Introduction
Sphingosine-1-phosphate (S1P) regulates a variety of
critical cellular processes, including apoptosis, proliferation,
and cellular motility. Furthermore, it supports the survival of
glioblastoma (GBM) stem cells (1-3) and
it is also involved in the regulation of neurogenesis and
angiogenesis (4). In recent years,
S1P has been recognized as an important oncogenic factor in several
solid malignancies (5).
The concentration of the bio-active sphingolipid S1P
is strongly regulated by the concerted action of synthesizing and
catabolizing enzymes (6).
Sphingosine kinases 1 and 2 (SphK1 and SphK2) synthesize S1P from
membranous sphingosine, whereas the S1P-phosphatases
(S1P-phosphatase 1 and 2) dephosphorylate S1P back to sphingosine
(6).
Additionally, S1P lyase mediates the irreversible
cleavage/division to hexadecenal, phosphoethanolamine and, finally,
phosphatidylethanolamine (7).
Additionally, the metabolism of ceramide is also a source for
sphingolipids and S1P (8). The
balance between S1P phosphorylation and degradation is important
for the regulation of cell growth, and plays a crucial role in
carcinogenesis and other pathological processes (9). Mora et al reported that, in
glioma cells, sphingosine was mainly used for the synthesis of
survival-promoting S1P (10).
Neurons and astrocytes, as well as GBM cells, can
synthesize and export S1P (11,12),
which acts as a second messenger for the activation of pathways and
the crucial regulation of cellular processes (5).
Our previous study reported that SphK1 and all S1P
receptors were expressed at different levels in primary,
recur-rent, and secondary GBM when compared to healthy brain tissue
(1). The elevated levels of S1P
and SphK1 in GBM tissue were found to be correlated with a shorter
survival rate of patients affected by GBM (1). Moreover, S1P was found to stimulate
the motility and invasiveness of GBM cells by triggering
S1P-producing enzymes and S1P receptors (13). The S1P G-coupled receptors are
responsible for the activation of the Ras/extracellular
signal-regulated kinase (ERK), the phosphoinositide 3-kinase
(PI3K)/AKT, and the Rho/Ras (ROCK) signaling pathways (14). These intracellular pathways are
associated with the growth and survival of GBM cells as well as the
mutation of the tumor-suppressor genes p21, p16, phosphatase and
tensin homolog (PTEN) and TP53 (15).
Bektas et al (16), reported that the inhibition of the
S1P pathway induces apoptosis; therefore, new GBM therapy
approaches should focus on the sphingosine pathway using
S1P-analogues (4,16-18).
FTY720 (fingolimod) is a S1P-analogue which was
found to modulate Rho-associated kinase-1 (ROCK1),
epithelial-mesenchymal transition (EMT)-related factors, and the
PI3K/protein kinase B (AKT)/mammalian target of the rapamycin
(mTOR)/p70S6 kinase signaling pathway (19). AKT is active in 70% of GBM
patients, particularly in those with PTEN loss (20). The ability of FTY720 to cross the
blood-brain barrier and the fact that it is well-tolerated in human
patients, makes it an excellent candidate for cancer therapy
(7,21-23).
FTY720 has also shown an immunomodulating activity by causing
immunosuppression in patients affected by glioblastoma (24).
Therefore, the aim of the present study was to
investigate the effects of FTY720 on the viability and the
proliferation of GBM cells and to compare its cytotoxicity with the
effects of temozolomide (TMZ), which is currently used as the
standard treatment for patients affected by GBM.
Materials and methods
Cell culture
Human GBM cell lines A172, G28 [obtained from the
American Type Culture Collection (ATCC)], U87 (glioblastoma of
unknown origin) as well as primary #33, #367, and #391 GBM cells
isolated from patient tissues were used for this study. The present
study was approved by the local ethics committee (Ethics Committee
of Medicine Faculty, Justus Liebig University Giessen),
(application no. AZ 07/09). All patients provided signed consent
for the collection of tumor tissue. All patients were female and
were surgically treated at Department of Neurosurgery in Justus
Liebig University Giessen between 2013-2020. Their ages were 67
(#33), 45 (#391) and 68 years (#367). The diagnosis of GBM was
established at the Department of Neuropathology in Justus Liebig
University Giessen.
For the primary cell cultures, tissue specimens were
freshly collected from surgery and immediately minced in Dulbecco's
modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc.) with
a sterile disposable scalpel and trypsinized with trypsin/EDTA
0.05% for several minutes. Trypsinization was stopped with 10 ml
DMEM and the mince was passed through a cell strainer (60
µm, Sigma Aldrich; Merck KGa) to obtain a single cell
suspension. Cells were washed with PBS and the pellet was
resuspended with DMEM and transferred to a 25 cm2
culture flask (Greiner, Bio-One) and grown at 37°C and 5%
CO2 saturation. Cells were passaged 2-3 times and
expanded to 175 cm2 flasks (Greiner, Bio-One) before
washing in PBS, resuspended with DMSO (Merck KGa) and snap frozen
in liquid nitrogen. For this study, only cell cultures from
patients with confirmed diagnosis of glioblastoma at our Department
of Neuropathology, Justus Liebig University Giessen were
chosen.
For our experiments cells were defrosted in a 37°C
water bath and cultured as an adherent monolayer in 25
cm2 flasks as previously described (25).
All cells were cultured in DMEM (Thermo Fisher
Scientific, Inc.), supplemented with 10% fetal bovine serum (FBS;
Biochrom AG) and 1% penicillin-streptomycin (Biochrom AG) at 37°C
and 5% CO2 saturation. The cells were washed with
phosphate-buffered saline (PBS; Biochrom AG) and trypsinized with
trypsin/EDTA 0.05% (Biochrom AG) after reaching 80% confluence. PBS
and trypsin/EDTA were pre-heated to 37°C.
Drugs and treatment
FTY720 (Cayman Chemical Co.) and TMZ were dissolved
in dimethyl sulfoxide (DMSO; Sigma Aldrich; Merck KGa) at a
concentration of 1 mM. FTY720 stock solution (1 mM) was further
diluted in culture medium (DMEM) to yield the concentrations of 1,
10, 25, 50, and 100 µM. Aliquots of the FTY720 stock and the
final diluted concentrations were stored at −20°C.
Cells were treated for 24, 48, or 72 h with FTY720.
The control samples refer to cells incubated with DMSO only.
xCELLigence
Impedance-based measurements of cell proliferation
and measurements of half maximal inhibitory concentration
(IC50) were performed with the xCELLigence Real-Time
Cell Analyzer (RTCA) in E-plates (Roche Applied Science) under
standard culture conditions as described above. The readout
recorded by the RTCA is a dimensionless cell index (CI) value that
correlates with cell number/quantity/amount. The cells were
continuously monitored every 15 min and allowed to adhere and
proliferate for 24 h in a drug-free medium. For the treatment, the
medium was completely replaced with a FTY720-containing medium in
serial concentrations of 5, 10 and 25 µM. For the controls,
a drug-free, DMSO-containing medium was used. Readings were
performed for at least 72 h after the treatment. The samples were
analyzed at the minimum in triplicate. The RTCA-software v1.2.2
(Roche Applied Science) was used to analyze the data. Curves were
normalized to the time-point just prior to adding the drug, to
which a CI value of 1 was assigned.
MTT assay
To assess cell proliferation and survival, a MTT
assay was performed with varying doses of FTY720 and TMZ.
Briefly/firstly, 104 cells per well were seeded into
96-well plates. The cell lines were treated with 5, 10, 25, and 50
µM of FTY720. Positive control cells were treated with TMZ
at concentrations of 25, 50, 75, and 100 µM, while negative
control cells were treated with 0.05% DMSO. Measurements were
performed after 24, 48, and 72 h. Cells were incubated for 4 h with
10 µl of MTT solution (Roche Applied Science). Finally, 100
µl of solution containing 10% sodium dodecyl sulfate (SDS)
in a 0.01 µM hydrochloride solution was added. The cells
were then incubated overnight at 37°C in an atmosphere with 5%
CO2 saturation. Absorbance at a wavelength of 550 nm was
measured, with 650 nm serving as a reference wavelength.
IC50 values for FTY720 were calculated at the 72 h
observation time point and compared to the effects of TMZ.
Flow cytometry (FACS)
Cell lines were cultured in 6-well plates and
prepared in triplicates. Cells were left to adhere to the bottom of
the culture flask overnight and subsequently treated with FTY720 in
the previously-mentioned concentrations for 24, 48, and 72 h,
respectively. Negative controls were prepared with DMSO in equal
concentrations. Thereafter, the cells were washed with PBS,
trypsinized and centrifuged for 5 min at 252 × g. The pellet was
resuspended in 1.5 ml of ice cold PBS. Cells were fixed with 3.5 ml
of ethanol (Carl Roth GmbH & Co. KG) while being carefully
vortexed, and subsequently, stored at -20°C for 24 h.
Before measurement, the cells were centrifuged for
10 min at 252 × g and the pellet was then resuspended in 1 ml of
PBS. Ten microliters of an RNAse solution (concentration 100
µg/ml) was added and the solution was incubated at 37°C on a
thermocycler at 18 × g for 10 min. Fifty microliters of a propidium
iodide (PI) solution (concentration 50 µg/ml) was then
added. Incubation took place in darkness for 5 min at room
temperature, and analysis was performed by using a BD FACSCalibur™
flow cytometer. The data was analyzed with CellQuest™ Pro software
(BD Biosciences). The rate of apoptosis (sub-G1 phase), and the G1
and G2 phases were determined using histogram analysis.
RNA isolation, cDNA synthesis, and
quantitative real-time PCR (qPCR)
RNA isolation was performed using the peqGOLD Total
RNA kit (PeqLab, VWR) following the manufacturer's instructions.
The obtained RNA concentrations were measured photometrically using
the NanoDrop™ ND-1000 spectrophotometer (Thermo Fisher Scientific,
Inc.). cDNA synthesis was performed using the
QuantiTect® Reverse Transcription kit (Qiagen GmbH)
following the manufacturer's instructions.
qPCR was performed using the TaqMan™ Gene Expression
Master Mix (Biosystems, Darmstadt, Germany). The following
commercially available primers were used (all purchased from Thermo
Fisher Scientific, Inc.): Actin β (Hs99999903_m1), Akt1
(Hs00920512_m1), MAPK1 (Hs01046828_m1), Rac1 (Hs01588892_g1), Roc1
(Hs01127701_m1), PKCE (Hs00942879_m1), and TP53 (Hs01034249_m1).
The oligos were previously tested by the manufacturer for their
specific binding. Sequences are not provided.
Analysis was performed in quadruplicate. StepOne™
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and the StepOne™ software v2.1 (Applied Biosystems; Thermo
Fisher Scientific, Inc.) were used for the amplification and data
analysis.
Statistical analysis
Statistical analysis was performed by the Student's
t-test, Man-Whitney U, and Wilcoxon tests. A two-sided P<0.05
was considered to indicate a statistically significant difference.
The analysis was performed using Excel 2010
(Microsoft®), Graphpad Prism v6 software (GraphPad
Software Inc.), and SPSS® statistics v24 (IBM Corp.).
Significant differences between the effect of treatment with FTY720
and TMZ were determined statistically by performing the Student's
t-test. The effect of FTY720 on the cell cycle was analyzed by
performing the Mann-Whitney U. The overexpression and
downregulation of cell cycle regulator genes after treatment with
FTY720 compared to untreated cells and time-dependent changes were
investigated in all the genes and analyzed by ANOVA (post hoc test:
Bonferroni).
Results
Effect of FTY720 on glioblastoma cell
proliferation
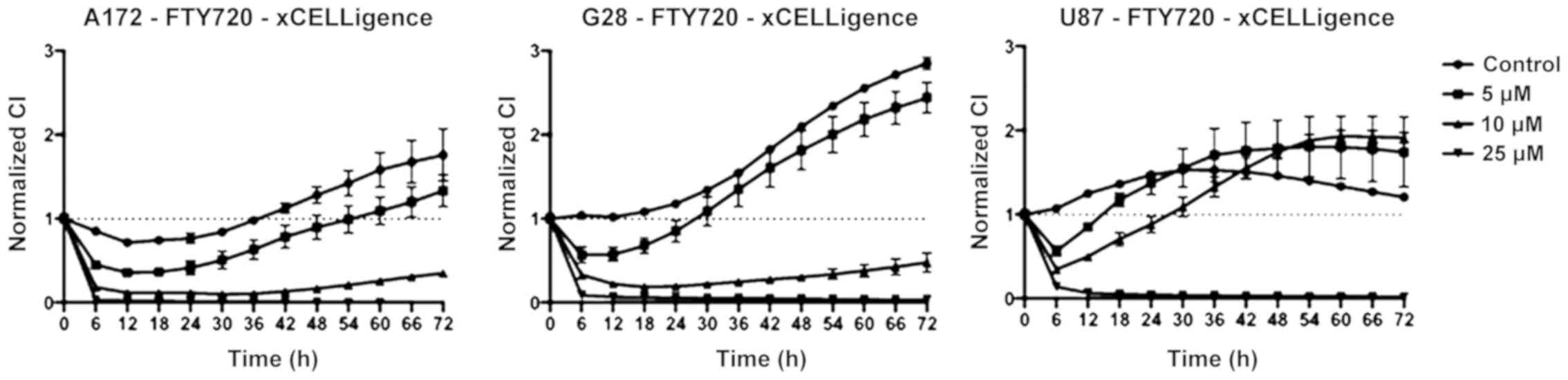
The treatment with FTY720 caused cell growth
inhibition, with an IC50 value of 4.6 µM in A172
cells, 17.3 µM in G28 cells and 25.2 µM in U87 cells
(Fig. 1).
A172 showed a decrease in cell proliferation after 6
h of incubation with 5 µM of FTY720. The cell viability
remained below 50% over 72 h of treatment with 10 µM of
FTY720. Cell growth arrest was observed after treatment with 25
µM of FTY720. A similar effect was observed in U87 cells,
where 5 µM of FTY720 produced a decrease in cell viability
as well. FTY720 at 10 µM achieved a stable reduction of
viability below 50% over 72 h. Again, 25 µM of FTY720 caused
cell growth arrest in G28 cells.
Comparison between FTY720 and
temozolomide
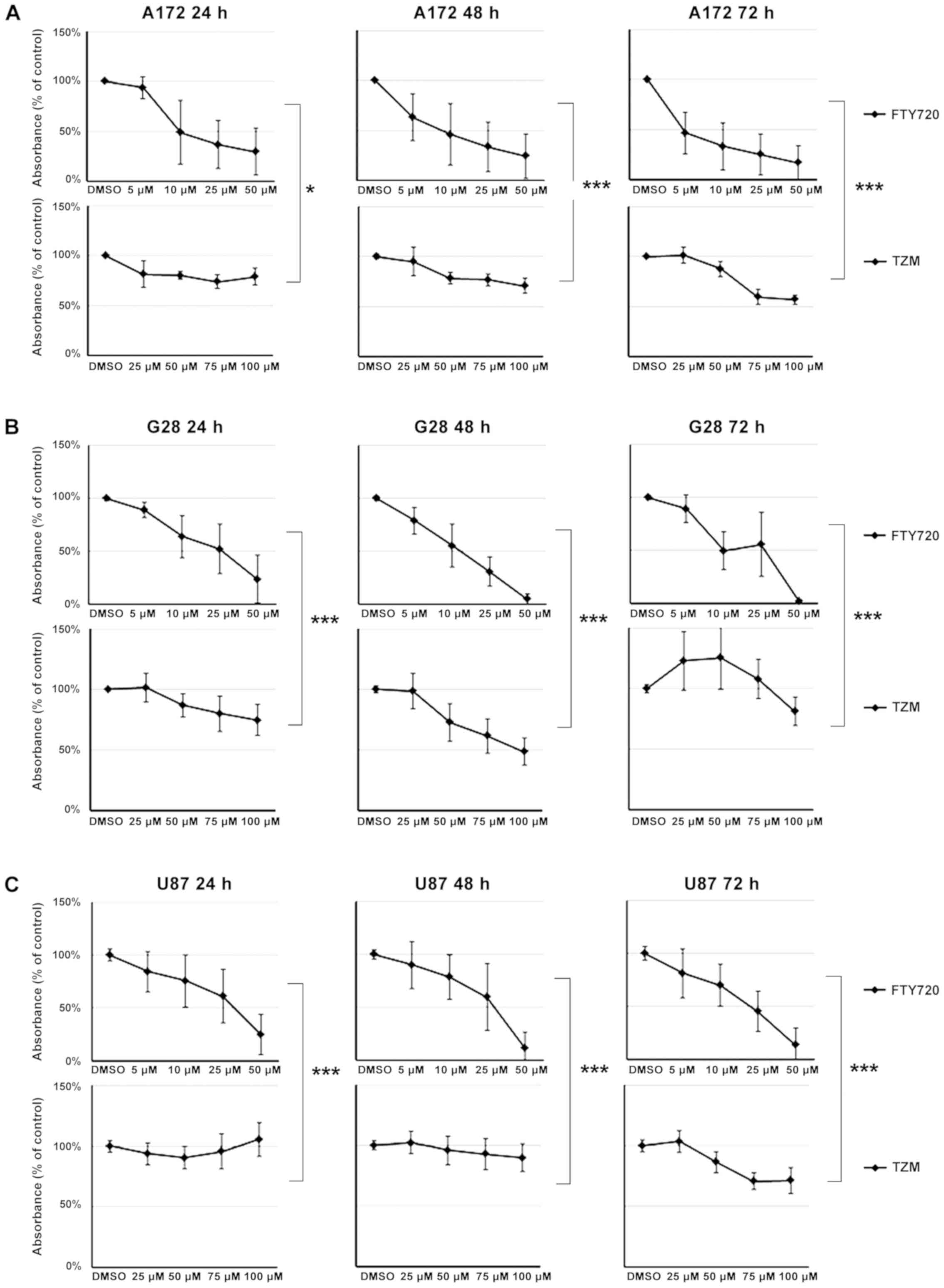
A reduction of 50% in cell viability was observed in
A172 cells after 24 h of treatment with 10 µM of FTY720. A
cell proliferation rate lower than 50% was observed after 72 h of
treatment with all concentrations of FTY720. The effect of FTY720
was stronger than the effect of TMZ after 24 h (P=0.011), 48 h
(P<0.001), and 72 h (P<0.001). While G28 cell proliferation
increased after treatment with TMZ, it was strongly suppressed by
treatment with FTY720, analogous to A172 cells. FTY720 at 25
µM caused a reduction of cell viability below 50% after 24
h, and below 5% after 48 h of treatment. Reduction of cell
viability (below 2.5%) could be induced by the administration of 50
µM of FTY720. In G28 cells, FTY720 had a significantly
greater decreasing effect than TMZ (P<0.001). FTY720 at 50
µM caused a significant reduction of cell viability (over
50%). Prolonged treatment (72 h) with 25 µM of FTY720 was
sufficient to decrease cell viability below 50%. Contrarily, TMZ
caused an increase in cell viability after 24 h and only a slight
reduction after 48 and 72 h.
Overall, A172, G28, and U87 cell lines showed a
significant dose-dependent decrease in cells after 72 h of
treatment with FTY720 compared to the TMZ-treated cells (P=0.001;
Fig. 2A-C).
Cell cycle distribution of GBM cells
treated with FTY720
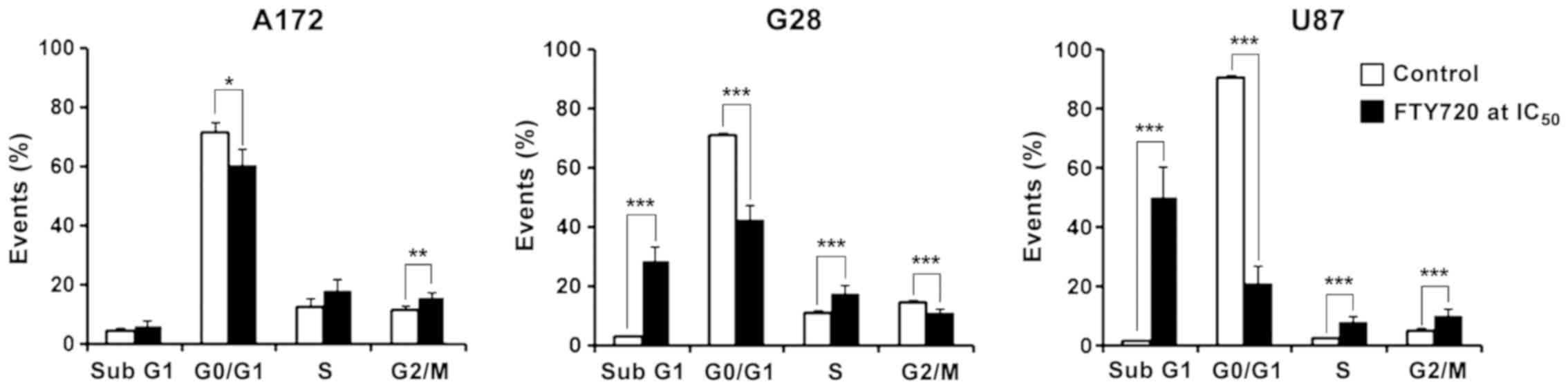
A172 cells, treated with FTY720, showed a
significant decrease in G0/G1-phase events (P=0.0015). An increase
in the percentage of events detected at the S phase (18.1%) and the
G2/M phase (15.6%) was observed. G28 cells showed significant
variation in the distribution of the events in all cell cycle
phases compared to the control cells. In the sub-G1 phase, there
was an increase of 26% (P<0.001) and in the S phase, an increase
of 6% (P<0.001). A decrease of 28% was observed in the G0/G1
phase. The strongest effect of FTY720 was detected in U87 cells.
The number of events detected in the sub-G1 phase increased up to
50% after treatment with FTY720 (P<0.001) and the G0/G1 events
simultaneously decreased to 21.3% (P<0.001; Fig. 3).
Expression of cell cycle regulator genes
after treatment with FTY720
A172 cells showed a significant increase in
AKT1 (2.1-fold, P=0.003), MAPK1 (2.6-fold,
P<0.001) and PKCE (4-fold, P<0.001) transcripts after
24 h of treatment with FTY720; RAC1 and ROCK1
transcripts were stable in comparison to the untreated cells.
Longer time exposure to FTY720 (72 h) caused a significant
downregulation of ROCK1 (0.4-fold, P=0.007) and a further
significant overexpression of PKCE (18.2-fold, P<0.001)
transcript. All other analyzed transcripts were stable (Fig. 4).
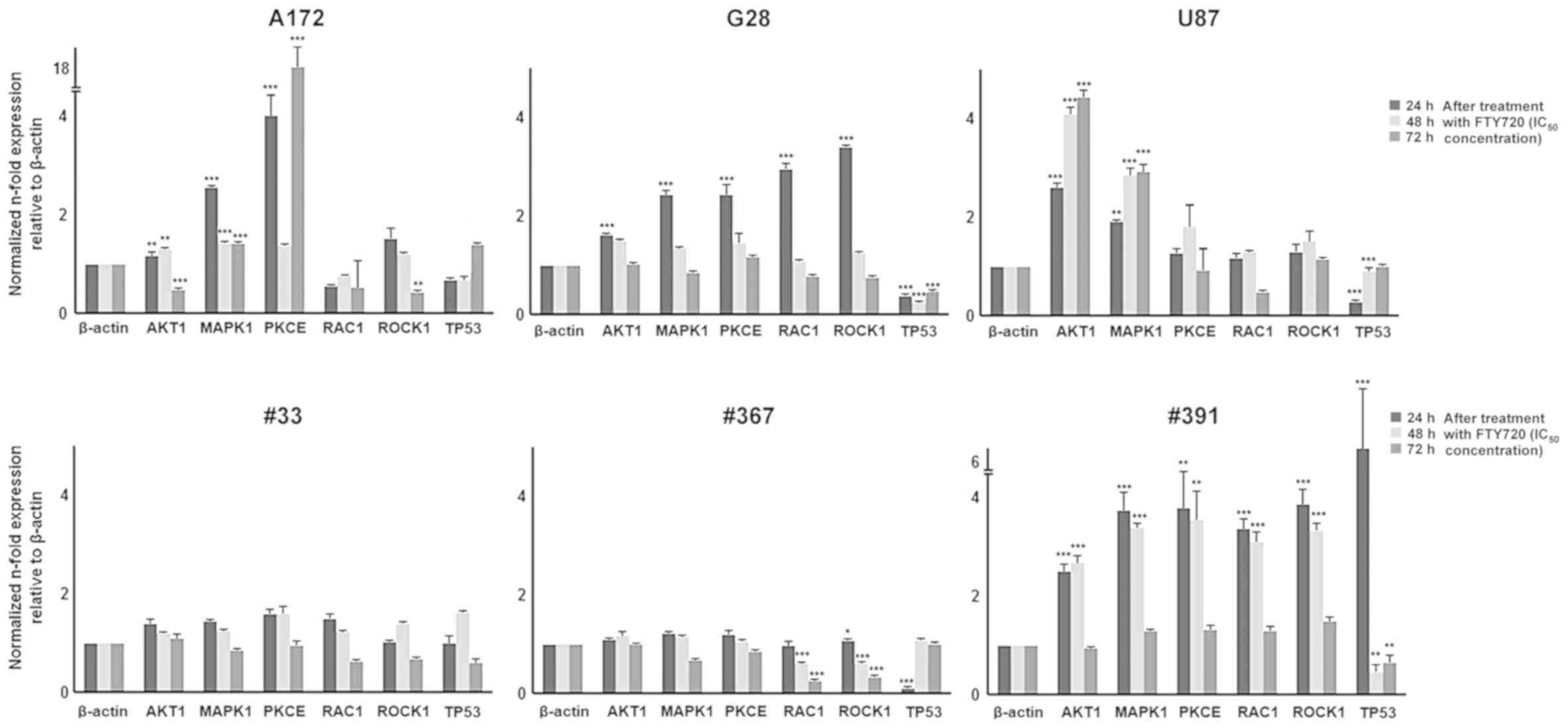 | Figure 4qPCR analysis of cell cycle
regulators AKT1, MAPK1, PKCE, RAC1, ROCK1 and TP53 in
A172, G28, U87, #33, #367 an #391 cells after 24, 48 and 72 h of
treatment with FTY720 (IC50 concentration). The values
were normalized in n-fold expression relative to β-actin. Measured
dose- and time-dependent changes were investigated in all the genes
and analyzed by ANOVA (post hoc test: Bonferroni). P-values show
the significant overexpression or downregulation after 24, 48 and
72 h of treatment with FTY720 compared to untreated cells. Shown
are means ± SEM of three independent experiments performed in
triplicates. FTY720, fingolimod; IC50, half maximal
inhibitory concentration. *P<0.05,
**P<0.01 and ***P<0.001, compared with
β-actin. |
G28 cells showed a significant overexpression of
MAPK1 (3.4-fold, P<0.001), PKCE (3-fold,
P<0.001), RAC1 (2.4-fold, P=0.003) and ROCK1
(2.4-fold, P<0.001) transcripts and a stable expression of the
AKT1 transcript after 24 h of treatment with FTY720. The
exposure to FTY720 for 48 and 72 h did not alter the expression of
the transcripts, which remained stable.
U87 cells expressed a significant upregulation of
the AKT1 transcript (P<0.001) at all time points of
exposure to FTY720 and a significant overexpression of MAPK1
after 48 and 72 h of treatment with FTY720 (P<0.001).
PKCE, RAC1 and ROCK1 were stably
expressed.
The GBM-derived cells #33 and #367 showed a stable
expression of AKT1, MAPK1 and PKCE transcripts
after treatment with FTY720. Interestingly, the transcription
levels of RAC1 and ROCK1 were significantly
downregulated (P<0.001) in the #367 cells, whereas they remained
stable in #33 cells after treatment with FTY720. In contrast, the
cells #391 showed a significant overexpression (P<0.001) of
MAPK1, PKCE, RAC1 and ROCK1 transcripts after 24 and
48 h of treatment with FTY720. Longer exposure to FTY720 (72 h)
caused no variation in the expression of these markers in
comparison to the untreated cells. The level of AKT1
transcript was stable at all treatment time points (Fig. 4).
Expression of the tumor-suppressor gene TP53
was detected by RT-qPCR in A172, G28, U87, and the three
GBM-derived cells (#33, #367 and #391) after 24, 48, and 72 h of
treatment with FTY720. The A172 cells showed no significant change
in TP53 transcript level. U87 cells showed a significant
down-regulation (P<0.001) of the TP53 transcription after
24, 48 and 72 h of treatment with FTY720. G28 cells were
characterized by a significant downregulation of TP53
(0.3-fold, P<0.001) after short time exposure (24 h) to FTY720,
whereas its transcript level remained stable after 48 and 72 h of
exposure. The #33 cells showed a stable expression of TP53
transcript after treatment with FTY720. The #367 cells were
characterized by a significant downregulation of the TP53
transcript (0.1-fold, P<0.001) after 24 h of administration of
FTY720 and a stable expression after longer time exposure (48 and
72 h). The #391 cells showed a significant overexpression of the
TP53 transcript after 24 h (6.3-fold, P<0.001), a
downregulation of the TP53 transcript after 48 h and a
stable expression after 72 h of treatment with FTY720 (Fig. 4).
Discussion
The discovery of factors that correlate with
glioblastoma (GBM) resistance to conventional chemotherapy is still
ongoing. It would be valuable to select and analyze the patients
who are potentially resistant to temozolomide (TMZ) and in need of
development of novel therapeutic strategies for the treatment of
GBM (22,26).
The S1P pathway, sphingosine kinase 1 and
S1P-receptor 1 are involved in cell survival, growth, migration,
angiogenesis and affect the survival of the patients affected by
glioblastoma (1,27).
The present study demonstrated that treatment with
fingolimod (FTY720) induced a significant reduction in cell
viability in A172, G28, and U87 cells. Treatment with 10 µM
of FTY720 reduced the percentage of viable A172 cells to less than
5%. Similar results were obtained for G28 and U87 cells, reaching a
viability percentage below 2% after 72 h of incubation with 50
µM of FTY720; thus, supporting the previous study of Zhang
and colleagues (21). Conversely,
the reduction in viable cells caused by the administration of TMZ,
even at 50 µM concentration, was not comparable to that of
FTY720; inducing only 40% of a decrease in cell viability in all
cell lines used in the study.
Synergy has already been observed between FTY720 and
other substances, including doxorubicin, etoposide, and cetuximab
for the treatment of colorectal cancer; or in combination with
cisplatin for the treatment of melanoma and hepatocellular
carcinoma (27-30). Interestingly, the combination of
FTY720 and cisplatin has been responsible for the inhibition of
autophagy, resulting in an additive cytotoxic effect (29,30).
Previous studies have shown that FTY720 can induce
apoptosis, and inhibit the migration and invasion of GBM cells by
inhibiting the activity of matrix metalloproteases (21,31).
Rac1, RhoA, Ras, and Cdc42 are members of the Rho family of GTPases
involved in cell migration. Rac1 and RhoA are both sufficient for
the generation of cell polarity. Inhibition of ROCK restored the
migration of GBM cells, but S1P2-dependent inhibition of Rac1
activity was not involved in the S1P-mediated inhibition of
migration in human GBM (32). In
the present study, we confirmed that RAC1, ROCK1, and
MAPK1 transcripts were modulated by treatment with
FTY720.
A decreased level of the RAC1 transcript was
observed in #367 cells after treatment with FTY720. Therefore, its
inhibition could further impede the epithelial-mesenchymal
transition of GBM cells; thus, blocking tumor growth and metastasis
(33,34).
Because of the interaction between Ras and the
abundant S1P, an oncogenic process in cancer cells could be
inhibited by the downregulation of S1P (13). However, FTY720 induced, in the
majority of the cells included in this study, a stable or
overexpressed level of the transcripts of the genes implicated in
cell cycle progression which include AKT1, MAPK1, PKCE, RAC1
and ROCK1.
A172 cells bear wild-type TP53, while mutated
TP53 is present in the G28, #33, and #391 cells (35). Interestingly, TP53
expression, after incubation with FTY720, was stable or
significantly downregulated in all cell lines involved in the
study. Only the #391 cells showed a significant overexpression of
the TP53 transcript after short time exposure to FTY720;
thereby showing that TP53, independently of its status,
could not be triggered by treatment with FTY720, with the only
exception represented by the #391 cells. Future investigations at
the protein level are needed to identify the status of p53 and its
cellular localization after treatment with FTY720.
Drug resistance to chemotherapy is still an unsolved
issue for the treatment of GBM. Many patients relapse and develop
resistance to TMZ-based chemotherapy (36). Consequently, a combination of TMZ
with other anticancer mediators, such as FTY720, which can overcome
drug resistance, could be one of the main approaches for achieving
a better outcome for GBM patients.
Estrada-Bernal et al showed that FTY720, in
combination with TMZ, appeared to decrease the invasiveness of
brain tumor stem cells in xenograft models (3). Furthermore, mice (with brain tumor
stem cells derived from human glioblastoma tissue intracranial)
treated with both FTY720 and TMZ showed increased survival compared
to those treated with a single compound (3).
FTY270 has also shown immunomodulatory effects and
could lead, at a high dosage, to the immunosuppression of patients
affected by GBM (37). FTY720 was
found to prevent peripheral lymphocytes from migrating into the
central nervous system (CNS) and directly inhibited the activation
of microglia and astrocytes in the brain (38-40).
Inhibition of innate and adaptive immune/inflammation responses is
probably triggered by the treatment with FTY720.
In conclusion, FTY720 exerted a cytotoxic effect in
GBM cells that significantly surpasses that of TMZ. It was
responsible for the reduction in cell viability, cell cycle arrest
and cell death of GBM cells, even after the overexpression of the
genes indicated in the regulation of cell cycle process. The lack
of the protein level of the factors involved in the mechanisms
modulated by FTY720 was a limitation of the study that needs to be
further expanded in order to better clarify the effect of FTY720 on
the molecular pathways responsible for proliferation and survival
of glioblastoma cells. In addition, it is essential to consider
phosphorylated AKT, which was not investigated in our study. These
findings suggest that FTY720 could be included in further
investigations, either in combination with TMZ or alone, for
patients with TMZ drug resistance.
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FTY720
|
fingolimod
|
|
GBM
|
glioblastoma
|
|
MTT
|
3-(4,5-diemethytiazol-2-yl)-2,5diphenyl tetrazolium bromide
|
|
TMZ
|
temozolomide
|
|
qPCR
|
real-time quantitative PCR
|
|
PCR
|
polymerase chain reaction
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
ROCK1
|
Rho-associated kinase-1
|
|
S1P
|
sphingosine-1-phosphate
|
|
SphK
|
sphingosine kinase
|
|
µM
|
micromolar
|
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The data and materials that support the findings are
available from the corresponding author upon reasonable
request.
Authors' contributions
MAK was responsible for the study and manuscript
design, analysis and review. BAB carried out the data collection
and analysis. JN carried out the statistical analysis. MAW and EU
reviewed the manuscript. FU carried out data analysis and review.
PDF carried out data analysis, review and statistical analysis. FPS
and MS carried out the study design and review. All authors read
and approved the manuscript and agree to be accountable for all
aspects of the research in ensuring that the accuracy or integrity
of any part of the work are appropriately investigated and
resolved.
Ethics approval and consent to
participate
The present study was approved by the local ethics
committee (Ethics Committee of Medicine Faculty, Justus Liebig
University Giessen), (application no. AZ 07/09) and the performed
research followed international and national regulations in
accordance with the Declaration of Helsinki. Patients consent was
obtained before treatment and is available from the corresponding
author, MAK, upon reasonable request.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
References
|
1
|
Quint K, Stiel N, Neureiter D, Schlicker
HU, Nimsky C, Ocker M, Strik H and Kolodziej MA: The role of
sphingosine kinase isoforms and receptors S1P1, S1P2, S1P3, and
S1P5 in primary, secondary, and recurrent glioblastomas. Tumour
Biol. 35:8979–8989. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Strub GM, Maceyka M, Hait NC, Milstien S
and Spiegel S: Extracellular and intracellular actions of
sphingosine-1-phosphate. Adv Exp Med Biol. 688:141–155. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Estrada-Bernal A, Palanichamy K, Ray
Chaudhury A and Van Brocklyn JR: Induction of brain tumor stem cell
apoptosis by FTY720: A potential therapeutic agent for
glioblastoma. Neuro Oncol. 14:405–415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ocker M, Tackenberg B and Strik H:
Fingolimod. Nervenheilkunde. 30:345–349. 2011. View Article : Google Scholar
|
|
5
|
Ponnusamy S, Meyers-Needham M, Senkal CE,
Saddoughi SA, Sentelle D, Selvam SP, Salas A and Ogretmen B:
Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in
the regulation of cell death and drug resistance. Future Oncol.
6:1603–1624. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bassi R, Anelli V, Giussani P, Tettamanti
G, Viani P and Riboni L: Sphingosine-1-phosphate is released by
cerebellar astrocytes in response to bFGF and induces astrocyte
proliferation through Gi-protein-coupled receptors. Glia.
53:621–630. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Spiegel S and Milstien S:
Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat Rev Mol
Cell Biol. 4:397–407. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Obinata H and Hla T: Sphingosine
1-phosphate in coagulation and inflammation. Semin Immunopathol.
34:73–91. 2012. View Article : Google Scholar
|
|
9
|
Mendelson K, Evans T and Hla T:
Sphingosine 1-phosphate signalling. Development. 141:5–9. 2014.
View Article : Google Scholar
|
|
10
|
Mora R, Dokic I, Kees T, Hüber CM, Keitel
D, Geibig R, Brügge B, Zentgraf H, Brady NR and Régnier-Vigouroux
A: Sphingolipid rheostat alterations related to transformation can
be exploited for specific induction of lysosomal cell death in
murine and human glioma. Glia. 58:1364–1383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anelli V, Gault CR, Snider AJ and Obeid
LM: Role of sphin-gosine kinase-1 in paracrine/transcellular
angiogenesis and lymphangiogenesis in vitro. FASEB J. 24:2727–2738.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riccitelli E, Giussani P, Di Vito C,
Condomitti G, Tringali C, Caroli M, Galli R, Viani P and Riboni L:
Extracellular sphingo-sine-1-phosphate: A novel actor in human
glioblastoma stem cell survival. PLoS One. 8:e682292013. View Article : Google Scholar
|
|
13
|
Mahajan-Thakur S, Bien-Möller S, Marx S,
Schroeder H and Rauch BH: Sphingosine 1-phosphate (S1P) signaling
in glioblastoma multiforme-A systematic review. Int J Mol Sci.
18:24482017. View Article : Google Scholar
|
|
14
|
Rosen H and Goetzl EJ: Sphingosine
1-phosphate and its receptors: An autocrine and paracrine network.
Nat Rev Immunol. 5:560–570. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Inda MM, Bonavia R and Seoane J:
Glioblastoma multiforme: A look inside its heterogeneous nature.
Cancers (Basel). 6:226–239. 2014. View Article : Google Scholar
|
|
16
|
Bektas M, Johnson SP, Poe WE, Bigner DD
and Friedman HS: A sphingosine kinase inhibitor induces cell death
in temozolomide resistant glioblastoma cells. Cancer Chemother
Pharmacol. 64:1053–1058. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia X, Li X, Li F, Wu X, Zhang M, Zhou H,
Huang N, Yang X, Xiao F, Liu D, et al: A novel tumor suppressor
protein encoded by circular AKT3 RNA inhibits glioblastoma
tumorigenicity by competing with active phosphoinositide-dependent
Kinase-1. Mol Cancer. 18:1312019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chin YR, Yuan X, Balk SP and Toker A:
PTEN-deficient tumors depend on AKT2 for maintenance and survival.
Cancer Discov. 4:942–955. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen WS: Growth retardation and increased
apoptosis in mice with homozygous disruption of the akt1 gene.
Genes Dev. 15:2203–2208. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Degtyarev M, De Mazière A, Orr C, Lin J,
Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al: Akt
inhibition promotes autophagy and sensitizes PTEN-null tumors to
lysosomotropic agents. J Cell Biol. 183:101–116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Wang H, Ding K and Xu J: FTY720
induces autophagy-related apoptosis and necroptosis in human
glioblastoma cells. Toxicol Lett. 236:43–59. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Brinkmann V: Sphingosine 1-phosphate
receptors in health and disease: Mechanistic insights from gene
deletion studies and reverse pharmacology. Pharmacol Ther.
115:84–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vessey DA, Kelley M, Zhang J, Li L, Tao R
and Karliner JS: Dimethylsphingosine and FTY720 inhibit the SK1
form but activate the SK2 form of sphingosine kinase from rat
heart. J Biochem Mol Toxicol. 21:273–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Patil SM, Beck PP, Arora N, Acevedo BA and
Dandachi D: Primary cutaneous cryptococcal infection due to
fingolimod- Induced lymphopenia with literature review. IDCases.
21:e008102020. View Article : Google Scholar
|
|
25
|
Mullins CS, Schneider B, Stockhammer F,
Krohn M, Classen CF and Linnebacher M: Establishment and
characterization of primary glioblastoma cell lines from fresh and
frozen material: A detailed comparison. PLoS One. 8:2013.
View Article : Google Scholar
|
|
26
|
Van Brocklyn JR, Jackson CA, Pearl DK,
Kotur MS, Snyder PJ and Prior TW: Sphingosine kinase-1 expression
correlates with poor survival of patients with glioblastoma
multiforme: Roles of sphingosine kinase isoforms in growth of
glioblastoma cell lines. J Neuropathol Exp Neurol. 64:695–705.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosa R, Marciano R, Malapelle U, Formisano
L, Nappi L, D'Amato C, D'Amato V, Damiano V, Marfè G, Del Vecchio
S, et al: Sphingosine kinase 1 overexpression contributes to
cetuximab resistance in human colorectal cancer models. Clin Cancer
Res. 19:138–147. 2013. View Article : Google Scholar
|
|
28
|
Chiba K, Matsuyuki H, Maeda Y and Sugahara
K: Role of sphingosine 1-phosphate receptor type 1 in lymphocyte
egress from secondary lymphoid tissues and thymus. Cell Mol
Immunol. 3:11–19. 2006.PubMed/NCBI
|
|
29
|
Ishitsuka A, Fujine E, Mizutani Y, Tawada
C, Kanoh H, Banno Y and Seishima M: FTY720 and cisplatin
synergistically induce the death of cisplatin-resistant melanoma
cells through the downregulation of the PI3K pathway and the
decrease in epidermal growth factor receptor expression. Int J Mol
Med. 34:1169–1174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stafman LL, Williams AP, Marayati R, Aye
JM, Stewart JE, Mroczek-Musulman E and Beierle EA: PP2A activation
alone and in combination with cisplatin decreases cell growth and
tumor formation in human HuH6 hepatoblastoma cells. PLoS One.
14:e02144692019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sonoda Y, Yamamoto D, Sakurai S, Hasegawa
M, Aizu-Yokota E, Momoi T and Kasahara T: FTY720, a novel
immunosuppressive agent, induces apoptosis in human glioma cells.
Biochem Biophys Res Commun. 281:282–288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lepley D, Paik JH, Hla T and Ferrer F: The
G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to
inhibit tumor cell migration. Cancer Res. 65:3788–3795. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stallings-Mann ML, Waldmann J, Zhang Y,
Miller E, Gauthier ML, Visscher DW, Downey GP, Radisky ES, Fields
AP and Radisky DC: Matrix metalloproteinase induction of Rac1b, a
key effector of lung cancer progression. Sci Transl Med.
4:142ra952012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang WH, Lan HY, Huang CH, Tai SK, Tzeng
CH, Kao SY, Wu KJ, Hung MC and Yang MH: RAC1 activation mediates
Twist1-induced cancer cell migration. Nat Cell Biol. 14:366–374.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sato Y, Kurose A, Ogawa A, Ogasawara K,
Traganos F, Darzynkiewicz Z and Sawai T: Diversity of DNA damage
response of astrocytes and glioblastoma cell lines with various p53
status to treatment with etoposide and temozolomide. Cancer Biol
Ther. 8:452–457. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Davis ME: Glioblastoma: Overview of
disease and treatment. Clin J Oncol Nurs. 20(Suppl 5): S2–S8. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sharim J, Tashjian R, Golzy N and
Pouratian N: Glioblastoma following treatment with fingolimod for
relapsing-remitting multiple sclerosis. J Clin Neurosci.
30:166–168. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hyland MH and Cohen JA: Fingolimod. Neurol
Clin Pract. 1:61–65. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rothhammer V, Kenison JE, Tjon E, Takenaka
MC, de Lima KA, Borucki DM, Chao CC, Wilz A, Blain M, Healy L, et
al: Sphingosine 1-phosphate receptor modulation suppresses
pathogenic astrocyte activation and chronic progressive CNS
inflammation. Proc Natl Acad Sci USA. 114:2012–2017. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kappos L, Radue EW, O'Connor P, Polman C,
Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M,
Zhang-Auberson L, et al: A Placebo-controlled trial of oral
fingolimod in relapsing multiple sclerosis. N Engl J Med.
362:387–401. 2010. View Article : Google Scholar : PubMed/NCBI
|