Introduction
Colorectal cancer (CRC) is the third most common
malignancy and the fourth highest cause of cancer-related
mortality, accounting for approximately 1.36 million new cases and
694,000 deaths worldwide in 2012 (1,2).
Several risk factors are associated with the development of CRC,
including familial history, inherited genetic mutations and food
habits (3). A main cause of CRC,
epigenetic alteration, can drive the transformation from normal
cells to cancer (4). Therefore,
such alterations are commonly perceived to be good molecular
targets for future CRC treatments.
Histone deacetylases (HDACs) are a family of crucial
epigenetic enzymes that play an important role in the regulation of
gene expression. The 18 isoforms of this family are classified into
4 groups: The zinc-dependent HDACs comprise class I (HDAC1, 2, 3
and 8), class II (HDAC4, 5, 6, 7, 9 and 10) and class IV (HDAC 11),
and the NAD+-dependent HDACs which belong to class III
(SIRT1-7) (5-7). HDAC1 and HDAC2, members of HDAC class
I, are found in mammalian cell nuclei and are located in 3 major,
stable, multiprotein co-repressor complexes: Sin3, nucleosome
remodeling and deacetylation (NuRD) and co-repressor for
element-1-silencing transcription factor (CoREST) (8,9).
The effects of HDACs on cell behavior were recently
demonstrated in several studies. HDAC1 and 2 antagonize tumor
suppressor p53 in the regulation of the cyclin-dependent kinase
(CDK) inhibitor p21, a key component in cell cycle control and
apoptosis (10-12). The knockdown of HDAC1 and 2 induces
the expression of CDK inhibitors, which leads to a cell cycle block
in G1 and apoptotic cell death (13). Furthermore, the aberrant
upregulation of HDAC1/2 is involved in the progression of various
types of cancer. Therefore, the inhibition of HDAC1/2 has emerged
as an effective anticancer treatment.
In CRC cells, several studies have revealed that the
upregulation pf HDAC1/2 found at the beginning of colon
carcinogenesis is implicated in cell tumorigenicity via chromatin
structure remodeling (14,15). The epithelial-mesenchymal
transition (EMT) process, which plays a role in the growth of
several types of CRC, is involved in aberrant HDAC expression
through the SNAIL transcription factors (16-18).
In addition, a decline in HDAC1 and 2 is considered to inhibit the
proliferation and induce the death of several CRC cell lines by
activating specific tumor-suppressor genes (19,20).
Thus, the development of CRC-targeted HDAC inhibitors is
potentially valuable in the search for new selective chemotherapy
agents. Several types of HDAC inhibitors have recently been
developed for the treatment of CRC (21-26).
Hydroxamic acid-based HDAC inhibitors have been used in the
treatment of CRC. For example, suberoylanilide hydroxamic acid
(SAHA), approved by the FDA for the treatment of T-cell lymphoma,
has been tested on solid tumors, including CRC (27,28).
However, unselective HDAC inhibition (SAHA inhibits approximately
11 isoforms of HDAC) could have undesirable side-effects (29,30).
Benzamide-type HDAC inhibitors also potentially inhibit several
cancer cell lines (26,31). The benzamide-type structure has a
higher affinity to zinc ions than hydroxamate-type HDAC inhibitors,
which produces highly selective class I HDAC inhibition and thereby
significantly reduces the side-effects of HDAC inhibition (32). Several previous studies have
modified benzamide-type HDAC inhibitors by adding an aryl group at
the aniline to bind to the 14-Å-long internal cavity, known to be
where the acetate byproduct is released after enzymatic hydrolysis.
The resulting increase in potency indicates that the modification
produces benefits for benzamide-type HDAC inhibitors (33-35).
The present study wished to discover a novel, potent
HDAC inhibitor that could serve as an antitumor agent via its
benzamide moiety. From previous studies on benzamide-type HDAC
inhibitors, it was expected that structural modification of the
internal cavity and a cap group would enhance the binding potency
of a benzamide-type HDAC inhibitor and improve its inhibition
activity and antitumor activity (32-35).
The present study reports the design and synthesis of a novel
fluorinated aminophenyl-benzamide-based compound, CBUD-1001, along
with biological evaluations of CBUD-1001 as a novel HDAC inhibitor
with potent antitumor agent against CRC.
Materials and methods
HDAC inhibitory assay
The HDAC inhibition assay of CBUD-1001 was conducted
by the Reaction Biology Corporation. CBUD-1001 was suspended in 10
mM DMSO stock solution. The compound was tested in singlet 10-dose
inhibitory concentration (IC50) mode with 3-fold serial
dilution starting at 1 µM against HDAC1 and 2. A fluorogenic
peptide from p53 residues 379-382 [RHKK(Ac)AMC] was used as the
substrate for HDAC1 and 2. IC50 values were calculated
using the GraphPad Prism 4 program based on a sigmoidal
dose-response equation. The blank (DMSO) value was entered as a
concentration of 1.00E−12 for curve fitting.
Molecular docking analysis
A docking model of CBUD-1001 was calculated on an
Intel® Core™ i7-8700 CPU @ 3.20 GHz (12 CPUs), using
Schrödinger Maestro 11.9 software (Schrödinger, LLC) and an HDAC
model (4LY1) available on a protein data bank (36). A grid box was created with the
following specifications: VdW radii of protein atoms scaled by 1.0;
charged cut-off for polarity 0.25; receptor setup (nsite, nx, ny,
nz, bsite)=(125, 19, 19, 19, 1.0); Dockman after grid: (nx, ny,
nz)=(56, 56, 56). The inhibitory ability of the
compound was expected to follow the calculated docking score. 2D
and 3D images of the interactions were also recorded.
Cell lines and cell culture
Human CRC cell lines (HCT116 and DLD-1) were
purchased from the American Type Culture Collection. The 3 human
CRC cell lines were validated by short-tandem repeat (STR) DNA
fingerprinting using the Promega PowerPlex 18D System and analyzed
by GeneMapper Software 5 at Cosmo Genetech Korea. Human intestinal
epithelial cells (IECs) were purchased from Lonza as a normal
control. The CRC cells were cultured in RPMI-1640 medium
supplemented with 10% heat-inactivated fetal bovine serum (FBS),
100 units of penicillin, and 100 units of streptomycin. Human IEC
cells were cultured in SmGM™-2 medium (Lonza Group, Ltd.),
containing various supplements and growth factors (insulin, hFGF-B,
hEGF, FBS and gentamicin/amphotericin-B). All cells were maintained
at 37°C in a humidified incubator with 5% CO2/95%
air.
Cell viability assay
The effects of CBUD-1001 on the proliferation of
human IEC, HCT116 and DLD-1 cells were evaluated using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT,
Sigma-Aldrich; Merck KGaA), and cell viability was associated with
the production of formazan. The cells were plated at a density of
1.0×104 cells per well in 96-well plates. Following
treatment with 0, 0.05, 0.1, 1, 10, 50, 100 and 200 µM of
CBUD-1001 for 24 h, 20 µl of MTT (5 mg/ml) were added to
each well. Following incubation for 4 h at 37°C, the culture medium
containing MTT was removed, and 200 µl of DMSO were added.
This was followed by shaking until the crystals were dissolved.
Viable cells were detected by measuring the absorbance at 570 nm
using a microplate reader (Molecular Devices, LLC). The
IC50 value of CBUD-1001 was then calculated in
comparison to the untreated controls.
Detection of cell apoptosis
Following treatment with 0, 1 and 3 µM of
CBUD-1001 for 24 h, cells were trypsinized, collected and washed
with ice-cold PBS. The following steps were based on the
manufacturer's instructions for the Annexin V-FITC/Propidium Iodide
(PI) Apoptosis Detection kit (BD Biosciences). After staining the
cells with 5 µl of Annexin V-FITC and 5 µl of PI for
15 min at room temperature in the dark, the fluorescence was
measured on a BD LSR flow cytometer (BD Biosciences) and processed
using Cell Quest software (BD Biosciences) for analysis. The
apoptotic features of the cancer cells were assessed by DNA
condensation using Hoechst 33258. The cells were treated with 1 and
3 µM of CBUD-1001 for 24 h and then stained with Hoechst
33258 (1 µg/ml) at 37°C for 10 min. Nuclear morphology was
examined under a confocal laser scanning microscope (Carl Zeiss AG)
to identify cells undergoing apoptosis.
Colony formation assay
For the colony formation assay, cells
(1×102 cells/well) were seeded into a 6-well plate and
then treated with the 0, 1 and 3 µM of CBUD-1001 for 24 h.
The cells were then washed with cold 1X PBS, and fresh growth
medium was added. Following 14 days of culture, the colonies were
fixed with 3.8% formaldehyde for 20 min and stained with 0.1%
crystal violet (Sigma-Aldrich; Merck KGaA) for 10 min at room
temperature.
Protein extraction and western blot
analysis
Cells were harvested by resolving them in RIPA
buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 1% sodium
deoxycholate, 0.1% SDS, and protease inhibitors) with a strong
vortex followed by centrifugation at 15,800 × g and 4°C for 30 min.
Following centrifugation, the supernatants were used as whole cell
extracts. The protein concentration in the cell lysates was
measured using a protein quantification kit from Bio-Rad
Laboratories, Inc. A total of 50 or 30 µg of protein per
lane was loaded onto an SDS-polyacrylamide gel. Following transfer
and blocking with 3% bovine serum albumin, the polyvinylidene
difluoride membrane was probed with various antibodies
[anti-Ac-histone H3 (1:1,000, sc-56616; Santa Cruz Biotechnology,
Inc.), anti-Ac-histone H4 (1:1,000, sc-515319, Santa Cruz
Biotechnology, Inc.), anti-caspase 8 (1:1,000, sc-73526, Santa Cruz
Biotechnology, Inc.), anti-tBid (1:1,000, sc-34325, Santa Cruz
Biotechnology, Inc.), anti-Bcl-2 (1:2,000, #2872, Cell Signaling
Technology, Inc.) anti-Bcl-xL (1:1,000, sc-8392, Santa Cruz
Biotechnology, Inc.), anti-Bax (1:1,000, sc-7480, Santa Cruz
Biotechnology, Inc.), anti- cytochrome c (1:1,000, sc-13156,
Santa Cruz Biotechnology, Inc.), anti-X-linked inhibitor of
apoptosis protein (XIAP, 1:2,000, #14334, Cell Signaling
Technology, Inc.), anti-caspase 3 (1:1,000, sc-7148, Santa Cruz
Biotechnology, Inc.), anti-poly(ADP-ribose) polymerase (PARP,
sc-7150, 1:1,000, Santa Cruz Biotechnology, Inc.), anti-E-cadherin
(1:2,000, #3195, Cell Signaling Technology, Inc.), anti-β-catenin
(1:2,000, #9582, Cell Signaling Technology, Inc.), anti- vimentin
(1:2,000, #5741, Cell Signaling Technology, Inc.), anti-matrix
metalloproteinase (MMP)2 (1:2,000, #13132, Cell Signaling
Technology, Inc.), anti-MMP9 (1:2,000, ab76003, Abcam), anti-SNAIL
(1:2,000, ab180714, Abcam), anti-SLUG (1:2,000, ab27568, Abcam) and
anti-actin (1:2,000, A2066, Sigma-Aldrich; Merck KGaA)] at 4°C for
overnight. Membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies [anti-mouse IgG
(1:1,000, sc-2005, Santa Cruz Biotechnology, Inc.), anti-rabbit IgG
(1:1,000, sc-2004, Santa Cruz Biotechnology, Inc.) anti-goat IgG
(1:1,000, sc-2354, Santacruz Biotechnology, Inc.)] for 1 h at room
temperature. Antibody-to-antigen binding was detected using an
Enhanced ECL Prime (GE Healthcare) and was captured and analyzed by
a Las-3000 luminescent image analyzer (Fuji Film).
Measurement of HDAC concentration
A colorimetric HDAC activity assay kit was used to
determine the in vitro HDAC concentration (BioVision, Inc.).
The cells were plated at a density of 1.0×106 cells in a
10-cm dish. Following treatment with CBUD-1001, total cell lysates
were resolved using RIPA buffer. The assay was performed according
to the manufacturer's protocol.
Mitochondrial transmembrane potential
(MMP, Δψm)
The mitochondrial membrane was monitored using
Rhodamine 123 fluorescent dye (Ex/Em=485 nm/535 nm; Sigma-Aldrich;
Merck KGaA), a cell-permeable cationic dye that preferentially
enters the mitochondria due to the highly negative Δψm. The
depolarization of the Δψm results in the loss of Rhodamine 123 from
the mitochondria and a decrease in intracellular fluorescence. In
brief, cells were incubated with 0, 1 and 3 µM of CBUD-1001
for 24 h. The cells were then washed twice with PBS and incubated
with Rhodamine 123 (0.1 µg/ml) at 37°C for 30 min. The
intensity of Rhodamine 123 staining was determined using a BD LSR
flow cytometer (BD Biosciences).
Wound healing assay and morphological
analysis
Cells (1×105) were seeded in 6-cm culture
plates and allowed to form a confluent monolayer. The monolayer was
scraped with a P200 pipette tip to generate a wound ~1 mm wide, and
the cells were then treated with 0, 1 and 3 µM of CBUD-1001.
Images of the wounds were captured at 0, 48 h, and the wound area
was determined using an inverted micro-scope (Olympus IX71, Olympus
Corporation). The ability of the cells to close the wound, as a
measure of motility, was evaluated by determining the healed area.
In addition, morphological alterations of the CRC cells were
observed using an inverted microscope (Olympus IX71, Olympus
Corporation).
Statistical analysis
For data analysis for the in vitro
experiments, one-way ANOVA (for differences between multiple
groups) assuming equality of variance with Tukey's multiple
comparison test, were used. The data are presented as the means ±
SD of at least 3 independent experiments. All data were entered
into Microsoft Excel 5.0, and GraphPad Prism 5.0 was used. A
probability (P)-value <0.05 was considered to indicate a
statistically significant difference.
Results
Synthesis and in vitro HDAC inhibitory
assay of CBUD-1001
The synthesis of the novel HDAC inhibitor
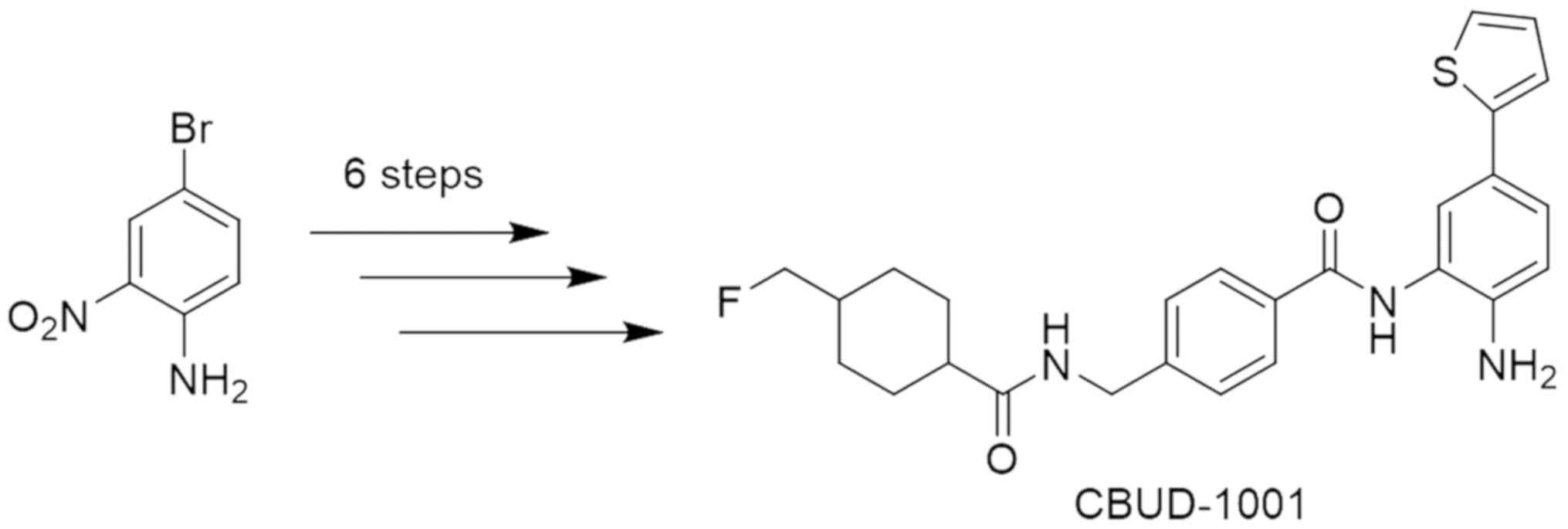
(CBUD-1001) used in the present study was achieved via 6 step
reactions from 4-bromo-2-nitroaniline (Figs. 1, S1
and S2, and Data S1). The newly synthesized compound
(CBUD-1001) was examined for its HDAC1, HDAC2 and HDAC3 inhibitory
activity using a fluorogenic peptide from p53 residues 379-382
[RHKK(Ac)AMC] as the HDAC substrate. The IC50 values of
CBUD-1001 exhibited nanomolar inhibition against HDAC1, and
sub-micromolar inhibition against HDAC2 and HDAC3:
IC50=28.1 nM against HDAC1, IC50=158 nM
against HDAC2, and IC50=404 nM against HDAC3. For the
positive control, the IC50 values of SAHA against HDAC1,
HDAC2 and HDAC3 were 115, 162 and 181 nM. Thus, CBUD-1001 exhibited
potent HDAC inhibitory activity (data not shown).
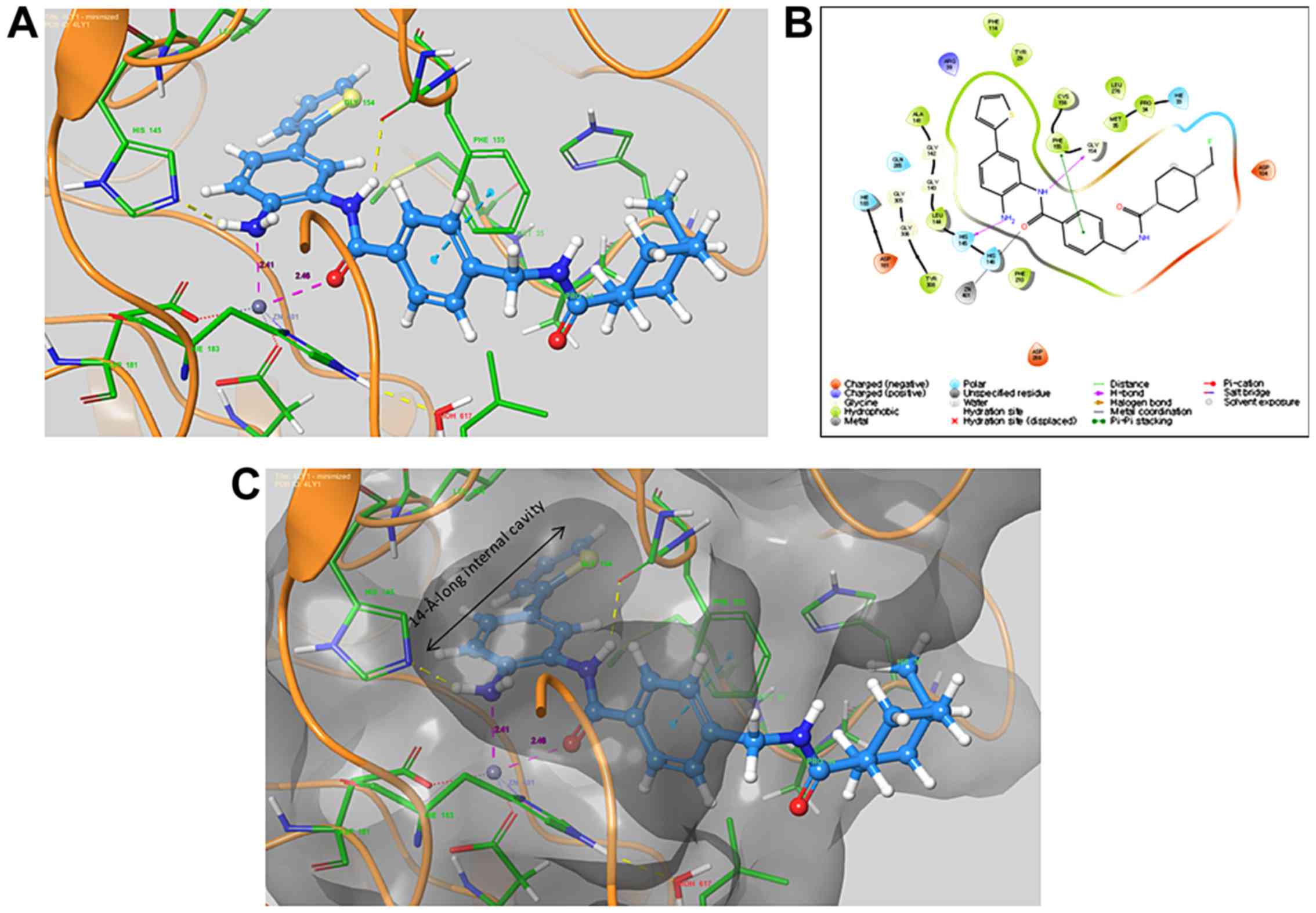
Docking analysis
To better understand the high HDAC inhibitory
activity of CBUD-1001, it was docked into the active sites of HDAC
using the Glide module of Schrödinger software with the Maestro
interface. The docking analysis produced a docking score of -13.283
(kcal/mol). Both the nitrogen in the ortho-NH2 group and
the carbonyl group in the core structure were observed to chelate
with Zn401 (Fig. 2). The
ortho-NH2 group also interacted with His145, and the NH
amide established a hydrogen bond with the C=O on Gly154. There was
a π-π stacking interaction between the phenyl group of the linker
and Phe155. The thiophenyl group was adequately located, with a
14-Å-long internal cavity. The presence of the thiophenyl group was
deemed to significantly increase the potential inhibition toward
HDAC.
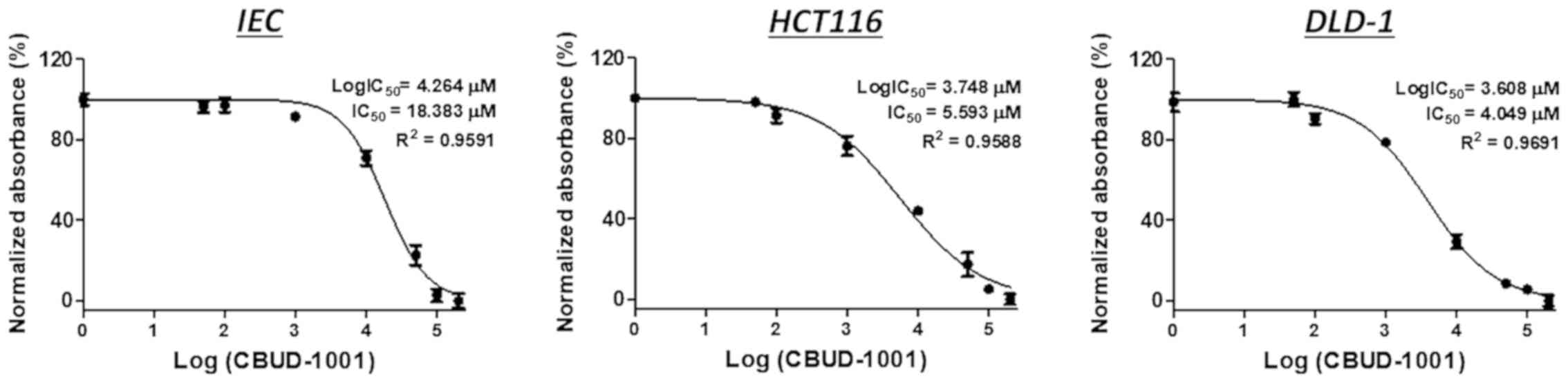
CBUD-1001 sensitively affects human CRC
cell viability
To evaluate the biological effects of CBUD-1001,
cell viability was examined following exposure to CBUD-1001 using
human CRC cells and normal IECs. The cells were treated with
various concentrations of CBUD-1001 (0.05, 0.1, 1, 10, 50, 100 and
200 µM) for 24 h. As revealed by the MTT assay, CBUD-1001
inhibited the growth of both the CRC cells and normal IECs in a
concentration-dependent manner (Fig.
3). However, in calculating the IC50 value of
CBUD-1001, it was confirmed that a higher concentration of
CBUD-1001 was required for the IECs than for the CRC cells. The
IC50 values of CBUD-1001 for 24 h in CRC cells viability
were 5.593 µM in the HCT116 cells and 4.049 µM in the
DLD-1 cells. For the positive control, the IC50 value of
CBID-1001 in the human IECs was 18.383 µM. Thus, the CRC
cells were more sensitive to CBUD-1001 than the normal IECs.
However, the effect of CBUD-1001 on the long-term viability of CRC
cells was not observed; thus, this is a limitation of the present
study.
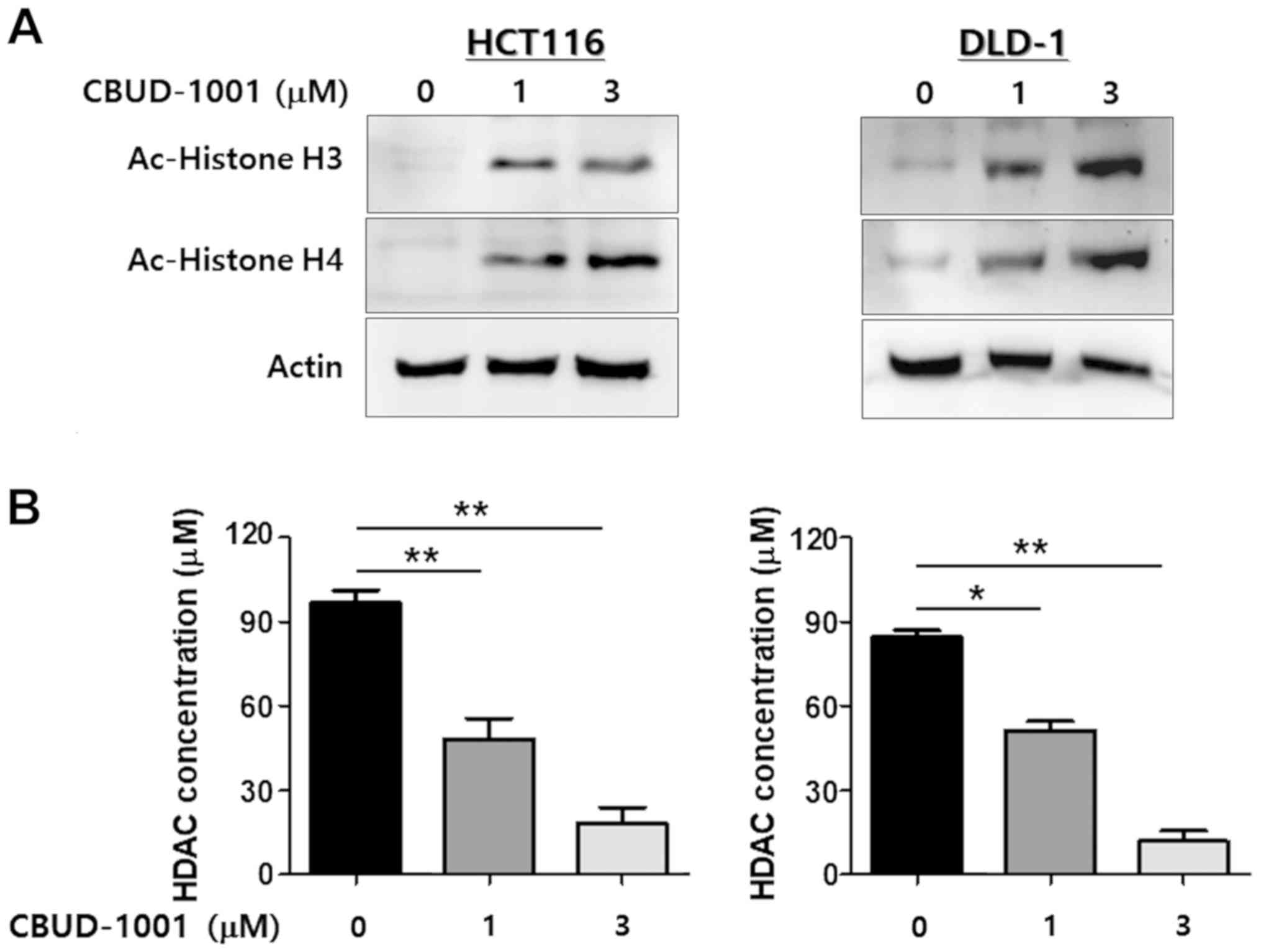
CBUD-1001 suppresses HDAC enzyme activity
in CRC cells
As CBUD-1001 was designed to be an HDAC inhibitor,
western blot analysis and a colorimetric HDAC activity assay with
total cell lysates were used to evaluate its ability to inhibit
HDACs. To confirm the ability of CBUD-1001 to inhibit the
deacetylation of HDACs, western blot analysis was performed to
analyze the acetylation of histones H3 and H4 in CRC cells. As was
revealed by the results, 24 h of CBUD-1001 treatment
dose-dependently increased the acetylation of histones H3 and H4
(Fig. 4A).
HDAC enzymatic activity also was evaluated using an
HDAC activity assay kit, and the exact concentration of HDAC was
calculated with a deacetylated standard (Ac-Lys-pNA 10-100
µM). As shown in Fig. 4B,
the dose-dependent inhibition of HDAC activity by CBUD-1001 was
observed in the CRC cell lines. Thus, these results suggest that
CBUD-1001 is a selective HDAC inhibitor that inactivates HDAC1 in
human CRC cells.
CBUD-1001 triggers the apoptotic death of
human CRC cells
As shown by the results presented in Figs. 3 and 4, it was confirmed that the HCT116 cells
exhibited marked changes in cell viability and HDAC activity by
CBUD-1001 treatment. Moreover, the HCT116 cells exhibited a
spindle-like morphology, the typical characteristics of mesenchymal
cells; thus, these cells can be more easily used for apoptosis and
EMT-related analyses. For this reason, the HCT116 cells were
selected as a representative CRC cell line for all the remaining
in vitro experiments.
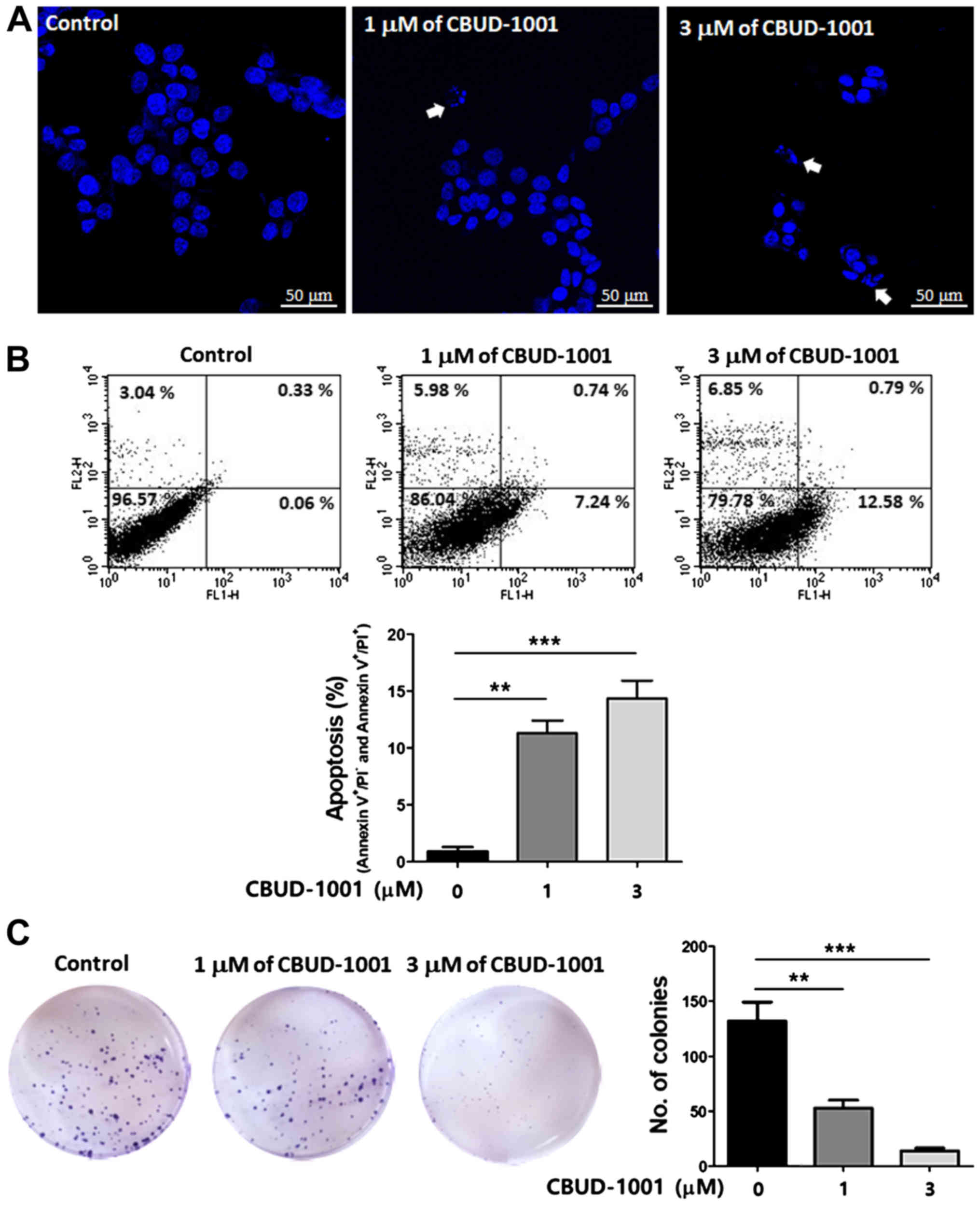
To verify whether CBUD-1001-induced cell death was
caused by apoptosis, the apoptotic effect of CBUD-1001 was examined
using various in vitro experiments. Hoechst 33258 staining
was used to observe the apoptotic nuclear morphology to determine
whether the cell death caused by CBUD-1001 in HCT116 cells was due
to apoptosis. Following treatment with 1 µM of CBUD-1001 for
24 h, the HCT116 cells began to exhibit apoptotic characteristics,
such as cell shrinkage, nuclear condensation and fragmentation.
With 3 µM of CBUD-1001, cell shrinkage was observed in the
majority of the cells, and DNA fragments were found on the surface
of the glass plate. In the control group, the cells continued to
exhibit a regular in morphology, grew fully in patches, and were
confluent, rarely sloughing off (Fig.
5A).
The rate of apoptotic cells was detected through a
FACS analysis of Annexin V-FITC/PI staining (Fig. 5B). As was revealed by the results,
the HCT116 cells treated with CBUD-1001 exhibited a higher rate of
apoptotic cell death than the control cells. Moreover, apoptotic
cell death was quantified by counting the cell populations in the
lower right and upper right quadrants. The results revealed that
the cell population in the 1 µM-treated group increased from
0.91±0.382 to 11.28±1.134%, and that in the 3 µM-treated
group increased to 14.31±1.602%.
Subsequently, it was further investigated whether
CBUD-1001 affects the growth and survival of CRC cells in long-term
culture. Following 24 h of treatment with the indicated
concentrations of CBUD-1001, clones of the cells were cultured and
then stained by crystal violet after 14 days. Compared with the
control group, clonogenic growth assay revealed that the
clonogenicity of the HCT116 cells was significantly decreased by
CBUD-1001 in a dose-dependent manner (Fig. 5C). Taken together, these results
indicate that CBUD-1001 potently suppresses the proliferation of
human CRC cells.
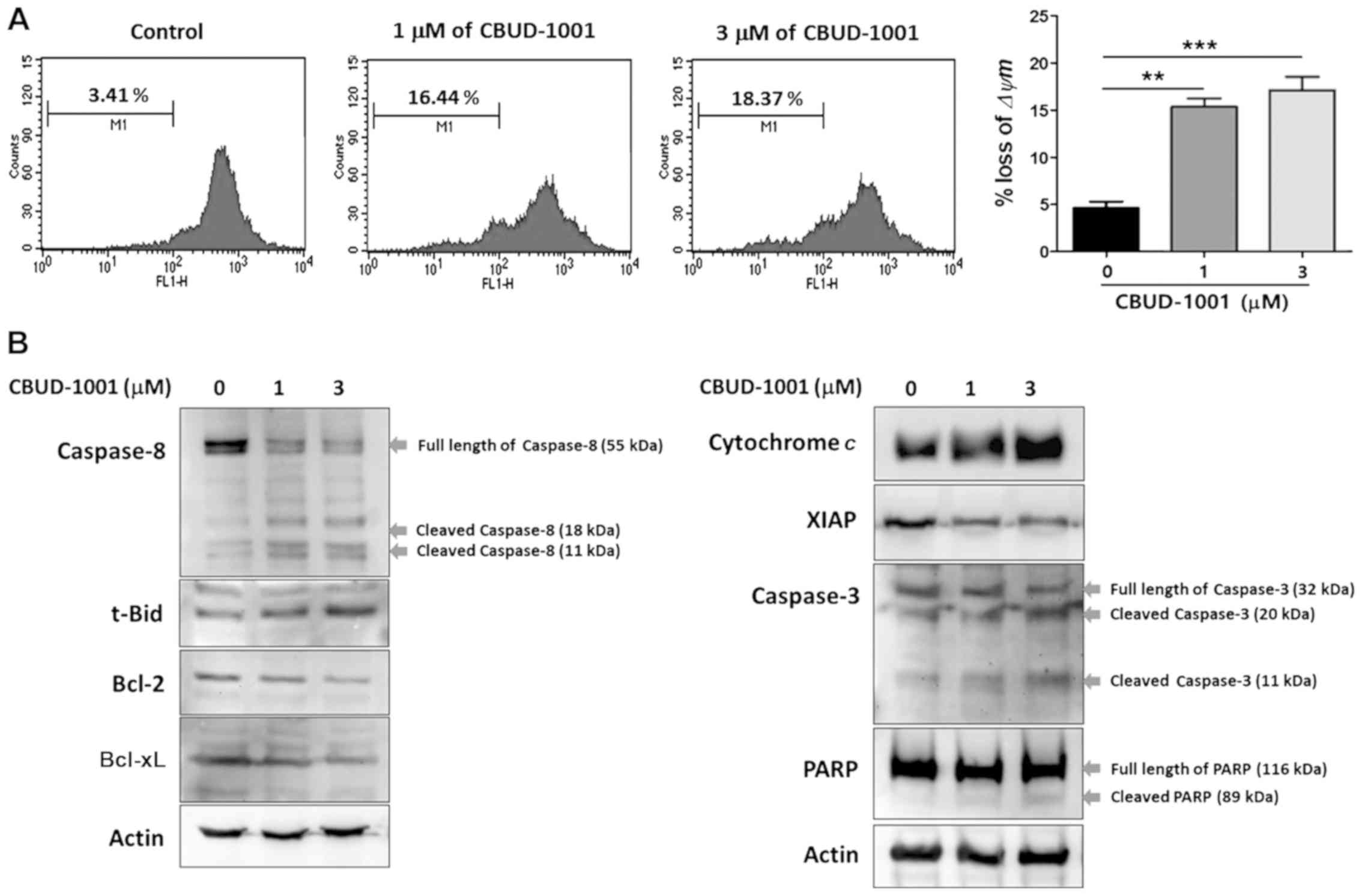
CBUD-1001 activates the intrinsic
apoptotic pathway
Mitochondrial dysfunction has been shown to
participate in the early stages of apoptosis and has even been
suggested to be central to the intrinsic apoptotic pathway
(37). Thus, the Δψm was
determined in the cells using Rhodamine 123 dye following 24 h of
CBUD-1001 treatment (Fig. 6A). As
was demonstrated, CBUD-1001 treatment triggered a significant loss
of Δψm in the HCT116 cells; the ratio of the Δψm peak was
4.6±1.208% (control), 15.34±1.530% (1 µM) and 17.11±2.459%
(3 µM).
To further investigate the mechanisms responsible
for the apoptosis induced by CBUD-1001, western blot analysis was
performed to detect the levels of apoptosis-related proteins in the
HCT116 cells following 24 h of treatment (Fig. 6B). The results revealed that
CBUD-1001 increased the cleavage of capase-8 (initiator of
apoptosis) and Bid truncation (initiator of the intrinsic apoptotic
pathway), whereas it decreased the expression of Bcl-2 and Bcl-xL
(anti-apoptotic proteins). Moreover, the release of cytochrome
c, accompanied by changes in the Bcl-2 family, was also
detected in CBUD-1001-treated cells. Subsequently, whether
CBUD-1001 regulates the protein levels of XIAP, which binds
directly to caspases to prevent apoptosis, was examined. The
results revealed that the level of XIAP was also decreased, and
subsequent increases in caspase-3 activation was detected following
CBUD-1001 treatment. The activation of caspase-3 leads to the
cleavage of their downstream molecular targets, including PARP, a
terminal factor of apoptosis. As was shown by the results, the
levels of cleaved PARP were increased by treatment with CBUD-1001.
Collectively, these results suggest that the major mechanism of
apoptosis induced by CBUD-1001 was the activation of the intrinsic
apoptotic pathway.
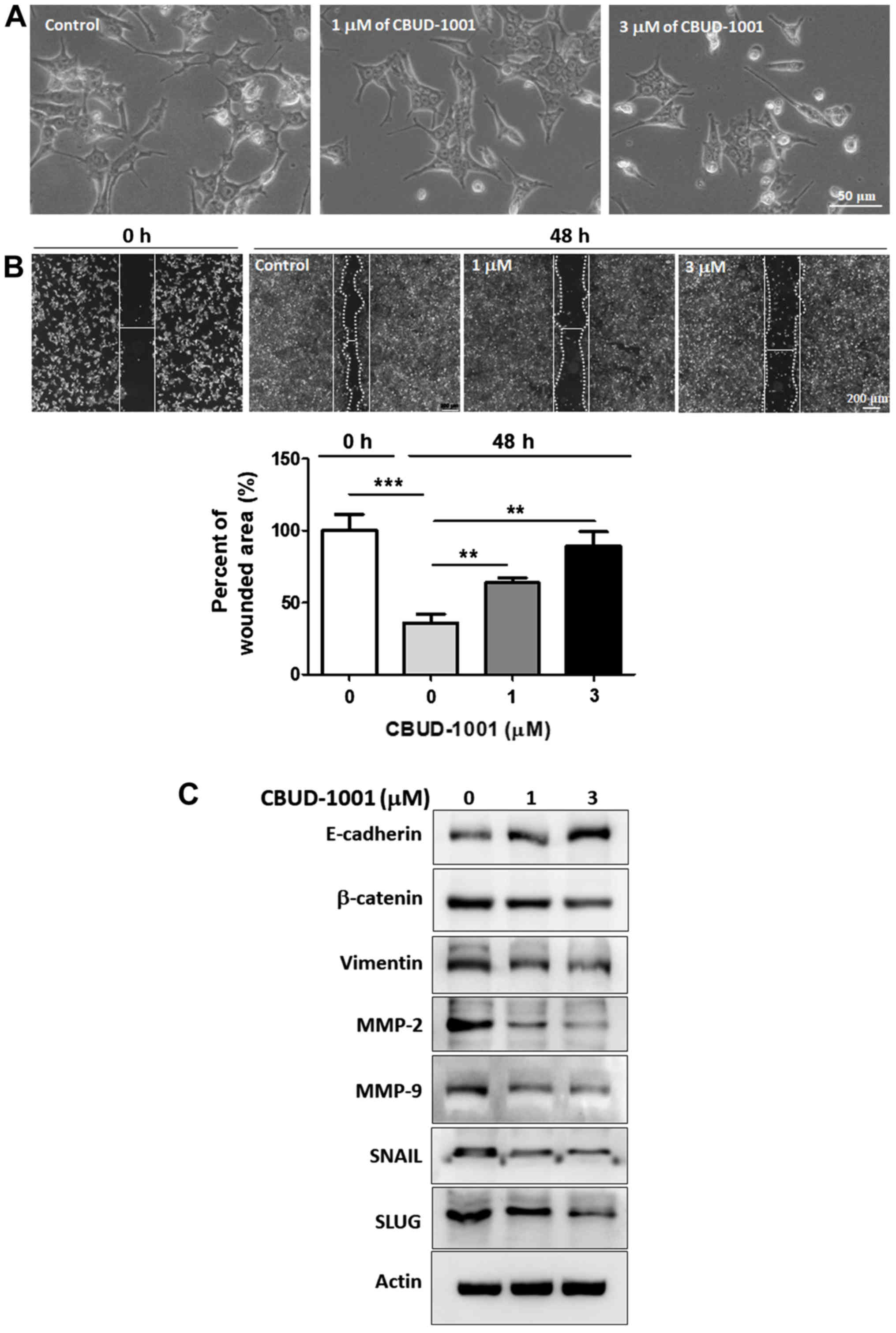
CBUD-1001 suppresses cell motility by
downregulating the EMT
During the in vitro experiments, it was
observed that following treatment with CBUD-1001, the cell
morphology altered and cells exhibited characteristics typically
associated with EMT. As shown in Fig.
7A, the cells exhibited a less spindle-like morphology in the
CBUD-1001-treated group than in the control group.
Subsequently, whether CBUD-1001 affects the motility
of CRC cells was examined to verify its effect on the EMT process.
Following 48 h of treatment with the indicated concentrations of
CBUD-1001, the distance between the wound areas in HCT116 cells was
markedly longer than in the control cells (Fig. 7B). The quantification of the wound
area in the scratch tests revealed that the wound area in the
control cells decreased by 35.87±6.33% after 48 h, whereas
following treatment with 3 µM CBUD-1001, it increased by
88.94±10.30%.
To elucidate the mechanismS of CBUD-1001 in EMT, the
protein levels of EMT-associated markers in THE HCT116 cells were
examined (Fig. 7C). First, the
level of E-cadherin, a representative epithelial marker, was
examined by western blot analysis. Following treatment with
CBUD-1001, the E-cadherin level recovered in a dose-dependent
manner. By contrast, the expression levels of mesenchymal markers,
such as β-catenin, vimentin, MMP-2, MMP-9, SNAIL, and SLUG,
decreased in a concentration-dependent manner, indicating that
CBUD-1001 altered the morphological phenotype and suppressed cell
motility by downregulating the EMT pathway.
Discussion
Over the past several decades, epigenetic
alterations have been shown to be hallmarks of cancer (13). Histone acetylation, a well-studied
epigenetic abnormality, is tightly controlled by a balance between
the opposing activities of histone acetyltransferases and HDACs
(38). Unusually, a high HDAC
activity modulates gene expression through epigenetic mechanisms
and leads to tumorigenesis. The elevated expression and activity of
HDACs has been reported in several types of cancer, including CRC
(20,39). Recent studies have demonstrated
that cancers are associated with abnormal cell functions, including
apoptosis, cell motility and DNA repair. These cell functions are
regulated at least in part by HDACs (40,41).
Therefore, HDACs have emerged as an important target in the
development of anticancer agents, and a number of HDAC inhibitors
have been developed (42). In the
present study, the HDAC1-specific inhibitor, CBUD-1001, was
developed and its potential utility to attenuate the tumorigenic
properties of CRC cells was examined.
A previously reported study on benzamide-type HDAC
inhibitors demonstrated that both the nitrogen of the
ortho-NH2 group and the carbonyl group on the benzamide
chelated with zinc. Moreover, this benzamide-type inhibitor
selectively inhibited class I HDAC isoforms, which significantly
reduced the side-effects of HDAC inhibition (43). Thus, the present study designed a
novel fluorinated aminophenyl-benzamide based compound (CBUD-1001)
as a potent HDAC inhibitor with high antitumor efficacy against
CRC.
The synthesis of the novel HDAC inhibitor
(CBUD-1001) involved three parts. In the first part, the benzamide
scaffold, which includes the zinc-binding and linker parts, was
synthesized from 4-bromo-2-nitroaniline. Boc-protection, the Suzuki
cross-coupling reaction, hydrogenation, amide bond formation, and
Gabriel synthesis successfully provided 4-(aminomethyl) benzamide
compounds with a 12% overall yield. In the second part, a
fluorinated cyclic moiety was prepared as a cap group to attach the
benzamide scaffold. 4-(Hydroxymethyl)cyclo-hexanecarboxylic acid
was used as the starting material. The protection of the carboxylic
acid group, tosylation, fluorination, and deprotection of the Cbz
group were conducted to yield 4-(fluoromethyl)cyclohexanecarboxylic
acid at a 50% overall yield. In the final part of the synthesis, an
EDC coupling reaction of 4-(aminomethyl)benzamide compound (the
benzamide scaffold) and 4-(fluoromethyl)cyclohexanecarboxylic acid
(the cap group) was followed by the deprotection of the Boc group
to yield target compound (CBUD-1001) at a 56% overall yield. The
synthesis of CBUD-1001 used facile synthetic chemistry.
The efficacy of the newly prepared compound
(CBUD-1001) against HDAC activity was then examined. The result of
in vitro HDAC assay indicated that the novel CBUD-1001
possessed good inhibitory activity against HDAC1 and it also
exhibited greater inhibitory activity against HDAC1 than SAHA, the
non-specific HDAC inhibitor. In particular, its nanomolar
inhibition suggested that the biaryl benzamide structure and
fluorinated cyclic moiety were suitable for HDAC1 inhibition.
The general pharmacophore indicates that the
structure of HDAC inhibitors includes 3 important components: The
zinc binding group (ZBG) for chelating the zinc atom in the
metal-binding site, the cap group for interacting with the external
surface, and the linker for attaching the ZBG to the cap group and
allowing the ZBG to access the active site (5,32).
The nanomolar inhibition of CBUD-1001 indicated that
the biaryl benzamide structure was compatible with the binding site
of HDAC. A molecular docking analysis of the active HDAC sites was
performed to investigate the binding mode of CBUD-1001. The results
indicated that both the nitrogen of the ortho-NH2 group
and the carbonyl group of CBUD-1001 could chelate with the Zn401
(Fig. 2). We observed an
inter-action between the ortho-NH2 group and His145, a
hydrogen bond with C=O on Gly154, and a π-π stacking interaction of
the phenyl group in the linker with Phe155. In addition, it was
found that the thiophenyl group was suitably fitted, with a
14-Å-long internal cavity suggesting the enhancement of HDAC
inhibition by CBUD-1001. Thus, the results of docking analysis
rationalize the high inhibitory activity and potency of CBUD-1001
and suggest that CBUD-1001 may be a promising HDAC inhibitor with
potential for use as an antitumor agent.
The class I HDACs (particularly HDAC1 and 2) are
ubiquitously expressed and overexpressed in several tumor types,
which renders them promising targets for HDAC inhibitor-mediated
tumor therapy (5). The
upregulation of HDAC1 has been observed in polyps and CRC compared
with normal mucosa (44).
Moreover, HDAC1 has been confirmed to be overexpressed in CRC by
exploratory tissue-based expression analysis on small sets of
tumors and to be significantly associated with CRC patient survival
(44,45). Important insights as to the
functional effects of HDAC1 on the regulation of proliferation,
apoptosis and tumorigenesis have been provided by in vitro
and in vivo studies. In 2007, Senese et al reported
that the silencing HDAC1 in osteosarcoma and breast cancer cells
affected the transcription of specific target genes involved in
proliferation and apoptosis (46).
The knockdown of HDAC1 also caused a decrease in the proliferation
of cervix adenocarcinoma cells (47). In CRC cells, Weichert et al
demonstrated that the selective knockdown of HDAC1 using siRNA
resulted in a significant reduction in the CX-2 cell population
(45). Of note, another study
demonstrated that the knockdown of HDAC1 produced the development
of hematological malignancy, suggesting that HDAC1 has an ambiguous
identity in the process of tumorigenesis (48). In the present study, the
anti-cancer role of HDAC1 was examined by treating CRC cells with
CBUD-1001. The results provide clear evidence that HDAC1 positively
modulates CRC cell proliferation and survival.
HDAC inhibitors have been reported to activate
either an extrinsic or intrinsic pathway in several types of cancer
(49,50). The mitochondrial apoptotic pathway
(intrinsic pathway) is activated by stress stimuli
(chemotherapeutic agents) and is regulated by the Bcl-2 family
(51,52). Biological events by Bcl-2 family
members in the apoptotic process can trigger mitochondrial
dysfunction and the loss of ΔΨm (53). Correspondingly, it was observed
that CBUD-1001 induced the loss of ΔΨm in CRC cells and regulated
the protein level of Bcl-2 family members, suggesting that
CBUD-1001 induced apoptosis via the intrinsic pathway. Following
mitochondrial dysfunction, cytochrome c in the mitochondrial
membranes is released into the cytoplasm (54,55).
XIAP is considered to be the most potent apoptotic regulator in
mammalian cells (56), and it
inhibits caspase activity by binding directly to caspase-3, -7 and
-9 in the apoptotic pathway (57).
Downstream of cytochrome c, caspase-3 is activated and leads
to the cleavage of downstream molecular targets, including PARP, a
hallmark of apoptosis (58). In
the present study, the results of western blot analysis revealed a
consistent alteration of apoptotic molecules, and these
observations indicate that CBUD-1001 induces apoptosis via the
intrinsic pathway by causing mitochondrial dysfunction in CRC
cells.
EMT is a crucial process in cancer progression that
provides cancer cells with the ability to escape from the primary
site, invade stromal tissues and migrate to distant regions. As a
result of EMT, epithelial cells lose their defined cell-
cell/cell-substratum contacts and their structural/functional
polarity and become spindle shaped and morphologically similar to
activated fibroblasts (59,60).
Recently, several studies reported that an HDAC inhibitor
suppressed the EMT in various cells, including cancer cells.
Sakamoto et al demonstrated that vorinostat (SAHA)
suppressed the TGF-β1-induced EMT and attenuated chemo-resistance
in biliary tract cancer (61). A
synergistic effect on the EMT from the use of silibinin and an HDAC
inhibitor (trichostatin A) was also investigated in non-small cell
lung cancer cells (62). Moreover,
the suppressive effect of HDAC inhibitors on EMT in CRC was
reported using valproic acid and trichostatin A (24,63).
In the present study, it was demonstrated that CBUD-1001 negatively
regulated EMT in CRC cells, as shown through changes in cell
morphology, motility and the expression level of various EMT
markers (E-cadherin, β-catenin, Vimentin, MMP-2, MMP-9, SNAIL and
SLUG). These observations suggest that CBUD-1001 may play a
therapeutic role in CRC and its metastasis by inhibiting EMT. Taken
together, the results indicate that CBUD-1001 has antitumor
activity and may be a highly potent HDAC inhibitor for CRC cancer
therapy. However, this requires further investigation, in order to
fully elucidate the mechanisms through which CBUD-1001 regulates
HDAC-mediated EMT signaling and metastasis using animal models.
In conclusion, even though several drugs are
currently being used for CRC therapy, effective therapeutic agents
are still needed. Several studies have suggested that HDAC
inhibitors exhibit promise for cancer treatment (21-28).
In the present study, a novel benzamide-based compound, CBUD-1001,
that can inhibit HDACs was reported. Molecular docking analysis
confirmed the high potency of CBUD-1001 as an HDAC1 inhibitor, and
CBUD-1001 exhibited potent inhibitory activity against HDAC enzyme
activity in CRC. Additionally, the results demonstrated that
CBUD-1001 triggered the apoptotic death of human CRC cells, and
suppressed cell motility by downregulating EMT. Therefore, the
present results suggest that CBUD-1001 may be a novel potent
therapeutic agent for CRC therapy.
Supplementary Data
Acknowledgments
Not applicable.
Funding
The present study was supported by the Fund of the
Biomedical Research Institute, Jeonbuk National University Hospital
and the CBNU fund overseas research 2007.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
HKK and SWK participated in the design of the study.
HKK participated in the data analysis in the synthesis of the study
and wrote the original draft. MTL synthesized, performed the
docking analysis and wrote the original draft of the synthesis of
the study. SLK and MWS carried out the molecular biology
experiment. SLK wrote the draft of the molecular biology section
and performed the statistical analysis. SWK edited the draft and
supervised all experimental procedures. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar
|
|
2
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar
|
|
3
|
Haggar FA and Boushey RP: Colorectal
cancer epidemiology: Incidence, mortality, survival, and risk
factors. Clin Colon Rectal Surg. 22:191–197. 2009. View Article : Google Scholar
|
|
4
|
Lao VV and Grady WM: Epigenetics and
colorectal cancer. Nat Rev Gastroenterol Hepatol. 8:686–700. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bertrand P: Inside HDAC with HDAC
inhibitors. Eur J Med Chem. 45:2095–2116. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jackson MD and Denu JM: Structural
identification of 2′- and 3′-O-acetyl-ADP-ribose as novel
metabolites derived from the Sir2 family of beta-NAD+-dependent
histone/protein deacetylases. J Biol Chem. 277:18535–18544. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lombardi PM, Cole KE, Dowling DP and
Christianson DW: Structure, mechanism, and inhibition of histone
deacetylases and related metalloenzymes. Curr Opin Struct Biol.
21:735–743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haberland M, Montgomery RL and Olson EN:
The many roles of histone deacetylases in development and
physiology: Implications for disease and therapy. Nat Rev Genet.
10:32–42. 2009. View
Article : Google Scholar
|
|
9
|
Kelly RD and Cowley SM: The physiological
roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with
multiple leading parts. Biochem Soc Trans. 41:741–749. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lagger G, Doetzlhofer A, Schuettengruber
B, Haidweger E, Simboeck E, Tischler J, Chiocca S, Suske G,
Rotheneder H, Wintersberger E and Seiser C: The tumor suppressor
p53 and histone deacetylase 1 are antagonistic regulators of the
cyclin-dependent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell
Biol. 23:2669–2679. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ropero S and Esteller M: The role of
histone deacetylases (HDACs) in human cancer. Mol Oncol. 1:19–25.
20072007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hill R, Bodzak E, Blough MD and Lee PW:
p53 Binding to the p21 promoter is dependent on the nature of DNA
damage. Cell Cycle. 7:2535–2543. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y and Seto E: HDACs and HDAC inhibitors
in cancer development and therapy. Cold Spring Harb Perspect Med.
6:a0268312016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu P, Martin E, Mengwasser J, Schlag P,
Janssen KP and Göttlicher M: Induction of HDAC2 expression upon
loss of APC in colorectal tumorigenesis. Cancer Cell. 5:455–463.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stypula-Cyrus Y, Damania D, Kunte DP, Cruz
MD, Subramanian H, Roy HK and Backman V: HDAC up-regulation in
early colon field carcinogenesis is involved in cell tumorigenicity
through regulation of chromatin structure. PLoS One. 8:e646002013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Spaderna S, Schmalhofer O, Hlubek F, Berx
G, Eger A, Merkel S, Jung A, Kirchner T and Brabletz T: A
transient, EMT-linked loss of basement membranes indicates
metastasis and poor survival in colorectal cancer.
Gastroenterology. 131:830–840. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: Implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peinado H, Ballestar E, Esteller M and
Cano A: Snail mediates E-cadherin repression by the recruitment of
the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell
Biol. 24:306–319. 2004. View Article : Google Scholar
|
|
19
|
Jurkin J, Zupkovitz G, Lagger S,
Grausenburger R, Hagelkruys A, Kenner L and Seiser C: Distinct and
redundant functions of histone deacetylases HDAC1 and HDAC2 in
proliferation and tumorigenesis. Cell Cycle. 10:406–412. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mariadason JM: HDACs and HDAC inhibitors
in colon cancer. Epigenetics. 3:28–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun PC, Tzao C, Chen BH, Liu CW, Yu CP and
Jin JS: Suberoylanilide hydroxamic acid induces apoptosis and
sub-G1 arrest of 320 HSR colon cancer cells. J Biomed Sci.
17:762010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang TY, Jia YL, Zhang X, Sun QL, Li YC,
Zhang JH, Zhao CP, Wang XY and Wang L: Treating colon cancer cells
with FK228 reveals a link between histone lysine acetylation and
extensive changes in the cellular proteome. Sci Rep. 5:184432015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhijun H, Shusheng W, Han M, Jianping L,
Li-Sen Q and Dechun L: Pre-clinical characterization of 4SC-202, a
novel class I HDAC inhibitor, against colorectal cancer cells.
Tumour Biol. 37:10257–10267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ji M, Lee EJ, Kim KB, Kim Y, Sung R, Lee
SJ, Kim DS and Park SM: HDAC inhibitors induce
epithelial-mesenchymal transition in colon carcinoma cells. Oncol
Rep. 33:2299–2308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Patel MM and Patel BM: Repurposing of
sodium valproate in colon cancer associated with diabetes mellitus:
Role of HDAC inhibition. Eur J Pharm Sci. 121:188–199. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bracker TU, Sommer A, Fichtner I, Faus H,
Haendler B and Hess-Stumpp H: Efficacy of MS-275, a selective
inhibitor of class I histone deacetylases, in human colon cancer
models. Int J Oncol. 35:909–920. 2009.PubMed/NCBI
|
|
27
|
Jin JS, Tsao TY, Sun PC, Yu CP and Tzao C:
SAHA inhibits the growth of colon tumors by decreasing histone
deacetylase and the expression of cyclin D1 and survivin. Pathol
Oncol Res. 18:713–7120. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chou CW, Wu MS, Huang WC and Chen CC: HDAC
inhibition decreases the expression of EGFR in colorectal cancer
cells. PLoS One. 6:e180872011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Khan N, Jeffers M, Kumar S, Hackett C,
Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, et al:
Determination of the class and isoform selectivity of
small-molecule histone deacetylase inhibitors. Biochem J.
409:581–589. 2008. View Article : Google Scholar
|
|
30
|
Kelly WK, O'Connor OA, Krug LM, Chiao JH,
Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP,
Schwartz L, et al: Phase I study of an oral histone deacetylase
inhibitor, suberoylanilide hydroxamic acid, in patients with
advanced cancer. J Clin Oncol. 23:3923–3931. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Flis S, Gnyszka A, Flis K and Spławiński
J: MS275 enhances cytotoxicity induced by 5-fluorouracil in the
colorectal cancer cells. Eur J Pharmacol. 627:26–32. 2010.
View Article : Google Scholar
|
|
32
|
Zhang L, Zhang J, Jiang Q, Zhang L and
Song W: Zinc binding groups for histone deacetylase inhibitors. J
Enzyme Inhib Med Chem. 33:714–721. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu H, Chen YD, Yang B and You QD: Design,
synthesis and biological evaluation of novel histone deacetylase
inhibitors based on virtual screening. Acta Pharmaceutica Sinica B.
1:240–247. 2011. View Article : Google Scholar
|
|
34
|
Vaisburg A, Paquin I, Bernstein N,
Frechette S, Gaudette F, Leit S, Moradei O, Raeppel S, Zhou N,
Bouchain G, et al:
N-(2-Amino-phenyl)-4-(heteroarylmethyl)-benzamides as new histone
deacetylase inhibitors. Bioorg Med Chem Lett. 17:6729–6733. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Methot JL, Chakravarty PK, Chenard M,
Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE,
Harrington P, et al: Exploration of the internal cavity of histone
deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:
2). Bioorg Med Chem Lett. 18:973–978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lauffer BEL, Mintzer R, Fong RN, Mukund S,
Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R,
et al: Histone deacetylase (HDAC) inhibitor kinetic rate constants
correlate with cellular histone acetylation but not transcription
and cell viability. J Biol Chem. 288:26926–26943. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zamzami N, Marchetti P, Castedo M, Zanin
C, V Vayssière JL, Petit PX and Kroemer G: Reduction in
mitochondrial potential constitutes an early irreversible step of
programmed lymphocyte death in vivo. J Exp Med. 181:1661–1672.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seto E and Yoshida M: Erasers of histone
acetylation: The histone deacetylase enzymes. Cold Spring Harb
Perspect Biol. 6:a0187132014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Spurling CC, Godman CA, Noonan EJ,
Rasmussen TP, Rosenberg DW and Giardina C: HDAC3 overexpression and
colon cancer cell proliferation and differentiation. Mol Carcinog.
47:137–147. 2008. View Article : Google Scholar
|
|
40
|
Chen HP, Zhao YT and Zhao TC: Histone
deacetylases and mechanisms of regulation of gene expression. Crit
Rev Oncog. 20:35–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Audia JE and Campbell RM: Histone
modifications and cancer. Cold Spring Harb Perspect Biol.
8:a0195212016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
De Souza C and Chatterji BP: HDAC
inhibitors as novel anti-cancer therapeutics. Recent Pat Anticancer
Drug Discov. 10:145–162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bozorgi AH, Bagheri M, Aslebagh R and
Rajabi MS: A structure-activity relationship survey of histone
deacetylase (HDAC) inhibitors. Chemometr Intell Lab. 125:132–138.
2013. View Article : Google Scholar
|
|
44
|
Huang BH, Laban M, Leung CH, Lee L, Lee
CK, Salto-Tellez M, Raju GC and Hooi SC: Inhibition of histone
deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression,
independent of histone deacetylase 1. Cell Death Differ.
12:395–404. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Weichert W, Roske A, Niesporek S, Noske A,
Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T and Denkert
C: Class I histone deacetylase expression has independent
prognostic impact in human colorectal cancer: Specific role of
class I histone deacetylases in vitro and in vivo. Clin Cancer Res.
14:1669–1677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Senese S, Zaragoza K, Minardi S, Muradore
I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser
C and Chiocca S: Role for histone deacetylase 1 in human tumor cell
proliferation. Mol Cell Biol. 27:4784–4795. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Glaser KB, Li J, Staver MJ, Wei RQ, Albert
DH and Davidsen SK: Role of class I and class II histone
deacetylases in carcinoma cells using siRNA. Biochem Biophys Res
Commun. 310:529–536. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Santoro F, Botrugno OA, Dal Zuffo R,
Pallavicini I, Matthews GM, Cluse L, Barozzi I, Senese S, Fornasari
L, Moretti S, et al: A dual role for Hdac1: Oncosuppressor in
tumorigenesis, oncogene in tumor maintenance. Blood. 121:3459–3468.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang L, Sowa Y, Sakai T and Pardee AB:
Activation of the p21WAF1/CIP1 promoter independent of p53 by the
histone deacetylase inhibitor suberoylanilide hydroxamic acid
(SAHA) through the Sp1 sites. Oncogene. 19:5712–5719. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rikiishi H: Autophagic and apoptotic
effects of HDAC inhibitors on cancer cells. J Biomed Biotechnol.
2011:8302602011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yang E and Korsmeyer SJ: Molecular
thanatopsis: A discourse on the BCL2 family and cell death. Blood.
88:386–401. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lencz T, Guha S, Liu C, Rosenfeld J,
Mukherjee S, DeRosse P, John M, Cheng L, Zhang C, Badner JA, et al:
Genome-wide association study implicates NDST3 in schizophrenia and
bipolar disorder. Nat Commun. 4:27392013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Eckelman BP, Salvesen GS and Scott FL:
Human inhibitor of apoptosis proteins: Why XIAP is the black sheep
of the family. EMBO Rep. 7:988–994. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Grutter MG: Caspases: Key players in
programmed cell death. Curr Opin Struct Biol. 10:649–655. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: New insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang Y and Weinberg RA:
Epithelial-to-mesenchymal transition in cancer: Complexity and
opportunities. Front Med. 12:361–373. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sakamoto T, Kobayashi S, Yamada D, Nagano
H, Tomokuni A, Tomimaru Y, Noda T, Gotoh K, Asaoka T, Wada H, et
al: A Histone deacetylase inhibitor suppresses
epithelial-mesenchymal transition and attenuates chemoresistance in
biliary tract cancer. PLoS One. 11:e01459852016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Mateen S, Raina K, Agarwal C, Chan D and
Agarwal R: Silibinin synergizes with histone deacetylase and DNA
methyltransferase inhibitors in upregulating E-cadherin expression
together with inhibition of migration and invasion of human
non-small cell lung cancer cells. J Pharmacol Exp Ther.
345:206–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Feng J, Cen J, Li J, Zhao R, Zhu C, Wang
Z, Xie J and Tang W: Histone deacetylase inhibitor valproic acid
(VPA) promotes the epithelial mesenchymal transition of colorectal
cancer cells via up regulation of Snail. Cell Adh Migr. 9:495–501.
2015. View Article : Google Scholar : PubMed/NCBI
|