Introduction
Glucocorticoids (GCs) are steroid hormones produced
by the adrenal glands in response to stress (1). Dexamethasone, a synthetic GC, has
been used to treat patients suffering from numerous diseases such
as leukaemia, inflammatory bowel disease, arthritis, asthma and
allergies due to its regulatory role in cellular inflammatory
responses, which include stimulating immunosuppression, inhibiting
cell cycle progression and inducing apoptosis (2).
Acute lymphoblastic leukaemia (ALL) is a type of
cancer of white blood cells that mainly affects children and
teenagers (3). Several gene
mutations and hazardous environmental factors (including radiation
and toxic chemical exposure) are associated with ALL in children
(4,5). Factors affecting the development of
resistance to ALL treatment include genetic variability in
xenobiotic metabolism (6),
prevalence of a GC receptor (GR)β splicing variant (7), upregulation of the antiapoptotic
Bcl-2 family proteins (8),
differential phosphorylation of GR in Dex-induced
apoptosis-resistant vs. sensitive ALL cells (9), alterations in DNA repair pathways
(10) and cell cycle checkpoint
defects (11).
Reactive oxygen species (ROS) serve an important
role in the regulation of GR function (12) and the GC-mediated apoptosis in
lymphoma cells (13). ROS mediate
several cell signalling pathways (14) including cell cycle control
(15), mitochondrial energy
metabolism (16) and endoplasmic
reticulum (ER) stress (17). ER
stress is activated in rapidly proliferating cancer cells by the
accumulation of misfolded or unfolded proteins as a consequence of
high cellular ROS levels (18).
The interplay among the mitochondria, ER and ROS levels, as well as
the connection between ROS-mitochondrial membrane potential
(MMP)-autophagy and ER stress have been extensively studied
(19-24).
ER chaperone glucose-regulated protein (GRP)78 and
GRP94 identify and bind to misfolded proteins, promote
ER-associated protein degradation and contribute to the initiation
of the immune response (25,26).
Induction of ER stress can lead to cell survival or death (27,28).
The pro-survival outcome of ER stress is in part due to the
induction of its inositol-requiring enzyme 1 arm, which activates
Jun N-terminal kinase (JNK) (29)
and stimulates Bcl-2 phosphorylation to regulate autophagy through
the modulation of the beclin-1 function and gene expression
(30). In addition, diverse ER
stress inducing conditions that involve the PKR-like endoplasmic
reticulum kinase pathway and eukaryotic initiation factor 2α
phosphorylation regulate the conversion of the pro-survival
autophagy mediating microtubule-associated protein 1 light chain 3α
(LC3) (31). However, if ER stress
persists and the refolding of unfolded proteins is not resolved,
unfolded protein response (UPR) ensues; UPR is primarily a
pro-survival process, but prolonged stress may result in the
induction of cell death (27,28,32)
via stimulation of CCAAT/enhancer-binding protein homologous
protein (CHOP) (33).
The communication between the mitochondria and the
endoplasmic reticulum (34) and
the regulation of Ca2+ homeostasis by the integration of
the function of Bcl-2 family members (35), endoplasmic reticulum chaperones
(36) and their adjustment to the
cellular redox state (37) is
another mechanism controlling the pro-survival and pro-death
pathways. Our bioinformatics analysis of Ca2+ signalling
and autophagy pathways indicated that out of the 11,320 genes
associated with Ca2+ signalling and 5,207 associated
with autophagy, 1,109 genes are associated with both pathways
(unpublished data). These genes include the inositol
1,4,5-trisphosphate receptor, ryanodine receptor, mitochondrial
calcium uniporter, two pore segment channels, transient receptor
potential channels, store-operated calcium channels, GRP78 and
GRP94, the functions of which have been previously described
(38-41).
T-cell ALL (T-ALL) and B-ALL differ in terms of
prognostic factors and risk classification such as age, white blood
cell count, hyperdiploidy and the presence of translocations
(42,43). Similar therapeutic schemes are used
for the treatment of T-ALL and B-ALL, although significant toxicity
associated with chemotherapy has been reported for B-ALL, as adults
do not always benefit from therapeutic schemes used for paediatric
patients (42,43).
The present study aimed to investigate the molecular
mechanisms mediating the resistance of T-ALL cells to Dex
treatment. Established T-ALL CEM-C7-14 and CEM-C1-15 cell lines,
which are sensitive and resistant, respectively, to Dex-induced
apoptosis, were used to determine the crosstalk of multiple
pathways involved in the response of T-ALL cells to chemotherapy.
In particular, the potential consequences of the differential ROS
detoxification in the T-ALL cells resistant or sensitive to
Dex-induced apoptosis (44) and
the ensuing changes in ER stress, autophagy and MMP were
investigated. Emphasis was placed on understanding how changes in
oxidative stress may be translated through autophagy, ER stress,
UPR and the function of the ER chaperone proteins GRP94 and GRP78
to GC-mediated cell death.
Materials and methods
Cell culture
A previously established T-ALL in vitro
system to study resistance to steroid response, consisting of the
CEM-C1-15 and CEM-C7-14 cell lines (45) derived from lymphoblastic cells of a
patient with ALL (46), was used
in the present study. The T-ALL MOLT4 (cat. no. TCP-1010; ATCC)
cells were used to explore whether the conclusions derived from the
experiments using CEM cells were relevant to other types of T-ALL
cells. Cells were cultured with 5% CO2 at 37°C in
RPMI-1640 medium (Sigma-Aldrich; Merck KGaA) supplemented with 10%
foetal bovine serum (Sigma-Aldrich; Merck KGaA), 1%
penicillin/streptomycin (Lonza Group, Ltd.) and 1% L-glutamine
(Lonza Group, Ltd.). Dextran-coated charcoal-treated serum
(Sigma-Aldrich; Merck KGaA) was used in all experiments prior to
the addition of drugs.
Drugs
The concentration of 1 µM dexamethasone (Dex;
Enzo Life Sciences, Inc.) was chosen for the treatment of the cells
at the indicated time-points based on previous studies (47-49).
The optimal treatment duration to observe differential effects of
glucocorticoids in the sensitive vs. resistant cells was 48 h
(49). The concentrations of
chloroquine (CLQ; 100 µM; Sigma-Aldrich; Merck KGaA),
thapsigargin (TG; 20 µM; Sigma-Aldrich; Merck KGaA) and
rotenone (ROT; 20 µM; Sigma-Aldrich; Merck KGaA) were
determined through optimization of experimental conditions (data
not shown) and literature search (47,48).
All drugs were dissolved in dimethyl sulfoxide (chloroquine,
thapsigargin and rotenone) or ethanol (dexamethasone) and used as
indicated. The control groups were treated with DMSO.
Cell proliferation assay
CEM-C1-15, CEM-C7-14 and MOLT4 cells
(1×106 cells/ml) were seeded in 96-well plates prior to
drug treatment for 48 h at 37°C. Once the drug incubation was
complete, 20 µl CellTiter 96 Aqueous MTS reagent working
solution (cat. no. G1112; Promega Corporation) was added to the
wells and incubated at 37°C for 4 h. The absorbance of the samples
was read at 490 nm using a microplate reader (Multiskan Ascent
Thermo Labsystems 354 with Ascent software version 2.6) with 690 nm
background compensation.
MMP assay
CEM-C1-15, CEM-C7-14 and MOLT4 cells
(1×106 cells//ml) were seeded in 6-well plates prior to
combination treatments (Dex, CLQ, TG, ROT, Dex + CLQ, Dex + TG or
Dex + ROT) for 48 h at 37°C. Subsequently, the cells were suspended
in 1 ml medium, and 6.25 µl solution 7 (JC-1 dye; ChemoMetec
A/S) was added to each sample, followed by 20-min incubation at
37°C. The stained cells were centrifuged at 400 × g for 5 min at
room temperature, the supernatant was removed, and the samples were
washed twice with PBS. The pellets were resuspended in 250
µl solution 8 (DAPI), and the samples were analysed using a
NucleoCounter NC-3000 with NucleoView software version 1
(ChemoMetec A/S).
ROS assay
A carboxy-H2DCADA probe (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to detect ROS generated in ALL cells
treated with the aforementioned drugs. A probe containing
2′,7′-dichlorofluorescein and calcein was used to detect oxidation
by determination of the increase in fluorescence signalling using a
flow cytometer, with excitation sources and filters for FITC
channel. CEM-C1-15, CEM-C7-14 and MOLT4 cells (1×106
cells/ml) were seeded in 6-well plates before combination
treatments (Dex, CLQ, TG, ROT, Dex + CLQ, Dex + TG or Dex + ROT)
for 24 h at 37°C. The cells were centrifuged at 753 × g for 3 min
at 4°C, and the supernatants were discarded. The cell pellets were
resuspended in 1 ml cold PBS and centrifuged at 753 × g for 3 min
at 4°C. Carboxy-H2DCFDA dye (100 µl) was added to the
pellets, and the cells were incubated for 30-60 min at 37°C with 5%
CO2 in the dark. The dye was discarded by centrifuging
the pellets at 753 × g for 3 min at 4°C. The cells were washed with
cold PBS and analysed by flow cytometry (BD FACSVerse™ with BD
FACSuite software version 1.0; BD Biosciences) using the FITC
channel.
Immunoblotting
CEM-C1-15, CEM-C7-14 and MOLT4 cells treated with
Dex, CLQ, TG or ROT alone or a combination of Dex with CLQ, TG or
ROT were harvested and lysed with RIPA buffer (cat. no. R0278;
Sigma-Aldrich; Merck KGaA). Protein concentration was determined
using the Bradford assay, and 40 µg of total protein lysates
were subjected to 12% sodium dodecyl sulphate polyacrylamide gel
electrophoresis and transferred to a polyvinylidene difluoride
membrane (EMD Millipore). Following blocking with 5% skimmed milk
in 1X PBS for 1 h at room temperature, the membrane was incubated
with rabbit polyclonal anti-LC3A/B (cat. no. 4108; 1:1,000; Cell
Signalling Technology, Inc.), rabbit monoclonal anti-beclin-1 (cat.
no. 3495; 1:1,000; Cell Signalling Technology, Inc.), mouse
monoclonal anti-GRP78 (cat. no. sc-376768; 1:500; Santa Cruz
Biotechnology, Inc.), mouse monoclonal anti-GRP94 (cat. no.
sc-53929; 1:500; Santa Cruz Biotechnology, Inc.) and rabbit
polyclonal anti-β-actin (cat. no. ab8227; 1:2,000; Abcam)
antibodies at 4°C overnight, followed by 1-h incubation at 4°C with
secondary antibodies conjugated with horseradish peroxidase
(anti-rabbit cat. no. NA934; anti-mouse cat. no. NA931; 1:4,000;
Cytiva). Protein bands were detected using an enhanced
chemiluminescence solution (Thermo Fisher Scientific, Inc.) and an
X-ray film (Fujifilm Corporation). The band intensities were
estimated using ImageJ version 1.51 (National Institutes of
Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNAs were isolated from 3×106
CEM-C7-14, CEM-C1-15 and MOLT4 cells using a High Pure RNA
Isolation kit (Roche Molecular Systems, Inc.) and the concentration
of the total RNA for each sample was measured using a NanoDrop™
spectrophotometer (Thermo Fisher Scientific, Inc.). Complementary
DNA was reverse-transcribed using a High-Capacity RNA-to-cDNA™ kit
(Applied Biosystems; Thermo Fisher Scientific, Inc.) at 37°C for 1
h. QPCR was performed using GRP94, GRP78 and RPL19 (internal
control) primers (Eurofins Scientific) mixed with SensiFAST
SYBR® No-ROX kit (Bioline) as previously described
(50,51). The primer sequences were as
follows: GRP78 forward, 5′-TGCCGTTCAAGGTGGTTG-3′ and reverse,
5′-CCA AATAAGCCTCAGCGG-3′; GRP94 forward, 5′-CTGGGTCCA
GCAGAAAAGAG-3′ and reverse, 5′-CACTCCTTCCTTGGC AACAT-3′; RPL19
forward, 5′-ATGTATCACAGCCTGTAC CTG-3′ and reverse,
5′-TTCTTGGTCTCTTCCTCCTTG-3′. The data were analysed using the
2−ΔΔCq method (52).
Cell surface staining assay
CEM-C1-15, CEM-C7-14 and MOLT4 cells
(1×106 cells/ml) were seeded in 6-well plates and
combination treatments (Dex, CLQ, TG, ROT, Dex + CLQ, Dx + TG or
Dex + ROT) were applied for 48 h at 37°C. Cells were washed with
cold (4°C) 2% FBS in PBS before incubation with an anti-GRP94
antibody (9G10) conjugated to Alexa Fluor® 488 (cat. no.
sc-32249 AF488; Santa Cruz Biotechnology, Inc.) for 2 h at 4°C. The
cell pellet was fixed with 4% paraformaldehyde in FBS/PBS at 4°C
for 15 min, washed twice with 500 µl PBS (4°C) and flow
cytometry (BD FACSVerse™ with BD FACSuite software) was used to
detect surface GRP94 fluorescence intensity.
Statistical analysis
Data are presented as the mean ± standard error of
the mean of data from three independent experiments. Statistical
analysis was conducted using GraphPad Prism 8 Software (GraphPad
Software, Inc.). One-way ANOVA and Dunnett's post hoc test were
used for multiple comparisons. P<0.05 was considered to indicate
a statistically significant difference.
Results
Cytotoxic effects of individual and
combined drug treatments
The present study analysed the autophagy pathway
using CLQ, which is an autophagy inhibitor (53), ER stress using TG, which regulates
Ca2+ homeostasis and activates ER stress (54), and ROS generation using ROT, which
induces mitochondrial oxidative stress by inhibiting complex I of
the mitochondrial respiratory chain and modulates autophagy
(55) in GC-treated ALL cells.
Time course and drug concentration studies were performed to
determine the optimal experimental conditions (data not shown).
To assess ALL cell viability upon treatment with
Dex, CLQ, TG and ROT alone or in combinations, MTS assay was
performed in CEM-C7-14, CEM-C1-15 and MOLT4 cells treated with Dex
alone or in combination with CLQ, TG or ROT (Fig. 1). Dex induced a less pronounced
reduction in CEM-C1-15 cell viability compared with that of the
CEM-C7-14 cells, whereas the viability of MOLT4 cells treated with
Dex was 50% of that in untreated cells (Fig. 1). CLQ, TG and ROT alone or in
combination with Dex inhibited cell proliferation in all cells
compared with that of untreated cells (Fig. 1). These results suggest that GCs
alone mostly affected CEM-C7-14 cells, whereas CEM-C1-15 cells
weakly responded to Dex, indicating a resistant phenotype, and
MOLT4 cells appeared to exhibit a stronger response to Dex compared
with CEM-C1-15 cells, but weaker compared with CEM-C7-14 cells
(Fig. 1). A potent cytotoxic
effect was observed in the CEM-C1-15 cells treated with CLQ alone
or in combination with Dex (Fig.
1B). These results suggested that Dex treatment, autophagy
inhibition, ER stress and induction of ROS generation exerted
different cytotoxic effects in the studied ALL cells.
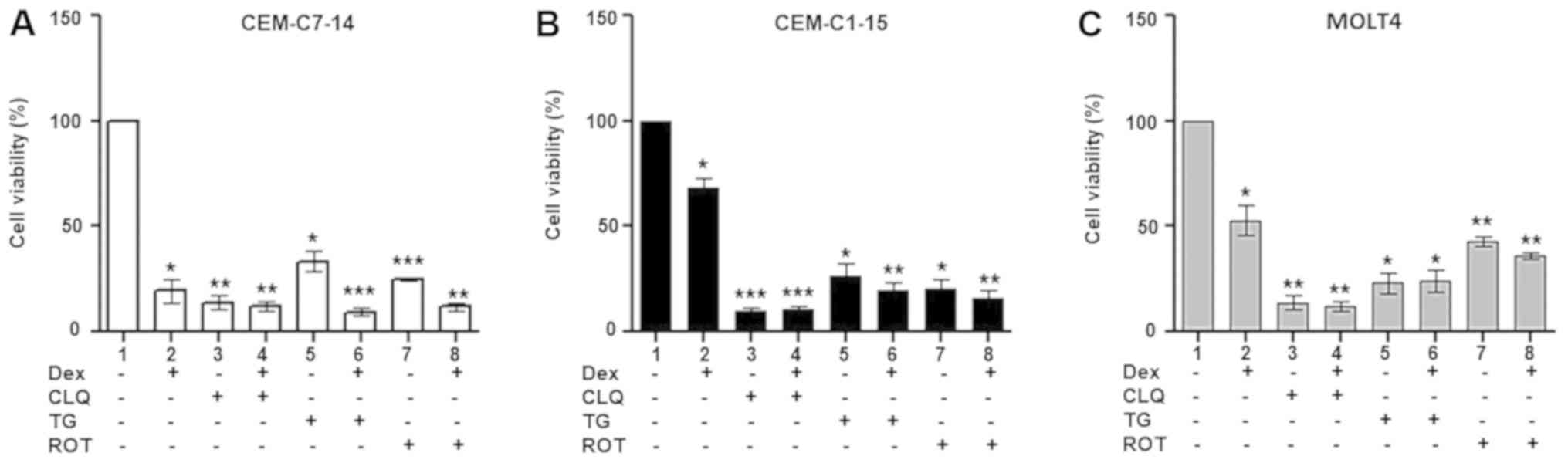 | Figure 1Cytotoxic effects of Dex and
anticancer agents on ALL cells. Dex (10 µM), CLQ (100
µM), TG (20 µM) and ROT (20 µM) were used to
treat (A) CEM-C7-14, (B) CEM-C1-15 and (C) MOLT4 cells for 48 h
individually or in combination to assess the cell viability using
MTS assay. Data are presented as the mean ± SEM.
*P<0.05, **P<0.01,
***P<0.001 vs. untreated control. ALL, acute
lymphoblastic leukaemia; Dex, dexamethasone; CLQ, chloroquine; TG,
thapsigargin; ROT, rotenone. |
ROS generation in ALL cells treated with
Dex, CLQ, TG and ROT alone or in combination
To investigate the potential involvement of ROS
generation in response to GC treatment of ALL cells, CEM-C1-15,
CEM-C7-14 and MOLT4 cells were treated with Dex, CLQ, TG or ROT
alone or in combination, and the ROS levels were determined by flow
cytometry. No significant difference in the ROS levels was observed
in Dex-treated compared with untreated CEM-C1-15 cells (Figs. 2B and S1), whereas significantly decreased ROS
levels were detected in Dex-treated compared with untreated
CEM-C7-14 cells (Figs. 2A and
S1). No significant effect of Dex
treatment on the ROS levels in MOLT4 cells was observed (Figs. 2C and S1). CLQ alone or in combination with Dex
slightly decreased the ROS levels in CEM-C1-15 cells, although the
changes were not statistically significant (Figs. 2B and S1). No significant effects of CLQ alone
or in combination with Dex on the ROS levels of CEM-C7-14 and MOLT4
cells were observed (Figs. 2A, C
and S1). Treatment of CEM-C7-14
cells with TG alone (Figs. 2A and
S1) and of MOLT4 cells with TG
alone or in combination with Dex (Figs. 2C and S1) significantly decreased ROS
generation. By contrast, ROT alone or in combination with Dex
substantially increased ROS production in CEM-C1-15 and CEM-C7-14
cells compared with that in the respective untreated cells
(Figs. 2A and B and S1). This effect appeared to be more
pronounced in CEM-C7-14 compared with CEM-C1-15 cells. The ROS
levels were not affected in MOLT4 cells by ROT alone, whereas
ROT-Dex co-treatment resulted in a significant decrease of ROS
generation in these cells (Figs.
2C and S1).
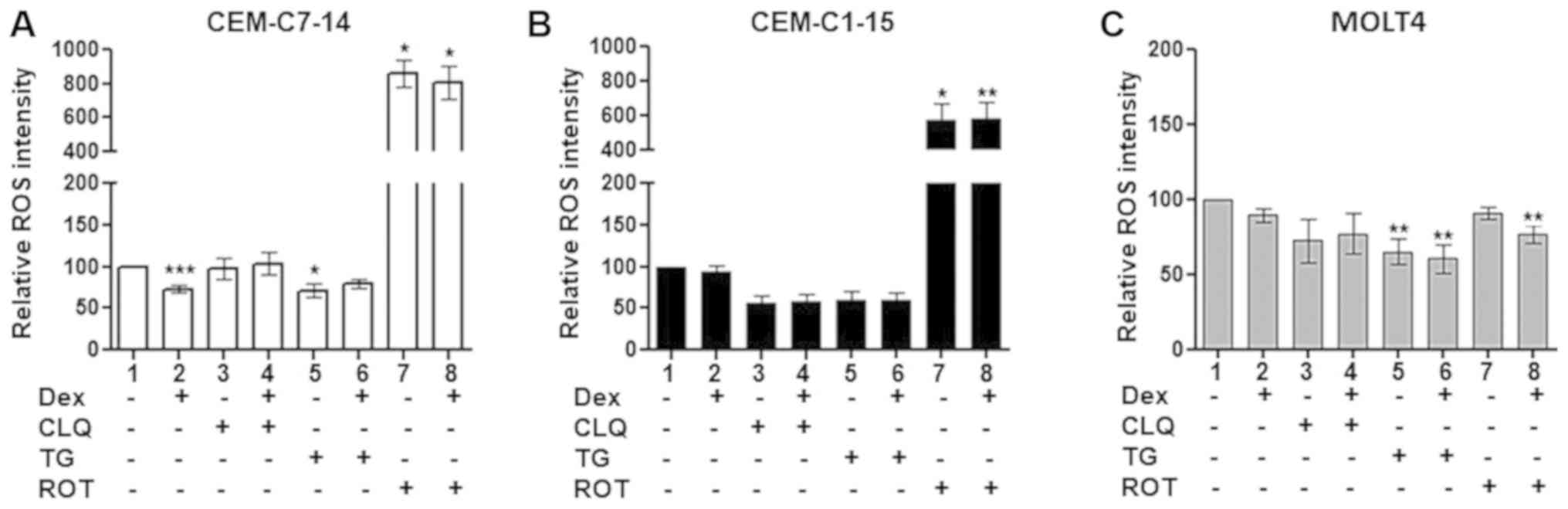 | Figure 2Effects of Dex and anticancer agents
on ROS production. The relative ROS intensity was recorded in (A)
CEM-C7-14, (B) CEM-C1-15 and (C) MOLT4 cells using a
2',7'-dichlorofluorescein probe and flow cytometry. Cells were
treated with 1 µM Dex, 50 µM CLQ, 10 µM TG and
10 µM ROT as indicated. Data are presented as the mean ±
SEM. *P<0.05, **P<0.01,
***P<0.001 vs. untreated control. Dex, dexamethasone;
CLQ, chloroquine; TG, thapsigargin; ROT, rotenone; ROS, reactive
oxygen species. |
In summary, lower ROS levels were recorded in Dex-
and TG-treated CEM-C7-14 cells compared with those in untreated
cells, whereas CLQ had no effect. In MOLT4 cells, TG reduced the
ROS levels. ROT increased the ROS levels in CEM-C7-14 and CEM-C1-15
cells compared with those in the respective untreated cells,
whereas it did not lead to an increase in the ROS levels in MOLT4
cells.
MMP in ALL cells treated with Dex, CLQ,
TG and ROT alone or in combination
Considering the differential effects of individual
Dex, CLQ, TG and ROT treatment on ROS generation in CEM-C7-14,
CEM-C1-15 and MOLT4 cells, and the interplay between mitochondrial
function and cellular ROS levels (56), the effects of Dex, CLQ, TG and ROT
alone or combined with Dex on the MMP of ALL cells were
investigated. Dex treatment alone significantly increased the MMP
in CEM-C7-14 cells compared with that in untreated cells (Figs. 3A and S2A), whereas limited effects were
observed in CEM-C1-15 and MOLT4 cells under the same conditions
(Figs. 3B, C, S2B and C). Increased MMP was observed in
the MOLT4 cell line treated with CLQ alone compared with that in
untreated cells (Figs. 3C and
S2C), as well as in CEM-C7-14 and
CEM-C1-15 cells treated with the combination of CLQ and Dex
compared with that in untreated cells (Figs. 3A, B, S2A and B). TG and ROT treatment alone
increased MMP in CEM-C7-14 and CEM-C1-15 cells compared with that
in untreated cells (Figs. 3A, B,
S2A and B). Increased MMP was
also observed in all studied cell lines treated with ROT or the
combination of ROT and Dex compared with that in untreated cells
(Figs. 3, S2A, B and C).
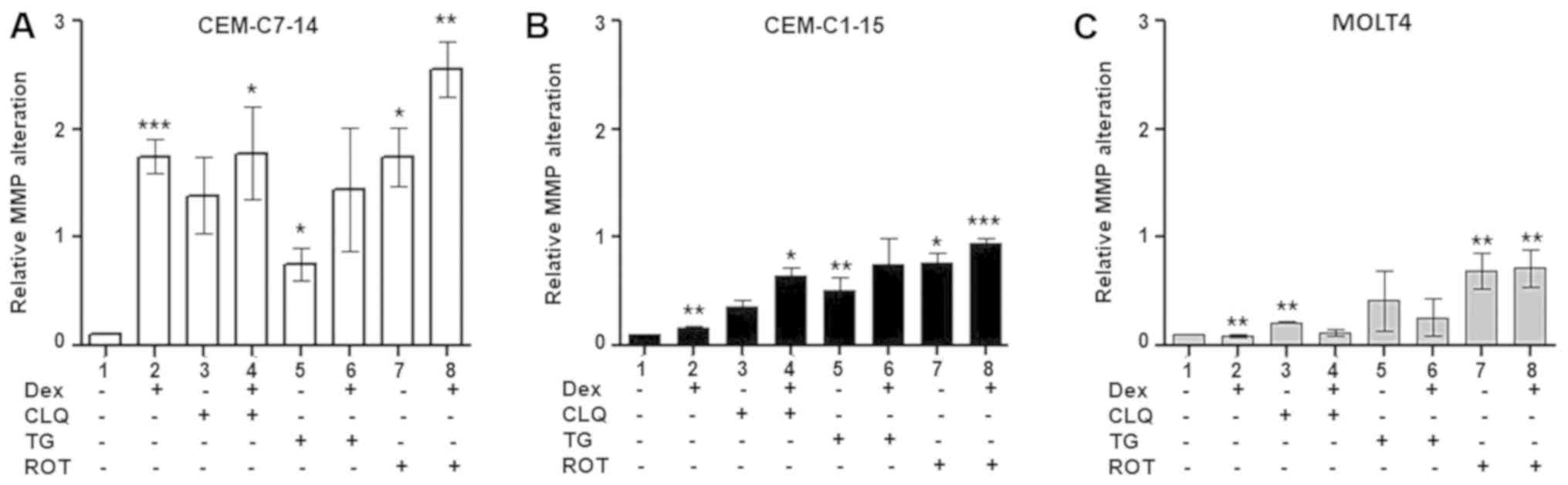 | Figure 3Effects of Dex and anticancer agents
on MMP alteration. MMP was measured (A) in CEM-C7-14, (B) CEM-C1-15
and (C) MOLT4 cells treated with 1 µM Dex, 50 µM CLQ,
10 µM TG and 10 µM ROT for 48 h and using the JC-1
dye. Data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001 vs. untreated
control. Dex, dexamethasone; CLQ, chloroquine; TG, thapsigargin;
ROT, rotenone; MMP, mitochondrial membrane potential. |
It appeared that most of the tested treatments
increased MMP in CEM-C7-14 and CEM-C1-15 cells with higher effects
in CEM-C7-14 cells, whereas in MOLT4 cells, CLQ and ROT increased
MMP compared with that in untreated cells. Addition of Dex to
TG-treated cells abolished the MMP increase in CEM-C7-14 and
CEM-C1-15 cells.
Effects of individual and combined drug
treatments on protein expression levels in ALL cells
To analyse the role of autophagy and ER stress in
GC-mediated ALL cell survival or death, the protein levels of
autophagy (beclin-1 and LC3-II) and ER stress (GRP78 and GRP94)
indicators were analysed by western blotting in CEM-C7-14,
CEM-C1-15 and MOLT4 cells (Figs.
4, S3A, B and C). A small but
significant increase in beclin-1 protein levels, which mark the
early stages of autophagy, was observed in TG-treated MOLT4 cells
compared with untreated cells (Figs.
4A and S3B), whereas a
decrease was observed in ROT and Dex co-treated CEM-C1-15 cells
(Figs. 4A and S3C). LC3-II, is an indicator of
late-stage autophagy, was increased in CEM-C1-15 and MOLT4 cells,
but marginally decreased in CEM-C7-14 cells treated with Dex alone
compared with the respective untreated cells (Figs. 4B, S3A, B and C). Higher LC3-II protein
levels were observed in cells treated with CLQ and TG alone and in
combination with Dex in all tested cell lines (Figs. 4B and S3B). No significant effects on LC3-II
were observed in ROT-treated CEM-C7-14 and MOLT4 cells, whereas in
CEM-C1-15 cells, downregulation of LC3-II was observed compared
with untreated cells (Figs. 4B and
S3C).
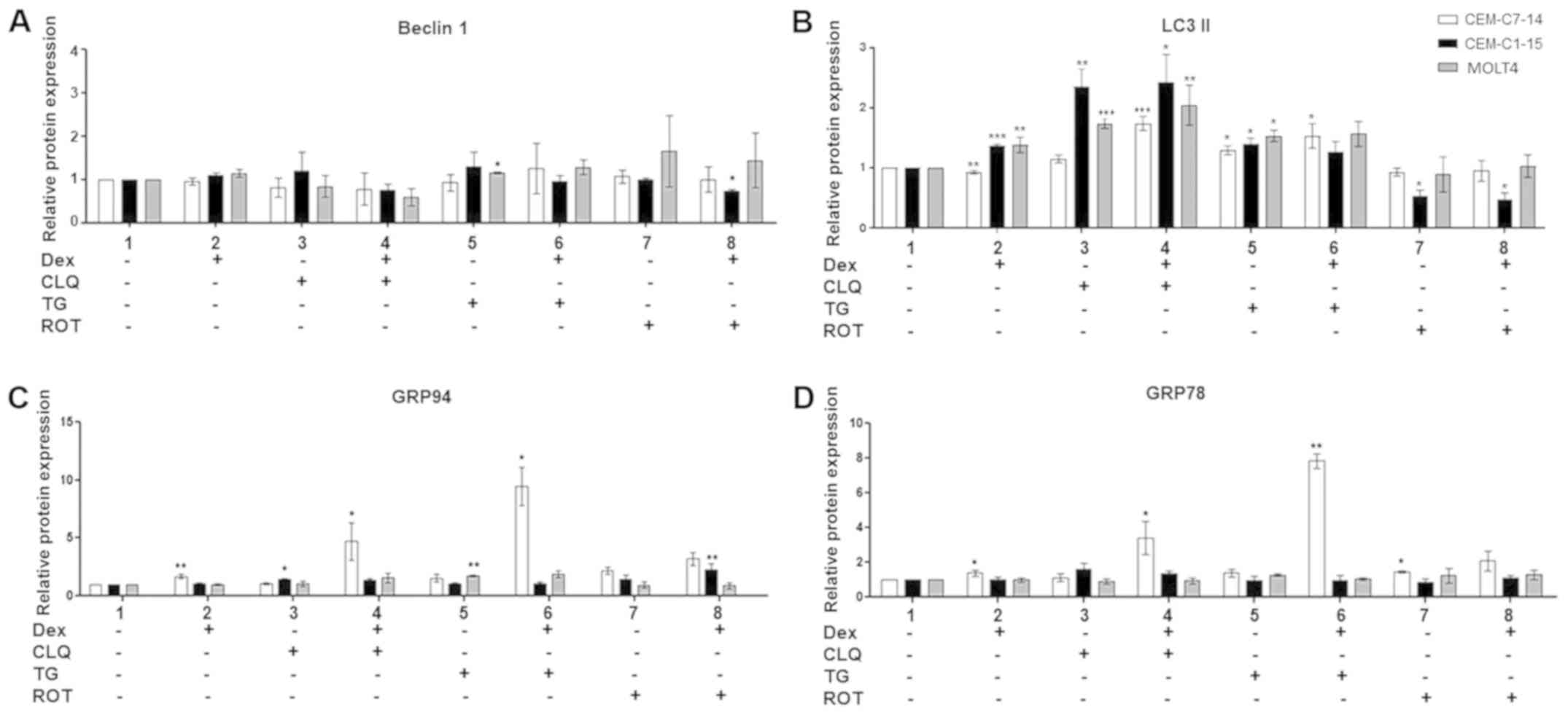 | Figure 4Protein expression in ALL following
combination drug treatments. The protein levels of (A) beclin-1,
(B) LC3-II, (C) GRP94 and (D) GRP78 were detected by western
blotting in CEM-C7-14 (white bars), CEM-C1-15 (black bars) and
MOLT4 (grey bars) cells after 48-h incubation with 1 µM Dex,
50 µM CLQ, 10 µM TG and 10 µM ROT individually
or in combination as indicated. The values were normalised to the
corresponding actin loading control. Data are presented as the mean
± SEM. *P<0.05, **P<0.01,
***P<0.001 vs. untreated control. ALL, acute
lymphoblastic leukaemia; Dex, dexamethasone; CLQ, chloroquine; TG,
thapsigargin; ROT, rotenone; LC3-II, microtubule-associated protein
1 light chain 3α; GRP, glucose-regulated protein. |
Dex treatment alone significantly increased GRP94
and GRP78 protein levels in CEM-C7-14 cells compared with those in
untreated cells and there were no changes in CEM-C1-15 and MOLT4
cells (Figs. 4C, D and S3). A significant increase of GRP94
protein levels was also evident in CEM-C7-14 cells co-treated with
Dex and CLQ or TG compared with those in untreated cells (Figs. 4C and S3A and B). In CEM-C1-15 cells treated
with CLQ alone or with a combination of Dex and ROT and in MOLT4
cells treated with TG alone, increased levels of GRP94 were
observed compared with those in untreated cells (Figs. 4C, S3A, B and C). Increased GRP78 protein
levels were observed in CEM-C7-14 cells treated with Dex alone, ROT
alone or a combination of CLQ or TG with Dex compared with those in
untreated cells (Figs. 4D,
S3A, B and C). The most
substantial increase in both chaperone cellular levels was observed
in GC-sensitive ALL cells (CEM-C7-14) co-treated with Dex and TG
(Fig. S3B).
To determine whether the changes in GRP94 and GRP78
protein levels were at the gene expression level, the effects of
Dex on GRP94 and GRP78 gene expression were studied using RT-qPCR.
GRP94 and GRP78 mRNA levels were not affected in Dex-treated CEM
cells, whereas decreased GRP94 mRNA levels were observed in MOLT4
cells compared with those in untreated cells (Fig. S4).
To summarise, compared with those in untreated
cells, the levels of GRP94 and GRP78 markers of ER stress were
increased in Dex-treated CEM-C7-14 cells, whereas the autophagy
marker LC3-II was decreased in CEM-C7-14 and increased in CEM-C1-15
and MOLT4 cells.
GRP94 surface expression in ALL
The extracellular surface GRP94 levels in ALL cells
treated with Dex, CLQ, TG and ROT alone or in combination were
investigated. TG and ROT treatment alone increased GRP94 surface
levels in both the GC-sensitive and resistant CEM cells compared
with those in the untreated control group (Figs. 5, S5A
and B). Combination of TG and ROT with Dex increased the GRP94
surface levels in the CEM-C7-14 and CEM-C1-15 cells compared with
those in untreated cells (Figs. 5,
S5A and B). TG-Dex co-treatment
exhibited a strong effect in CEM-C7-14 cells, whereas ROT effects
individually or in combination with Dex was prominent in CEM-C1-15
cells. Surface GRP94 levels did not change in MOLT4 cells in any
conditions (Figs. 5 and S5C).
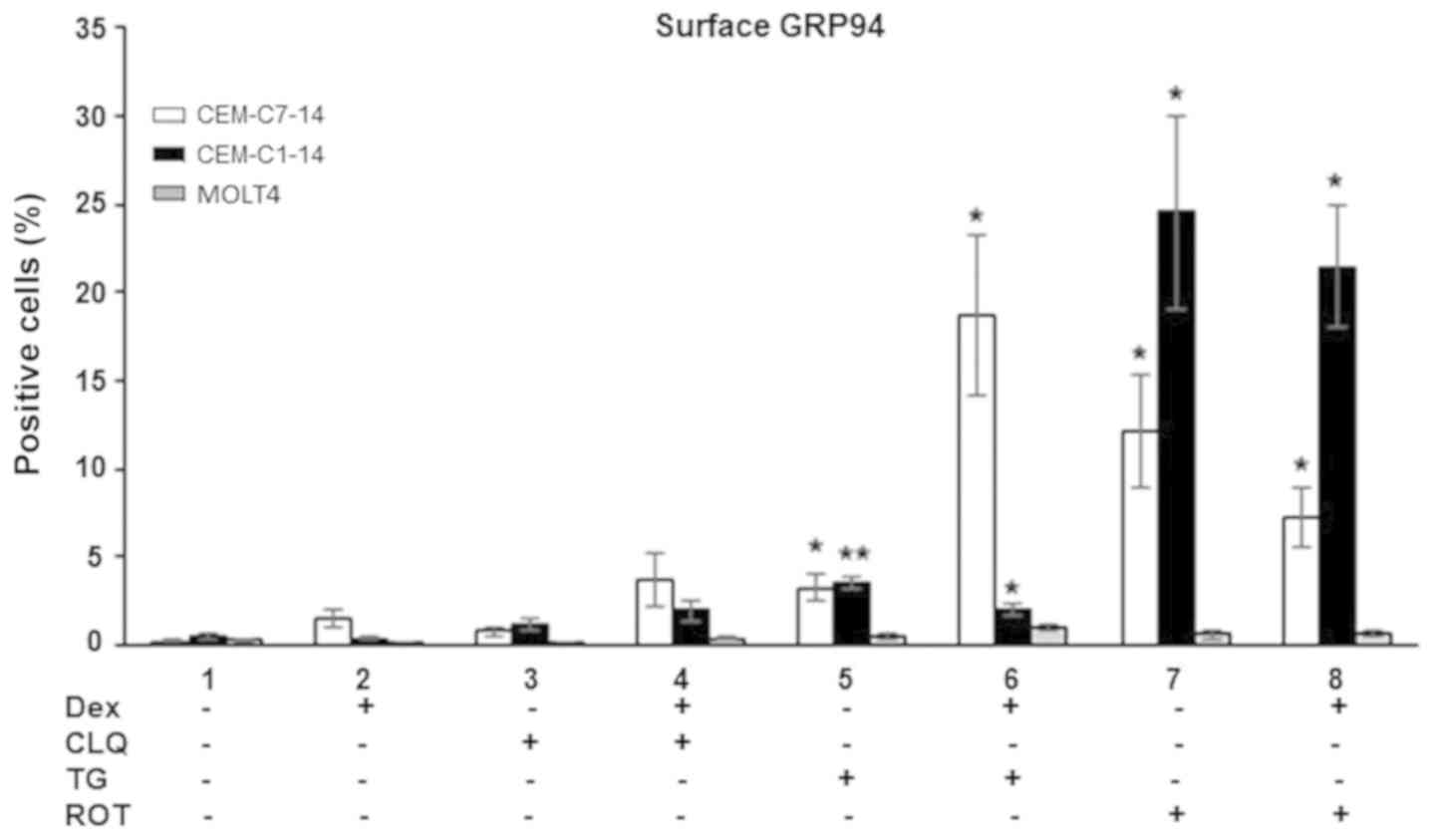 | Figure 5GRP94 surface expression levels on
ALL cells. The CEM-C7-14 (white bars), CEM-C1-15 (black bars) and
MOLT4 (grey bars) cells stained positive for GRP94 surface
expression were detected by flow cytometry after 48-h treatments
with 1 µM Dex, 50 µM CLQ, 10 µM TG and 10
µM ROT alone or in combination as indicated. Data are
presented as the mean ± SEM. *P<0.05,
**P<0.01 vs. untreated control. ALL, acute
lymphoblastic leukaemia; Dex, dexamethasone; CLQ, chloroquine; TG,
thapsigargin; ROT, rotenone; GRP, glucose-regulated protein. |
These results suggested that GC or CLQ treatment
alone did not significantly affect GRP94 surface location, whereas
ER stress and ROS generation increased the GRP94 surface levels in
CEM-C7-14 and CEM-C1-15 cells. Dex potentiated the effects of TG in
CEM-C7-14 cells and inhibited TG effect in CEM-C1-15 cells, and ROT
displayed more potent effects in the GC-resistant CEM-C1-15 cells
compared with those in CEM-C7-14 cells.
Discussion
The main therapeutic options for patients with ALL
are based on treatment with GC hormones, which exert their effects
by inducing apoptosis of malignant T cells through intrinsic and
extrinsic pathways (50). As a
result of greater potency and CNS penetration, dexamethasone is
frequently selected as the treatment of choice for T-ALL (42,43).
A major drawback of GC treatment is the development of resistance
(57). Inhibition of cell death
and resistance of T cells to GC treatment has been attributed to a
variety of molecular mechanisms (58), including autophagy (59). Autophagy is important for numerous
physiological and pathological processes and for the function of
immune system cells from antigen presentation to inflammatory
signalling and metabolism (60).
Various forms of autophagy triggered by distinct stimuli, such as
ER stress, Ca2+ homeostasis and ROS signalling, control
T cell survival (61). Since ER
stress and UPR contribute to inflammatory signalling by activating
a number of stress-responsive kinases including ERK, p38
mitogen-activated protein kinase and JNK (62), which have been demonstrated to
induce GR post-translational modifications that affect its
transcriptional activity (51,63,64),
it is possible that these mechanisms lead to resistance to
GC-induced apoptosis in ALL by modulating the function of the GR
(51,62-64).
In the present study, Dex, CLQ, TG and ROT were used
to treat GC-sensitive (CEM-C7-14) and resistant (CEM-C1-15) as well
as MOLT4 cells to investigate the roles of autophagy, ER stress,
UPR and oxidative stress in the mechanisms of GC-induced ALL
apoptosis, development of resistance and potential ways to overcome
it. Characterisation of the CEM genetic alterations has been
reported in the literature (47-49,65).
Shedding light on this topic may facilitate the improvement of the
current therapeutics and the development of novel anti-ALL
treatments.
Autophagy has been reported to promote or suppress
the proliferation of ALL cells (66). Recent studies have indicated that
autophagy and ROS may serve a significant role in the determination
of effects of GC treatment on ALL cells (59,67-69).
Treatment of the GC-sensitive and resistant leukaemia cells with
the autophagy inhibitor CLQ reduced their viability, and this
reagent appeared to be the most effective in inducing GC- resistant
ALL cell death. These findings suggested that autophagy may be
involved in the GR-induced cell death (70) and it may facilitate the activation
of the pro-survival mechanisms in GC-resistant cells, which was in
agreement with previous studies indicating cytoprotective effects
of autophagy in T-ALL cells treated with PI3K/mTOR or Akt
inhibitors (71).
Accumulating evidence indicates an association
between ROS generation and autophagy with alterations in the
mitochondrial permeability transition pore (MPTP), ER stress,
Ca2+ homeostasis and apoptosis (20,72).
In addition, the mitochondria-ER contact sites regulate immune cell
survival/death decisions (73).
The results of the present study demonstrated that GR-induced
apoptosis in GC-sensitive ALL cells was associated with the
increase in MMP disruption, suggesting that the regulation of the
MPTP opening is a potential mechanism underlying the effects of Dex
in the determination of ALL cell survival or death. A previous
studies has demonstrated that spliced x-box binding protein 1 and
GRP78 are upregulated in Ph+ leukaemia cell lines,
leading to the activation of the UPR-related apoptosis protein CHOP
(74). Although various cell type-
and treatment duration-dependent effects were observed in the
present study, definite conclusions regarding the chronological
order of the occurrence of ROS generation, ER stress and autophagy
in Dex-induced apoptosis-resistant vs. sensitive T-ALL cells cannot
be drawn based on the experimental approaches used.
In the present study, Dex treatment of the
GC-sensitive cells, but not the GC-resistant cells, led to
increased expression levels of GRP78 and GRP94 compared with those
in untreated cells. High GRP expression has been reported to be
associated with cancer cell aggressiveness and metastatic potential
in several cancer cell lines such as breast carcinoma, prostate
adenocarcinoma, liver cancer, colorectal cancer, multiple myeloma
and leukaemia (75). The results
of the present study demonstrated that Dex stimulated GRP78 and
GRP94 expression in GC-sensitive cells in a
transcription-independent manner. GRP78 inhibits the translocation
function on the ER membrane, preventing Ca2+ leakage
from the ER to the cytoplasm (36)
and altering the Ca2+-dependent mitochondrial apoptosis
(73), which may explain the
differential response of CEM-C7-14 and CEM-C1-15 cells to Dex
treatment.
The results of the present study which indicated
that Dex induced GRP94 expression in the GC-sensitive ALL cells
were unexpected, as GRP78 and GRP94 upregulation is associated with
a negative prognosis in various types of cancer such as prostate,
oesophageal, gastric, breast and lung cancer (75). The role of GRP94 in the stimulation
of T cells and regulation of immunity as well as induction of
antitumour activity has been indicated (76). Surface localization of GRP94 is an
important level of control of its function (77-79).
In the present study, GCs induced the intracellular protein levels
of these chaperones, but did not affect the mRNA or surface GRP94
expression levels; however, when Dex was combined with TG, the
GRP94 surface expression increased in the GC-sensitive cells and
decreased in the GC-resistant cells, suggesting that selective
mechanisms determining the subcellular location of GRP94 may
operate in the GC-resistant and sensitive cells. The molecular
mechanisms involved in the regulation of resistance or sensitivity
of ALL cells to Dex treatment through the induction of GRP94 are
novel observations described in the present manuscript. In
addition, considering the role of GRP proteins in the process of
immunosurveillance and immune response (26,80),
it may be speculated that the changes occurring in the GRP levels
and localization may directly or indirectly affect the immune
response to the drug treatment. Therefore, it is possible that by
differentially modulating the GRP94 surface levels in resistant and
sensitive ALL cells co-treated with Dex and TG or ROT, GCs
facilitate their recognition and elimination by the immune system,
highlighting the need for further investigation of the secreted GRP
levels and their effects on the immune system.
Limitations of the present study include the lack of
an additional control cell line to consolidate the findings, and
images demonstrating the morphology of the cells in culture were
not provided. The effects of Dex alone or in combination with TG or
ROT in B-ALL cells were not investigated, which should be addressed
in the future. Of note, in vivo and in vitro drug
doses are not comparable (81).
Future in vivo studies with patients are required to verify
the findings of the present study.
In conclusion, the drugs tested in the present study
decreased the viability of CEM-C7-14 cells via mitochondria-ER
communication mechanisms, upregulating GRP78 and GRP94 and inducing
cell death (Fig. 6). These
observations, if confirmed in clinical settings, may be used to
increase the visibility of ALL cells to the immune system for the
stratification of patients for immunotherapy and increased
therapeutic success.
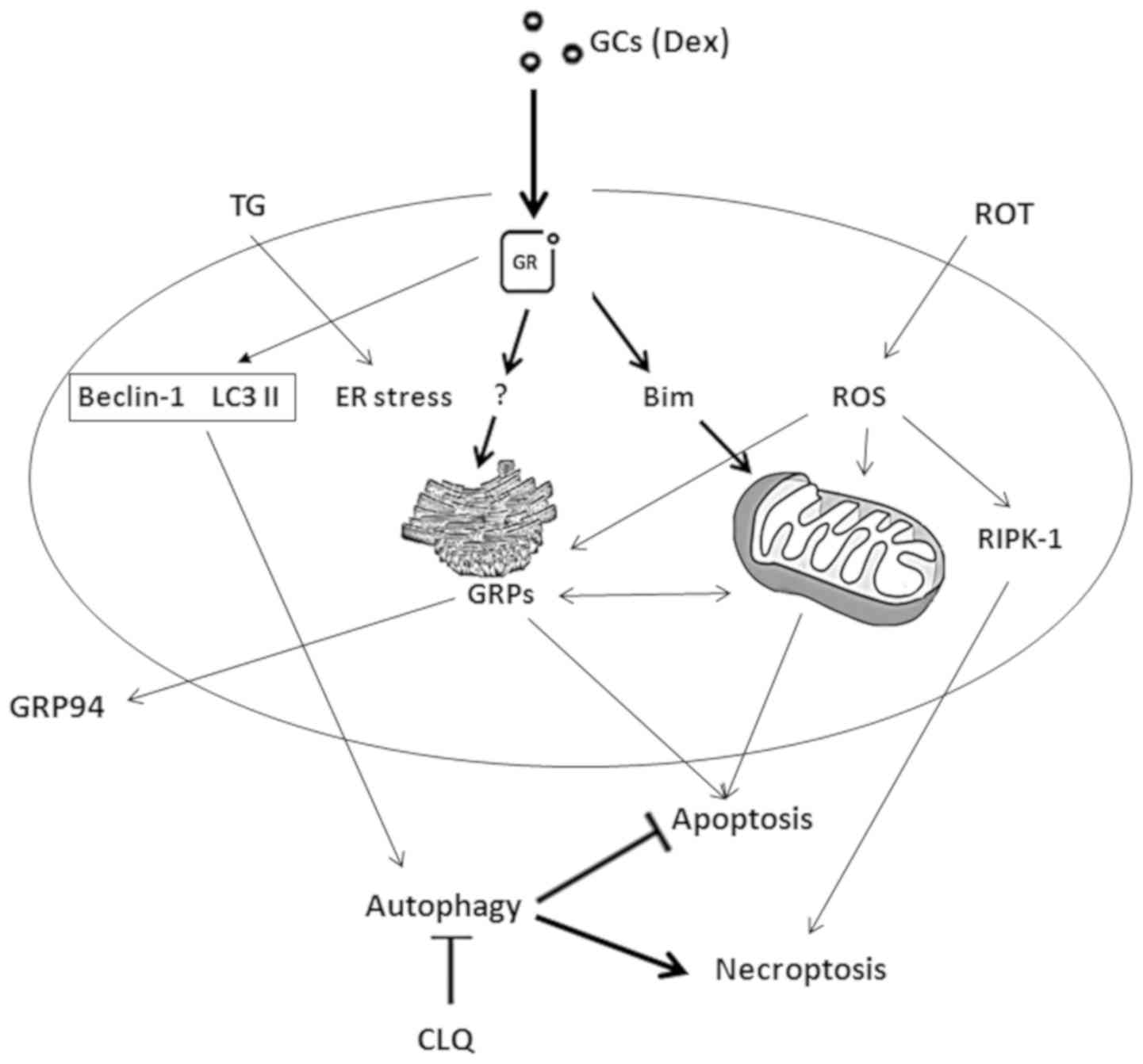 | Figure 6Summary of the hypothetical
GC-mediated pathways leading to T-ALL cell death. GCs induce cell
death in the sensitive CEM-C7-14 cells by inducing mitochondria and
ER stress-mediated cell death through the induction of Bim and
GRPs, respectively. Alteration of mitochondrial membrane potential
may be involved in mitochondria-mediated apoptosis induced by ROT.
GRP activation triggered by TG possibly regulates ER
stress-mediated cell death. CLQ-mediated inhibition of autophagy in
CEM-C7-14 and CEM-C1-15 or induction of ROS generation by ROT in
these cells may stimulate other types of cell death (e.g.
necroptosis). ER stress and induction of ROS generation affect
GRP94 plasma membrane relocalisation; the magnitude of this effect
is differentially regulated in CEM-C7-14 and CEM-C1-15 and may be
hormone-dependent. T-ALL, T-cell acute lymphoblastic leukaemia;
GCs, glucocorticoids; GRP, glucose-regulated protein; Dex,
dexamethasone; CLQ, chloroquine; TG, thapsigargin; ROT, rotenone;
ROS, reactive oxygen species; ER, endoplasmic reticulum. |
Supplementary Data
Acknowledgments
Part of this study was included in S. Sudsaward's
PhD thesis.
Funding
This study was supported by the Royal Thai
Government Scholarships, Ministry of Science and Technology,
Thailand (to SS), the Staff Development Fund, Naresuan University
(SK) the Thailand Research Fund (TRF; grant no. IRG5980006 to PY),
the TRF-International Research Network (grant no. IRN58W001 to PY),
the Siriraj Research Fund (grant no. R016034008 to PY), the Siriraj
Chalermprakiat Grant (to PY and TL) and a TRF-Royal Golden Jubilee
Ph.D. Scholarship (no. PHD/0044/2556 to CT).
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
SS planned and performed experiments, analysed the
results and prepared the draft of the manuscript. SK, CT and AO
performed the experiments, analysed the results and reviewed the
manuscript. TL, PTY and LM analysed the results and reviewed the
manuscript. MKD and CD formulated the hypothesis and supervised the
study, interpreted the results and prepared the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arlt W and Stewart PM: Adrenal
corticosteroid biosynthesis, metabolism, and action. Endocrinol
Metab Clin North Am. 34:293–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galon J, Franchimont D, Hiroi N, Frey G,
Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP and
Bornstein SR: Gene profiling reveals unknown enhancing and
suppressive actions of glucocorticoids on immune cells. FASEB J.
16:61–71. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith MA, Seibel NL, Altekruse SF, Ries
LA, Melbert DL, O'Leary M, Smith FO and Reaman GH: Outcomes for
children and adolescents with cancer: Challenges for the
twenty-first century. J Clin Oncol. 28:2625–2634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greaves M: Infection, immune responses and
the aetiology of childhood leukaemia. Nat Rev Cancer. 6:193–203.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Greaves MF and Wiemels J: Origins of
chromosome translocations in childhood leukaemia. Nat Rev Cancer.
3:639–649. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krajinovic M, Sinnett H, Richer C, Labuda
D and Sinnett D: Role of NQO1, MPO and CYP2E1 genetic polymorphisms
in the susceptibility to childhood acute lymphoblastic leukemia.
Int J Cancer. 97:230–236. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Webster JC, Oakley RH, Jewell CM and
Cidlowski JA: Proinflammatory cytokines regulate human
glucocorticoid receptor gene expression and lead to the
accumulation of the dominant negative beta isoform: A mechanism for
the generation of glucocorticoid resistance. Proc Natl Acad Sci
USA. 98:6865–6870. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alves NL, Derks IA, Berk E, Spijker R, van
Lier RA and Eldering E: The Noxa/Mcl-1 axis regulates
susceptibility to apoptosis under glucose limitation in dividing T
cells. Immunity. 24:703–716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berrou I, Demonacos C and Krstic-Demonacos
M: Molecular mechanisms conferring resistance/sensitivity to
glucocorticoid-induced apoptosis. Glucocorticoids - New Recognition
of Our Familiar Friend. Qian X: InTech; pp. 151–174. 2012
|
|
10
|
Rosen DB, Putta S, Covey T, Huang YW,
Nolan GP, Cesano A, Minden MD and Fantl WJ: Distinct patterns of
DNA damage response and apoptosis correlate with Jak/Stat and
PI3kinase response profiles in human acute myelogenous leukemia.
PLoS One. 5:e124052010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Healy J, Bélanger H, Beaulieu P, Larivière
M, Labuda D and Sinnett D: Promoter SNPs in G1/S checkpoint
regulators and their impact on the susceptibility to childhood
leukemia. Blood. 109:683–692. 2007. View Article : Google Scholar
|
|
12
|
Nicolaides NC, Galata Z, Kino T, Chrousos
GP and Charmandari E: The human glucocorticoid receptor: Molecular
basis of biologic function. Steroids. 75:1–12. 2010. View Article : Google Scholar :
|
|
13
|
Tome ME, Jaramillo MC and Briehl MM:
Hydrogen peroxide signaling is required for glucocorticoid-induced
apoptosis in lymphoma cells. Free Radic Biol Med. 51:2048–2059.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu
Y and Dong W: ROS and ROS-mediated cellular signaling. Oxid Med
Cell Longev. 2016:43509652016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Burhans WC and Heintz NH: The cell cycle
is a redox cycle: Linking phase-specific targets to cell fate. Free
Radic Biol Med. 47:1282–1293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liemburg-Apers DC, Willems PH, Koopman WJ
and Grefte S: Interactions between mitochondrial reactive oxygen
species and cellular glucose metabolism. Arch Toxicol.
89:1209–1226. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsiotra PC and Tsigos C: Stress, the
endoplasmic reticulum, and insulin resistance. Ann NY Acad Sci.
1083:63–76. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kato H and Nishitoh H: Stress responses
from the endoplasmic reticulum in cancer. Front Oncol. 5:932015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cao SS and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress in cell fate decision and
human disease. Antioxid Redox Signal. 21:396–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zeeshan HM, Lee GH, Kim HR and Chae HJ:
Endoplasmic reticulum stress and associated ROS. Int J Mol Sci.
17:3272016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Almanza A, Carlesso A, Chintha C,
Creedican S, Doultsinos D, Leuzzi B, Luís A, McCarthy N,
Montibeller L, More S, et al: Endoplasmic reticulum stress
signalling - from basic mechanisms to clinical applications. FEBS
J. 286:241–278. 2019. View Article : Google Scholar
|
|
22
|
Llanos-González E, Henares-Chavarino ÁA,
Pedrero-Prieto CM, García-Carpintero S, Frontiñán-Rubio J,
Sancho-Bielsa FJ, Alcain FJ, Peinado JR, Rabanal-Ruíz Y and
Durán-Prado M: Interplay between mitochondrial oxidative disorders
and proteostasis in Alzheimer's disease. Front Neurosci.
13:14442020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Delprat B, Crouzier L, Su TP and Maurice
T: At the crossing of ER stress and MAMs: A key role of Sigma-1
receptor. Calcium Signaling. Islam MS: Springer International
Publishing; Cham: pp. 699–718. 2020, View Article : Google Scholar
|
|
24
|
Ferro F, Servais S, Besson P, Roger S,
Dumas JF and Brisson L: Autophagy and mitophagy in cancer metabolic
remodelling. Semin Cell Dev Biol. 98:129–138. 2020. View Article : Google Scholar
|
|
25
|
Brown MK and Naidoo N: The endoplasmic
reticulum stress response in aging and age-related diseases. Front
Physiol. 3:2632012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Binder RJ: Functions of heat shock
proteins in pathways of the innate and adaptive immune system. J
Immunol. 193:5765–5771. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Merksamer PI and Papa FR: The UPR and cell
fate at a glance. J Cell Sci. 123:1003–1006. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Corazzari M, Gagliardi M, Fimia GM and
Piacentini M: Endoplasmic reticulum stress, unfolded protein
response, and cancer cell fate. Front Oncol. 7:782017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar
|
|
32
|
Kharabi Masouleh B, Chevet E, Panse J,
Jost E, O'Dwyer M, Bruemmendorf TH and Samali A: Drugging the
unfolded protein response in acute leukemias. J Hematol Oncol.
8:872015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sano R and Reed JC: ER stress-induced cell
death mechanisms. Biochim Biophys Acta. 1833:3460–3470. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Csordás G, Weaver D and Hajnóczky G:
Endoplasmic reticulum-mitochondrial contactology: Structure and
signaling functions. Trends Cell Biol. 28:523–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Akl H, Vervloessem T, Kiviluoto S,
Bittremieux M, Parys JB, De Smedt H and Bultynck G: A dual role for
the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus
endoplasmic reticulum. Biochim Biophys Acta. 1843:2240–2252. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hammadi M, Oulidi A, Gackière F,
Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P,
Lepage G, Lotteau S, et al: Modulation of ER stress and apoptosis
by endoplasmic reticulum calcium leak via translocon during
unfolded protein response: Involvement of GRP78. FASEB J.
27:1600–1609. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xia M, Zhang Y, Jin K, Lu Z, Zeng Z and
Xiong W: Communication between mitochondria and other organelles: A
brand-new perspective on mitochondria in cancer. Cell Biosci.
9:272019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kondratskyi A, Kondratska K, Skryma R,
Klionsky DJ and Prevarskaya N: Ion channels in the regulation of
autophagy. Autophagy. 14:3–21. 2018. View Article : Google Scholar :
|
|
39
|
Sun F, Xu X, Wang X and Zhang B:
Regulation of autophagy by Ca(2). Tumour Biol. 37:15467–15476.
2016. View Article : Google Scholar
|
|
40
|
Bootman MD, Chehab T, Bultynck G, Parys JB
and Rietdorf K: The regulation of autophagy by calcium signals: Do
we have a consensus? Cell Calcium. 70:32–46. 2018. View Article : Google Scholar
|
|
41
|
Kania E, Pająk B and Orzechowski A:
Calcium homeostasis and ER stress in control of autophagy in cancer
cells. BioMed Res Int. 2015:3527942015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Raetz EA and Teachey DT: T-cell acute
lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program.
2016:580–588. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Terwilliger T and Abdul-Hay M: Acute
lymphoblastic leukemia: A comprehensive review and 2017 update.
Blood Cancer J. 7:e5772017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Irwin ME, Rivera-Del Valle N and Chandra
J: Redox control of leukemia: From molecular mechanisms to
therapeutic opportunities. Antioxid Redox Signal. 18:1349–1383.
2013. View Article : Google Scholar :
|
|
45
|
Harmon JM and Thompson EB: Isolation and
characterization of dexamethasone-resistant mutants from human
lymphoid cell line CEM-C7. Mol Cell Biol. 1:512–521. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Foley GE, Lazarus H, Farber S, Uzman BG,
Boone BA and McCarthy RE: Continuous culture of human lymphoblasts
from peripheral blood of a child with acute leukemia. Cancer.
18:522–529. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Saenz GJ, Hovanessian R, Gisis AD and Medh
RD: Glucocorticoid-mediated co-regulation of RCAN1-1, E4BP4 and BIM
in human leukemia cells susceptible to apoptosis. Biochem Biophys
Res Commun. 463:1291–1296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jiang L, Xu L, Xie J, Li S, Guan Y, Zhang
Y, Hou Z, Guo T, Shu X, Wang C, et al: Inhibition of autophagy
overcomes gluco-corticoid resistance in lymphoid malignant cells.
Cancer Biol Ther. 16:466–476. 2015. View Article : Google Scholar :
|
|
49
|
Norman MR and Thompson EB:
Characterization of a gluco-corticoid-sensitive human lymphoid cell
line. Cancer Res. 37:3785–3791. 1977.PubMed/NCBI
|
|
50
|
Lynch JT, Rajendran R, Xenaki G, Berrou I,
Demonacos C and Krstic-Demonacos M: The role of glucocorticoid
receptor phosphorylation in Mcl-1 and NOXA gene expression. Mol
Cancer. 9:382010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Qattan MY, Bakker EY, Rajendran R, Chen
DW, Saha V, Liu J, Zeef L, Schwartz JM, Mutti L, Demonacos C, et
al: Differential regulation of cell death pathways by the
microenvironment correlates with chemoresistance and survival in
leukaemia. PLoS One. 12:e01786062017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
53
|
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr
M, Hijlkema KJ, Coppes RP, Engedal N, Mari M and Reggiori F:
Chloroquine inhibits autophagic flux by decreasing
autophagosome-lysosome fusion. Autophagy. 14:1435–1455. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sehgal P, Szalai P, Olesen C, Praetorius
HA, Nissen P, Christensen SB, Engedal N and Møller JV: Inhibition
of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by
thapsigargin analogs induces cell death via ER Ca2+
depletion and the unfolded protein response. J Biol Chem.
292:19656–19673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Y, McMillan-Ward E, Kong J, Israels
SJ and Gibson SB: Mitochondrial electron-transport-chain inhibitors
of complexes I and II induce autophagic cell death mediated by
reactive oxygen species. J Cell Sci. 120:4155–4166. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zorova LD, Popkov VA, Plotnikov EY,
Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD,
Balakireva AV, Juhaszova M, et al: Mitochondrial membrane
potential. Anal Biochem. 552:50–59. 2018. View Article : Google Scholar :
|
|
57
|
Wilkinson L, Verhoog NJ and Louw A:
Disease- and treatment-associated acquired glucocorticoid
resistance. Endocr Connect. 7:R328–R349. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bakker E, Qattan M, Mutti L, Demonacos C
and Krstic-Demonacos M: The role of microenvironment and immunity
in drug response in leukemia. Biochim Biophys Acta. 1863:414–426.
2016. View Article : Google Scholar
|
|
59
|
Djavaheri-Mergny M, Giuriato S, Tschan MP
and Humbert M: Therapeutic modulation of autophagy in leukaemia and
lymphoma. Cells. 8:E1032019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Pua HH and He YW: Autophagy and lymphocyte
homeostasis. Curr Top Microbiol Immunol. 335:85–105.
2009.PubMed/NCBI
|
|
61
|
Botbol Y, Guerrero-Ros I and Macian F: Key
roles of autophagy in regulating T-cell function. Eur J Immunol.
46:1326–1334. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hotamisligil GS and Davis RJ: Cell
signaling and stress responses. Cold Spring Harb Perspect Biol.
8:a0060722016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Davies L, Karthikeyan N, Lynch JT, Sial
EA, Gkourtsa A, Demonacos C and Krstic-Demonacos M: Cross talk of
signaling pathways in the regulation of the glucocorticoid receptor
function. Mol Endocrinol. 22:1331–1344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen DW, Saha V, Liu JZ, Schwartz JM and
Krstic-Demonacos M: Erg and AP-1 as determinants of glucocorticoid
response in acute lymphoblastic leukemia. Oncogene. 32:3039–3048.
2013. View Article : Google Scholar
|
|
65
|
Medh RD, Webb MS, Miller AL, Johnson BH,
Fofanov Y, Li T, Wood TG, Luxon BA and Thompson EB: Gene expression
profile of human lymphoid CEM cells sensitive and resistant to
glucocorticoid-evoked apoptosis. Genomics. 81:543–555. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Evangelisti C, Evangelisti C, Chiarini F,
Lonetti A, Buontempo F, Neri LM, McCubrey JA and Martelli AM:
Autophagy in acute leukemias: A double-edged sword with important
therapeutic implications. Biochim Biophys Acta. 1853:14–26. 2015.
View Article : Google Scholar
|
|
67
|
Auberger P and Puissant A: Autophagy, a
key mechanism of oncogenesis and resistance in leukemia. Blood.
129:547–552. 2017. View Article : Google Scholar
|
|
68
|
Takahashi H, Inoue J, Sakaguchi K, Takagi
M, Mizutani S and Inazawa J: Autophagy is required for cell
survival under L-asparaginase-induced metabolic stress in acute
lymphoblastic leukemia cells. Oncogene. 36:4267–4276. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Rothe K, Porter V and Jiang X: Current
outlook on autophagy in human leukemia: Foe in cancer stem cells
and drug resistance, friend in new therapeutic interventions. Int J
Mol Sci. 20:E4612019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Laane E, Tamm KP, Buentke E, Ito K,
Kharaziha P, Oscarsson J, Corcoran M, Björklund AC, Hultenby K,
Lundin J, et al: Cell death induced by dexamethasone in lymphoid
leukemia is mediated through initiation of autophagy. Cell Death
Differ. 16:1018–1029. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Simioni C, Neri LM, Tabellini G, Ricci F,
Bressanin D, Chiarini F, Evangelisti C, Cani A, Tazzari PL,
Melchionda F, et al: Cytotoxic activity of the novel Akt inhibitor,
MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia.
26:2336–2342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Verfaillie T, Salazar M, Velasco G,
Agostinis P and Linking ER: Linking ER stress to autophagy:
Potential implications for cancer therapy. Int J Cell Biol.
2010:9305092010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Martinvalet D: The role of the
mitochondria and the endoplasmic reticulum contact sites in the
development of the immune responses. Cell Death Dis. 9:3362018.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dengler MA, Staiger AM, Gutekunst M,
Hofmann U, Doszczak M, Scheurich P, Schwab M, Aulitzky WE and van
der Kuip H: Oncogenic stress induced by acute hyper-activation of
Bcr-Abl leads to cell death upon induction of excessive aerobic
glycolysis. PLoS One. 6:e251392011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lee AS: Glucose-regulated proteins in
cancer: Molecular mechanisms and therapeutic potential. Nat Rev
Cancer. 14:263–276. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Biswas C, Sriram U, Ciric B, Ostrovsky O,
Gallucci S and Argon Y: The N-terminal fragment of GRP94 is
sufficient for peptide presentation via professional
antigen-presenting cells. Int Immunol. 18:1147–1157. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ansa-Addo EA, Thaxton J, Hong F, Wu BX,
Zhang Y, Fugle CW, Metelli A, Riesenberg B, Williams K, Gewirth DT,
et al: Clients and oncogenic roles of molecular chaperone
gp96/grp94. Curr Top Med Chem. 16:2765–2778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Duan XF and Xin YW: Overexpression of
molecule GRP94 favors tumor progression in lung adenocarcinoma by
interaction with regulatory T cells. Thorac Cancer. 11:704–712.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wiersma VR, Michalak M, Abdullah TM,
Bremer E and Eggleton P: Mechanisms of translocation of ER
chaperones to the cell surface and immunomodulatory roles in cancer
and autoimmunity. Front Oncol. 5:72015. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Binder RJ: Immunosurveillance of cancer
and the heat shock protein-CD91 pathway. Cell Immunol.
343:1038142019. View Article : Google Scholar
|
|
81
|
Sundman-Engberg B, Tidefelt U, Gruber A
and Paul C: Intracellular concentrations of mitoxantrone in
leukemic cells in vitro vs. in vivo Leuk Res. 17:347–52. 1993.
View Article : Google Scholar
|














