Introduction
Non-small cell lung cancer (NSCLC) is the most
common type of cancer and accounts for 85% of the total number of
lung cancer cases. The delayed diagnosis of NSCLC renders treatment
more difficult using surgical interventions and also renders the
tumor insensitive to conventional chemotherapy (1). Over the past two decades, different
expression patterns have been detected for various proteins,
including epidermal growth factor receptor (EGFR) (2,3) and
KRAS (4,5), which are associated with various
types of lung cancer. The most common altered expression pattern is
observed for EGFR, a membrane receptor tyrosine kinase in NSCLC
tumors. Since this observation was made, the detection of EGFR
mutations in patients with NSCLC has become an important test to
predict the treatment response to the Food and Drug Administration
(FDA)-approved tyrosine kinase inhibitors (TKIs), gefitinib and
erlotinib. Patients with mutations in exon 19 and 21 of the
receptor tyrosine kinase domain tend to have an improved outcome
following treatment with these drugs (2,3,6).
Nonetheless, the majority of patients develop secondary mutation.
T790M in the ATP binding site of exon 20 confers drug resistance.
Since the discovery of T790M, this amino acid change has become a
major determinant of acquired resistance to first generation drugs
(7-9). The 2nd generation TKIs targeting
sensitive and resistant mutant receptors have exhibited promising
results; however, they are associated with dose-limiting toxicity
due to the inhibition of wild-type (Wt) EGFR signaling. Similarly,
3rd generation TKIs selectively and irreversibly target both
resistant and sensitive mutant receptors with no effect or limited
effects on the Wt receptor. Osimertinib, a 3rd generation TKI, has
been approved by the FDA and the European Medicines Agency for the
treatment of patients with NSCLC with EGFR-T790M mutations
following the failure of 1st and 2nd generation TKIs (10-12).
In spite of the availability of receptor targeted drugs and the
consideration of receptor mutations for the treatment prediction,
the prognosis of patients with advanced stage lung tumors remains
poor.
Somatic mutations in the tyrosine kinase domain
maintain the receptor constitutively active in a ligand-dependent
and -independent manner, promoting several proliferative pathways
which contribute to lung tumor cell proliferation and the evasion
of programmed cell death (13,14).
Tumor progression is generally associated with the invasion and
metastasis of cancer cells, which is a complex process. The
majority of studies on lung cancer-associated EGFR mutations are
limited to the importance of kinase domain mutations for the
prediction of the response of patients to TKIs. However, the
functional dependence of signaling pathways recruited in the
response to activated mutant receptor kinases for tumor progression
and its connection with the metastatic potential of cells harboring
EGFR mutations remains unclear. In the advanced stages of cancer,
tumor cells leave the site of origin and invade various parts of
the body. Most commonly, the invasion of cancer cells to the liver,
lungs, brain and bones is the main reason for the mortality of
patients with cancer. Thus, the diagnosis and treatment of
metastasis varies with the original cancer type and is vital for
treating the patient.
In the present study, the signaling pathways
recruited in response to mutant receptor kinases, and their
functional role in the invasive potential of cells expressing NSCLC
associated EGFR mutants using 293 cells was investigated. Cells
stably expressing receptor mutants sensitive to FDA-approved drugs,
L858R and L861Q of exon 21, and 293 cells expressing resistant
mutants, T790M of exon 20 and double-mutant-harboring L858R/T790M,
were used in the present study. Furthermore, the regulatory role of
mutant receptor-mediated phospholipase (PLC)γ1 in the invasive and
migratory potential that signifies metastasis, was ascertained. The
present study demonstrated an increased phosphorylation of PLCγ1
and other signaling molecules in cells expressing L858R, L861Q and
L858R/T790M, compared to the Wt and T790M receptor. The inhibition
of PLCγ1 phosphorylation following the reduction of receptor
phosphorylation with TKI treatment confirms that PLCγ1 activation
is driven by mutant receptors. A significant reduction in the in
vitro migration and invasion of cells following TKI or PLC
inhibitor treatment suggests that EGFR-mediated PLCγ1 activation
may be crucial in maintaining the invasive phenotype of NSCLC tumor
cells harboring EGFR mutations.
Materials and methods
Generation of EGFR mutants and stable
cell lines
EGFR mutants; L861Q, L858R, T790M and double-mutant
L858R/T790M were generated by introducing point mutations at
desired locations in the full-length EGFR coding sequence in the
CMV promoter driven eukaryotic expression vector, pcDNA3.1, as
previously described (15). Clones
were confirmed by sequencing. The 293 cell line was obtained from
the National Centre for Cell Science (NCCS), India and 293 cells
(with no endogenous EGFR expression) stably expressing mutant
receptors were also generated using Lipofectamine 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) following the manufacturer's
instructions. Stable clones were selected with the neomycin
analogue, G418 (Invitrogen; Thermo Fisher Scientific, Inc.) at a
concentration of 600 µg/ml. Stable cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (HiMedia Laboratories) and 300 µg of
G418. The expression of Wt and mutant receptors in 293 cells was
confirmed by western blot analysis using anti-EGFR anti-bodies
(Santa Cruz Biotechnology, Inc.).
Western blot analysis
For western blot analysis, 293 cells stably
expressing Wt and mutant receptors were grown to 80-90% confluency,
followed by an overnight serum starvation. The following day, the
cells were stimulated with 10 ng/ml recombinant human EGF
(Sigma-Aldrich; Merck KGaA) for varying time points up to 30 min.
Following treatment, the cells were lysed using 1X lysis buffer
containing 150 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1 mM
Na2EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1%
Triton X-100, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM
PMSF and 1 µg/ml leupeptin (Cell Signaling Technology,
Inc.). The protein concentration was determined by Bradford assay;
20 µg of total protein from each sample was resolved using
10 % SDS-PAGE followed by western blot analysis. Blocking of
non-specific proteins was performed by incubating the blots in 5%
BSA or 5% skimmed milk in 1X TBST for 2 h at room temperature. The
blots were incubated with anti-bodies against phospho-EGFR (Y1068)
(cat. no. 3777, diluted 1:5,000), phospho-PLCγ1 (cat. no. 14008,
diluted 1:5,000), phospho-Akt (cat. no. 4060, diluted 1:1,000),
phospho-c-Cbl (cat. no. 8869, diluted 1:2,000), phospho-Erk1/2
(cat. no. 4370, diluted 1:5,000), phospho-Shc (cat. no. 2434,
diluted 1:2,000), phospho-Gab1 (cat. no. 3233, diluted 1:2,000)
(all from Cell Signaling Technology, Inc.) and β-actin (sc-47778,
Santa Cruz Biotechnology, Inc.; diluted 1:6,000) in blocking
buffer. Protein expression was detected using HRP-conjugated
anti-rabbit (cat. no. A0545, diluted 1:10,000) or anti-mouse (cat.
no. A4416, diluted 1:10,000) secondary antibodies (both from
Sigma-Aldrich; Merck KGaA). Protein bands were visualized using ECL
substrate (cat. no. 170-5060) obtained from Bio-Rad Laboratories
Inc. The blots were stripped using a stripping buffer composed of
62.5 mM Tris-HCl (pH 6.8), 100 mM β-mercaptoethanol and 2% SDS at
55°C for 45 min and re-probed with antibodies against total PLCγ1
(cat. no. 5690, diluted 1:2,000; Cell Signaling Technology, Inc.),
EGFR (sc-373746, diluted 1:2,000; Santa Cruz Biotechnology, Inc.)
and c-Cbl (cat. no. 9794, diluted 1:1,000; Cell Signaling
Technology, Inc.).
Treatment with TKI and U73122
The first-generation TKIs, gefitinib and erlotinib,
the 2nd generation TKI, afatinib, and the 3rd generation drug,
osimertinib (Biovision, Inc.), were used in the present study. TKIs
were reconstituted in 99.5% DMSO and desired dilutions were made in
fresh DMEM prior to use. The PLC inhibitor, U73122 (Sigma-Aldrich;
Merck KGaA), was used to block PLC activity. U73122 was first
dissolved in chloroform at 10 mg/ml. This was then aliquoted and
purged with nitrogen gas to leave a thin, dry film and was stored
at −20°C. Immediately prior to the experiment, one vial was removed
from −20°C and reconstituted with 99.5% DMSO to make a 5 mM stock
solution. Working dilutions were made in serum-free medium. For
inhibitor experiments, 293 cells expressing Wt and mutant receptors
were grown to 90% confluency, serum-starved overnight and treated
with TKIs (erlotinib, 100 nm; gefitinib, 100 nm; afatinib, 50 nm;
and osimertinib, 50 nm) for 2 h or for 25 min with PLC inhibitor at
concentrations of 0, 10 and 20 µm. This was followed by
stimulation with 10 ng/ml recombinant human EGF for 10 min. Equal
amounts of total protein from each sample was analyzed for EGFR and
PLCγ1 phosphorylation as described above in the western blot
analysis section.
Wound healing assay
Cells expressing Wt or mutant receptors were plated
in 12-well plates and grown to 90% confluency. Cells were then
starved overnight and a wound was generated by scratching the
confluent monolayer using a sterile micropipette tip. Media was
replaced with fresh medium containing 0.1% FBS, 200 nM TKI or 3
µM U73122 in the absence or presence of EGF (10 ng/ml). DMSO
was used as a control. Images were captured at 0 and 24 h
post-treatment with a digital microscopic camera, DC5 (Magnus)
connected to an inverted microscope (Nikon ECLIPSE, TS-100, Nikon
Healthcare Japan Inc.) and wound closure was measured using ImageJ
software (ImageJ 1.46r, National Institute for Health) (16). A total of 6 measurements per image
were acquired and the average gap distance was calculated. Results
are presented as a mean of two different experiments.
Boyden chamber assay
Boyden chambers with 6.5 mm diameter polycarbonate
filters of 8 µm pore size (BD Biosciences) were used in the
present study. Wt or mutant receptor-expressing cells were grown as
mentioned above and serum starved. Following serum starvation, the
cells were trypsinized and 4×104 cells were suspended in
serum-free DMEM containing 200 nM TKI or 3 µM U73122 or DMSO
and EGF (10 ng/ml). A total of 200 µl of the cell suspension
was added to the upper chamber of a Transwell insert, placed in a
24-well plate and cells were allowed to settle down for 5 min.
Subsequently, 700 µl of 10% FBS was added to the lower
chamber of the Transwell insert as a chemoattractant and the cells
were incubated at 37°C in 5% CO2. At 24 h
post-incubation, the Transwell inserts were removed from 24-well
plates and non-migrated cells from the upper chamber were removed
using a cotton swab. The cells that had migrated through the
membrane were stained with 0.1% crystal violet (Thermo Fisher
Scientific, Inc.) at room temperature for 1 h. Stained cells were
counted at a higher magnification from 6 different representative
fields using a phase contrast inverted microscope (Nikon Healthcare
Japan Inc.). Data are represented as the means ± SEM from 3
independent experiments.
Matrigel invasion assay
For invasion assays, stock solution of 8.5 mg/ml
Matrigel was thawed overnight on ice in 4°C refrigerator. The
following day, Matrigel was diluted to a concentration of 0.3 mg/ml
in serum-free DMEM using pre-chilled tips and tubes. A total of 40
µl of Matrigel was added to the upper chamber of a Transwell
insert in 24-well plate and allowed to solidify for 1 h at 37°C in
5% CO2 to form a thin Matrigel layer. The cell
suspension containing 4×104 cells was added on top of
the Matrigel layer to facilitate invasion. Following 24 h of
incubation at 37°C in 5% CO2, invading cells were
stained and counted as described above. Data are shown as the means
± SEM from 3 independent experiments.
MTT assay
Cell viability was measured using MTT assays. In
brief, 293 cells stably expressing Wt and mutant receptors were
cultured in 96-well plates in triplicates at a density of 10,000
cells/well. After 24 h, growth medium was replaced with maintenance
medium containing either PLC inhibitor, U73122 or DMSO (vehicle
control). At 48 h post-drug treatment, the cells were incubated
with 20 µl of 5 mg/ml MTT at 37°C in 5% CO2 for 3
h. MTT reagent was then aspirated off and formazan crystals were
dissolved in 200 µl of DMSO. The absorbance was recorded at
a 560 and 670 nm wavelength on a SpectroMax Plus 384 microplate
reader (Molecular Devices, LLC). The percentage cell viability was
calculated using the following formula: OD of treated cells −
blank/OD of untreated cells − blank ×100 and plotted against the
concentration of the drug. Data are presented as the means ± SD
using GraphPad Prism software (GraphPad Software, Inc.).
Soft agar colony formation assay
The anchorage-independent growth of cells expressing
mutant receptors was determined using soft agar assays. The bottom
layer of 0.8% agarose was created by mixing equal volumes of 1.6%
low melting agarose, 2X DMEM supplemented with 20% FBS and 2X
antibiotics to give a solution of 0.8% agar + 1X medium + 10% FBS +
1X antibiotics. A total of 1.5 ml of the solution was poured into
each well of 6-well plates and allowed to solidify for 30 min at
room temperature. Cells expressing Wt or mutant receptors were
trypsinized and were suspended at the rate of 5,000 cells/well into
750 µl of 2X culture medium containing 1 µM TKI or 10
µM U73122, 10 ng/ml EGF and G418. This solution was then
mixed with 750 µl of 0.8% low melting agarose to make the
concentration of agarose 0.4%. Immediately, the cell suspension was
poured over the solidified bottom layer and the plate was incubated
at 37°C in 5% CO2 for 9-10 days. Cells were fed with 200
µl of culture medium twice a week to prevent the desiccation
and deprivation of nutrients. After 10 days, colonies were stained
with 0.005% crystal violet (Thermo Fisher Scientific, Inc.) for 30
min at room temperature. Images were captured using an inverted
microscope (Nikon Healthcare Japan Inc.) and colonies were counted
using ImageJ software (ImageJ 1.46r, NIH).
Statistical analysis
Data analysis was carried out using GraphPad Prism
v.5.0 software (GraphPad Software, Inc.). One-way ANOVAs (Tukey's
post hoc test for multiple comparisons) were applied to assess the
significance of differences between samples and a P-value <0.05
was considered to indicate a statistically significant
difference.
Results
Activation of mutant EGFR induces the
increased phosphorylation of PLCγ1 and other signaling
molecules
Cells stably expressing TKI sensitizing mutants,
L861Q, L858R; resistant mutants, T790M, L858R/T790M (double-mutant)
and Wt receptor were analyzed for their activation and recruited
signaling molecules in response to receptor phosphorylation. The
activation of mutant receptors following EGF stimulation at
different time points resulted in an increased phosphorylation of
PLCγ1 in 293 cells. The absence of EGF also increased the
phosphorylation of mutant receptor and PLCγ1. The phosphorylation
of PLCγ1 in cells expressing receptor mutants independent of each
other, exhibited an association with the corresponding receptor
phosphorylation. These experiments confirmed the activation of
mutant receptors in a ligand-dependent/independent manner and
mutant receptor-driven PLCγ1 phosphorylation (Fig. 1). The total PLCγ1, EGFR and β-actin
levels remained unaltered in all samples. Furthermore, signaling
molecules, such as Akt, Erk1/2 and c-Cbl, and the adaptor proteins,
Gab1 and Shc, also exhibited an increased phosphorylation following
EGF stimulation (Fig. S1A and
B).
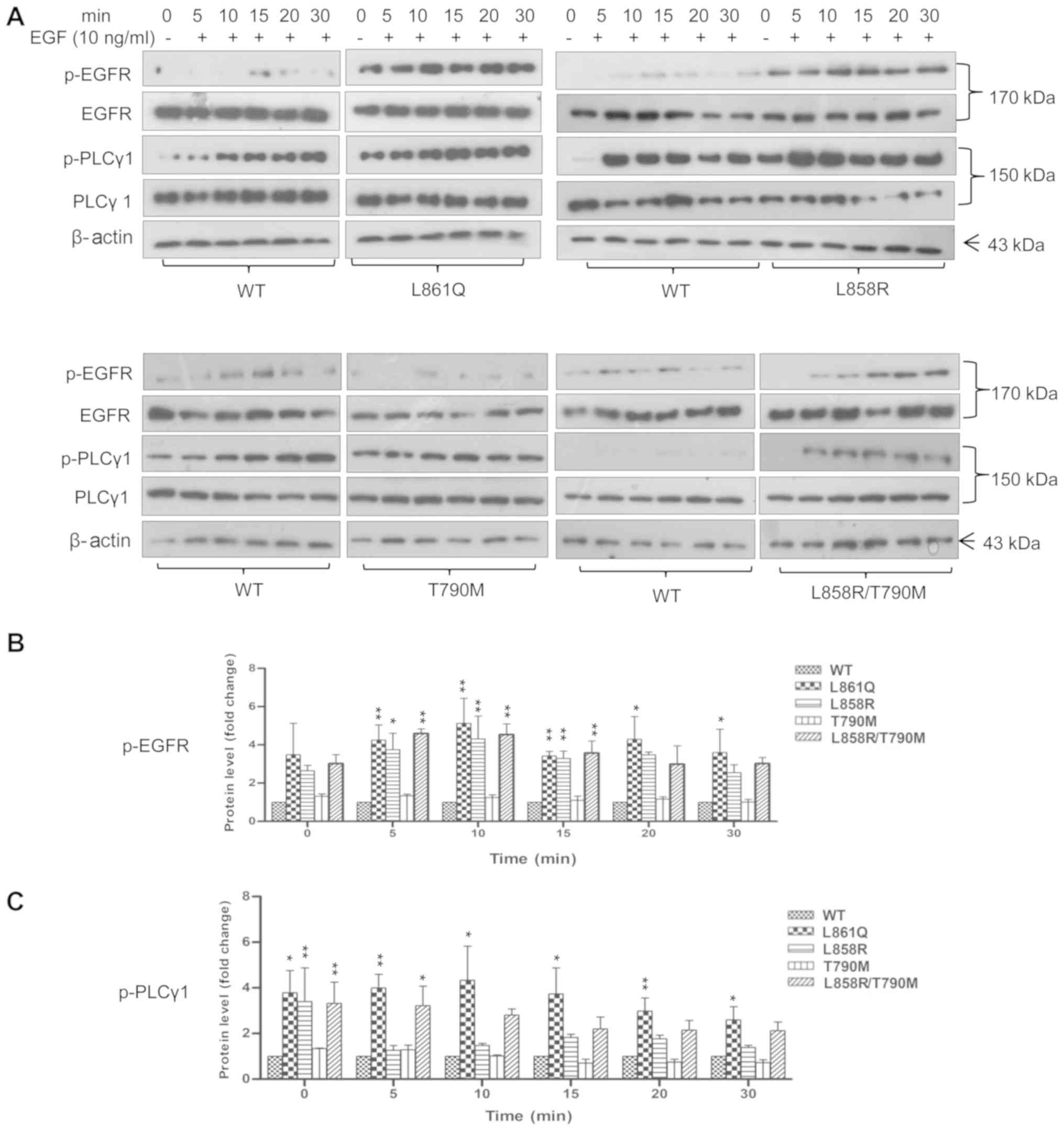 | Figure 1Mutant EGFR induces PLCγ1
phosphorylation. Cells stably expressing Wt or mutant receptors
were serum-starved overnight and stimulated with 10 ng/ml of EGF
for different periods of time. Following stimulation, cells were
harvested and lysates were prepared. An equal amount of each
protein sample was subjected to SDS-PAGE followed by western blot
analysis using anti-p-EGFR (1068), anti-p-PLCγ1, total EGFR and
PLCγ1 antibodies. β-actin was used as a loading control. (A)
TKI-sensitive mutants, L861Q, L858R, double-mutant (L858R/T790M)
exhibited an increased phosphorylation of PLCγ1 as compared to Wt
and resistant mutant receptor, T790M-expressing cells corresponding
to receptor phosphorylation. Total protein levels of EGFR and PLCγ1
remained unaltered. Expression of proteins from the western blots
was quantified using ImageJ software and normalized to the total
protein levels. Values are represented as bar graphs from 3
independent experiments and the statistical significance of
phosphoprotein expression levels between wild-type and mutants was
indicated on graphs (*P<0.05,
**P<0.01). Bar graphs showing the quantification of
(B) p-EGFR vs. EGFR (C) p-PLCγ1 vs. PLCγ1. EGFR, epidermal growth
factor receptor; PLCγ1, phospholipase γ1; TKI, tyrosine kinase
inhibitor; Wt, wild-type. |
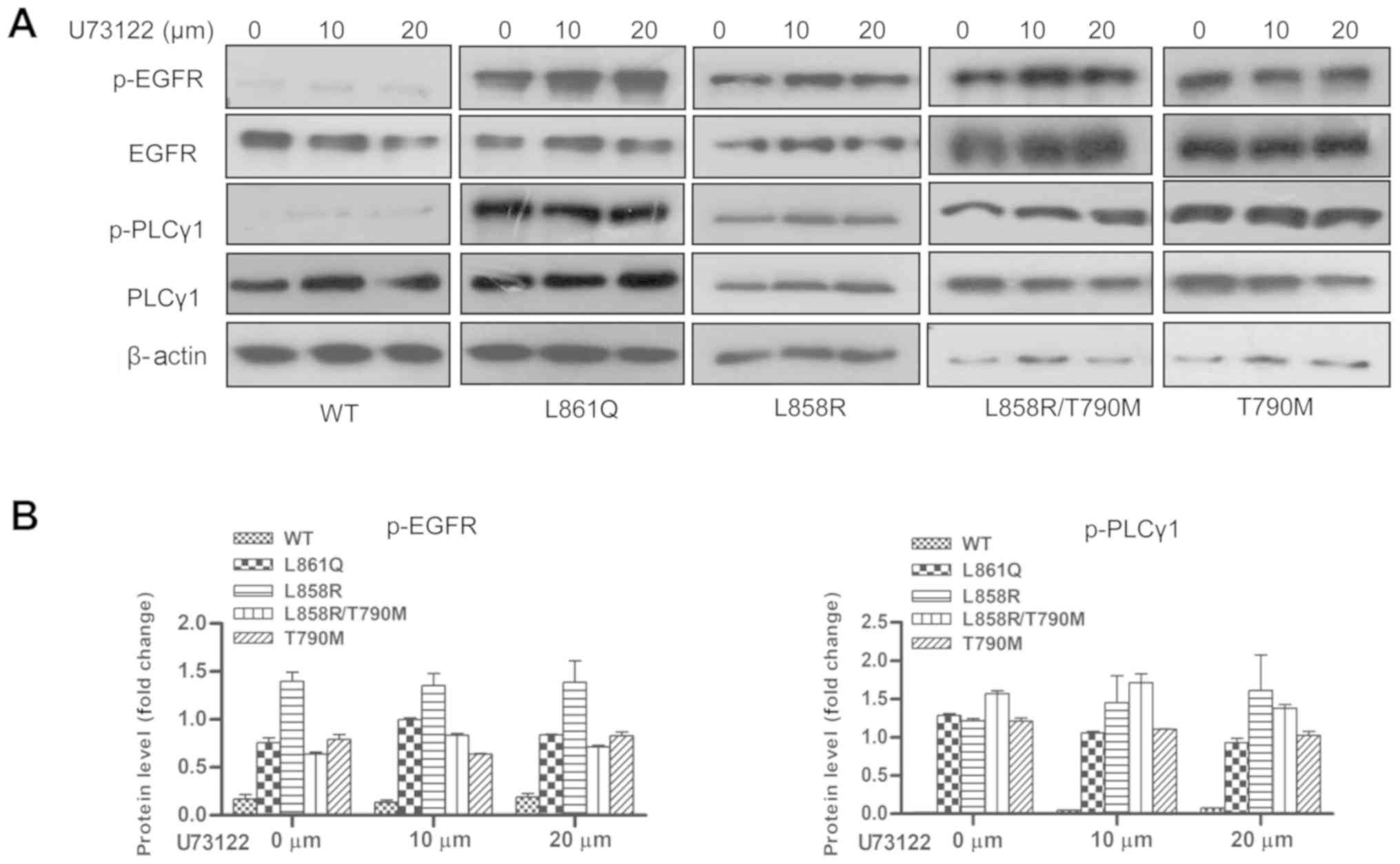
Inhibition of EGFR activity by TKIs
abrogates PLCγ1 phosphorylation
Mutant receptor-mediated PLCγ1 phosphorylation was
measured in the presence of FDA-approved EGFR-targeted drugs
(TKIs). Cells expressing mutant receptors were treated with the
desired concentrations of 1st, 2nd and 3rd generation TKIs. Cells
were serum-starved overnight followed by EGF stimulation for 10 min
and TKI treatment for 2 h. The phosphorylation of PLCγ1, as
examined by western blot analysis, was shown to be reduced
following the inhibition of receptor phosphorylation by TKIs,
demonstrating that PLCγ1 activation is mutant receptor-driven
(Fig. 2). Conversely, treatment of
the cells with the PLC inhibitor, U73122, had no marked effect on
EGFR and PLCγ1 phosphorylation (Fig.
3). Treatment with TKIs resulted in the inhibition of the
aforementioned signaling molecules, suggesting that these are
recruited in response to mutant receptor activation (data not
shown).
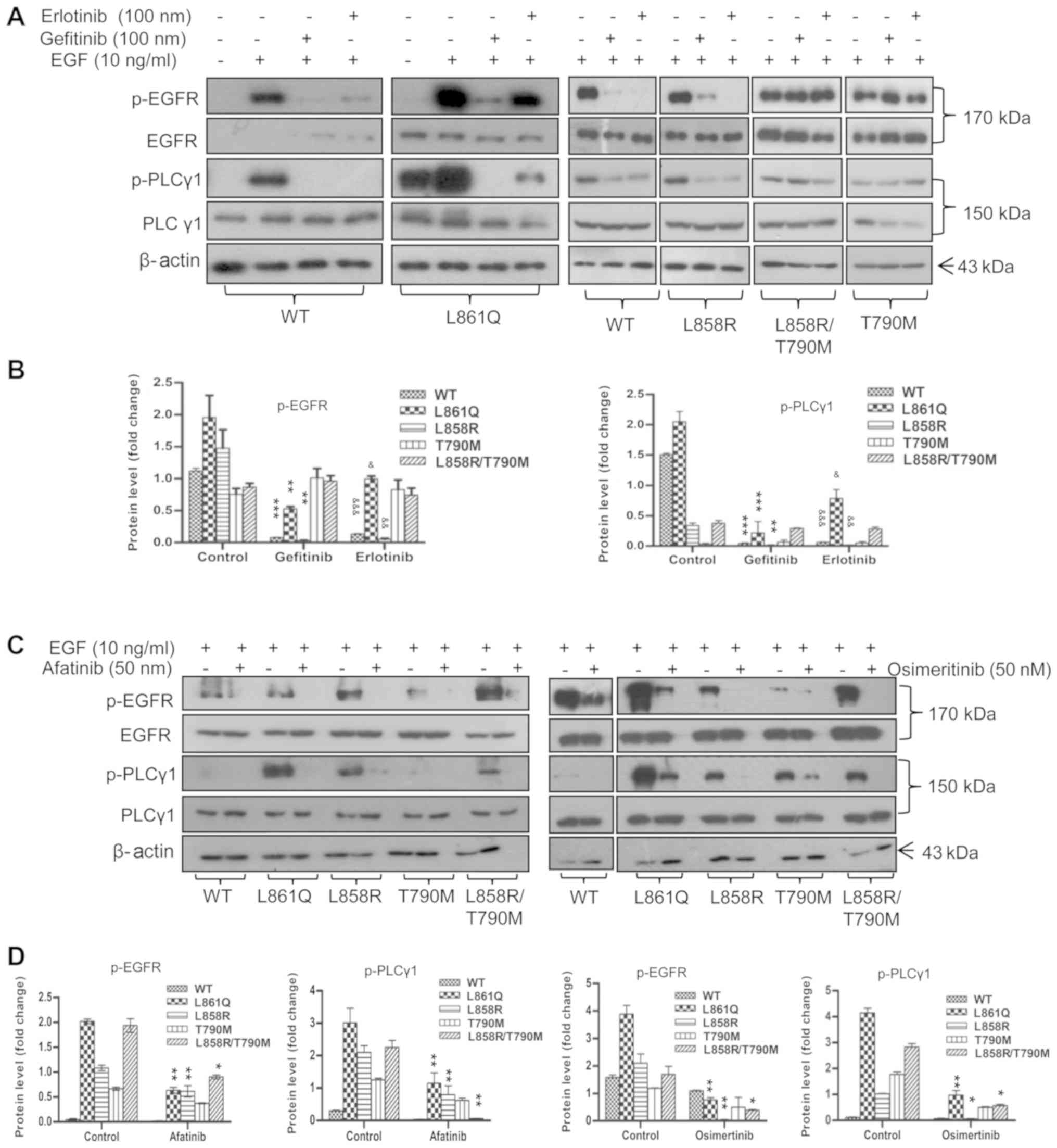 | Figure 2Inhibition of EGFR activity by TKIs
abrogates PLCγ1 phosphorylation. Cells expressing Wt or mutant
receptor were serum-starved overnight. The following day, the cells
were pre-treated with the desired concentrations of 1st, 2nd and
3rd generation TKIs prior to EGF stimulation. Post-stimulation,
cells were harvested and subjected to SDS-PAGE followed by western
blot analysis using anti-p-EGFR (1068), anti-p-PLCγ1, total PLCγ1
and total EGFR antibodies. β-actin was used as a loading control.
(A) Treatment of cells with gefitinib and erlotinib (B)
quantification of p-EGFR and p-PLCγ1 in gefitinib- and
erlotinib-treated cells (C) treatment of cells with afatinib and
osimertinib (D) quantification of p-EGFR and p-PLCγ1 in afatinib-
and osimertinib-treated cells. Densitometric values are represented
as bar graphs of 3 independent experiments and the statistical
significance of p-EGFR and p-PLCγ1 expression between TKI-treated
and untreated cells expressing receptor mutants is indicated on the
graphs. For gefitinib treatment, significance is indicated with
asterisk symbols (*P<0.05, **P<0.01,
***P<0.001); and for erlotinib, ampersand (&)
symbols were used (&P<0.05,
&&P<0.01,
&&&P<0.001). EGFR, epidermal growth
factor receptor; PLCγ1, phospholipase γ1; TKI, tyrosine kinase
inhibitor; Wt, wild-type. |
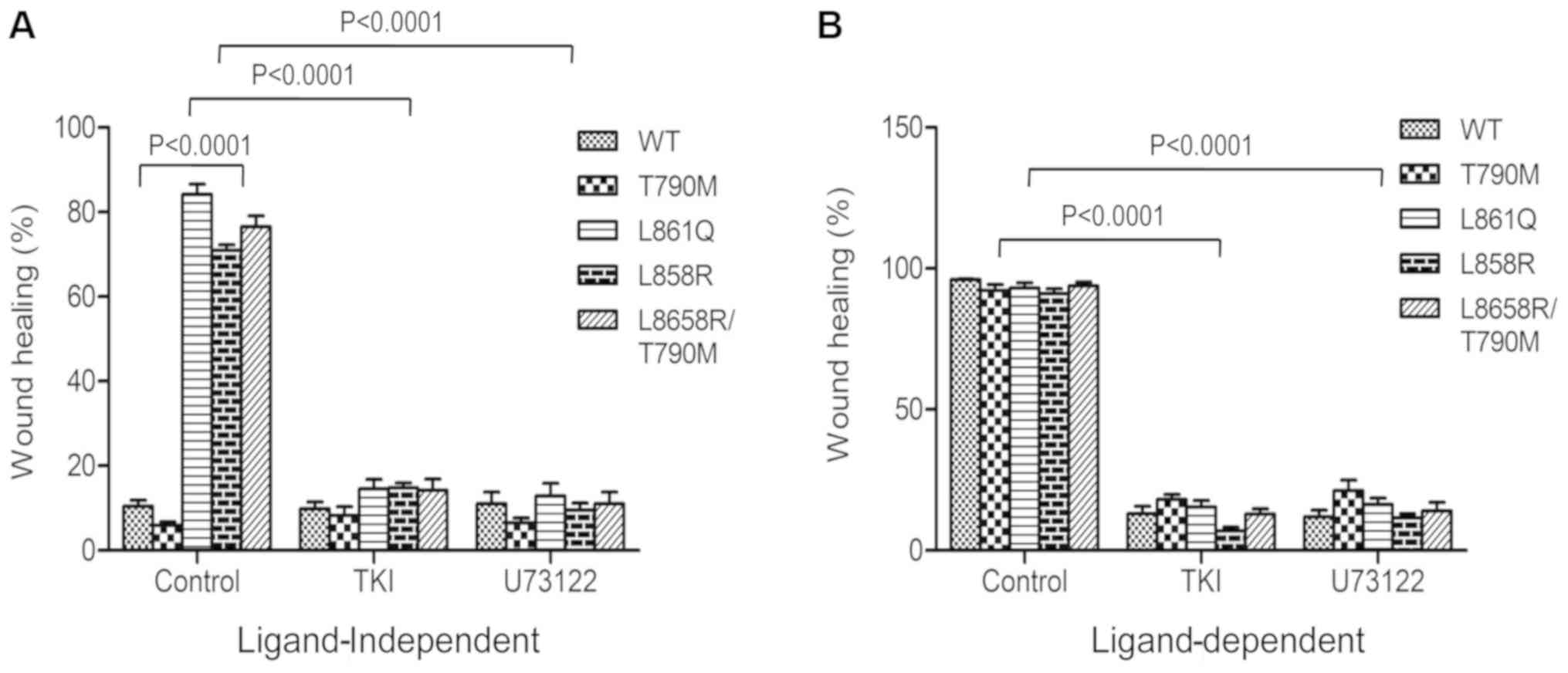
PLCγ1-mediated migration and invasion of
cells expressing mutant receptors
PLCγ1 is a known regulator of tumor progression and
cell migration activated by receptor tyrosine kinase signaling
(17,18). The role of PLCγ1 in the migration
and invasion of cells expressing NSCLC-associated receptor mutants
was determined using wound healing and Boyden chamber assays. Wound
healing assays were carried out in cells expressing L861Q, L858R,
L858R/T790M, T790M and Wt receptor treated with TKIs or the PLC
inhibitor, U73122 with or without EGF stimulation. Afatinib, a 2nd
generation TKI, was only used to inhibit receptor phosphorylation
in this experiment, as it has been approved for the treatment of
NSCLC tumors with resistant mutations. It has also been shown to be
effective against tumors harboring both common and uncommon EGFR
mutations in vitro (19)
and in clinical settings (20).
Wounds were generated using a micropipette tip and
wound closure was measured using ImageJ software at 0 and 24 h
post-incubation. In the absence of EGF, cells expressing L861Q,
L858R and L858R/T790M mutants migrated out to fill the gap, thus
closing the wound. Cells expressing Wt or T790M mutant did not
exhibit any marked migration, leaving the gap unfilled. The extent
of the migration of the cells is illustrated in Fig. 4A. EGF stimulation at the
concentration of 10 ng/ml induced the migration of cells expressing
Wt and resistant mutant T790M as well, subsequently leading to
wound closure (Fig. 4B). However,
treatment with TKIs and PLC inhibitor blocked cell migration,
irrespective of EGF stimulation (Figs. S2-S6).
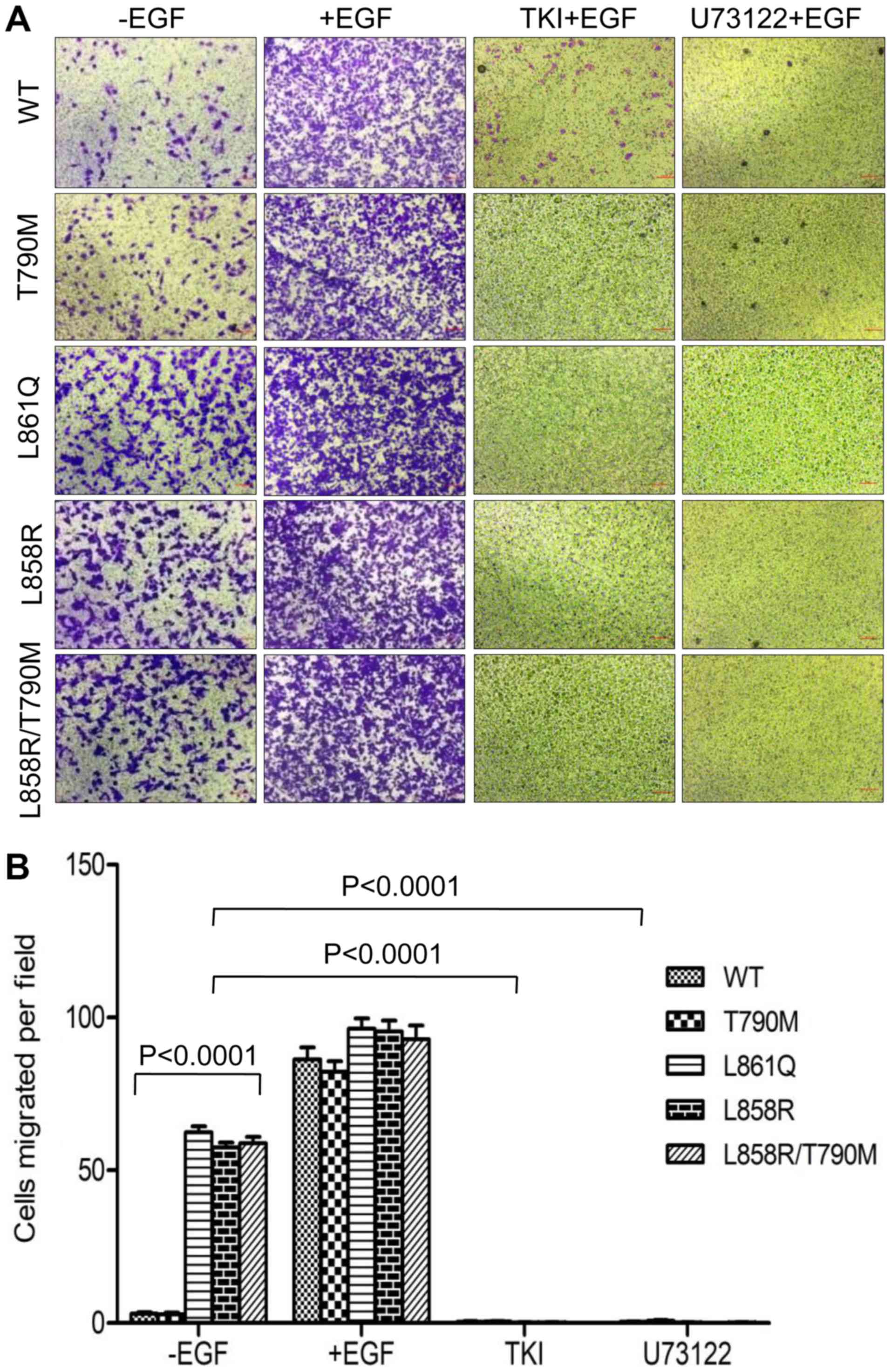
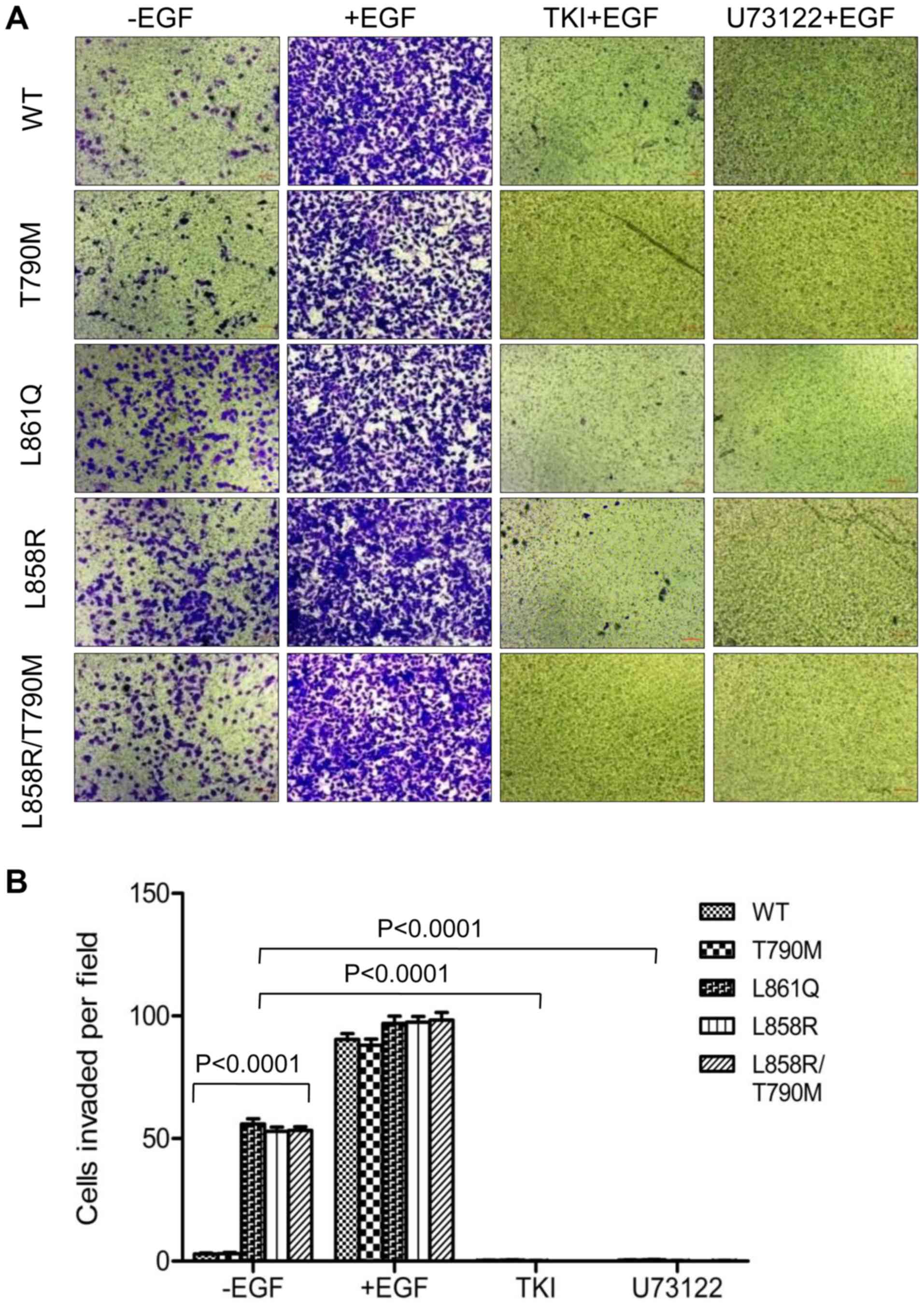
Similar results were obtained with Boyden chamber
migration and invasion assays. In the absence of EGF, the migration
of cells expressing mutant receptors through Transwell inserts
further confirmed the role of the ligand-independent activation of
mutant EGFR and PLCγ1 signaling in cell migration and invasion.
Treatment with afatinib at a concentration of 200 nM and U73122 at
3 µM followed by EGF stimulation, inhibited the migration
(Fig. 5) and invasion (Fig. 6) of cells expressing Wt and mutant
receptors. Taken together, these data demonstrate that mutant
receptor driven PLCγ1 is a potential regulator of invasive and
migratory potential of NSCLC cells harboring EGFR mutations.
PLC inhibitor, U73122, does not inhibit
the proliferation of cells expressing EGFR mutants
The inhibition of proliferation of NSCLC tumors
harboring EGFR mutations with TKI treatment is a well-established
fact. In the present study, the role of PLCγ1 in tumor cell
proliferation and invasion was determined with the PLC inhibitor,
U73122, following EGF stimulation. Treatment of cells with U73122
for 25 min significantly reduced the invasion and migration of 293
cells expressing both TKI sensitive and resistant mutants as
aforementioned. Nonetheless, no effect of PLC inhibitor treatment
on cell viability was observed. Cells were observed to be 100%
viable after 48 h following U73122 treatment, as assessed by MTT
assays (Fig. 7A). Upon validation
of the signaling pathways, U73122 treatment for 25 min at 10-20
µM inhibited Akt and Erk1/2 phosphorylation transiently,
whereas it increased the phosphorylation of both these signaling
molecules was observed following 48 h of drug treatment. This
suggested that cells can maintain their proliferative potential
independent of PLC activation (Fig.
7B).
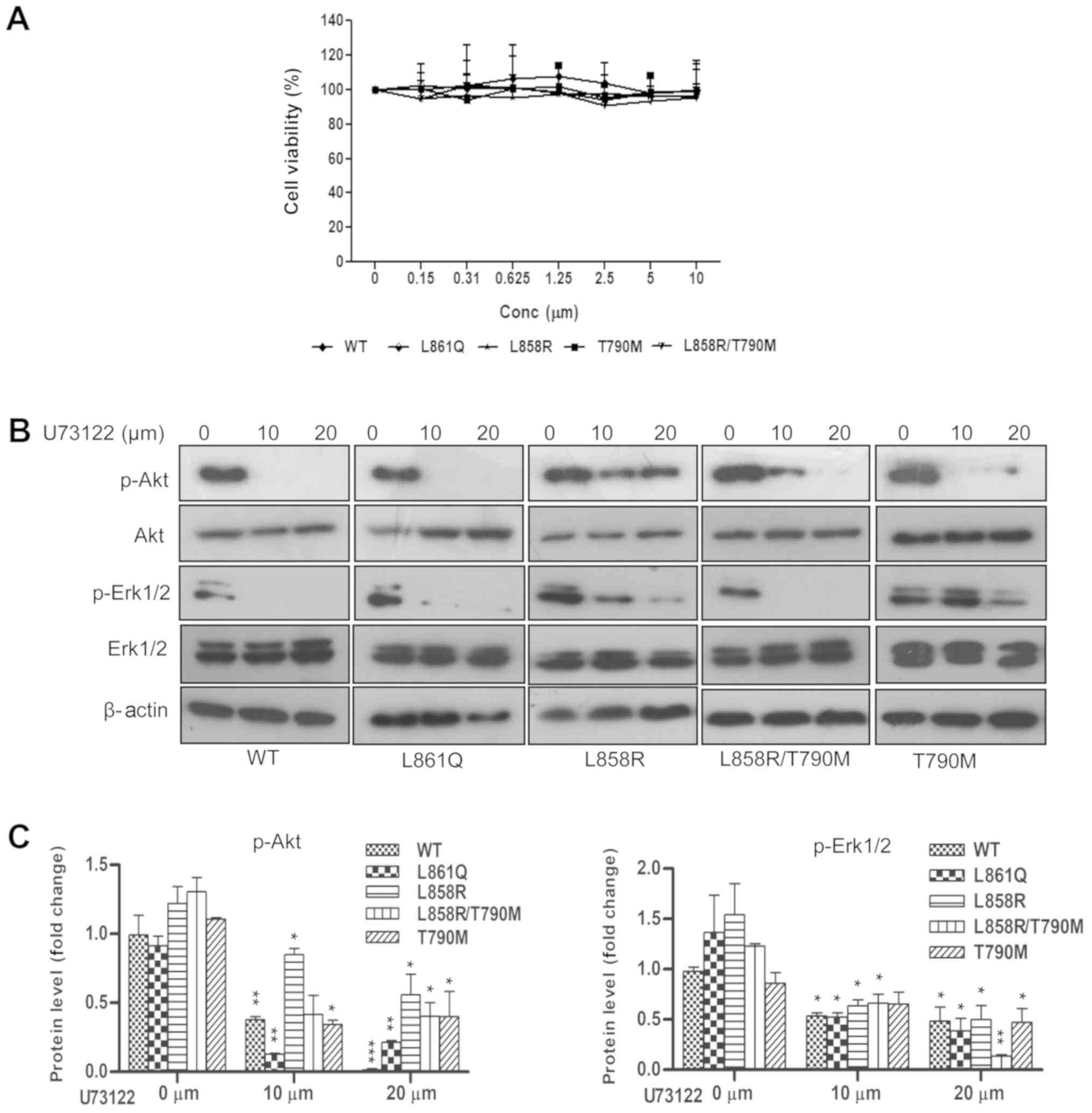 | Figure 7Downregulation of Akt and Erk1/2
signaling following inhibition of PLC activity. Cells expressing
mutants were plated in 96- and 12-well plates. At the desired
confluency, cells were serum-starved and treated with various
concentrations of the PLC inhibitor, U73122, for 25 min followed by
EGF stimulation. Lysates were prepared from 12-well plates
post-drug treatment and subjected to SDS-PAGE followed by western
blot analysis with specific antibodies to anti-p-Akt,
anti-p-Erk1/2, anti β-actin. Cells in the 96-well plate were
incubated in U73122-containing medium and the percentage of
viability was measured at 48 h by MTT assay. (A) Graph representing
the percentage of viability in cells treated with U73122, (B)
expression of p-Akt, total Akt, p-Erk1/2, total Erk1/2 and β-actin,
(C) quantification of p-Akt vs. Akt and p-Erk1/2 vs. Erk1/2 levels.
Densitometric values are represented as 3 independent experiments
and the statistical significance of p-Akt and p-Erk1/2 expression
in U73122-treated vs. untreated cells expressing receptor mutants
is indicated on the graphs (*P<0.05,
**P<0.01, ***P<0.001). EGFR, epidermal
growth factor receptor; PLCγ1, phospholipase γ1; TKI, tyrosine
kinase inhibitor; WT, wild-type. |
The effect of PLC inhibitor on cell proliferation
was validated using soft agar colony formation assays. Cells
expressing Wt and mutant receptors treated with 10 µM
U73122, a higher concentration than the concentration used for the
invasion and migration assays, exhibited no significant reduction
in colony size compared to the untreated cells (Fig. S7). This experiment confirmed that
PLCγ1 may not have a direct regulatory role on the proliferation of
cells expressing NSCLC associated EGFR mutants, irrespective of EGF
stimulation. Cells treated with 1µM TKIs were included in
this assay as a positive control. The anchorage independent colony
forming ability of 293 cells expressing receptor mutants was
reduced following treatment with TKIs. Treatment with U73122 did
not inhibit the formation of colonies, further identifying that
PLCγ1 may be a downstream component of activated EGFR. In addition,
no significant effect of U73122 on Akt and Erk1/2, cell survival
signaling pathways was observed up to 48 h post-treatment (Fig. S8). However, this experiment was
performed once, as it was already confirmed that U73122 exerted no
inhibitory effect on cell proliferation by soft agar assay.
Discussion
Receptor tyrosine kinase activity, induced upon
ligand binding, recruits various signaling pathways and downstream
molecules which are regulators of cell survival and growth. The
dysregulated activity of receptors, due to mutations in its kinase
domain, is one of the mechanisms involved in the progression of a
number of tumors, including lung cancer types. In the present
study, the signaling molecules recruited in response to
NSCLC-associated EGFR mutations in a ligand-dependent and
-independent manner in the 293 cell line were evaluated. It is a
non-endogenous EGFR-expressing cell line and is considered
EGFR-negative. It was used as a negative control for the comparison
of EGFR expression in lung cancer cell lines expressing EGFR
(21). Thus, the 293 cell line
with a EGFR null background was used to investigate the effects of
EGFR mutants on molecular signaling. The present study demonstrated
an increased phosphorylation of the signaling molecules, PLCγ1,
c-Cbl, Stat, Erk1/2, Akt, Shc and Gab1, in response to altered EGFR
activity and the abrogation of their phosphorylation following
treatment with TKIs. These signaling molecules are known to be
involved in a number of biological processes that are essential for
tumor cell proliferation, survival, migration and invasion. Both
Gab1 and Shc are adaptor proteins, essential for the
receptor-activated Erk1/2, Akt and Stat pathways (22,23).
c-Cbl is an ubiquitin ligase that plays an important role in
ligand-dependent receptor ubiquitination (24). The results from the present study
are in agreement with those of earlier reports, in which the
induction of signaling pathways was demonstrated in response to
NSCLC-associated EGFR mutant kinase activity (13,14).
The mutant receptor remains constitutively active and leads to
malignant cell survival, tumor progression, migration and
metastasis.
The induction of tumor cell migration is a foremost
step in tumor metastasis, and it is evident from a number of
studies that PLCγ1 is highly expressed in invasive tumors, such as
metastatic colorectal cancer cells (25) and breast carcinoma (26). PLCγ1 plays a regulatory role in
cell proliferation, invasion and metastasis in various other cancer
types (17,18). PLCγ1 is a lipase and isozyme of the
phosphoinositide specific PLC family. It is a multi-domain protein
consisting of two pleckstrin homology (PH), two Src homology 2
(SH2), one Src homology 3 (SH3) and two catalytic domains. PLCγ1
signaling is induced by growth factors, such as EGF (27), PDGF (28) and IGF (29) following the increased activity of
their respective receptors, and subsequently contributes to
enhanced cell motility (30,31).
PLCγ1 binds directly with activated EGFR through its SH2 domain
(32). EGFR then phosphorylates
PLCγ1 at its tyrosine residues and activates its lipase activity.
Activated PLCγ1 hydrolyzes phosphatidylinositol 4, 5-bispho-sphate
(PIP2) to give two secondary messengers, inositol 1, 4,
5-triphosphate (IP3) and diacylglycerol (DAG). IP3 is important for
the transient increase in intracellular Ca2+, while DAG
activates protein kinase C (PKC). These processes are essential for
cell proliferation, invasion and migration.
Previous studies have demonstrated a fundamental
role of the EGFR family members for PLCγ1 activation in tumor cell
motility and metastasis control (33). However, NSCLC-associated EGFR
mutant-driven PLCγ1 activation and its role in cell invasion and
migration is currently unknown. The migration of tumors from the
site of origin is the primary cause of the majority of
cancer-related mortality. In the present study, NSCLC-associated
EGFR mutant-driven PLCγ1 phosphorylation and its functional role in
cell invasion and migration was demonstrated. Following EGF
stimulation, 293 cells expressing L861Q, L858R and double-mutant
L858R/T790M exhibited an increased phosphorylation of PLCγ1, as
compared to the drug-resistant mutant, T790M and Wt receptor.
Phospho-PLCγ1 levels were associated with the corresponding mutant
receptor phosphorylation levels in 293 cells. Receptor
mutant-recruited PLCγ1-mediated cell invasion and migration was
validated using wound healing assays, Transwell cell migration
assays and invasion assays using TKIs and the PLC inhibitor,
U73122. These experiments together demonstrated NSCLC-associated
EGFR mutant-driven PLCγ1 activation in 293 cells. NSCLC patients
commonly develop TKI resistance due to a variety of reasons. The
most commonly reported is the development of secondary mutations in
the exon 20 region of receptor kinase domain (7-9).
Patients not presenting T790M mutations develop resistance due to
the modulation of other proliferative signal pathways (34-38)
resulting in enhanced tumor progression.
PLCγ1 is the downstream signaling molecule of
activated receptor kinase by growth factors and plays a significant
role in the intracellular signaling to modulate cell proliferation
and growth (39). Its role in the
induction of proliferation was investigated by inhibiting its
lipase activity in 293 cells expressing receptor mutants. Treatment
of the cells with U73122 inhibited cell migration and invasion with
no notable effect on EGFR and PLCγ1 phosphorylation. Similarly,
following U73122 treatment, no effect on cell proliferation was
observed. This may be due to the fact that cell proliferation
depends upon interactions between PLCγ1 protein and other effector
molecules instead of its lipase activity as the PLC inhibitor,
U73122, is known to inhibit its lipase activity (40). A study by Xie et al also
reported that the catalytic activity of PLCγ1 is required for the
EGF-induced migration of squamous cell carcinoma cells (41).
Akt and Erk1/2 pathways were inhibited within 25 min
of U73122 treatment followed by EGF stimulation, although the cells
remained viable even after 48 h of drug treatment. In association
with cell viability, the increased phosphorylation of survival
signaling molecules, Akt and Erk1/2 were recorded. This indicates
that the inhibition of PLCγ1 catalytic activity by U73122 may
attenuate the recruitment of survival signals soon after its
addition; however, this effect may be nullified by other
mechanisms, which warrant further investigation. EGFR directly
activates PLCγ1 and indirectly activates Akt. Both these signaling
molecules are important in intracellular signaling. One is involved
in metastasis and the other one in proliferation, respectively The
interaction of phospho-PLCγ1 with Akt in EGF-stimulated receptor
tyrosine kinase activation in metastasis has been previously
reported (32). In the present
study, the interaction between phospho-PLCγ1 and Akt in 293 cells
expressing receptor mutants was not examined; however, whether
these two proteins associate or not and whether their association
has any significance in the regulation of cell proliferation is
another aspect that warrants investigation. It is evident from the
present study that PLC inhibitors do not impose any negative effect
on mutant receptor activity; therefore, it remains constitutively
active and recruits survival signals. This allows cells to enter
the proliferative mode within a short period even in the presence
of PLC inhibitor. As discussed by Jang et al, phospho-PLCγ1
likely interacts with other signaling molecules to maintain the
cells in a proliferative mode, as PLC inhibitor has no effect on
PLCγ1 phosphorylation (40).
Similar effects have been reported in HNSCC and
prostate cancer cells where the inactivation of PLCγ1 inhibited
cell invasion without affecting the cell growth and tumor volume
in vitro and in vivo, respectively (42,43).
On the contrary, in human gastric adenocarcinoma cells,
PLCγ1-dependent proliferation was reported beyond its role in cell
migration (44), although the
upregulation of PLCγ1 was negatively associated with the survival
of patients with hepatocellular carcinoma. It exerts an oncogenic
effect and induces HCC tumorigenesis through Erk1/2 and NF-kB
pathways (45). The suppression of
PLCγ1 prevents the phosphorylation of NF-κB and Erk1/2 activation.
It was has also been demonstrated to be involved in colorectal
tumorigenesis (46). PLCγ1 has
been established as a key modulator of migration and invasion of
various tumor types. However, the detailed mechanism of action for
PLCγ1 mediated tumor proliferation needs to be explored in various
cancer types including lung cancer.
In conclusion, the results of the present study
demon-strated that mutant receptor-recruited PLCγ1 activation
mediates the invasion and migration of 293 cells expressing
NSCLC-associated EGFR mutants. Nonetheless, constitutively active
mutant receptor overcomes the effects induced by the inhibition of
PLCγ1 lipase activity, recruiting proliferative signals to keep the
cells viable. Considering the aforementioned results, PLCγ1 may be
considered as a drug target upon validation and confirmation in
more lung cancer cell lines with NSCLC-associated EGFR mutations in
combination with other therapeutics to increase the disease-free
survival of patients with NSCLC.
Supplementary Data
Acknowledgments
The authors acknowledge the University Grants
Commission for fellowship of the first author and Jawaharlal Nehru
University for providing infrastructure and facilities.
Funding
The present study was supported by the Indian
Council of Medical Research (Project No. 5/13/94/2008-NCD- III) and
DST-PURSE, New Delhi, India.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MSR designed the study and supervised the work. SM,
AK, AMJ performed the experiments. MSR and SM analyzed the data and
prepared the manuscript. All the authors read approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong K-K: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eberhard DA, Johnson BE, Amler LC, Goddard
AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson
DH, et al: Mutations in the epidermal growth factor receptor and in
KRAS are predictive and prognostic indicators in patients with
non-small-cell lung cancer treated with chemotherapy alone and in
combination with erlotinib. J Clin Oncol. 23:5900–5909. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lièvre A, Bachet J-B, Le Corre D, Boige V,
Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, et al:
KRAS mutation status is predictive of response to cetuximab therapy
in colorectal cancer. Cancer Res. 66:3992–3995. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Raymond E, Faivre S and Armand JP:
Epidermal growth factor receptor tyrosine kinase as a target for
anticancer therapy. Drugs. 60(Suppl 1): 15–23; discussion 41–42.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nguyen K-SH, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oxnard GR, Arcila ME, Sima CS, Riely GJ,
Chmielecki J, Kris MG, Pao W, Ladanyi M and Miller VA: Acquired
resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung
cancer: Distinct natural history of patients with tumors harboring
the T790M mutation. Clin Cancer Res. 17:1616–1622. 2011. View Article : Google Scholar
|
|
10
|
Liao B-C, Lin C-C and Yang JC-H: Second
and third-generation epidermal growth factor receptor tyrosine
kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin
Oncol. 27:94–101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jänne PA, Yang JC-H, Kim D-W, Planchard D,
Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, et al: AZD9291
in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J
Med. 372:1689–1699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cross DAE, Ashton SE, Ghiorghiu S,
Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA,
Mellor MJ, et al: AZD9291, an irreversible EGFR TKI, overcomes
T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer
Discov. 4:1046–1061. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim Y, Apetri M, Luo B, Settleman JE and
Anderson KS: Differential effects of tyrosine kinase inhibitors on
normal and oncogenic EGFR signaling and downstream effectors. Mol
Cancer Res. 13:765–774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamath A, Joseph AM, Gupta K, Behera D,
Jaiswal A, Dewan R and Rajala MS: Proteomic analysis of HEK293
cells expressing non small cell lung carcinoma associated epidermal
growth factor receptor variants reveals induction of heat shock
response. Exp Hematol Oncol. 4:162015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rasband WS: US National Institutes of
Health, MD, USA. http://imagejnih.gov/ij/uri.
2011, Accessed August 28, 2008.
|
|
17
|
Piccolo E, Innominato PF, Mariggio MA,
Maffucci T, Iacobelli S and Falasca M: The mechanism involved in
the regulation of phospholipase Cgamma1 activity in cell migration.
Oncogene. 21:6520–6529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wells A and Grandis JR: Phospholipase C-γ1
in tumor progression. Clin Exp Metastasis. 20:285–290. 2003.
View Article : Google Scholar
|
|
19
|
Li D, Ambrogio L, Shimamura T, Kubo S,
Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A,
Himmelsbach F, et al: BIBW2992, an irreversible EGFR/HER2 inhibitor
highly effective in preclinical lung cancer models. Oncogene.
27:4702–4711. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kobayashi Y and Mitsudomi T: Not all
epidermal growth factor receptor mutations in lung cancer are
created equal: Perspectives for individualized treatment strategy.
Cancer Sci. 107:1179–1186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang F, Wang S, Yin L, Yang Y, Guan Y,
Wang W, Xu H and Tao N: Quantification of epidermal growth factor
receptor expression level and binding kinetics on cell surfaces by
surface plasmon resonance imaging. Anal Chem. 87:9960–9965. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yart A, Laffargue M, Mayeux P, Chretien S,
Peres C, Tonks N, Roche S, Payrastre B, Chap H and Raynal P: A
critical role for phosphoinositide 3-kinase upstream of Gab1 and
SHP2 in the activation of ras and mitogen-activated protein kinases
by epidermal growth factor. J Biol Chem. 276:8856–8864. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashimoto A, Kurosaki M, Gotoh N, Shibuya
M and Kurosaki T: Shc regulates epidermal growth factor-induced
activation of the JNK signaling pathway. J Biol Chem.
274:20139–20143. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shtiegman K, Kochupurakkal BS, Zwang Y,
Pines G, Starr A, Vexler A, Citri A, Katz M, Lavi S, Ben-Basat Y,
et al: Defective ubiquitinylation of EGFR mutants of lung cancer
confers prolonged signaling. Oncogene. 26:6968–6978. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nomoto K, Tomita N, Miyake M, Xhu DB,
LoGerfo PR and Weinstein IB: Expression of phospholipases γ1, β1,
and δ1 in primary human colon carcinomas and colon carcinoma cell
lines. Mol Carcinog. 12:146–152. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arteaga CL, Johnson MD, Todderud G, Coffey
RJ, Carpenter G and Page DL: Elevated content of the tyrosine
kinase substrate phospholipase C-gamma 1 in primary human breast
carcinomas. Proc Natl Acad Sci USA. 88:10435–10439. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen P, Xie H, Sekar MC, Gupta K and Wells
A: Epidermal growth factor receptor-mediated cell motility:
Phospholipase C activity is required, but mitogen-activated protein
kinase activity is not sufficient for induced cell movement. J Cell
Biol. 127:847–857. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kundra V, Escobedo JA, Kazlauskas A, Kim
HK, Rhee SG, Williams LT and Zetter BR: Regulation of chemotaxis by
the platelet-derived growth factor receptor-β. Nature. 367:474–476.
1994. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bornfeldt KE, Raines EW, Nakano T, Graves
LM, Krebs EG and Ross R: Insulin-like growth factor-I and
platelet-derived growth factor-BB induce directed migration of
human arterial smooth muscle cells via signaling pathways that are
distinct from those of proliferation. J Clin Invest. 93:1266–1274.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wells A, Kassis J, Solava J, Turner T and
Lauffenburger DA: Growth factor-induced cell motility in tumor
invasion. Acta Oncol. 41:124–130. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Polk DB: Epidermal growth factor
receptor-stimulated intestinal epithelial cell migration requires
phospholipase C activity. Gastroenterology. 114:493–502. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Y, Wu J and Wang Z: Akt binds to and
phosphorylates phospholipase C-gamma1 in response to epidermal
growth factor. Mol Biol Cell. 17:2267–2277. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Appert-Collin A, Hubert P, Crémel G and
Bennasroune A: Role of ErbB receptors in cancer cell migration and
invasion. Front Pharmacol. 6:2832015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sos ML, Koker M, Weir BA, Heynck S,
Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P,
et al: PTEN loss contributes to erlotinib resistance in EGFR-mutant
lung cancer by activation of Akt and EGFR. Cancer Res.
69:3256–3261. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bidkhori G, Moeini A and Masoudi-Nejad A:
Modeling of tumor progression in NSCLC and intrinsic resistance to
TKI in loss of PTEN expression. PLoS One. 7:e480042012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kanda R, Kawahara A, Watari K, Murakami Y,
Sonoda K, Maeda M, Fujita H, Kage M, Uramoto H, Costa C, et al:
Erlotinib resistance in lung cancer cells mediated by integrin
β1/Src/Akt-driven bypass signaling. Cancer Res. 73:6243–6253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pao W, Wang TY, Riely GJ, Miller VA, Pan
Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG and Varmus HE: KRAS
mutations and primary resistance of lung adenocarcinomas to
gefitinib or erlotinib. PLoS Med. 2:e172005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wahl M and Carpenter G: Selective
phospholipase C activation. BioEssays. 13:107–113. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jang H-J, Suh P-G, Lee YJ, Shin KJ, Cocco
L and Chae YC: PLCγ1: Potential arbitrator of cancer progression.
Adv Biol Regul. 67:179–189. 2018. View Article : Google Scholar
|
|
41
|
Xie Z, Peng J, Pennypacker SD and Chen Y:
Critical role for the catalytic activity of phospholipase C-gamma1
in epidermal growth factor-induced cell migration. Biochem Biophys
Res Commun. 399:425–428. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Davies G, Martin TA, Ye L, Lewis-Russell
JM, Mason MD and Jiang WG: Phospholipase-C gamma-1 (PLCγ-1) is
critical in hepatocyte growth factor induced in vitro invasion and
migration without affecting the growth of prostate cancer cells.
Urol Oncol. 26:386–391. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thomas SM, Coppelli FM, Wells A, Gooding
WE, Song J, Kassis J, Drenning SD and Grandis JR: Epidermal growth
factor receptor-stimulated activation of phospholipase Cgamma-1
promotes invasion of head and neck squamous cell carcinoma. Cancer
Res. 63:5629–5635. 2003.PubMed/NCBI
|
|
44
|
Dai L, Zhuang L and Zhang B, Wang F, Chen
X, Xia C and Zhang B: DAG/PKCδ and IP3/Ca2+/CaMK IIβ
operate in parallel to each other in PLCγ1-driven cell
proliferation and migration of human gastric adenocarcinoma cells,
through Akt/mTOR/S6 pathway. Int J Mol Sci. 16:28510–28522. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tang W, Zhou Y, Sun D, Dong L, Xia J and
Yang B: Oncogenic role of phospholipase C-γ1 in progression of
hepatocellular carcinoma. Hepatol Res. 49:559–569. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang P, Zhao Y, Zhu X, Sedwick D, Zhang X
and Wang Z: Cross-talk between phospho-STAT3 and PLCγ1 plays a
critical role in colorectal tumorigenesis. Mol Cancer Res.
9:1418–1428. 2011. View Article : Google Scholar : PubMed/NCBI
|