Introduction
Non-small cell lung cancer (NSCLC) is the most
common histological subtype of lung cancer, accounting for
approximately 80% of all lung cancer (1). Erlotinib remarkably prolongs the
survival time of NSCLC patients with epidermal growth factor
receptor (EGFR) mutations, yet following 8-16 months, treatment is
often accompanied by drug resistance and disease progression
(2). To date, several resistance
mechanisms have been explored, including the secondary T790M
mutation in EGFR, small cell transition, MET or human epidermal
growth factor receptor 2 (HER2) amplification (3), and epithelial-mesenchymal transition
(EMT) (4). However, as these
events do not adequately elucidate for all NSCLC cases (5), the strategies to overcome erlotinib
resistance deserve further study.
Guanylate-binding protein-1 (GBP1) is a
guanosine-5′-tri-phosphate-binding protein in the dynamin
superfamily, and regulates multiple cell functions (6). A report showed that GBP1 modulated
the migration and invasion of oral cavity squamous carcinoma cells
in vitro (7). Yamakita
et al, discovered that GBP1 promoted lung adenocarcinoma
invasiveness (8). GBP1 was also
reported to be related to paclitaxel resistance in ovarian cancer
cell lines (9). Therefore GBP1 is
associated with tumor progression and chemotherapy drug resistance.
However, the relationship between GBP1 and epidermal growth factor
receptor tyrosine kinase inhibitor (EGFR-TKI) resistance is
unclear. In the present study, we found that GBP1 was expressed at
higher levels in resistant cells than in sensitive cells, thus we
further aimed to ascertain whether GBP1 plays a role in erlotinib
resistance.
To investigate the role of GBP1, we used a small
hairpin RNA (shRNA) targeting GBP1 to knock down GBP1 expression in
erlotinib-resistant cells. Cell Counting Kit-8 (CCK-8) experiment
showed that erlotinib-resistant cells were more sensitive to
erlotinib after GBP1 knockdown. Then, we overexpressed GBP1 in
erlotinib-sensitive cells by transfected with an overexpression
plasmid and found that upregulation of GBP1 contributed to a higher
erlotinib IC50 than in control cells. Our data showed
that GBP1 overexpression inhibited apoptosis and caused a G1 to S
phase transition, and the opposite effects were observed in
GBP1-knockdown cells. In addition, we confirmed that GBP1 knockdown
in resistant cells markedly reduced the tumor volume in
vivo. In addition, high GBP1 expression predicted poor
prognosis in a survival analysis of clinical samples. The above
results indicated that GBP1 promotes erlotinib resistance.
To further study the mechanism by which GBP1
regulates EGFR-TKI resistance, we performed mass spectrometry
analysis of GBP1 in cells. After screening by mass spectrometry, we
identified PGK1 as a GBP1-interacting protein. PGK1, an important
metabolic enzyme, is overexpressed in many cancers, such as
glioblastoma (10) and
triple-negative breast cancer (11). Moreover, PGK1 promotes the
migration and invasion of NSCLC cells via the AKT/mTOR signaling
pathway (12). Here, we found that
PGK1 is involved in erlotinib resistance in NSCLC by activating the
EMT pathway, causing the loss of epithelial markers and the gain of
mesenchymal markers in cancer cells. Subsequently, our results
showed that GBP1 modulates erlotinib resistance through EMT
mediated by PGK1.
In summary we found that GBP1 caused resistance to
erlotinib. The knockdown of GBP1 expression significantly
sensitized NSCLC cells to erlotinib in vitro and in
vivo. Our discoveries provide support for the feasibility of
targeting GBP1 to overcome erlotinib resistance.
Materials and methods
Differential gene selection
Firstly, we selected 3 data-sets concerning human
NSCLC samples from the Gene Expression Omnibus (GEO) database
(http://www.ncbi.nlm.nih.gov/geo/), in
which GSE64322 (http://www.ncbi.nlm.nih.gov/geo/qyery/acc.cgi?acc=GSE64322)
(13) showed molecular changes
between EGFR-TKI-sensitive and EGFR-TKI-resistant NSCLCs by
transforming to NSCLC cells, GSE38310 (http://www.ncbi.nlm.nih.gov/geo/qyery/acc.cgi?acc=GSE38310)
(14) displays gene expression
differences between HCC827 and HCC827ER cells and GSE34228
(http://www.ncbi.nlm.nih.gov/geo/qyery/acc.cgi?acc=GSE34228)
(15) contains genome expression
differences of PC9 and PC9ER cells. Then, the R limma package
(16) was used to determine which
genes were different in the various gene sets. The results
indicated that GBP1, TGM2, PAPPA, HAS3, CCD2A, HMGA2, INSL3,
LDB3, PMIL and SLC35B3 were significantly differentially
expressed between EGFR-TKI-resistant and EGFR-TKI-sensitive lung
cancer cell lines, based with a P value <0.05 and |log2(FC)|
value >0.50. Then the relationship between the above genes and
erlotinib resistance was explored.
Reagents and antibodies
Erlotinib was purchased from Shanghai Anpu
Experiment Technology Co., dissolved in DMSO and maintained at
-20°C. Antibodies against GAPDH (cat. no. 60004-1-lg), BAX (cat.
no. 50599-2-lg), Bcl-2 (cat. no. 12789-1-AP), P21 (cat. no.
60214-1-lg), CDK4 (cat. no. 66950-1-lg), CDK6 (cat. no.
66278-1-lg), Cyclin D1 (cat. no. 60186-1-lg), E-cadherin (cat. no.
60335-1-lg), N-cadherin (cat. no. 66219-1-lg), GBP1 (cat. no.
15303-1-AP) and β-actin (cat. no. 60008-1-lg) were purchased from
ProteinTech Group, Inc. and diluted at 1:500-1,000. Antibodies
against cleaved PARP1 (cat. no. ab32561), Twist (cat. no. ab187008)
and vimentin (cat. no. ab92547) were purchased from Abcam and a
PGK1 (cat. no. sc-130335) antibody was purchased from Santa Cruz
Biotechnology, Inc. All antibodies were diluted 1:500 or
1:1,000.
Establishment of cell lines
The human NSCLC cell lines PC9 (EGFR del19) and
HCC827 (EGFR del19) were purchased from the American Type Culture
Collection (ATCCP and cultured in RPMI-1640 (Gibco; Thermo Fisher
Scientific, Inc.) medium containing 10% or 20% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.). Erlotinib-resistant
cell lines (PC9ER and HCC827ER) were generated via exposure to
gradually increasing doses of erlotinib and were maintained in the
presence of erlotinib after 6 months. In addition, the T790M
mutation was not detected in the PC9ER and HCC827ER cells using a
T790M test kit (HGN-eg02; Wuhan Haijili Biological Technology Co.,
China; Fig. S1A).
shRNA construction and transfection
Firstly, pCDH-CMV- MCS-EF1-copGFP-Puro (4
µg), pLP1 (3 µg), pLP2 (2 µg), and pLP-VSVG (3
µg) were cotransfected into 293T cells, which were purchased
from Addgene Co., and the virus titer was 0.98×108
TU/ml. After PC9ER and HCC827ER cells reached 70% confluence
(3×106 cells), lentiviral particles of GBP1 were used to
transduce cells at a multiplicity of infection (MOI) of 50 in the
presence of polybrene (2 µg/ml; Sigma-Aldrich; Merck KGaA).
Afterwards, shRNA-trans-duced cells were screened with puromycin
(0.5 g/ml; Sigma-Aldrich; Merck KGaA) for 72 h. Finally, the
derived cell clones were successfully cultivated. In overexpression
lentivirus, pGC-FU-3FLAG-SV40-puromycin-library (6 µg),
pHelper1.0 (3 µg) and pHelper2.0 (2 µg) were
cotransfected into 293T cells, which were purchased from Genechem
Co.; the virus titer was 1.5E+9 TU/ml. After collection by
centrif-ugation for 1 h at 30,000 rpm, overexpression lentiviruses
were transduced into erlotinib-resistant cells with 20 MOI and used
in subsequent experiments. The inserted sequences of two shRNAs and
overexpression lentivirus are listed in Fig. S1E.
RT-qPCR
Total RNA was extracted from tumor cells with TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.). The RNA concentration
was detected by measuring the absorbance at 260/280 nm. Then, cDNA
was synthesized with a reverse transcription reagent kit
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. qPCR was performed with SYBR Green PCR
Mater Mix (Takara Bio, Inc.). Finally, PCR was performed, under the
following conditions: 10 min at 95°C, followed by 40 cycles of 10
sec at 94°C, 30 sec at 60°C and 30 sec at 72°C. The results were
analyzed with the 2−ΔΔCq method (17). The sequences of primers for GBP1,
GAPDH and E-cadherin are presented in Fig. S1B.
Drug sensitivity tests
Cell viability was measured by CCK-8 assays
(Beyotime Institute of Biotechnology). Erlotinib-sensitive and
erlotinib-resistant cells were placed into 96-well microplates, and
erlotinib was added after 24 h. After 48 h, CCK-8 reagent was added
to each well. After incubation with CCK-8 reagent at 37°C for 10
min, the absorbance values in the microplate were detected by using
a microplate reader. Three independent experiments were carried
out.
Western blot (WB) analysis
After cells were collected in RIPA buffer, the
protein was analyzed with a Pierce BCA protein assay (cat. no.
23227; Thermo Fisher Scientific, Inc.), and transferred onto
polyvinylidene difluoride membranes (Bio-Rad Laboratories, Inc.).
After blocking, the membranes were probed with primary antibodies
overnight at 4℃. After washing, the membranes were incubated with
goat anti-rabbit immunoglobulin G (IgG) (cat. no. A0208; Beyotime
Institute of Biotechnology) or goat anti-mouse IgG (cat. no. 7074
or 7076; Cell Signaling Technology, Inc.) for 1 h at 37℃. Finally,
an enhanced chemiluminescence detection reagent (Pierce; Thermo
Fisher Scientific, Inc.) was applied to visualize the protein
bands. Antibodies against GAPDH and β-actin served as controls. In
Table SIA-E, the optimal density
value of all WB bands are displayed, in which all comparisons
between the two groups were statistically significant.
Apoptosis and cell cycle detection
After centrifugation for 5 min at 1000 rpm and
resuspension, 2×105 cells were stained with Annexin-V
FITC and propidium iodide (PI) in the dark. Then, stained cells
were resuspended in 400 µl of binding buffer, and the
samples were analyzed with a BD FACSVerse flow cytometer (BD
Biosciences). Finally, the data were analyzed by FlowJo software
v7.6.5 (Treestar Co.). In the cell cycle experiment,
1×106 cells were used and resuspended in ice-cold
phosphate-buffered saline (PBS). Cold 100% ethanol was slowly added
to each tube. Then, the cells were incubated at 4°C overnight.
After centrifugation for 5 min at 800 rpm and washing, the cells
were stained with 5 µl of FITC-conjugated Annexin-V and 5
µl of PI for 1 h and were analyzed. The data were processed
by MODFIT software (Verity Software House).
Mouse xenograft study
Athymic nude mice (4 weeks old, female, n=32, 4 mice
per group) were purchased from Southern Medical University
Experimental Animal Research Center and housed under specific
pathogen-free conditions; 5×106 PC9-overexpressing cells
were subcutaneously injected into the flanks of nude mice on day 1
of the experiment. When the tumors reached 450 mm3 on
day 7, erlotinib was intragastrically administered to 4 groups of
mice once a day for 2 weeks. Tumor volume (Length x
Width2 / 2) and body weight were measured and recorded.
When the largest tumor approached 816 mm3 in the
PC9-NC+PBS group on day 40 after resistant cell inoculation, all
mice were euthanized by cervical dislocation. Next, the tumors were
resected and analyzed by immunohistochemistry (IHC). None of the
animals were found dead. According to humane spirit, the maximum
volume of mice was limited to 4000 mm3. These studies
were conducted in compliance with the guidelines from the Animal
Care and Use Committee of Southern Medical University.
IHC
Tumor tissues were immediately fixed in 4%
paraformaldehyde (Wuhan Boster Biological Technology, Ltd.) and
embedded in paraffin. Samples were carefully sectioned, serially
deparaffinized, rehydrated, and subjected to antigen retrieval.
Monoclonal primary antibodies were incubated with samples for 1 h
at 37℃. Subsequently, after incubation with a suitable HRP-labeled
secondary antibody for 30 min, sections were stained with
diaminobenzidine (OriGene Technologies, Inc.). Finally, sections
were dehydrated and mounted. Meanwhile, the immunostained tumor
cells were scored according to the following evaluation (0,=
negative; 1, weak staining; 2, moderate; 3, strong), and the
results are showed in Table SIIA and
B, in which the comparison between the two groups were
significant at P<0.01.
Immunofluorescence and confocal
microscopy
A total of 5×105 PC9ER or
2×105 HCC827ER cells were fixed with cold methanol for
10 min, permeabilized in 0.2% Triton X-100 for 15 min and blocked
with sheep serum for 30 min. After incubation with primary
antibodies at 4°C overnight, cells were incubated with secondary
antibodies for 1 h at 37°C, and stained with
4′,6-diamino-2-phenyl-indole (DAPI; cat. no. C1005; Beyotime
Institute of Biotechnology). Images were captured on a confocal
microscope with a ×63 objective (TCS SP8; Leica) and analyzed by
Leica application suite software (Leica Microsystems).
Coimmunoprecipitation (CoIP)
For the CoIP experiments, 3×106 PC9ER
cells were lysed in ice-cold Pierce lysis buffer (Thermo Fisher
Scientific, Inc.) for 10 min, followed by centrifugation at 13,000
× g for 10 min. Then, cell lysates were incubated with
antibody-crosslinked magnetic beads (Beyotime Institute of
Biotechnology) for 1 h. Normal IgG served as the control according
to the instruction manual. Beads were washed with lysis buffer, and
protein complexes were eluted in the next step. The input and CoIP
samples were detected by WB analysis with primary antibodies
against a variety of proteins.
Statistical analysis
We used a two-tailed Student's t-test or one-way
ANOVA with GraphPad Prism 7 software (v8; GraphPad Software, Inc.)
for statistical analysis and performed least significance
difference (LSD) to obtain P-value for the one-way ANOVA.
Statistical significance is indicated by P-value with symbols as
defined in the legends (*P<0.05, **P<0.01 and
***P<0.001). Based on the median follow-up in months,
the results of survival analysis were displayed using Kaplan-Meier
survival curves by the log rank test and P<0.05 indicated
statistical significance. In addition, we utilized univariable and
multivariate Cox regression to identify the variables affecting
overall survival (OS) and clinical features in lung adenocarcinoma
(LUAD). Image J software v1.8.0 (National Institutes of Health,
Bethesda, MD, USA) was used to analyze gray value of the WB bands
and the average optimal density of IHC.
Results
GBP1 is upregulated in
erlotinib-resistant cells
First, we screened different genes from datasets in
the GEO database. Then, to investigate whether these genes were
related to erlotinib resistance, we established erlotinib-resistant
PC9 and HCC827 cell lines (PC9ER and HCC827ER) through stepwise
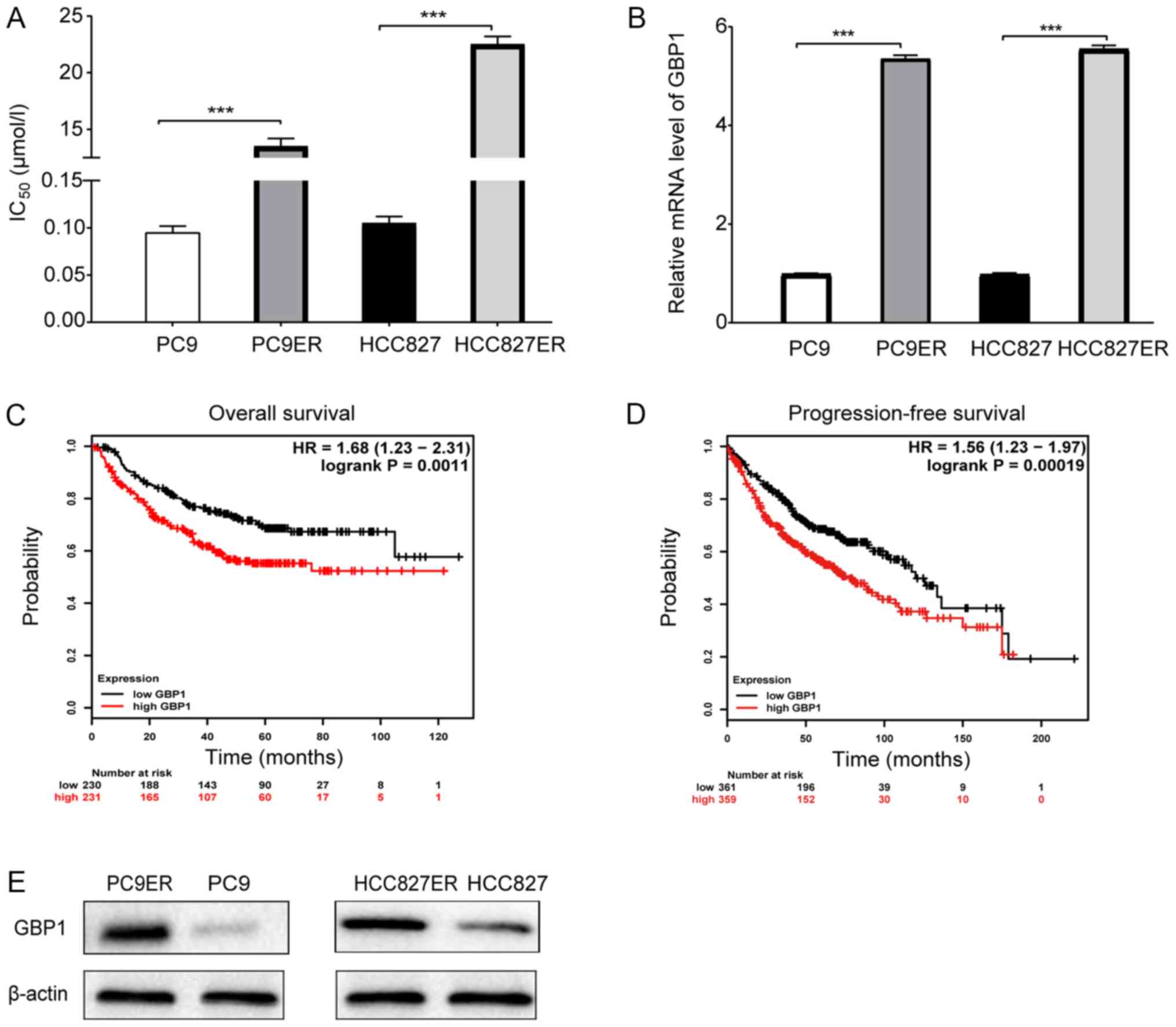
induction. The half maximal inhibitory concentration
(IC50) value of erlotinib was significantly higher in
the erlotinib-resistant cells than that noted in the corresponding
erlotinib-sensitive cells as shown in Fig. 1A. Next, we analyzed the mRNA levels
of these genes in erlotinib-resistant and erlotinib-sensitive
cells. Finally, GBP1 (Fig.
1B) and TGM2 (Fig.
S1C) were selected due to the significant differences in their
expression. However, there have been many studies on TGM2
and drug resistance, thus we chose to focus on GBP1 in this
study. Subsequently, we explored the relationship of these genes
with prognosis through Kaplan-Meier Plotter analysis. As shown in
Fig. 1C and D, patients were
divided into a high-level group and a low-level GBP1 group
according to the median GBP1 expression value (10.4684), and
overall survival (OS) and progression-free survival (PF) were
shorter in the high-level group than in the low-level group among
patients with lung adenocarcinoma. Fig. 1C includes 230 lung adenocarcinoma
patients with low GBP1 expression and 231 patients with high GBP1
expression from The Cancer Genome Atlas (TCGA) database. A total of
620 lung adenocarcinoma patients with PF time data were analyzed,
as shown in Fig. 1D. Moreover, we
found that high GBP1 expression was an independent prognostic
factor of OS in LUAD (Table I),
and related to tumor stage in 234 patients with LUAD from the
Oncomine database (https://www.oncomine.org/resource/login.html; Fig. S1D). In addition, GBP1 protein
levels were differentially expressed in the erlotinib-sensitive and
erlotinib-resistant cells (Fig.
1E). Overall, GBP1 was found to be related to erlotinib
resistance and worthy of further study.
 | Table IUnivariate and multivariate analyses
of overall survival. |
Table I
Univariate and multivariate analyses
of overall survival.
| Features | Overall survival
|
|---|
Univariate analysis
| Multivariate
analysis
|
|---|
| Hazard ratio (95%
CI) | P-value | Hazard ratio (95%
CI) | P-value |
|---|
| Age (years) | | | | |
| <65 | 1 (reference) | | 1 (reference) | |
| ≥65 | 0.773
(0.505-1.181) | 0.233 | 0.613
(0.373-1.009) | 0.054 |
| Sex | | | | |
| Male | 1 (reference) | | 1 (reference) | |
| Female | 1.137
(0.746-1.732) | 0.552 | 0.892
(0.561-1.418) | 0.629 |
| Pathologic
stage | | | | |
| Stage I | 1 (reference) | | 1 (reference) | |
| Stage II | 0.276
(0.126-0.604) | 0.001a | 0.469
(0.191-1.149) | 0.098 |
| Stage III | 0.570
(0.255-1.274) | 0.171 | 0.686
(0.274-1.714) | 0.420 |
| Stage IV | 1.160
(0.518-2.598) | 0.719 | 1.941
(0.740-5.092) | 0.178 |
| Anatomic neoplasm
subdivision | | | | |
| L-upper | 1 (reference) | | 1 (reference) | |
| L-lower | 1.001
(0.482-2.077) | 0.998 | 1.021
(0.468-2.228) | 0.958 |
| R-upper | 0.756
(0.439-1.302) | 0.313 | 0.607
(0.340-1.082) | 0.090 |
| R-lower | 1.390
(0.792-2.438) | 0.251 | 0.786
(0.413-1.496) | 0.463 |
| R-middle | 1.213
(0.286-5.143) | 0.793 | 0.247
(0.050-1.233) | 0.088 |
| Primary therapy
outcome | | | | |
| Progressive
disease | 1 (reference) | | 1 (reference) | |
| Partial
response | 2.331
(1.120-4.851) | 0.024a | 2.703
(1.253-5.830) | 0.011a |
| Complete
response | 0.000
(0.000-1.528E+239) | <0.001a | 0.000
(0.000-1.693E+238) | 0.965 |
| Stable
disease | 0.477
(0.231-0.984) | 0.045a | 1.689
(0.760-3.754) | 0.198 |
| Radiation
therapy | | | | |
| Yes | 1 (reference) | | 1 (reference) | |
| No | 3.136
(2.001-4.915) | <0.001a | 1.535
(0.854-2.760) | 0.152 |
| Neoplasm cancer
status | | | | |
| Tumor-free | 1 (reference) | | 1 (reference) | |
| With tumor | 0.149
(0.090-0.246) | <0.001a | 0.160
(0.087-0.293) | <0.001a |
| GBP1 expression
level | | | | |
| Low
expression | 1 (reference) | | 1 (reference) | |
| High
expression | 1.810
(1.179-2.777) | 0.007a |
2.136(1.303-3.501) | 0.003a |
GBP1 regulates the sensitivity of
erlotinib-resistant cells in vitro
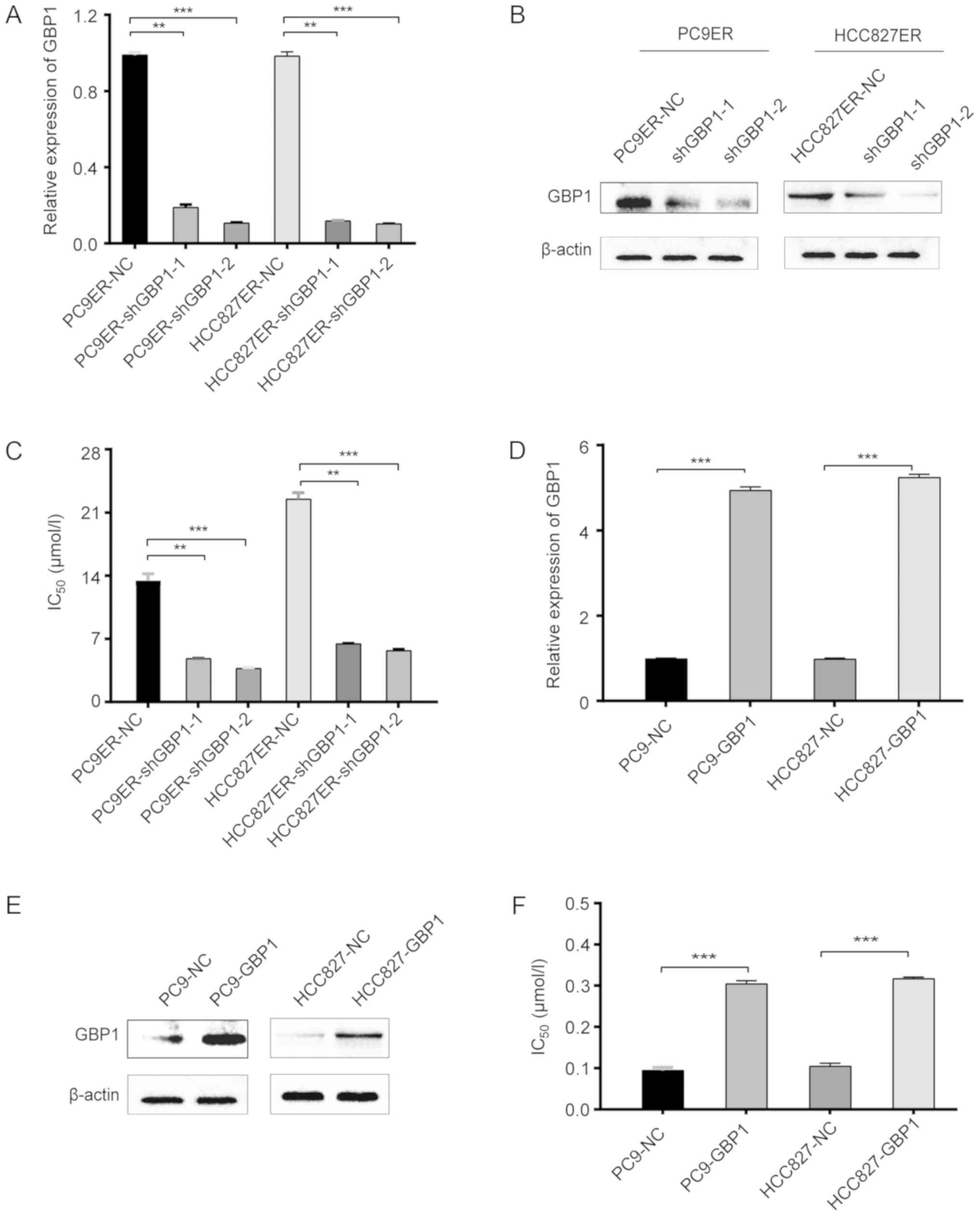
To confirm the role of GBP1, gain- and
loss-of-function experiments were performed. After lentivirus
transfection, shGBP1-transfected PC9ER cells had significantly
lower GBP1 mRNA and protein levels than these levels in the control
cells (Fig. 2A and B). In
addition, Fig. 2C shows that the
IC50 value for erlotinib was several times higher in the
PC9ER-NC cells than this value in the shGBP1-transfected PC9ER
cells. Similar to the results in PC9ER cells, the results in the
HCC827ER cells showed the same trend (Fig. 2A-C). In the overexpression assay,
resistance to erlotinib was higher in the GBP1-overexpressing PC9
(PC9-GBP1) and HCC827 (HCC827-GBP1) cells than in the corresponding
negative control-transfected PC9 (PC9-NC) and HCC827 (HCC827-NC)
cells (Fig. 2D-F), demonstrating
that GBP1 overexpression contributed to erlotinib resistance as
indicated by a notable change in the IC50 of erlotinib.
Based on these data, PC9ER and HCC827ER cells transfected with
shRNA became sensitive to erlotinib, but the overexpression of GBP1
in PC9 and HCC827 cells conferred erlotinib resistance, indicating
that GBP1 regulates erlotinib resistance in vitro.
Knockdown of GBP1 induces cell apoptosis
in erlotinib-resistant cells
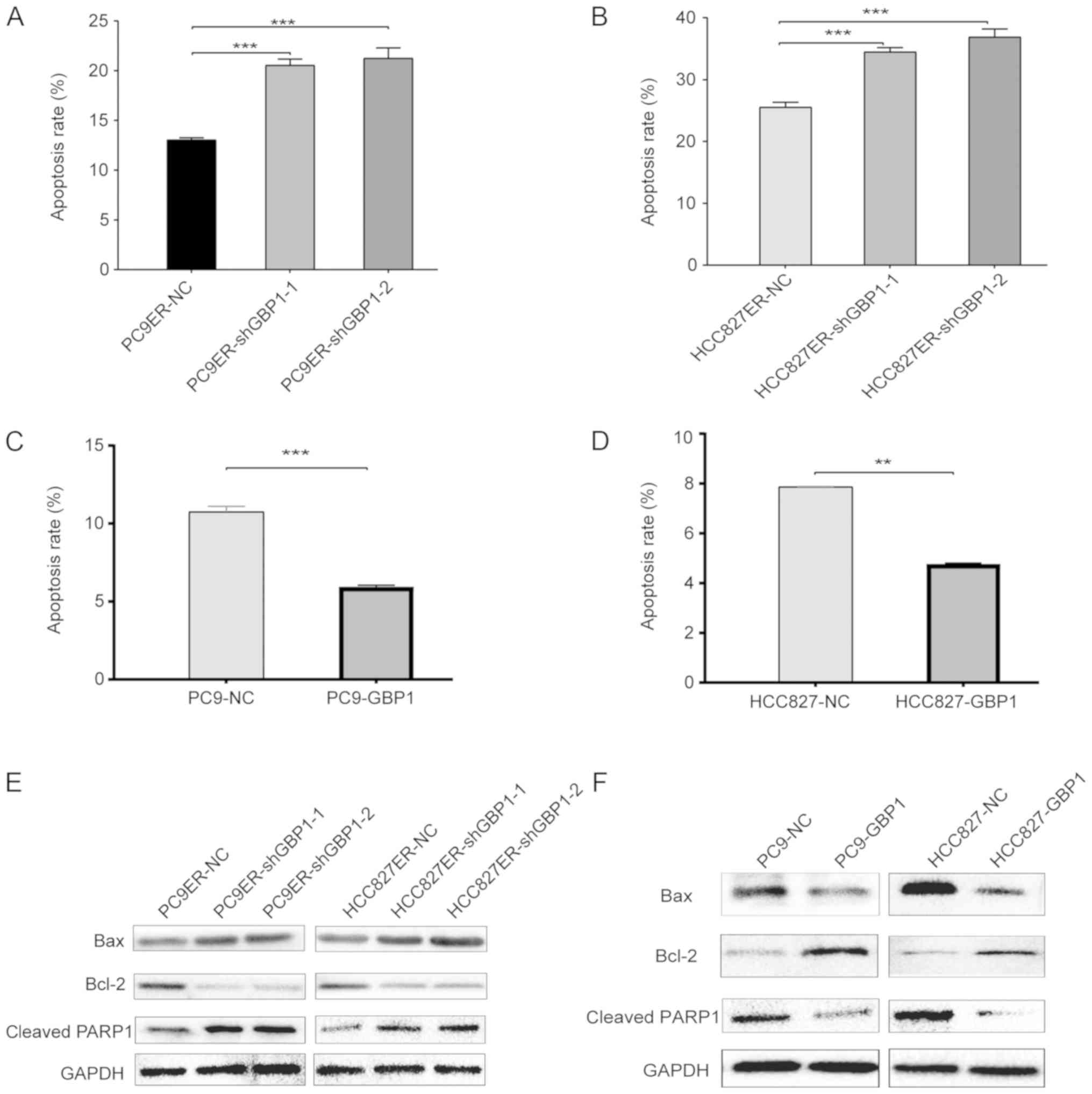
After cells had been treated for 48 h with erlotinib
at a concentration close to its IC50, flow cytometry
analysis revealed that the proportion of late apoptotic cells was
13.1, 20.1 and 20.5% in the PC9ER-NC, PC9ER-shGBP1-1 and
PC9ER-shGBP1-2 cells, respectively (Fig. 3A). Unlike HCC827ER-NC cells, the
ratio of late apoptotic cells among HCC827ER-shGBP1-1 and
HCC827ER-shGBP1-2 cells was 34.5 and 36.9% respectively (Fig. 3B). Moreover, the overexpression of
GBP1 in erlotinib-sensitive cells (PC9-GBP1 and HCC827-GBP1)
decreased the percentage of late apoptotic cells compared with that
in the NC cells (Fig. 3C and D).
Using WB analyses, we determined the levels of the apoptotic
markers, Bax, Bcl-2 and cleaved PARP1. When GBP1 was knocked down
in erlotinib-resistant cells, cleaved PARP1 and Bax protein levels
were increased, and the change in Bcl-2 protein levels showed
opposite trends (Fig. 3E). After
GBP1 overexpression, the opposite effects were observed (Fig. 3F). All of the above results showed
that GBP1 is involved in erlotinib resistance via the apoptotic
pathway.
GBP1 induces a G1 phase transition
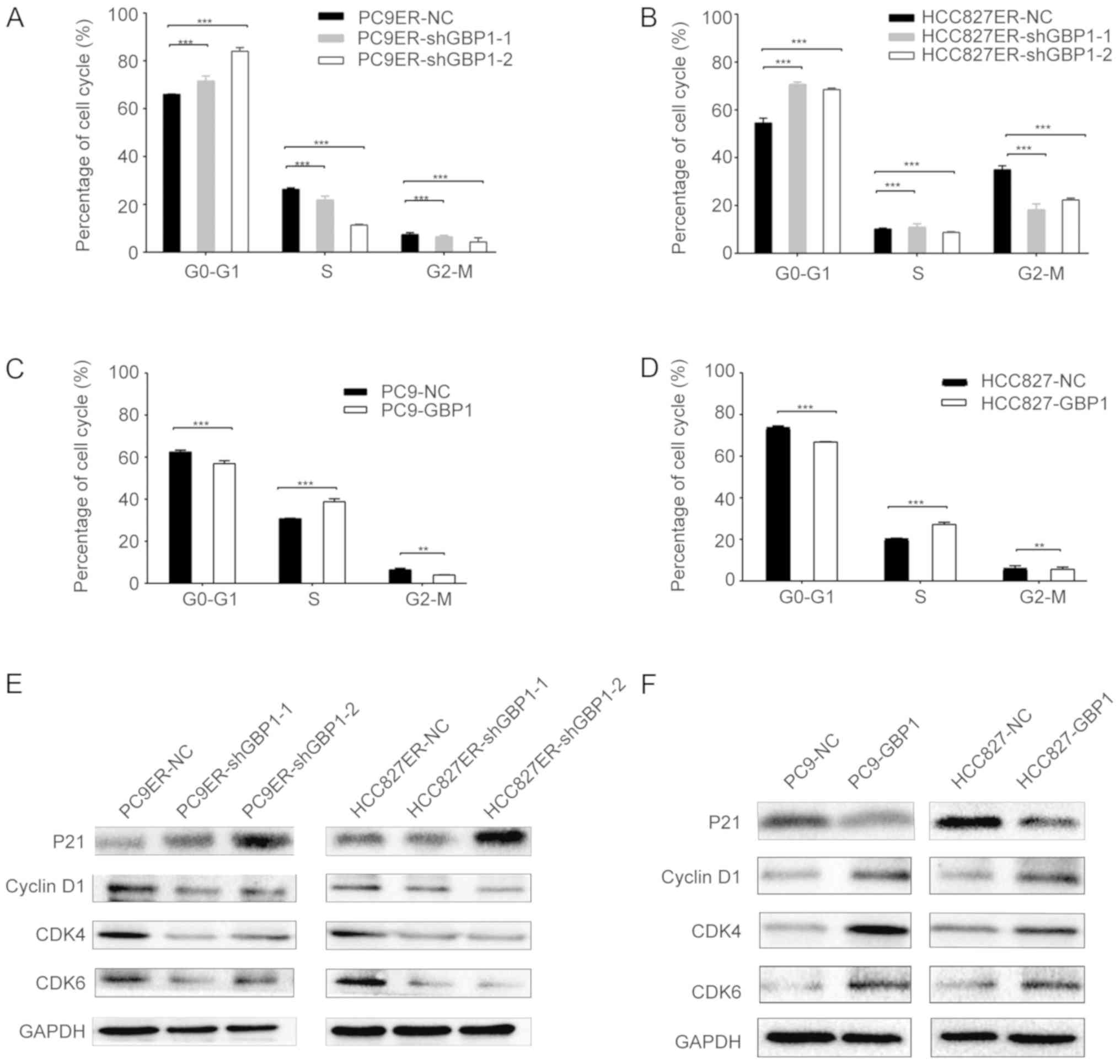
Then, we explored the role of GBP1 in the cell
cycle. By performing flow cytometry analysis, we discovered that
the proportions of shGBP1-1 and shGBP1-2-transfected PC9ER cells in
the G1 phase were 73.1 and 85.1%, respectively (Fig. 4A), while the proportion of PC9-GBP1
cells in the G1 phase was 57.0%, which significantly differed from
the respective control cells (Fig.
4C). There was also a significant difference in the proportion
of shGBP1-transfected HCC827ER cells and control cells in the G1
phase (Fig. 4B), and the
proportion of HCC827-GBP1 cells and control cells in the G1 phase
(Fig. 4D). P21 is an important
regulator of the G1 cell cycle checkpoint (18), and cyclin D1, CDK4 and CDK6
regulate the G0/G1 transition in the cell cycle (19). The P21 protein level was elevated
when GBP1 was downregulated in PC9ER or HCC827ER cells (Fig. 4E), and the other three markers
exhibited an opposite effect. But when GBP1 was overexpressed, the
P21 protein level was decreased (Fig.
4F). All of these results regarding cell cycle proteins showed
that GBP1 regulates the cell cycle and leads to a G1 phase
transition.
GBP1 is associated with erlotinib
resistance in mouse xenografts
To further verify the role of GBP1 in erlotinib
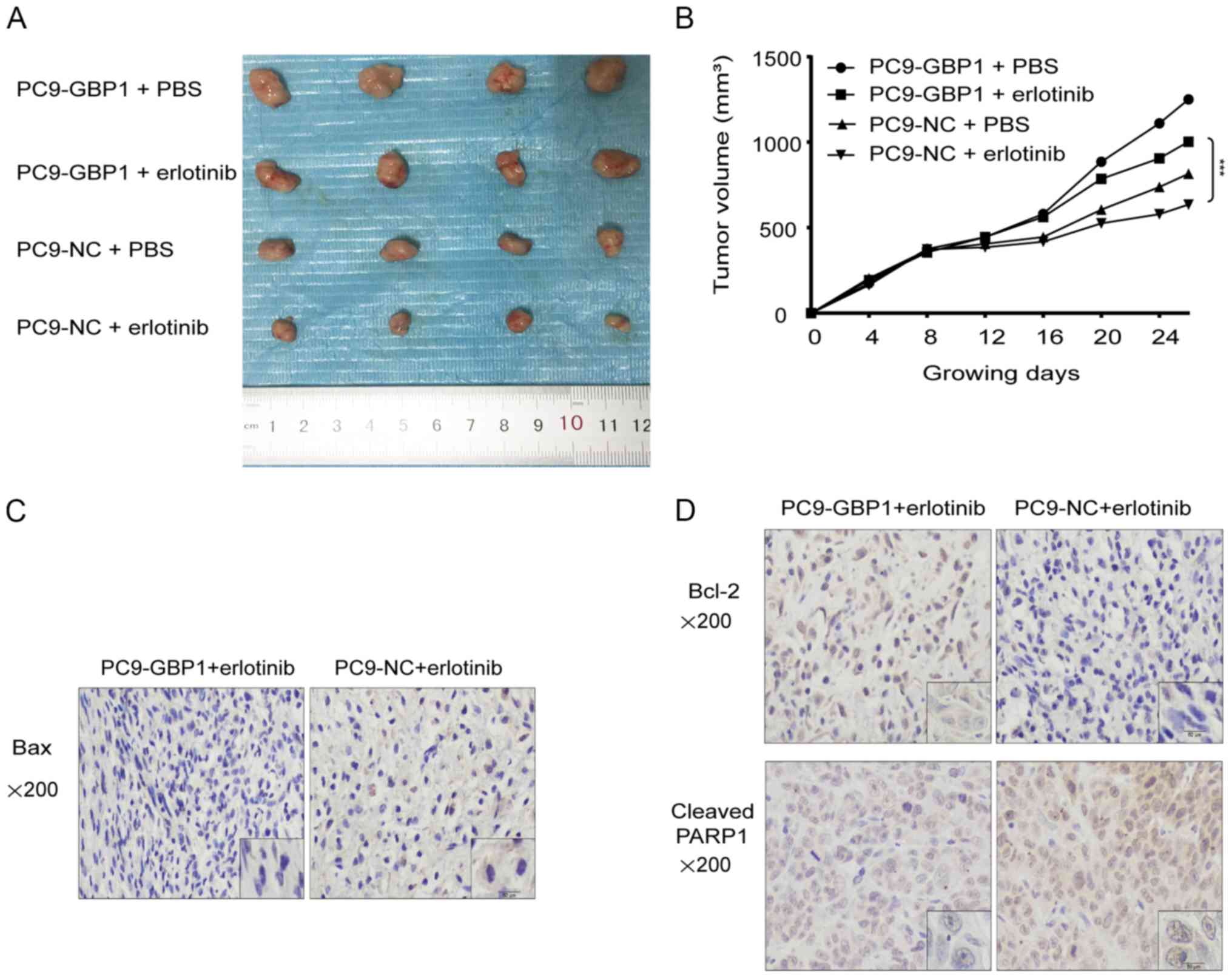
resistance, we performed subcutaneous tumor formation experiments
in nude mice. The tumor size in mice injected with PC9-GBP1 cells
and treated with erlotinib was significantly larger than that in
mice injected with PC9-NC and treated with erlotinib (Fig. 5A and B), which showed that the
overexpression of GBP1 made the tumors resistant to erlotinib.
Overall, the results of animal experiments showed that erlotinib
sensitivity was regulated by either GBP1 knockout or
overexpression, which once again verified the relationship between
GBP1 and erlotinib resistance. In addition, the expression of
apoptotic proteins was examined using IHC (Fig. 5C and D), and the results were
consistent with those of the in vitro experiments. Compared
to PC9-NC + erlotinib, the expression of cleaved PARP1 in PC9-GBP1
+ erlotinib was decreased (Fig.
5D). Overall, the in vivo studies showed that GBP1 can
regulate erlotinib resistance.
GBP1 contributes to erlotinib resistance
by interacting with PGK1
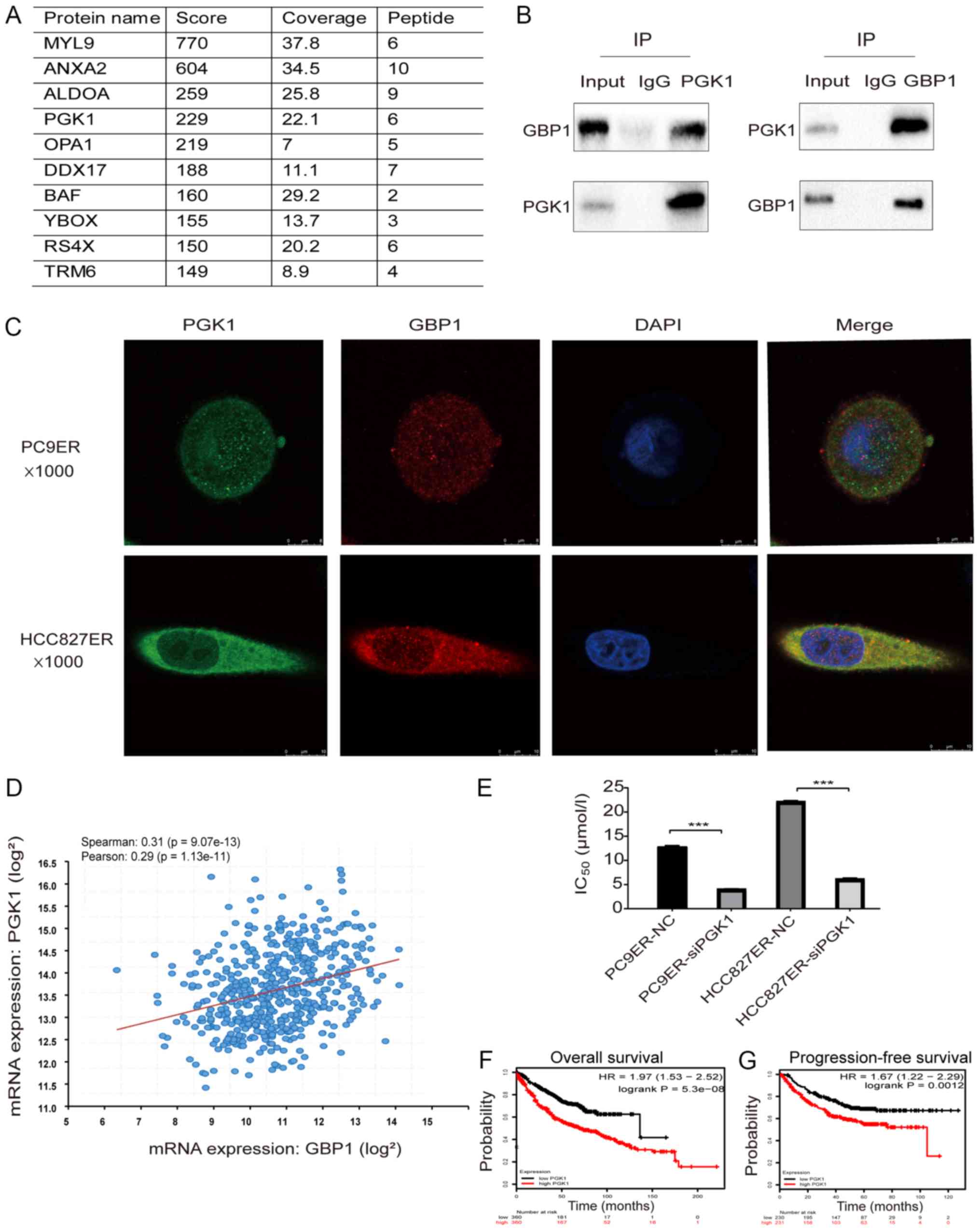
To explore GBP1-interacting proteins, we performed
mass spectrometry, which can predict hundreds of possible
interacting proteins (Fig. 6A).
After analyzing the results, we selected PGK1, myosin light chain 9
(MYL9), annexin A2 (ANXA2) and aldolase A (ALDOA) as our research
objects based on a total score >200 with >5 qualitative
peptides. As PGK1 may be involved in erlotinib resistance (20), we chose PGK1 for further analysis.
As shown in Fig. 6B, CoIP
experiments showed that PGK1 interacted with the GBP1 protein.
Next, the results of immunofluorescence staining showed the
colocalization of GBP1 and PGK1 in the two resistant cell lines
(Fig. 6C). Analysis of the cBio
Cancer Genomics Portal, showed that the mRNA expression level of
PGK1 was positively correlated with that of GBP1 (P<0.05) and
the Spearman correlation coefficient between GBP1 and PGK1
expression in lung adenocarcinoma was 0.31 (Fig. 6D). Taken together, these results
showed that GBP1 interacts with PGK1. To determine whether PGK1
affects erlotinib resistance in NSCLC, we used CCK-8 assays and
found that PGK1 downregulation reduced the IC50 of
erlotinib (Fig. 6E). Finally, the
relationship between PGK1 and prognosis was examined through
Kaplan-Meier Plotter analysis; high PGK1 expression predicted poor
OS and PF in lung adenocarcinoma (Fig.
6F and G). In conclusion, GBP1 regulates erlotinib resistance
via PGK1.
EMT mediated by PGK1 plays an important
role in GBP1-regulated erlotinib resistance
To determine which downstream pathways are involved
in resistance, we examined the literature and found that PGK1 can
interact with HIF-2 to promote EMT in breast cancer (20). In addition, EMT is closely related
to EGFR-TKI resistance in many types of tumors, so we explored the
relationship between GBP1, PGK1 and EMT with erlotinib resistance.
WB data showed that the knockdown of GBP1 induced a marked increase
in E-cadherin levels and decreased N-cadherin and vimentin levels.
However increased PGK1 expression reversed changes in the protein
levels of E-cadherin, N-cadherin and vimentin (Fig. 7A). PGK1 down-regulation after GBP1
overexpression reversed the effect of GBP1 on EMT-related proteins
(Fig. 7B). Additionally, Twist, as
a key factor in EMT, was decreased at the protein level when PGK1
was inhibited using siRNA (Fig.
7C), further supporting that PGK1 regulated EMT. In Fig. 7D, the mRNA level of E-cadherin in
resistant cells was decreased when compared to that in sensitive
cells. In addition, we further analyzed the change in the
IC50 caused by E-cadherin downregulation in
erlotinib-sensitive cells and found that reduced E-cadherin led to
erlotinib resistance (Fig. 7E).
These results indicated that EMT participates in the regulation of
erlotinib resistance. Finally, as shown in Fig. 7F and G, we found that the results
of histochemical staining for PGK1 and EMT proteins were consistent
with those of WB. Moreover, the immunostaining score of PGK1 and
EMT proteins are displayed in Table
SIIA. Changes in protein levels of E-cadherin, N-cadherin and
vimentin indicated again that GBP1 could activate EMT. The above
experiments showed that GBP1 induces erlotinib resistance via the
EMT pathway, which is activated by PGK1 in NSCLC. In the future,
further analysis of the relationship between EMT and erlotinib
resistance through EMT inhibitors or agonists must be
performed.
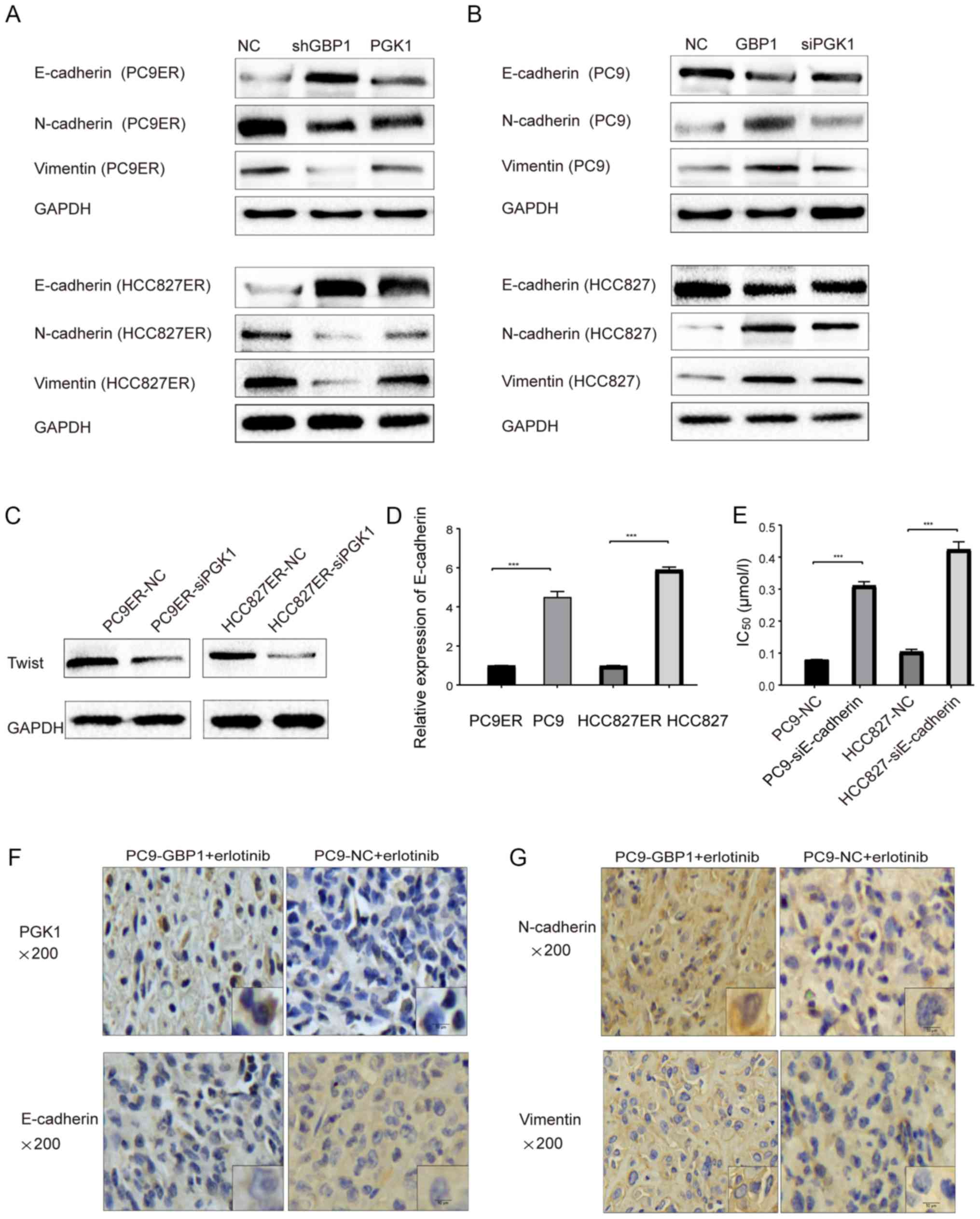 | Figure 7EMT pathway mediated by PGK1 plays an
important role in GBP1-modulated erlotinib resistance. (A and B)
PGK1 restores the effect of GBP1 on the levels of EMT-related
proteins, E-cadherin, N-cadherin and vimentin, in both
erlotinib-resistant (PC9ER and HCC827ER) (A) and sensitive (PC9 and
HCC827) cells (B). (C) Change in Twist protein was displayed after
downregulation of PGK1 (-siPGK1) in erlotinib-resistant (PC9ER and
HCC827ER) cells compared with the NC groups. (D) E-cadherin mRNA
levels were determined in sensitive (PC9 and HCC827) and resistant
(PC9ER and HCC827ER) cells. (E) IC50 values were
determined in erlotinib-sensitive cells following E-cadherin
downregulation (-siE-cadherin). ***P<0.001, when
compared with the relevant negative control (NC) group. (F and G)
Expression of PGK1, E-cadherin, N-cadherin and vimentin by
immunohistochemistry (IHC) are showed in animal tumor samples. ETM,
epithelial-mesenchymal transition; GBP1, guanylate-binding
protein-1; PGK1, phosphoglycerate kinase 1; IC50, half
maximal inhibitory concentration. |
Discussion
By interrupting the epidermal growth factor receptor
(EGFR) signaling pathway, erlotinib is highly successful as an
initial treatment for non-small cell lung cancer (NSCLC), but
resistance limits the clinical utility of erlotinib. A detailed
understanding of its mechanism is critical for patients to benefit
from an optimal therapeutic response. There are many reports
concerning epidermal growth factor receptor tyrosine kinase
inhibitor (EGFR-TKI) resistance. Kobayashi et al, reported
that the T790M mutation is a common mechanism of EGFR-TKI
resistance (21). Oser et
al found that a histological transition from NSCLC to small
cell lung cancer (SCLC) is a mechanism of EGFR-TKI resistance
(22). However, many mechanisms of
EGFR-TKI resistance remain unclear.
In the present study, we found that
guanylate-binding protein-1 (GBP1) expression was increased in
erlotinib-resistant cells. Previous research has shown that GBP1
expression is high in lung adenocarcinoma (8) and colorectal cancer (23). Wadi et al reported that
elevated GBP1 was correlated to a shorter progression-free time in
ovarian cancer (24). In addition,
studies have reported that GBP1 overexpression is associated with
paclitaxel resistance (25).
However, it remains unknown whether GBP1 is involved in the
regulation of EGFR-TKI resistance. Our experiments showed that the
downregulation of GBP1 in erlotinib-resistant cells increased
erlotinib sensitivity, while GBP1 overexpression in
erlotinib-sensitive cells promoted erlotinib resistance. Moreover,
our study revealed that GBP1 regulates apoptosis and the cell
cycle. In vivo models also indicated that GBP1 is associated
with erlotinib resistance. Furthermore, survival analysis showed
that high GBP1 levels predict poor prognosis. Overall, these
results demonstrate that GBP1 induces erlotinib resistance in
NSCLC.
To explore the mechanism by which GBP1 regulates
erlotinib resistance, we performed mass spectrometry to identify
GBP1 interacting proteins. By CoIP experiments, we identified
phosphoglycerate kinase 1 (PGK1) as a GBP1-interacting protein.
Several studies reported an association between PGK1 and
chemotherapeutic resistance. Zhou et al observed that PGK1
induced cisplatin resistance in endometrioid adeno-carcinoma
(26). In a recent study, the
blockade of PGK1 was shown to overcome chemotherapeutic resistance
in gastric cancer (27), and
elevated PGK1 was found to promote paclitaxel resistance in breast
cancer (28). However, a
relationship between PGK1 and erlotinib resistance has not been
reported. In the present study, we found that PGK1 regulates
erlotinib resistance through CCK-8 assays. Therefore, inhibiting
the GBP1-PGK1 axis may be an effective way to overcome erlotinib
resistance.
Several studies have suggested that PGK1 facilitates
cell invasion via EMT in breast cancer (29). Epithelial-mesenchymal transition
(EMT) is a biological process that is involved in cell
proliferation invasion, migration and chemotherapy resistance in
NSCLC. Specifically, EMT induces erlotinib resistance in NSCLC
(30), and Lou et al also
reported that EMT generates cellular erlotinib resistance via the
STAT3 pathway in NSCLC (31). In
addition, the EMT status was found to influence EGFR-TKI resistance
via the Wnt or Notch pathway in EGFR-mutant NSCLC cancer (32). Weng et al also showed that
EMT is a common mechanism of EGFR-TKI resistance (33). Therefore, we further evaluated the
importance of the EMT signaling pathway in mediating erlotinib
resistance via PGK1 and discovered that GBP1 and PGK1 strongly
depend on the EMT pathway to regulate erlotinib resistance.
Additionally, the downregulation of E-cadherin in
erlotinib-sensitive cells led to a change in erlotinib sensitivity.
All the above results showed that EMT is a key step in erlotinib
resistance and that the activation of the EMT pathway by PGK1
promotes erlotinib resistance mediated by GBP1 in NSCLC.
In summary, the present study found that GBP1 is a
critical modulator of erlotinib resistance and is expected to
provide basic experimental evidence for 'targeting GBP1 to reduce
EGFR-TKI resistance'.
Supplementary Data
Acknowledgments
The authors thank the staff involved in the present
study.
Funding
This research was supported by the National Natural
Science Foundation of China (grant nos. 81772457 and 81871859), The
Province Medical Scientific Research Foundation of Guangdong (grant
no. A2018246), and The Province Natural Science Foundation of
Guangdong (grant no. 2018A030310471).
Availability of data and materials
All data are included in this article and its
supplementary information files.
Authors' contributions
LC collected the data and wrote the manuscript. LG
reviewed the manuscript. TW analyzed the results. JZ contributed to
the conception and design of the research. All authors were
involved in some aspect of conducting the experiments. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Southern Medical University (Guangzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jänne PA, Wang X, Socinski MA, Crawford J,
Stinchcombe TE, Gu L, Capelletti M, Edelman MJ, Villalona-Calero
MA, Kratzke R, et al: Randomized phase II trial of erlotinib alone
or with carboplatin and paclitaxel in patients who were never or
light former smokers with advanced lung adenocarcinoma: CALGB 30406
trial. J Clin Oncol. 30:2063–2069. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Suda K, Rivard CJ, Mitsudomi T and Hirsch
FR: Overcoming resistance to EGFR tyrosine kinase inhibitors in
lung cancer, focusing on non-T790M mechanisms. Expert Rev
Anticancer Ther. 17:779–786. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Westover D, Zugazagoitia J, Cho BC, Lovly
CM and Paz-Ares L: Mechanisms of acquired resistance to first- and
second-generation EGFR tyrosine kinase inhibitors. Ann Oncol.
29(suppl_1): i10–i19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Britzen-Laurent N, Lipnik K, Ocker M,
Naschberger E, Schellerer VS, Croner RS, Vieth M, Waldner M,
Steinberg P, Hohenadl C, et al: GBP-1 acts as a tumor suppressor in
colorectal cancer cells. Carcinogenesis. 34:153–162. 2013.
View Article : Google Scholar
|
|
7
|
Yu CJ, Chang KP, Chang YJ, Hsu CW, Liang
Y, Yu JS, Chi LM, Chang YS and Wu CC: Identification of
guanylate-binding protein 1 as a potential oral cancer marker
involved in cell invasion using omics-based analysis. J Proteome
Res. 10:3778–3788. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamakita I, Mimae T, Tsutani Y, Miyata Y,
Ito A and Okada M: Guanylate binding protein 1 (GBP-1) promotes
cell motility and invasiveness of lung adenocarcinoma. Biochem
Biophys Res Commun. 518:266–272. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tipton AR, Nyabuto GO, Trendel JA, Mazur
TM, Wilson JP, Wadi S, Justinger JS, Moore GL, Nguyen PT and Vestal
DJ: Guanylate-binding Protein-1 protects ovarian cancer cell lines
but not breast cancer cell lines from killing by paclitaxel.
Biochem Biophys Res Commun. 478:1617–1623. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ji X, Zhu H, Dai X, Xi Y, Sheng Y, Gao C,
Liu H, Xue Y, Liu J, Shi J, et al: Overexpression of GBP1 predicts
poor prognosis and promotes tumor growth in human glioblastoma
multiforme. Cancer Biomark. 25:275–290. 2019. View Article : Google Scholar
|
|
11
|
Quintero M, Adamoski D, Reis LMD, Ascenção
CFR, Oliveira KRS, Gonçalves KA, Dias MM, Carazzolle MF and Dias
SMG: Guanylate-binding protein-1 is a potential new therapeutic
target for triple-negative breast cancer. BMC Cancer. 17:7272017.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu T, Zhao Y, Hu Z, Li J, Chu D, Zhang J,
Li Z, Chen B, Zhang X, Pan H, et al: MetaLnc9 facilitates lung
cancer metastasis via a PGK1-activated AKT/mTOR pathway. Cancer
Res. 77:5782–5794. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Niederst MJ, Sequist LV, Poirier JT,
Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN,
Moran T, et al: RB loss in resistant EGFR mutant lung
adenocarcinomas that transform to small-cell lung cancer. Nat
Commun. 6:63772015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nakata A, Yoshida R, Yamaguchi R, Yamauchi
M, Tamada Y, Fujita A, Shimamura T, Imoto S, Higuchi T, Nomura M,
et al: Elevated β-catenin pathway as a novel target for patients
with resistance to EGF receptor targeting drugs. Sci Rep.
5:130762015. View Article : Google Scholar
|
|
16
|
Smyth GK: Limma: linear models for
microarray. Bioinformatics and Computational Biology Solutions
Using R and Bioconductor. Gentleman R, Carey VJ, Huber W, Irizarry
RA and Dudoit S: Springer; New York, NY: pp. 397–420. 2005,
View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Xiong Y, Hannon GJ, Zhang H, Casso D,
Kobayashi R and Beach D: p21 is a universal inhibitor of cyclin
kinases. Nature. 366:701–704. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Massagué J: G1 cell-cycle control and
cancer. Nature. 432:298–306. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang H, Geng YH, Wang P, Zhou YT, Yang H,
Huo YF, Zhang HQ, Li Y, He HY, Tian XX, et al: Extracellular ATP
promotes breast cancer invasion and epithelial-mesenchymal
transition via hypoxia-inducible factor 2α signaling. Cancer Sci.
110:2456–2470. 2019.PubMed/NCBI
|
|
21
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oser MG, Niederst MJ, Sequist LV and
Engelman JA: Transformation from non-small-cell lung cancer to
small-cell lung cancer: Molecular drivers and cells of origin.
Lancet Oncol. 16:e165–e172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thomas H: Colorectal cancer: CRC
endothelial regulation. Nat Rev Gastro Hepat. 13:6822016.
View Article : Google Scholar
|
|
24
|
Wadi S, Tipton AR, Trendel JA, Khuder SA
and Vestal DJ: hGBP-1 expression predicts shorter progression-free
survival in ovarian cancers, while contributing to paclitaxel
resistance. J Cancer Ther. 7:994–1007. 2016. View Article : Google Scholar
|
|
25
|
Duan Z, Foster R, Brakora KA, Yusuf RZ and
Seiden MV: GBP1 overexpression is associated with a paclitaxel
resistance phenotype. Cancer Chemother Pharmacol. 57:25–33. 2006.
View Article : Google Scholar
|
|
26
|
Zhou JW, Tang JJ, Sun W and Wang H: PGK1
facilities cisplatin chemoresistance by triggering HSP90/ERK
pathway mediated DNA repair and methylation in endometrial
endometrioid adeno-carcinoma. Mol Med. 25:112019. View Article : Google Scholar
|
|
27
|
Schneider CC, Archid R, Fischer N, Bühler
S, Venturelli S, Berger A, Burkard M, Kirschniak A, Bachmann R,
Königsrainer A, et al: Metabolic alteration–Overcoming therapy
resistance in gastric cancer via PGK-1 inhibition in a combined
therapy with standard chemotherapeutics. Int J Surg. 22:92–98.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun S, Liang X, Zhang X, Liu T, Shi Q,
Song Y, Jiang Y, Wu H, Jiang Y, Lu X, et al: Phosphoglycerate
kinase-1 is a predictor of poor survival and a novel prognostic
biomarker of chemoresistance to paclitaxel treatment in breast
cancer. Br J Cancer. 112:1332–1339. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fu D, He C, Wei J, Zhang Z, Luo Y, Tan H
and Ren C: PGK1 is a potential survival biomarker and invasion
promoter by regulating the HIF-1alpha-mediated
epithelial-mesenchymal transition process in breast cancer. Cell
Physiol Biochem. 51:2434–2444. 2018. View Article : Google Scholar
|
|
30
|
Byers LA, Diao L, Wang J, Saintigny P,
Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al: An
epithelial-mesenchymal transition gene signature predicts
resistance to EGFR and PI3K inhibitors and identifies Axl as a
therapeutic target for overcoming EGFR inhibitor resistance. Clin
Cancer Res. 19:279–290. 2013. View Article : Google Scholar
|
|
31
|
Lou W, Chen Y, Zhu KY, Deng H, Wu T and
Wang J: Polyphyllin I overcomes EMT-associated resistance to
erlotinib in lung cancer cells IL-6/STAT3 pathway inhibition. Biol
Pharm Bull. 40:1306–1313. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jakobsen KR, Demuth C, Sorensen BS and
Nielsen AL: The role of epithelial to mesenchymal transition in
resistance to epidermal growth factor receptor tyrosine kinase
inhibitors in non-small cell lung cancer. Transl Lung Cancer Res.
5:172–182. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weng CH, Chen LY, Lin YC, Shih JY, Lin YC,
Tseng RY, Chiu AC, Yeh YH, Liu C, Lin YT, et al:
Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per
se is a common mechanism for acquired resistance to EGFR TKI.
Oncogene. 38:455–468. 2019. View Article : Google Scholar
|