Introduction
Endometrial carcinoma (EC) is one of the most common
types of gynecologic malignancy, with an estimated 61,880 diagnosed
cases, accounting for 6.94% of all new female cancer cases and
12,160 mortalities, leading to 4.26% of all mortalities in females
in 2019 according to the American Cancer Society (1). With the development of medical
technologies, the majority of ECs are diagnosed at the early stage
(stage I-II), according to staging standards of the International
Federation of Gynecology and Obstetrics, and hysterectomy is the
recommended treatment (2).
However, 30% of EC cases are not diagnosed until metastasis is
identified (stage III-IV), which is associated with low survival
and worse prognosis (3,4). Furthermore, particularly for patients
with EC who are nulliparous, hysterectomy is not a primary
treatment option (5). Currently,
the global incidence of EC is on the rise. In 2013, there were an
estimated 49,560 cases in the USA, while by 2018, the number of new
cases had risen to 63,230 individuals (6); this increase may be attributed to an
increased prevalence of patients who are overweight, obese and
physically inactive (7,8). Therefore, therapeutic strategies
focusing on inhibiting the proliferation, migration and invasion of
EC are critical to the treatment and prognosis of patients with EC.
However, the molecular and cellular mechanisms responsible for the
carcinogenesis, progression and survival prognosis of ECs have
remained elusive.
EC is classified into two subtypes: Type I,
accounting for 85% of total cases, 20-50% of which have acquired
p53-mutant status; and type II, accounting for 15% of total cases,
>90% of which have p53-mutant status (9-11).
Previous studies have reported that activating transcription factor
3 (ATF3) binds to the C terminus of wild-type (WT) p53, preventing
p53 from ubiquitin-mediated degradation in response to DNA damage
(12). Moreover, ATF3 is able to
bind to mutant p53 to inhibit the migration and invasion of
p53-mutated cancer cells, suppressing the oncogenic function of
mutant p53 proteins (13).
Therefore, ATF3 may be a potential therapeutic target for
carcinomas closely linked to p53 mutations, including ECs. To the
best of our knowledge, the present study was the first to
investigate the role of ATF3 in EC. Its implication in cell
proliferation, invasion, signaling pathway markers, matrix
metalloproteinases (MMPs), tissue inhibitors of metalloproteinases
(TIMPs), protein interaction and DNA binding were assessed to
identify the regulatory function of ATF3 in ECs and the potential
underlying mechanisms.
Materials and methods
Ethics statement
The protocol (approval no. 0101N16032) of the
present study was approved by the Human Investigation Ethical
Committee of Shanghai First People's Hospital Affiliated Shanghai
Jiao Tong University (Shanghai, China) and Renji Hospital
Affiliated to Shanghai Jiao Tong University School of Medicine
(Shanghai, China). The sampling methods in patients were performed
in accordance with the Operational Guidelines for Ethics Committees
That Review Biomedical Research (https://www.who.int/tdr/publications/documents/ethics.pdf).
All samples were obtained from patients who had provided written
informed consent for the use of their tissues for the purposes of
research after the operation.
The animal experiment was in accordance with the
recommendations in the Guidelines for the Care and Use of
Laboratory Animals of China (http://www.nsfc.gov.cn/nsfc/cen/pfzl/pufanew/20110801_02.pdf).
The protocol (approval no. 2013020346) was approved by the
Committee on the Ethics of Animal Experiments of the School of
Pharmacy of Shanghai Jiao Tong University (Shanghai, China). All
efforts were made to minimize animal suffering.
Cell culture, plasmids and
transfections
The human EC cell lines HEC-1B and AN3CA were
cultured in DMEM/nutrient mixture F-12 (DMEM/F12; Hyclone; Cytiva)
supplemented with 10% heat-inactivated fetal bovine serum (FBS,
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.).
These cell lines were purchased from the Chinese Academy of
Sciences Committee Type Culture Collection and were routinely
maintained in the Fangyuan Wang's laboratory. Cell cultures were
maintained at 37°C in a humidified incubator with 5%
CO2. The plasmid pShuttle Vector containing the open
reading frame (ORF) of ATF3 was obtained from GeneCopoeia, Inc.
For transfection, the ATF3 ORF was inserted to the
multiple cloning sites of pCMV-script (Agilent Technologies, Inc.)
and transfected using Lipofectamine® 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.). To stably express ATF3 in HEC-1B
and AN3CA cells, the EC cells were transfected with
pCMV-script-ATF3 or the negative control (NC) vector pCMV-script (5
µg) and then selected with G418 (800 µg/ml;
Sigma-Aldrich; Merck KGaA) after 6 weeks to obtain the stable
transfection cell lines. The cell lines with ATF3 overexpression
were referred to as 'HEC-1B-ATF3' and 'AN3CA-ATF3', respectively,
with their control cells referred to as 'HEC-1B-NC' and 'AN3CA-NC',
respectively.
Tissue collection
EC samples (n=50; age, 29-67 years) and the healthy
samples adjacent to tumor tissues (n=6; age, 35-62 years) were
obtained from patients who received surgical therapy at the
Department of Obstetrics and Gynecology, Shanghai First People's
Hospital Affiliated to Shanghai Jiao Tong University (Shanghai,
China), between February 2010 and December 2013. The healthy
tissues were from the same group of patients for the EC tissues. In
brief, a vaginal speculum was used to expose the cervix after
general or local anesthesia, followed by disinfection of the cervix
using 0.5% povidone iodine (Qingdao Hainuo Biology Engineering Co.,
Ltd.) and paralysis of the cervical canal via a multiple-point
injection of 1% lidocaine 20-100 mg (Tianjin Zhongxin
Pharmaceutical Group Co., Ltd.). The cervix was then dilated, and
the uterine cavity was penetrated using a long and thin metal rod
with a curette at the end to perform curettage of the endometrium.
Tumor staging and histological grading were performed according to
the International Federation for Obstetrics and Gynecology criteria
(2009) (14). Clinical and
pathological data of the cohort are presented in Table I.
 | Table IClinicopathological summary of the
samples from patients with endometrial carcinoma. |
Table I
Clinicopathological summary of the
samples from patients with endometrial carcinoma.
| Clinical
pathological variables | Number | Ratio (%) |
|---|
| Age (years) | | |
| ≥55 | 40 | 80 |
| <55 | 10 | 20 |
| FIGO stage | | |
| Stage I | 31 | 62 |
| Stage II | 11 | 22 |
| Stage III | 8 | 16 |
| Grade | | |
| G1 | 26 | 52 |
| G2 | 15 | 30 |
| G3 | 9 | 18 |
| Myometrial
invasion | | |
| <1/2 | 40 | 80 |
| ≥1/2 | 10 | 20 |
| Nodal
metastasis | | |
| Positive | 9 | 18 |
| Negative | 41 | 82 |
| Total | 50 | 100 |
Primary cell culture
Fresh clinical samples of the tumor (1 cm) were
minced into 2-mm fragments and then transferred to a solution of
trypsin 0.25% in medium (containing 0.1 mM EDTA), followed by
incubation at 37°C for 20 min. Isolated EC cells were recovered in
0.5% trypsin inhibitor (Merck KGaA) and 1.0% albumin
(Sigma-Aldrich; Merck KGaA) in DMEM/F12 medium and then isolated by
centrifugation at 400 × g for 10 min at 4°C. The cells obtained
were manually counted using a hemocytometer under a light
microscope (magnification, ×40; Eclipse Ts2; Nikon Corporation) and
transferred to 75-cm2 culture flasks and incubated in
medium supplemented with 10% FBS in an incubator at 37°C with a
humidified atmosphere containing 5% CO2.
Immunohistochemical staining
EC tissues, adjacent healthy tissues or xenografts
were fixed using 10% neutral paraformaldehyde at room temperature
for 24 h and then sections with a thickness of 5 µm were
prepared. After deparaffinization and dehydration, sections were
boiled in 10 mM tris(hydroxymethyl)
aminomethane-Ethylenediaminetetraacetic acid (Tris-EDTA) buffer to
retrieve antigens and then blocked in 5% normal goat serum
(Sigma-Aldrich; Merck KGaA) at room temperature for 2 h. After
incubation with primary antibodies overnight at 4°C, sections and
secondary antibodies at room temperature for 1 h, the sections were
stained using an ABC Elite kit (cat. no. PK6200; Vector
Laboratories, Inc.) and a diaminobenzidine kit (cat. no. SK4100;
Vector Laboratories, Inc.) according to the manufacturer's
protocol. The images of sections were captured under a light
microscope (magnification, ×40; Eclipse Ts2; Nikon Corporation).
Primary antibodies used in the present study were rabbit polyclonal
anti-ATF3 antibody (cat. no. ab87213; 1:200; Abcam), mouse
monoclonal anti-JunB antibody (cat. no. sc8051; 1:200; Santa Cruz
Biotechnology, Inc.) anti-proliferating cell nuclear antigen
antibody (PCNA; cat. no. ab29; 1:100; Abcam) and rabbit polyclonal
anti-Ki67 antibody (cat. no. ab15580; 1:100; Abcam). For the
biotinylated horseradish peroxidase anti-rabbit or anti-mouse
secondary antibodies were included in the ABC Elite kit, and a
dilution of 1:3,000 was used.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from HEC-1B-NC, HEC-1B-ATF3,
AN3CA-NC and AN3CA-ATF3 cells with TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) and reverse
transcribed at 42°C for 30 min to cDNA using the Prime Script RT
reagent kit (Takara Bio, Inc.). RT-qPCR was performed using SYBR
Premix Ex Taq (Takara Bio, Inc.) and analyzed with an ABI Prism
7000 Sequence Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions were as follows:
Initial denaturation at 95°C for 10 min, followed by 42 cycles at
95°C for 15 sec, 60°C for 1 min and 72°C for 1.5 min. The
oligonucleotide primer sequences used for RT-qPCR were as follows:
ATF3 forward, 5′-CAT CTT TGC CTC AAC TCC AG-3′ and reverse, 5′-GAC
ACT GCT GCC TGA ATC CT-3′; JunB forward, 5′-AGC CAC CTC CCG TTT ACA
-3′ and reverse, 5′-TCT GCG GTT CCT CCT TGA -3′; and GAPDH forward,
5′-AGG TCG GAG TCA ACG GAT TTG -3′ and reverse, 5′-GTG ATG GCA TGG
ACT GTG GT-3′. All the qPCR analyses were performed following the
procedures reported previously (15). Values on the y-axis equal to
2−∆∆Cq were selected, where ∆Cq is the difference
between the target gene Cq and the normalizer gene Cq. Cq
represents the cycle of the threshold at which the fluorescence
increases above the baseline with statistical significance. Gene
expression data were obtained from three independent experiments.
With regard to the RT-qPCR analysis of MMPs and TIMPs, the
experiment was conducted as aforementioned and all oligos (Table II) were supplied in the
RT2 qPCR First Strand kit (cat. no. 330404; Qiagen,
Inc.).
| Table IIPrimer sequences used for
quantitation of MMPs and TIMPs. |
Table II
Primer sequences used for
quantitation of MMPs and TIMPs.
A, MMPs.
|
|---|
| Gene | Direction | Sequences |
|---|
| MMP2 | Forward |
5′-AACTACGATGACGACCGCAAGT-3′ |
| Reverse |
5′-AGGTGTAAATGGGTGCCATCA-3′ |
| MMP3 | Forward |
5′-TTCCGCCTGTCTCAAGATGATAT-3′ |
| Reverse |
5′-AAAGGACAAAGCAGGATCACAGTT-3′ |
| MMP7 | Forward |
5′-CTTTGCGCGAGGAGCTCA-3′ |
| Reverse |
5′-CAGGCGCAAAGGCATGA-3′ |
| MMP9 | Forward |
5′-AGGCGCTCATGTACCCTATGTAC-3′ |
| Reverse |
5′-GCCGTGGCTCAGGTTCA-3′ |
| MMP10 | Forward |
5′-GGACCTGGGCTTTATGGAGATAT-3′ |
| Reverse |
5′-CCCAGGGAGTGGCCAAGT-3′ |
| MMP11 | Forward |
5′-GGGTGCCCTCTGAGTCGA-3′ |
| Reverse |
5′-TCACAGGGTCAAACTTCCAGTAGA-3′ |
| MMP13 | Forward |
5′-AAATTATGGAGGAGATGCCCATT-3′ |
| Reverse |
5′-TCCTTGGAGTGGTCAAGACCTAA-3′ |
|
| B, TIMPs. |
|
| Gene | Direction | Sequences |
|
| TIMP1 | Forward |
5′-GACGGCCTTCTGCAATTCC-3′ |
| Reverse |
5′-GTATAAGGTGGTCTGGTTGACTTCTG-3′ |
| TIMP2 | Forward |
5′-GAGCCTGAACCACAGGTACCA-3′ |
| Reverse |
5′-AGGAGATGTAGCACGGGATCA-3′ |
| TIMP3 | Forward |
5′-CCAGGACGCCTTCTGCAA-3′ |
| Reverse |
5′-CCCCTCCTTTACCAGCTTCTTC-3′ |
| TIMP4 | Forward |
5′-CACCCTCAGCAGCACATCTG-3′ |
| Reverse |
5′-GGCCGGAACTACCTTCTCACT-3′ |
Western blot (WB) analysis
The primary EC cells, HEC-1B and AN3CA cells were
lysed using Extraction and Quantification ProteoJET Mammalian Cell
Lysis reagent (Fermentas; Thermo Fisher Scientific, Inc.) with
protease inhibitor cocktail (Roche Diagnostics). Total protein
concentrations were determined using the bicinchoninic acid (BCA)
method (Pierce; Thermo Fisher Scientific, Inc.). A total of 50
µg protein was separated by 12% SDS-PAGE and transferred to
a polyvinylidene difluoride membrane. Membranes were blocked using
5% non-fat milk powder for 1 h at room temperature, incubated with
primary antibodies overnight at 4°C and then washed three times in
Tris-buffered saline (pH 7.4). Subsequently, membranes were
incubated for 2 h with secondary antibodies at room temperature.
The primary antibodies used were as follows: Rabbit anti-ATF3
(1:200; cat. no. ab216569; Abcam), mouse anti-JunB (1:200; cat. no.
sc-8051; Santa Cruz Biotechnology, Inc.) and rabbit anti-GAPDH
(1:20,000; cat. no. ab199553; Abcam). Secondary horseradish
peroxidase-conjugated goat anti-rabbit (1:1,000; cat. no. sc-2004;
Santa Cruz Biotechnology, Inc.) and goat anti-mouse antibodies
(1:4,000; cat. no. SA00001-1; ProteinTech Group, Inc.) were
visualized with the Super Signal West Pico chemiluminescent
substrate (Thermo Fisher Scientific, Inc.) using a gel imaging
system with a preconfigured Image Lab software 5.2.1 (ChemiDoc
XRS+; Bio-Rad Laboratories, Inc.).
For the apoptosis marker detection, HEC-1B-NC,
HEC-1B-ATF3, AN3CA-NC or AN3CA-ATF3 cells were lysed and the
protein concentrations were determined using BCA assay. The cell
lysates were subject to WB analysis as aforementioned using the
primary antibodies anti-poly(ADP-ribose) polymerase (PARP; 1:500;
cat. no. 614302; Biolegend, Inc.), anti-Bcl2 (1:1,000; cat. no.
658702; Biolegend, Inc.), anti-Bcl-x (1:500; cat. no. 633902;
Biolegend, Inc.), anti-Bax (1:200; cat. no. 633602; Biolegend,
Inc.), anti-p38 (1:500; cat. no. 622402; Biolegend, Inc.) and
anti-phosphorylated (p)-p38 antibodies (1:200; cat. no. 690202;
Biolegend, Inc.).
Yeast two-hybrid (Y2H) assay
The Y2H assay was performed using a Matchmaker
Gal4-based two-hybrid assay kit (Clontech Laboratories, Inc.)
according to the manufacturer's instructions. A bait protein (ATF3)
was expressed in the yeast strain Y2HGold as a fusion to the Gal4
DNA-binding domain (DNA-BD) using the pGBKT7 vector with the ORF of
ATF3 inserted, while the prey proteins from the the high-complexity
EC library, which are provided in yeast strain Y187, were expressed
as fusions to the Gal4 activation domain (AD) using the pGADT7
vector, according to manufacturer's protocols. When cultures of the
two transformed strains were mixed together for 20-24 h at 30°C
with slow shaking at 1-5 × g, they mated to create diploids. Thus,
the DNA-BD and AD were guided into proximity to activate the
transcription of four independent reporter genes (AUR1-C, ADE2,
HIS3 and MEL1, coding for enzyme inositol phosphoryl ceramide
synthase, histidine, adenine and α-galactosidase, respectively).
Via streaking with toothpicks onto high-stringency selective
synthetic dropout media supplemented with 20 µg/ml X-α-Gal,
screening was performed based on whether bait (ATF3) and prey
(library) fusion proteins interacted with each other. The
interactions between the baits and preys were determined 24 h after
culturing at 30°C. The positive control mating between Y2HGold
[pGBKT7-53] and Y187 [pGADT7-T], and the NC mating between Y2HGold
[pGBKT7-Lam] and Y187 [pGADT7-T] were performed simultaneously.
Autoactivation of reporter genes in Y2HGold by the bait (ATF3) was
confirmed via streaking the plasmid-harbored yeast cells on the
selective synthetic dropout media prior to the two-hybrid screen,
in the absence of a prey protein.
Polyhistidine (His) tag-fused ATF3
(His-ATF3) expression and purification
The ORF of ATF3 was subcloned into the pET32a vector
(Novagen; Merck KGaA) and introduced into Escherichia coli BL21
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's protocol. His-ATF3 was induced in the host bacteria
using 0.2 µM isopropyl-β-D-1-t hiogalactopyranoside (IPTG;
Sigma-Aldrich; Merck KGaA) at 37°C for 4 h and the soluble protein
of His-ATF3 was purified using a His-Tag purification column
(Sigma-Aldrich; Merck KGaA) according to the manufacturer's
instructions.
BIAcore assay
The affinity and binding kinetics were measured
using BIAcore assays. The dissociation rate constant (Kd) between
His-ATF3 and JunB (Origene Technologies, Inc.) was determined using
BIAcore 3000 (BIAcore, Inc.) and the data were analyzed using
BIAEVALUTION software version 4.1 (BIAcore, Inc.). Standard
ethyl(dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide
coupling was used to covalently immobilize JunB to CM5 sensor chips
(BIAcore, Inc.) according to the manufacturer's instructions, and
the interaction was detected in HBS-EP buffer (10 mM HEPES, pH 7.4,
0.15 M NaCl, 3 mM EDTA, 0.005% v/v surfactant P20). Flow cell 1 was
left blank as a NC. Association rates were measured under a
continuous flow of 10 µl/min using His-ATF3 at
concentrations ranging between 30-150 nM plus a concentration of 0
nM, and the data were fitted using 1:1 Langmuir binding with no
bulk refractive shift.
In vitro His-tag pulldown assay
Cell lysates containing His-ATF3 were prepared from
the bacteria harboring pET32a-ATF3 plasmid after induction by IPTG
for 4 h via sonication (0°C, 20 KHz, output 3; 50% duty cycle; 15
min) and incubated with 100 µl Ni-NTA agarose resin
(Sigma-Aldrich; Merck KGaA) at 4°C overnight. After washing twice
with PBS, bound proteins were boiled in the loading buffer,
separated using 12% SDS-PAGE and detected by WB as
aforementioned.
Co-immunoprecipitation (Co-IP) assay
Plasmids pCMV-Script-HA-ATF3 and
pCMV-Script-Myc-JunB were prepared for the assay via inserting
hemagglutinin (HA) or Myc sequence before the ORFs of ATF3 and
JunB, respectively. For the Co-IP assay, HEC-1B-NC, HEC-1B-ATF3 or
293T cells (Chinese Academy of Sciences Committee Type Culture
Collection) were transfected with 2.5 µg pCMV-Script-HA-ATF3
or pCMV-Script-Myc-JunB vector as aforementioned, respectively.
After 24 h, cells were collected and then lysed in IP assay lysis
buffer [10 mM Tris-HCl (pH 7.3), 100 mM NaCl, 1 mM EDTA, 0.2%
Triton X-100, 0.2 mM dithiothreitol (DTT), 10% glycerol and
protease inhibitors]. Cell lysates (1 mg) were incubated with 1
µg JunB antibody (1:50; cat. no. sc-8051; Santa Cruz
Biotechnology, Inc.) or 1 µg immunoglobulin G1 (1:50; IgG1;
cat. no. 401401; Biolegend, Inc.) as a control at 4°C overnight.
The immunocomplex was precipitated with 25 µl Protein A
Sephasrose 6MB (Cytiva), pelleted at 800 × g, 4°C for 5 min, washed
with 1 ml IP assay buffer supplemented with 300 mM NaCl three times
and then detected using 12% SDS-PAGE and WB as aforementioned with
anti-ATF3 antibody and anti-JunB antibody.
Co-localization assay
HEC-1B cells were seeded at a confluency of 80% on a
glass slip and co-transfected with 5 µg pCMV-Script-HA-ATF3
and pCMV-Script-Myc-JunB plasmid, respectively, with
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The plasmids were prepared as aforementioned.
After 24 h, cells were fixed in 4% formaldehyde for 15 min at 4°C,
permeabilized with 0.2% Triton X-100 for 5 min at room temperature
and blocked with 5% bovine serum albumin (cat. no. A7906;
Sigma-Aldrich; Merck KGaA) for 50 min at room temperature. Cells
were subsequently incubated with ATF3 (C-19) antibody (1:200; cat.
no. sc-188; Sant Cruz Biotechnology. Inc.) and JunB antibody
(1:200; cat. no. sc-8051; Santa Cruz Biotechnology, Inc.) at 4°C
overnight and then labeled with FITC-conjugated anti-rabbit IgG
(1:20,000; cat. no. 111-095-003; Jackson ImmunoResearch
Laboratories, Inc.) and rhodamine (TRITC)-conjugated anti-mouse IgG
(1:20,000; cat. no. 115-025-146; Jackson ImmunoResearch
Laboratories, Inc.) for 1 h at room temperature. After washing with
PBS three times, 30 sec each time, cells were counterstained with
DAPI at room temperature for 10 min. After washing with PBS, cells
were covered with Vectashield mounting media (Vector Laboratories,
Inc.). Images were acquired with a laser scanning confocal
microscope (Leica Microsystems, Inc.) using a ×60 oil immersion
lens.
Cell proliferation and colony formation
assays
To assess cell proliferation, cells were seeded into
96-well plates at 2,000 cells per well. At the time points of 1, 2,
3, 4 and 5 days, the number of metabolically active cells was
measured using a MTT assay (Sigma-Aldrich; Merck KGaA). With the
formed formazan dissolved in 100 µl dimethyl sulfoxide, the
absorbance values were measured at 490 nm with a Spectra Max 190
microplate reader (model 680; Bio-Rad Laboratories, Inc.).
For colony formation assays, cells were seeded in
6-well plates at 200 cells/well and grown in normal culture medium
(DMEM/F12 with 10% FBS) at 37°C and 5% CO2 for 9 days
for colony formation. Colonies were fixed in 4% formaldehyde for 30
min at room temperature and then counted under a light microscope
(magnification, ×40; Eclipse Ts2; Nikon Corporation) after staining
with 0.5% crystal violet for 20 min at room temperature. The
experiments were repeated independently ≥3 times.
Chromatin IP (ChIP) assay
Cells with or without overexpression of ATF3 were
fixed at room temperature in 0.5% formaldehyde for 20 min. After
sonication (0°C; 20 KHz; output 2; 20% duty cycle; 5 min) using a
sonifier (Model 450; Branson; Thermo Fisher Scientific, Inc.), IP
was performed at 4°C overnight using 5 µg anti-ATF3 (1:50;
cat. no. sc-87213; Santa Cruz Biotechnology, Inc.), anti-JunB
antibody (1:50, cat. no. sc-8051; Santa Cruz Biotechnology, Inc.)
or control mouse IgG1 (1:50; cat. no. 401401; Biolegend, Inc.).
Pulled-down chromatins were denatured at 65°C overnight and the DNA
was purified using the QIAquick purification column (size, 0.6 ml;
Cytiva). The resulting AP-1 site included DNA was quantified by
qPCR as aforementioned using the following primer pairs: Forward,
5'-TAA CTG CTC GGA AGT CCC AC-3' and reverse, 5′-ACC CGA CTA TCT
GCC AGG TC-3′. Fluorescence intensities were calculated using the
following formula: Fluorescence intensity=(IP average ± IgG
average)/(Input average).
Transwell migration and invasion
assays
For Transwell migration assays, the lower surface of
polycarbonate membranes of the inserts (8-µm pore size;
Corning, Inc.) was coated with 100 µg fibronectin
(Sigma-Aldrich; Merck KGaA). A total of 2×105 cells
suspended in 100 µl DMEM/F12 medium (without FBS) were
dispensed into the upper chambers of the Transwells (with 0.6 ml
DMEM/F12 medium supplemented with 10% FBS in the lower chamber) and
incubated at 37°C for 24 h. Cells migrated to the lower membrane
surface were fixed at room temperature for 20 min with 4%
paraformaldehyde, stained with crystal violet (20 min at room
temperature) and counted under a light microscope (magnification,
×40; Eclipse Ts2; Nikon Corporation). For invasion assays,
106 cells were plated in Transwells coated with Matrigel
(1:6; BD Biosciences) at room temperature for 1 h and cultured at
37°C for 24 h. Invaded cells were stained and counted as
aforementioned.
Apoptosis assay and cell cycle
analysis
The apoptotic-related proteins were detected using
WB as aforementioned. To measure the apoptosis ratios using flow
cytometry, a total of 1×106 HEC-1B-NC or -ATF3 cells
were digested with 0.25% trypsin (without EDTA) and stained with
FITC-Annexin V in fluorescence-assisted cell sorting buffer (cat.
no. 556547; BD Biosciences) for 30 min at room temperature after
washing with PBS. The total apoptotic (early and late apoptosis)
cells were then analyzed with the software Kaluza 1.3 (Beckman
Coulter, Inc.) on a Beckman Gallios flow cytometer (Beckman
Coulter).
For the cell cycle analysis, cells were fixed with
4% para-formaldehyde at room temperature for 2 h and stained with
7-aminoactinomycin D at room temperature for 10 min. After the
cells were detected by flow cytometry (Beckman Gallios; Beckman
Coulter, Inc.), cell cycle analysis was performed with the software
Kaluza version 1.3 (Beckman Coulter, Inc.). Experiments were
performed in as three independent replicates.
Caspase enzymatic activity
measurement
The activities of Caspase-3/7, Caspase-8 and
Caspase-9 were determined using the Caspase-Glo 3/7 Assay kit,
Caspase-Glo 8 Assay kit and the Caspase-Glo 9 Assay kit (Promega
Corporation), respectively, according to the manufacturer's
protocol. HEC-1B-NC, HEC-1B-ATF3, AN3CA-NC and AN3CA-ATF3 cells
were seeded at a density of 4×105 cells/well in 6-well
plates and cultured overnight. Following digestion,
~6×104 cells per sample were collected in 1.5-ml tubes.
After 100 µl Caspase-Glo reagent (relative caspase
substrate) was added to each sample, the cells were thoroughly
mixed and incubated for 1 h in the dark at 37°C. Luminescence
values of each sample were measured using an IVIS Kinetic Imaging
System (Caliper Life Sciences; PerkinElmer, Inc.). The quantitative
analysis was performed based on the relative caspase activities and
normalized to the raw luminescence units of the untreated
control.
JunB reporter assay
JunB promoter constructs driving the expression of
the luciferase reporter gene were used. A DNA fragment of the WT or
mutant (MT) 5'untranslated region (UTR) of JunB was cloned into the
pRL-CMV luciferase reporter plasmid (50 ng; cat. no. E2261; Promega
Corporation) and the resultant vectors were designated as
'pRL-CMV-JunB-5'UTR-WT' and 'pRL-CMV-JunB-5'UTR-MT', respectively.
HEC-1B-NC or HEC-1B-ATF3 cells were transiently transfected with 1
µg Renilla constructs (as an internal control) or
plasmids pRL-CMV-JunB-5'UTR-WT or pRL-CMV-JunB-5'UTR-MT using
Lipofectamine® 3000 as aforementioned. Cell lysates were
prepared after 24 h and the luciferase activities of cell lysates
were then determined using a luminometer (Glomax 96; Promega
Corporation). The relative luciferase activity was calculated as
firefly/Renilla. All experiments were performed as ≥3
independent replicates.
DNA precipitation (DNAP) assay
Nuclear extracts (100 µg) in 500 µl
nuclear extraction buffer (Thermo Fisher Scientific, Inc.) from
HEC-1B-NC or HEC-1B-ATF3 cells were mixed with a biotinylated
activator protein (AP)-1 site probe (50 pmol; cat. no. D3118;
Beyotime Institute of Biotehcnology) in a buffer [HEPES-KOH (pH
7.9), 80 mM KCl, 1 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 10% (w/v)
glycerol, 0.1% Triton X-100 and 1 µg poly(dI-dC)] at 0°C for
30 min. Then, 100 µl Streptavidin agarose beads
(Sigma-Aldrich; Merck KGaA) were added and the mixture (0.6 ml) was
gently agitated for 1 h at room temperature. Beads were then
pelleted at 800 × g for 5 min at room temperature and washed three
times with PBS, and the bound proteins were analyzed using WB as
aforementioned. The sequence of the biotinylated AP-1 site used was
as follows: 5′-TAT GGA GAT GAC TCA AAG GGG GCG TGC A-3′.
Xenograft assay in nude mice
A total of 12 BALB/c nude mice (age, 4-5 weeks;
weight 16-18 g) were purchased from Shanghai Laboratory Animal
Research Center. The mice were housed in individually ventilated
cages on a ventilated rack (Model GH; Suzhou Fengshi Laboratory
Animal Equipment Co., Ltd.) in a specific pathogen-free facility
with 12 h cycle of light/dark. The temperature, humidity and air
flow of the ventilating air was set at 22°C, 40% and 25 cm/sec,
respectively. The animals were able to move freely with unlimited
access to water and food. A total of 5×106 cells
suspended in 100 µl 1X PBS were injected into the
interscapular area subcutaneously of the mice. A group of mice
(n=8) received HEC-1B-ATF3 (referred to as exATF3). Another NC
group (n=4) received HEC-1B-NC cells transfected with control
plasmid (cloning vector only, referred to as exNC). The sizes of
tumors were measured daily over 4 weeks. Mice were sacrificed at 30
days post-injection by CO2 exposure at an air
displacement rate of 30% per min. Tumors were excised and measured.
The tumor volume (cm3) was calculated by using the
following formula: Volume=(longest diameter) × (shortest
diameter)2 ×0.5.
Statistical analysis
All statistical analyses were performed using SPSS
16.0 (SPSS, Inc.) or GraphPad Prism 5.0 (GraphPad Software, Inc.).
Each experiment was performed ≥3 times and data are presented as
the mean ± SD. A one-tailed, unpaired Student's t-test or
Mann-Whitney U-test were utilized to determine significant
differences between the treatment groups. P<0.05 was considered
to indicate a statistically significant difference.
Results
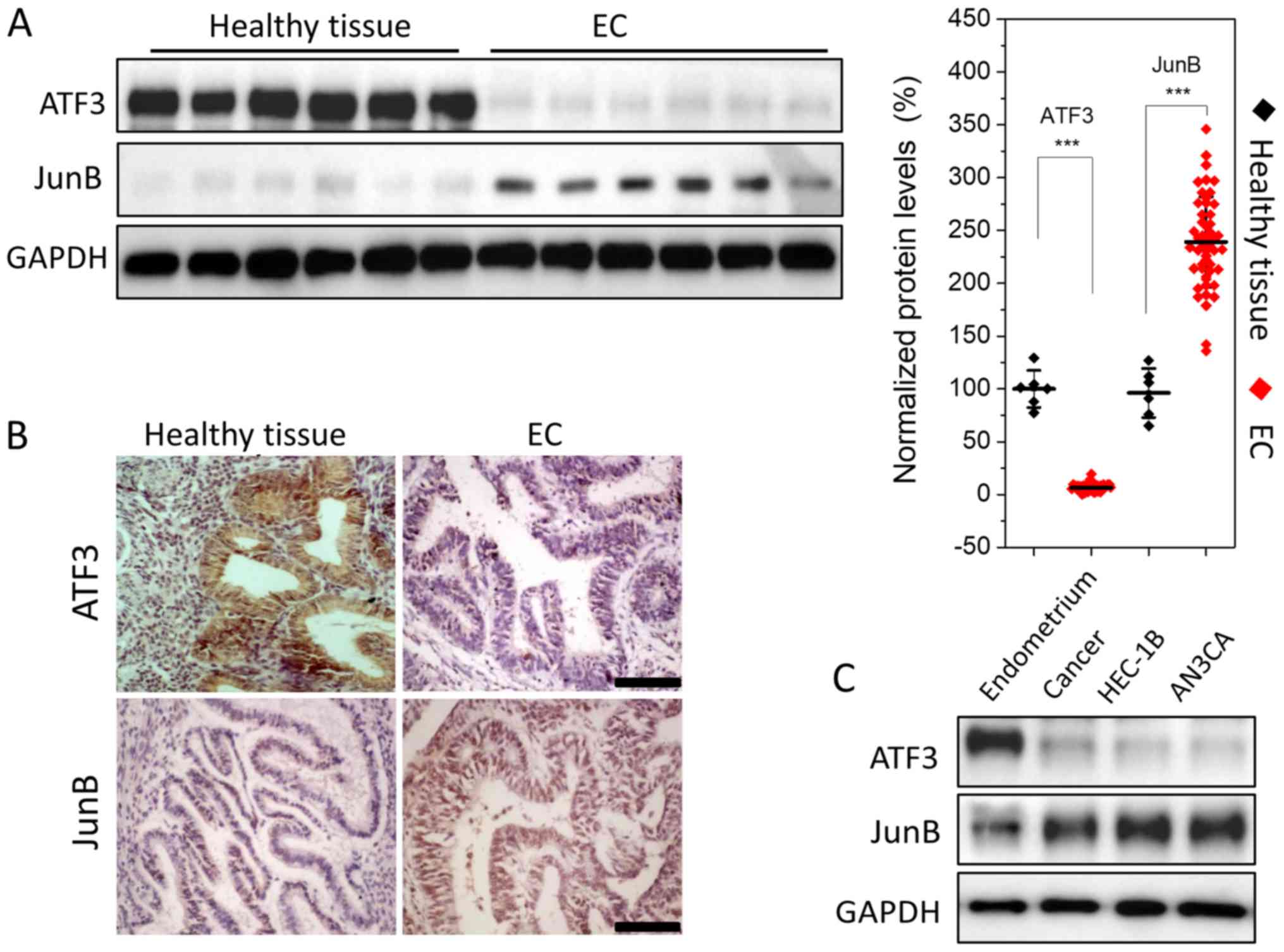
ATF3 is downregulated and JunB is
upregulated in EC
The expression levels of ATF3 and JunB were
semi-quantitatively assessed using WB, indicating that ATF3
expression was low in EC tissues; it was >20-fold lower compared
with healthy endometrium. However, JunB expression was relatively
high, with a ~2-fold increase compared with healthy tissues
(Fig. 1A). To further evaluate
this result, healthy endometrial and EC tissues were subjected to
immunohistochemical analysis. Strong staining for JunB and weak
staining for ATF3 were observed in EC tissues (Fig. 1B). Thus, the results indicated that
ATF3 was downregulated, while JunB was upregulated in EC.
Next, ATF3 and JunB expression levels were assessed
in EC cell lines. WB analysis demonstrated that JunB was expressed
in the type I and type II EC cell lines HEC-1B and AN3CA, while the
expression of ATF3 was low, which was consistent with the WB and
immunohistochemistry results (Fig.
1C).
ATF3 overexpression impacts the
proliferation, cell cycle, apoptosis, migration and invasion of
tumor cells in vitro
Following the identification of the low expression
of ATF3 in ECs, HEC-1B cells were transfected to stably overexpress
ATF3 in order to demonstrate the function of ATF3 in endome-trial
tumorigenesis. Stable transfection significantly increased the mRNA
transcription of ATF3 by >180-fold (Fig. S1) and its protein expression by
~9-fold in HEC-1B cells (Fig. 2A).
After transfection, the proliferation and colony-forming ability of
HEC-1B cells was assessed using a MTT assay and a colony formation
assay, respectively. The results suggested that over-expression of
ATF3 in HEC-1B cells significantly inhibited cell proliferation
(Fig. 2B) and colony formation
(Fig. 2C) compared with exNC
cells.
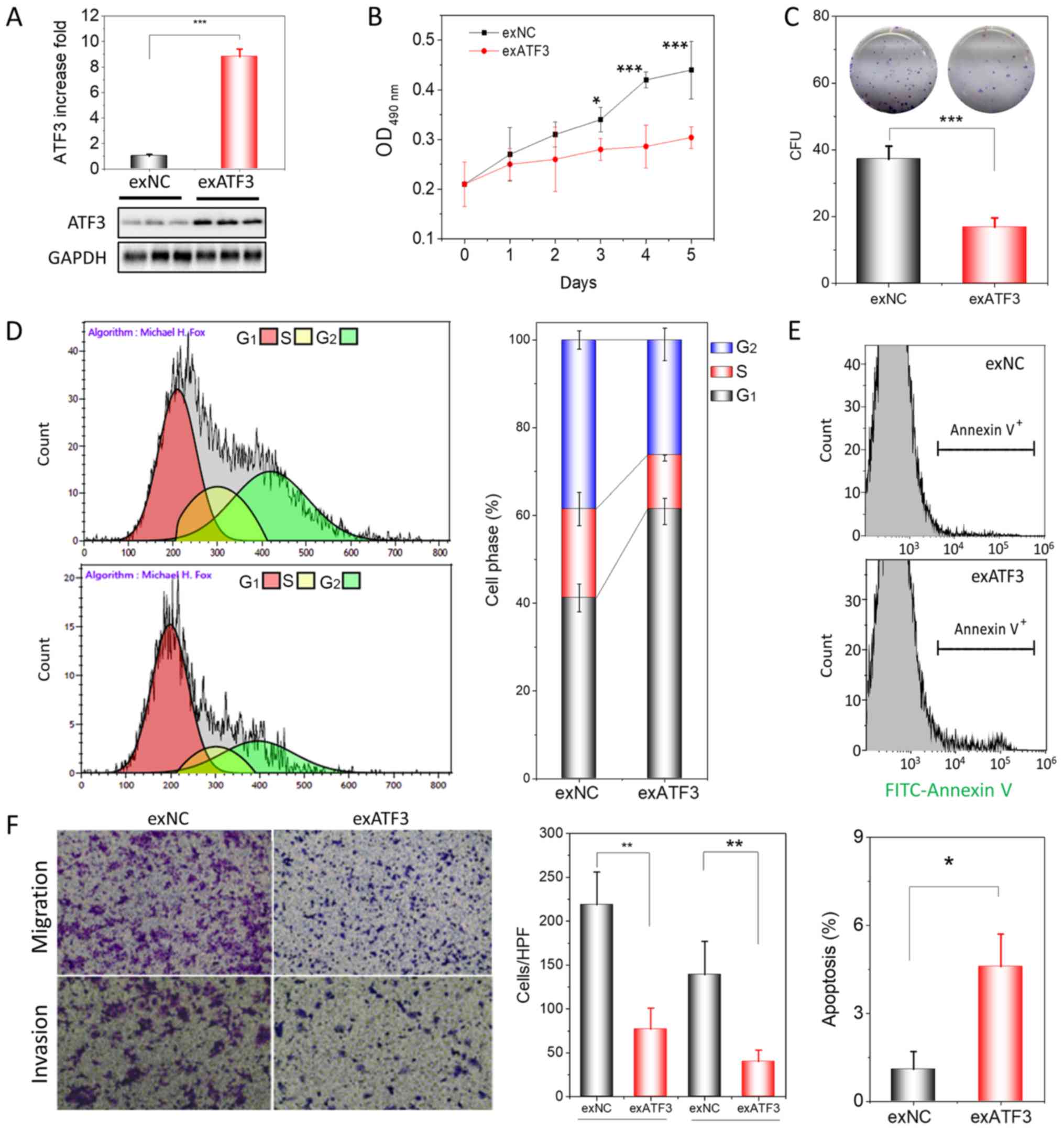 | Figure 2ATF3 overexpression inhibits
proliferation, migration and invasion of HEC-1B cells. (A)
Assessment of ATF3 protein expression in exATF3 cells after stable
transfection. (B) Assessment of the cell proliferation of exNC and
exATF3 cells. (C) Colony-formation assays of exATF3 and exNC cells,
and graphical representation of the numbers of colonies in three
independent experiments. (D) Cell cycle determined using flow
cytometry and the quantitative analysis. (E) Representative images
of flow cytometry analysis of apoptotic rates of exNC and exATF3
cells. The apoptotic cells were stained with FITC-Annexin V. (F)
Cell migration and invasion assay of exNC and exATF3 cells, and
graphical representation of the numbers of migrated and invaded
cells in three independent experiments (magnification, ×40). Data
are presented as the mean ± SD (n=3) and were analyzed using
one-tailed Student's t-test. *P<0.05, **P<0.01,
***P<0.001 vs. controls. ATF3, activating
transcriptional factor 3; exATF3, exogeneous ATF3 high expression
cells; OD, optical density; NC, negative control; exNC, negative
control cells of exATF3; HPF, high-power field; CFU, colony forming
unit. |
ATF3 is closely associated with the cell cycle,
apoptosis, invasion and metastasis in certain tumor types (16-18).
Therefore, it was investigated whether overexpression of ATF3 was
able to change the cell cycle profile and trigger apoptosis in
exATF3 cells. The flow cytometry results demonstrated that exATF3
cells had a markedly increased G1 phase population (60.9±3.0%;
n=3), while the S (12.2±0.7%; n=3) and G2 phase (25.9±3.7%, n=3)
populations were decreased, compared with those in exNC cells
(41.2±3.2, 20.3±3.8 and 38.5±2.1%, respectively; n=3; Fig. 2D).
The cells of exNC and exATF3 were further analyzed
with FITC-Annexin V staining and were subjected to analysis of
early and late apoptotic cell percentages using flow cytometry. The
results indicated that a greater percentage of exATF3 cells
(4.6±1.1%) were apoptotic compared with exNC cells (1.0±0.6%;
Fig. 2E). In addition, cells
transfected with ATF3 demonstrated a significantly reduced
migratory and invasive abilities compared with the control cells
(Fig. 2F). Collectively, the
results suggested that ATF3 had a suppressive function in ECs.
Overexpression of ATF3 regulates the
expression of apoptotic markers, MMPs and TIMPs
After the inhibitory effects of ATF3 on the
proliferation, transmigration and invasion of ECs were identified,
it was further investigated whether apoptotic signaling pathways
were activated; thus, changes in the levels of apoptotic markers
after overexpression of ATF3 in ECs were detected. The enzymatic
activity of Caspases is regarded as an important indicator of
apoptosis (19). Therefore, the
activities of Caspase-3/7, Caspase-8 and Caspase-9 were first
measured, and it was found that the activities of all of these
enzymes increased after ATF3 was overexpressed in the cells
(Fig. 3A). In addition, the
expression levels apoptotic-associated proteins, including cleaved
PARP, Bax and p-p38 increased, while Bcl2, and Bcl-x decreased in
the two cell lines (Fig. 3B).
Thus, these results may explain the increased apoptotic ratios of
host cells after overexpression of ATF3. Moreover, the relative
protein levels of p-p38 and p38 were found to be significantly
increased, indicating that apoptosis may be mediated via a
p38-mitogen-activated protein kinase signaling pathway (Fig. 3B).
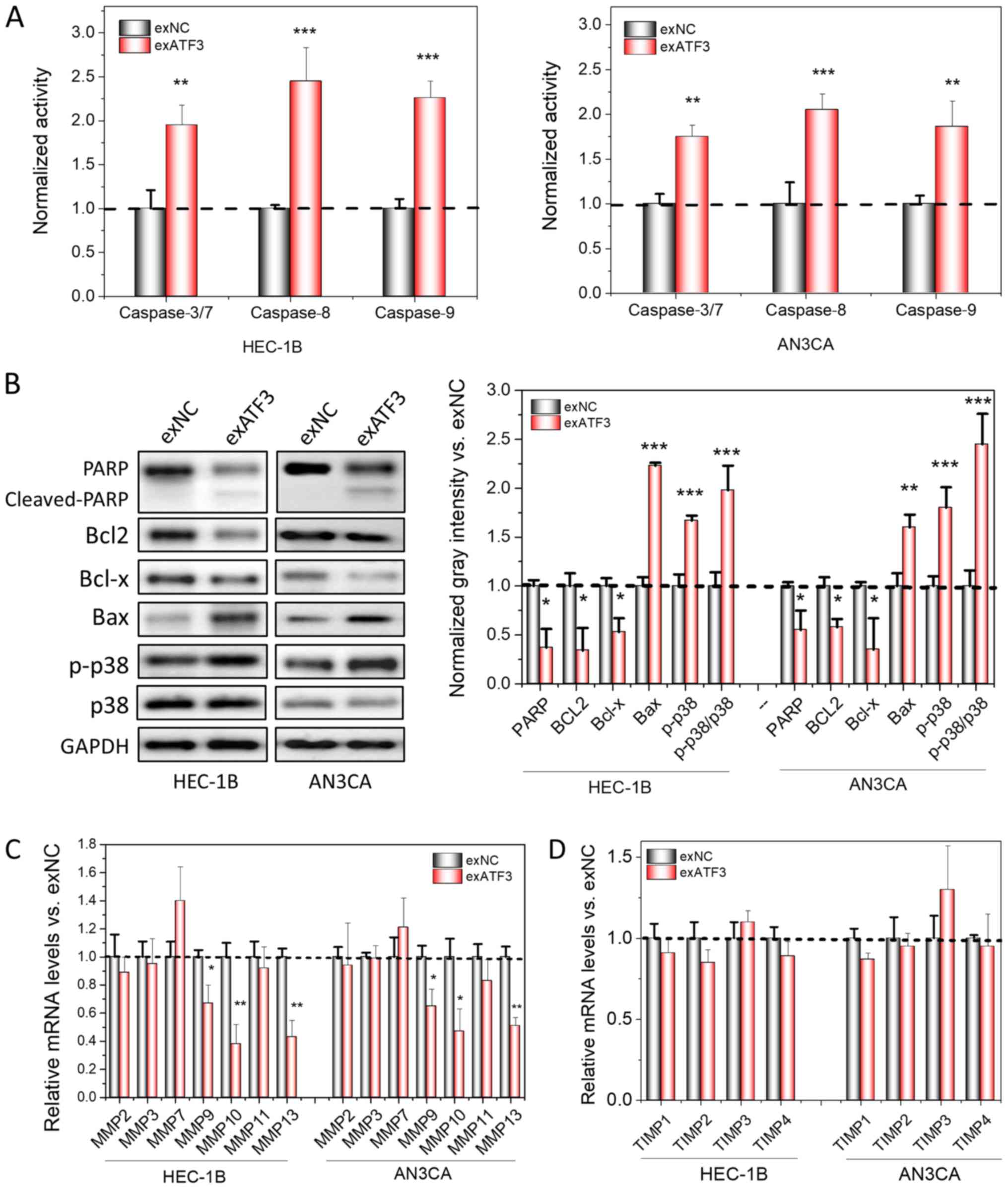 | Figure 3Overexpression of ATF3 induces
apoptosis and loss of MMPs in cancer cell lines. (A) Measurement of
apoptotic-related Caspase 3/7, 8 and 9 activities using
spectrometry in ATF3-overexpressed HEC-1B and AN3CA cells,
normalized to the activities of control. (B) Apoptosis marker
detection using western blotting. The semi-quantitation analysis
was performed based the gray intensities, normalized to GAPDH. mRNA
levels of MMPs (C) and TIMPs (D) were determined by reverse
transcription-quantitative PCR. The quantitation analysis was
normalized to control. Data are presented as the mean ± SD (n=3)
and were analyzed using one-tailed Student's t-test.
*P<0.05, **P<0.01,
***P<0.001 vs. exNC. ATF3, activating transcriptional
factor 3; MMP, matrix metallopeptidase; TIMP, tissue inhibitors of
metalloproteinase; PARP, poly(ADP-ribose) polymerase; p-,
phosphorylated. |
MMPs and TIMPs have prominent roles in cancer
invasion and metastasis. TIMPs, binding to MMPs at a 1:1 molar
ratio, are able to block the access of substrates to the catalytic
domain of endopeptidases and eventually inhibit the bioactivities
of MMPs (20). Furthermore,
increases in the MMP/TIMP ratio have been reported to affect
invasion and metastasis, and are widely considered as a marker for
cancer invasion (21-23). Therefore, the mRNA expression
levels of MMPs and TIMPs in both cell lines with and without ATF3
overexpression were compared. It was identified that certain MMPs,
including MMP9, MMP10 and MMP13, were significantly down-regulated
following overexpression of ATF3 (Fig.
3C and D); MMP7 was slightly upregulated, but was not
significantly different compared with the control. However, the
expression levels of all TIMPs were not significantly different
after ATF3 was overexpressed and remained stable.
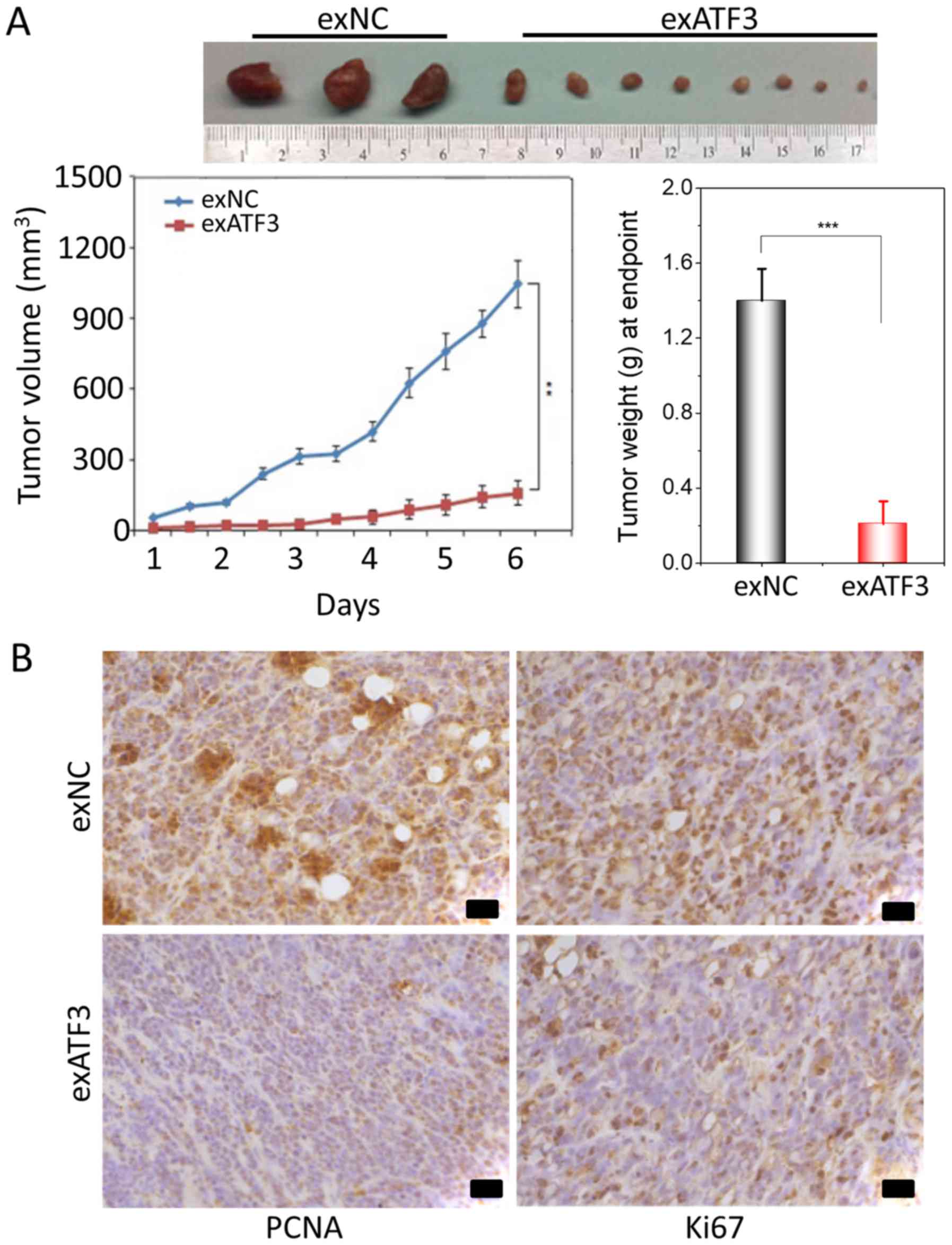
Overexpression of ATF3 decreases the
tumor size in a xenograft model
To further study the anti-tumor potential of ATF3, a
xenograft tumor growth assay in nude mice subcutaneously injected
with stably transfected exATF3 or exNC cells was performed. The
results indicated that overexpression of ATF3 significantly
decreased the volume and weight of the xenograft tumors compared
with the exNC group (Fig. 4A).
To determine the effect of ATF3 on proliferation and
apoptosis, immunohistochemical of staining of the tumor tissues for
Ki67 and PCNA was performed. The expression of Ki67, indicative of
tumor cell proliferation, was decreased by exATF3 in the xenograft
tumors. Moreover, notably weaker PCNA staining was detected in the
exATF3 group compared with the exNC group (Fig. 4B).
Overexpressed ATF3 directly interacts
with JunB and binds to AP-1 sites in ECs
The expression of ATF3 and JunB (Fig. S2) in EC was visualized by confocal
microscopy. ATF3 and JunB were identified to be predominantly
localized in the nucleus of EC cells (Fig. 5A). Previously, the binding partners
of ATF3 had been screened in a protein library of ECs using Y2H
analysis and JunB was identified as one of its interactors
(Fig. S3A and B). In addition,
their interaction was demonstrated in vitro using Co-IP
(Fig. S3C). To prepare the
recombinant protein of ATF3, a prokaryotic expression plasmid,
pET32a-ATF3, was constructed and His-ATF3 was expressed in
bacteria. The bioactive His-ATF3 was obtained after affinity
purification and denaturation. Using the His-ATF3, the interaction
was assessing with a His-tag pulldown assay (Fig. S3D).
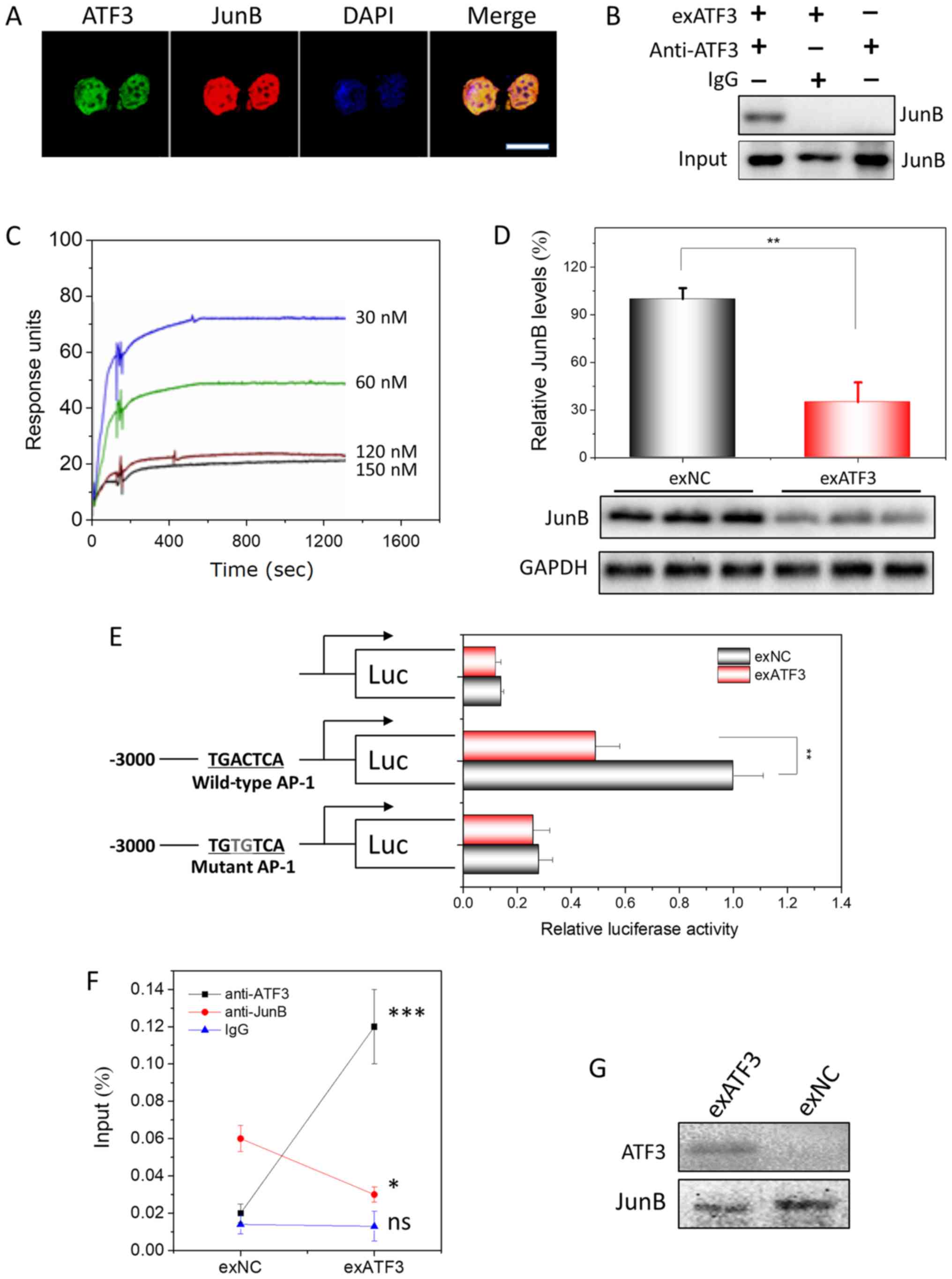 | Figure 5ATF3 interacts to JunB in endometrial
carcinoma cells and their binding to AP-1 site. (A) Subcellular
colocalization of ATF3 and JunB in exATF3 cells. Nuclei were
stained using DAPI. Scale bar, 7.5 µm. (B) Interaction
between ATF3 and JunB in exATF3 was assessed using
co-immunoprecipitation. (C) BIAcore assay of the interaction
between His-ATF3 and JunB. The values for association rate constant
and disassociation rate constant (Kd) were calculated from
sensorgrams using four concentrations of His-ATF3. The binding
affinity of Kd=1.9×10−8 M. (D) JunB was downregulated by
ATF3 overexpression in HEC-1B cells. The semi-quantitation was
performed based on band gray intensities, normalized to the
intensities before overexpression. (E) The cells of exATF3 orexNC
cells were transfected with reporter plasmids, pCMV-RL for 24 h.
Luciferase activity was normalized to Renilla luciferase
activity and expressed as fold change vs. controls. (F) ATF3 and
JunB chromatin immunoprecipitation was performed in exATF3 and exNC
cells. As a control, an isotype of anti-ATF3 and anti-JunB
monoclonal antibodies was used to measure the background. (G) DNA
precipitation was performed in exATF3 and control cells to extract
biotinylated DNA fragment (containing an AP-1 site) bound ATF3 and
JunB, detected using western blotting. Data are presented as the
mean ± SD (n=3) and were analyzed using one-tailed Student's
t-test. *P<0.05, **P<0.01,
***P<0.001 vs. the control group unless otherwise
specified. ATF3, activating transcriptional factor 3; ns, not
significant; NC, negative control; AP-1, activator protein 1; Kd,
dissociation constant. |
The presence of an ATF3/JunB interaction in ECs
after ATF3 overexpression was assessed using Co-IP with monoclonal
anti-ATF3 antibodies. In the native HEC-1B cells, no interaction
between ATF3 and JunB was detected. However, from HEC-1B cells
stably overexpressing ATF3, JunB was precipitated, indicating that
the ATF3-JunB interaction only occurs in the ATF3-overexpressing
cells (Fig. 5B).
To measure the affinity capacity between ATF3 and
JunB, which represents the minimum concentrations of His-ATF3 or
JunB required for binding, the association rate constant (Ka) and
Kd of the interaction between His-ATF3 and JunB were determined
with a BIAcore instrument. Fitting the data to a 1:1 binding model
yielded an apparent binding affinity with the equilibrium
dissociation constant KD=1.9×10−8 M, obtained from the
ratio of the rate constants kd/ka (Fig. 5C).
Similar to the composition of the promoters of most
MMPs (24,25), an AP-1 site is located in the
promoter of JunB. WB analysis indicated that JunB was downregulated
~3-fold by overexpression of ATF3 (Figs. 5D and S4). To demonstrate that the
downregulation of JunB was exerted by ATF3, lucif-erase plasmids
with JunB promoter containing a WT AP-1 site or a MT AP-1 site at
-2,322 to -2,315 were constructed (Fig. 5E). The highest luciferase activity
was observed in the HEB-1C cells transfected with the plasmid
containing the WT AP-1 binding site, and only slight or background
bioactivities were measured when no AP-1 sites or a MT AP-1 site
were included. In addition, the luciferase activity exhibited a
significant decrease when ATF3 overexpression plasmid was
co-transfected with reporter plasmid containing the WT AP-1 binding
site. Furthermore, no difference in luciferase activity was
identified between the groups transfected with exATF3 or exNC
co-transfected with plasmid containing MT AP-1 biding sites or
without promoter. Thus, the results indicated that the
overexpression of ATF3 regulated JunB transcription and translation
via binding to the AP-1 site in the promoter of JunB.
To demonstrate that the binding of ATF3 requires the
presence of JunB, ChIP was performed. The JunB promoter DNA
sequence was precipitated when either anti-ATF3 or anti-JunB
antibodies were utilized for precipitation (Fig. 5F). In addition, the levels of
precipitated DNA were in parallel with the expression levels of
ATF3 or JunB. When DNA was precipitated with the AP-1 binding site,
ATF3 and JunB were precipitated together alone with the DNA,
indicating that ATF3 regulated AP-1 signaling by interacting with
JunB (Fig. 5G).
Discussion
EC is one of the most common cancer types of the
female genital tract worldwide (1). While it is known that in the majority
of cases, estrogen and progesterone have a significant role in the
pathogenesis of EC, the basic mechanism is yet to be fully
elucidated. ATF3 has been reported to participate in the
carcinogenesis of various types of cancer, such as prostatic cancer
(26), but the roles of ATF3 in EC
remain unknown. The present results demonstrated that ATF3
inhibited the proliferation, migration and invasion of EC cells, at
least in part by interacting with JunB.
JunB is an important transcriptional factor
regulating the cell cycle, thus reflecting cell proliferation in
ECs (27,28). JunB has been shown to have an
oncogenic role and can promote cancer progression, invasion and
metastasis in uveal carcinoma (29), multiple myeloma (30), breast cancer (31) and squamous cell carcinoma (32,33).
Furthermore, JunB acts as an oncogene and induces abnormal
proliferation of previously quiescent cells (34). JunB, as a member of the AP-1
family, is considered to exert its function via AP-1 signaling
(35). Various mechanisms
underlying the AP-1-mediated regulation of tumor progression,
particularly invasion and metastasis, have been reported. For
instance, JunB regulates several genes, including MMP2, MMP9 and
C-C motif chemokine ligand-2, to enhance tumor invasion and
angiogenesis in renal cell carcinomas (36). With AP-1 sites in the promoters,
MMPs are considered to be regulated by AP-1, formed by AP-1 family
members, including JunB (24,25).
Therefore, JunB may be a promising therapeutic target for various
cancer types.
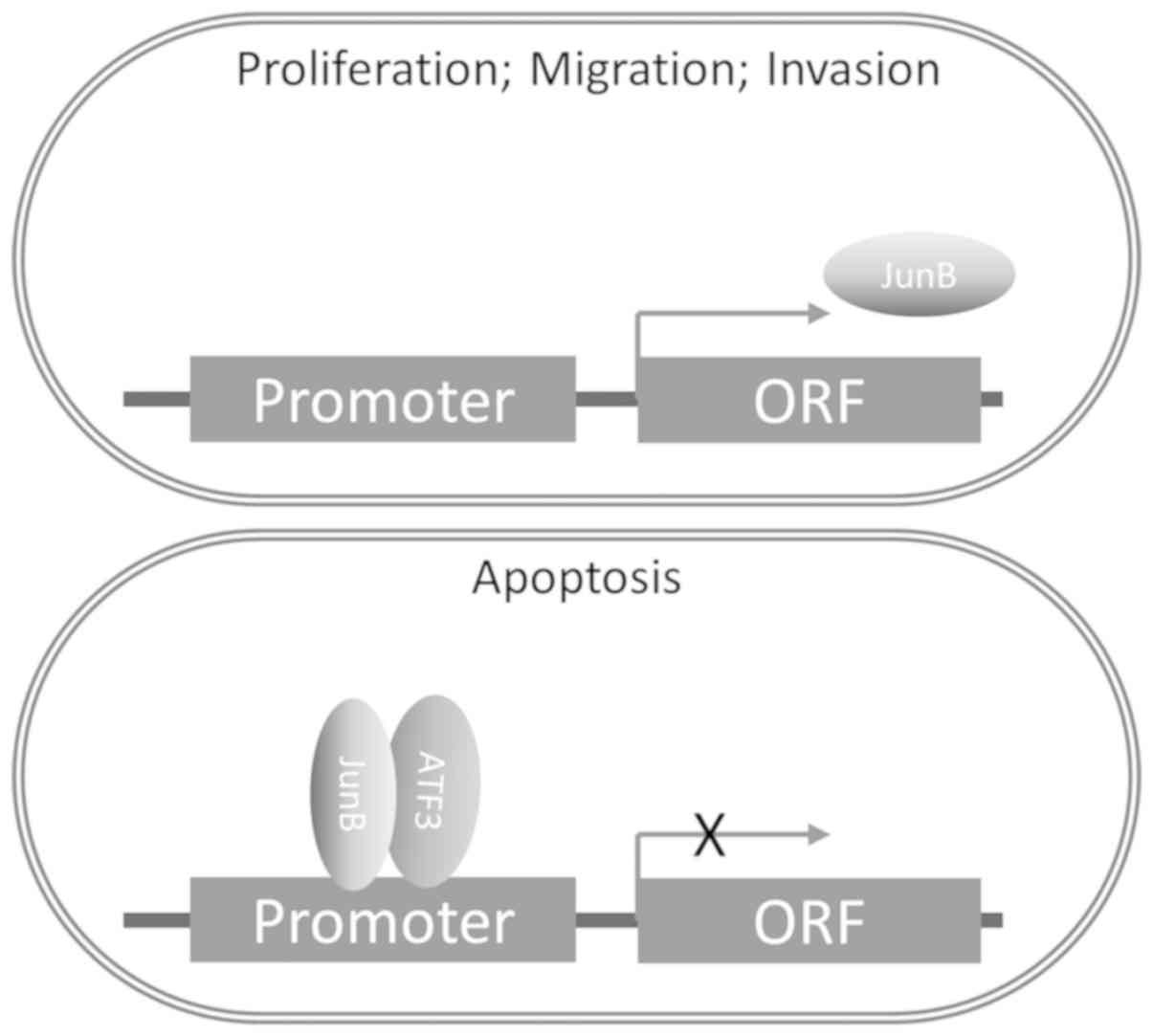
In the present study, it was demonstrated that ATF3
was able to impair the function of AP-1 and JunB to regulate MMP by
forming a complex with JunB. In addition, ATF3 suppressed the
transcription and translation of JunB (Fig. 6). To the best of our knowledge,
this is a novel mechanism regarding the regulation of the function
of JunB in carcinomas. In addition, the minimum concentration
requirements for the interaction between ATF3 and JunB were
examined, and it was speculated that the possible reason as to why
ATF3 did not bind to JunB in native ECs may be the low
concentration of ATF3, which impedes the affinity between ATF3 and
JunB.
Similar to JunB, ATF3 is a member of the AP-1
family, but the role of ATF3 in tumors is more complex and its
influence on cells is context-dependent (37). ATF3 regulates gene expression in
various conditions via binding to the different ATF3/cyclic
adenosine monophosphate-responsive element-binding protein
cis-regulatory elements (38-40).
Moreover, the expression of ATF3 is activated by transforming
growth factor-β binding to its receptor, resulting in the
phosphorylation of SMAD3. The stress signal, acting via p38 kinase,
is also able to induce ATF3 expression (41).
ATF3 can inhibit apoptosis induced by stimuli in
cardiac myocytes (42), but it
also induces apoptosis in tumors (16). Moreover, in varying tumor types,
the functions of ATF3 may be different or even the opposite. Under
certain circumstances, ATF3 has been shown to promote oncogenesis
and migration in skin, breast, prostate and colon cancer (17,43-46).
ATF3 expression may be associated with increased metastasis in
melanoma and in breast cancer cells (47,48),
and knockdown of ATF3 reduces the ability of HT29 colon cancer
cells to invade (46), suggesting
that ATF3 acts as a cancer promoter. However, previous studies on
ATF3 have also revealed a tumor suppressor role in colon, breast,
prostate, bladder, glioblastoma and lung cancer by inhibiting cell
proliferation and metastasis, and promoting cell death and
apoptosis (47-53). Furthermore, a previous study
reported that ATF3 may function as a tumor suppressor in
Ras-mediated tumorigenesis (54).
It has been suggested that the various and context-dependent roles
of ATF3 in cancer are due to the complex ATF3-associated
protein-protein interaction networks. Thus, ATF3 may be capable of
interacting with numerous important and critical proteins to
regulate their functions, in addition to its transcriptional
regulation (55).
The ATF3/JunB interaction was previously identified
to exert an inhibitory effect on human prostate cancer (45). In line with this finding, the
present results demonstrated that ATF3 was able to inhibit the
progression, invasion and metastasis of ECs via binding to the AP-1
sites in the promoters of various genes, including JunB and MMPs.
However, it remains elusive whether ATF3, independent of the
ATF3/JunB complex, is able to help degrade MMPs via a
proteasome-dependent pathway, as has been reported in esophageal
cancer (56). The present findings
may explain why ECs lost their transmigration and invasion ability
after ATF3 overexpression, as the ratio of MMPs/TIMPs, regulated by
ATF3, decrease significantly. Moreover, these results are in line
with a previous study, which revealed that ATF3 was able to reduce
the ability of human glioblastoma to migrate by regulating MMPs and
TIMPs (21).
In our previous study, Y2H mating experiments of
ATF3 were performed and >274 interactions were identified in
human healthy ovary tissues, of which JunB is one of the most
positive interacting partners of ATF3 (data not yet published).
This finding is consistent with the result of the present study and
the earlier studies, which demonstrated that the Fos/Jun and
ATF/cAMP response element binding protein families of transcription
factors function in dimerization (57), which is mediated by the
'leucinezipper' motif, and bind to DNA to alter the expression of
specific target genes. While the interaction between ATF3 and JunB
has been revealed in other tissues or cell types, to the best of
our knowledge, the present study was the first to assess this
interaction in ECs. The results suggested that the inhibition of
ECs by ATF3 may be used as a novel therapeutic strategy for EC.
In conclusion, analysis of EC tissue samples from 50
cases identified a weak expression of ATF3, but upregulation of
JunB expression in EC tissues. It was found that overexpression of
ATF3 resulted in downregulation of JunB expression. Further
investigation demonstrated that the ATF3/JunB complex bound to the
promoter and inhibited the expression of JunB, thus resulting in
the remodeling of EC cells and mitigated developments of EC in
vitro and in vivo. Therefore, JunB may serve as a
potential therapeutic target for EC, and ATF3 as an effective
therapeutic tool.
Supplementary Data
Acknowledgments
The authors would like to thank Dr Hao Ye (School of
Pharmacy, Shanghai Jiao Tong University) for his technical
assistance.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81502233 and 31770922), the
Science and Technology Commission of Shanghai Municipality (grant
no. 134119a8100) and the Young Scientific Research Project of
Shanghai Municipal Health Bureau (grant no. 20134Y176).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
FW, JG, HW and JL conducted the experiments and
analyzed the results. FW and JG conceived the experiments. FZ and
JG supervised the project and interpreted the data. FW, FZ and JG
wrote the article. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The protocol (approval no. 0101N16032) of the
present study was approved by the Human Investigation Ethical
Committee of Shanghai First People's Hospital Affiliated Shanghai
Jiao Tong University (Shanghai, China) and Renji Hospital
Affiliated to Shanghai Jiao Tong University School of Medicine
(Shanghai, China). All samples were obtained from patients who had
provided written informed consent for the use of their tissues for
the purposes of research after the operation. The animal experiment
was in accordance with the recommendations in the Guidelines for
the Care and Use of Laboratory Animals of China (http://www.nsfc.gov.cn/nsfc/cen/pfzl/pufanew/20110801_02.pdf).
The protocol (approval no. 2013020346) was approved by the
Committee on the Ethics of Animal Experiments of the School of
Pharmacy of Shanghai Jiao Tong University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clini. 69:7–34. 2019. View Article : Google Scholar
|
|
2
|
Sorosky JI: Endometrial cancer. Obstet
Gynecol. 120:383–397. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Salvesen HB, Haldorsen IS and Trovik J:
Markers for individualised therapy in endometrial carcinoma. Lancet
Oncol. 13:e353–e361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dedes KJ, Wetterskog D, Ashworth A, Kaye
SB and Reis-Filho JS: Emerging therapeutic targets in endometrial
cancer. Nature reviews Clin Oncol. 8:261–271. 2011. View Article : Google Scholar
|
|
5
|
Gressel GM, Parkash V and Pal L:
Management options and fertility-preserving therapy for
premenopausal endometrial hyperplasia and early-stage endometrial
cancer. Int J Gynaecol Obstet. 131:234–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brooks RA, Fleming GF, Lastra RR, Lee NK,
Moroney JW, Son CH, Tatebe K and Veneris JL: Current
recommendations and recent progress in endometrial cancer. CA
Cancer J Clin. 69:258–279. 2019.PubMed/NCBI
|
|
7
|
Wang D, Zheng W, Wang SM, Wang JB, Wei WQ,
Liang H, Qiao YL and Boffetta P: Estimation of cancer incidence and
mortality attributable to overweight, obesity, and physical
inactivity in China. Nutr Cancer. 64:48–56. 2012. View Article : Google Scholar
|
|
8
|
Lee YC, Lheureux S and Oza AM: Treatment
strategies for endometrial cancer: Current practice and
perspective. Curr Opin Obstet Gynecol. 29:47–58. 2017. View Article : Google Scholar
|
|
9
|
Bokhman JV: Two pathogenetic types of
endometrial carcinoma. Gynecol Oncol. 15:10–17. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lax SF, Kendall B, Tashiro H, Slebos RJ
and Hedrick L: The frequency of p53, K-ras mutations, and
microsatellite instability differs in uterine endometrioid and
serous carcinoma: Evidence of distinct molecular genetic pathways.
Cancer. 88:814–824. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Norimatsu Y, Ohsaki H, Yanoh K, Kawanishi
N and Kobayashi TK: Expression of immunoreactivity of nuclear
findings by p53 and cyclin a in endometrial cytology: Comparison
with endometrial glandular and stromal breakdown and endometrioid
adenocarcinoma grade 1. Diagn Cytopathol. 41:303–307. 2013.
View Article : Google Scholar
|
|
12
|
Yan C, Lu D, Hai T and Boyd DD: Activating
transcription factor 3, a stress sensor, activates p53 by blocking
its ubiquitination. EMBO J. 24:2425–2435. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wei S, Wang H, Lu C, Malmut S, Zhang J,
Ren S, Yu G, Wang W, Tang DD and Yan C: The activating
transcription factor 3 protein suppresses the oncogenic function of
mutant p53 proteins. J Biol Chem. 289:8947–8959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Corrigendum to 'Revised FIGO staging for
carcinoma of the cervix uteri' (Int J Gynecol Obstet 145(2019)
129-135). Int J Gynecol Obstet. 147:279–280. 2019. View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
16
|
Song HM, Park GH, Eo HJ and Jeong JB:
Naringenin-mediated ATF3 expression contributes to apoptosis in
human colon cancer. Biomol Ther (Seoul). 24:140–146. 2016.
View Article : Google Scholar
|
|
17
|
Wu ZY, Wei ZM, Sun SJ, Yuan J and Jiao SC:
Activating transcription factor 3 promotes colon cancer metastasis.
Tumour Biol. 35:8329–8334. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Zhou X, Li Y, Zu L, Pan H, Liu B,
Shen W, Fan Y and Zhou Q: Activating transcription factor 3
promotes malignance of lung cancer cells in vitro. Thorac Cancer.
8:181–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fan TJ, Han LH, Cong RS and Liang J:
Caspase family proteases and apoptosis. Acta Biochim Biophys Sin
(Shanghai). 37:719–727. 2005. View Article : Google Scholar
|
|
20
|
Inoue M, Uchida Y, Edagawa M, Hirata M,
Mitamura J, Miyamoto D, Taketani K, Sekine S, Kawauchi J and
Kitajima S: The stress response gene ATF3 is a direct target of the
Wnt/β-catenin pathway and inhibits the invasion and migration of
HCT116 human colorectal cancer cells. PLoS One. 13:e01941602018.
View Article : Google Scholar
|
|
21
|
Guenzle J, Wolf LJ, Garrelfs NW, Goeldner
JM, Osterberg N, Schindler CR, Saavedra JE and Weyerbrock A: ATF3
reduces migration capacity by regulation of matrix
metalloproteinases via NFkappaB and STAT3 inhibition in
glioblastoma. Cell Death Discov. 3:170062017. View Article : Google Scholar
|
|
22
|
Yang HK, Jeong KC, Kim YK and Jung ST:
Role of matrix metalloproteinase (MMP) 2 and MMP-9 in soft tissue
sarcoma. Clin Orthop Surg. 6:443–454. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jujo T, Sakao S, Tsukahara M, Kantake M,
Maruoka M, Tanabe N, Masuda M and Tatsumi K: The role of matrix
metalloproteinase in the intimal sarcoma-like cells derived from
endarterectomized tissues from a chronic thromboembolic pulmonary
hypertension patient. PLoS One. 9:e874892014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Clark IM, Swingler TE, Sampieri CL and
Edwards DR: The regulation of matrix metalloproteinases and their
inhibitors. Int J Biochem Cell Biol. 40:1362–1378. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Page-McCaw A, Ewald AJ and Werb Z: Matrix
metalloproteinases and the regulation of tissue remodelling. Nature
reviews Mol Cell Biol. 8:221–233. 2007. View Article : Google Scholar
|
|
26
|
Wang Z, Kim J, Teng Y, Ding HF, Zhang J,
Hai T, Cowell JK and Yan C: Loss of ATF3 promotes hormone-induced
prostate carcinogenesis and the emergence of CK5(+)CK8(+)
epithelial cells. Oncogene. 35:3555–3564. 2016. View Article : Google Scholar
|
|
27
|
Papoudou-Bai A, Goussia A, Batistatou A,
Stefanou D, Malamou-Mitsi V and Kanavaros P: The expression levels
of JunB, JunD and p-c-Jun are positively correlated with tumor cell
proliferation in diffuse large B-cell lymphomas. Leuk Lymphoma.
57:143–150. 2016. View Article : Google Scholar
|
|
28
|
Bamberger AM, Milde-Langosch K, Rossing E,
Goemann C and Loning T: Expression pattern of the AP-1 family in
endometrial cancer: Correlations with cell cycle regulators. J
Cancer Res Clin Oncol. 127:545–550. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gong C, Shen J, Fang Z, Qiao L, Feng R,
Lin X and Li S: Abnormally expressed JunB transactivated by
IL-6/STAT3 signaling promotes uveal melanoma aggressiveness via
epithelial-mesenchymal transition. Biosci Rep. 38:BSR201805322018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan F, Bashari MH, Morelli E, Tonon G,
Malvestiti S, Vallet S, Jarahian M, Seckinger A, Hose D, Bakiri L,
et al: The AP-1 transcription factor JunB is essential for multiple
myeloma cell proliferation and drug resistance in the bone marrow
microenvironment. Leukemia. 31:1570–1581. 2017. View Article : Google Scholar
|
|
31
|
Sundqvist A, Morikawa M, Ren J, Vasilaki
E, Kawasaki N, Kobayashi M, Koinuma D, Aburatani H, Miyazono K,
Heldin CH, et al: JUNB governs a feed-forward network of TGFβ
signaling that aggravates breast cancer invasion. Nucleic Acids
Res. 46:1180–1195. 2018. View Article : Google Scholar
|
|
32
|
Sun Y, Wang J, Pan S, Yang T, Sun X, Wang
Y, Shi X, Zhao X, Guo J and Zhang X: LINC00657 played oncogenic
roles in esophageal squamous cell carcinoma by targeting miR-615-3p
and JunB. Biomed Pharmacother. 108:316–324. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hyakusoku H, Sano D, Takahashi H, Hatano
T, Isono Y, Shimada S, Ito Y, Myers JN and Oridate N: JunB promotes
cell invasion, migration and distant metastasis of head and neck
squamous cell carcinoma. J Exp Clin Cancer Res. 35:62016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pei H, Guo Z, Wang Z, Dai Y, Zheng L, Zhu
L, Zhang J, Hu W, Nie J, Mao W, et al: RAC2 promotes abnormal
proliferation of quiescent cells by enhanced JUNB expression via
the MAL-SRF pathway. Cell Cycle. 17:1115–1123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rao GN, Katki KA, Madamanchi NR, Wu Y and
Birrer MJ: JunB forms the majority of the AP-1 complex and is a
target for redox regulation by receptor tyrosine kinase and G
protein-coupled receptor agonists in smooth muscle cells. J Biol
Chem. 274:6003–6010. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kanno T, Kamba T, Yamasaki T, Shibasaki N,
Saito R, Terada N, Toda Y, Mikami Y, Inoue T, Kanematsu A, et al:
JunB promotes cell invasion and angiogenesis in VHL-defective renal
cell carcinoma. Oncogene. 31:3098–3110. 2012. View Article : Google Scholar
|
|
37
|
Rohini M, Haritha Menon A and Selvamurugan
N: Role of activating transcription factor 3 and its interacting
proteins under physiological and pathological conditions. Int J
Biol Macromol. 120:310–317. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen BP, Liang G, Whelan J and Hai T: ATF3
and ATF3 delta Zip. Transcriptional repression versus activation by
alternatively spliced isoforms. J Biol Chem. 269:15819–15826.
1994.PubMed/NCBI
|
|
39
|
Hashimoto Y, Zhang C, Kawauchi J, Imoto I,
Adachi MT, Inazawa J, Amagasa T, Hai T and Kitajima S: An
alternatively spliced isoform of transcriptional repressor ATF3 and
its induction by stress stimuli. Nucleic Acids Res. 30:2398–2406.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai Y, Zhang C, Nawa T, Aso T, Tanaka M,
Oshiro S, Ichijo H and Kitajima S: Homocysteine-responsive ATF3
gene expression in human vascular endothelial cells: Activation of
c-Jun NH(2)-terminal kinase and promoter response element. Blood.
96:2140–2148. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Y, Li Z, Zhang C, Li P, Wu Y, Wang C,
Bond Lau W, Ma XL and Du J: Cardiac Fibroblast-specific activating
transcription factor 3 protects against heart failure by
suppressing MAP2K3-p38 signaling. Circulation. 135:2041–2057. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nobori K, Ito H, Tamamori-Adachi M, Adachi
S, Ono Y, Kawauchi J, Kitajima S, Marumo F and Isobe M: ATF3
inhibits doxorubicin-induced apoptosis in cardiac myocytes: A novel
cardioprotective role of ATF3. J Mol Cell Cardiol. 34:1387–1397.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu X, Nguyen BC, Dziunycz P, Chang S,
Brooks Y, Lefort K, Hofbauer GF and Dotto GP: Opposing roles for
calcineurin and ATF3 in squamous skin cancer. Nature. 465:368–372.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yin X, Wolford CC, Chang YS, McConoughey
SJ, Ramsey SA, Aderem A and Hai T: ATF3, an adaptive-response gene,
enhances TGF{beta} signaling and cancer-initiating cell features in
breast cancer cells. J Cell Sci. 123:3558–3565. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu W, Iiizumi-Gairani M, Okuda H,
Kobayashi A, Watabe M, Pai SK, Pandey PR, Xing F, Fukuda K, Modur
V, et al: KAI1 gene is engaged in NDRG1 gene-mediated metastasis
suppression through the ATF3-NFkappaB complex in human prostate
cancer. J Biol Chem. 286:18949–18959. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ishiguro T, Nagawa H, Naito M and Tsuruo
T: Inhibitory effect of ATF3 antisense oligonucleotide on ectopic
growth of HT29 human colon cancer cells. Jpn J Cancer Res.
91:833–836. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ishiguro T, Nakajima M, Naito M, Muto T
and Tsuruo T: Identification of genes differentially expressed in
B16 murine melanoma sublines with different metastatic potentials.
Cancer Res. 56:875–879. 1996.PubMed/NCBI
|
|
48
|
Iyengar P, Combs TP, Shah SJ, Gouon-Evans
V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C,
Lisanti MP, et al: Adipocyte-secreted factors synergisti-cally
promote mammary tumorigenesis through induction of anti-apoptotic
transcriptional programs and proto-oncogene stabilization.
Oncogene. 22:6408–6423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gargiulo G, Cesaroni M, Serresi M, de
Vries N, Hulsman D, Bruggeman SW, Lancini C and van Lohuizen M: In
vivo RNAi screen for BMI1 targets identifies TGF-beta/BMP-ER stress
pathways as key regulators of neural- and malignant glioma-stem
cell homeostasis. Cancer Cell. 23:660–676. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yuan X, Yu L, Li J, Xie G, Rong T, Zhang
L, Chen J, Meng Q, Irving AT, Wang D, et al: ATF3 suppresses
metastasis of bladder cancer by regulating gelsolin-mediated
remodeling of the actin cytoskeleton. Cancer Res. 73:3625–3637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hackl C, Lang SA, Moser C, Mori A,
Fichtner-Feigl S, Hellerbrand C, Dietmeier W, Schlitt HJ, Geissler
EK and Stoeltzing O: Activating transcription factor-3 (ATF3)
functions as a tumor suppressor in colon cancer and is up-regulated
upon heat-shock protein 90 (Hsp90) inhibition. BMC Cancer.
10:6682010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jan YH, Tsai HY, Yang CJ, Huang MS, Yang
YF, Lai TC, Lee CH, Jeng YM, Huang CY, Su JL, et al: Adenylate
kinase-4 is a marker of poor clinical outcomes that promotes
metastasis of lung cancer by downregulating the transcription
factor ATF3. Cancer Res. 72:5119–5129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bandyopadhyay S, Wang Y, Zhan R, Pai SK,
Watabe M, Iiizumi M, Furuta E, Mohinta S, Liu W, Hirota S, et al:
The tumor metastasis suppressor gene Drg-1 down-regulates the
expression of activating transcription factor 3 in prostate cancer.
Cancer Res. 66:11983–11990. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lu D, Wolfgang CD and Hai T: Activating
transcription factor 3, a stress-inducible gene, suppresses
Ras-stimulated tumorigenesis. J Biol Chem. 281:10473–10481. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Buganim Y, Madar S, Rais Y, Pomeraniec L,
Harel E, Solomon H, Kalo E, Goldstein I, Brosh R, Haimov O, et al:
Transcriptional activity of ATF3 in the stromal compartment of
tumors promotes cancer progression. Carcinogenesis. 32:1749–1757.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xie JJ, Xie YM, Chen B, Pan F, Guo JC,
Zhao Q, Shen JH, Wu ZY, Wu JY, Xu LY and Li EM: ATF3 functions as a
novel tumor suppressor with prognostic significance in esophageal
squamous cell carcinoma. Oncotarget. 5:8569–8582. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gazon H, Barbeau B, Mesnard JM and
Peloponese JM Jr: Hijacking of the AP-1 signaling pathway during
development of ATL. Front Microbiol. 8:26862017. View Article : Google Scholar
|