induction. The present findings suggested
that ATC cells or tumors harboring BRAF mutations may be a
more viable target of sorafenib treatment.
The secretion of VEGF from ATC cells leads to
vigorous tumor neovascularization to enable aggressive cancer
growth (25). VEGF also affects
existing vessels to increase the permeability of the vascular wall,
facilitating the migration of cancer cells into vessels (29), a fundamental step for metastasis.
These effects are mediated by the phosphorylation of VEGFR on
vascular endothelial cells. Indeed, VEGFR expression on cancer
cells has been reported (28,30),
which enables accelerating proliferation via autocrine growth
factor signaling cascades. However, VEGFR expression was not
detected in the ATC cell lines used in the present study,
suggesting that the VEGF-VEGFR autocrine cascade may be limited in
ATC.
Kim et al (28) investigated intratumoral
microvessels in an orthotopic ATC xenograft model to demonstrate
the antiangiogenic effect of sorafenib. They concluded that
sorafenib has more effective anticancer effects by impairing
modifications to the tumor vasculature rather than direct effects
on cancer cells, based on pathological comparisons of the damage to
microvessels and cancer cells. Previous reports suggested that
impairment of the VEGF-mediated proliferation of the tumor
neovasculature is the primary anticancer mechanism of sorafenib
(26,28). However, the effects of sorafenib on
the interactions between cancer cells and vascular endothelial
cells are yet to be fully determined.
In the present study, the stimulation of HUVEC
proliferation by humoral factors secreted from ATC cells was
demonstrated, which was partially blocked by anti-VEGF blockade,
but more markedly inhibited by sorafenib. Although additional
studies to determine the involvement of other humoral factors such
as PDGF are required, these results indicated that the antitumor
effects of sorafenib are primarily due to the impairment of tumor
vascularization stimulated by humoral factors, including VEGF, from
ATC cells.
Clinical trials have not yet demonstrated a clear
therapeutic effect of sorafenib alone on ATC (13,14).
Based on the present findings: i) Sorafenib requires high dose to
show significant direct inhibition of cell growth; ii) the effects
of sorafenib are detected in the form of cell cycle arrest but not
apoptosis; and iii) activation of PI3K/AKT/mTOR signaling pathway
in addition to RAS/RAF/MEK, often found in ATC, can involve in
sorafenib resistance. These properties may result in inadequate
clinical efficacy in treating ATC using sorafenib. Several
synergistic combinations have been reported to enhance the
therapeutic effect of sorafenib (31,32).
The potential synergistic effect of paclitaxel, an effective drug
recommended to manage ATC clinically (33,34),
combined with sorafenib were evaluated; however, no synergistic
effect was observed. A previous study reported enhancement of the
antitumor effects of paclitaxel against ATC cells by lenvatinib
both in vitro and in vivo (35). They suggested not only the cell
viability, but also the apoptosis and G2-M cell cycle arrest
induced by paclitaxel were significantly enhanced with concomitant
lenvatinib treatment. However, it is difficult to directly compare
these results with the present findings, as high doses of
lenvatinib (40-fold higher than sorafenib used in this study) and
paclitaxel (250-fold higher concentrations as used in this study)
were required to demonstrate synergistic effects in the previous
study. Future directions may include evaluations of molecular
targeted drugs (5,36) or immune-checkpoint inhibitors
(37) as possible partners to
strengthen the efficacy of sorafenib for its clinical application
in treating ATC.
The present study possesses several limitations.
First, the experiments focused on the investigation of the cancer
cell-damaging mechanisms of sorafenib in a sorafenib-sensitive ATC
cell line, OCUT-4. Conversely, the mechanisms in
sorafenib-resistant cell lines were not investigated in detail;
further studies comparing the present results to those obtained in
other cell lines are required to confirm the proposed mechanisms of
action of sorafenib. Second, the effect of sorafenib on VEGF
secretion from ATC cells was not studied. This should be studied in
future experiments to clarify the specific action of sorafenib on
VEGFR. Third, the concentration of only VEGF in the conditioned
medium was measured. The involvement of other humoral factors
should be studied further, as well as their secretion levels
compared with VEGF, in order to reveal the most important factor or
factors responsible for cancer progression and sorafenib
sensitivity.
In conclusion, the present study suggested that
sorafenib more effectively inhibited RAF-generated growth
signals in ATC cells compared to those generated by its upstream
gene, RAS. ATC cells stimulated the growth of endothelial
cells via the secretion of humoral factors, including VEGF; this
effect was inhibited by sorafenib. Although a suitable partner for
clinically effective combination therapy should be identified to
improve clinical response, the present observations indicated that
sorafenib has a certain degree of therapeutic potential for the
management of ATC.
Funding
Part of this study was supported by a research grant
on the ‘Investigation of the efficacy of multi-kinase inhibitor in
anaplastic thyroid cancer' awarded by Bayer Medical Affairs (Osaka,
Japan).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
NO was involved in the conception of the study, and
SI, NO, YA, YT, TM, SK, TT and MO were involved in the design of
the study. SI, NO, SN, SK and YA performed experiments and the
statistical analysis. SI and NO wrote the initial draft of the
manuscript. NO, SN, YT, TM, SK, TT and MO were involved in
reviewing and editing the manuscript. NO supervised the study and
provided funding.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
NO received honoraria from Eisai, Bayer, and Sanofi,
and research funding from Eisai and Bayer. SN received research
funding from Eisai. SK received honoraria and research funding from
Eisai. SI, YA, YT, TM, TT and MO have nothing to declare. The
funders had no role in the design of the study; in the collection,
analyses, or interpretation of data; in the writing of the
manuscript, or in the decision to publish the results.
Acknowledgments
The authors thank Yayoi Matsukiyo (Osaka City
University) for preparing the experiment.
References
|
1
|
Sugitani I, Miyauchi A, Sugino K, Okamoto
T, Yoshida A and Suzuki S: Prognostic factors and treatment
outcomes for anaplastic thyroid carcinoma: ATC Research Consortium
of Japan cohort study of 677 patients. World J Surg. 36:1247–1254.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haymart MR, Banerjee M, Yin H, Worden F
and Griggs JJ: Marginal treatment benefit in anaplastic thyroid
cancer. Cancer. 119:3133–3139. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chung SH, Onoda N, Ishikawa T, Ogisawa K,
Takenaka C, Yano Y, Hato F and Hirakawa K: Peroxisome
proliferator-activated receptor gamma activation induces cell cycle
arrest via the p53-independent pathway in human anaplastic thyroid
cancer cells. Jpn J Cancer Res. 93:1358–1365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nobuhara Y, Onoda N, Yamashita Y, Yamasaki
M, Ogisawa K, Takashima T, Ishikawa T and Hirakawa K: Efficacy of
epidermal growth factor receptor-targeted molecular therapy in
anaplastic thyroid cancer cell lines. Br J Cancer. 92:1110–1116.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurata K, Onoda N, Noda S, Kashiwagi S,
Asano Y, Hirakawa K and Ohira M: Growth arrest by activated BRAF
and MEK inhibition in human anaplastic thyroid cancer cells. Int J
Oncol. 49:2303–2308. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Onoda N, Nakamura M, Aomatsu N, Noda S,
Kashiwagi S, Kurata K, Uchino S and Hirakawa K: Significant
cytostatic effect of everolimus on a gefitinib-resistant anaplastic
thyroid cancer cell line harboring PI3KCA gene mutation. Mol Clin
Oncol. 3:522–526. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kasaian K, Wiseman SM, Walker BA, Schein
JE, Zhao Y, Hirst M, Moore RA, Mungall AJ, Marra MA and Jones SJ:
The genomic and transcriptomic landscape of anaplastic thyroid
cancer: Implications for therapy. BMC Cancer. 15:9842015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Molinaro E, Romei C, Biagini A, Sabini E,
Agate L, Mazzeo S, Materazzi G, Sellari-Franceschini S, Ribechini
A, Torregrossa L, et al: Anaplastic thyroid carcinoma: From
clinicopathology to genetics and advanced therapies. Nat Rev
Endocrinol. 13:644–660. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yi H, Ye X, Long B, Ye T, Zhang L, Yan F,
Yang Y and Li L: Inhibition of the AKT/mTOR pathway augments the
anticancer effects of sorafenib in thyroid cancer. Cancer Biother
Radiopharm. 32:176–183. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Escudier B, Eisen T, Stadler WM, Szczylik
C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA,
et al: Sorafenib in advanced clear-cell renal-cell carcinoma. N
Engl J Med. 356:125–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brose MS, Nutting CM, Jarzab B, Elisei R,
Siena S, Bastholt L, de la Fouchardiere C, Pacini F, Paschke R,
Shong YK, et al: Sorafenib in radioactive iodine-refractory,
locally advanced or metastatic differentiated thyroid cancer: A
randomised, double-blind, phase 3 trial. Lancet. 384:319–328. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Savvides P, Nagaiah G, Lavertu P, Fu P,
Wright JJ, Chapman R, Wasman J, Dowlati A and Remick SC: Phase II
trial of sorafenib in patients with advanced anaplastic carcinoma
of the thyroid. Thyroid. 23:600–604. 2013. View Article : Google Scholar :
|
|
14
|
Ito Y, Onoda N, Ito KI, Sugitani I,
Takahashi S, Yamaguchi I, Kabu K and Tsukada K: Sorafenib in
japanese patients with locally advanced or metastatic medullary
thyroid carcinoma and anaplastic thyroid carcinoma. Thyroid.
27:1142–1148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tahara M, Kiyota N, Yamazaki T, Chayahara
N, Nakano K, Inagaki L, Toda K, Enokida T, Minami H, Imamura Y, et
al: Lenvatinib for anaplastic thyroid cancer. Front Oncol.
7:252017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sugitani I, Onoda N, Ito KI and Suzuki S:
Management of anaplastic thyroid carcinoma: The fruits from the ATC
research consortium of Japan. J Nippon Med Sch. 85:18–27. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brose MS, Worden FP, Newbold KL, Guo M and
Hurria A: Effect of age on the efficacy and safety of lenvatinib in
radio-iodine-refractory differentiated thyroid cancer in the phase
III SELECT trial. J Clin Oncol. 35:2692–2699. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zaman A, Wu W and Bivona TG: Targeting
oncogenic BRAF: Past, present, and future. Cancers (Basel).
11:E11972019. View Article : Google Scholar
|
|
19
|
Sanz-Garcia E, Argiles G, Elez E and
Tabernero J: BRAF mutant colorectal cancer: Prognosis, treatment,
and new perspectives. Ann Oncol. 28:2648–2657. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ascierto PA, Kirkwood JM, Grob JJ, Simeone
E, Grimaldi AM, Maio M, Palmieri G, Testori A, Marincola FM and
Mozzillo N: The role of BRAF V600 mutation in melanoma. J Transl
Med. 10:852012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schreuer M, Jansen Y, Planken S, Chevolet
I, Seremet T, Kruse V and Neyns B: Combination of dabrafenib plus
trametinib for BRAF and MEK inhibitor pretreated patients with
advanced BRAFV600-mutant melanoma: An open-label, single arm,
dual-centre, phase 2 clinical trial. Lancet Oncol. 18:464–472.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Planchard D, Besse B, Groen HJM, Souquet
PJ, Quoix E, Baik CS, Barlesi F, Kim TM, Mazieres J, Novello S, et
al: Dabrafenib plus trametinib in patients with previously treated
BRAF(V600E)-mutant metastatic non-small cell lung cancer: An
open-label, multicentre phase 2 trial. Lancet Oncol. 17:984–993.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pozdeyev N, Gay LM, Sokol ES, Hartmaier R,
Deaver KE, Davis S, French JD, Borre PV, LaBarbera DV, Tan AC, et
al: Genetic analysis of 779 advanced differentiated and anaplastic
thyroid cancers. Clin Cancer Res. 24:3059–3068. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang HM, Huang YW, Huang JS, Wang CH, Kok
VC, Hung CM, Chen HM and Tzen CY: Analastic carcinoma of the
thyroid arising more often from follicular carcinoma than papillary
carcinoma. Ann Surg Oncol. 14:3011–3018. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Onoda N, Nakamura M, Aomatsu N, Noda S,
Kashiwagi S and Hirakawa K: Establishment, characterization and
comparison of seven authentic anaplastic thyroid cancer cell lines
retaining clinical features of the original tumors. World J Surg.
38:688–695. 2014. View Article : Google Scholar
|
|
26
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43-9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Costache MI, Ioana M, Iordache S, Ene D,
Costache CA and Săftoiu A: VEGF expression in pancreatic cancer and
other malignancies: A review of the literature. Rom J Intern Med.
53:199–208. 2015.PubMed/NCBI
|
|
28
|
Kim S, Yazici YD, Calzada G, Wang ZY,
Younes MN, Jasser SA, El-Naggar AK and Myers JN: Sorafenib inhibits
the angiogenesis and growth of orthotopic anaplastic thyroid
carcinoma xenografts in nude mice. Mol Cancer Ther. 6:1785–1792.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
D'Haene N, Koopmansch C, Van Eycke YR,
Hulet F, Allard J, Bouri S, Rorive S, Remmelink M, Decaestecker C,
Maris C and Salmon I: The prognostic value of the combination of
low VEGFR-1 and High VEGFR-2 expression in endothelial cells of
colorectal cancer. Int J Mol Sci. 19:pii: E35362018. View Article : Google Scholar
|
|
31
|
Chen G, Nicula D, Renko K and Derwahl M:
Synergistic anti-proliferative effect of metformin and sorafenib on
growth of anaplastic thyroid cancer cells and their stem cells.
Oncol Rep. 33:1994–2000. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Zhang C, Ning Z, Xu L, Zhu X and
Meng Z: Bufalin enhances anti-angiogenic effect of sorafenib via
AKT/VEGF signaling. Int J Oncol. 48:1229–1241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Higashiyama T, Ito Y, Hirokawa M,
Fukushima M, Uruno T, Miya A, Matsuzuka F and Miyauchi A: Induction
chemotherapy with weekly paclitaxel administration for anaplastic
thyroid carcinoma. Thyroid. 20:7–14. 2010. View Article : Google Scholar
|
|
34
|
Onoda N, Sugino K, Higashiyama T, Kammori
M, Toda K, Ito K, Yoshida A, Suganuma N, Nakashima N, Suzuki S, et
al: The safety and efficacy of weekly paclitaxel administration for
anaplastic thyroid cancer patients: A nationwide prospective study.
Thyroid. 26:1293–1299. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jing C, Gao Z, Wang R, Yang Z, Shi B and
Hou P: Lenvatinib enhances the antitumor effects of paclitaxel in
anaplastic thyroid cancer. Am J Cancer Res. 7:903–912.
2017.PubMed/NCBI
|
|
36
|
Subbiah V, Kreitman RJ, Wainberg ZA, Cho
JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME,
Urbanowitz G, et al: Dabrafenib and trametinib treatment in
patients with locally advanced or metastatic BRAF V600-mutant
anaplastic thyroid cancer. J Clin Oncol. 36:7–13. 2018. View Article : Google Scholar
|
|
37
|
Gunda V, Gigliotti B, Ndishabandi D, Ashry
T, McCarthy M, Zhou Z, Amin S, Freeman GJ, Alessandrini A and
Parangi S: Combinations of BRAF inhibitor and anti-PD-1/PD-L1
antibody improve survival and tumour immunity in an immunocompetent
model of orthotopic murine anaplastic thyroid cancer. Br J Cancer.
119:1223–1232. 2018. View Article : Google Scholar : PubMed/NCBI
|