Introduction
Liver cancer is one of the most frequent malignant
tumors with a high prevalence in Eastern and South-Eastern Asia,
and rising incidence rates in the Western world. It is the fourth
leading cause of cancer-related mortality worldwide, due to high
rates of late-stage diagnosis and limited treatment options
(1,2). Hence, novel therapeutic strategies
targeting oncogenic drivers are warranted.
Proviral integration site for Moloney murine
leukemia virus 2 (PIM2) belongs to a family of constitutively
active serine/threonine kinases comprising 3 members, PIM1, PIM2
and PIM3. These proto-oncogenes are involved in several cellular
processes, including cell survival/apoptosis, cell cycle
progression, migration and invasion (3-5).
While in some cases they exhibit redundancy and can compensate for
one another (6,7), they also have isoenzyme-specific
functions (8,9). PIM1 and PIM2 are most frequently
deregulated in hematological malignancies, whereas PIM3 is often
found to be overexpressed in solid tumors (10). From a therapeutic viewpoint, PIM1
has been explored most extensively so far, with PIM1 inhibitors
being tested in clinical studies as a monotherapy or in combined
treatment strategies, predominantly in patients with hematological
cancers (11).
PIM2 and its role in cancer progression has not yet
been studied in detail. It has been shown to synergize with c-MYC
to drive lymphomagenesis (6,12,13)
in an NF-κB-dependent manner (14)
and is overexpressed in multiple myeloma (7,15,16).
In ovary and breast tumors, PIM2 has been found to be upregulated
and to be associated with poor survival rates (17). In tumor entities other than liver
cancer, PIM2 has been described to directly interact with and
phosphorylate cell cycle regulators, p21WAF1/CIP1
(18) and p27KIP1
(19), as well as the
pro-apoptotic protein, BAD (20),
thus facilitating cell survival by maintaining mitochondrial
potential (21,22). Furthermore, PIM2 has been found to
be involved in the negative regulation of the DNA damage response
pathway (23) and mediate drug
resistance (24,25), as well as invasion (26,27).
In liver cancer, there is evidence to suggest that
PIM2 expression is upregulated (28-30),
e.g., by inhibiting apoptosis (29,30).
As demonstrated in a previous study, upon the stable PIM2
overexpression in normal L02 liver cells, this otherwise
non-tumorigenic cell line underwent malignant transformation to
form tumor xenografts in a mouse model (31). These observations encouraged us to
further explore PIM2 as a potential target for the treatment of
liver cancer. However, no specific PIM2 inhibitors are available
for functional, mechanistic and therapeutic studies. Thus, we used
RNAi-mediated specific PIM2 knockdown for further assessing its
functional relevance in liver cancer, and by employing a
nanoparticle-based strategy for siRNA delivery in vivo, we
also explored its therapeutic potential in a preclinical mouse
model.
Materials and methods
siRNAs and plasmids
A panel of three pre-designed siRNAs was purchased
from Thermo Fisher Scientific. The cells were transfected with
siRNA targeting PIM2 [siPIM2A: ACC UUC UUC CCG ACC CUC AdTdT
(sense) and UGA GGG UCG GGA AGA AGG UdTdT (antisense), siPIM2B: CUU
GGU UUU ACA GGU CAU UdTdT (sense) and AAU GAC CUG UAA AAC CAA GdCdT
(antisense), siPIM2C: GCC GGG AUU GUC CAA UUA CdTdT (sense) and GUA
AUU GGA CAA UCC CGG CdTdC (antisense) or negative control siRNA
(targeting Firefly luciferase mRNA, siCtrl: CUU ACG CUG AGU ACU UCG
AdTdT (sense) and UCG AAG UAC UCA GCG UAA GdTdT (antisense)]. In
addition, expression plasmids coding for p21WAF1/CIP1,
pcDNA3.1+p21wt and pcDNA3.1+p21mut were a kind gift from Dr Kurt
Engeland and Dr Gerd Müller (Department of Gynecology, University
Clinic of Leipzig).
Cell culture and transfection
The liver cancer cell lines, HepG2 and Huh-7, were
purchased from the American Type Culture Collection (ATCC). The
cell lines were authenticated by STR profiling. The cells were
maintained in a humidified incubator under standard conditions
(37°C, 5% CO2) in IMDM (PAA Laboratories supplemented
with 10% fetal calf serum (FCS).
For siRNA transfection, 5×102 (HepG2) or
3×102 (Huh-7) cells/well were seeded in a 96-well plate,
or 3×105 (HepG2) or 2×105 (Huh-7) cells in a
24-well plate, respectively, and incubated under standard
conditions unless stated otherwise. Using the INTERFERin™ siRNA
transfection reagent (Polyplus), 2.5 nM siRNA (for sequences, see
Table SI) for proliferation
assays or 5 nM siRNA for all other assays were transfected
according to the manufacturer’s instructions and incubated for the
time periods as indicated in the figures. The cells were then
analyzed in the well plates or harvested by trypsinization and
subsequent transfer to Eppendorf cups and used as described
below.
Transfection of 400 ng plasmid DNA per 24 well was
performed using FuGENE® transfection reagent (Promega)
according to the manufacturer’s protocol and incubated at 37°C for
6 h, prior to media change and siRNA transfection.
RNA preparation and mRNA detection by
qPCR
Total cellular RNA was prepared by phenol/chloroform
extraction using 250 µl TRI-Reagent (Sigma-Aldrich)
according to the manufacturer’s protocol. cDNA was transcribed from
800 ng RNA using the RevertAid™ H Minus First Strand cDNA Synthesis
Kit (Fermentas). Quantitative PCR was performed in an Applied
Biosystems StepOnePlus Real-Time PCR System (Thermo Fisher
Scientific) using the PerfeCTa SYBR®-Green FastMix ROX
(Quantabio). All procedures were conducted according to the
manufacturers’ protocols with 4 µl cDNA (diluted 1:10 with
nuclease-free water), 1 µl primers (5 µM; see
Table SI) and 5 µl
SYBR-Green master mix. A pre-incubation for 15 sec at 95°C was
followed by 40 amplification cycles: 10 sec at 95°C, 10 sec at 55°C
and 10 sec at 72°C. The melting curve for PCR product analysis was
determined by rapid cooling down from 95°C to 65°C, and incubation
at 65°C for 15 sec prior to heating to 95°C. To normalize for equal
mRNA/cDNA amounts, PCR reactions with target-specific and with
actin-specific primer sets were always run in parallel for each
sample, and target levels were determined by the ΔΔCq method
(32).
Western blot analysis
For protein analysis of cell cultures, the cells
were seeded and transfected in 24-well plates as described above.
At 72 h after transfection, the medium was removed and the cells
were washed once with PBS. 80 µl RIPA lysis buffer [50 mM
Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.5% sodium
deoxycholate, 0.1% SDS, 2.5 mM sodium pyrophosphate, 1 mM EDTA, 10
mM NaF, Protease Inhibitor Cocktail Set III (EDTA-free, Merck,
Darmstadt, Germany), PhosSTOP Phosphatase Inhibitor Cocktail
(Roche)] was added per well and plates were incubated on ice for 10
min. The lysate was centrifuged in Eppendorf tubes (9,600 × g, 4°C,
10 min) and the supernatant was transferred to a new cup.
To analyze the protein of the tumor samples, small
sections of tumor (~100 mg) were homogenized in 15 ml round bottom
tubes with 1 ml RIPA lysis buffer, using an Ultra-Turrax (IKA,
Staufen, Germany). Lysates were incubated 10 min on ice prior to
transfer to Eppendorf tubes. After centrifugation (9,600 × g, 4°C,
10 min), supernatants were transferred to fresh cups, centrifuged
once more and supernatants were, again, transferred to fresh cups.
The protein concentration was determined using the Bio-Rad DC™
Protein-Assay (Bio-Rad) according to manufacturer’s protocol.
Subsequently, 4X loading buffer (0.25 mM Tris-HCl, pH 6.8, 20%
glycerol, 10% β-mercaptoethanol, 8% SDS, 0.08% bromophenol blue)
was added to the samples to yield a 1X concentration. A volume
equivalent to 20 µg protein was loaded onto a 10%
polyacrylamide gel, separated by SDS-PAGE, and transferred onto
0.45 µm Immobilon™-P Transfer PVDF Membranes (Millipore).
The membranes were blocked with a protein-free blocking buffer
(Pierce), washed in TBST (10 mM Tris-HCl, pH 7.6, 150 mM NaCl, 0.1%
Tween-20), and incubated with primary antibodies diluted in
blocking buffer: Anti-PIM2 (monoclonal, rabbit anti-human, 1:1,000,
#4730, Cell Signaling Technology), anti-survivin (monoclonal,
rabbit anti-human, 1:5,000, GTX62039, GeneTex), anti-PLK1
(monoclonal, rabbit anti-human, 1:1,000, #4513, Cell Signaling
Technology), anti-vinculin (monoclonal, mouse anti-human, 1:5,000,
V9131, Sigma-Aldrich) and anti-tubulin (monoclonal, mouse
anti-human, 1:2,000, T5168, Sigma-Aldrich). The blots were
incubated overnight at 4°C, washed in TBST and incubated for 1 h at
room temperature with horseradish peroxidase-coupled goat
anti-rabbit IgG (1:2,000; Cell Signaling Technology, #4414) or
horseradish peroxidase-coupled goat anti-mouse IgG (1:5,000; Thermo
Fisher Scientific, #31430) in blocking buffer. After washing again,
bound antibodies were visual-ized by enhanced chemiluminescence
[ECL kits SignalFire™ (Cell Signaling) or SuperSignal®
West Femto (Thermo Fisher Scientific)]. Scanned bands were
quantitated using ImageJ software (National Institutes of
Health).
Proliferation assay
At the time points indicated, numbers of viable
cells were determined using a colorimetric assay. Briefly, the
medium was aspirated from the wells and 50 µl of a 1:10
dilution of cell proliferation Reagent WST-1 (Roche Molecular
Biochemicals) in serum-free medium was added to the cells, followed
by incubation for 1 h at 37°C. The absorbance at 450 nm was
measured using an ELISA reader (Asys Hightech).
Colony formation assay
Cell numbers were determined and 400 cells per well
were seeded in 6-well plates in triplicates. At the indicated time
points, the medium was aspirated and cells were fixed and stained
with a crystal violet staining solution [0.4% crystal violet
(Roth), 50% methanol] for 20 min at room temperature. The staining
solution was removed and plates were washed twice with
dH2O. The plates were then left to dry overnight and the
number of colonies (a colony defined as ≥50 cells) was counted by
the naked eye.
Colony spread assay
The cells were counted and 20 µl drops of
cell suspension containing 5,000 cells were set in the middle of a
well of a new 6-well plate; 3 wells were loaded for each treatment.
The cells were allowed to attach for 2 h at 37°C before aspirating
excess media and carefully adding 2 ml of fresh media. Following
incubation for the time periods indicated in the figures, the
medium was removed and the cells were fixed and stained with a
crystal violet staining solution for 20 min at room temperature.
After aspirating the staining solution, the plates were washed
twice with dH2O and allowed to dry overnight. The
density of the main colony in the center, as well as the number of
distant colonies were determined using ImageJ software (version
1.48).
Spheroid assay
A total of 5,000 cells per well were seeded in
ultra-low attachment U-bottom plates (Nexcelom Bioscience) and the
spheroid was allowed to form. At the indicated time points, the
spheroid size was documented and the spheroid area was measured
using a Celigo Imaging Cytometer (Nexcelom Bioscience) and the
corresponding software. To determine the amount of dead cells,
spheroids were stained with propidium iodide (PI; 10 µg/ml)
and Hoechst 33342 (3 µg/ml) for 30 min at 37°C. The
fluorescence intensity was quantitated with the Celigo Imaging
Cytometer (Nexcelom).
Apoptosis analysis
To determine the rate of apoptosis by flow
cytometry, the cell suspension was spun down by centrifugation (200
× g, room temperature, 5 min). The supernatant was aspirated, the
cells were washed with ice-cold PBS and cell numbers were
determined. A total of 1×105 cells were transferred to
fresh Eppendorf tubes and centrifuged (200 × g, room temperature, 5
min). After resuspending the cells in 100 µl Annexin
V-binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2), 5 µl Annexin V-FITC (Thermo Fisher
Scientific) was added and the cells were incubated at room
temperature for 15 min in the dark. Subsequently, 2 µl of a
2 mg/ml PI solution was added, as well as another 400 µl of
Annexin V-binding buffer. The cells were analyzed using an Attune™
Flow Cytometer (Thermo Fisher Scientific) in the BL1 and BL3
channel.
To determine the activity of effector caspase 3 and
caspase 7, the Caspase-Glo® 3/7 Assay (Promega) was
used. A total of 2×103 cells were seeded in a 96-well
plate, transfected as described above, and incubated at 37°C for 72
h. The medium was aspirated and caspase substrate was diluted 1:5
in serum-free medium, prior to the addition of 50 µl to the
cells. Following incubation for 1 h at room temperature in the
dark, luminescence was measured using a Fluostar Optima reader (BMG
Labtec). A WST-1 assay was performed in parallel on the same plate
to normalize for differences in cell density.
Cell cycle analysis
At 48 h following transfection, the cells were
treated with nocodazole (100 ng/ml; Roth) to induce a G2/M block.
After 20 h, raw cells were trypsinized, transferred to Eppendorf
tubes and centrifuged (200 × g, room temperature, 5 min). The
supernatant was discarded and the cell pellet resuspended in ice
cold 70% ethanol. The cells were fixed for at least 1 h at −20°C
prior to centrifugation as above and were then subjected to RNase
digestion (50 µg/ml RNase A in PBS) for 30 min at 37°C. PI
solution was added to yield a final concentration of 50
µg/ml. Cell cycle distribution was analyzed in the BL2
channel using an Attune™ Flow Cytometer (Thermo Fisher
Scientific).
Mouse xenograft models
Twelve female athymic nude mice (Crl:NU-Foxn1nu,
Charles River Laboratories; age, 10 weeks; average weight, ~20 g)
were housed at 23°C in a humidified atmosphere and a
12-h-light/dark cycle, with standard rodent chow and water ad
libitum. For tumor establishment, 5×106 HepG2 cells
in 150 µl PBS were injected subcutaneously (s.c.) into both
flanks of the mice. When solid tumor xeno-grafts reached a volume
of ~50 mm3, the mice were randomly divided into specific
treatment and negative control treatment groups (6 mice each, with
n=8 tumors per group). For polymeric nanoparticle formation, siRNAs
were complexed with PEI F25-LMW essentially as previously described
(33). Briefly, one treatment, 10
µg siRNA was dissolved in 75 µl complexation buffer
(10 mM HEPES, 150 mM NaCl, pH 7.4) and incubated for 10 min. 50
µg PEI F25-LMW was dissolved in the same buffer, incubated
at room temperature for 10 min and then mixed with the siRNA
solution. Aliquots sufficient for treating all mice of each group
at a given time point were prepared and stored frozen at -80°C.
Prior to use, the complexes were thawed and incubated at room
temperature for 15-30 min. Every 2-3 days, 150 µl complex
equivalent to 10 µg PEI-complexed siRNA was administered by
intraperitoneal (i.p.) injection. Tumor volumes were monitored
every 2-3 days. The mice were sacrificed one day after the final
treatment, and tumors were removed. Sections of the tumor tissue
where snap-frozen in liquid nitrogen for RNA or protein
preparation.
Statistical analysis
Statistical analyses were performed using the
Student’s t-test or one-way ANOVA with Tukey’s multiple comparison
post-test. Values are presented as the means ± SEM, with 'n’ in the
figure legends indicating the number of independent experiments.
All independent experiments were performed at least in triplicates.
A value of P<0.05 was considered to indicate a statistically
significant difference.
Results
RNAi-mediated knockdown of PIM2 in HepG2
and Huh-7 liver cancer cell lines
As a specific PIM2 inhibitor is not yet available,
we selected an RNAi-based knockdown strategy to target PIM2. Three
PIM2-specific siRNAs were tested in the liver cancer cell lines,
HepG2 and Huh-7. RT-qPCR revealed that all three siRNAs
substantially reduced the mRNA levels of PIM2 to comparable extents
in both cell lines (Fig. S1A).
Transfection with the most efficient siRNA, siPIM2A, reduced the
PIM2 mRNA levels by almost 90%, as compared to the negative control
siRNA (siCtrl) targeting an unrelated gene (Firefly luciferase). In
the case of siPIM2C, residual PIM2 mRNA levels were observed at 30%
(HepG2 cells) and 13% (Huh-7 cells), while the least efficient
siPIM2B still resulted in a >60% knockdown in both cell lines.
Similar results were obtained at the protein level, as determined
by western blot analysis, confirming the substantial downregulation
of PIM2 with all PIM2-specific siRNAs and identifying siPIM2A as
the most efficient (Fig. 1B). For
further experiments, two of the three siRNAs were selected:
siPIM2A, as it achieved the most efficient knockdown and siPIM2B,
which exhibited a lower knockdown efficiency and was thus able to
resolve the association of the PIM2 expression levels with
phenotypic effects.
PIM2 knockdown leads to decreased
proliferation and impaired colony formation
For the initial assessment of the relevance of PIM2
as a possible target gene in liver cancer therapy, we analyzed the
effect of an RNAi-mediated knockdown on cell proliferation in
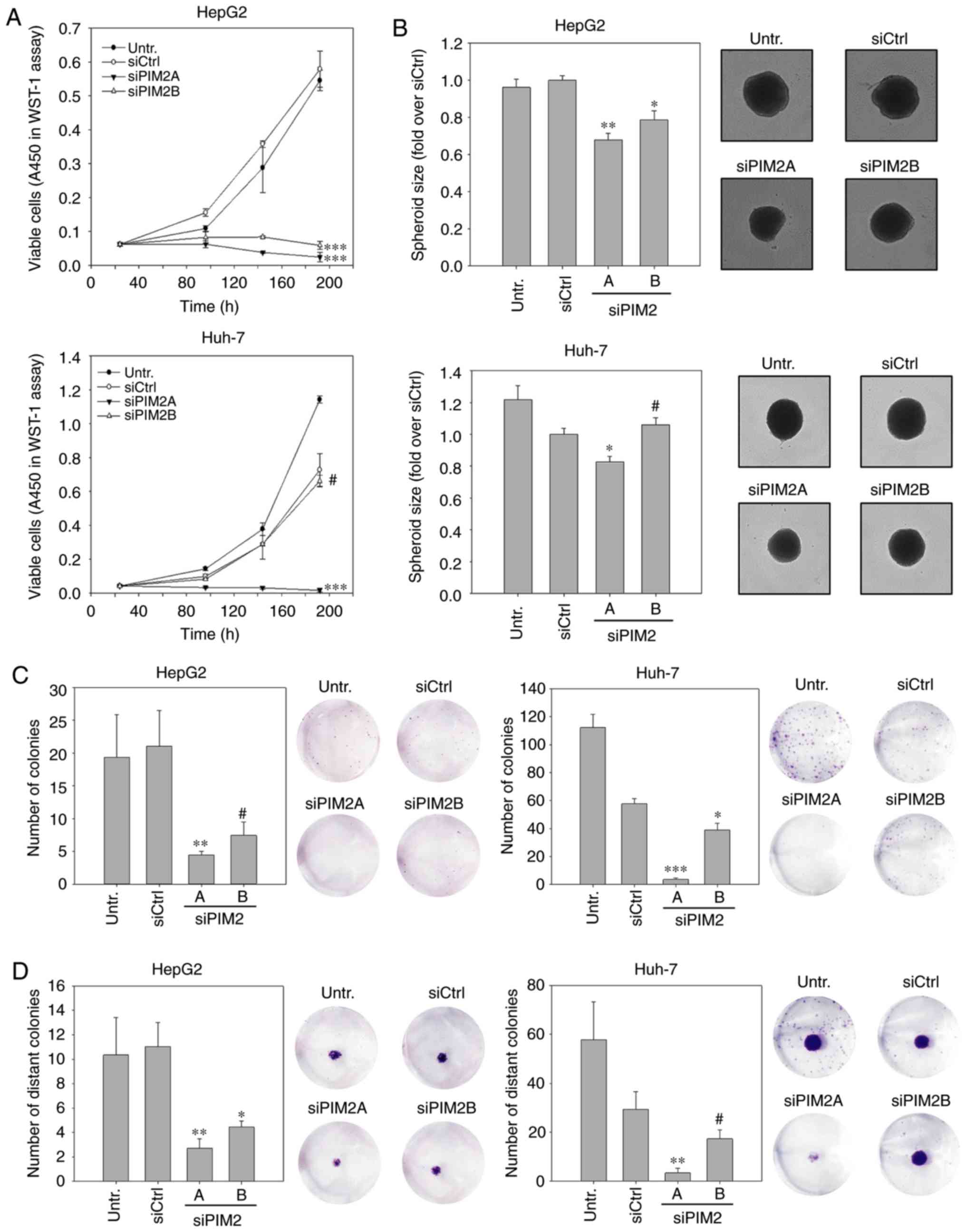
vitro. WST-1 assays in HepG2 cells revealed a profound decrease
in cell proliferation in the case of both PIM2-specific siRNAs. In
fact, while no non-specific siRNA transfection effects on the PIM2
level were observed (compare siCtrl vs. untreated), PIM2 knockdown
essentially led to an arrest in cell proliferation (Fig. 1A, upper graph). By contrast, a
similar abolishment of Huh-7 cell proliferation was only observed
in the case of the more potent siRNA, siPIM2A, whereas the less
potent siRNA, siPIM2B, was not able to inhibit cell proliferation
more than the siCtrl (Fig. 1A,
lower graph).
The anti-proliferative effects were subsequently
tested in a more complex 3D cell culture system. Compared with 2D
cultures, spheroid cultures more closely resemble the in
vivo situation with regard to cell-cell and cell-matrix
contacts, gradient access to oxygen and nutrient supply. In this
experiment, the HepG2 or Huh-7 cells were transfected prior to the
generation of spheroids, which were then allowed to grow for 7
days. Compared to the negative controls, the siRNA- mediated
knockdown of PIM2 did not alter the shape or formation kinetics
(e.g., more rapid or delayed formation; data not shown), but led to
significantly smaller HepG2 spheroids. The comparison between the
two specific siRNAs also revealed a gene-dose effect, with size
reductions of 32% (siPIM2A) and 21% (siPIM2B) as compared to the
control spheroids (Fig. 1B, upper
panel). Similar to the 2D proliferation assay, spheroid sizes of
the Huh-7 cells only decreased upon transfection with the more
efficient siRNA, siPIM2A (17% reduction compared to the siCtrl;
Fig. 1B, lower panel).
Colony numbers and sizes were also profoundly
reduced in the HepG2 cells, with a >80% inhibition for both
PIM2-specific siRNAs over the siCtrl. As expected, siPIM2A was
slightly more efficient than siPIM2B (Fig. 1C, left panels). Again, the siRNA
knockdown efficiency was more variable in the Huh-7 cells where, in
addition to some rather profound non-specific effects, an almost
complete abolishment of colony formation was observed for siPIM2A.
The less efficient siPIM2B reduced the colony number by only ~30%
as compared to siCtrl (Fig. 1C,
right panels). To investigate this further, we performed colony
spread assays. In this experiment, a colony is transferred to the
middle of an empty well, is allowed to grow for a specified time
period and the establishment of distant colonies is then assessed.
Similar to the above-mentioned experiments, it was observed that
the primary colony sizes were smaller in the siRNA-treated HepG2
(both siRNAs) and Huh-7 cultures (siPIM2A only; Fig. 1D, cell staining images).
Additionally, decreases in the number of distant colonies were also
observed (Fig. 1D, bar diagrams).
It should also be noted that the densities of the primary colonies
were decreased in the siPIM2-treated cells compared to the control
treatment. This was observed for the HepG2 cells treated with both
PIM2 siRNAs and in the Huh-7 cells exposed to the more potent
siRNA, siPIM2A, while the less potent siRNA, siPIM2B, again exerted
no marked effect (Fig. S2).
The combined observations of this experiment suggest
that Huh-7 cells are less sensitive to PIM2 knockdown, with higher
reductions in PIM2 expression were required in this cell line to
obtain inhibitory effects. Due to the observed non-specific
transfection effects, it was not possible to further increase the
siRNA amounts. This emphasizes the need for high efficiency siRNAs
in Huh-7 cells, while this was found to be less critical for the
HepG2 cells.
Rate of apoptosis is not affected by
knockdown of PIM2
Subsequently, we examined whether the inhibitory
effects of PIM2 knockdown may at least in part be due to elevated
cell death, since the evasion of apoptosis is one of the hallmarks
of cancer cells, and PIM2 kinase has been described to be involved
in this process (16,21). To this end, we first examined
changes in the proportion of apoptotic cells in the cell
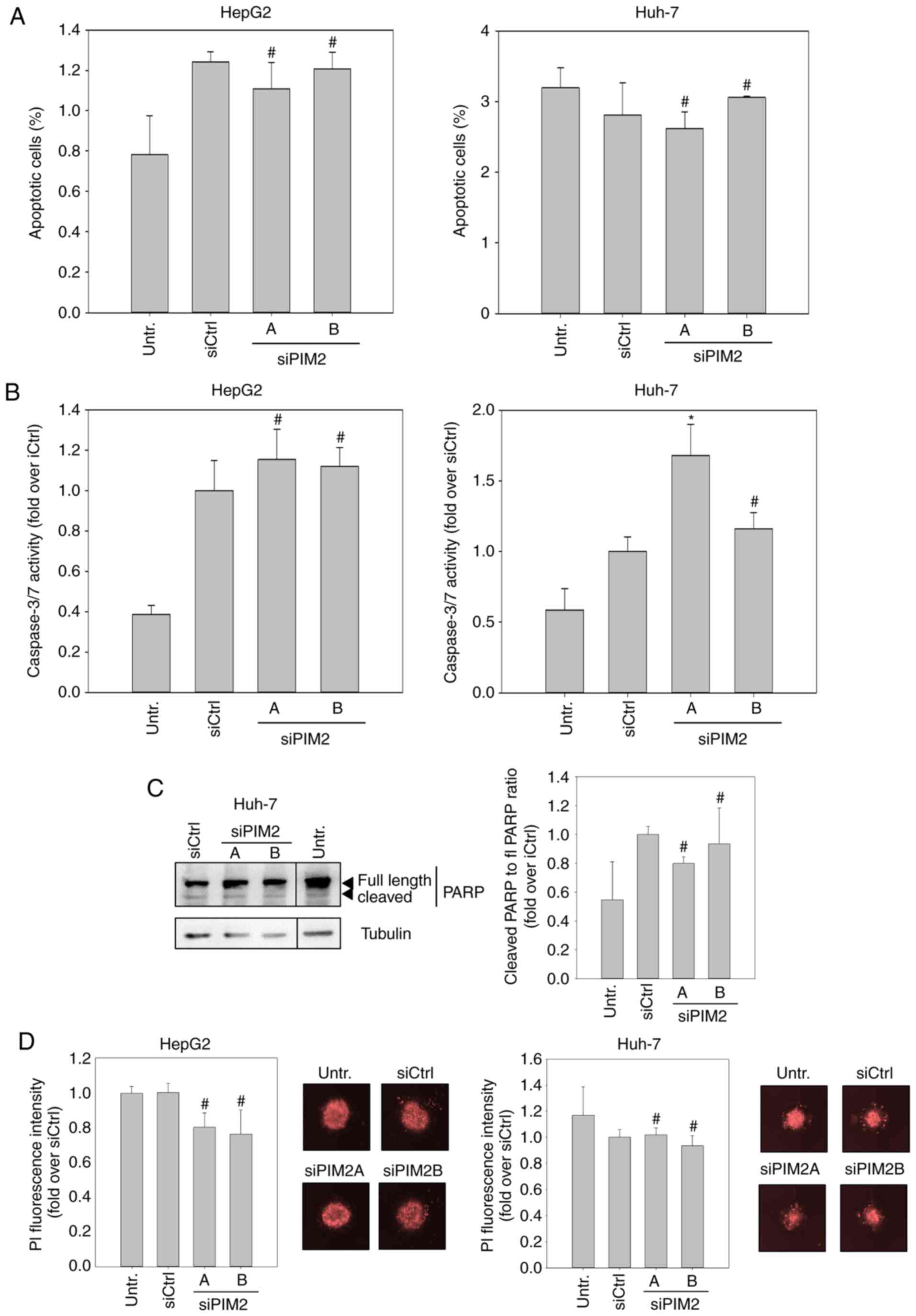
population. Using flow cytometry, no significant elevation in the
numbers of Annexin-V-positive and PI-negative cells was detected
upon siPIM2 transfection in both cell lines (Figs. 2A and S3). When examining the effects of PIM2
knockdown in HepG2 cells on the effector caspases of intrinsic and
extrinsic apoptosis pathways, caspases 3 and 7, we only found a
marginal increase in caspase activity upon siPIM2 transfection in
comparison to the siCtrl (Fig. 2B,
left graph). However, siPIM2A transfection in the Huh-7 cells led
to a significant (~30%) induction of caspase 3/7 activity (Fig. 2B, right graph). To address the
discrepancy between the unaltered numbers of apoptotic Huh-7 cells
and the elevated caspase 3/7 activity upon siPIM2A transfection, we
performed a western blot analysis of poly(ADP-ribose)-polymerase
(PARP), a downstream target of caspases 3 and 7. Notably, no
increase in PARP cleavage was detected following PIM2 knockdown,
indicating that the measured elevation in caspase activity was not
sufficient to mediate apoptosis in Huh-7 cells (Fig. 2C). In spheroid cultures stained
with PI for the detection of dead cells, again no effect of PIM2
knockdown on cell death was found (Fig. 2D).
Collectively, these data suggest that the induction
of apoptosis is not a key mechanism of cell inhibition upon PIM2
knockdown, which in turn indicates that the main functional
consequence of PIM2 overexpression in liver cancer is not the
increase of cell survival/resistance to cell death.
Cell cycle block in G0/G1 phase upon PIM2
knockdown
In view of the absence of major effects of PIM2
knockdown on apoptosis, the observed reduction in cell
proliferation following treatment could be due to the deceleration
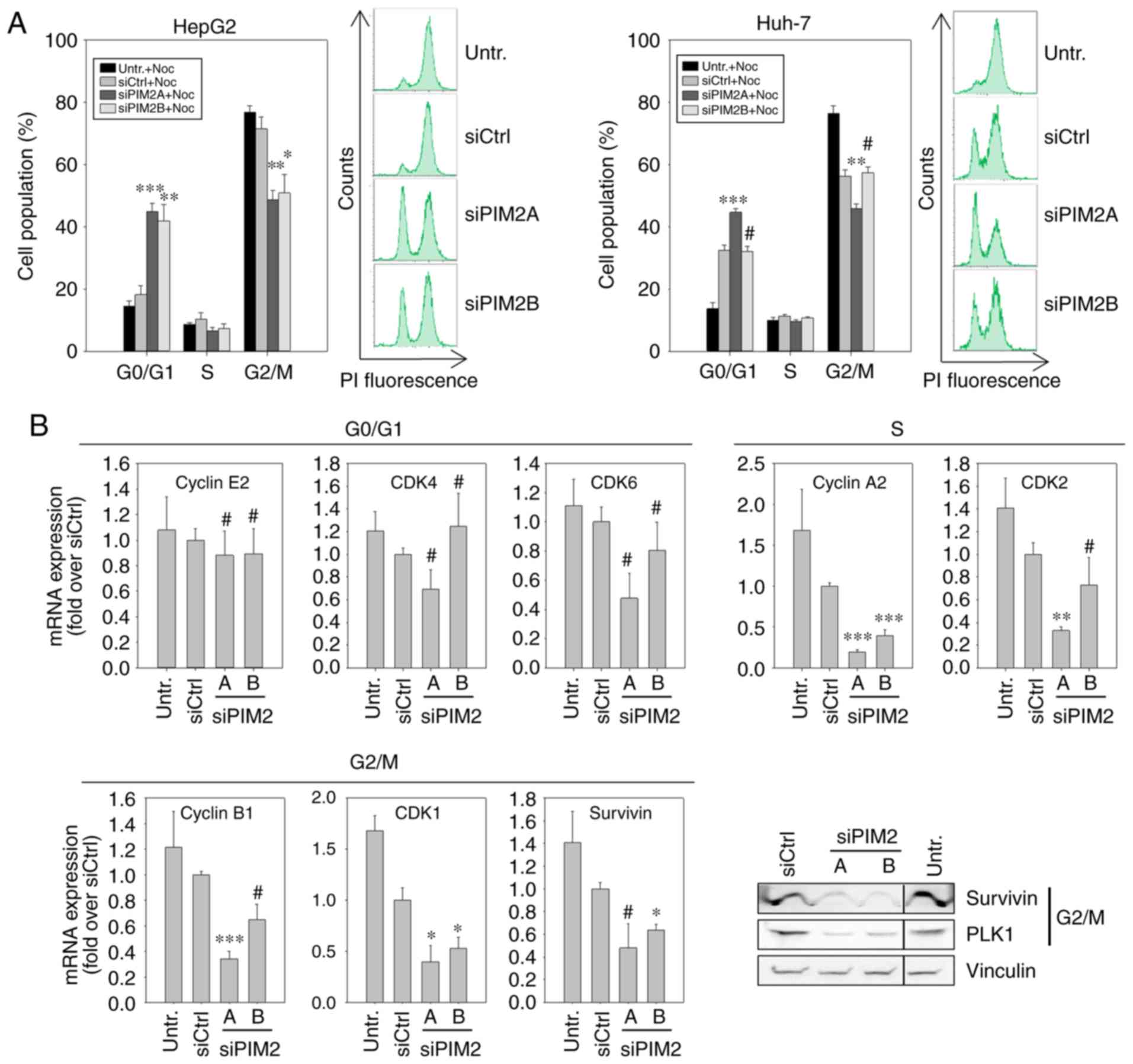
of the cell cycle. To assess this, 48 h following siRNA
transfection, the cells were treated with nocodazole, which
interferes with the polymerization of microtubules and thus blocks
the cells in the G2/M phase. By determining cell cycle distribution
at a defined time point (20 h) following the addition of
nocodazole, the proportion of cells in the G2/M phase was
quantified. Higher percentages indicated a more rapid cell cycle
progression, while lower proportions denoted a decelerated cell
cycle progression. In the case of untransfected cells or those
transfected with a negative control, almost 80% of the cell
population were found to be in the G2/M phase, with <20%
remaining in the G0/G1 phase (Fig.
3A, left panels). By contrast, following the knockdown of PIM2,
only ~50% of the cells had progressed to the G2/M phase, while a
similar percentage was still in the G0/G1 phase. This was true for
both PIM2-specific siRNAs in HepG2 cells, while in the Huh-7 cells,
a reduced cell cycle progression (i.e., the number of cells in
G2/M<G0/G1) was only observed following transfection with the
more potent siRNA, siPIM2A (Fig.
3A, right panels).
To investigate the underlying molecular effects of
the reduced cell cycle progression upon PIM2 knockdown and possible
differences between the two cell lines, we quantified the mRNA
levels of proteins known to play a critical role in cell cycle
regulation. More specifically, we performed RT-qPCR for G0/G1
phase-[cyclin E2, cyclin-dependent kinase (CDK)4 and CDK6] S
phase-(cyclin A2 and CDK2) and G2/M phase-related genes (cyclin B1,
CDK1 and Survivin). Additionally, the expression the of G2/M
phase-related genes, Survivin and Polo-like kinase 1 (PLK1), was
examined at the protein level by western blot analysis. In the
HepG2 cells, PIM2 knockdown resulted in a decreased expression of
all S phase- and G2/M phase-related genes examined, with the mRNA
levels reflecting the respective knockdown efficiencies of siPIM2A
and siPIM2B, as also observed above (Fig. 3B). At the protein level, a marked
downregulation in the levels of Survivin and PLK1 was observed in
the HepG2 cells (Fig. 3B, lower
right panel). By contrast, the levels of the G0/G1 phase-related
genes in the Huh-7 cells were only marginally affected by PIM2
knockdown or not at all, with some statistically non-significant
minor reductions in the mRNA levels of CDK4 and CDK6 upon
transfection with the more potent siRNA, siPIM2A. A significant
decrease in mRNA expression was only observed for the S
phase-related genes, cyclin A2 and CDK2 (Fig. 3C). Western blot analysis of the
G2/M phase-related genes, Survivin and PLK1, revealed only minor
reductions in expression levels compared to control treatments, and
again only for the more potent siRNA, siPIM2A.
It was hypothesized that the differences between the
cell lines, HepG2 and Huh-7, may be dependent on the expression of
the cell cycle regulator, p21WAF1/CIP1, since HepG2 and
Huh-7 cells differ in their status of p53, a known transcriptional
regulator of p21WAF1/CIP1 (HepG2, p53 wild type; Huh-7,
p53 mutated). Indeed, we found that the p21WAF1/CIP1
mRNA levels were 30-fold higher in the HepG2 cells as compared to
the Huh-7 cells (Fig. S4A).
Recently, Liu et al reported that PIM2 induced cell cycle
arrest in the G0/G1 phase via p53-independent, but
p21WAF1/CIP1-dependent signaling in lung cancer cell
lines (34). Thus, in this study,
we examined whether the restoration of p21WAF1/CIP1
expression in the Huh-7 cells would enable a more sensitive cell
cycle regulation following PIM2 knockdown. Expression plasmids
encoding either wild-type (wt) or mutated, non-functional (mut)
p21WAF1/CIP1 were transfected into the Huh-7 cells
followed by siRNA transfection, nocodazole treatment and the
analysis of cell cycle distribution as described above. The ectopic
expression of wt p21WAF1/CIP1 attenuated cell cycle
progression, as compared to the cells trans-fected with
p21WAF1/CIP1 mut (Fig.
S4B, black bars indicating more cells in the G0/G1 phase and
fewer cells in the G2/M phase). This is in line with findings in
the study by Yew et al in multipotent stroma cells, where
p21WAF1/CIP1 knockdown cells exhibited an accelerated
cell cycle progression compared to the control cells (35). Upon PIM2 knockdown in the Huh-7
cells, the p21WAF1/CIP1 wt-transfected cells exhibited a
minor further decrease in cell cycle progression, while the effect
in the p21WAF1/CIP1 mut transfected counterparts was
weaker and observed exclusively in the siPIM2A-transfected cells.
This finding indicates that the effects of PIM2 knockdown on the
cell cycle are only marginally dependent on p21WAF1/CIP1
in liver cancer.
PIM2 knockdown leads to antitumor effects
in vivo
Finally, to assess whether the profound
anti-proliferative, tumor cell-inhibitory and cell cycle blocking
effects of PIM2 knockdown in HepG2 cells are applicable to the
in vivo situation, we explored a therapeutic PIM2 knockdown
in a murine tumor xenograft model. Following the establishment of
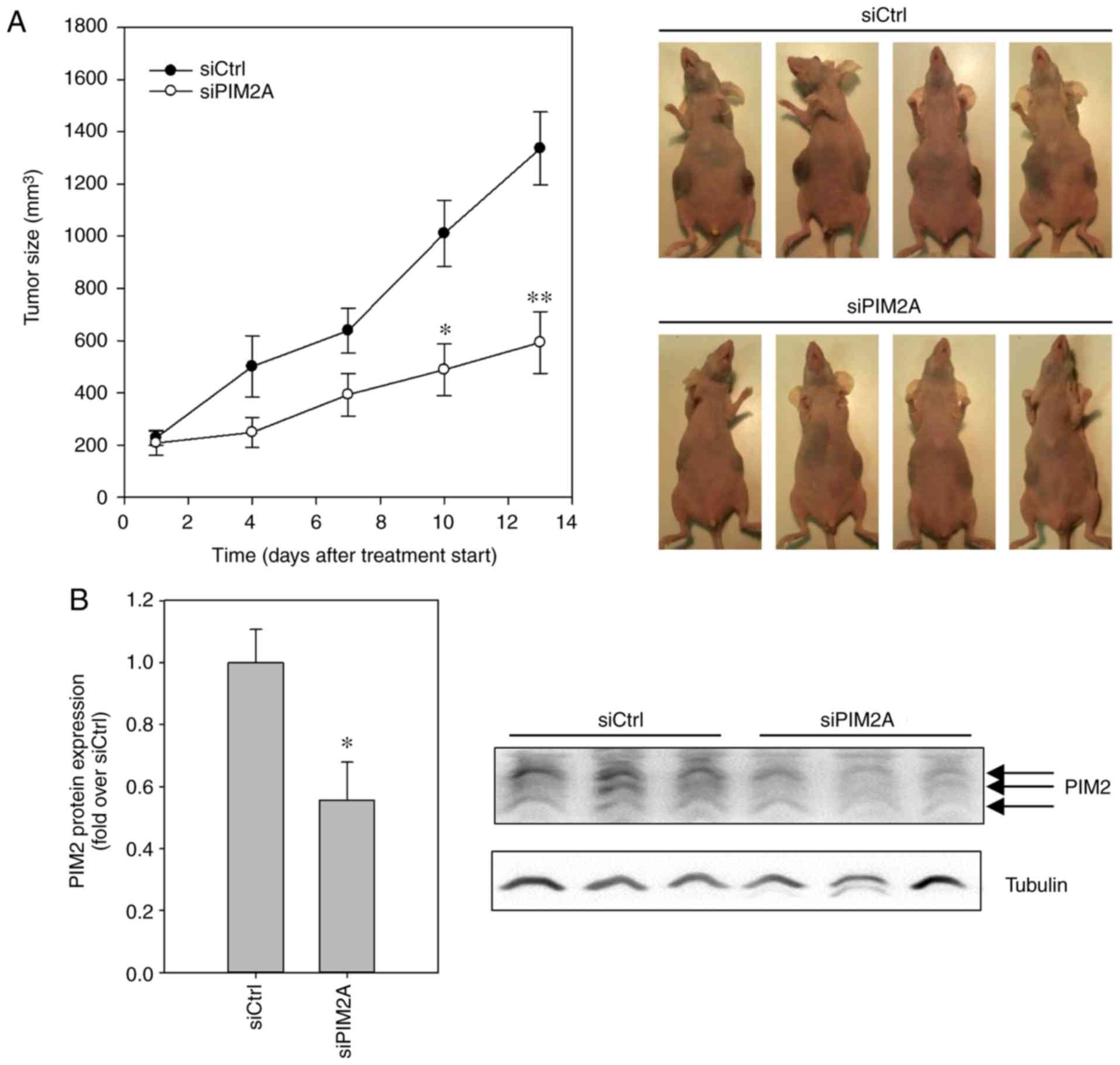
subcutaneous (s.c.) HepG2 tumor xenografts in nude mice, the
animals were randomly divided into groups and treated systemically
by i.p. injection 3×/week with either polyethylenimine
(PEI)-complexed siPIM2A (specific treatment) or PEI-complexed
siCtrl (negative control). Over a period of 13 days, tumor
xenografts in the mice treated with negative control PEI/siRNA
complexes grew by a factor of ~7, from ~200 mm3 to a
mean tumor volume of almost 1,400 mm3. By contrast, the
administration of PEI/siPIM2A complexes substantially decreased
tumor growth rates, with mean tumor volumes only reaching 600
mm3 in the same time period (Fig. 4A). The effects on tumor growth were
already visible at day 4 after commencing treatment and reached
statistical significance at day 10. The treatment, involving
repeated injection and systemic delivery of the PEI/siRNA
nanoplexes, was well-tolerated, with no obvious side-effects (e.g.,
alterations in mouse body weight or behavior, or other signs of
discomfort). Upon termination of the experiment, the analysis of
PIM2 expression levels by western blot analysis revealed a
significant ~50% reduction of PIM2 levels in the tumors of the mice
treated with PEI/siPIM2A nanoplexes, as compared to the tumor
xenografts from the mice in the negative control group (Fig. 4B). This confirmed that the observed
antitumor effect was indeed caused by the efficient knockdown of
PIM2.
Discussion
The overexpression of the survival kinase, PIM2, is
found in hematopoietic malignancies and solid tumors, and has been
associated with a poor prognosis (10,36).
In liver cancer, PIM2 is overexpressed as well, but little is known
about the functional relevance of PIM2 in this tumor entity. This
study demonstrates that a specific knockdown of PIM2 exerts tumor
cell-inhibitory effects in various in vitro settings, as
well as in an animal in vivo model.
The evasion of apoptosis has been described in
various tumor entities as one of the mechanisms through which
members of the PIM protein family promote cell survival (5,10).
PIM2 has mostly been described as an anti-apoptotic mediator
(7,16,21,22,29,30),
but controversially, in some reports, positive (18,37)
or no effects on apoptosis have been described as well (15,38,39).
This indicates that the role of PIM2 in apoptosis is
context-dependent. In HepG2 cells, two studies have described
higher apoptosis rates, as well as lower p-4E-BP and p-BAD levels
upon siRNA-mediated PIM2 knockdown (29,30).
By contrast, in this study, we did not observe the induction of
apoptosis or cell death upon PIM2 reduction, neither in 2D nor in
3D cell cultures. These contradictory findings may be due to
different cell culture conditions in terms of media composition and
FCS in particular, since differences in growth factor and cytokine
levels may exist between different brands and even batches of FCS.
In support of this hypothesis, Gong et al demonstrated that
HepG2 cells are less sensitive to apoptosis mediated by PIM2
knockdown in the presence of IL-3 (30), a finding shared by Fox et
al, who reported that PIM2 maintained a hyperphosphorylated
state of 4E-BP upon IL-3 withdrawal in the murine pro-B cell line
FL5.12 (21).
The potent negative effect on cell proliferation by
PIM2 knockdown observed in this study may be attributed to a
substantial block of the cell cycle in the G0/G1 phase in HepG2
cells. In line with these findings, Kreuz et al reported
that PIM2 knockdown in Burkitt’s lymphona cells was mainly
associated with a cell cycle block in G0/G1 rather than induction
of apoptosis (39). Accompanying
this cell cycle block, in this study, we observed a decrease in the
expression of S phase- and G2/M phase-related genes. To the best of
our knowledge, this is the first time that a broad influence of
PIM2 on the expression of cyclins, CDKs, and other cell
cycle-related genes is described. One of these genes is the mitosis
and cytokinesis regulator, PLK1, that functions as an oncogene in
liver cancer and is overexpressed in this tumor entity (40,41).
In this study, we found PLK1 to be downregulated upon PIM2
knockdown, whereas Adam et al (38) recently described the colocalization
of PLK1 and PIM2 following combined PIM1/PIM2 knockdown in HeLa
cells, as well as the dephosphorylation of PLK1 that led neither to
decreased PLK1 levels nor affected cell cycle distribution. The
results of this study underscore the potential of PIM2 to regulate
the cell cycle and to interfere with PLK1 oncogenic pathways via
different mechanisms.
The two liver cancer cell lines that were
investigated in this study, HepG2 and Huh-7, exhibited varying
sensitivities towards the knockdown of PIM2, as determined by
decreased proliferation rates and a decelerated cell cycle
progression. One explanation we considered relies on the
differential expression of the cell cycle inhibitor,
p21WAF1/CIP1, which is dependent on the p53 status of
the cells and seems to make HepG2 cells with wt p53 and thus higher
p21WAF1/CIP1 expression more sensitive to PIM2 knockdown
than Huh-7 with mutant p53 and a low p21WAF1/CIP1
expression. This is in line with the findings of Liu et al,
who described a p21WAF1/CIP1-dependent cell cycle block
in G0/G1 upon PIM2 knockdown in the non-small cell lung cancer cell
lines A549 and H1299 (34).
However, our findings in the Huh-7 cells also suggest an
additional, p21WAF1/CIP1-independent mechanism of the
cell cycle regulation by PIM2 in liver cancer cells, since the
ectopic overexpression of wt p21WAF1/CIP1 in this cell
line led to an only slight increase in sensitivity towards PIM2
knockdown. Another difference between the HepG2 and Huh-7 cells is
in their β-catenin status. There is increasing evidence that the
deregulation of the Wnt/β-catenin signaling pathway is involved in
liver cancer tumorigenesis (42,43).
Approximately 50% of liver cancers are characterized by the nuclear
accumulation of active β-catenin. HepG2 carries mutated β-catenin
heterozygously (one wild-type and one mutated allele), with the
concomitant deletion of aa25-140 resulting in the activation of the
protein and a strong nuclear accumulation (44), but also in a further decrease of
its already low expression levels. By contrast, Huh-7 cells carry
stabilized wild-type β-catenin and express higher levels of total
and activated β-catenin (45).
Nuclear, active β-catenin is involved in cell cycle progression and
its expression peaks in G2/M (46). Possibly, the lower amount of active
β-catenin may render HepG2 cells by a yet unknown crosstalk more
sensitive to cell cycle deceleration upon PIM2 knockdown,
suggesting that the PIM2-mediated cell cycle block in liver cancer
cells may be β-catenin-dependent. Thus, future studies are
warranted to explore and extensively characterize a possible
synergistic effect between PIM2 and β-catenin in a combined
knockdown strategy.
Another explanation as to why Huh-7 cells exhibit
less cell inhibition and effects on cell cycle upon PIM2 knockdown
compared to the HepG2 cells may related to the expression patterns
of the three PIM2 isoforms. They arise from three different
translation start sites on the same mRNA. In murine cells, isoforms
1 (40 kDa) and 2 (37 kDa) result from the utilization of
non-classical CUG translation initiation codons. The first AUG
codon gives rise to isoform 3 (34 kDa), but is not embedded in a
Kozak sequence and thus translated with low efficiency (12). For isoform 3, pro-apoptotic and
cell cycle inhibiting effects upon its ectopic expression were
described in cancer cell lines (18,37),
antagonizing the otherwise oncogenic activity of total PIM2
protein. In this study, by western blot analyses, we found a higher
proportion of expression of the 34 kDa isoform in Huh-7 cells than
in HepG2 cells, which may result in the less pronounced
antiproliferative effects of PIM2 knockdown in Huh-7 cells observed
in our experiments. By contrast, however, Yan et al found
the anti-apoptotic effects of the 34 kDa PIM2 isoform in the
non-cancerous murine hematopoietic cell line, FDCP1, upon IL-3
withdrawal (20), suggesting the
distinction between the roles of the different isoforms to be more
complex. Further studies are required, in which single isoform
knockdown strategies may help elucidate the individual roles of the
three PIM2 isoforms.
The profound effects of PIM2 knockdown on the number
of distant colonies in our colony spread assays gives rise to the
notion that PIM2 not only influences colony formation, but may
enable cells to disseminate to form new colonies, which is crucial
for metastasis. Cellular alterations in the potential to detach,
disseminate, migrate and/or re-attach at a new site may have
contributed to the phenotypic observations. A deeper understanding
of the mechanisms of migration and invasion leading to metastasis
in liver cancer, and the possible role of PIM2 in these processes,
is crucial, since the disease is often diagnosed at late stages
when patients have already developed extrahepatic metastases
predominantly located in the lung, lymph nodes, and bone, and
treatment options are thus limited (47). In contrast to PIM1 and PIM3, very
little is known regarding the impact of PIM2 on migration and
metastasis. There is evidence to suggest that PIM2 regulates the
invasion of breast cancer cells. It promotes signal transducer and
activator of transcription 3 (STAT3) activity via a
cytokine-dependent feedback loop, leading to STAT3 Ser727
phosphorylation and eventually to epithelial-mesenchymal transition
(EMT), increased migration and invasion (26). While having observed this feedback
loop previously in colorectal carcinoma cells for PIM1 (48), we did not observe any changes in
STAT3 Ser272 phosphorylation in liver cancer cell lines upon PIM2
knockdown. Instead, we found a decreased STAT3 phosphorylation at
Tyr705 (data not shown), which also modulates STAT3 transcriptional
activity and has been described to be associated with migration and
invasion (49). Further studies
are warranted to reveal the role of PIM2 in liver cancer migration
and invasion and the underlying molecular pathways.
In our mouse model of subcutaneous HepG2 xenografts,
PIM2 knockdown mediated substantial anti-tumor effects, confirming
that our findings from cell culture could be transferred to a
complex in vivo situation. Our polymeric, PEI-based
nanoparticle system for siRNA or miRNA delivery [(33,50; reviewed
in ref. 51)] allowed us to
specifically study in vivo effects of PIM2 knockdown, with
other PIM kinases remaining unaffected. The formulation of siRNAs
in a nanoparticle system addresses several shortcomings of small
RNAs as drugs, including insufficient stability, delivery and cell
uptake, as well as rapid elimination. In addition, the fact that
the first siRNA drug has obtained market approval most recently
(52) clearly indicates the
potential of siRNAs for oncogene-targeting therapies.
Taken together, the findings of this study highlight
the potential of targeted therapies in liver cancer that are based
on the specific inhibition of PIM2. This provides the basis for
further studies, aiming at a deeper exploration and understanding
of the underlying molecular effects. This should be done in
particular with regard to their dependency on the genetic
background of the liver cancer cells, thus identifying biomarkers
(e.g., p53 and β-catenin) for the efficacy of a PIM2 knockdown
therapy. Furthermore, our results encourage the idea of synergistic
combination therapies, utilizing PIM2 siRNAs together with other
cytostatics, for example those affecting the cell cycle as
well.
Supplementary Data
Funding
This study was funded by a grant from the Deutsche
Krebshilfe (Az: 110992) to AA, AG and RKH.
Availability of data and materials
The datasets used during the current study are
available from the corresponding author on reasonable request.
Authors’ contributions
UW and AA were involved in the conception and design
of the study. PK and UW were involved in the experimental
procedures. All authors (PK, AG, RKH, AA and UW) were involved in
the discussion of the experiments and the interpretation of the
data. UW was involved in the writing of the manuscript. AA, AG, RKH
and PK were involved in the revision of the manuscript. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
This article does not contain any studies with human
participants performed by any of the authors. All applicable
international, national, and/or institutional guidelines for the
care and use of animals were followed. Animal experiments were
approved by the Landesdirektion Sachsen, Germany (TVV 38/16).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Gerd Müller
(University Clinics Leipzig, Department of Gynecology) for
providing assistance with the interpretation of the cell cycle
data, Markus Böhlmann and Kathrin Krause (Rudolf-Boehm-Institute
for Pharmacology and Toxicology, University of Leipzig) for taking
care of the animals used in this study, as well as Professor Lea
Ann Dailey (Martin Luther-University Halle) for extensively
proofreading the manuscript.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2018. CA Cancer J Clin. 68:7–30. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mondello P, Cuzzocrea S and Mian M: Pim
kinases in hematological malignancies: Where are we now and where
are we going? J Hematol Oncol. 7:952014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Warfel NA and Kraft AS: PIM kinase (and
Akt) biology and signaling in tumors. Pharmacol Ther. 151:41–49.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Narlik-Grassow M, Blanco-Aparicio C and
Carnero A: The PIM family of serine/threonine kinases in cancer.
Med Res Rev. 34:136–159. 2014. View Article : Google Scholar
|
|
6
|
Zhang Y, Wang Z, Li X and Magnuson NS: Pim
kinase-dependent inhibition of c-Myc degradation. Oncogene.
27:4809–4819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nair JR, Caserta J, Belko K, Howell T,
Fetterly G, Baldino C and Lee KP: Novel inhibition of PIM2 kinase
has significant anti-tumor efficacy in multiple myeloma. Leukemia.
31:1715–1726. 2017. View Article : Google Scholar :
|
|
8
|
Grundler R, Brault L, Gasser C, Bullock
AN, Dechow T, Woetzel S, Pogacic V, Villa A, Ehret S, Berridge G,
et al: Dissection of PIM serine/threonine kinases in
FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of
CXCL12-CXCR4-mediated homing and migration. J Exp Med.
206:1957–1970. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Adam M, Pogacic V, Bendit M, Chappuis R,
Nawijn MC, Duyster J, Fox CJ, Thompson CB, Cools J and Schwaller J:
Targeting PIM kinases impairs survival of hematopoietic cells
transformed by kinase inhibitor-sensitive and kinase
inhibitor-resistant forms of Fms-like tyrosine kinase 3 and
BCR/ABL. Cancer Res. 66:3828–3835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brault L, Gasser C, Bracher F, Huber K,
Knapp S and Schwaller J: PIM serine/threonine kinases in the
pathogenesis and therapy of hematologic malignancies and solid
cancers. Haematologica. 95:1004–1015. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keane NA, Reidy M, Natoni A, Raab MS and
O’Dwyer M: Targeting the pim kinases in multiple myeloma. Blood
Cancer J. 5:e3252015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van der Lugt NM, Domen J, Verhoeven E,
Verhoeven E, Linders K, van der Gulden H, Allen J and Berns A:
Proviral tagging in E mu-myc transgenic mice lacking the Pim-1
proto-oncogene leads to compensatory activation of Pim-2. EMBO J.
14:2536–2544. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Allen JD, Verhoeven E, Domen J, van der
Valk M and Berns A: Pim-2 transgene induces lymphoid tumors,
exhibiting potent synergy with c-myc. Oncogene. 15:1133–1141. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hammerman PS, Fox CJ, Cinalli RM, Xu A,
Wagner JD, Lindsten T and Thompson CB: Lymphocyte transformation by
Pim-2 is dependent on nuclear factor-kappaB activation. Cancer Res.
64:8341–8348. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu J, Zavorotinskaya T, Dai Y, Niu XH,
Castillo J, Sim J, Yu J, Wang Y, Langowski JL, Holash J, et al:
Pim2 is required for maintaining multiple myeloma cell growth
through modulating TSC2 phosphorylation. Blood. 122:1610–1620.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Asano J, Nakano A, Oda A, Amou H, Hiasa M,
Takeuchi K, Miki H, Nakamura S, Harada T, Fujii S, et al: The
serine/threo-nine kinase Pim-2 is a novel anti-apoptotic mediator
in myeloma cells. Leukemia. 25:1182–1188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jimenez-Garcia MP, Lucena-Cacace A,
Robles-Frias MJ, Ferrer I, Narlik-Grassow M, Blanco-Aparicio C and
Carnero A: Inflammation and stem markers association to PIM1/PIM2
kinase-induced tumors in breast and uterus. Oncotarget.
8:58872–58886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Z, Zhang Y, Gu JJ, Davitt C, Reeves R
and Magnuson NS: Pim-2 phosphorylation of p21(Cip1/WAF1) enhances
its stability and inhibits cell proliferation in HCT116 cells. Int
J Biochem Cell Biol. 42:1030–1038. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Morishita D, Katayama R, Sekimizu K,
Tsuruo T and Fujita N: Pim kinases promote cell cycle progression
by phosphorylating and down-regulating p27Kip1 at the
transcriptional and posttran-scriptional levels. Cancer Res.
68:5076–5085. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan B, Zemskova M, Holder S, Chin V, Kraft
A, Koskinen PJ and Lilly M: The PIM-2 kinase phosphorylates BAD on
serine 112 and reverses BAD-induced cell death. J Biol Chem.
278:45358–45367. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fox CJ, Hammerman PS, Cinalli RM, Master
SR, Chodosh LA and Thompson CB: The serine/threonine kinase Pim-2
is a transcriptionally regulated apoptotic inhibitor. Genes Dev.
17:1841–1854. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hospital MA, Jacquel A, Mazed F, Saland E,
Larrue C, Mondesir J, Birsen R, Green AS, Lambert M, Sujobert P, et
al: RSK2 is a new Pim2 target with pro-survival functions in
FLT3-ITD-positive acute myeloid leukemia. Leukemia. 32:597–605.
2018. View Article : Google Scholar
|
|
23
|
Ramachandran J, Santo L, Siu KT, Panaroni
C and Raje N: Pim2 is important for regulating DNA damage response
in multiple myeloma cells. Blood Cancer J. 6:e4622016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang T, Ren C, Qiao P, Han X, Wang L, Lv
S, Sun Y, Liu Z, Du Y and Yu Z: PIM2-mediated phosphorylation of
hexokinase 2 is critical for tumor growth and paclitaxel resistance
in breast cancer. Oncogene. 37:5997–6009. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Musiani D, Hammond DE, Cirillo L, Erriquez
J, Olivero M, Clague MJ and Di Renzo MF: PIM2 kinase is induced by
cispl-atin in ovarian cancer cells and limits drug efficacy. J
Proteome Res. 13:4970–4982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Uddin N, Kim RK, Yoo KC, Kim YH, Cui YH,
Kim IG, Suh Y and Lee SJ: Persistent activation of STAT3 by
PIM2-driven positive feedback loop for epithelial-mesenchymal
transition in breast cancer. Cancer Sci. 106:718–725. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Narlik-Grassow M, Blanco-Aparicio C,
Cecilia Y, Peregrina S, Garcia-Serelde B, Muñoz-Galvan S, Cañamero
M and Carnero A: The essential role of PIM kinases in sarcoma
growth and bone invasion. Carcinogenesis. 33:1479–1486. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen J and Yang X: The role of Pim-2 in
apoptotic signal transduction pathway of hepatocellular carcinoma.
Int Surg J. 4:13802017. View Article : Google Scholar
|
|
29
|
Ren K, Zhang W, Shi Y and Gong J: Pim-2
activates API-5 to inhibit the apoptosis of hepatocellular
carcinoma cells through NF-kappaB pathway. Pathol Oncol Res.
16:229–237. 2010. View Article : Google Scholar
|
|
30
|
Gong J, Wang J, Ren K, Liu C, Li B and Shi
Y: Serine/threonine kinase Pim-2 promotes liver tumorigenesis
induction through mediating survival and preventing apoptosis of
liver cell. J Surg Res. 153:17–22. 2009. View Article : Google Scholar
|
|
31
|
Ren K, Duan W, Shi Y, Li B, Liu Z and Gong
J: Ectopic overexpression of oncogene Pim-2 induce malignant
transformation of nontumorous human liver cell line L02. J Korean
Med Sci. 25:1017–1023. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Hobel S, Koburger I, John M, Czubayko F,
Hadwiger P, Vornlocher HP and Aigner A: Polyethylenimine/small
interfering RNA-mediated knockdown of vascular endothelial growth
factor in vivo exerts anti-tumor effects synergistically with
Bevacizumab. J Gene Med. 12:287–300. 2010.PubMed/NCBI
|
|
34
|
Liu Z, Liu H, Yuan X, Wang Y, Li L, Wang
G, Song J, Shao Z and Fu R: Downregulation of Pim-2 induces cell
cycle arrest in the G0/G1 phase via the p53-non-dependent p21
signaling pathway. Oncol Lett. 15:4079–4086. 2018.PubMed/NCBI
|
|
35
|
Yew TL, Chiu FY, Tsai CC, Chen HL, Lee WP,
Chen YJ, Chang MC and Hung SC: Knockdown of p21(Cip1/Waf1) enhances
proliferation, the expression of stemness markers, and osteogenic
potential in human mesenchymal stem cells. Aging Cell. 10:349–361.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nawijn MC, Alendar A and Berns A: For
better or for worse: The role of Pim oncogenes in tumorigenesis.
Nat Rev Cancer. 11:23–34. 2011. View Article : Google Scholar
|
|
37
|
Levy D, Davidovich A, Zirkin S, Frug Y,
Cohen AM, Shalom S and Don J: Activation of cell cycle arrest and
apoptosis by the proto-oncogene Pim-2. PLoS One. 7:e347362012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Adam K, Cartel M, Lambert M, David L, Yuan
L, Besson A, Mayeux P, Manenti S and Didier C: A PIM-CHK1 signaling
pathway regulates PLK1 phosphorylation and function during mitosis.
J Cell Sci. 131:pii: jcs213116. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kreuz S, Holmes KB, Tooze RM and Lefevre
PF: Loss of PIM2 enhances the anti-proliferative effect of the
pan-PIM kinase inhibitor AZD1208 in non-Hodgkin lymphomas. Mol
Cancer. 14:2052015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Pellegrino R, Calvisi DF, Ladu S, Ehemann
V, Staniscia T, Evert M, Dombrowski F, Schirmacher P and Longerich
T: Oncogenic and tumor suppressive roles of polo-like kinases in
human hepatocellular carcinoma. Hepatology. 51:857–868.
2010.PubMed/NCBI
|
|
41
|
Liu Z, Sun Q and Wang X: PLK1, A potential
target for cancer therapy. Transl Oncol. 10:22–32. 2017. View Article : Google Scholar
|
|
42
|
Waisberg J and Saba GT: Wnt-/-β-catenin
pathway signaling in human hepatocellular carcinoma. World J
Hepatol. 7:2631–2635. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vilchez V, Turcios L, Marti F and Gedaly
R: Targeting Wnt/β-catenin pathway in hepatocellular carcinoma
treatment. World J Gastroenterol. 22:823–832. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
de La Coste A, Romagnolo B, Billuart P,
Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C,
Kahn A and Perret C: Somatic mutations of the beta-catenin gene are
frequent in mouse and human hepatocellular carcinomas. Proc Natl
Acad Sci USA. 95:8847–8851. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ding Z, Shi C, Jiang L, Tolstykh T, Cao H,
Bangari DS, Ryan S, Levit M, Jin T, Mamaat K, et al: Oncogenic
dependency on β-catenin in liver cancer cell lines correlates with
pathway activation. Oncotarget. 8:114526–114539. 2017. View Article : Google Scholar
|
|
46
|
Schmucker S and Sumara I: Molecular
dynamics of PLK1 during mitosis. Mol Cell Oncol. 1:e9545072014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Katyal S, Oliver JH III, Peterson MS,
Ferris JV, Carr BS and Baron RL: Extrahepatic metastases of
hepatocellular carcinoma. Radiology. 216:698–703. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Weirauch U, Beckmann N, Thomas M,
Grünweller A, Huber K, Bracher F, Hartmann RK and Aigner A:
Functional role and therapeutic potential of the pim-1 kinase in
colon carcinoma. Neoplasia. 15:783–794. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 2013:4218212013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ibrahim AF, Weirauch U, Thomas M,
Grunweller A, Hartmann RK and Aigner A: MicroRNA replacement
therapy for miR-145 and miR-33a is efficacious in a model of colon
carcinoma. Cancer Res. 71:5214–5224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hobel S and Aigner A: Polyethylenimines
for siRNA and miRNA delivery in vivo. Wiley Interdiscip Rev Nanomed
Nanobiotechnol. 5:484–501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wood H: FDA approves patisiran to treat
hereditary transthyretin amyloidosis. Nat Rev Neurol.
14:5702018.PubMed/NCBI
|