Introduction
Head and neck squamous cell carcinoma (HNSCC) is one
of the leading types of cancer by incidence worldwide (1). Although the prevalence of this type
of cancer is in a steady decline and treatment outcomes have
improved (2,3), the prognosis of patients with
advanced stages of HNSCC remains poor. Tumor resistance and drug
toxicity impair the clinical validity of available therapeutics,
particularly in advanced stages of the disease (4,5).
Tumor necrosis factor-α (TNF-α) is a potent tumor
suppressor cytokine that is a potential viable agent for tumor
biotherapy (6). TNF-α mediates
both pro- and anti-tumoral effects in the form of distinct or
overlapping functions via two major receptors, 55 kDa TNFR1 and 75
kDa TNFR2 (7). While TNFR1 is
ubiquitously expressed in normal and tumor tissues, the expression
of TNFR2 is restricted to immune cells (8). In addition to its apoptotic
proficiency, TNF-α is able to trigger the activity of the nuclear
transcription factor, nuclear factor-κB (NF-κB), which in turn
mediates the regulation of apoptotic inhibitor genes, such as
Bcl-2, Bcl-xL, cellular inhibitors of apoptosis (cIAPs), X-linked
inhibitor of apoptosis (xIAP) and cFLIP (9,10).
While TNF-α shows promise as a cancer therapeutic agent, and
clinical trials have shown that treatment with recombinant TNF
exerts antitumor effects, there is an alarming induction of
endotoxic shock and systemic toxicity following TNF-α
administration (11,12).
Apoptosis is the orderly process of programmed cell
death that occurs in multicellular organisms. This tightly
orchestrated process represents a vital component of cellular
functioning, and is necessary for normal cell turnover and
embryonic development. While apoptosis is a natural process that
can block cancer development, its deregulation is implicated in the
progression of tumorigenesis (13). The apoptotic process is driven by
three common pathways: i) The death receptor-mediated extrinsic
pathway; ii) the mitochondria-mediated intrinsic pathway; and iii)
the endoplasmic reticulum (ER) stress-mediated apoptotic pathway
(14,15). The extrinsic pathway is one of the
two major apoptotic pathways whose activation is initiated by
transmembrane receptor(s) through ligation to the corresponding
ligand(s) or agonist(s). These receptors include FasL/FasR,
TNF-α/TNFR1, Apo3L/DR3, Apo2L/DR4 and Apo2L/DR5, with the most
well-characterized receptors being the members of the TNF receptor
gene superfamily (16). The key
components of these apoptotic pathways are potential targets for
genetic and/or epigenetic alteration, which can lead to failure in
the cellular death machinery that is thought to be the main cause
of tumor progression and resistance (17,18).
Thus, the activation of apoptosis-associated pathways is a feasible
strategy for tumor prevention and treatment.
In the present study, we provide insight into the
mechanisms of TNF-α-induced opposing signals in HNSCC cells, and
describe in detail the mechanism through which TNF-α induces
apoptosis, with the aim to develop therapeutic strategies that can
minimize the systemic toxicity of this cytokine.
Materials and methods
Cells, cell culture and treatment
The HNSCC cell lines used in this study were the
following: i) The human oral squamous cell carcinoma cell line,
CLS-3540, obtained from CLS Cell Lines Service GmbH (Eppelheim,
Germany); and ii) the human nasal septum squamous cell carcinoma
cell line, RPMI2650, obtained from the American Tissue Culture
Collection (ATCC, Manassas, VA, USA). The CLS-354 cells were
cultivated and maintained in DMEM supplemented with 10% fetal
bovine serum and 2 mM L-glutamine, while RPMI2650 cells were
cultivated and maintained in EMEM media supplemented with 10% fetal
bovine serum and 2 mM L-glutamine. The treatment of the cells with
TNF-α (#C-63722; PromoCell GmbH, Heidelberg, Germany) was performed
at a concentration of 10 ng/ml for 48 h. In addition, 1 µM
inhibitor of ASK1 (thioredoxin), 5 µM inhibitor of NF-κB
(Bay-11-7082), 10 µM inhibitor of JNK (SP600125) (all from
Biomol GmbH, Loerrach, Germany), 25 µM of inhibitor of IRE1α
(irestatine; Axon Medchem, Reston, VA, USA) and 20 µM
inhibitor of caspase-8 (Z-IETD-FMK; R&D Systems, Minneapolis,
MN, USA) were added to the cell culture 1 h prior to exposure to
TNF-α for the indicated time period of 48 h.
Assessment of cell survival
The cells were exposed to the recommended
concentration of TNF-α (10 ng/ml) before the measurement of cell
viability using MTT assays (Roche, Bâle, Switzerland), as
previously described (19-21). Briefly, the HNSCC cell lines were
allowed to grow for 24 h prior to exposure to TNF-α (10 ng/ml) for
the indicated periods of time. The treated and control cells were
then incubated with 50 µl of MTT substrate (5 mg/ml) per
well for 3 h under normal cell culture conditions. Following the
removal of the culture media, the cells were lysed in 300 µl
of MTT lysis buffer and the relative cell number was assessed by an
ELISA reader (VersaMax ELISA Microplate Reader; Molecular Devices,
San Jose, CA, USA) at 570-590 nm.
RNA interference
siRNA specific to protein kinase RNA-like
endoplasmic reticulum kinase (PERK; SC-36213), siRNA to human Noxa
(SC-37306), as well as the scramble siRNA (included with each
respective siRNA) were obtained from Santa Cruz Biotechnology
(Santa Cruz, CA, USA). Knockdown experiments were carried out as
recommended by the manufacturer's instructions. The cells were
transfected with Lipofectamine 2000 as previously described
(19,22).
Western blot analysis
Western blot analysis was performed according to the
standard procedures. The treated and control cells were washed
twice with cold PBS and lysed with lysis buffer containing 25 mM
HEPES (pH 7.5), 0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA,
0.5 mM DTT, 20 mM β-glycerolphosphate, 0.1 mM sodium orthovanadate,
0.1% Triton X-100 and a protease inhibitor cocktail (Roche
Diagnostics, Mannheim, Germany). The protein concentration was
determined using Bradford Protein Assay (Bio-Rad, Hercules, CA,
USA). The separation of proteins (20 g per lane) was carried out by
12% of SDS-polyacrylamide gel electrophoresis (Bio-Rad). The
transfer of proteins to nitrocellulose membranes (Schleicher &
Schuell, Dassel, Germany) from SDS-PAGE was accomplished in a
Hoefer TE 62X Transphor II unit. The membranes were blocked with
Tris-buffered saline (TBS) buffer with 5% non-fat dry milk
(Bio-Rad) overnight at 4°C. The blots were incubated with the
following antibodies at the indicated dilutions: Anti-apoptosis
signaling regulating kinase 1 (ASK1; SC-7931), 1:500; anti-p-ASK1
(SC-109911), 1:1,000; anti-c-jun N-terminal kinase (JNK; SC-474),
1:1,000; anti-p-JNK (SC-6254), 1:1,000; anti-p38 (SC-535), 1:1,000;
anti-p-p38 (SC-7973), 1:1,000; anti-Noxa (SC-2697), 1:1,000;
anti-actin (SC-1615), 1:5,000; anti-Tom20 (SC-11415), 1:100;
anti-Bap31 (SC-18579), 1:500; anti-IRE1α (SC-20790), 1:500;
anti-PERK (SC-9477), 1:1,000; anti-activating transcription factor
4 (ATF-4; SC-200), 1:1,000; anti-IκB-α (SC-203), 1:500; anti-
p-IκB-α (SC-8404), 1:500 (all from Santa Cruz Biotechnology);
anti-p-IRE1α (PA1-16927; Thermo Fisher Scientific, West Palm Beach,
FL, USA), 1:1,000; anti-CHOP (#2895), 1:1,000; anti-caspase 3
(#7190), 1:1,000; anti-caspase-9 (#9501), 1:1,000; 1:500; anti-Fas
associated via death domain (FADD; #2782), 1:1,000;
anti-phospho-FADD (#2785), 1:500 anti-poly(ADP-ribose) polymerase
(PARP; #9542), 1:500; caspase-8 (#9496), 1:1,000;
anti-phospho-TRAF2 (#13908), 1:1,000 (all from Cell Signaling
Technology Inc., Danvers, MA, USA); anti-TRAF2 antibody (ab37118),
1:1,000; Abcam anti-p-IRE1 (ab48187), 1:1,000; anti-p-ATF-4
(ab28830), 1:1,000 (from Abcam, Cambridge, MA, USA), anti-eIF2α
antibody (LS-C285898EIF2), 1:1,000 and anti-phospho-eIF2α antibody
(LS-C191237), 1:1,000 (from LSBio, Seattle, WA, USA).
Immunofluorescence staining
The RPMI2650 cells were allowed to grow for 24 h
prior to exposure to TNF-α for the indicated periods of time. The
cells were then subjected to immunofluorescence staining as
described previously (19).
Primary antibodies, including anti-Noxa (SC-2697), 1:200;
anti-Tom20 (SC-11415), 1:200; anti-Bap31 (SC-18579), 1:200 (all
from Santa Cruz Biotechnology) were allowed to bind for 2 h at room
temperature. Subsequently, the cells were washed 3 times in PBS and
incubated with Alexa Fluor-labeled secondary antibodies, including,
Alexa Fluor 488 Dye and Alexa Fluor 555 Dye and Alexa Fluor 647 Dye
(all from Thermo Fisher Scientific) for 2 h at room temperature.
Following an additional 3 washes in PBS, the cells were mounted
using DAKO mounting medium. Photomicrographs were acquired on a
Leica fluorescence microscope (Leica, Wetzlar, Germany).
Electrophoretic mobility shift assay
(EMSA)
The details of EMSA have been described elsewhere
(19,23). Briefly, the nuclear extracts of the
treated and control cells were prepared by the addition of cell
lysis buffer [20 mM HEPES (pH 7.9), 10 mM NaCl, 0.2 EDTA, 2 mM DTT,
1 mM Na Vanadate, and 1 mM proteinase inhibitor], and the nuclei
were precipitated by centrifugation at 10,000 × g for 3 min and the
subsequent addition of nuclear lysis buffer [20 mM HEPES (pH 7.9),
1.5 mM MgCl2, 1 mM Na Vanadate, 420 mM NaCl, 0.2 EDTA, 2
mM DTT, 25% glycerol and 1 mM protease inhibitor]. The
double-stranded synthetic oligonucleotides carrying a defined
binding site for AP-1, p53 and NF-κB (Santa Cruz Biotechnology),
ATF-3 (5′-GTGACGT[AC] [AG]-3′) were then end-labeled with
[y32P] ATP (Hartmann Analytika, Munich, Germany) in the
presence of T4 polynucleotide kinase (GeneCraft, Münster, Germany).
Briefly, a reaction volume of 20 µl containing 10 pmol (2
µl) of oligonucleotides carrying a defined binding site of
the above mentioned transcription factors, 5 µCi of
[y32P] ATP (5 µl), 4 µl T4 kinase buffer
(5X) and 1 µl T4 kinase and 3 µl H2O.
Following incubation at 37°C for 30 min, the labeled
oligonucleotides were purified using the oligonucleotide
purification kit (Qiagen, Hilden, Germany) and stored at -20°C
until use. Approximately 4 µg of nuclear extracts were bound
to a labeled probe in a total volume of 30 µl for 30 min at
room temperature in binding buffer (10 mM Tris, pH 7.5; 50 mM NaCl,
1 mM EDTA; 1 mM MgCl2; 0.5 mM DTT and 4% glycerol). The
specificity of the binding was analyzed by competition with an
unlabeled oligonucleotide assay. The competition assay was
performed in the same manner, except that unlabeled probes
containing oligonucleotide sequences (binding sites) were incubated
with nuclear extracts for 20 min at room temperature before adding
the labeled probes. Electrophoresis was performed for 3 h at 100 V
in 0.5 X Tris-borate-EDTA running buffer at room temperature. The
dried gel was visualized by exposure to high performance
autoradiography film.
Detection of apoptosis using Annexin
V/propidium iodide
The analysis of the TNF-α-induced apoptosis of the
CLS-354 and RPMI2650 cells was performed by flow cytometry using
Annexin V-FITC/propidium iodide (PI); (Vybrant; Invitrogen,
Karlsruhe, Germany). Following the incubation of treated and
control cells with Annexin/PI for 15 min at room temperature with
protection from light, the cells were measured as previously
described (19,21). Cells stained with Annexin were
identified as early apoptotic cells, while cells stained with both
Annexin and PI were identified as late apoptotic cells.
Measurement of mitochondrial membrane
potential (Δψm) using JC-1
The CLS-354 and RPMI2650 cells were stained with 10
µM JC-1 for 30 min at room temperature in the dark. The
intensities of red (>550 nm) and green (520-530 nm) fluorescence
of 50,000 individual cells were analyzed by flow cytometry as
previously described (19,24). At a low Δψm, JC-1 is predominantly
a monomer that yields green fluorescence with the emission of
530±15 nm, while at a high Δψm, the dye aggregates yielding a red
to orange colored emission (590±17.5 nm). Therefore, a decrease in
the aggregate fluorescent count is indicative of depolarization,
whereas an increase is indicative of hyperpolarization.
Statistical analysis
Data were expressed as the means ± SD. Statistical
comparisons were performed with one-way ANOVA followed by Dunnett's
test. Statistical differences between 2 groups were determined
using the unpaired Student's t-test. P<0.05 was considered to
indicate a statistically significant difference. All statistical
analyses were performed using SPSS statistical software (version
16.0; SPSS, Chicago, IL, USA).
Results
TNF-α-induced apoptosis of HNSCC cells is
mediated by mitochondrial-dependent mechanisms
Initially, we determined the IC50 value
of TNF-α in the HNSCC cells. The CLS-354 and RPMI2650 cell lines
were allowed to grow for 24 h prior to exposure to various
concentrations of TNF-α (1.0, 2.0, 3.0, 4.0, 5.0, 10, 15 and 20
ng/ml) for a time period of 24 h or 48 h, and the viability of both
treated and control cells was then measured by cell viability assay
following incubation with MTT substrate. MTT assay revealed an
IC50 value of TNF-α (10 ng/ml) which was able to induce
a 50% decrease in the cell number after 48 h in both the CLS-354
and RPMI2650 cells, when compared to the untreated controls (data
not shown). We then performed a time course experiment to confirm
the effect of the determined IC50 value of TNF-α on the
viability of HNSCC cells by MTT assay. The CLS-354 and RPMI2650
cell lines were cultured for 24 h prior to exposure to TNF-α (10
ng/ml) for regulated time intervals up to 72 h. The data obtained
from MTT assay (Fig. 1A) revealed
a time-dependent reduction in cell viability in both the CLS-354
and RPMI2650 cells in response to treatment with TNF-α.
TNF-α-induced cell growth inhibition was noted first at 12 h
post-treatment and increased thereafter to reach a maximum by 72 h.
After the optimal concentration of TNF-α (10 ng/ml) and time (48 h)
had been determined, we set out to perform all functional
experiments under the same conditions.
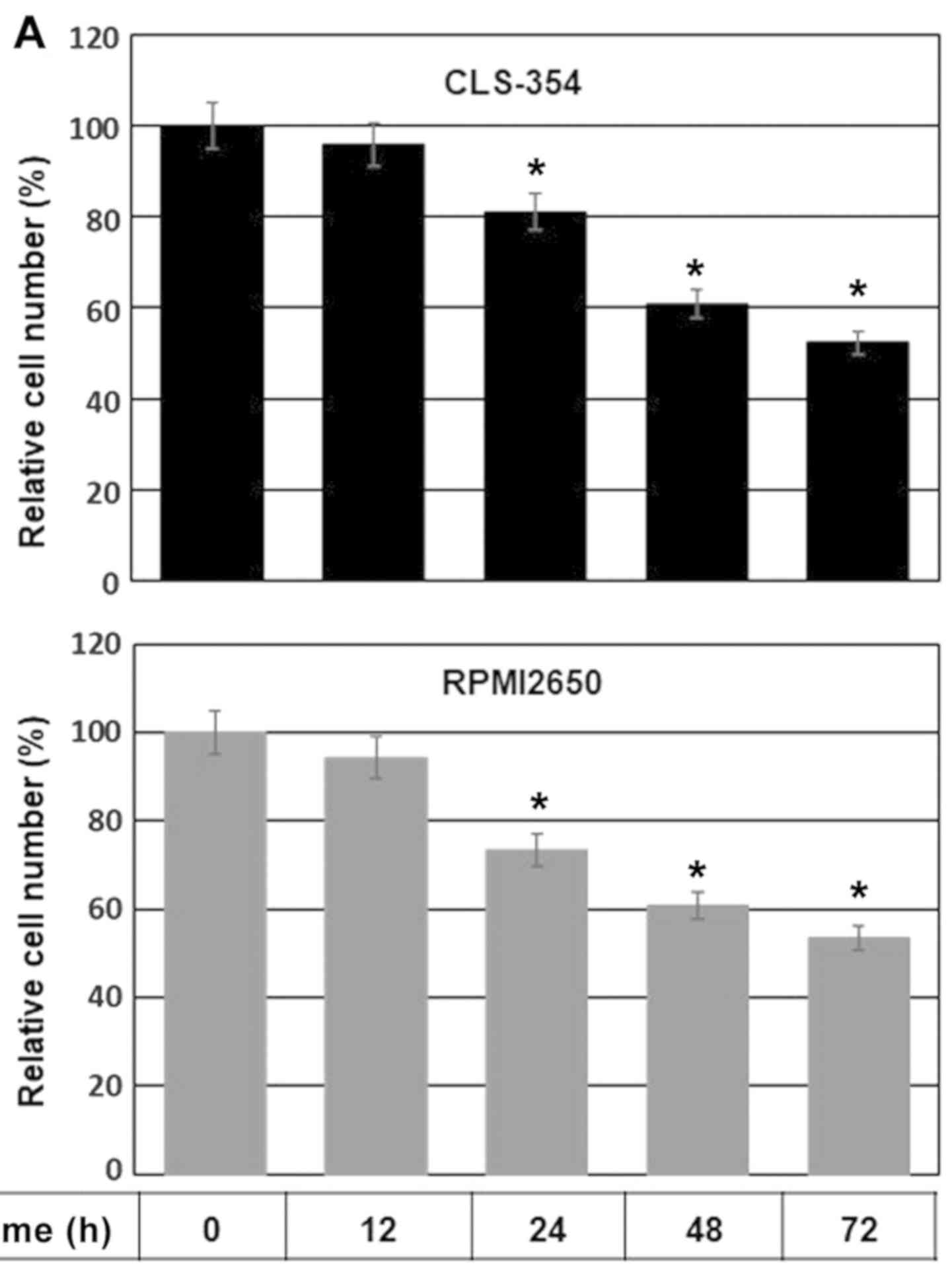 | Figure 1(A) Time course-dependent inhibition
of the growth rate of head and neck squamous cell carcinoma (HNSCC)
cells in response to exposure to TNF-α. Relative cell number (%)
assessed by MTT assay following the exposure of the HNSCC cell
lines, CLS-345 and RPMI2650, to TNF-α (10 ng/ml) for regulated time
intervals up to 72 h. Data are presented as the means ± SD (n=4).
*P<0.05, significantly different from the control as
shown by ANOVA and Dunnett's test. (B) Flow cytometric analysis
using Annexin V/propidium iodide (PI) staining demonstrating the
TNF-α-induced apoptosis of the HNSCC cell lines, CLS-345 and
RPMI2650. (C) Data of of Annexin V/PI are presented as the means ±
SD (n=3). *P<0.05, significantly different from the
control as shown by ANOVA and Dunnett's test. (D) Flow cytometric
analysis using JC-1 staining demonstrating the loss of
mitochondrial membrane potential (Δψm) in TNF-α-treated cells. (E)
Data of JC-1 staining are presented as the means ± SD (n=3). In
each treatment group, the first one of the two bars represents the
portion of the cells that do not show loss of mitochondrial
membrane potential, while the second bar represents the portion of
cells that show the loss of mitochondrial membrane potential.
*P<0.05, significantly different from the control as
shown by ANOVA and Dunnett's test. (F) Western blot analysis
demonstrating the induction of Noxa expression, the release of
cytochrome c (Cyt.c), and the cleavage of caspase-9,
caspase-3 and PARP in response to the treatment of the HNSCC cell
lines, CLS-345 and RPMI2650, with TNF-α for 48 h. Actin was used as
an internal control for loading and transfer. (G) Analyses of band
intensity on films are presented as the relative ratio of Noxa to
actin, released Cyt.c to actin, cleaved caspase-9 (Cl.Casp.9) to
actin, cleaved caspase-3 (Cl.casp.3) to actin and cleaved PARP
(Cl.PARP) to actin. Bars represent the means ± SD (n=3).
*P<0.05 vs. control. |
Subsequently, we investigated whether the
TNF-α-induced death of HNSCC cells is mediated by an apoptotic
mechanism. Accordingly, we analyzed the TNF-α-induced cell death of
both the CLS-354 and RPMI2650 cells using an apoptosis-specific
assay. According to apoptosis-specific protocol, early apoptotic
cells are Annexin V-positive and PI-negative, while late apoptotic
cells are Annexin V-positive and PI-positive. In accordance with
the protocol, our results revealed a significant increase in the
percentage of early cell death in both the CLS-354 (16.33%) and
RPMI5026 (15.77%) cells, while the level of late apoptotic cells
was ~3.42% in the CLS-354 cells and 2.62% in the RPMI5026 cells
following treatment with TNF-α for 48 h (Fig. 1B). However, statistical analysis of
the 3 independent experiments demonstrated a significant elevation
of early apoptosis in both cell lines (P<0.05; Fig. 1C), an evidence of the involvement
of an apoptotic mechanism in the regulation of the TNF-α-induced
death of HNSCC cells.
To determine whether TNF-α-induced apoptosis is
associated with mitochondrial dysregulation, the RPMI2650 and
CLS-354 cells were treated with TNF-α for 48 h, and the treated and
control cells were then subjected to flow cytometric analysis using
JC-1 staining. As expected, the results of flow cytometric analysis
(Fig. 1D and E) demonstrated the
loss of Δψm in both cell lines when compared to the control cells,
suggesting that the TNF-α-induced apoptosis of HNSCCs is mediated
by a mitochondrial dysregulation-dependent mechanism. Based on its
role in the modulation of mitochondrial dysregulation (21), we set out to analyze the expression
of the pro-apoptotic protein, Noxa, in the treated and control
cells. The analysis of the total cell lysates of the RPMI2650 and
CLS-354 cells using western blot analysis (Fig. 1F and G) revealed the induction of
Noxa protein in response to treatment with TNF-α, suggesting an
important role for Noxa protein in the TNF-α-induced apoptosis of
HNSCC cells. We then confirmed the TNF-α-induced apoptosis of HNSCC
cells at the molecular level. Accordingly, we analyzed hallmarks of
apoptosis, such as cytochrome c, caspase-9, caspase-3, and
PARP by western blot analysis. Treatment of both the RPMI2650 and
CLS-354 cells with TNF-α was found to enhance the release of
mitochondrial cytochrome c into the cytoplasm, as well as
the cleavage of caspas-9, caspase-3 and PARP (Fig. 1F and G).
TNF-α triggers the activation of pro- and
anti-apoptotic-dependent pathways in HNSCC cells
To further investigate the mechanisms associated
with the TNF-α-induced effects on HNSCC-derived cell lines, the
RPMI2650 and CLS-354 cells were treated with TNF-α for 48 h. Total
cell lysate and nuclear extracts were prepared for western blot
analysis and EMSA, respectively. We analyzed the possible proteins
that can be targeted by the activation of the TNF receptor, such as
FADD, TRAF2 and caspase-8, as well as the regulatory proteins of
the NF-κB and MAP kinase pathways. Treatment of both the RPMI2650
and CLS-354 cells with TNF-α enhanced the phosphorylation levels of
FADD and TRAF2, the degradation of IκBα, and the cleavage of
caspase-8, without any alterations in the expression levels of FADD
or TRAF2 (Fig. 2A and B). In
addition, treatment of both the RPMI2650 and CLS-354 cells with
TNF-α induced the phosphorylation of ASK1, JNK and p38, without
influencing their basal expression (Fig. 2C and D). The analysis of the
nuclear extracts of the TNF-α-treated and control cells using EMSA
revealed the activation of AP-1 (Fig.
2E), ATF-2 (Fig. 2F), p53
(Fig. 2G) and NF-κB (Fig. 2H), suggesting an important role for
these transcription factors in the modulation of TNF-α-induced
effects in HNSCC derived cell lines.
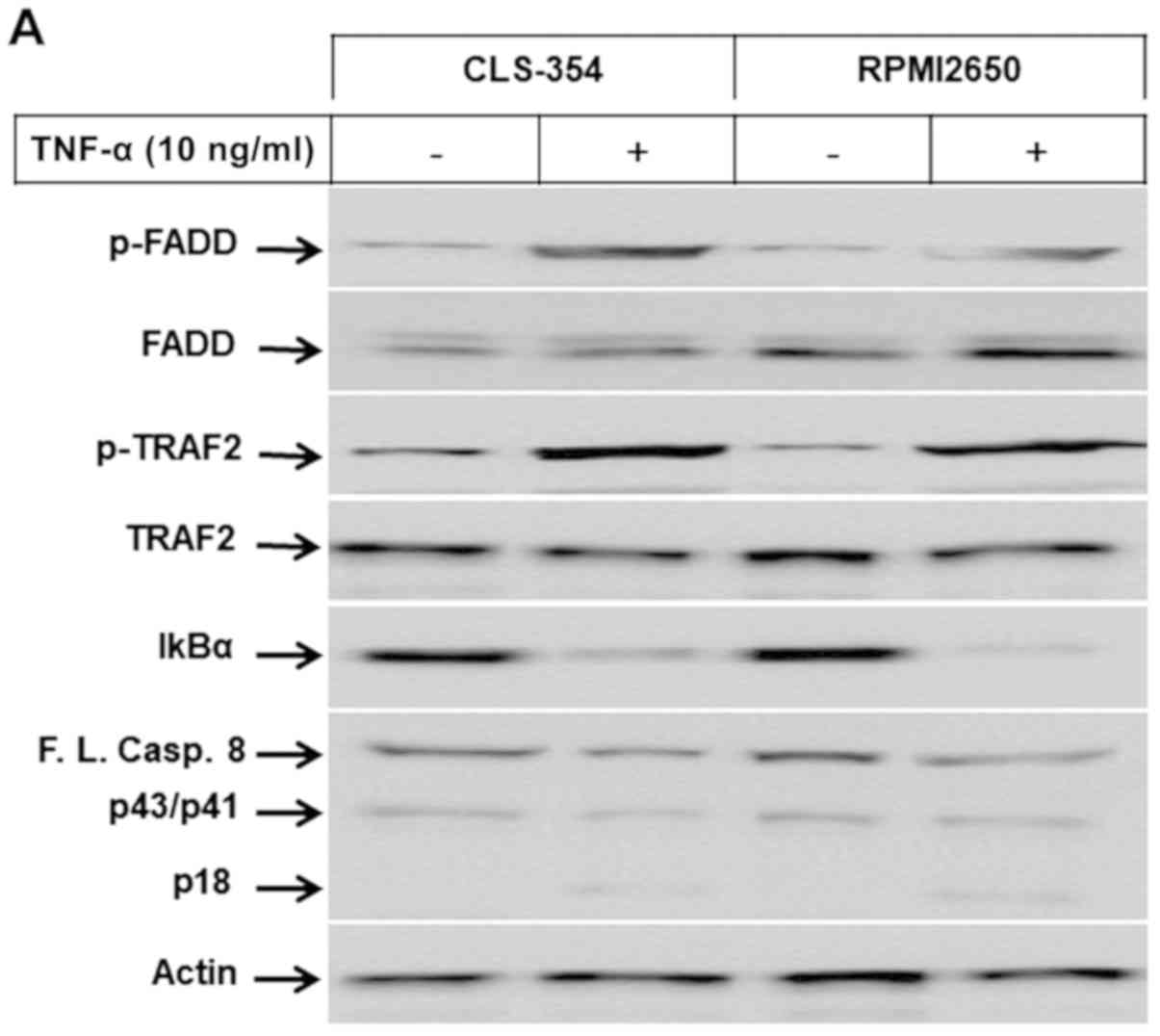 | Figure 2(A) Western blot analysis
demonstrating the phosphorylation of FADD and TRAF2 proteins, and
the degradation of IκBα and the cleavage of caspase-8 in response
to the exposure of head and neck squamous cell carcinoma (HNSCC)
cell lines, CLS-345 and RPMI2650, to TNF-α. Actin was used as an
internal control for loading and transfer. (B) Analyses of band
intensity on films are presented as the relative ratio of p-FADD to
actin, p-TRAF2 to actin, IκBα to actin and cleaved caspase-8/p18
(Cl.casp.8/p18) to actin. Bars represent the means ± SD (n=3).
*P<0.05 vs. control. (C) Western blot analysis
demonstrating the phosphorylation of ASK1, JNK and p38 kinase in
response to the exposure of the HNSCC cell lines, CLS-345 and
RPMI2650, to TNF-α. Actin was used as an internal control for
loading and transfer. (D) Analyses of band intensity on films are
presented as the relative ratio of p-ASK1 to actin, p-JNK to actin
andp-p38 to actin. Bars represent the means ± SD (n=3).
*P<0.05 vs. control. EMSA demonstrates the
enhancement of the DNA-binding activity of the transcription
factor, (E) AP-1. EMSA demonstrates the enhancement of the
DNA-binding activity of the transcription factors, (F) ATF-2, (G)
p53 and (H) NF-κB in response to the treatment of the HNSCC cell
lines, CLS-345 and RPMI2650, with TNF-α. Data are representative of
3 independent experiments. |
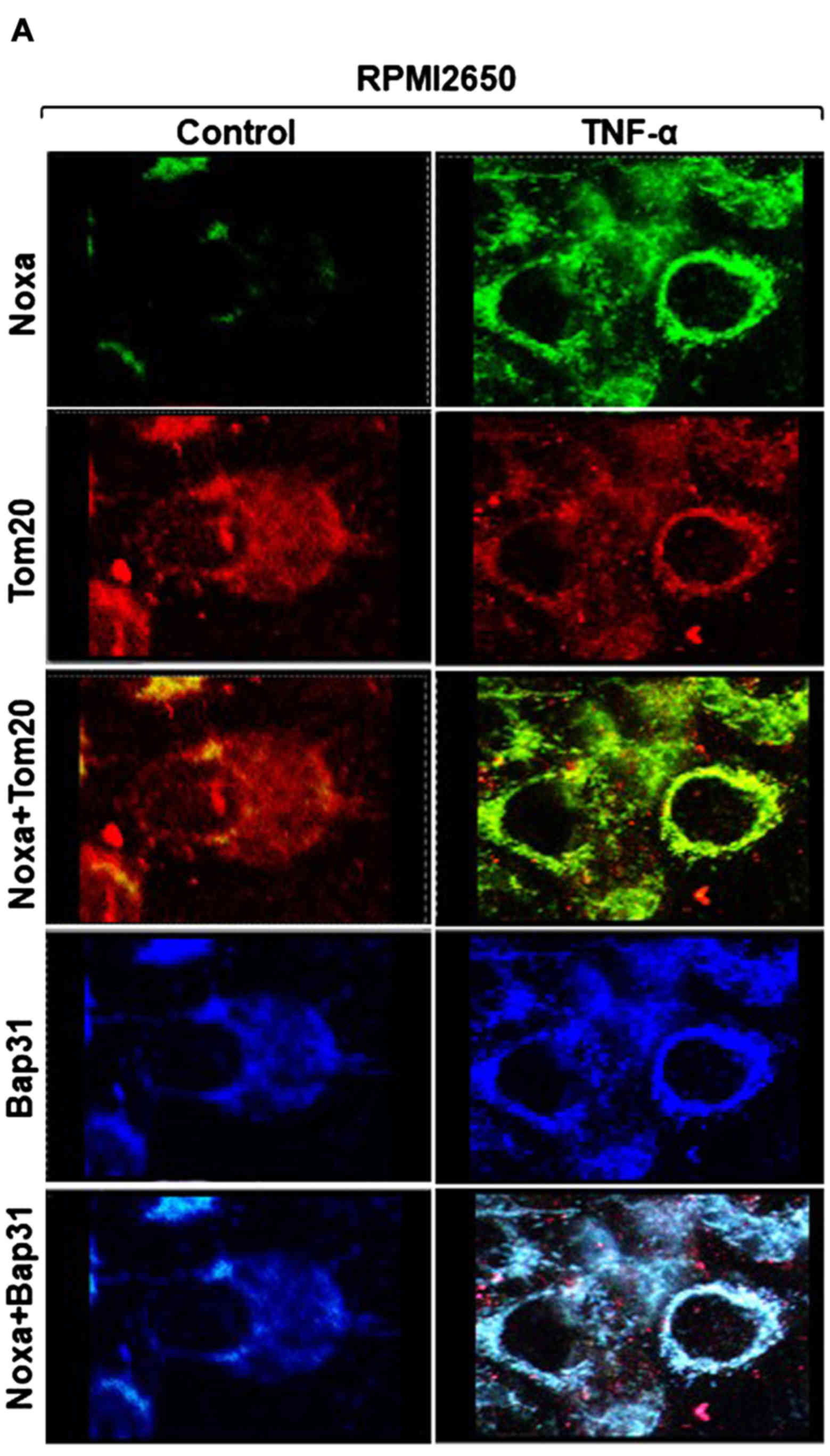
Subcellular localization of TNF-α-induced
Noxa protein to both the mitochondria and ER
To investigate whether TNF-α-induced Noxa protein is
localized to the mitochondria and/or the ER, the RPMI2650 and
CLS-354 cells were treated with the indicated concentration of
TNF-α for 48 h, and the subcellular localization of Noxa protein
was then analyzed in both treated and control cells using
immunofluorescence staining and western blot analysis of
mitochondrial and ER fractions. Immunofluorescence staining using
anti-Noxa, anti-Tom20 (marker for mitochondria) and anti-Bap31
(marker for ER) antibodies revealed the expression of Noxa protein
(green) in the treated RPMI2650 cells, whereas the expression of
Tom20 and Bap31 proteins was observed in both the treated and
control cells (Fig. 3A). As
expected, the merge of Noxa and Tom20 revealed the subcellular
localization of Noxa protein to the mitochondria, and the merge of
Noxa and Bap31 confirmed the subcellular localization of Noxa
protein to ER (Fig. 3A). We
further confirmed the subcellular localization of TNF-α-induced
Noxa protein via the analysis of mitochondrial and ER fractions of
the treated and control cells (Fig. 3B
and C). First, we assessed the purity of both mitochondrial and
ER fractions by western blot analysis using anti-Tom20 and
anti-Bap31, respectively. The detection of Noxa protein in the
mitochondrial fractions of TNF-α-treated cells confirmed the
subcellular localization of Noxa protein to the mitochondria, while
the detection of Noxa proteins in the ER fraction of TNF-α-treated
cells confirmed the subcellular localization of Noxa protein to the
ER (Fig. 3B and C). These data
suggest an important role for the subcellular localization of Noxa
in the modulation of TNF-α-induced apoptosis via a mechanism
mediated by mitochondrial dysregulation and ER stress-dependent
pathways.
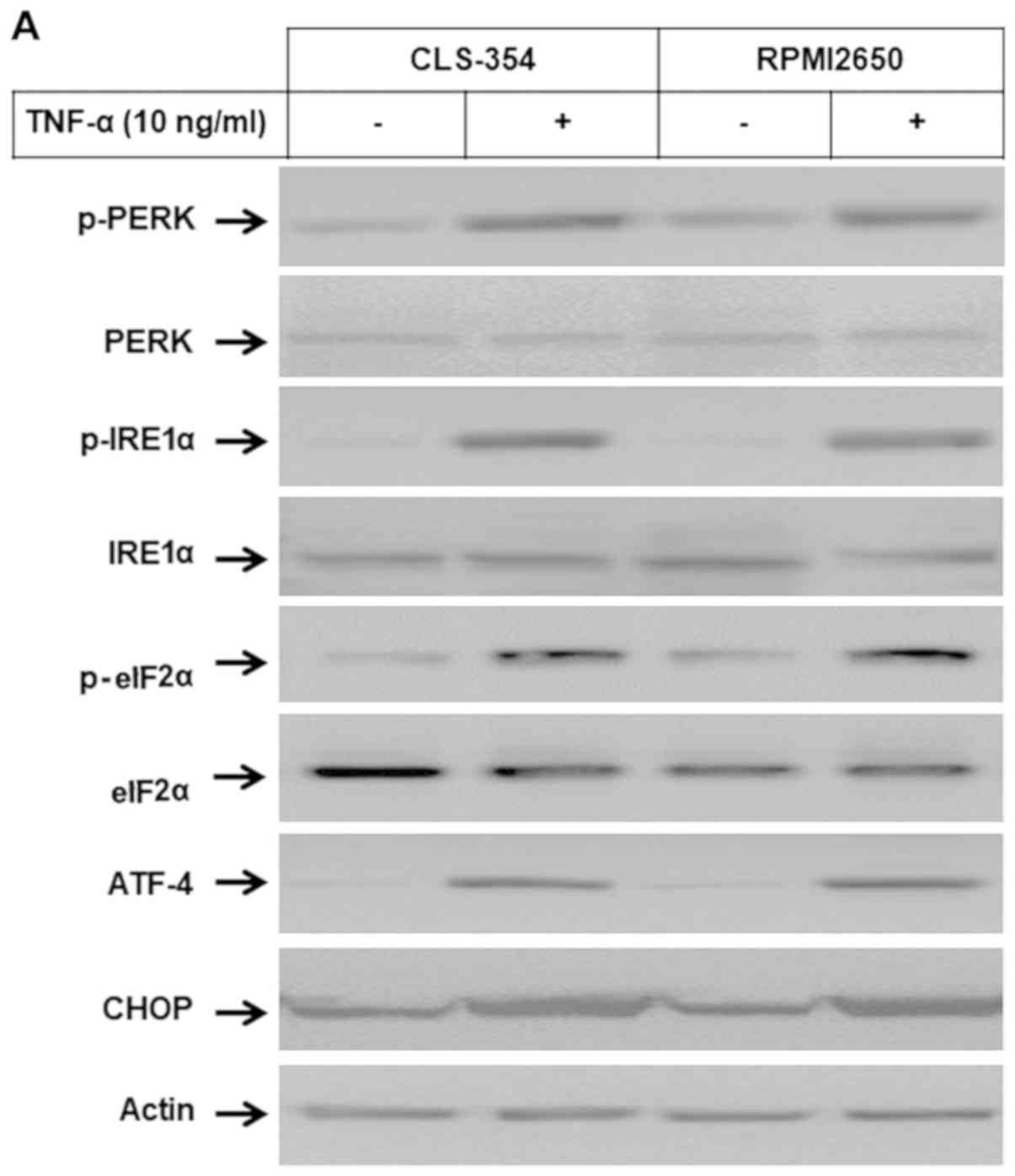
Induction of ER stress-associated
pathways by TNF-α in HNSCC cells
Western blot analysis and EMSA of the total cell
lysates and nuclear extracts of the TNF-α-treated and control cells
were used to examine whether the subcellular localization of Noxa
proteins to ER influences the ER stress-dependent pathways. Western
blot analysis (Fig. 4A and B)
demonstrated the ability of TNF-α to enhance the phosphorylation of
PERK, IRE1α, eIf2α and ATF-4 proteins together with the expression
of CHOP, without influencing the expression levels of PERK, IRE1α
eIf2α or ATF-4 proteins. EMSA (Fig.
4C) revealed the enhancement of the DNA-binding activity of the
transcription factor ATF-3 in response to treatment of the HNSCC
cells with TNF-α. Taken together, these data address an essential
role for Noxa protein in the modulation of TNF-α-induced ER stress
and subsequent apoptosis.
TNF-α-induced apoptosis is mediated by
mitochondrial- and non-mitochondrial-dependent pathways and is
enhanced by the inhibition of NF-κB
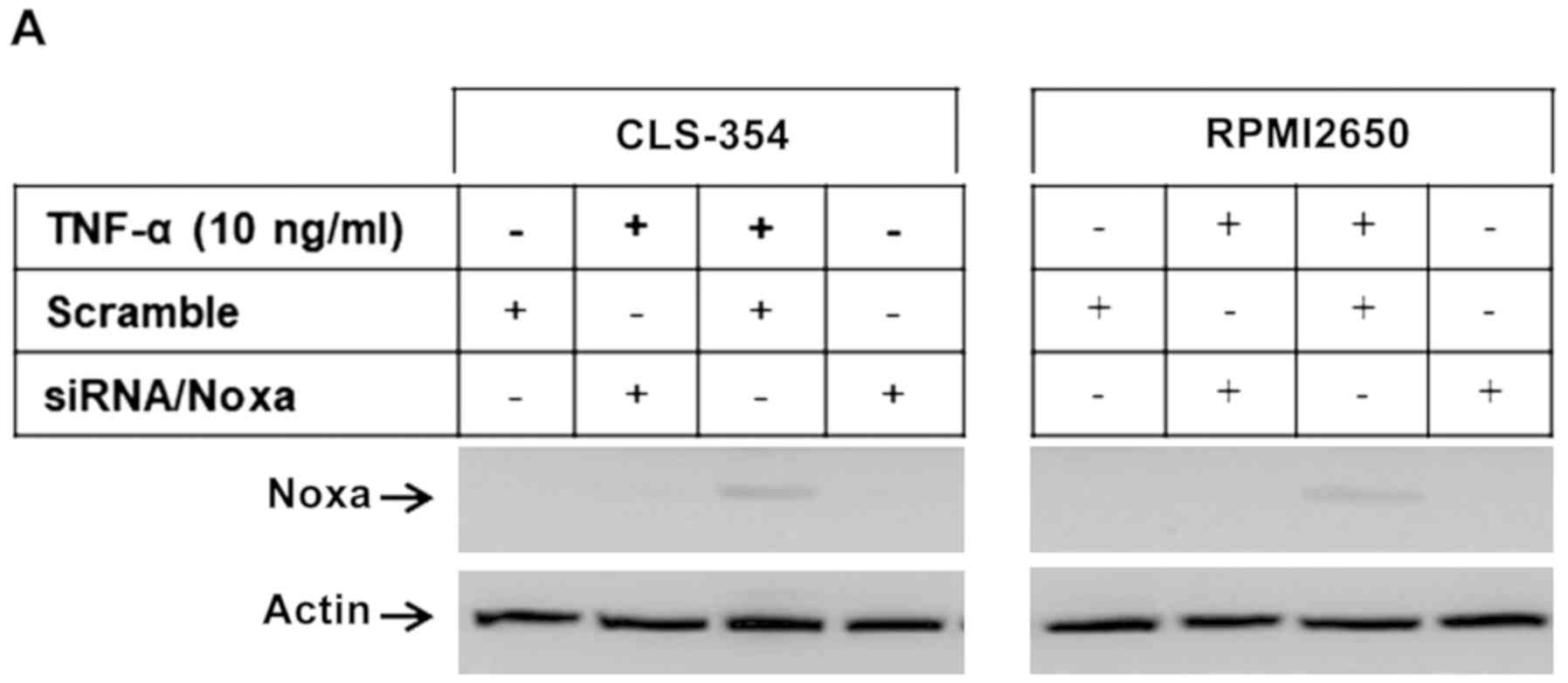
We then wished to elucidate the pathways essential
for the modulation of TNF-α-induced apoptosis. The RPMI2650 and
CLS-354 cell lines were transfected with PERK-specific or
Noxa-specific siRNAs, or treated with the inhibitors of caspase-8
(I.Casp.8), ASK1 (thioredoxin), JNK (SP600125), IRE1α (Irestatin),
or NF-κB (Bay11-7082) prior to exposure to TNF-α for 48 h. First,
we examined the efficiency of the specific siRNAs against Noxa and
PERK using western blot analysis of the transfected and control
cells before and after exposure to TNF-α. Western blot analysis
revealed the abrogation of Noxa (Fig.
5A and B) and PERK (Fig. 5C and
D) and TNF-α-induced Noxa expression. We then examined the
viability of the treated and control cells by MTT assay (Fig. 5E). There was a marked inhibition of
the viability of both the RPMI2650 and CLS-354 cells in response to
treatment with TNF-α, while the knockdown of Noxa protein was found
to mostly, although not completely block the TNF-α-induced decrease
in cell viability (Fig. 5E). By
contrast, the knockdown of PERK by its specific siRNA, the
inhibition of caspase-8 by its specific inhibitor, the inhibition
of ASK1 by thioredoxin, the inhibition of JNK by SP600125, and the
inhibition of IRE1α by irestatin only partially blocked the
TNF-α-induced decrease in the viability of both cell lines
(Fig. 5E). Conversely, the
inhibition of the NF-κB pathway by Bay11-7082 enhanced the
suppressive effects of TNF-α on the viability of both the RPMI2650
and CLS-354 cells (Fig. 5E).
However, the complete abrogation of the decrease in viability was
noted by the combination of the inhibitors of caspase-8 with ASK1,
JNK, or IRE1α inhibitors (Fig.
5E). In addition, the combination of caspase-8 inhibitor
together with the knockdown of PERK by its specific siRNA, or the
knockdown of Noxa by the corresponding specific siRNA was found to
block TNF-α -induced apoptosis (Fig.
5E). Taken together, these data suggest that the TNF-α-induced
decrease in cell viability is mediated by different pathways,
including the FADD-caspase-8 and TRAF-ASK1-JNK-p53-Noxa axis, while
TNF-α-induced survival is mediated by the TRAF-NF-κB axis.
The possible pathways which are
implicated in the modulation of TNF-α-induced opposing signals
Based on the outcomes of this study, we proposed
model for the possible of TNF-α-induced opposing signals in HNSCC
cells (Fig. 6). One of these
signals is associated with cell death/apoptosis and the other with
cell survival. TNF-α-induced apoptosis is mediated via the
FADD-caspase-8 and TRAF2-ASK-JNK-p53-Noxa axes, while TNF-α-induced
cell survival is mediated via the TRAF2-NF-κB axis. The inhibition
of the NF-κB pathway is expected to be a feasible option with which
to improve the killing efficiency and to minimize the systemic
toxicity of TNF-α in HNSCCs.
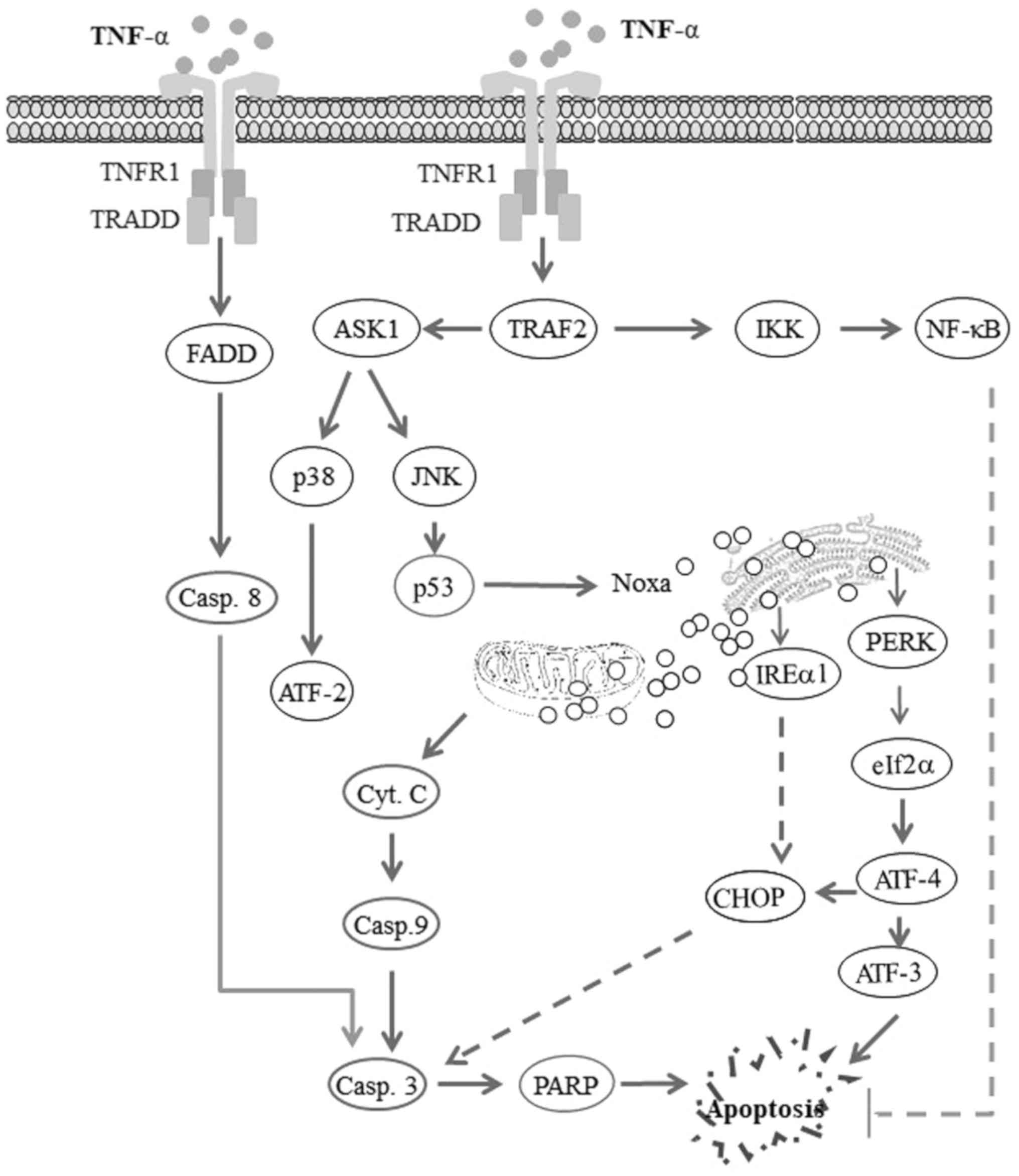 | Figure 6Proposed model for the TNF-α-induced
apoptosis of head and neck squamous cell carcinoma (HNSCC) cells.
The activation of tumor necrosis factor receptor1 (TNFR1) by tumor
necrosis factor-α (TNF-α) triggers opposing biological signals. One
of these signals drives the processes of cell survival via a
TRAF2-IKK-NF-κB pathway-dependent mechanism, while the other signal
drives mitochondrial and non-mitochondrial-dependent cell death via
TRAF2-ASK1 and FADD-caspase-8 mediated pathways. Thus, the
TNF-α-induced activation of NF-κB pathway results in the
transcriptional activation of anti-apoptotic genes, leading to the
inhibition of apoptosis. By contrast, the TNF-α-induced activation
of the FADD-caspase-8 pathway results in the cleavage of caspase-3
and PARP, leading to apoptosis. TNF-α-induced TRAF2-ASK1-JNK
activation results in the activation of ASK1, the subsequent
activation of the kinase JNK and p38, and leads to the enhancement
of the DNA-binding activity of the transcription factors p53 and
ATF-2, respectively. The transcription factor p53 then enhances the
expression of the pro-apoptotic protein Noxa, whose localization to
mitochondria and endoplasmic reticulum (ER) results in
mitochondrial dysregulation and ER stress. Mitochondrial
dysregulation results in the release of cytochrome c
(Cyt.c), which in turn leads to the cleavage of caspase-9,
caspase-3 and PARP, and finally apoptosis. ER stress results in the
activation of PERK, which in turn mediates the phosphorylation of
IRE1α, activating the transcription factor ATF-4 which triggers the
activation of ATF-3 and the expression of CHOP, ultimately leading
to the induction of apoptosis. |
Discussion
In the present study, we demonstrated the molecular
mechanisms through which TNF-α induces opposing signals in HNSCC
cells. One of these signals leads to cell death and the other leads
to cell survival. The TNF-α-induced apoptosis of HNSCC cell lines
is mediated by extrinsic and intrinsic-dependent mechanisms. The
extrinsic apoptotic signal is modulated via the following: i) the
FADD-mediated activation of caspase-8, which in turn, triggers the
activation of caspase-3, leading to PARP cleavage and subsequent
apoptosis; and ii) the activation of the TRAF2-ASK1-JNK-p53-Noxa
axis, leading to ER stress-dependent apoptosis. The intrinsic
apoptotic signal is mediated via the activation of the
TRAF-2-ASK-JNK-p53-Noxa axis, leading to mitochondrial
dysregulation-dependent apoptosis. The observed mitochondrial
dysregulation and ER stress are the consequences of the subcellular
localization of Noxa protein to both mitochondria and ER,
respectively.
TNF-α is able to trigger opposing signals in normal
and tumor cells; however, the imbalance between survival and
apoptotic signals determines whether the cell will survive or die
(25-27). In some cell types TNF-α can
activate NF-κB, leading to cell survival, while in other cell types
TNF-α can trigger cell death (15).
In the present study, the induction of opposing
signals was noted following the exposure of HNSCC cells to TNF-α.
The apoptotic signal was mediated by FADD-caspase-8 and the
TRAF2-ASK1-JNK axis, while the survival signal was mediated via the
TRAF2-NF-κB axis.
As shown by our data, the TNF-α-induced activation
of the NF-κB pathway was involved in the inhibition of
TNF-α-induced apoptosis. The TNF-α-induced activation of the NF-κB
pathway was involved in the inhibition of TNF-α-induced apoptosis,
since the inhibition of the NF-κB pathway was found to enhance the
TNF-α-induced apoptosis of HNSCC cells.
The binding of TNF-α to tumor necrosis factor
receptor 1 (TNFR1) has been shown to trigger opposing biological
responses, leading to cell survival via the NF-κB pathway and cell
death via the FADD-caspase-8 pathway (28-30).
The FADD-caspase 8 and TRADD/TRAF2-ASK1-JNK pathways are involved
in the modulation of TNF-α-induced apoptotic signals (31-33),
whereas the TRADD/TRAF2-NF-κB pathway mediates TNF-α-induced
survival signals (34,35).
The role of ASK1 in the modulation of apoptotic
signals induced by different apoptotic stimuli has been reported in
several studies (36-38). ASK1 is a member of the MAPK family,
as well as an upstream activator of the JNK and p38-MAPK signaling
cascades (39). Our data revealed
that the activation of ASK1 by TNF-α via TRAF2 resulted in the
activation of JNK, and subsequently the DNA-binding activities of
the transcription factor p53 that play an essential role in the
transcriptional regulation of the pro-apoptotic mediator Noxa. Our
findings demonstrate that the localization of Noxa protein to both
the mitochondria and ER is essential to trigger mitochondrial
dysregulation and ER stress. Noxa-induced mitochondrial
dysregulation is characterized by the loss of Δψm, cytochrome
c release, and the cleavage of caspase-3, caspase-9 and
PARP. Noxa-induced ER stress is associated with the activation of
both the PERK and IRE1α pathways, including the downstream
activation of eIf2α and ATF-3, ATF-4 induction and CHOP expression.
However, the role of mitochondrial-dependent (19,40)
and ER stress-dependent (19)
pathways in the modulation of the apoptosis of HNSCC cells has not
been reported to date, at least to the best of our knowledge.
TRAF2 has been recognized for its crucial role in
the regulation of the downstream signaling pathways that are
implicated in the modulation of TNF-α signals, leading to either
cell growth or cell death. The induction of apoptosis by TNF-α is
mediated by TRAF2 via the activation of the ASK1-JNK pathway, while
TNF-α-induced cell growth is mediated via the TRAF2-IKK-NF-κB
pathway. The lack of TRAF2 results in the suppression of the
TNF-α-induced activation of the ASK1-JNK and NF-κB pathways
(41,42).
In the present study, we demonstrated that the
TNF-α-induced activation of NF-κB attenuated TNF-α-induced
apoptosis, while the inhibition of the NF-κB pathway was associated
with the enhancement of TNF-α-induced apoptosis. By contrast, the
TNF-α-induced activation of the ASK1-JNK pathway is responsible for
the enhancement of the DNA-binding activity of the transcription
factor p53, leading to the transcriptional activation of the
pro-apoptotic gene Noxa. As consequence, the development of a
therapeutic strategy based on the combination of TNF-α and the
inhibitor of NF-κB may be a good option for the treatment of
HNSCC.
In this study, TNF-α-induced opposite signals
triggered both cell death through extrinsic and intrinsic apoptotic
pathways, and cell survival through the NF-κB pathway. The
extrinsic apoptotic pathway is mediated by TRADD/FADD/caspase-8,
while the activation the intrinsic apoptotic pathway the
consequence of TRADD/TRAF axis-induced ASK1 activation. The
activation of ASK1 results in the activation of JNK that, in turn,
enhances the DNA binding activity of the transcription factors such
as p53 to promote transcription and subsequently the translation of
the apoptotic mediator Noxa. As consequence, the binding of Noxa
protein to both the mitochondria and ER results in the induction of
mitochondrial dysregulation and ER stress-dependent apoptotic
pathways, while the TNF-α-induced survival/inhibition of apoptosis
is mediated by the TRADD/TRAF2/IKK/NF-κB axis.
In a previous study by our group [El Jamal et
al (19)], we demonstrated
that the production of IFNγ by immune effector cells as a
consequence of tumor-induced immune response. IFNγ triggers the
activation of the corresponding receptor that in turn activates the
JAK1/STAT1 pathway to enhance the DNA-binding activity of the
transcription factor, interferon regulatory factor-1 (IRF-1) to
enhance the transcription of the indolamine-2,3-dioxygenase (IDO)
that functions as inhibitor of the anti-oxidant protein heme
oxygenase-1 (HO-1). As consequence, the uncontrolled tumor
oxidation processes increases the accumulation of the reactive
oxygen species (ROS), leading to the induction cellular stress-
dependent mechanisms in the form of ASK1 activation. Activated ASK1
then is able to trigger the activation of different pathways,
leading to the enhancement of the DNA-binding activity of
transcription factors NF-κB, p53 and AP-1 that are essential for
the transcriptional activation of Noxa gene. The binding of Noxa
protein to both mitochondria and ER results in the induction of
mitochondrial dysregulation and ER stress leading the induction of
pro-and anti-apoptotic-dependent pathways.
Although the role of Noxa protein is specific for
both studies, Noxa protein in the study by El Jamal et al
(19) is the main mediator of
IFNγ-induced apoptosis of HNSCC-derived cell lines. While in the
present study, Noxa protein contributes in part in the modulation
of TNF-α-induced apoptosis.
It should be noted herein that, although there are
certain similarities, our previous study [El Jamal et al
(19)] and the present study, do
have some differences. The present study addresses the opposing
signaling that can be induced as consequence for the treatment of
HNSCC with TNF-α with the aim of reducing the adverse effects of
TNF-α as an anticancer agent. In this study, the TNF-α-induced
opposing signals trigger both cell death through extrinsic and
intrinsic apoptotic pathways, and the survival pathway through
NF-κB. The extrinsic apoptotic pathway is mediated by
TRADD/FADD/caspase-8, while the activation the intrinsic apoptotic
pathway is the consequence of TRADD/TRAF axis-induced ASK1
activation. The activation of ASK1 results in the activation of JNK
that has the ability to enhance the DNA binding activity of the
transcription factors, such as p53 for the transcriptional
activation of the apoptotic mediator, Noxa. As consequence, the
binding of Noxa protein to both the mitochondria and ER results in
the induction of mitochondrial dysregulation and ER
stress-dependent apoptotic pathways, while the TNF-α-induced
survival/inhibition of apoptosis is mediated by the
TRADD/TRAF2/IKK/NF-κB axis.
In conclusion, based on our findings, we proposed a
model for the TNF-α-induced effects on HNSCC cells (Fig. 6). Our model outlines the possible
pathways involved in the modulation of TNF-α-induced opposing
signals in HNSCC cells. One of these signals is associated with
cell death/apoptosis and the other is associated with cell
survival. TNF-α-induced apoptosis is mediated via the
FADD-caspase-8 and TRAF2-ASK-JNK-p53-Noxa axes, while TNF-α-induced
cell survival is mediated via the TRAF2-NF-κB axis. The inhibition
of the NF-κB pathway is expected to be a feasible option with which
to improve the killing efficiency and to minimize the systemic
toxicity of TNF-α in HNSCCs. Thus, the confirmation of our in
vitro findings in a preclinical model may help in the
development of a clinically relevant protocol for the treatment of
HNSCC with TNF-α.
Acknowledgments
The authors would like to thank Miss Sarah-Lilly
Hassan for providing technical assistance.
Funding
This study was supported by grants from the German
Research Foundation (HA5081/3-1), from L'Alsace contre le cancer,
France, German cancer research Foundation (10-2202-Ha1) to MH.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
DS, ER and SYH carried out the cell culture, MTT
assay, flow cytometry analysis, western blot analysis and
immunofluorescence staining experiments; TWF performed the
immunofluorescence experiment, and was involved in the flow
cytometric analysis, as well as in manuscript editing; DS, SS and
MH conceived the study design and designed the experiments; DS and
MH carried out EMSA; SS, RA, PF, YH, RUW, EK, MM and MH contributed
to the design of the study, as well as to the analysis and
discussion of the results, and the writing of the manuscript. All
authors read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Aderhold C, Faber A, Umbreit C, Birk R,
Weiss C, Sommer JU, Hörmann K and Schultz JD: Targeting mTOR and
AREG with everolimus, sunitinib and sorafenib in HPV-positive and
-negative SCC. Anticancer Res. 35:1951–1959. 2015.PubMed/NCBI
|
|
2
|
Pauzie A, Gavid M, Dumollard JM,
Timoshenko A, Peoc'h M and Prades JM: Infracentimetric cervical
lymph node metastasis in head and neck squamous cell carcinoma:
Incidence and prognostic value. Eur Ann Otorhinolaryngol Head Neck
Dis. 133:307–311. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zumsteg ZS, Cook-Wiens G, Yoshida E, Shiao
SL, Lee NY, Mita A, Jeon C, Goodman MT and Ho AS: Incidence of
Oropharyngeal Cancer Among Elderly Patients in the United States.
JAMA Oncol. 2:1617–1623. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kurzweg T, Kimmeyer J, Knecht R, Hoffmann
TK, Busch CJ, Lörincz BB, Schuler PJ and Laban S: Curative
treatment of head and neck squamous cell carcinoma: Organ
preservation strategies in clinical routine in German-speaking
countries. HNO. 64:501–507. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Braig F, Kriegs M, Voigtlaender M, Habel
B, Grob T, Biskup K, Blanchard V, Sack M, Thalhammer A, Ben Batalla
I, Braren I, Laban S, et al: Cetuximab Resistance in Head and Neck
Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res.
77:1188–1199. 2017. View Article : Google Scholar
|
|
6
|
Baetu TM and Hiscott J: On the TRAIL to
apoptosis. Cytokine Growth Factor Rev. 13:199–207. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Harry GJ, Lefebvre d'Hellencourt C,
McPherson CA, Funk JA, Aoyama M and Wine RN: Tumor necrosis factor
p55 and p75 receptors are involved in chemical-induced apoptosis of
dentate granule neurons. J Neurochem. 106:281–298. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fiers W: Tumor necrosis factor.
Characterization at the molecular, cellular and in vivo level. FEBS
Lett. 285:199–212. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elkady AI, Hussein RA and El-Assouli SM:
Mechanism of action of Nigella sativa on human colon cancer cells:
The suppression of AP-1 and NF-κB transcription factors and the
induction of cyto-protective genes. Asian Pac J Cancer Prev.
16:7943–7957. 2015. View Article : Google Scholar
|
|
10
|
Moujalled DM, Cook WD, Lluis JM, Khan NR,
Ahmed AU, Callus BA and Vaux DL: In mouse embryonic fibroblasts,
neither caspase-8 nor cellular FLICE-inhibitory protein (FLIP) is
necessary for TNF to activate NF-κB, but caspase-8 is required for
TNF to cause cell death, and induction of FLIP by NF-κB is required
to prevent it. Cell Death Differ. 19:808–815. 2012. View Article : Google Scholar
|
|
11
|
Van Hauwermeiren F, Vandenbroucke RE,
Grine L, Lodens S, Van Wonterghem E, De Rycke R, De Geest N, Hassan
B and Libert C: TNFR1-induced lethal inflammation is mediated by
goblet and Paneth cell dysfunction. Mucosal Immunol. 8:828–840.
2015. View Article : Google Scholar
|
|
12
|
Lans TE, Bartlett DL, Libutti SK, Gnant
MF, Liewehr DJ, Venzon DJ, Turner EM and Alexander HR: Role of
tumor necrosis factor on toxicity and cytokine production after
isolated hepatic perfusion. Clin Cancer Res. 7:784–790.
2001.PubMed/NCBI
|
|
13
|
Frączek N, Bronisz I, Pietryka M, Kępińska
D, Strzała P, Mielnicka K, Korga A and Dudka J: An outline of main
factors of drug resistance influencing cancer therapy. J Chemother.
28:457–464. 2016. View Article : Google Scholar
|
|
14
|
Hassan M, Feyen O and Grinstein E:
Fas-induced apoptosis of renal cell carcinoma is mediated by
apoptosis signal-regulating kinase 1 via mitochondrial
damage-dependent caspase-8 activation. Cell Oncol. 31:437–456.
2009.PubMed/NCBI
|
|
15
|
Cetindere T, Nambiar S, Santourlidis S,
Essmann F and Hassan M: Induction of indoleamine 2, 3-dioxygenase
by death receptor activation contributes to apoptosis of melanoma
cells via mitochondrial damage-dependent ROS accumulation. Cell
Signal. 22:197–211. 2010. View Article : Google Scholar
|
|
16
|
Rauert H, Stühmer T, Bargou R, Wajant H
and Siegmund D: TNFR1 and TNFR2 regulate the extrinsic apoptotic
pathway in myeloma cells by multiple mechanisms. Cell Death Dis.
2:e1942011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ridinger-Saison M, Evanno E, Gallais I,
Rimmelé P, Selimoglu-Buet D, Sapharikas E, Moreau-Gachelin F and
Guillouf C: Epigenetic silencing of Bim transcription by Spi-1/PU.1
promotes apoptosis resistance in leukaemia. Cell Death Differ.
20:1268–1278. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chung SK, Lee MG, Ryu BK, Lee JH, Han J,
Byun DS, Chae KS, Lee KY, Jang JY, Kim HJ, et al: Frequent
alteration of XAF1 in human colorectal cancers: Implication for
tumor cell resistance to apoptotic stresses. Gastroenterology.
132:2459–2477. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
El Jamal SM, Taylor EB, Abd Elmageed ZY,
Alamodi AA, Selimovic D, Alkhateeb A, Hannig M, Hassan SY,
Santourlidis S, Friedlander PL, et al: Interferon gamma-induced
apoptosis of head and neck squamous cell carcinoma is connected to
indoleamine-2,3-dioxygenase via mitochondrial and ER
stress-associated pathways. Cell Div. 11:112016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
El-Khattouti A, Selimovic D, Hannig M,
Taylor EB, Abd Elmageed ZY, Hassan SY, Haikel Y, Kandil E, Leverkus
M, Brodell RT, et al: Imiquimod-induced apoptosis of melanoma cells
is mediated by ER stress-dependent Noxa induction and enhanced by
NF-κB inhibition. J Cell Mol Med. 20:266–286. 2016. View Article : Google Scholar
|
|
21
|
Hassan M, Alaoui A, Feyen O,
Mirmohammadsadegh A, Essmann F, Tannapfel A, Gulbins E,
Schulze-Osthoff K and Hengge UR: The BH3-only member Noxa causes
apoptosis in melanoma cells by multiple pathways. Oncogene.
27:4557–4568. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Selimovic D, Ahmad M, El-Khattouti A,
Hannig M, Haïkel Y and Hassan M: Apoptosis-related protein-2
triggers melanoma cell death by a mechanism including both
endoplasmic reticulum stress and mitochondrial dysregulation.
Carcinogenesis. 32:1268–1278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
El-Khattouti A, Sheehan NT, Monico J,
Drummond HA, Haikel Y, Brodell RT, Megahed M and Hassan M:
CD133+ melanoma subpopulation acquired resistance to
caffeic acid phenethyl ester-induced apoptosis is attributed to the
elevated expression of ABCB5: Significance for melanoma treatment.
Cancer Lett. 357:83–104. 2015. View Article : Google Scholar
|
|
24
|
Selimovic D, Badura HE, El-Khattouti A,
Soell M, Porzig BB, Spernger A, Ghanjati F, Santourlidis S, Haikel
Y and Hassan M: Vinblastine-induced apoptosis of melanoma cells is
mediated by Ras homologous A protein (Rho A) via mitochondrial and
non-mitochondrial-dependent mechanisms. Apoptosis. 18:980–997.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lo SZ, Steer JH and Joyce DA: Tumor
necrosis factor-alpha promotes survival in methotrexate-exposed
macrophages by an NF-kappaB-dependent pathway. Arthritis Res Ther.
13:R242011. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Benedetti G, Ramaiahgaris S, Herpers B,
van de Water B, Price LS and de Graauw M: A screen for apoptotic
synergism between clinical relevant nephrotoxicant and the cytokine
TNF-α. Toxicol In Vitro. 27:2264–2272. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gozzelino R, Sole C, Llecha N, Segura MF,
Moubarak RS, Iglesias-Guimarais V, Perez-Garcia MJ, Reix S, Zhang
J, Badiola N, et al: BCL-XL regulates TNF-alpha-mediated cell death
independently of NF-kappaB, FLIP and IAPs. Cell Res. 18:1020–1036.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kitagawa M, Shiozaki A, Ichikawa D,
Nakashima S, Kosuga T, Konishi H, Komatsu S, Fujiwara H, Okamoto K
and Otsuji E: Tumor necrosis factor-α-induced apoptosis of gastric
cancer MKN28 cells: Accelerated degradation of the inhibitor of
apoptosis family members. Arch Biochem Biophys. 566:43–48. 2015.
View Article : Google Scholar
|
|
29
|
Kim H and Ray R: Evasion of TNF-α-mediated
apoptosis by hepatitis C virus. Methods Mol Biol. 1155:125–132.
2014. View Article : Google Scholar
|
|
30
|
Liu L, Yim H, Choi JH, Kim ST, Jin Y and
Lee SK: ATM kinase promotes both caspase-8 and caspase-9 activation
during TNF-α-induced apoptosis of HeLa cells. FEBS Lett.
588:929–935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng M, Wu Z, Wu A, Huang Z, He N and Xie
X: MiR-145 promotes TNF-α-induced apoptosis by facilitating the
formation of RIP1-FADDcaspase-8 complex in triple-negative breast
cancer. Tumour Biol. 37:8599–8607. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fukuyo Y, Kitamura T, Inoue M, Horikoshi
NT, Higashikubo R, Hunt CR, Usheva A and Horikoshi N:
Phosphorylation-dependent Lys63-linked polyubiquitination of Daxx
is essential for sustained TNF-{alpha}-induced ASK1 activation.
Cancer Res. 69:7512–7517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Min W, Lin Y, Tang S, Yu L, Zhang H, Wan
T, Luhn T, Fu H and Chen H: AIP1 recruits phosphatase PP2A to ASK1
in tumor necrosis factor-induced ASK1-JNK activation. Circ Res.
102:840–848. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jackson-Bernitsas DG, Ichikawa H, Takada
Y, Myers JN, Lin XL, Darnay BG, Chaturvedi MM and Aggarwal BB:
Evidence that TNF-TNFR1-TRADD-TRAF2-RIP-TAK1-IKK pathway mediates
constitutive NF-kappaB activation and proliferation in human head
and neck squamous cell carcinoma. Oncogene. 26:1385–1397. 2007.
View Article : Google Scholar
|
|
35
|
Zhang J, Liang Y, Lin Y, Liu Y, You You
and Yin W: IRE1α-TRAF2-ASK1 pathway is involved in CSTMP-induced
apoptosis and ER stress in human non-small cell lung cancer A549
cells. Biomed Pharmacother. 82:281–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
World C, Spindel ON and Berk BC:
Thioredoxin-interacting protein mediates TRX1 translocation to the
plasma membrane in response to tumor necrosis factor-α: A key
mechanism for vascular endothelial growth factor receptor-2
transactivation by reactive oxygen species. Arterioscler Thromb
Vasc Biol. 31:1890–1897. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Guo Y, Lin D, Zhang M, Zhang X, Li Y, Yang
R, Lu Y, Jin X, Yang M, Wang M, et al: CLDN6-induced apoptosis via
regulating ASK1-p38/JNK signaling in breast cancer MCF-7 cells. Int
J Oncol. 48:2435–2444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bieerkehazhi S, Chen Z, Zhao Y, Yu Y,
Zhang H, Vasudevan SA, Woodfield SE, Tao L, Yi JS, Muscal JA, et
al: Novel Src/Abl tyrosine kinase inhibitor bosutinib suppresses
neuroblastoma growth via inhibiting Src/Abl signaling. Oncotarget.
8:1469–1480. 2017. View Article : Google Scholar :
|
|
39
|
Mantzaris MD, Bellou S, Skiada V, Kitsati
N, Fotsis T and Galaris D: Intracellular labile iron determines
H2O2-induced apoptotic signaling via sustained activation of
ASK1/JNK-p38 axis. Free Radic Biol Med. 97:454–465. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhai X, Yang Y, Wan J, Zhu R and Wu Y:
Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and
increases radio-sensitivity in nasopharyngeal carcinoma cells.
Oncol Rep. 30:2983–2991. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nishitoh H, Saitoh M, Mochida Y, Takeda K,
Nakano H, Rothe M, Miyazono K and Ichijo H: ASK1 is essential for
JNK/SAPK activation by TRAF2. Mol Cell. 2:389–395. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cabal-Hierro L, Rodríguez M, Artime N,
Iglesias J, Ugarte L, Prado MA and Lazo PS: TRAF-mediated
modulation of NF-κB and JNK activation by TNFR2. Cell Signal.
26:2658–2666. 2014. View Article : Google Scholar : PubMed/NCBI
|