Introduction
Metastasis remains a major challenge in the clinical
management of ovarian cancer, and an improved mechanistic
understanding of ovarian cancer metastasis and more effective
therapeutic approaches for metastatic disease are urgently required
(1).
Before the epithelial ovarian cancer (EOC) cells
detach and begin metastasizing, they commonly undergo
epithelial-mesenchymal transition (EMT), a process through which
epithelial cells lose their cell polarity and gain invasive
properties to become mesenchymal-like cells (2). Typically, in cancer cells, EMT can be
triggered by transforming growth factor-β (TGF-β), which
facilitates tumorigenesis and metastasis in the late stages of
cancer progression, including in EOC (2-6).
TGF-β binds to serine/threonine kinase receptors
(TβR1/TβR2) at the cell membrane, and activates a signaling cascade
by phosphorylating specific receptor-regulated SMADs, namely, SMAD2
and SMAD3 (7). Phosphorylated
SMAD2/3 forms a complex with SMAD4 and shuttles to the nucleus to
activate the transcription of downstream effectors (7,8).
Furthermore, SMAD7 acts as a bridge protein by recruiting SMAD
specific E3 ubiquitin protein ligase 2 (SMURF2), an E3 ubiquitin
ligase, to the TGF-β receptor complex, which subsequently results
in the proteasomal-mediated degradation of TβR1, thereby
attenuating TGF-β signaling (9,10).
FXYD domain-containing ion transport regulator 5
(FXYD5) has been identified as a cancer-associated protein whose
expression inhibits E-cadherin and promotes metastasis (11-13).
As a single span type I membrane protein and an auxiliary subunit
of the Na+/K+-ATPase, FXYD5 also modulates
cellular junctions and adhesion through the regulation of the
β-Na+-K+-ATPase subunit (12-16).
Additionally, Nam et al (17) have reported that C-C motif
chemokine ligand mediates the pro-metastatic effect of FXYD5 in
human breast cancer cells.
However, to date, few studies have systematically
examined the functional significance of FXYD5 in a clinical setting
or in the molecular behavior of ovarian cancer (12,18,19).
In addition, to the best of our knowledge, there are no studies
which have linked FXYD5 to the TGF-β signaling pathway. The present
study demonstrated that FXYD5 forms a positive feedback loop with
TGF-β to drive EMT and promote metastasis in ovarian cancer in
vitro and in vivo.
Materials and methods
Bioinformatics analysis
All The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) data and figures
were accessed, analyzed and generated using the Ovarian Serous
Cystadenocarcinoma (TCGA, Provisional) database from cBio Cancer
Genomics Portal (http://cbioportal.org) (20). All data included in this manuscript
are in agreement with the TCGA publication guidelines. The present
study utilized datasets (including Bonome Ovarian, Bittner Ovarian
2005, Badea Pancreas 2008, DErrico Gastric 2009, Gumz Renal 2007,
He Thyroid 2005, Korkola Seminoma 2006 and TCGA Colorectal 2011)
from the Oncomine database (http://www.oncomine.org), an online microarray
database and web-based data-mining platform that comprises
transcriptome data, to compare the FXYD5 mRNA expression
differences between normal and tumor tissues of multiple types of
human cancer. The Kaplan-Meier Plotter (http://kmplot.com/analysis/) was utilized to calculate
the probability of disease progression, and the analysis included
1,648 patients with ovarian cancer with a mean follow-up period of
40 months (21). Using TFSEARCH
(http://www.cbrc.jp/research/db/TFSEARCH), four
putative transcriptional factor binding sites, including
SMAD-binding element (SBE)1, SBE2, SBE3 and SBE4, which were
located in the -1439/+4 FXYD5 promoter region, were identified.
Study population
The present study included 58 patients who were
diagnosed with pathologically high-grade and stage III serous
ovarian cancer and 22 patients who were diagnosed with a benign
ovarian tumor or other benign uterine lesions and underwent
prophylactic adnexectomy between March 2015 and October 2016 at the
Obstetrics and Gynecology Hospital of Fudan University (Shanghai,
China). All patients were female, and aged between 27 and 67 years
(average age, 41.8 years). Two experienced and independent
pathologists from the Pathology Department at the Obstetrics and
Gynecology Hospital of Fudan University (Fudan, China) verified the
diagnoses. Approval was obtained from the Human Research Ethics
Committee of the Obstetrics and Gynecology Hospital of Fudan
University for the use of all samples by using a protocol that
conforms to the provisions of the Declaration of Helsinki (as
revised in Seoul, 2008; reference no. [2015] 27).
In vivo experiments
A total of 50 female BALB/c nude mice (age, 4-6
weeks; weight, 13-15 g; n=6-10 mice/group) were used in the present
study according to a standard protocol that was approved by the
Institutional Animal Care and Use Committee of Fudan University.
The mice were purchased from SLAC Laboratory Animal Co., Ltd.
(SCXK-2007-004) and maintained at 22±2°C under a 12-h light/dark
cycle in a pathogen-free environment. All mice were freely accessed
autoclaved standard food and water. For nude mouse xenograft
assays, SKOV3 cells (3×106 per mouse) transfected with
the FXYD5 overexpression lentivirus or control plasmid vector were
suspended in 100 μl PBS and injected subcutaneously into
mice on the right side of their backs. The body weight and tumor
volume (V) (calculated using the formula V=length x width x
thickness in mm3 to estimate the actual volume of the
tumors) were monitored 3 times per week. For intraperitoneal
metastasis assays, SKOV3-IP cells (7×106/0.2 ml PBS per)
transfected with the FXYD5 silencing lentivirus or control plasmid
vector were injected intraperitoneally into each mouse. After 3-4
weeks, tumors were surgically excised.
Cell lines
Ovarian cancer cell lines, including SKOV3, OVCAR3,
OVCA433, A2780, HEY and CAOV3, were obtained from the American Type
Culture Collection. The metastatic human serous ovarian cancer cell
line, SKOV3-IP, was obtained from the M.D. Anderson Cancer Center
(Houston, TX, USA). The 293T cell line, which was authenticated by
applying the short tandem repeat profiling method each year, was
obtained from the Shanghai Cell Bank, Type Culture Collection
Committee of Chinese Academy of Science. All preserved cell lines
in the laboratory underwent routine cell quality examinations by HD
Biosciences Co., Ltd. to achieve a high-quality cellular standard.
The ovarian cancer cell lines were maintained in RPMI-1640
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.),
100 IU/ml penicillin, and 100 mg/ml streptomycin. 293T cells were
grown in DMEM (Corning Inc.) supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.), 100 IU/ml penicillin, and 100
mg/ml streptomycin. All cells were cultured in an incubator at 37°C
with 5% CO2.
Cellular treatment
TGF-β (0-10 ng/ml; cat. no. AF-100-21C; PeproTech,
Inc.) was used to activate the TGF-β pathway in control SKOV3 cells
and in SKOV3 cells overexpressing FXYD5. Cyclohexane (CHX; cat. no.
5087390001; Sigma-Aldrich; Merck KGaA) was used to inhibit protein
synthesis and MG132 (10 μM; cat. no. M8699-1MG;
Sigma-Aldrich; Merck KGaA) was used to inhibit protein degradation
through the proteasome pathway in control and FXYD5-overexpressing
SKOV3 cells.
Immunohistochemistry (IHC) and IHC
variable evaluation
Ovarian cancer and normal ovarian tissues were fixed
in 10% formalin at room temperature for 24 h, dehydrated in an
ascending series of alcohol (70, 85, 95 and 100%) and xylene,
embedded in paraffin and sliced into 3-μm sections.
Subsequently, sections of paraffin-embedded tissues (3-μm
thick) were deparaffinized in dimethylbenzene and rehydrated in a
descending series of alcohol (100, 95, 85 and 70%) and distilled
water. Endogenous peroxidase activity was quenched using 10-30%
hydrogen peroxide in methanol at room temperature for 15 min.
Subsequently, antigen retrieval was conducted using citric acid
buffer (pH 6.0) at 99°C for 30 min followed by cooling for 20 min,
and blocking in 5% BSA (cat. no. SW3015; Beijing Solarbio Science
& Technology Co., Ltd.) for 1 h at room temperature. The tissue
was incubated in primary anti-FXYD5 antibody (1:500; cat. no.
12166-1-AP; ProteinTech Group, Inc.), anti-β-catenin (1:400; cat.
no. 8480; Cell Signaling Technology, Inc.) and vimentin (1:500;
cat. no. 5741; Cell Signaling Technology, Inc.) overnight at 4°C.
Incubation with the biotinylated secondary antibody (rabbit IgG;
1:1,000-3,000; cat. no. 7074; Cell Signaling Technology, Inc.) was
followed by the addition of horseradish peroxidase. Counterstaining
was performed using hematoxylin at room temperature for 1 min.
Images were acquired using a Leica TCS SP2 confocal laser-scanning
microscope (Leica Microsystems, Inc.). For quantification, overall
immunostaining scores were calculated using the H-Score system
(22). The same procedure was
applied for tissues from the mouse in situ and metastatic
tumors.
Plasmids and short hairpin RNA
(shRNA)
Human FXYD5 cDNA was subcloned from the SKOV3
ovarian cancer cell line into the pCDH-CMV-MCS-EF1-Puro lentiviral
vector. The cloned primer sequence is shown in Table SI. Human FXYD5 shRNA and the
negative control, which were expressed in the GV248 backbone, were
obtained from GeneChem, Inc. Among five identified shRNAs, the two
most effective were used for further experiments, and the target
sequences are presented in Table
SI.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cultured cells using
the TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and RT-qPCR was performed with GAPDH as an
internal control. Total RNA was then reverse transcribed into cDNA
using the Prime-Script RT Reagent kit (Takara Bio, Inc.). RT was
performed using 2.0 μl 5X gDNA Eraser Buffer, 1.0 μl
gDNA Eraser, 1 μg Total RNA and RNase Free dH2O,
to a total volume of 10 μl. The volume was maintained at
42°C for 2 min and was then rapidly cooled to 4°C. Subsequently,
the aforementioned 10.0 μl reaction solution was mixed with
1.0 μl PrimeScript RT Enzyme Mix 1, 1.0 μl RT Primer
Mix, 4.0 μl 5X PrimeScript Buffer 2 and 4.0 μl RNase
Free dH2O. The reaction conditions were as follows: 37°C
for 15 min, 85°C for 5 sec, followed by cooling to 4°C and storage
at -20°C. qPCR was then performed using SYBR Premix Ex Taq (Takara
Bio, Inc.) according to the manufacturer's instructions. The
thermocycling conditions were as follows: Denaturation for 10 sec
at 95°C, followed by 40 cycles of amplification at 95°C for 15 sec
and 60 sec at 34°C, and a final extension step at 72°C for 1 min.
qPCR was performed on the ABI Prism 7500 instrument (Applied
Biosystems; Thermo Fisher Scientific, Inc.). A fluorescence-based
qPCR method was performed using 2 μl cDNA, 10 μl SYBR
Green, 0.6 μl PCR forward primer (10 μM), 0.6
μl PCR reverse primer (10 μM) and 6.8 μl
dH2O in a 20-μl PCR reaction volume. GAPDH was
used as a reference gene, and the data were normalized using the
standard comparative Cq method (23). The specific primers that were used
in the present study are listed in Table SI.
Lentivirus packaging and infection
Briefly, the 293T cells (cells growing to 80%
confluence) were co-transfected with lentiviral vectors (PCDH or
GV248) and the packaging vectors (psPAX2 and pMD2G), as described
previously (24).
RNA interference
The SKOV3 and SKOV3-IP cells (cells growing to
30-50% confluence) were transfected with 50 nM small interfering
RNA (siRNA) that targeted corresponding genes or 50 nM scrambled
negative control (Shanghai GenePharma Co., Ltd.) using the
HiPerFect Transfection reagent (Qiagen GmbH) according to the
manufacturer's instructions. Following 48 h of transfection, the
cells were collected for use in further experiments. The targeting
sequences of the siRNAs and negative control are listed in Table SI.
RNA sequencing analysis
Total RNA was extracted from FXYD5-overexpressing
and control SKOV3 cells using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). Quantified total RNA
was further purified using the RNeasy Micro kit (cat. no. 74004;
Qiagen GmbH) and RNase-Free DNase Set (cat. no. 79254; Qiagen GmbH)
and then used for Solexa/Illumina sequencing (Shanghai
Biotechnology Corp.). Hg38 RefSeq (RNA sequences, GRCh38) was
downloaded from the UCSC Genome Browser (http://genome.ucsc.edu). Kyoto Encyclopedia of Genes
and Genomes and Gene Ontology enrichment analyses were performed on
the differentially expressed genes, which were defined as genes
with changes in expression of >2 or <0.5 and a false
discovery rate Q value (adjusted P-value) <0.05. Data analysis
was performed utilizing FunRich (version 3), an open access
standalone functional enrichment and interaction network analysis
tool (25).
Western blot analysis
Briefly, whole cell extracts were prepared in
chilled RIPA lysis buffer (Beyotime Institute of Biotechnology)
containing protease inhibitor cocktail (Roche Diagnostics), as well
as phosphatase inhibitors (Sangon Biotech Co., Ltd.). The protein
concentration of the supernatants was then measured using a
bicinchoninic acid (BCA) assay reagent kit (Thermo Fisher
Scientific, Inc.). For western blot analysis, 40 μg total
cell lysate, either from ovarian cancer cell culture or from
ovarian tumor tissues, was subjected to SDS-PAGE (8-12% gels) and
transferred to 0.45 or 0.22-μm PVDF membranes (EMD
Millipore). The membranes were blocked with 5% milk or 5% BSA at
room temperature for 60 min or at 4°C overnight, followed by
incubation with the indicated primary antibodies overnight at 4°C.
The membranes were then incubated with the appropriate horseradish
peroxidase-conjugated secondary antibodies (Cell Signaling
Technology, Inc.) at room temperature for 2 h, and finally
identified using an ECL Western Blotting system (Pierce; Thermo
Fisher Scientific, Inc.). Densitometric analyses of the immunoblots
were conducted using ImageJ software (version 1.50i; National
Institutes of Health). All antibodies used in the present study are
presented in Table SII.
Transwell assays
Briefly, 4×104 ovarian cancer cells were
plated in the top chamber with the non-coated membrane (8-μm
pore size; Corning Life Sciences) for migration assays and with
Matrigel-coated membrane (8-μm pore size; BD Biosciences)
for invasion assays in 200 μl serum-free DMEM. DMEM
containing 20% FBS was used as a chemo-attractant in the lower
chamber. Following incubation for 10-24 h at 37°C, the cells which
did not traverse through the pores in the top layer were removed
with a sterile cotton swab, whereas the remaining cells on the
lower surface of the membrane were fixed with methanol and stained
with 0.1% crystal violet (Sigma-Aldrich; Merck KGaA) at room
temperature for 30 min. The number of migratory and invasive cells
was counted in five randomly selected fields of each chamber under
a light microscope (Nikon Corporation; magnification, ×200), and an
average number of cells was then calculated.
Clonogenic assay
After 7 days of growth, cell survival was evaluated
by the addition of 500 cells/well to 6-well plates, fixing the
cells with methanol and staining of the cells with 1% crystal
violet at room temperature for 30 min. Colonies were recorded using
ImageJ software (version 1.50i; National Institutes of Health).
ELISA
The TGF-β levels in the conditioned media were
measured using the human TGF-β1 ELISA kit (cat. no. ELH-TGFb1-1;
RayBiotech, Inc.) according to the manufacturer's protocol.
Immunofluorescence
SKOV3 stably transfected cell lines grown on glass
culture slides were fixed with 4% paraformaldehyde for 10 min at
room temperature, followed by permeabilization with 0.5% Triton
X-100 for 5 min. Subsequently, the cells were blocked with 5% BSA
at room temperature for 1 h, and then incubated with primary
antibodies against N-cadherin, E-cadherin and vimentin (1:200
dilution; Cell Signaling Technology, Inc.) overnight at 4°C. The
slides were incubated with Alexa Fluor 488-conjugated secondary
antibody (1:500 dilution; Invitrogen; Thermo Fisher Scientific,
Inc.) for 40 min at room temperature. The immunofluorescence of the
cytoskeleton was performed by incubation with rhodamine-conjugated
phalloidin (Sigma-Aldrich; Merck KGaA) at room temperature for 40
min. Following counterstaining with DAPI for 10 min at room
temperature; images were captured under a confocal microscope
(Leica TCS SP5 II; Leica Microsystems, Inc.). All antibodies used
in the present study are presented in Table SII.
Immunoprecipitation
Control and FXYD5-overexpressing SKOV3 cells
(4×106 per group) were treated with MG132 (10 μM;
cat. no. M8699-1MG; Sigma-Aldrich; Merck KGaA) at room temperature
for 4 h to block proteasome activity. They were then lysed with
RIPA lysis buffer (Beyotime Institute of Biotechnology) containing
protease inhibitor cocktail (Roche Diagnostics) and phosphatase
inhibitors (Sangon Biotech Co., Ltd.), and the lysates were
centrifuged at 12,000 × g for 10 min at 4°C. The protein
concentrations were measured using a bicinchoninic acid assay
reagent kit (Thermo Fisher Scientific, Inc.), and equal amounts of
the lysates were used for immunoprecipitation. Thereafter, the cell
lysates were immunoprecipitated overnight at 4°C with an anti-TβR1
antibody, and the protein A/G beads were mixed with the
immunoprecipitates, followed by incubation at 4°C for 3 h. The
precipitates were collected by centrifugation at 10,000 × g at 4°C
for 2 min and washed three times with washing buffer. The
immunoprecipitated protein complex was separated by SDS-PAGE (8-12%
gels), followed by immunoblotting overnight at 4°C with
anti-ubiquitin antibody to detect polyubiquitinated TβR1
proteins.
The present study utilized co-immunoprecipitation to
test the formation of the SMURF2-SMAD7-TβR1 complex. As
aforedescribed, cell lysates were incubated overnight at 4°C with
an anti-TβR1 antibody, the complex was conjugated to protein A/G
sepharose beads, and the beads were collected and washed with lysis
buffer and subjected to SDS-PAGE. Where indicated, the cell lysates
were immunoprecipitated overnight at 4°C with the anti-SMURF2 and
anti-SMAD7 antibodies (Santa Cruz Biotechnology, Inc.). All
antibodies used in the present study are presented in Table SII.
Luciferase reporter assay
The promoter sequence of FXYD5 was synthesized by
GeneCopoeia, Inc. A series of truncated FXYD5-promoter luciferase
constructs were generated according to the predicted
SMAD2-SMAD3-SMAD4 complex binding sites for FXYD5 promoter in
Jaspar (http://jaspar.genereg.net). In the
luciferase reporter assays, 293T cells were seeded on 24-well
plates at a density of 1×105 cells/well and transfected
with PGL3-Promoter 0, 1, 2, 3 or 4 (P0, P1, P2, P3 or P4), together
with the Renilla pRL-TK plasmid (a normalizing control;
Promega Corporation), using FuGENE HD Transfection Reagent (Promega
Corporation), according to the manufacturer's protocol. The cells
were then treated with or without 10 ng/ml TGF-β for 24 h.
Sequences, such as P0, P1, P2, P3 and P4, that contained truncated
promoter regions of FXYD5 were amplified and subcloned into the
PGL3-Basic Vector (Promega Corporation). After 48 h, cells were
harvested, and the luciferase activities were determined using the
Dual-Glo® Luciferase Assay system with a Modulus™ single
tube multimode reader (Turner BioSystems; Thermo Fisher Scientific,
Inc.) at 595 nm. The relative firefly luciferase activities were
obtained by normalizing the firefly luciferase activity level to
the Renilla luciferase activity level. Promoter and primer
sequences for the construction of the specific plasmid are listed
in Tables SIII and SIV.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the EZ-Magna ChIPTM
Chromatin Immunoprecipitation kit (EMD Millipore) according to the
manufacturer's protocol. Briefly, immunoprecipitation was conducted
by mixing the samples with the anti-p-SMAD3 and anti-SMAD4
antibodies (Cell Signaling Technology, Inc.) (incubating overnight
at 4°C). Rabbit IgG was used as a negative control. Purified ChIP
DNA segments were used as templates for qPCR, which was conducted
using SYBR Premix Ex Taq (Takara Bio, Inc.) and the ABI Prism 7500
instrument (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The RT-qPCR conditions were as follows: Pre-heating for 10 sec at
95°C, followed by 40 cycles at 95°C for 15 sec and 60 sec at 34°C,
and final extension at 72°C for 1 min. qPCR was used to amplify the
FXYD5 promoter regions, SBE1-4. The specific ChIP primers that were
used to measure the enrichment of the putative SMAD3/4 binding
sites in the FXYD5 promoter are listed in Table SI. All antibodies used in the
present study are presented in Table
SII.
Statistical analysis
Unless otherwise specified, data are presented as
the mean ± SD of at least three independent experiments.
Statistical analyses were performed using PRISM v6.0 (GraphPad
Software, Inc.) and SPSS v16.0 software (SPSS, Inc.). Student's
t-test was used to compare quantitative data between two groups,
and one-way analysis of ANOVA with Dunnett's or Least-Significant
Difference post hoc tests were used to compare the means among
multiple groups. Spearman's correlation analysis was used to
analyze the correlation between FXYD5 and TGFB1, and FXYD5 and
TGF-β-induced transcript 1 (TGFB1I1) expression. The Kaplan-Meier
method and log-rank test were used to plot the survival curves.
P<0.05 was considered to indicate a statistically significant
difference.
Results
FXYD5 is upregulated in advanced-stage
EOC and predicts poor survival in patients with ovarian cancer
First, the present study confirmed that FXYD5
expression was upregulated in the SKOV3-IP cell line, which is an
in vivo passaged variant of SKOV3 cells established by Yu
et al that exhibits a more malignant phenotype, with higher
cell growth and DNA synthesis rates, when compared with its
parental SKOV3 cell line (26)
(Fig. 1B). Notably, in a subset of
patients (stage III EOC, n=58; normal, n=22) treated at the
Obstetrics and Gynecology Hospital of Fudan University, who were
diagnosed with a benign ovarian tumor or other benign uterine
lesions and underwent prophylactic adnexectomy from March 2015 to
October 2016. it was observed that 45 (77.6%) of the tumor samples
scored as moderate-strong, whereas 3 (13.6%) normal samples scored
as moderate-strong for FXYD5 protein expression (Fig. 1C and D).
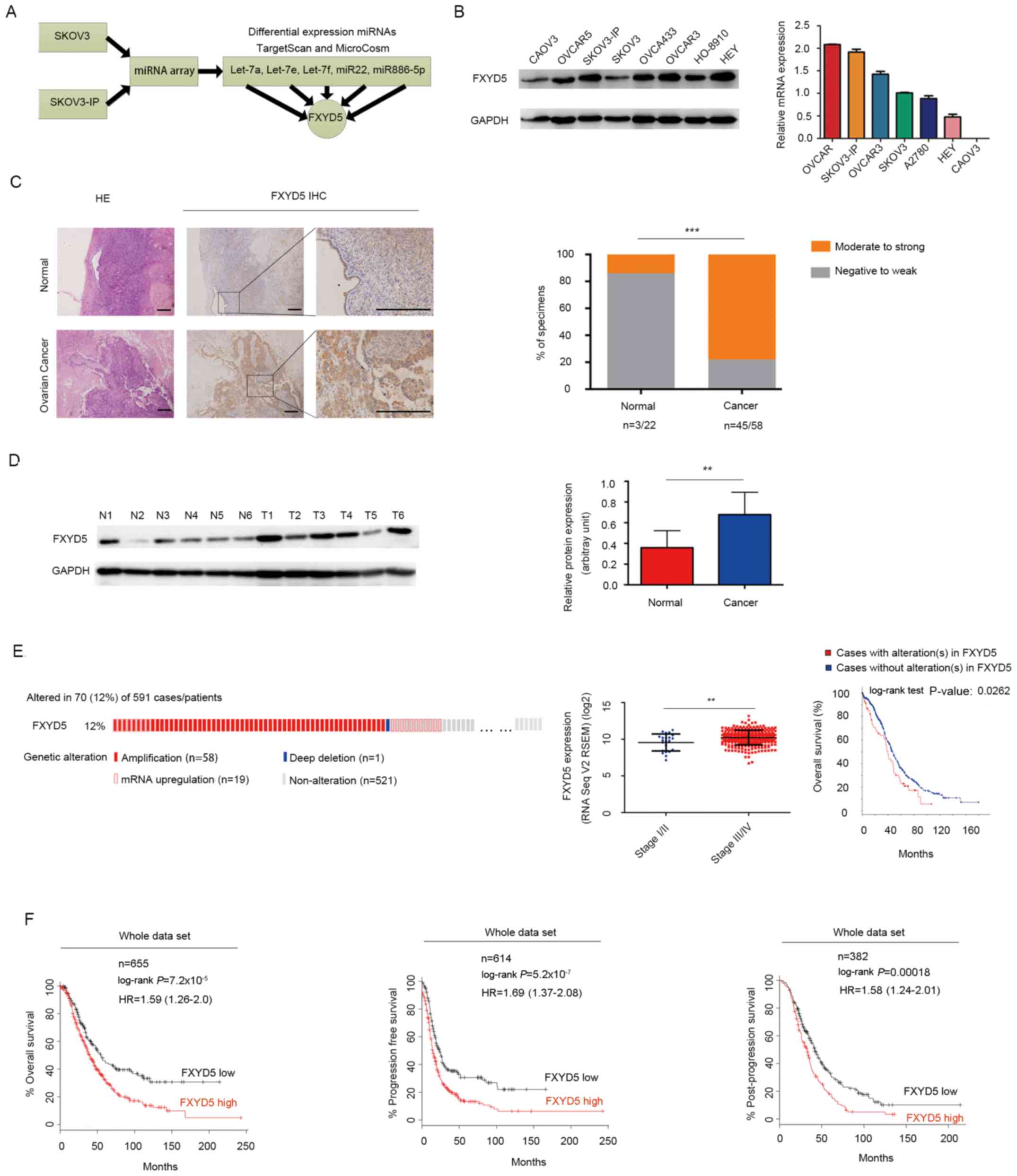 | Figure 1FXYD5 is upregulated in advanced
stage EOC and predicts poor survival in patients with ovarian
cancer. (A) FXYD5 was the common target gene of the screened miRNAs
that were potential suppressors of EOC metastasis. (B) Protein and
mRNA expression of FXYD5 in EOC cell lines. (C) Representative
images of FXYD5 immunohistochemistry are shown in the large and
small images. The percentages of specimens displaying
negative-to-weak and moderate-to-strong FXYD5 protein expression
are presented on the right. Scale bar, 20 μm. (D) Western
blot analysis of FXYD5 protein expression in ovarian cancer and
normal tissues. Histogram comparing FYXD5 protein expression
between tumor and normal samples. (E) FYXD5 gene alterations in the
cohort of patients with ovarian cancer from the TCGA dataset
(n=591), including amplification (n=58; 9.81% of total cases), mRNA
upregulation (n=19; 3.2% of total cases) and gene deletion (n=1;
0.17% of total cases). FXYD5 mRNA expression was significantly
higher in the later than the earlier stages of EOC. Patients
harboring FXYD5 alterations exhibited poor overall survival. (F)
Expression and survival analysis of FXYD5 mRNA in patients with
ovarian cancer using an online Kaplan-Meier plotter. Statistical
analysis was performed using Student's t-test (n≥3). The error bars
represent the SD. **P<0.01; ***P<0.001;
ns, no significant difference. EOC, epithelial ovarian cancer;
FXYD5, FXYD domain-containing ion transport regulator 5; HE,
hematoxylin and eosin; miRNA, microRNA; N, normal; T, tumor. |
Among the 591 patients from the TCGA database, 70
cases (12%) presented with FXYD5 alterations, including gene
amplification (n=58), mRNA upregulation (n=19), and gene deletions
(n=1; Fig. 1E). One mechanism for
the induction of high FXYD5 expression in EOC cases could be copy
number aberrations (Fig. S1A).
Additionally, genomic alterations of FXYD5 were associated with a
poor survival rate (Fig. 1E).
Furthermore, large-scale data analysis using the
Kaplan-Meier plotter indicated that FXYD5 mRNA upregulation was
associated with poor overall, relapse-free and post-progression
survival in patients with EOC [hazard ratio (HR)=1.59, 95% CI,
1.26-2.0, P=7.2×10-5, n=655; HR=1.69, 95% CI, 1.37-2.08,
P=5.2×10-7, n=614; and HR=1.58, 95% CI, 1.24-2.01,
P=1.8×10-4, n=382, respectively; Fig. 1F]. An intrinsic subtype and
clinicopathological feature analysis revealed that the effect of
FXYD5 mRNA expression on patient survival may be influenced by the
histological subtype, clinical stage and debulking surgery
(Fig. S1F; Tables SV-SVII). Notably, for patients
treated with chemotherapy (single or combination treatment with
platinum and Taxol; Tables
SV-SVII) or other types of malignant tumors (Fig. S1G), high FXYD5 expression
indicated a poor survival.
Subsequently, to examine whether FXYD5 was
upregulated specifically in EOC, an analysis of additional
independent datasets in the TCGA and Oncomine platforms was
performed, and it was revealed that FXYD5 expression was elevated
in various types of human cancer (Fig. S1B-E).
Overall, these data suggested that FXYD5 may serve
oncogenic and metastasis-promoting roles in ovarian cancer, making
FXYD5 an interesting target for further investigation.
FXYD5 promotes ovarian cancer metastasis
and tumor growth in vitro and in vivo
To investigate the role of FXYD5 in tumor migration
and metastasis, three EOC cell line models, in which FXYD5 was
overexpressed or knocked down, were established for in vitro
and in vivo experiments (Fig.
2A).
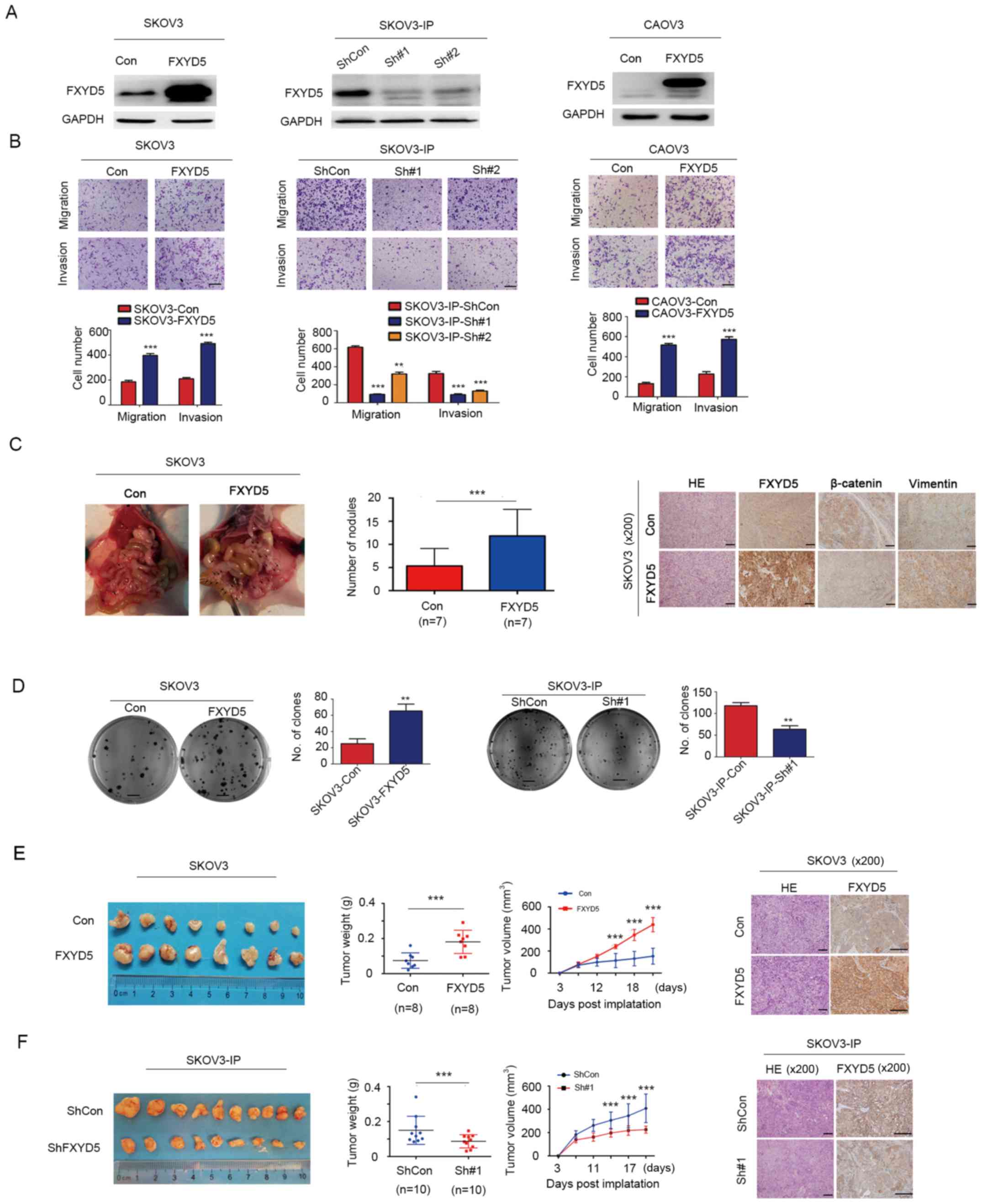 | Figure 2FXYD5 promotes ovarian cancer
metastasis and tumor formation in vitro and in vivo.
The migration and invasion abilities of each cell line were
evaluated by Transwell assays in vitro. (A) FXYD5 protein
overexpression or knockdown effects in cell lines, including SKOV3,
SKOV3-IP and CAOV3. (B) Upper panel, images of representative
fields of invasive cells. Magnification, ×10. Scale bar, 10
μm. Lower panel, histograms of the results.
**P<0.01; ***P<0.001 vs.
SKOV3-IP-ShCon. (C) Whole-enterocoelia images and quantification of
metastasis nodules in the peritoneal cavity on day 28 after
intraperi-toneally injection of SKOV3 cells (n=10 mice per group).
Arrows indicate metastasis nodules. HE and IHC staining for FXYD5,
β-catenin and vimentin in representative control and FXYD5 tumors.
Magnification, ×200. Scale bar, 40 μm. (D) Clonogenic assays
to assess cellular survival in ectopic FXYD5 expression SKOV3 cells
and endogenous FXYD5 silencing SKOV-IP cells. Fold changes in
number of colonies for ectopic FXYD5 cells vs. control and control
vs. FXYD5 silencing cells (right). Scale bar, 500 μm.
**P<0.01. (E and F) Ovarian tumors were removed and
collected from the control, ectopic FXYD5 and FXYD5 deletion mice
(n=8-10 per group) 28 days post-orthotopic implantation (left). The
estimated tumor weight (g) and tumor volume (mm3) were
measured twice per week (middle). HE and IHC staining for FXYD5 in
representative mouse tumors (right). Scale bar, 40 μm. (E)
***P<0.001 D12 vs. D15, D15 vs. D18 and D18 vs. D21;
(F) ***P<0.001 D11 vs. D14, D14 vs. D17 and D17 vs.
D20. Student's t-test was used to compare quantitative data between
two groups, and one-way ANOVA with Least-Significant Difference [E,
tumor volume, D15 compared with D18 and D18 compared with D21; F,
tumor volume, D14 compared with D17 and D17 compared with D20] or
Dunnett's (B, SKOV3-IP-ShCon compared with SKOV3-IP-Sh#1 and
SKOV3-IP-Sh#2) post hoc tests were used to compare the means among
multiple groups (n>3). The error bars represent the SD.
***P<0.001; ns, no significant difference. Con,
control; FXYD5, FXYD domain-containing ion transport regulator 5;
HE, hematoxylin and eosin; IHC, immunohistochemistry; sh, short
hairpin RNA; IP, immunoprecipitation. |
Notably, in the SKOV3 and CAOV3 cell lines with
relatively low levels of endogenous FXYD5 expression (Fig. 1B), FXYD5 overexpression
significantly promoted cell migration and invasion in Transwell
assays (Fig. 2A and B). Using a
complementary inverse approach, FXYD5 deletion suppressed the
migratory and invasive abilities of the SKOV3-IP cells in
vitro (Fig. 2A and B).
Subsequently, the present study examined the effects
of FXYD5 on cancer cell dissemination in vivo. Notably, the
FXYD5-overexpressing group formed more disseminated nodules (12 vs.
5 intraperitoneal nodules on average for the test and control
groups) than the control group (n=14; Fig. 2C). Additionally, IHC analysis of
the intraperitoneal nodules revealed that, compared with the
control tumors, the FXYD5 tumors exhibited a robust ectopic FXYD5
overexpression, with lower β-catenin expression and higher vimentin
expression (Fig. 2C).
Additionally, colony formation assays demonstrated
that FXYD5 overexpression could significantly promote colony
formation of SKOV3 cells, whereas the loss of FXYD5 expression
inhibited the colony formation of SKOV3-IP cells (Fig. 2D).
Furthermore, the present study established a
xenograft model of ovarian cancer to examine the effects of FXYD5
on tumor growth in vivo. The sizes of the tumors, which were
dissected from each mouse, were markedly larger in the FXYD5 group
than in the control group (Fig.
2E). Consistently, the average tumor weight was considerably
greater in the FXYD5 group, with a 2.5-fold change compared with
the control group (Fig. 2E). By
day 21, the estimated tumor volume in the FXYD5 group was almost
3-fold greater than that in the control group (Fig. 2E). Conversely, FXYD5 silencing
inhibited SKOV3-IP cell survival and tumor growth in vivo,
as shown in Fig. 2F. Collectively,
these data demonstrated that FXYD5 promoted EOC tumor growth and
metastasis in vitro and in vivo.
FXYD5 activates TGF-β/SMADs-induced
EMT
To investigate the specific mechanisms that drive
EOC metastasis, high-throughput RNA sequencing (Illumina) was
performed, using the SKOV3 and SKOV3-IP cell lines with FXYD5
over-expression and FXYD5 deletion, respectively, in combination
with a global transcriptome- and pathway-based analysis. Using a
minimum fold-change threshold of >2 [Q (adjusted P-value)
<0.05], differentially expressed genes were identified (Tables SVIII and SIX). Gene set
enrichment analysis indicated that the TGF-β (P=6.12×10-4; Q=0.015)
signaling pathway was the most significantly deregulated pathway
(according to the enriched gene number and Q value) (Figs. 3A and S2). Therefore, it was speculated that
overexpression of FXYD5 may activate the TGF-β signaling
pathway.
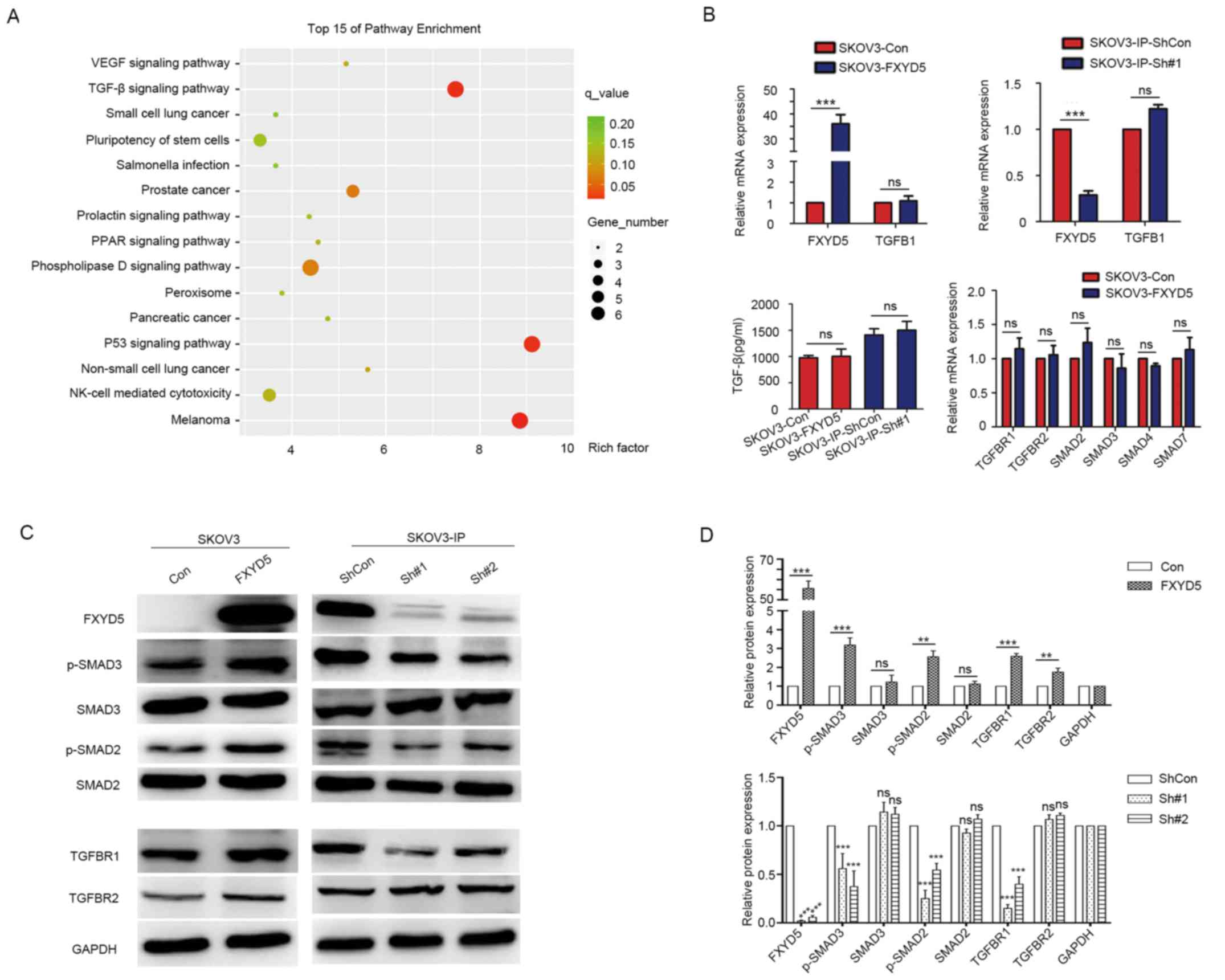 | Figure 3FXYD5 activates TGF-β/SMADs signaling
in vitro. (A) Global canonical pathway analysis.
Differential genes of the RNA-sequencing datasets from SKOV3-con
and SKOV3-FXYD5 cell lines were mapped to the Kyoto Encyclopedia of
Genes and Genomes pathways. Statistical significance is expressed
as a P-value calculated using the right-tailed Fisher's exact test.
(B) FXYD5 overexpression or FXYD5 silencing had no effects on TGF-β
mRNA expression (upper left and right). FXYD5 overexpression had no
effects on TGF-β protein secretion levels in the medium in the
SKOV3 cell line (lower left). FXYD5 overexpression had no effects
on the mRNA level of TGFBR1, TGFBR1, SMAD2, SMAD3 and
SMAD4 in the SKOV3 cell line (lower right). (C) Western
blotting for the FXYD5, TGF-β receptors and SMAD proteins assessing
TGF-β pathway activity in FXYD5-overexpressing and FXYD5-silenced
cells. (D) Histograms of the results from (A), (B) and (C).
Student's t-test was used to compare quantitative data between two
groups, and one-way ANOVA with Dunnett's (D, lower panel) post hoc
tests were used to compare the means among multiple groups
(n>3). The error bars represent the SD. **P<0.01;
***P<0.001; ns, no significant difference. Con,
control; FXYD5, FXYD domain-containing ion transport regulator 5;
p-, phosphorylated; TGF-β, transforming growth factor-β; VEGF,
vascular endothelial growth factor; PPAR, peroxisome proliferators
activated receptor; NK-cell, natural killer cell. |
Subsequently, it was observed that the upregulation
of FXYD5 slightly elevated the protein levels of TβR1, whereas it
notably increased the phosphorylation of SMAD2 and SMAD3 in the
SKOV3 cell line, with no alterations observed in the protein levels
of TβR2, SMAD2, SMAD3 and SMAD4 (Fig.
3C). Conversely, FXYD5 deletion notably decreased the TβR1
protein, and SMAD2 and SMAD3 phosphorylation levels in SKOV3-IP
cells (Fig. 3C). Additionally,
ectopic FXYD5 expression did not induced TGF-β gene transcription
or protein secretion, and no alterations were observed in the
transcription levels of these TGF-β signaling pathway components
(Fig. 3B). These findings
suggested that FXYD5 activated TGF-β signaling by elevating the
TβR1 protein level.
Notably, enrichment analysis of the data from the
RNA-sequencing and TCGA datasets indicated that EMT and
EMT-associated processes, including integrin family cell surface
interactions, extracellular matrix, intermediate filament and
others, were most associated with the FXYD5 alterations (Figs. S3 and 4; Tables SVIII-X). It has been confirmed
that TGF-β stimulates EMT, migration, invasion and the metastasis
of ovarian cancer cells (Fig. S5A and
B) (27). Therefore, it was
speculated that FXYD5 may induce EMT and promote EOC metastasis by
activating the TGF-β signaling pathway.
Subsequently, through western blotting and
immunofluorescence assays, it was demonstrated that the
overexpression of FXYD5 resulted in higher N-cadherin and vimentin
expression in the SKOV3 cell line (Fig. 4A and B). Among the other EMT
biomarkers, β-catenin, another epithelial marker, was
downregulated. However, the expression levels of the other
mesenchymal markers, including SNAG transcriptional repressor,
snail family transcriptional repressor 2, matrix metalloproteinase
(MMP)2 and MMP9, were all upregulated, in response to FXYD5
overexpression (Fig. 4A and B).
Additionally, in the more mesenchymal-like and metastatic SKOV3-IP
cell line, FXYD5 silencing reversed the expression trends of the
EMT markers (Fig. 4A) and
stimulated mesenchymal-epithelial transition in the cell line.
Morphologically, the FXYD5-overexpressing SKOV3 cells became
rounded in shape, which is another hallmark of EMT (Fig. 4B).
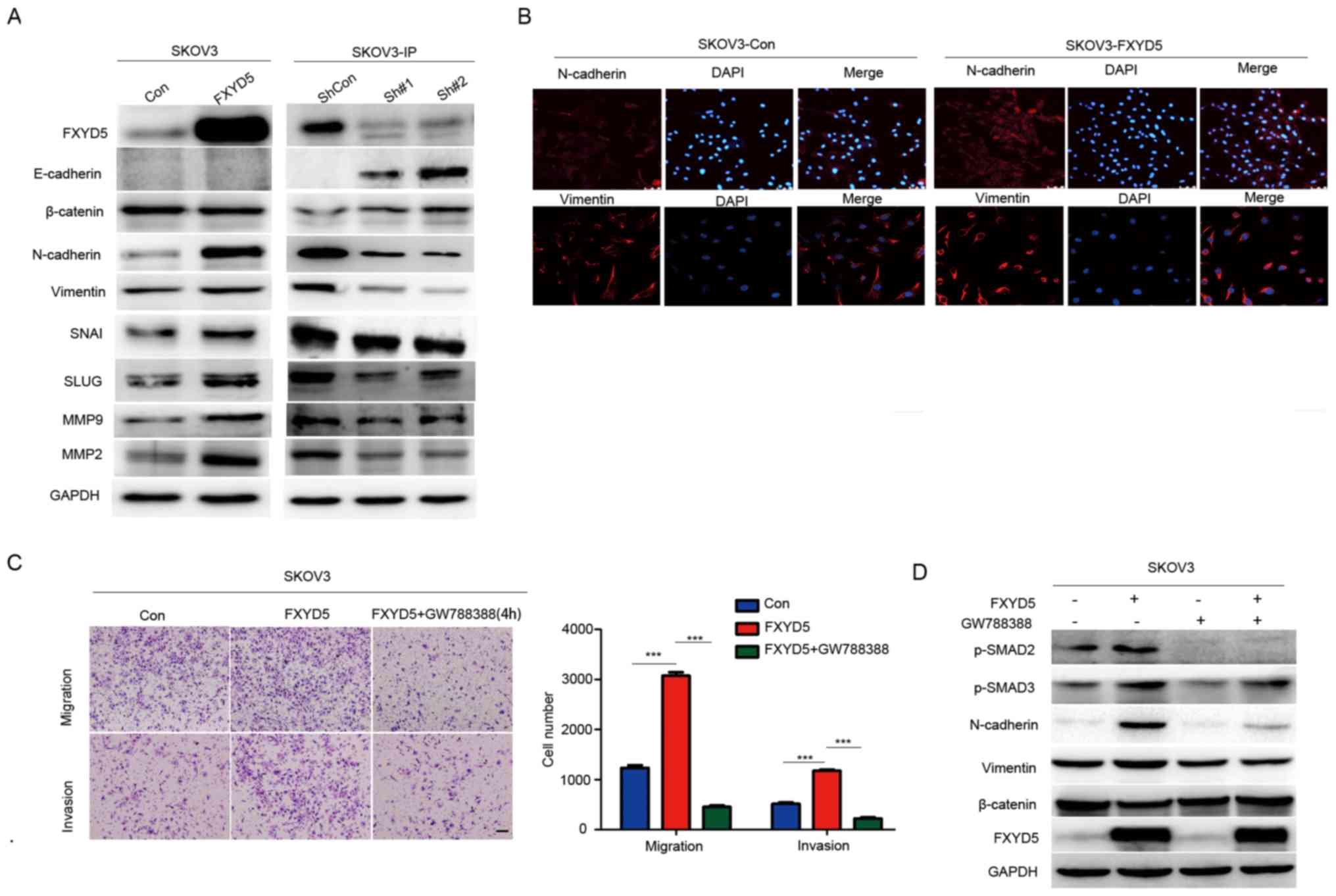 | Figure 4FXYD5 activates EMT by activating
TGF-β/SMADs signaling pathway. (A) Western blotting for epithelial
(E-cadherin and β-catenin) and mesenchymal (N-cadherin and
vimentin) markers to confirm EMT in SKOV3 cells with ectopic FXYD5
expression, and in SKOV3-IP cells with FXYD5 deletion. EMT
regulators, including SNAI1, SLUG, MMP9 and MMP2 were examined in
SKOV3 and SKOV3-IP cells. (B) Representative images showing
overexpressing FXYD5 in SKOV3 cells exhibited decreased levels of
epithelial markers, such as E-cadherin, and increased levels of
mesenchymal markers, including N-cadherin and vimentin. Scale bar,
10 μm. (C) Transwell assays of cells treated with GW788388
(10 μM) for 48 h and histogram of the results.
Magnification, ×10. Scale bar, 10 μm. (D) Key markers of the
TGF-β signaling pathway and EMT were examined in SKOV3 cells with
stable overexpression of FXYD5 treated with GW788388 (10 μM)
for 48 h. One-way ANOVA with Least-Significant Difference post hoc
tests were used to compare the means among multiple groups
(n>3). The error bars represent the SD.
***P<0.001; ns, no significant difference. Con,
control; EMT, epithelial-mesenchymal transition; FXYD5, FXYD
domain-containing ion transport regulator 5; sh, short hairpin RNA;
MMP, matrix metalloproteinase; SNAI, SNAG transcriptional
repressor; SLUG, snail family transcriptional repressor 2; TGF-β,
transforming growth factor-β. |
Additionally, it was revealed that FXYD5
overexpression or treatment of the SKOV3 cells with TGF-β promoted
cell migration and invasion, and the ectopic expression of FXYD5
enhanced the effects of TGF-β on cell movements (Fig. S5A and B). Notably, treatment with
GW788388, an inhibitor of TGF-β/SMADs signaling (28), reversed the effects of ectopic
FXYD5 on cell migration, invasion and EMT (Fig. 4C and D). Overall, these results
suggested that FXYD5 potentiated TGF-β/SMADs signaling and
TGF-β-induced EMT in ovarian cancer cells.
FXYD5 maintains the continuous activation
of TGF-β/SMAD signaling by suppressing TβR1 degradation
Notably, TβR1 protein, but not mRNA alterations,
were observed in response to FXYD5 alterations (Fig. 3B and C). Therefore, it was proposed
that the loss of FXYD5 would predominantly degrade the TβR1 protein
in a post-transcriptional manner. To confirm this hypothesis, the
SKOV3 cells overexpressing FYXD5 were incubated with CHX, an
inhibitor of protein biosynthesis. Compared with the control group
(SKOV3-Con), and according to the curve shown in Fig. 5A, TβR1 protein was markedly
degraded at a slower rate, and even increased within 6 h of the CHX
treatment in the FXYD5-overexpressing group (SKOV3-FXYD5; Fig. 5A). Furthermore, treatment of these
cells with MG132, a proteasome inhibitor, increased the stable TβR1
protein level (Fig. 5B).
Additionally, in the SKOV3-FXYD5 cell lines, poly-ubiquitination of
TβRI was reduced (Fig. 5C),
indicating that TβR1 protein degradation is directed by the
ubiquitin-proteasome system.
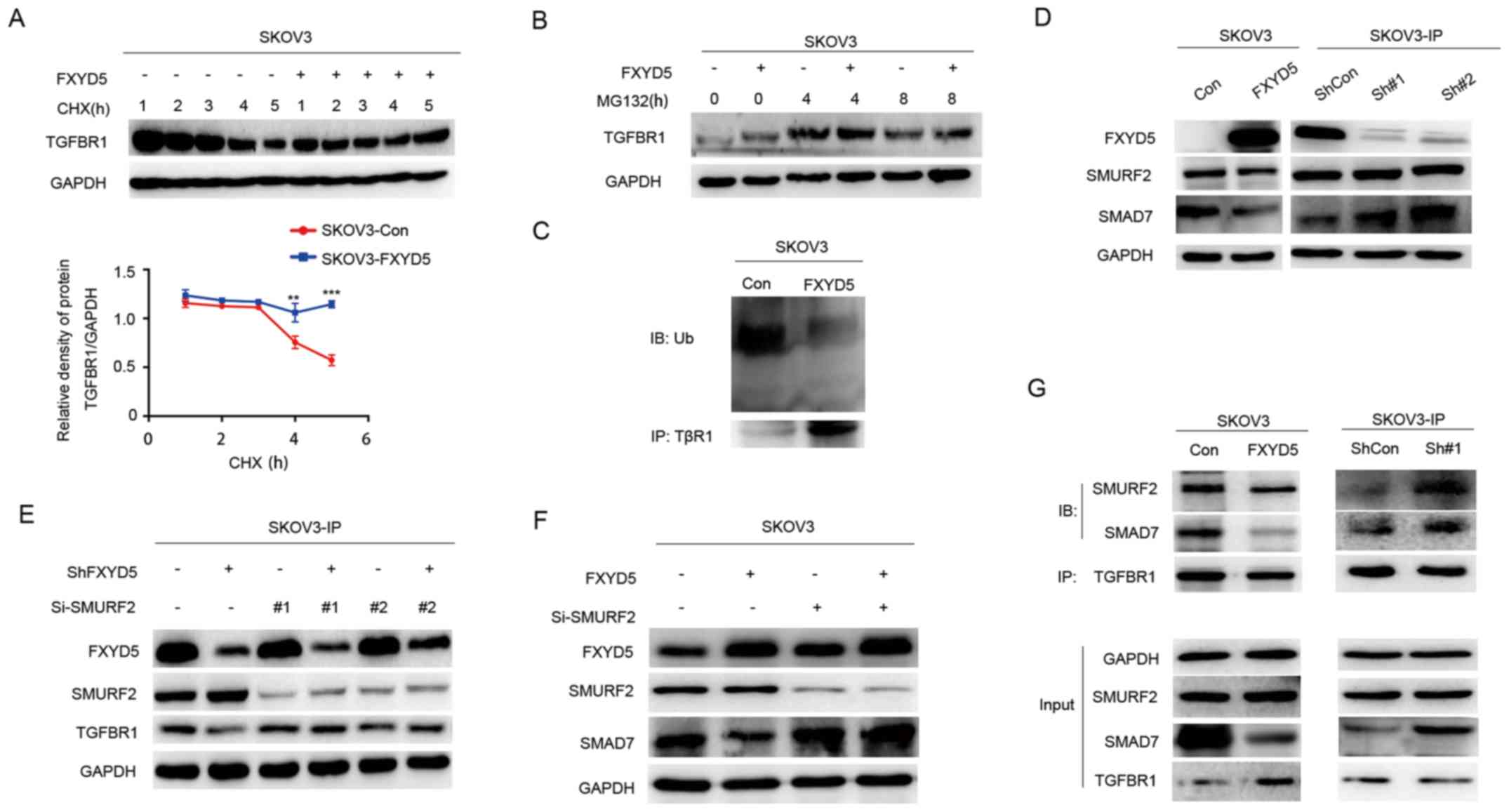 | Figure 5FXYD5 activates the TGF-β/SMADs
signaling pathway via suppressing TβR1 degradation. TGFBR1
expression by IB analysis in SKOV3-Con and SKOV3-FXYD5 cells
treated with (A) CHX (100 μg/ml) and (B) MG132 (10
μM) for the indicated lengths of time. (C) SKOV3-Con and
SKOV3-FXYD5 cells were treated with 10 μM MG132 for 4 h.
Following cell harvest, proteins were immunoprecipitated with an
anti-TβR1 antibody and detected using a polyubiquitin antibody. (D)
WB analysis of SMRF2 and SMAD7 in FXYD5-overexpressing SKOV3 and
FXYD5-silenced SKOV3-IP cells. WB analysis of TβR1 and SMAD7 in (E)
FXYD5-silenced SKOV3-IP and (F) FXYD5-overexpressing SKOV3 cells
after being treated with si-SMURF2 and Si-Control plasmid vectors
for 48 h. (G) Lysates of FXYD5-overexpressing SKOV3 and
FXYD5-silenced SKOV3-IP cells were subjected to anti-TβR1 IP
followed by IB with anti-SMAD7 and anti-SMURF2. Statistical
analysis was performed using Student's t-test (n≥3). The error bars
represent the SD. **P<0.01; ***P<0.001;
TGFBR1, TGF-β receptor 1; IB, immunoblotting; WB, western blotting;
IP, immunoprecipitation; Con, control; FXYD5, FXYD
domain-containing ion transport regulator 5; Ub, ubiquitin; sh,
short hairpin RNA; SMURF2, SMAD specific E3 ubiquitin protein
ligase 2; si, small interfering RNA. |
As shown in Fig.
5D, the ectopic expression of FXYD5 markedly decreased SMAD7
expression in the SKOV3 cells. Conversely, FXYD5 silencing led to a
marked increase in SMAD7 expression in the SKOV3-IP cells (Fig. 5D). However, FXYD5 silencing had no
effect on SMURF2 expression in the SKOV3-IP cell line (Fig. 5D). To further examine whether
SMURF2 was involved in this process, an effective small interfering
RNA (si-)SMURF2 construct was generated to knockdown SMURF2, and it
was revealed that si-SMURF2 significantly reversed the degrading
effects of FXYD5 deletion on TβR1 and the derogation of SMAD7
induced by FXYD5 overexpression (Fig.
5E and F), suggesting that SMURF2 was required for
FXYD5-mediated TβR1 and SMAD7 alterations. Furthermore, IP assays
demonstrated that sh-FXYD5 promoted the binding of SMURF2 and SMAD7
to TβR1, whereas FXYD5 inhibited these interactions (Fig. 5G). These results demonstrated that
FXYD5 upregulation dispersed the SMAD7-SMURF2-TβR1 complex,
promoted the deubiquitination and stabilization of TβR1, and
activated tTGF-β/SMADs signaling. The SMAD7 protein may also be
degraded by the ubiquitin-proteasome system in response to FXYD5
upregulation, which requires further investigation.
TGF-β-activated SMAD3/4 complex
upregulates FXYD5 expression
Notably, positive correlations were identified
between the FXYD5 and TGFB1 and the FXYD5 and TGFB1I1 mRNA levels
in the patients with ovarian cancer from the two datasets from TCGA
(Figs. 6A and S3D; Table
SX). Additionally, TGFB1 and TGFB1I1 were also positively
co-expressed in the two TCGA datasets (Fig. S5D). As aforementioned, FXYD5 did
not induce either TGF-β gene transcription or protein secretion
(Fig. 3B). Therefore, it was
proposed that activated TGF-β signaling could induce FXYD5
expression. SKOV3 cells were treated with various concentrations of
TGF-β (0-10 ng/ml) for 24 h (Fig.
S5E). As shown in Figs. S3E and
6B, the TGF-β treatment upregulated the FXYD5 mRNA and protein
levels in a dose- and time-dependent manner. Inversely, treatment
with GW788388, an inhibitor of TGF-β/Smad3 signaling, abolished the
effects of TGF-β on FXYD5 expression (Fig. S5F-H).
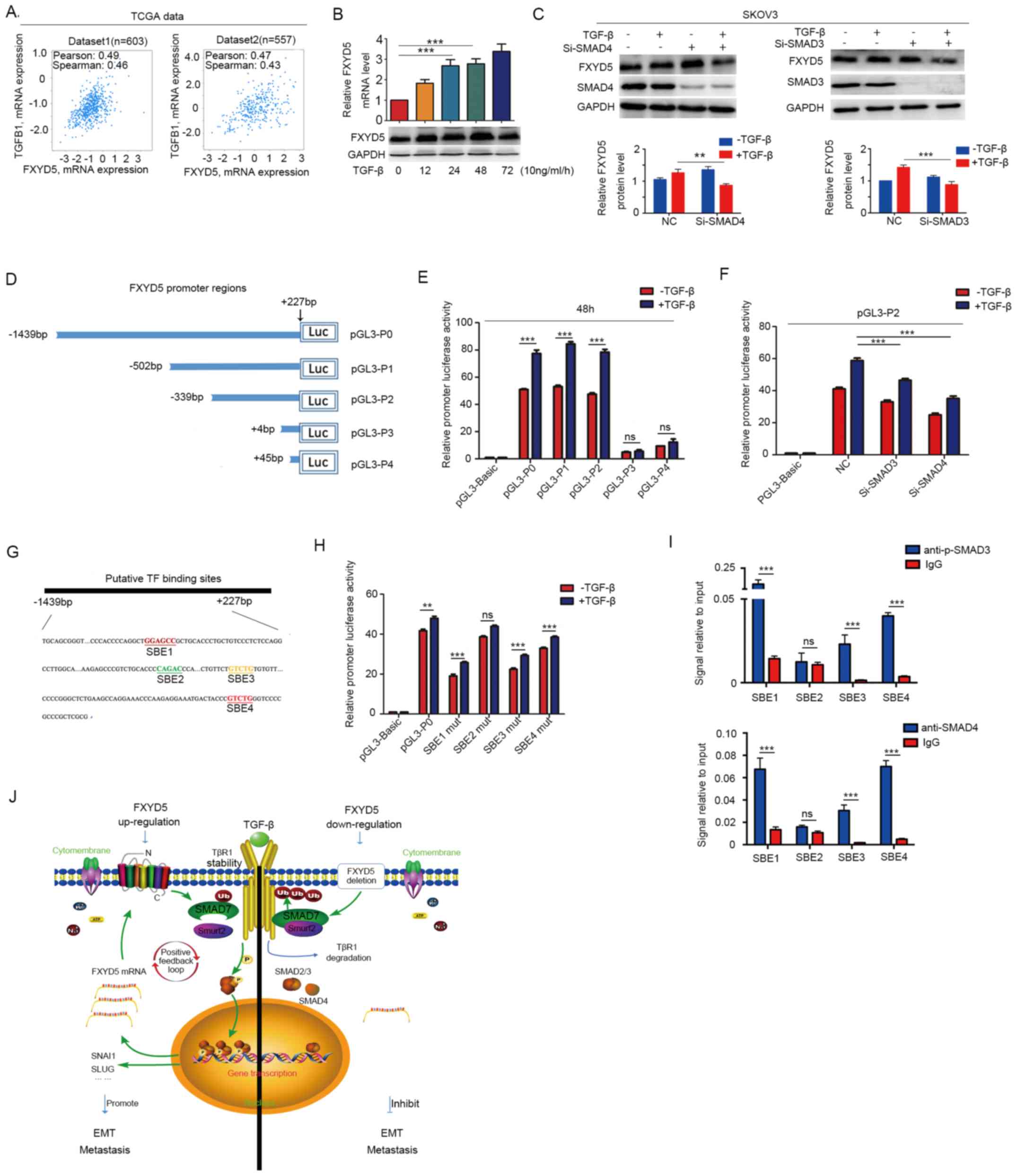 | Figure 6TGF-β-activated SMAD3/4 complex is
recruited to FXYD5 promoter and binds to SBEs to enhance
transcription. TGFB1 was co-expressed with FXYD5 in
two TCGA datasets. Correlation values and P-values were determined
using Spearman and Pearson's correlation. (B) FXYD5 mRNA and
protein levels in SKOV3 cells in the presence of TGF-β (10 ng/ml)
for the indicated times. (C) WB analysis of FXYD5 expression in
SKOV3 cells treated with si-SMAD4 and si-SMAD3, in the absence or
presence of TGF-β for 48 h. (D) A series of truncated
FXYD5-promoter luciferase constructs were generated according to
the predicted SMAD2-SMAD3-SMAD4 complex binding sites for the FXYD5
promoter in the Jaspar (http://jaspar.genereg.net). (E) 293T cells were
transiently transfected with the constructs indicated in (D), and
then treated with or without TGF-β (10 ng/ml) for 48 h.
Subsequently, their luciferase activities were tested. (F)
Luciferase activity of PGL-P2 promoter in 293T cells that were
transfected with NC, si-SMAD3 and si-SMAD4 in the absence or
presence of TGF-β for 48 h. (G) Computational algorithms by use of
TFSEARCH (http://www.cbrc.jp/research/db/TFSEARCH) predicted
that the PGL-P0 promoter region harbored four putative
protein-binding sites (SBE1-4), which were shown in different
colors. (H) 293T cells were transfected with the constructs
including the wild type or mutant type of the putative binding
sites, which were indicated in (G), and then the luciferase
activities were assessed in the absence or presence of TGF-β (10
ng/ml) for 48 h. (I) Chromatin immunoprecipitation assays using
antibodies against p-SMAD3 and SMAD4 were utilized to determine the
enrichment degree of the FXYD5 promoter containing SBEs in 293T
cells upon TGF-β stimulation. (J) A positive feedback loop for the
regulation of TGF-β-induced EMT by FXYD5. Student's t-test was used
to compare quantitative data between two groups, and one-way ANOVA
with Least-Significant Difference post hoc tests were used to
compare the means among multiple groups (n>3). The error bars
represent the SD. **P<0.01; ***P<0.001;
ns, no significant difference. SBE, SMAD-binding elements; TCGA,
The Cancer Genome Atlas; TGF-β, transforming growth factor-β;
FXYD5, FXYD domain-containing ion transport regulator 5; WB,
western blotting; si, small interfering RNA; NC, negative control;
p-, phosphorylated; IgG, immunoglobulin G; EMT,
epithelial-mesenchymal transition. |
To examine whether the SMAD signaling pathway is
required for TGF-β-induced FXYD5 upregulation in human ovarian
cancer cells, SMAD signaling transduction was blocked by the
siRNA-mediated knockdown of common SMAD4. As shown in Fig. 6C, the SMAD4 siRNA significantly
downregulated endogenous SMAD4 expression, and the TGF-β-induced
elevation of the FXYD5 protein levels was abolished by SMAD4
silencing. To further determine which SMAD was required for
TGF-β-induced FXYD5 expression, a specific siRNA was used to
knockdown SMAD2 or SMAD3 in SKOV3 cells. As shown in Figs. 6C and S3I, the silencing of SMAD3 alone, but
not that of SMAD2, attenuated TGF-β-induced upregulation of the
FXYD5 protein levels. Hence, it was hypothesized that the
SMAD3/SMAD4 complex mediates FXYD5 transcription by binding to the
promoter region of the FXYD5 gene.
Identification of the TGF-β response
regions that contain putative functional SBEs in the FXYD5
promoter
To identify the role of activated TGF-β signaling in
regulating FXYD5 promoter transcription, a DNA fragment between
-1,439 and +227 relative to the FXYD5 transcription start site
(TSS) was cloned into a pGL3-Basic plasmid to yield a pGL3-TGF-β
recombinant vector that could deliver the FXYD5 promoter (Fig. 6D and E). Sequence constructs P0, P1
and P2 increased the luciferase activity level in the 293T cells;
however, sequence constructs P3 and P4 exhibited luciferase
activity levels that were equivalent to those of the control
(Fig. 6E). This result indicated
that activated TGF-β signaling increased the transcriptional
activity of the FXYD5 promoter by binding to the region that was
located between -1439 and +4 relative to the TSS (Fig. 6D and E). Furthermore, it was
observed that the luciferase activity of the P2 promoter was
significantly decreased in the 293T cells in which SMAD3 and SMAD4
were silenced and which were treated with TGF-β (Fig. 6F). These results suggested that the
TGF-β-activated SMAD3/4 complex may have the capacity to bind to
the FXYD5 promoter and initiate FXYD5 transcription.
Furthermore, using TFSEARCH, four putative
transcriptional factor binding sites, including SBE1, SBE2, SBE3
and SBE4, that were located in the -1439/+4 FXYD5 promoter region,
were identified (Fig. 6G).
Subsequently, various luciferase reporter constructs containing the
wild-type and mutant forms of the four TF-binding sites were
transfected into the 293T cells. Upon TGF-β stimulation, three
constructs exhibited significantly decreased luciferase activities
compared with those of the wild-type construct; the mutant
construct of the SBE2-binding site was the exception (Fig. 6H). As the TGF-β-activated SMAD3/4
complex can be recruited to SBEs (29,30),
the findings suggested the possibility that the TGF-β-activated
SMAD3/4 complex may positively regulate the transcription of FXYD5
by binding to SBE1, SBE3 and SBE4.
Finally, the ChIP assays revealed that upon TGF-β
stimulation, the SMAD-binding elements in the FXYD5 promoter,
including SBE1, SBE3 and SBE4, were more enriched in the
immunoprecipitates that were obtained using the corresponding
antibodies (Fig. 6I).
Collectively, these results indicated that the
TGF-β-activated SMAD3/SMAD4 complex was directly recruited to the
FXYD5 promoter region and that the complex interacted with specific
SBEs, thus promoting FXYD5 transcription.
Discussion
Our previous study screened and identified a set of
miRNAs, including let-7a, let-7e, let-7f, miR-22 and miR-886-5p,
from the SKOV3 ovarian cancer cell line and from SKOV3-IP, the
metastatic subline of SKOV3, as the most significant potential
suppressor genes associated with ovarian cancer invasion and
metastasis (26,31,32).
The present study, utilizing bioinformatics analysis, identified
FXYD5 as the common target gene of these miRNAs. Therefore, it was
speculated that FXYD5 may serve a promotor role during EOC
progression.
The present study demonstrated that FXYD5 was
upregulated in metastatic ovarian cancer, and that it was
associated with a worse patient survival. Extensive functional
analyses confirmed its metastasis promoter role in vitro.
Mechanistically, by affecting the SMURF2-SMAD7 complex in
regulating the TβR1 protein level, FXYD5 positively regulated
TGF-β/SMAD signaling activities, which in turn induced FXYD5
expression via the activated SMAD3/4 complex, creating a positive
feedback loop and driving the cells to undergo the EMT mediated
metastasis in ovarian cancer (Fig.
6J).
The present study revealed that FXYD5 was
substantially upregulated in the more aggressive subline, SKOV3-IP,
which was developed from the parental SKOV3 cells by in vivo
selection in mice (26).
Accordingly, the SKOV3-IP cells exhibited an increased invasiveness
compared with the parental SKOV3 cells. Due to the similar genetic
background of these cells, they provide a unique model for
identifying candidate metastasis-associated biomarkers and
potential therapeutic targets for EOC.
Previous data from a large-scale high-throughput
analysis of numerous high-grade serous ovarian cancer samples
suggested that the high invasive propensity of ovarian cancer cells
is coupled with a TGF-β gene signature (3,29).
The results of the present study first linked FXYD5 to TGF-β
signaling. Positive feedback is renowned to magnify a signal and
facilitates a self-maintaining mode, which is autonomous to the
initial inducements. It was hypothesized that, once activated by
TGF-β, the FXYD5-mediated feedback loop would enable EOC cells to
become self-governing, which would reinforce the propensity of the
EOC cells to invade and metastasize to new microenvironments, and
would explain the co-expression and significant upregulation of
FXYD5 and TGF-β in late-stage EOC (4,5).
To date, to the best of our knowledge, no additional
partners for interaction have been described for FXYD5, apart from
for the Na+/K+-ATPase subunits (14,33).
Previous studies have demonstrated that the SMAD7-SMURF2 complex is
recruited to the TGF-β receptor complex, where it results in the
ubiquitination and degradation of the receptors as well as SMAD7
via the proteasome-mediated signaling pathway (9,34).
Kavsak et al (9) have
defined SMAD7 as an adaptor in an E3 ubiquitin ligase complex that
targets the TGF-ß receptor for degradation. The present study
demonstrated that FXYD5 not only downregulated the SMAD7 protein
expression levels, but also dispersed the SMAD7-SMURF2 complex,
which can be recruited to the TGF-β receptor, where it
de-ubiquitinates and stabilizes TβR1 (9,10,34,35).
Therefore, the post-transcriptional regulation of SMAD7 or
regulation of the stability of the SMAD7-SMURF2 complex by FXYD5
may serve an important role in the progression of ovarian cancer
and in TGF-β signaling. However, the detailed mechanisms though
which FXYD5 regulates SMAD7 and the SMAD7-SMURF2 complex, whether
through the Na+/K+-ATPase or not, requires
further investigation. Additionally, the problem that the TGFβR1
expression at 1-3 h in the FXYD5(-) group was significantly higher
than that in the FXYD5(+) group was noted. Since GAPDH expression
at 1-3 h in the FXYD5(-) group was also significantly higher than
that in the FXYD5(+) group; it was likely that the aforementioned
results were caused by the difference in the amount of the total
protein samples. Therefore, TGFBR1 protein expression was
normalized based on GAPDH protein expression. Furthermore,
according to the ratio of TGFBR1 to GAPDH over time, the curve was
made to compare the degradation rates of TGFBR1 protein between the
FXYD5(+) and FXYD5(-) groups. In the curve shown in Fig. 5A, there was no significant
difference at the initial 1-3 h; however, the FXYD5(+) group
exhibited a slower degradation rate at 4-6 h.
The TGF-β signaling pathway is currently viewed as
a therapeutic target in advanced tumors (36). To disrupt this intriguing feedback
loop, FXYD5 represents an ideal target. As a transmembrane protein
that is located in the cytomembrane, the unusually long
extracellular domain of FXYD5 (15) may enable the design of a homing
target for immunotoxins or cancer-directed imaging agents.
Additionally, the survival analysis indicated that for patients
undergoing chemotherapy, high FXYD5 expression as associated with
poor survival. Therefore, in addition to these chemotherapeutic
strategies, FXYD5 may be an alternative therapeutic target that can
extend the survival time of patients.
Due to the tumor heterogeneity, there are marked
differences in molecular biological behaviors between different
tumors. Whether this feedback loop exists in other tumors remains
to be explored. In the future it should be explored whether this
feedback loop could be applied to other types of cancer, including
cervical cancer, breast cancer and lung cancer.
In summary, the results identified FXYD5 as a novel
metastasis driver, thus elucidating the mechanisms that underlie
the TGF-β/SMADs signaling pathway in ovarian cancer, and providing
a promising therapeutic target for human ovarian cancer.
Supplementary Data
Funding
The present study was supported by grants from
Shanghai Sailing Program (grant nos. 16YF1401100 and 19YF1404300),
Natural Science Foundation of Shanghai (grant no. 17ZR1403500) and
Natural Science Foundation of China (grant no. 81802596).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
YB collected the clinical samples and patient
information, and conducted the majority of in vitro and
in vivo experiments. LDL conducted the bioinformatics
analysis, immunohisto-chemistry staining and survival analysis. JL,
RFC and HLY helped establishing the stable cell lines and
participated in in vitro experiments on functions. HFS and
JYW participated in western blot analysis and made constructs. YB,
LDL and XL conceived the project, designed most of the experiments
and wrote the manuscript. XL supervised the project. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Approval was obtained from the Human Research
Ethics Committee of the Obstetrics and Gynecology Hospital of Fudan
University for the use of all samples by using a protocol that
conforms to the provisions of the Declaration of Helsinki (as
revised in Seoul, 2008; reference no. [2015] 27). Written informed
consent was obtained from all patients. Animal experiments were
approved by the Institutional Animal Care and Use Committee of
Fudan University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Abbreviations:
|
EOC
|
epithelial ovarian cancer
|
|
TGF-β
|
transforming growth factor-β
|
|
TβR1
|
TGF-β receptor 1
|
|
EMT
|
epithelial-mesenchymal transition
|
|
FXYD5
|
FXYD domain-containing ion transport
regulator 5
|
|
SMAD7
|
SMAD family member 7
|
|
SMURF2
|
SMAD specific E3 ubiquitin protein
ligase 2
|
|
TCGA
|
The Cancer Genome Atlas
|
|
IHC
|
immunohistochemistry
|
|
SBEs
|
SMAD-binding elements
|
References
|
1
|
Lengyel E: Ovarian cancer development and
metastasis. Am J Pathol. 177:1053–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ye X and Weinberg RA:
Epithelial-mesenchymal plasticity: A central regulator of cancer
progression. Trends Cell Biol. 25:675–686. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang D, Sun Y, Hu L, Zheng H, Ji P, Pecot
CV, Zhao Y, Reynolds S, Cheng H, Rupaimoole R, et al: Integrated
analyses identify a master microRNA regulatory network for the
mesenchymal subtype in serous ovarian cancer. Cancer Cell.
23:186–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parikh A, Lee C, Joseph P, Marchini S,
Baccarini A, Kolev V, Romualdi C, Fruscio R, Shah H, Wang F, et al:
MicroRNA-181a has a critical role in ovarian cancer progression
through the regulation of the epithelial-mesenchymal transition.
Nat Commun. 5:29772014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsumura N, Huang Z, Mori S, Baba T,
Fujii S, Konishi I, Iversen ES, Berchuck A and Murphy SK:
Epigenetic suppression of the TGF-beta pathway revealed by
transcriptome profiling in ovarian cancer. Genome Res. 21:74–82.
2011. View Article : Google Scholar :
|
|
6
|
Wang Y, Shi J, Chai K, Ying X and Zhou BP:
The role of Snail in EMT and tumorigenesis. Curr Cancer Drug
Targets. 13:963–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ikushima H and Miyazono K: TGFbeta
signalling: A complex web in cancer progression. Nat Rev Cancer.
10:415–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kavsak P, Rasmussen RK, Causing CG, Bonni
S, Zhu H, Thomsen GH and Wrana JL: Smad7 binds to Smurf2 to form an
E3 ubiquitin ligase that targets the TGF beta receptor for
degradation. Mol Cell. 6:1365–1375. 2000. View Article : Google Scholar
|
|
10
|
Eichhorn PJ, Rodón L, Gonzàlez-Juncà A,
Dirac A, Gili M, Martínez-Sáez E, Aura C, Barba I, Peg V, Prat A,
et al: USP15 stabilizes TGF-β receptor I and promotes oncogenesis
through the activation of TGF-β signaling in glioblastoma. Nat Med.
18:429–435. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee YK, Lee SY, Park JR, Kim RJ, Kim SR,
Roh KJ and Nam JS: Dysadherin expression promotes the motility and
survival of human breast cancer cells by AKT activation. Cancer
Sci. 103:1280–1289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lubarski I, Asher C and Garty H: FXYD5
(dysadherin) regulates the paracellular permeability in cultured
kidney collecting duct cells. Am J Physiol Renal Physiol.
301:F1270–F1280. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lubarski I, Asher C and Garty H:
Modulation of cell polarization by the
Na+-K+-ATPase-associated protein FXYD5
(dysadherin). Am J Physiol Cell Physiol. 306:C1080–C1088. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lubarski-Gotliv I, Dey K, Kuznetsov Y,
Kalchenco V, Asher C and Garty H: FXYD5 (Dysadherin) may mediate
metastatic progression through regulation of the beta-
Na+-K+-ATPase subunit in 4T1 mouse breast
cancer model. Am J Physiol Cell Physiol. 313:C108–C117. 2017.
View Article : Google Scholar
|
|
15
|
Lubarski I, Pihakaski-Maunsbach K, Karlish
SJ, Maunsbach AB and Garty H: Interaction with the Na,K-ATPase and
tissue distribution of FXYD5 (related to ion channel). J Biol Chem.
280:37717–37724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lubarski I, Karlish SJ and Garty H:
Structural and functional interactions between FXYD5 and the
Na+-K+-ATPase. Am J Physiol Renal Physiol.
293:F1818–F1826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nam JS, Kang MJ, Suchar AM, Shimamura T,
Kohn EA, Michalowska AM, Jordan VC, Hirohashi S and Wakefield LM:
Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of
dysadherin in human breast cancer cells. Cancer Res. 66:7176–7184.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raman P, Purwin T, Pestell R and Tozeren
A: FXYD5 is a marker for poor prognosis and a potential driver for
metastasis in ovarian carcinomas. Cancer Inform. 14:113–119. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lubarski Gotliv I: FXYD5:
Na(+)/K(+)-ATPase regulator in health and disease. Front Cell Dev
Biol. 4:262016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gyorffy B, Lánczky A and Szállási Z:
Implementing an online tool for genome-wide validation of
survival-associated biomarkers in ovarian-cancer using microarray
data from 1287 patients. Endocr Relat Cancer. 19:197–208. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McCarty KS Jr, Miller LS, Cox EB, Konrath
J and McCarty KS Sr: Estrogen receptor analyses. Correlation of
biochemical and immunohistochemical methods using monoclonal
antireceptor antibodies. Arch Pathol Lab Med. 109:716–721.
1985.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Jiang HL, Sun HF, Gao SP, Li LD, Huang S,
Hu X, Liu S, Wu J, Shao ZM and Jin W: SSBP1 suppresses TGFβ-driven
epithelial-to-mesenchymal transition and metastasis in
triple-negative breast cancer by regulating mitochondrial
retrograde signaling. Cancer Res. 76:952–964. 2016. View Article : Google Scholar
|
|
25
|
Pathan M, Keerthikumar S, Ang CS, Gangoda
L, Quek CY, Williamson NA, Mouradov D, Sieber OM, Simpson RJ, Salim
A, et al: FunRich: An open access standalone functional enrichment
and interaction network analysis tool. Proteomics. 15:2597–2601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu D, Wolf JK, Scanlon M, Price JE and
Hung MC: Enhanced c-erbB-2/neu expression in human ovarian cancer
cells correlates with more severe malignancy that can be suppressed
by E1A. Cancer Res. 53:891–898. 1993.PubMed/NCBI
|
|
27
|
Vergara D, Merlot B, Lucot JP, Collinet P,
Vinatier D, Fournier I and Salzet M: Epithelial-mesenchymal
transition in ovarian cancer. Cancer Lett. 291:59–66. 2010.
View Article : Google Scholar
|
|
28
|
Petersen M, Thorikay M, Deckers M, van
Dinther M, Grygielko ET, Gellibert F, de Gouville AC, Huet S, ten
Dijke P and Laping NJ: Oral administration of GW788388, an
inhibitor of TGF-beta type I and II receptor kinases, decreases
renal fibrosis. Kidney Int. 73:705–715. 2008. View Article : Google Scholar
|
|
29
|
Yang H, Wang L, Zhao J, Chen Y, Lei Z, Liu
X, Xia W, Guo L and Zhang HT: TGF-β-activated SMAD3/4 complex
transcriptionally upregulates N-cadherin expression in non-small
cell lung cancer. Lung Cancer. 87:249–257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shi Y, Wang YF, Jayaraman L, Yang H,
Massagué J and Pavletich NP: Crystal structure of a Smad MH1 domain
bound to DNA: Insights on DNA binding in TGF-beta signaling. Cell.
94:585–594. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang SH, Li J, Al-beit M, Zhang J, Ma D
and Lu X: Screening and identification of potential miRNA involved
in ovarian cancer invasion and metastasis. Zhonghua Zhong Liu Za
Zhi. 32:650–654. 2010.In Chinese. PubMed/NCBI
|
|
32
|
Bai F, Feng J, Cheng Y, Shi J, Yang R and
Cui H: Analysis of gene expression patterns of ovarian cancer cell
lines with different metastatic potentials. Int J Gynecol Cancer.
16:202–209. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Antonov AV, Krestyaninova M, Knight RA,
Rodchenkov I, Melino G and Barlev NA: PPISURV: A novel
bioinformatics tool for uncovering the hidden role of specific
genes in cancer survival outcome. Oncogene. 33:1621–1628. 2014.
View Article : Google Scholar
|
|
34
|
Ogunjimi AA, Briant DJ, Pece-Barbara N, Le
Roy C, Di Guglielmo GM, Kavsak P, Rasmussen RK, Seet BT, Sicheri F
and Wrana JL: Regulation of Smurf2 ubiquitin ligase activity by
anchoring the E2 to the HECT domain. Mol Cell. 19:297–308. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang HL, Sun HF, Gao SP, Li LD, Hu X, Wu
J and Jin W: Loss of RAB1B promotes triple-negative breast cancer
metastasis by activating TGF-β/SMAD signaling. Oncotarget.
6:16352–16365. 2015.PubMed/NCBI
|
|
36
|
Akhurst RJ and Hata A: Targeting the TGFβ
signalling pathway in disease. Nat Rev Drug Discov. 11:790–811.
2012. View Article : Google Scholar : PubMed/NCBI
|














