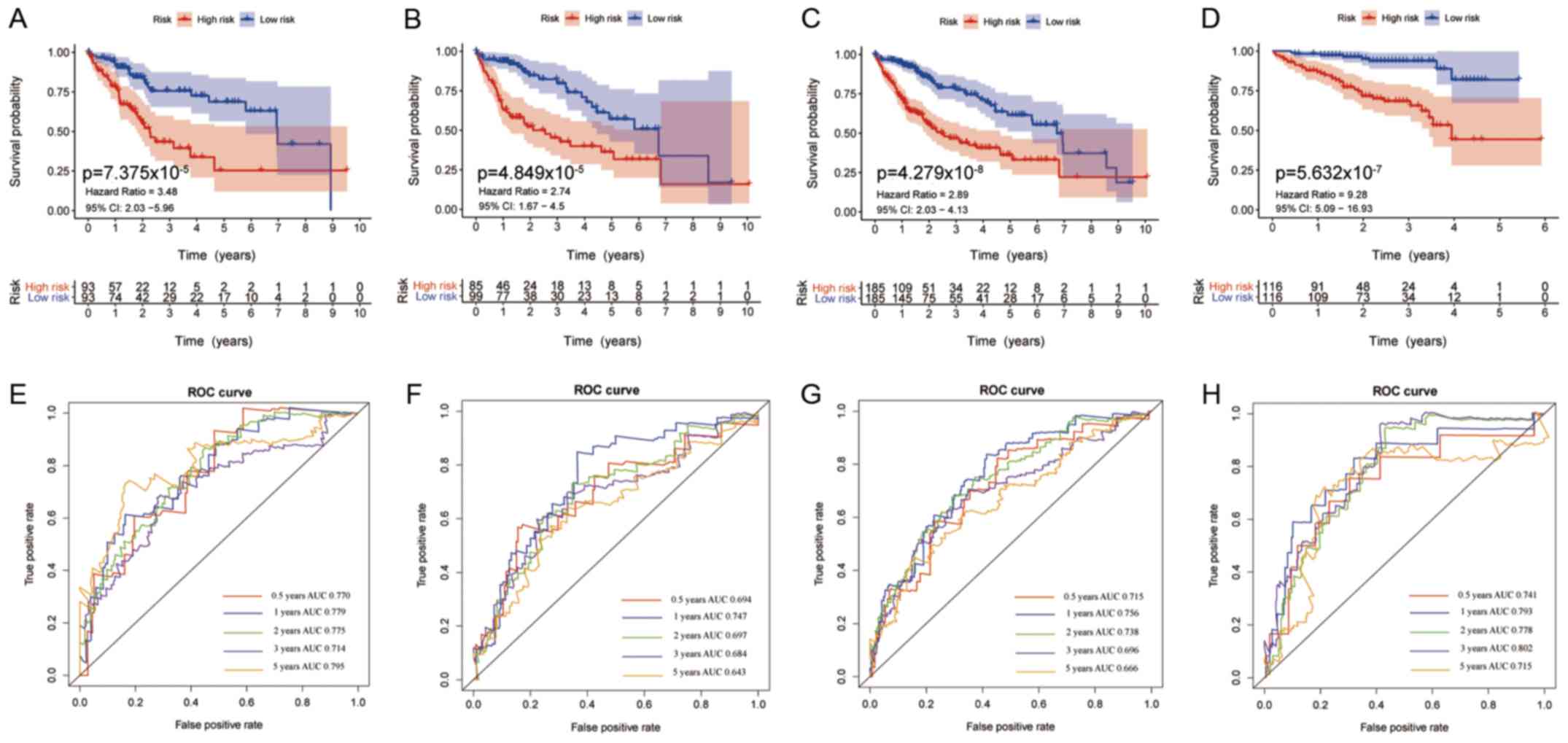
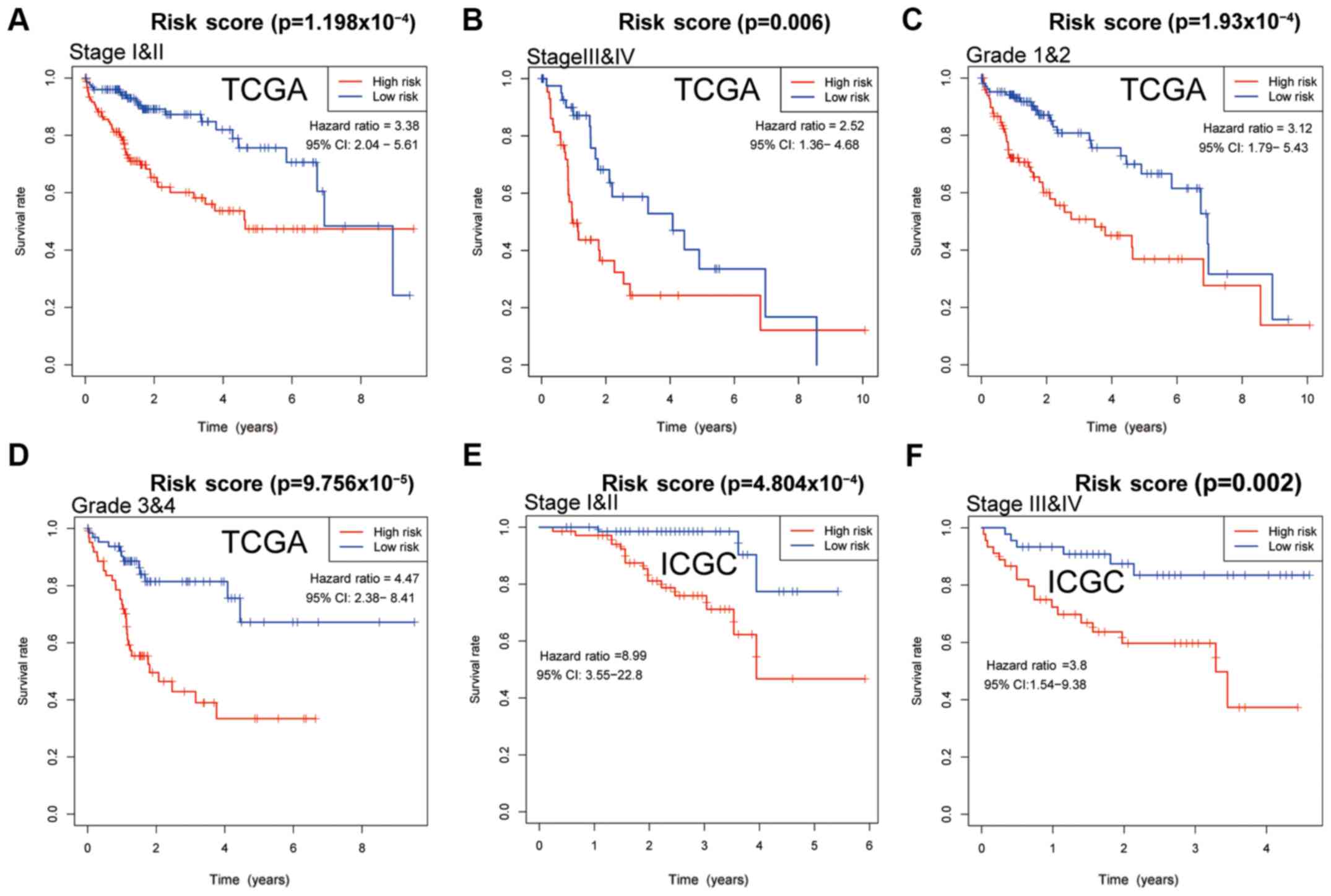
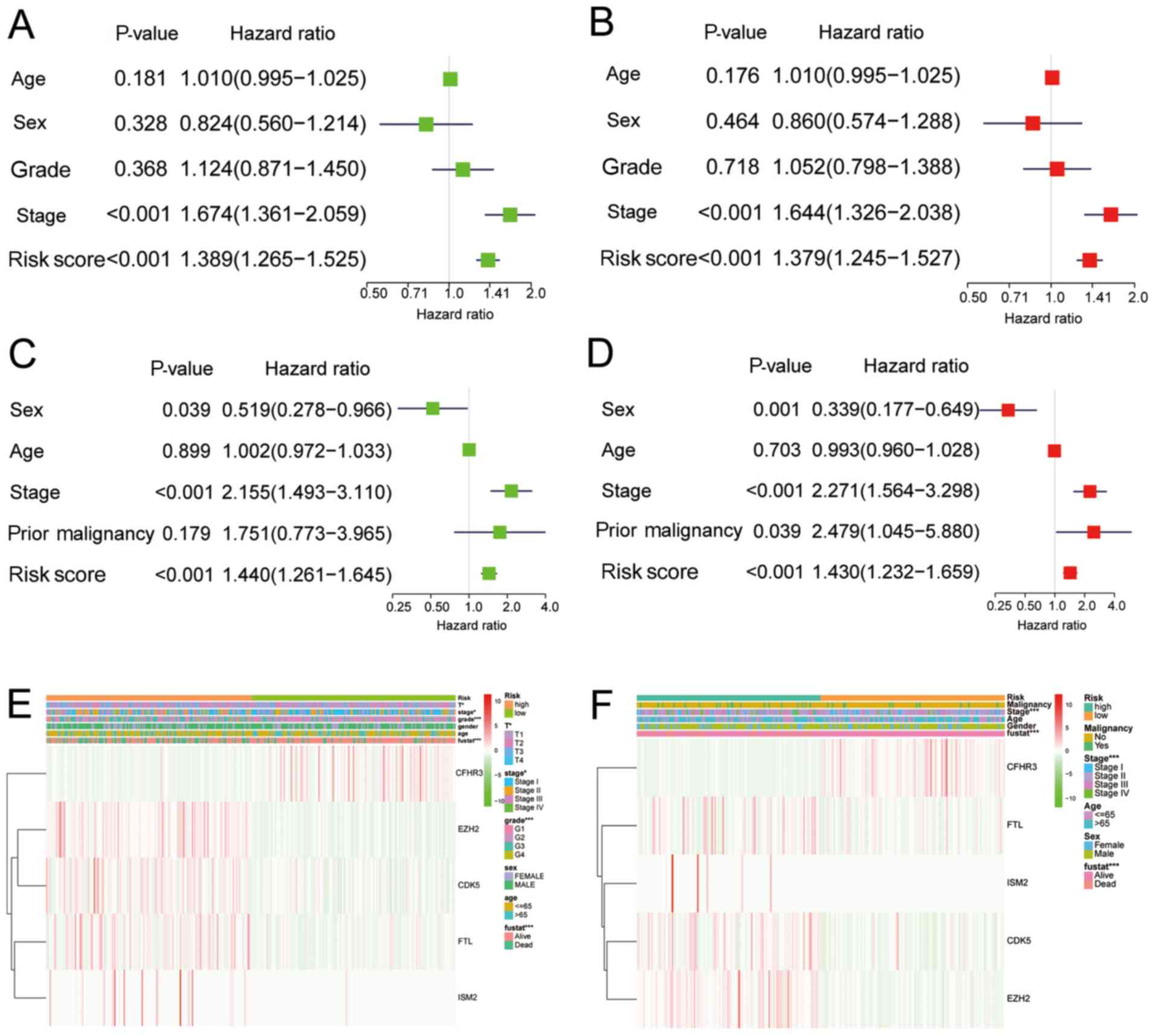
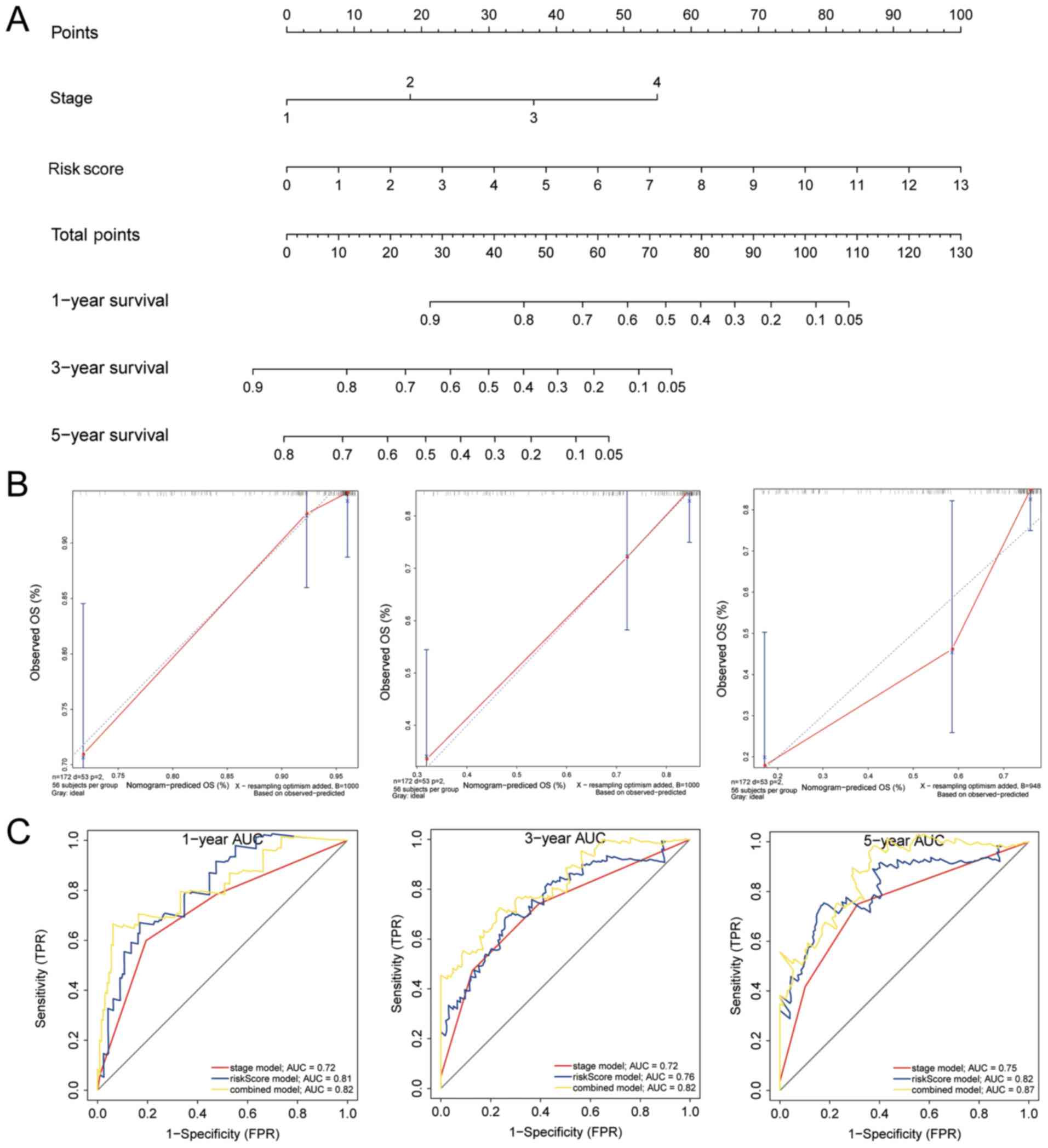
|
1
|
Singal AG, Lampertico P and Nahon P:
Epidemiology and surveillance for hepatocellular carcinoma: New
trends. J Hepatol. 72:250–261. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer sta-tistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA-Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar
|
|
3
|
Chen L and Han X: Anti-PD-1/PD-L1 therapy
of human cancer: Past, present, and future. J Clin Invest.
125:3384–3391. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun C, Mezzadra R and Schumacher TN:
Regulation and function of the PD-L1 checkpoint. Immunity.
48:434–452. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu G, Rui W, Zheng H, Huang D, Yu F,
Zhang Y, Dong J, Zhao X and Lin X: CXCR2-modified CAR-T cells have
enhanced trafficking ability that improves treatment of
hepatocellular carcinoma. Eur J Immunol. Jan 24–2020.Epub ahead of
print. View Article : Google Scholar
|
|
6
|
Batra SA, Rathi P, Guo L, Courtney AN,
Fleurence J, Balzeau J, Shaik RS, Nguyen TP, Wu MF, Bulsara S, et
al: Glypican-3-specific CAR T cells co-expressing IL15 and IL21
have superior expansion and antitumor activity against
hepato-cellular carcinoma. Cancer Immunol Res. Jan 17–2020.Epub
ahead of print. View Article : Google Scholar
|
|
7
|
Jindal A, Thadi A and Shailubhai K:
Hepatocellular carcinoma: Etiology and current and future drugs. J
Clin Exp Hepatol. 9:221–232. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zucman-Rossi J, Villanueva A, Nault JC and
Llovet JM: Genetic landscape and biomarkers of hepatocellular
carcinoma. Gastroenterology. 149:1226–1239.e4. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cancer Genome Atlas Research Network:
Electronic address simplewheeler@bcm.edu; Cancer
Genome Atlas Research Network: Comprehensive and integrative
genomic characterization of hepatocellular carcinoma. Cell.
169:1327–1341.e23. 2017. View Article : Google Scholar
|
|
10
|
Li B, Cui Y, Diehn M and Li R: Development
and validation of an individualized immune prognostic signature in
early-stage nonsquamous non-small cell lung cancer. JAMA Oncol.
3:1529–1537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen CH, Lu YS, Cheng AL, Huang CS, Kuo
WH, Wang MY, Chao M, Chen IC, Kuo CW, Lu TP and Lin CH: Disparity
in tumor immune microenvironment of breast cancer and prognostic
impact: Asian versus Western populations. Oncologist. 25:e16–e23.
2020. View Article : Google Scholar :
|
|
12
|
Ge P, Wang W, Li L, Zhang G, Gao Z, Tang
Z, Dang X and Wu Y: Profiles of immune cell infiltration and
immune-related genes in the tumor microenvironment of colorectal
cancer. Biomed Pharmacother. 118:1092282019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin P, Guo YN, Shi L, Li XJ, Yang H, He Y,
Li Q, Dang YW, Wei KL and Chen G: Development of a prognostic index
based on an immunogenomic landscape analysis of papillary thyroid
cancer. Aging (Albany NY). 11:480–500. 2019. View Article : Google Scholar
|
|
14
|
Wang J, Li Y, Fu W, Zhang Y, Jiang J,
Zhang Y and Qi X: Prognostic nomogram based on immune scores for
breast cancer patients. Cancer Med. 8:5214–5222. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Foerster F, Hess M, Gerhold-Ay A,
Marquardt JU, Becker D, Galle PR, Schuppan D, Binder H and Bockamp
E: The immune contexture of hepatocellular carcinoma predicts
clinical outcome. Sci Rep. 8:53512018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng M and Tian Z: Liver-mediated
adaptive immune tolerance. Front Immunol. 10:25252019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu LC, Hsu C, Shao YY, Chao Y, Yen CJ,
Shih IL, Hung YP, Chang CJ, Shen YC, Guo JC, et al: Differential
differential organ-specific tumor response to immune checkpoint
inhibitors in hepatocellular carcinoma. J Liver Cancer. 8:480–490.
2019. View Article : Google Scholar
|
|
18
|
Keenan BP, Fong L and Kelley RK:
Immunotherapy in hepatocel-lular carcinoma: The complex interface
between inflammation, fibrosis, and the immune response. J
Immunother Cancer. 7:2672019. View Article : Google Scholar
|
|
19
|
Charoentong P, Finotello F, Angelova M,
Mayer C, Efremova M, Rieder D, Hackl H and Trajanoski Z: Pan-cancer
immunoge-nomic analyses reveal genotype-immunophenotype
relationships and predictors of response to checkpoint blockade.
Cell Rep. 18:248–262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rooney MS, Shukla SA, Wu CJ, Getz G and
Hacohen N: Molecular and genetic properties of tumors associated
with local immune cytolytic activity. Cell. 160:48–61. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
International Cancer Genome Consortium;
Hudson TJ, Anderson W, Aretz A, Barker AD, Bell C, Bernabé RR, Bhan
MK, Calvo F, Eerola I, et al: International network of cancer
genome projects. Nature. 464:993–998. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tomczak K, Czerwinska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.
|
|
23
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu J, Koh H, He L, Liu M, Blaser MJ and Li
H: A two-stage microbial association map-ping framework with
advanced FDR control. Microbiome. 6:1312018. View Article : Google Scholar
|
|
25
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kanehisa M: Post-genome Informatics.
Oxford University Press; 2000
|
|
27
|
Heagerty PJ: Compute time-dependent ROC
curve from censored survival data using Kaplan-Meier (KM) or
Nearest Neighbor Estimation (NNE) method of Heagerty, Lumley &
Pepe. Biometrics. 56:337–344. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li B, Severson E, Pignon JC, Zhao H, Li T,
Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al: Comprehensive
analyses of tumor immunity: Implications for cancer immunotherapy.
Genome Biol. 17:1742016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li T, Fan J, Wang B, Traugh N, Chen Q, Liu
JS, Li B and Liu XS: TIMER: A web server for comprehensive analysis
of tumor-infiltrating immune cells. Cancer Res. 77:e108–e110. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Team RC: R: A language and environment for
statistical computing. R Foundation for Statistical Computing;
Vienna, Austria: 2012
|
|
31
|
Team RS: RStudio: Integrated Development
for R. RStudio, Inc; Boston, MA: 2015
|
|
32
|
A P, Xu X, Wang C, Yang J, Wang S, Dai J
and Ye L: EZH2 promotes DNA replication by stabilizing interaction
of POLδ and PCNA via methylation-mediated PCNA trimerization.
Epigenetics Chromatin. 11:442018. View Article : Google Scholar
|
|
33
|
Batool A, Jin C and Liu YX: Role of EZH2
in cell lineage determination and relative signaling pathways.
Front Biosci (Landmark Ed). 24:947–960. 2019. View Article : Google Scholar
|
|
34
|
Cheng T and Xu Y: Effects of enhancer of
zeste homolog 2 (EZH2) expression on brain glioma cell
proliferation and tumori-genesis. Med Sci Monit. 24:7249–7255.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mochizuki D, Misawa Y, Kawasaki H, Imai A,
Endo S, Mima M, Yamada S, Nakagawa T, Kanazawa T and Misawa K:
Aberrant epigenetic regulation in head and neck cancer due to
distinct EZH2 overexpression and DNA hypermethylation. Int J Mol
Sci. 19:pii: E3707. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Karlowee V, Amatya VJ, Takayasu T, Takano
M, Yonezawa U, Takeshima Y, Sugiyama K, Kurisu K and Yamasaki F:
Immunostaining of increased expression of enhancer of zeste homolog
2 (EZH2) in diffuse midline glioma H3K27M-mutant patients with poor
survival. Pathobiology. 86:152–161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Makk E, Bálint L, Cifra J, Tornóczky T,
Oszter A, Tóth A, Kálmán E and Kovács K: Robust expression of EZH2
in endocervical neoplastic lesions. Virchows Arch. 475:95–104.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Romanchikova N and Trapencieris P:
Wedelolactone targets EZH2-mediated Histone H3K27 methylation in
mantle cell lymphoma. Anticancer Res. 39:4179–4184. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou L, Wei E, Zhou B, Bi G, Gao L, Zhang
T, Huang J, Wei Y and Ge B: Anti-proliferative benefit of curcumol
on human bladder cancer cells via inactivating EZH2 effector.
Biomed Pharmacother. 104:798–805. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Böhm J, Muenzner JK, Caliskan A,
Ndreshkjana B, Erlenbach-Wünsch K, Merkel S, Croner R, Rau TT,
Geppert CI, Hartmann A, et al: Loss of enhancer of zeste homologue
2 (EZH2) at tumor invasion front is correlated with higher
aggressiveness in colorectal cancer cells. J Cancer Res Clin Oncol.
145:2227–2240. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Natsumeda M, Liu Y, Nakata S, Miyahara H,
Hanaford A, Ahsan S, Stearns D, Skuli N, Kahlert UD, Raabe EH, et
al: Inhibition of enhancer of zest homologue 2 is a potential
therapeutic target for high-MYC medulloblastoma. Neuropathology.
39:71–77. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cozzi A, Rovelli E, Frizzale G, Campanella
A, Amendola M, Arosio P and Levi S: Oxidative stress and cell death
in cells expressing L-ferritin variants causing
neuroferritinopathy. Neurobiol Dis. 37:77–85. 2010. View Article : Google Scholar
|
|
43
|
Vanarsa K, Ye Y, Han J, Xie C, Mohan C and
Wu T: Inflammation associated anemia and ferritin as disease
markers in SLE. Arthritis Res Ther. 14:R1822012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kwiatek-Majkusiak J, Dickson DW, Tacik P,
Aoki N, Tomasiuk R, Koziorowski D and Friedman A: Relationships
between typical histopathological hallmarks and the ferritin in the
hippocampus from patients with Alzheimer's disease. Acta Neurobiol
Exp (Wars). 75:391–398. 2015.
|
|
45
|
Döring M, Cabanillas Stanchi KM, Feucht J,
Queudeville M, Teltschik HM, Lang P, Feuchtinger T, Handgretinger R
and Müller I: Ferritin as an early marker of graft rejection after
allogeneic hematopoietic stem cell transplantation in pediatric
patients. Ann Hematol. 95:311–323. 2016. View Article : Google Scholar
|
|
46
|
Ojha NK and Lole KS: Hepatitis E virus
ORF1 encoded macro domain protein interacts with light chain
subunit of human ferritin and inhibits its secretion. Mol Cell
Biochem. 417:75–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yazar S, Franchina M, Craig JE, Burdon KP
and Mackey DA: Ferritin light chain gene mutation in a large
Australian family with hereditary hyperferritinemia-cataract
syndrome. Ophthalmic Genet. 38:171–174. 2017. View Article : Google Scholar
|
|
48
|
Chekhun VF, Lukyanova NY, Burlaka CA,
Bezdenezhnykh NA, Shpyleva SI, Tryndyak VP, Beland FA and Pogribny
IP: Iron metabolism disturbances in the MCF-7 human breast cancer
cells with acquired resistance to doxorubicin and cisplatin. Int J
Oncol. 43:1481–1486. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu T, Li Y, Liu B, Zhang S, Wu L, Zhu X
and Chen Q: Expression of ferritin light chain (FTL) is elevated in
glioblastoma, and FTL silencing inhibits glioblastoma cell
proliferation via the GADD45/JNK pathway. PLoS One.
11:e01493612016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Khanna V, Karjodkar F, Robbins S, Behl M,
Arya S and Tripathi A: Estimation of serum ferritin level in
potentially malignant disorders, oral squamous cell carcinoma, and
treated cases of oral squamous cell carcinoma. J Cancer Res Ther.
13:550–555. 2017.PubMed/NCBI
|
|
51
|
Liu J, Li W and Zhao H: CFHR3 is a
potential novel biomarker for hepatocellular carcinoma. J Cell
Biochem. Nov 10–2019.Epub ahead of print.
|
|
52
|
Hellwage J, Jokiranta TS, Koistinen V,
Vaarala O, Meri S and Zipfel PF: Functional properties of
complement factor H-related proteins FHR-3 and FHR-4: Binding to
the C3d region of C3b and differential regulation by heparin. FEBS
Lett. 462:345–352. 1999. View Article : Google Scholar
|
|
53
|
Spencer KL, Hauser MA, Olson LM, Schmidt
S, Scott WK, Gallins P, Agarwal A, Postel EA, Pericak-Vance MA and
Haines JL: Deletion of CFHR3 and CFHR1 genes in age-related macular
degeneration. Hum Mol Genet. 17:971–977. 2008. View Article : Google Scholar
|
|
54
|
Pouw RB, Gómez Delgado I, López Lera A,
Rodríguez de Córdoba S, Wouters D, Kuijpers TW and Sánchez-Corral
P: High complement factor H-related (FHR)-3 levels are associated
with the atypical hemolytic-uremic syndrome-risk allele CFHR3*B.
Front Immunol. 9:8482018. View Article : Google Scholar
|
|
55
|
Belotti D, Capelli C, Resovi A, Introna M
and Taraboletti G: Thrombospondin-1 promotes mesenchymal stromal
cell functions via TGFβ and in cooperation with PDGF. Matrix Biol.
55:106–116. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xing T, Wang Y, Ding WJ, Li YL, Hu XD,
Wang C, Ding A and Shen JL: Thrombospondin-1 production regulates
the inflammatory cytokine secretion in THP-1 cells through NF-κB
signaling pathway. Inflammation. 40:1606–1621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Firlej V, Mathieu JR, Gilbert C, Lemonnier
L, Nakhlé J, Gallou-Kabani C, Guarmit B, Morin A, Prevarskaya N,
Delongchamps NB and Cabon F: Thrombospondin-1 triggers cell
migration and development of advanced prostate tumors. Cancer Res.
71:7649–7658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jayachandran A, Anaka M, Prithviraj P,
Hudson C, McKeown SJ, Lo PH, Vella LJ, Goding CR, Cebon J and
Behren A: Thrombospondin 1 promotes an aggressive phenotype through
epithelial-to-mesenchymal transition in human melanoma. Oncotarget.
5:5782–5797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ioachim E, Damala K, Tsanou E, Briasoulis
E, Papadiotis E, Mitselou A, Charhanti A, Doukas M, Lampri L and
Arvanitis DL: Thrombospondin-1 expression in breast cancer:
Prognostic significance and association with p53 alterations,
tumour angiogenesis and extracellular matrix components. Histol
Histopathol. 27:209–216. 2012.
|
|
60
|
Lin XD, Chen SQ, Qi YL, Zhu JW, Tang Y and
Lin JY: Overexpression of thrombospondin-1 in stromal
myofibroblasts is associated with tumor growth and nodal metastasis
in gastric carcinoma. J Surg Oncol. 106:94–100. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Horiguchi H, Yamagata S, Rong Qian Z,
Kagawa S and Sakashita N: Thrombospondin-1 is highly expressed in
desmo-plastic components of invasive ductal carcinoma of the breast
and associated with lymph node metastasis. J Med Invest. 60:91–96.
2013. View Article : Google Scholar
|
|
62
|
Kashihara H, Shimada M, Yoshikawa K,
Higashijima J, Tokunaga T, Nishi M, Takasu C and Ishikawa D:
Correlation between thrombospondin-1 expression in non-cancer
tissue and gastric carcinogenesis. Anticancer Res. 37:3547–3552.
2017.PubMed/NCBI
|
|
63
|
Pozo K and Bibb JA: The emerging role of
Cdk5 in cancer. Trends Cancer. 2:606–618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lopes JP and Agostinho P: Cdk5:
Multitasking between physiological and pathological conditions.
Prog Neurobiol. 94:49–63. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liebl J, Weitensteiner SB, Vereb G, Takács
L, Fürst R, Vollmar AM and Zahler S: Cyclin-dependent kinase 5
regulates endothelial cell migration and angiogenesis. J Biol Chem.
285:35932–35943. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shupp A, Casimiro MC and Pestell RG:
Biological functions of CDK5 and potential CDK5 targeted clinical
treatments. Oncotarget. 8:17373–17382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu
DX, Feng J, Hou P, Yao R, Zhang Y, et al: CDK5 is essential for
TGF-β1-induced epithelial-mesenchymal transition and breast cancer
progression. Sci Rep. 3:29322013. View Article : Google Scholar
|
|
68
|
Yushan R, Wenjie C, Suning H, Yiwu D,
Tengfei Z, Madushi WM, Feifei L, Changwen Z, Xin W, Roodrajeetsing
G, et al: Insights into the clinical value of cyclin-dependent
kinase 5 in glioma: A retrospective study. World J Surg Oncol.
13:2232015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang X, Zhong T, Dang Y, Li Z, Li P and
Chen G: Aberrant expression of CDK5 infers poor outcomes for
nasopharyngeal carcinoma patients. Int J Clin Exp Pathol.
8:8066–8074. 2015.PubMed/NCBI
|
|
70
|
Pan DH, Zhu ML, Lin XM, Lin XG, He RQ,
Ling YX, Su ST, Wickramaarachchi MM, Dang YW, Wei KL and Chen G:
Evaluation and clinical significance of cyclin-dependent kinase5
expression in cervical lesions: A clinical research study in
Guangxi, China. Eur J Med Res. 21:282016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wei K, Ye Z, Li Z, Dang Y, Chen X, Huang
N, Bao C, Gan T, Yang L and Chen G: An immunohistochemical study of
cyclin-dependent kinase 5 (CDK5) expression in non-small cell lung
cancer (NSCLC) and small cell lung cancer (SCLC): A possible
prognostic biomarker. World J Surg Oncol. 14:342016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mandl MM, Zhang S, Ulrich M, Schmoeckel E,
Mayr D, Vollmar AM and Liebl J: Inhibition of Cdk5 induces cell
death of tumor-initiating cells. Br J Cancer. 116:912–922. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
NavaneethaKrishnan S, Rosales JL and Lee
KY: Loss of Cdk5 in breast cancer cells promotes ROS-mediated cell
death through dysregulation of the mitochondrial permeability
transition pore. Oncogene. 37:1788–1804. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ehrlich SM, Liebl J, Ardelt MA, Lehr T, De
Toni EN, Mayr D, Brandl L, Kirchner T, Zahler S, Gerbes AL and
Vollmar AM: Targeting cyclin dependent kinase 5 in hepatocellular
carcinoma-A novel therapeutic approach. J Hepatol. 63:102–113.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chen QF, Li W, Wu PH, Shen LJ and Huang
ZL: Significance of tumor-infiltrating immunocytes for predicting
prognosis of hepatitis B virus-related hepatocellular carcinoma.
World J Gastroenterol. 25:5266–5282. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lu J, Xu Y, Wu Y, Huang XY, Xie JW, Wang
JB, Lin JX, Li P, Zheng CH, Huang AM and Huang CM:
Tumor-infiltrating CD8+ T cells combined with tumor-associated
CD68+ macrophages predict postoperative prognosis and adjuvant
chemotherapy benefit in resected gastric cancer. BMC Cancer.
19:9202019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wang J, Li Z, Gao A, Wen Q and Sun Y: The
prognostic landscape of tumor-infiltrating immune cells in cervical
cancer. Biomed Pharmacother. 120:1094442019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kobayashi N, Hiraoka N, Yamagami W, Ojima
H, Kanai Y, Kosuge T, Nakajima A and Hirohashi S: FOXP3+ regulatory
T cells affect the development and progression of
hepatocarcino-genesis. Clin Cancer Res. 13:902–911. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Mathai AM, Kapadia MJ, Alexander J,
Kernochan LE, Swanson PE and Yeh MM: Role of Foxp3-positive
tumor-infiltrating lymphocytes in the histologic features and
clinical outcomes of hepatocellular carcinoma. Am J Surg Pathol.
36:980–986. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Brunner SM, Rubner C, Kesselring R, Martin
M, Griesshammer E, Ruemmele P, Stempfl T, Teufel A, Schlitt HJ and
Fichtner-Feigl S: Tumor-infiltrating, interleukin-33-producing
effector-memory CD8(+) T cells in resected hepatocellular carcinoma
prolong patient survival. Hepatology. 61:1957–1967. 2015.
View Article : Google Scholar : PubMed/NCBI
|