Introduction
Gastric cancer (GC) and colon cancer (CC) are the
most common gastrointestinal malignancies worldwide (1), accounting for 10 and 6% of all cancer
diagnoses, and also being the second and third leading cause of
cancer mortality, respectively (2,3). The
prevalence and poor prognosis of GC and CC as well as limited
treatment options necessitate the search for novel treatments.
Eph receptors belong to a large family of receptor
tyro-sine kinases (RTKs), and are key regulators of both normal
development and disease (4).
Perturbation of the Eph receptor and Ephrin ligand system has been
observed in various human cancers. Particularly, EphA2 (EPH
receptor A2) (erythropoietin-producing hepatocellular receptor
tyrosine kinase subtype A2) is the most frequently affected Eph
receptor in human cancers (5).
EphA2 is overexpressed in various types of cancers, and promotes
tumor growth, metastasis, and cancer stem properties through a
ligand-independent mechanism, and high EphA2 expression is also
associated with an aggressive phenotype and poor patient prognosis
(6-11). Thus, overexpression of EphA2 has
been considered as a promising target for the treatment of cancers.
Various approaches for downregulating EphA2, such as EphA2
antibody, Ephrin-A1 ligand, Ephrin-A1 mimic peptides and RNA
interference, have attracted considerable interest as anticancer
strategies (12,13).
We recently used immunoprecipitation and mass
spectrometry analysis (IP-MS) to search for proteins that interact
with EphA2 in nasopharyngeal carcinoma cells, and found that
Annexin A1 (ANXA1) is one of the proteins that interact with EphA2,
proteomic data of which are available via ProteomeXchange with
identifier PXD015242 (https://www.ebi.ac.uk/pride/archive/projects/PXD015242/).
ANXA1 is the first identified member of the annexin family of
Ca2+ and phospholipid-binding proteins (14). It plays a role in the inflammatory
and immune response, cell proliferation, apoptosis and
differentiation (15,16). ANXA1 expression is deregulated in
cancers, and has been linked to tumor development and metastasis
(17). Accumulated studies have
found that both ANXA1 and EphA2 are overexpressed, and promote
tumor growth and progression in GC(18-24)
and CC(25-29). However, the physiological and
pathological significances of ANXA1 and EphA2 interaction in GC and
CC are completely unclear.
Protein-protein interaction (PPI) controls various
cellular functions by modulating protein stability,
post-translational modification, and subcellular location. Previous
studies indicate that interaction of ANXA1 N-terminal with the
epidermal growth factor (EGF) receptor (EGFR) regulates the
abundance of the EGFR (30,31),
and promotes the oncogenicity of the EGFR (32). Interestingly, our present study
found that interaction of ANXA1 and EphA2 increased EphA2 stability
and tumor growth in GC and CC cells. Accumulative studies indicate
that abnormal PPI is associated with cancers, representing a
pivotal target for chemicobiological interventions (33-35).
Numerous studies have indicated that inhibition of PPI by peptides
is an efficient anticancer approach (36-38).
Moreover, peptides possess various advantages with respect to
chemotherapeutic drugs such as low toxicity, ease of synthesis,
high target specificity, feasibility of chemical modification, and
biocompatibility, making them suitable drug candidates (39,40).
In the present study, based on the fact that the
interaction of ANXA1 and EphA2 increase EphA2 stability, we
investigated the anti-GC and anti-CC effects of ANXA1-derived
peptides, and found that ANXA1 N-terminal-derived 3 and 11 amino
acid-long peptides disturbed EphA2-ANXA1 interaction, reduced EphA2
protein stability, and suppressed GC and CC cell growth in
vitro and in vivo. Our findings provide an important
basis to use the two ANXA1-derived peptides for the treatment of GC
and CC.
Materials and methods
Clinical specimens
Formalin-fixed and paraffin-embedded archival tissue
specimens from 30 GC, 30 CC, and 30 paired paracancerous tissues
were obtained from Xiangya Hospital, Central South University
between January 2019 and June 2019. All specimens were subjected to
hematoxylin and eosin (H&E) staining, and the diagnosis was
confirmed by two pathologists. The clinicopathological data of the
patients are presented in Tables SI
and SII.
Cell lines and culture
Human GC cell line AGS and human colon cancer cell
lines HCT116 and SW620, and 293 cells were purchased from the
American Type Culture Collection (ATCC). All cell lines were grown
in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS;
Thermo Fisher Scientific, Inc.) at 37°C in 5% CO2. The
presence of mycoplasma was detected by staining with
4,6-dimethylin-dole-2-phenylindole in the cell lines, and no
mycoplasma was detected.
Antibodies and reagents
The following antibodies were used in the present
study: EphA2 (sc-398832; Santa Cruz Biotechnology, Inc.), ANXA1
(ab137745; Abcam), Flag-tag (F1804; Sigma-Aldrich; Merck KGaA),
tubulin (E-AB-20036; Elabscience), goat anti-rabbit IgG-HRP
(ab6721; Abcam), and goat anti-mouse IgG-HRP (ab6789; Abcam).
Protein G/A-Sepharose™ 4B (82085), streptavidin agarose (20357) and
Lipofectamine 2000 (11668019) were purchased from Thermo Fischer
Scientific, Inc. pBabepuro-EphA2 expression plasmid, and lentiviral
vector GV101 expressing EphA2 shRNA have been previously described
by us (41). Lentiviral vector
GV112 expressing ANXA1 shRNA was constructed, and its target
sequence located in the 3′UTR of ANAX1 mRNA is 5′-AAC CCT ATA CAA
GTT GTT CTA-3′. The full-length and N-terminal deletion mutant
ANXA1 with Flag tag were constructed, the vector of which was
pcDNA3.1. All constructs were established by Genechem (Shanghai,
China), and were verified by DNA sequencing.
Peptides
Cell-penetrating peptide (YGRKKRRQRRR) (CPP),
CPP-ANXA1-derived 3-mer peptide (28-30aa) (SKG), CPP-ANXA1-derived
11-mer peptide (20-30aa) (EYVQTVKSSKG), FITC-labeled CPP,
FITC-labeled CPP-ANXA1-derived 3-mer peptide, FITC-labeled
CPP-ANXA1-derived 11-mer peptide, biotin-labeled ANXA1-derived
3-mer peptide, and biotin-labeled ANXA1-derived 11-mer peptide were
synthesized by ChinaPeptides (Suzhou, China).
Immunoblotting
Immunoblotting was performed to detect the
expression of proteins in the indicated cells as described
previously by us (41,42). Briefly, proteins were exacted from
cells using RIPA lysis buffer. An equal amount of protein in each
sample was subjected to SDS-PAGE separation, followed by blotting
onto a PVDF membrane. After blocking, blots were incubated with
primary antibodies overnight at 4°C, followed by incubation with
HRP-conjugated secondary antibody for 2 h at room temperature. The
signal was visualized with an enhanced chemiluminescence detection
reagent (Roche).
Immunoprecipitation and immunoblotting
(co-IP)
Co-IP was performed to detect protein and protein
interaction. In brief, whole cell lysates were incubated with
indicated antibodies and Protein G/A-Sepharose 4B overnight at 4°C.
After 5 times wash with RIPA buffer, beads were boiled in 2X
SDS-PAGE loading buffer for 5 min to elute protein complexes,
followed by SDS-PAGE separation and immunoblotting with specific
antibodies.
Biotin pull-down assay
Biotin pull-down assay was performed to detect the
interaction of ANXA1-derived 3-mer or 11-mer peptide and EphA2 as
previously described (43). In
brief, 1 mg of whole cell lysates was incubated with 30 nM peptide
over-night at 4°C, and then incubated with 30 µl
streptavidin agarose beads for 4 h at 4°C. After 5 times wash with
PBS buffer, the beads were boiled in 2X SDS-PAGE loading buffer for
6 min, followed by SDS-PAGE separation and immunoblotting with the
EphA2 antibody.
Immunohistochemistry
Immunohistochemical staining of ANXA1 and EphA2 was
performed on the formalin-fixed and paraffin-embedded tissue
sections as described previously by us (44). Briefly, tissue sections were
incubated with ANXA1 antibody (1:1,000 dilution) or EphA2 antibody
(1:100 dilution) overnight at 4°C, and then incubated with a
biotinylated secondary antibody at room temperature for 15 min, and
stained with DAB (3,3-diaminobenzidine). Finally, tissue sections
were counterstained with hematoxylin. In negative controls, primary
antibodies were replaced with a mouse or rabbit IgG.
Immunohistochemical staining was assessed and scored
by two independent pathologists who were blinded to the
clinicopathological data; discrepancies were resolved by consensus.
Positive reactions were defined as brown signals in the cytoplasm
and/or cell membrane. Staining intensity was categorized: Absent
staining as 0, weak as 1, moderate as 2, and strong as 3. The
percentage of stained cells (examined in at least 500 cells) was
categorized as no staining=0, <30% of stained cells=1, 30~60%=2,
and >60%=3. The staining score (ranging from 0-6) for each
tissue was calculated by adding the area score and the intensity
score. A combined staining score of ≤3 was considered to be low
expression, and >3 was considered to be high expression.
Quantitative PCR
qPCR was performed to detect the expression of ANXA1
and EphA2 in the indicated cells as described previously by us
(41,42). The primers are presented in the
Table SIII.
Molecular docking
The human ANXA1 structure was modelled on the
available porcine full-length ANXA1 structure (PDB code: 1hm6)
using the SWISS-MODEL server (45). The modelled ANXA1 structure was
then docked to the EphA2 structure (PDB code: 5ia2) using ClusPro
(46). The best docking result was
selected based on the related experimental results, which show that
the three amino acids (S28, K29 and G30) of ANXA1 are critical to
the binding. To further refine the binding model, molecular
dynamics simulations were conducted on the ANXA1 and EphA2 complex.
The modelled structure was firstly prepared with the Protein
Preparation Wizard implemented in the Schrödinger suite 2015
(47). This procedure added
hydrogen atoms of residues and optimized the orientation of polar
hydrogens and the protonated states of the proteins. The simulation
system was then built with AmberTools14. The prepared simulation
box contains 3,340 TIP3P water molecules and 8 chloride ions,
resulting in a total of 80,119 atoms. The proteins, water
molecules, and ions were modelled using the ff14SB force field
(48). The system was minimized
and equilibrated using Amber 14 in an NPT ensemble at 300 K and 1
bar. The production run lasted for 10 nsec.
MTT assay
Cells were treated with the treatment peptides or
control CPP for 24 h, and cell viability was tested by MTT assay as
previously described by us (49).
The cytotoxicity of peptides was calculated using the formula: %
viability=(A570-A630) treated/(A570-A630) control ×100%. The assay
was performed three times in triplicate.
Cell Counting Kit-8 (CCK-8) assay
Cells were treated with the treatment peptides or
control CPP, and the peptides were replenished every day. Cell
proliferation was measured using a CCK-8 kit as previously
described by us (50). The assay
was performed three times in triplicate.
Plate colony formation assay
Cells were treated with the treatment peptides or
control CPP, and the peptides were replenished every day. Cell
proliferation was measured by plate colony formation assay as
previously described by us (50).
The assay was performed three times in triplicate.
Soft agar colony formation assay
Cells were treated with the treatment peptides or
control CPP, and the peptides were replenished every day. Soft agar
colony formation assay was performed to detect cell anchorage
independent growth as previously described by us (50). Cells were allowed to grow in the
soft agar cultures for 12 days and colonies consisting of >50
cells were counted under a microscope (LEICA, ×50 magnification).
The assay was performed three times in triplicate.
Animal experiment
Thirty-six nude male mice (BALB/c nu/nu) (initial
weight 16~18 g; 4 weeks old) were obtained from the Laboratory
Animal Center of Central South University and maintained in
pathogen-free conditions. Mice were randomly divided into three
groups before inoculation, cancer cells (2×106) were
inoculated into the flank of mice by subcutaneous injection. After
xenografts grew for 6 days, CPP-ANXA1-derived 3-mer peptide,
CPP-ANXA1-derived 11-mer peptide or control CPP was
intraperitoneally injected into the mice at a dose of 10 mg/kg once
daily, tumors were measured using an electronic caliper daily, and
tumor volume was calculated using the formula (length ×
width2/2). The mice were sacrificed by cervical
dislocation at a time-defined endpoint, and their tumor and organs
(heart, liver, spleen, lung and kidney) were removed and measured
using double-blinded evaluation. The tissues were fixed with 4%
paraformaldehyde and embedding in paraffin, and subjected to
H&E and/or immunohistochemistry.
Statistical analysis
Statistical analysis was performed using IBM SPSS
statistical software package 22 (IBM Corp.). Data are presented as
means ± SD. For comparisons between two groups, a Student's t test
was used, and for analysis with multiple comparisons, one-way ANOVA
test followed by Turkey's post-hoc analysis was used.
Classification variables were compared by the Chi-square test.
Pearson test was used for correlation analysis. P-values <0.05
were considered statistically significant.
Results
ANXA1 is a protein that interacts with
EphA2, and expression of both proteins is positively correlated in
GC and CC
We recently used immunoprecipitation and mass
spectrometry analysis (IP-MS) to search for proteins that interact
with EphA2 in nasopharyngeal carcinoma cells, and found that ANXA1
is one of the proteins that interacts with EphA2, proteomic data of
which are available via ProteomeXchange with identifier PXD015242.
As both ANXA1 and EphA2 promote tumor growth and progression in GC
(18-24) and CC (25-29),
we aimed to ascertain whether ANXA1 interacts with EphA2 in GC and
CC cells. Co-IP showed that ANXA1 interacted with EphA2 in the GC
(AGS) and CC (HCT116 and SW620) cell lines (Fig. 1A). Next, we detected the expression
levels of ANXA1 and EphA2 in the 30 GC, 30 CC, and 30 paired
paracancerous tissues by immunohistochemistry, and observed that
the expression levels of both ANXA1 and EphA2 were significantly
higher in the GC and CC tissues than those in the paracancerous
tissues (Fig. 1B), and were
positively correlated in the GC and CC tissues (Fig. 1C). The interaction of ANXA and
EphA2, and positive correlation of their expression levels,
prompted us to investigate the function and significance of the
ANXA-EphA2 interaction in GC and CC.
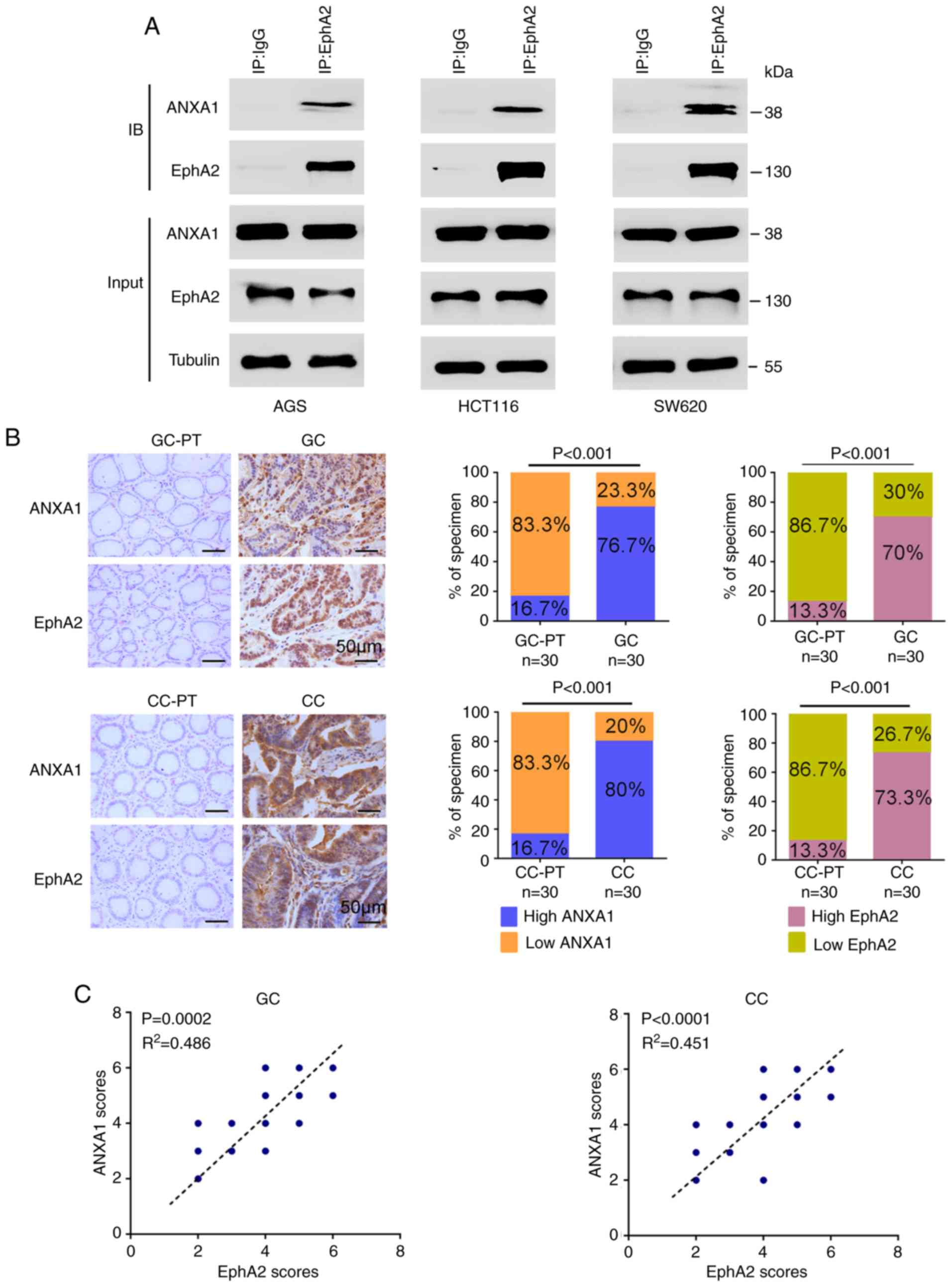 | Figure 1The interaction and expression
correlation of ANXA1 and EphA2 in GC and CC. (A) Co-IP showing the
interaction of endogenous ANXA1 and EphA2 in the GC (AGS) and CC
(HCT116 and SW620) cell lines. Total proteins from the cells were
prepared, and subjected to immunoprecipitation (IP) with anti-EphA2
antibody or control IgG followed by immunoblotting (IB) with
antibodies against ANXA1 or EphA2. (B) Immunohistochemistry (IHC)
showing the expression levels of ANXA1 and EphA2 in the 30 GC, 30
CC, and their paracancerous tissues (PT). Representative IHC images
are shown on the left, and quantitative data are presented on the
right. P<0.001, Chi-squared test. Scale bars, 50 µm. (C)
Positive correlation between ANXA1 and EphA2 expression in the 30
GC and 30 CC tissues. P<0.001, Pearson's correlation test. GC,
gastric cancer; CC, colon cancer; ANXA1, Annexin 1. |
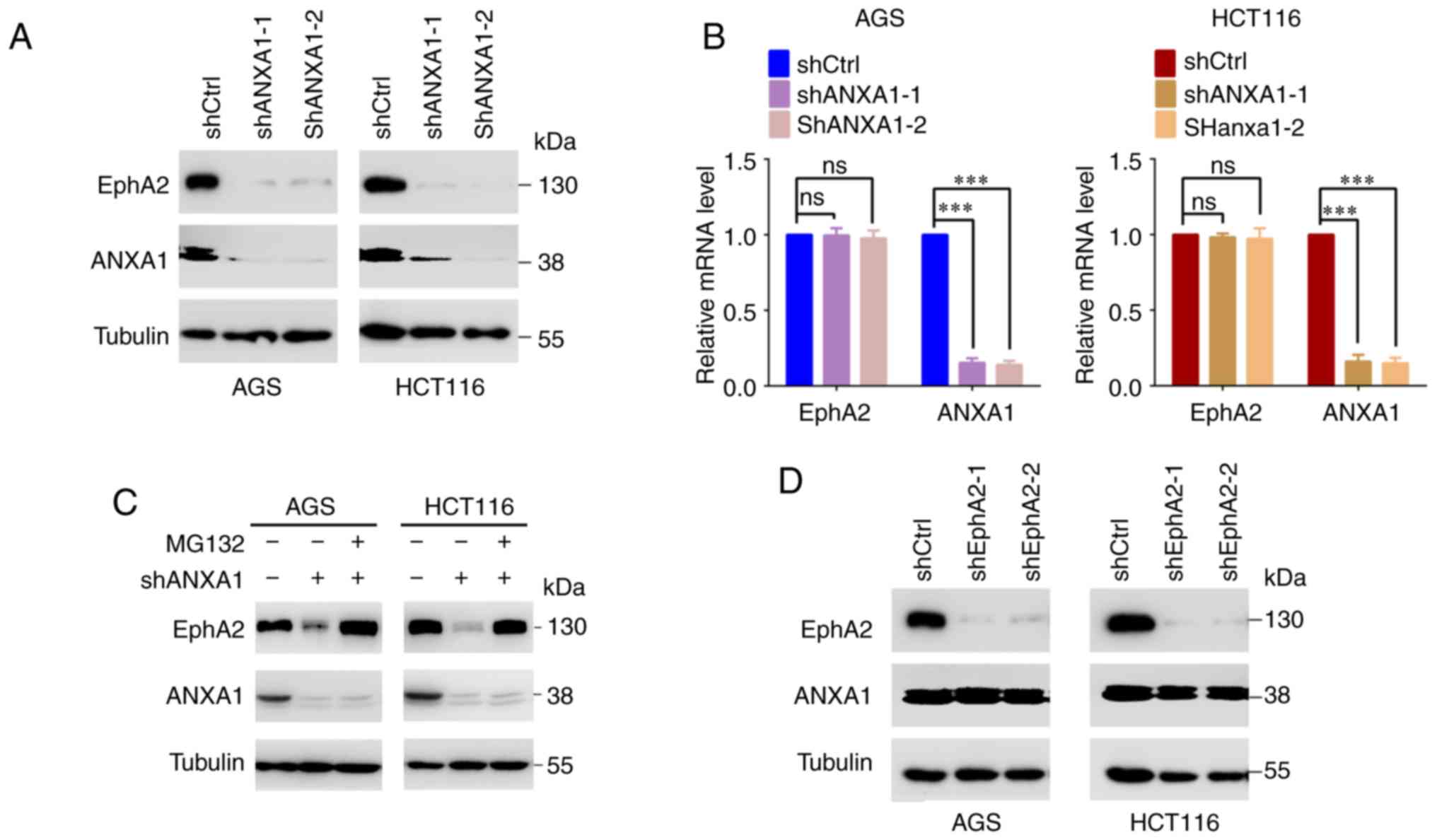
ANXA1 increases EphA2 stability in the GC
and CC cells
It has been reported that ANXA1 regulates the
stability of the EGFR (30,31).
Therefore, we analyzed the effect of ANXA1 on EphA2 protein
stability after blocking protein synthesis with cycloheximide
(CHX). The result showed that knockdown of ANXA1 by shRNA
dramatically decreased EphA2 levels in the AGS and HCT116 cells
(Fig. 2A), but had no effect on
its mRNA levels (Fig. 2B),
indicating that ANXA1 increased EphA2 protein stability. We also
observed that the decrease in EphA2 protein in the ANXA1-knockdown
AGS and HCT116 cells was reversed by treatment with the proteasome
inhibitor MG132 (Fig. 2C),
indicating that ANXA1 increases EphA2 stability by a
proteasome-dependent mechanism. However, knockdown of EphA2 had no
impact on ANXA1 protein stability in the AGS and HCT116 cells
(Fig. 2D).
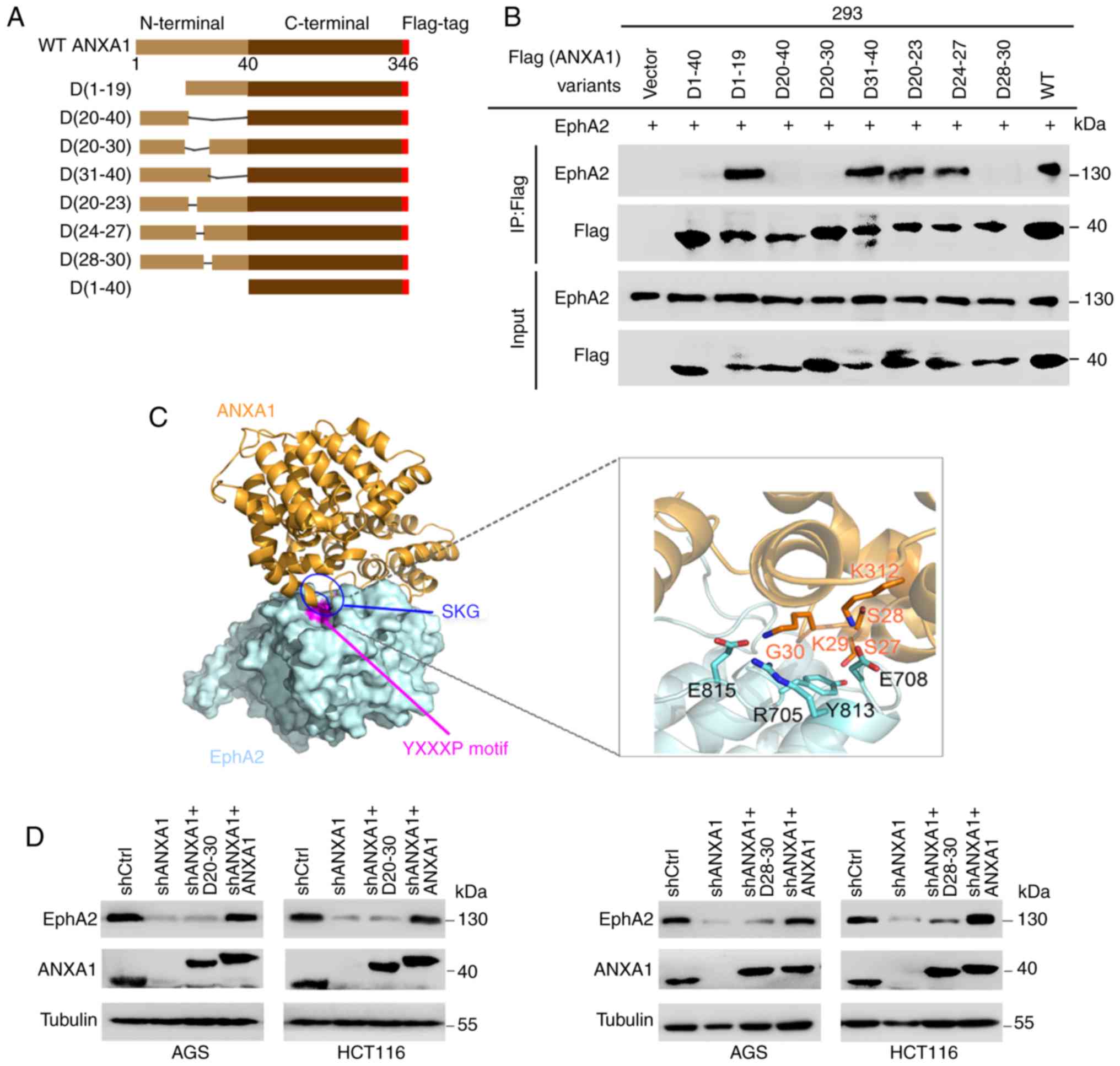
Identification of the ANXA1 region that
binds to EphA2
To map ANXA1 N-terminal responsible for binding
EphA2, we constructed a series of ANXA1 N-terminal deletion mutants
(Fig. 3A), and cotransfected each
of ANXA1 deletion mutants with EphA2 into 293 cells following co-IP
analysis. The results showed that N-terminal deletion ANXA1 (D1-40)
could not bind to EphA2, indicating that ANXA1 N-terminal was
responsible for binding EphA2 (Fig.
3B). We further mapped the ANXA1 N-terminal region responsible
for binding EphA2, and observed that D1-19 and D31-40 but not
D20-40 and D20-30 were able to bind to EphA2, indicating the 11
amino acid residues (20-30aa) responsible for binding EphA2
(Fig. 3B). Moreover, D28-30 could
not bind to EphA2, indicating the 3 amino acid residues (28-30aa)
responsible for binding EphA2 (Fig.
3B). Modeling of the structure of the ANXA1-EphA2 complex
showed that the three amino acid residues 28-30 (S28, K29 and G30)
of ANXA1 are critical to binding EphA2 (Fig. 3C), supporting our experimental
result. Next, we analyzed whether the amino acid residues (28-30aa)
and (20-30aa) of ANXA1 have functional relevance with EphA2
stability. We transfected the plasmid expressing shRNA-resistant
ANXA1, D28-30 or D20-30 into AGS and HCT116 cells with knockdown of
endogenous ANXA1 by shRNA, and observed that ANXA1 but not D28-30
and D20-30 could rescue EphA2 levels (Fig. 3D), indicating that the amino acid
residues (28-30aa) and (20-30aa) of ANXA1 are important for EphA2
stability.
ANXA1-dirived peptides block EphA2-ANXA1
interaction and target EphA2 degradation
To explore the effects of ANXA1-derived peptides on
EphA2-ANXA1 interaction and EphA2 stability, ANXA1-derived 3-mer
(28-30aa) (SKG) and 11-mer (20-30aa) (EYVQTVKSSKG) peptides were
synthesized in fusion to previously characterized cell-penetrating
peptide (CPP) (YGRKKRRQRRR) respectively (51), thereafter named as A1(28-30)
and A1(20-30) respectively, and CPP was used as
control. Efficient cellular uptake of the three peptides was
confirmed by immunofluorescent labeling with fluorescein
isothiocyanate (FITC) (Fig. 4A).
As compared to CPP, both A1(28-30)
and A1(20-30) dramatically decreased ANXA1 bound to
EphA2 (Fig. 4B), and efficiently
decreased EphA2 protein levels in the GC and CC cells (Fig. 4C). Moreover, biotin pull-down
assay, a method for detecting peptide-protein interaction, showed
that both A1(28-30) and A1(20-30)
could efficiently pull down EphA2 in the GC and CC cells (Fig. 4D), indicating that A1(28-30)
and A1(20-30) bind EphA2. Collectively, these
results demonstrate that both A1(28-30)
and A1(20-30) block ANXA1 binding EphA2, and target
EphA2 degradation.
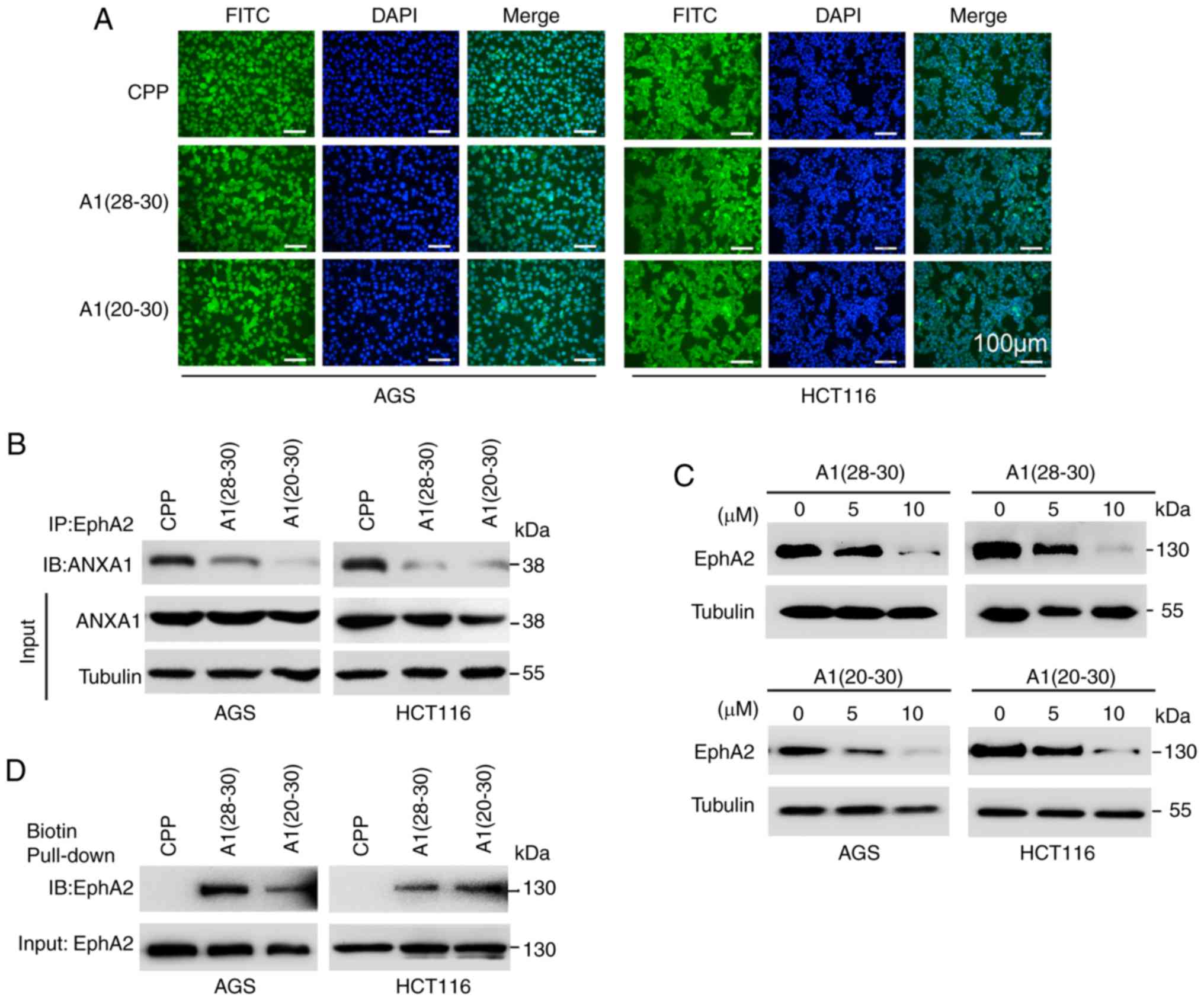 | Figure 4ANXA1-dirived peptides block
EphA2-ANXA1 interaction and target EphA2 for degradation in GC and
CC cells. (A) The subcellular distribution of FITC-labeled
CPP-A1(28-30), FITC-labeled CPP-A1(20-30)
and control FITC-labeled CPP in the AGS and HCT116 cells. Cells
were incubated with 5 µM FITC-labeled peptides for 1 h, then
observed by fluorescence microscopy. Cell nuclei were stained by
DAPI. Scale bars, 100 µm. (B) Co-IP showing that the effects
of A1(28-30) and A1(20-30)
on ANXA1 bound to EphA2 in the AGS and HCT116 cells. Total proteins
were prepared from the cells incubated with 10 µM peptides
for 24 h, and subjected to immunoprecipitation (IP) with anti-EphA2
antibody followed by immunoblotting with anti-ANXA1 antibody. (C)
Immunoblotting showing that the effects of A1(28-30)
and A1(20-30) on the protein levels of EphA2 in the
AGS and HCT116 cells. The cells were incubated with 5 and 10
µM peptides for 24 h respectively, and total cell proteins
were subjected to immunoblotting with anti-EphA2 antibody. (D)
Biotin pull-down showing A1(28-30)
and A1(20-30) binding endogenous EphA2. Total
proteins from AGS and HCT116 cells were incubated with the
biotin-labeled peptides and streptavidin-conjugated agarose.
Samples were electrophoresed and immunoblotted with against EphA2
antibody. A1(28-30), CPP-ANXA1-derived 3-mer (28-30aa)
(SKG); A1(20-30), CPP-11-mer (20-30aa) (EYVQTVKSSKG)
peptides; CPP, cell-penetrating peptide. GC, gastric cancer; CC,
colon cancer; ANXA1, Annexin 1. |
A1(28-30)
and A1(20-30) possess anti-GC and-CC effects in
vitro and in vivo
Overexpression of EphA2 has been considered as a
promising target for the treatment of cancers. Therefore, we tested
the tumor-suppression function of A1(28-30)
and A1(20-30). MTT assay showed that A1(28-30)
and A1(20-30) dramatically decreased the viability
of CC and GC cells (Fig. 5A),
CCK-8 and plate colony formation assay showed that A1(28-30)
and A1(20-30) dramatically inhibited CC and GC cell
proliferation (Fig. 5B and C), and
soft agar colony formation assay showed that A1(28-30)
and A1(20-30) dramatically inhibited CC and GC cell
anchorage-independent growth (Fig.
5D). Moreover, EphA2 overexpression was able to rescue the
proliferation and anchorage-independent growth of CC and GC cells
treated with the two peptides (Fig.
5B-D).
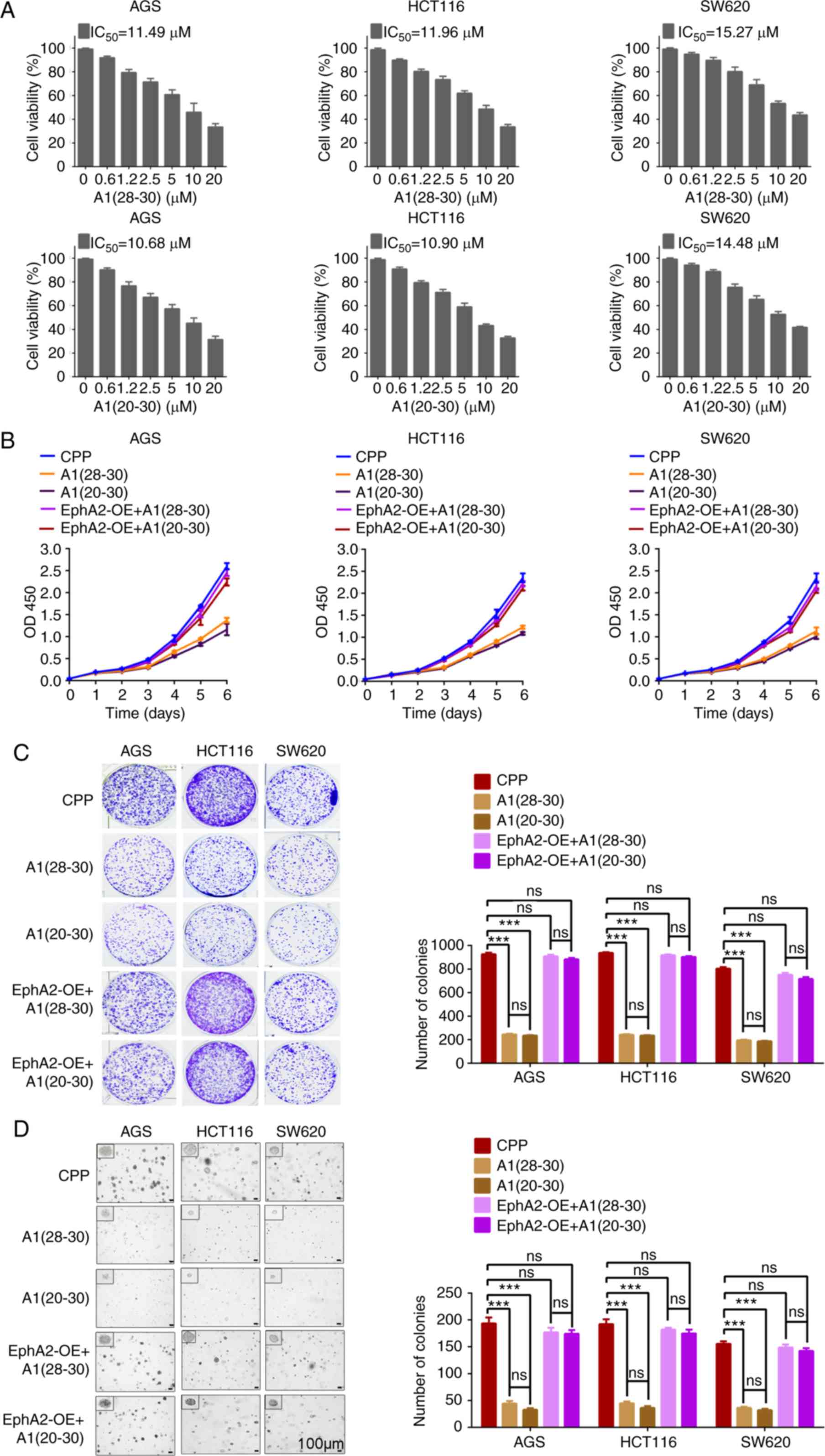 | Figure 5A1(28-30)
and A1(20-30) possess anti-GC and anti-CC effect
in vitro. (A) A1(28-30)
and A1(20-30) decrease the viability of GC (AGS)
and CC (HCT116 and SW620) cells. The cells were incubated with 0-20
µM peptides for 48 h, and cell viability was measured by MTT
assay. (B and C) A1(28-30) and A1(20-30)
decrease the proliferation of GC (AGS) and CC (HCT116 and SW620)
cells, and EphA2 overexpression rescues the effect of both peptides
on the proliferation of GC and CC cells. The cells were incubated
with 10 mM peptides that was replenished every 24 h, and cell
proliferation was detected by CCK-8 (B) and plate colony formation
(C) assay. (D) A1(28-30) and A1(20-30)
decrease the anchorage-independent growth of GC (AGS) and CC
(HCT116 and SW620) cells, and EphA2 overexpression rescues the
effect of both peptides on the anchorage-independent growth of GC
and CC cells. The cells were incubated with 10 µM peptides
that was replenished every 24 h, and cell anchorage-independent
growth was detected by soft agar colony formation assay.
Representative images are shown on the left, and quantitative data
are presented on the right. Scale bars, 100 µm. Error bars
indicate means ± SD. ***P<0.001; ns, not significant
as determined by Student's t-test. EphA2-OE, EphA2 overexpression;
GC, gastric cancer; CC, colon cancer. |
Next, we further tested the in vivo tumor
suppression function of A1(28-30)
and A1(20-30) via peritoneal injection into mice
carrying the xenograft tumors of GC and CC cells. Consistent with
our in vitro observations, not only the sizes and weights of
tumors (Fig. 6A), but also EphA2
expression in the tumors was markedly decreased in the mice
received A1(28-30) or A1(20-30)
(Fig. 6B). Moreover, H&E
staining showed that the morphology and structure of the heart,
lung, liver and kidney of mice receiving peptide A1(28-30)
or A1(20-30) were normal, indicating that
A1(28-30) and A1(20-30)
are not toxicity to mice (Fig.
6C).
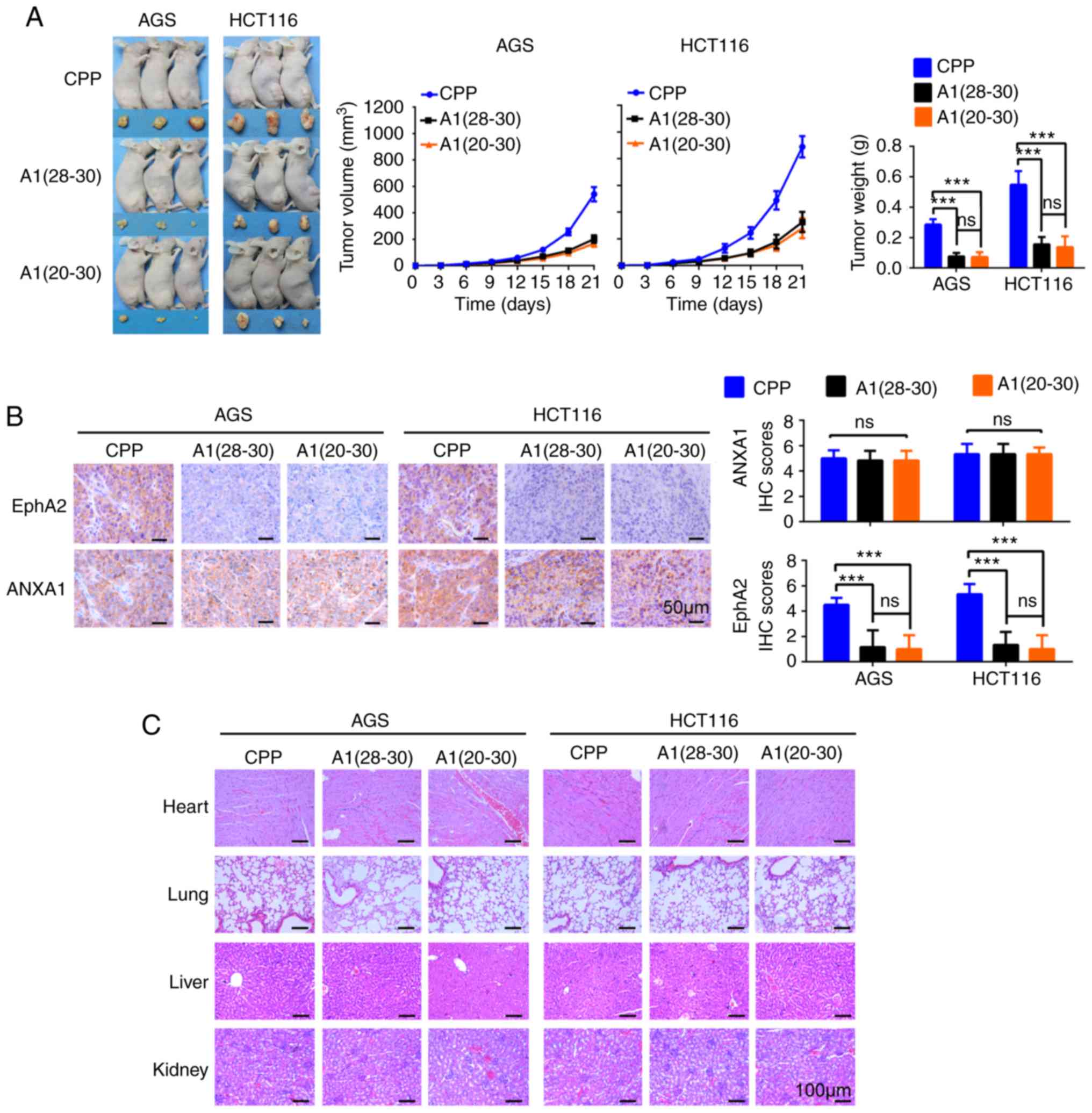 | Figure 6A1(28-30)
and A1(20-30) possess anti-GC and anti-CC effect
in vivo. (A) Tumor formation assay evaluating the effect of
A1(28-30) and A1(20-30)
on the oncogenicity of GC and CC cells in mice. (Left) The
subcutaneous xenografts harvested from each mouse intraperitoneally
injected with A1(28-30), A1(20-30)
or CPP were imaged before further processing. (Middle and right)
Tumor volume was periodically monitored, and tumor volume and
weight for each group (six mice) were plotted.
***P<0.001, ns, not significant as determined by
Student's t-test. (B) Immunohistochemistry (IHC) showing the
expression of EphA2 and ANXA1 in the xenograft tumors.
Representative IHC images are shown on the left, and quantitative
data are presented on the right. Scale bars, 50 µm. Error
bars indicate means ± SD. ***P<0.001, ns, not
significant as determined by one-way ANOVA test. (C) H&E
staining showing the morphology and structure of heart, lung, liver
and kidney from the mice received A1(28-30),
A1(20-30) or CPP. Scale bars, 100 µm.
GC, gastric cancer; CC, colon cancer; ANXA1, Annexin 1; CPP,
cell-penetrating peptide. |
Discussion
EphA2 is overexpressed, promotes tumor growth and
progression, and correlates with poor patient prognosis in gastric
cancer (GC) (18-20) and colon cancer (CC) (25,26).
Therefore, overexpression of EphA2 is a promising target for the
treatment of GC and CC. Abnormal protein-protein interaction (PPI)
is associated with cancer, representing a pivotal target for
chemico-biological interventions (33-35).
In the present study, we identified the interaction
of ANXA and EphA2, and demonstrated a positive correlation of the
expression levels of both proteins in CC and GC, which prompted us
to investigate the function and significance of the ANXA-EphA2
interaction. Our results revealed that ANXA1 obviously increased
EphA2 protein stability, and proteasome inhibitor MG132 was able to
reverse the decrease in EphA2 protein in the ANXA1-knockdown GC and
CC cells, indicating that ANXA1 stabilizes EphA2 possibly by
inhibiting its proteasomal degradation.
It has been reported that ANXA1 N-terminal regulates
the level and activity of the epidermal growth factor receptor
(EGFR) (30-32). Therefore, we analyzed whether ANXA1
N-terminal is responsible for binding EphA2, and observed that
ANXA1 N-terminal bound to EphA2. We further mapped the region of
ANXA1 binding to EphA2, and found the amino acid residues 20-30 and
28-30 of ANXA1 N-terminal were responsible for binding EphA2, which
functionally was correlated with EphA2 stability.
Protein-derived peptides and peptidomimetics have
the propensity to bind targeted protein surfaces and interfere with
PPIs (52,53). Numerous studies have indicated that
inhibition of PPI by peptides is an efficient anticancer approach
(36-38), possessing many advantages compared
with chemotherapeutic drug (39,40).
Since the EphA2-ANXA1 interaction stabilized EphA2, one potentially
effective approach for targeting EphA2 degradation is to disturb
EphA2-ANXA1 interaction. Based on the amino acid residues 20-30 and
28-30 of ANXA1 N-terminal responsible for binding EphA2, we
synthesized ANXA1-derived 3-mer (28-30aa) and 11-mer (20-30aa)
peptides, named A1(28-30) and A1(20-30),
respectively. With the help of cell-penetrating peptide (CPP),
A1(28-30) and A1(20-30)
could be uptaken by GC and CC cells, bound with EphA2, blocked
ANXA1 binding EphA2, and targeted EphA2 degradation.
Next, we tested the effect of A1(28-30)
and A1(20-30) on the oncogenicity of GC and CC
cells. The results showed that A1(28-30)
and A1(20-30) dramatically decreased the viability
of GC and CC cells, inhibited the proliferation and
anchorage-independent growth of GC and CC cells, and decreased the
growth of xenografts from GC and CC cells without toxicity. Our
results indicate that inhibition of ANXA1-EphA2 interaction by
A1(28-30) and A1(20-30)
may represent a promising strategy to impair the oncogenicity of
EphA2, and antagonize GC and CC growth.
Why do A1(28-30)
and A1(20-30) decrease EphA2 stability in the GC
and CC cells? It is reported that the levels of many RTKs are
regulated by ubiquitination degradation, and ubiquitination
represents a key mechanism driving proteasomal degradation of EphA2
(54-56). We believe that A1(28-30)
and A1(20-30) decrease EphA2 stability possibly by
inhibiting its ubiquitination degradation, the detailed mechanism
of which needs further study.
In summary, in the present study we demonstrated
that the interaction of ANXA1 and EphA2 stabilized EphA2 in GC and
CC cells, and we developed two ANXA1-derived peptides, which
blocked the interaction of ANXA1 and EphA2, and downregulated EphA2
with anti-GC and -CC effects in vitro and in
vivo.
Supplementary Data
Funding
This research was supported by the National Natural
Science Foundation of China (81874132, 81672687), the Natural
Science Foundation of Hunan Province of China (2019JJ40486), and
Shenzhen Science and Technology Program of China
(KQTD20170810160226082).
Availability of data and materials
The mass spectrometry proteomics data have been
deposited to the ProteomeXchange Consortium via the PRIDE partner
repository with the dataset identifier PXD015242 (https://www.ebi.ac.uk/pride/archive/projects/PXD015242/).
Authors' contributions
JF conceived the study, performed the experiments
and analyzed and interpreted the data. TX and SSL performed the
experiments and analyzed and interpreted data. XPH, HY and QYH
performed the experiments. WH and YYT collected and provided the
resources and supervised the study. ZQX designed and supervised
this project, and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The use of human tissues was approved by the Ethics
Committee, Xiangya Hospital of Central South University. As only
archived tumor specimens were included in this study, the ethics
committee waived the need for consent. All animal experimental
procedures were performed in accordance with the Guide for the Care
and Use of Laboratory Animals of Xiangya Hospital of Central South
University, with the approval of the Institutional Animal Ethics
Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors state that they have no competing
interests.
Acknowledgments
We thank Dr Xiaojing Yuan (Shanghai Institute of
Medicine, Chinese Academy of Sciences) for assistance with the
structural modelling of the ANXA1-EphA2 complex.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kelley JR and Duggan JM: Gastric cancer
epidemiology and risk factors. J Clin Epidemiol. 56:1–9. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pasquale EB: Eph-ephrin bidirectional
signaling in physiology and disease. Cell. 133:38–52. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markosyan N, Li J, Sun YH, Richman LP, Lin
JH, Yan F, Quinones L, Sela Y, Yamazoe T, Gordon N, et al: Tumor
cell-intrinsic EPHA2 suppresses anti-tumor immunity by regulating
PTGS2 (COX-2). J Clin Invest. 129:3594–3609. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou Y, Yamada N, Tanaka T, Hori T,
Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I and
Sakurai H: Crucial roles of RSK in cell motility by catalysing
serine phosphorylation of EphA2. Nat Commun. 6:76792015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miao H, Gale NW, Guo H, Qian J, Petty A,
Kaspar J, Murphy AJ, Valenzuela DM, Yancopoulos G, Hambardzumyan D,
et al: EphA2 promotes infiltrative invasion of glioma stem cells in
vivo through cross-talk with Akt and regulates stem cell
properties. Oncogene. 34:558–567. 2015. View Article : Google Scholar
|
|
9
|
Song W, Ma Y, Wang J, Brantley-Sieders D
and Chen J: JNK signaling mediates EPHA2-dependent tumor cell
proliferation, motility, and cancer stem cell-like properties in
non-small cell lung cancer. Cancer Res. 74:2444–2454. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Binda E, Visioli A, Giani F, Lamorte G,
Copetti M, Pitter KL, Huse JT, Cajola L, Zanetti N, DiMeco F, et
al: The EphA2 receptor drives self-renewal and tumorigenicity in
stem-like tumor-propagating cells from human glioblastomas. Cancer
Cell. 22:765–780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miao H, Li DQ, Mukherjee A, Guo H, Petty
A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, et al: EphA2
mediates ligand-dependent inhibition and ligand-independent
promotion of cell migration and invasion via a reciprocal
regulatory loop with Akt. Cancer Cell. 16:9–20. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wykosky J and Debinski W: The EphA2
receptor and ephrinA1 ligand in solid tumors: Function and
therapeutic targeting. Mol Cancer Res. 6:1795–1806. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tandon M, Vemula SV and Mittal SK:
Emerging strategies for EphA2 receptor targeting for cancer
therapeutics. Expert Opin Ther Targets. 15:31–51. 2011. View Article : Google Scholar :
|
|
14
|
Rescher U and Gerke V: Annexins-unique
membrane binding proteins with diverse functions. J Cell Sci.
117:2631–2639. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Perretti M and D'Acquisto F: Annexin A1
and glucocorticoids as effectors of the resolution of inflammation.
Nat Rev Immunol. 9:62–70. 2009. View Article : Google Scholar
|
|
16
|
Senchenkova EY, Ansari J, Becker F, Vital
SA, Al-Yafeai Z, Sparkenbaugh EM, Pawlinski R, Stokes KY, Carroll
JL, Dragoi AM, et al: Novel role for the AnxA1-Fpr2/ALX signaling
axis as a key regulator of platelet function to promote resolution
of inflammation. Circulation. 140:319–335. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo C, Liu S and Sun MZ: Potential role of
Anxa1 in cancer. Future Oncol. 9:1773–1793. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang J, Xiao D, Li G, Ma J, Chen P, Yuan
W, Hou F, Ge J, Zhong M, Tang Y, et al: EphA2 promotes
epithelial-mesenchymal transition through the Wnt/β-catenin pathway
in gastric cancer cells. Oncogene. 33:2737–2747. 2014. View Article : Google Scholar
|
|
19
|
Huang C, Yuan W, Lai C, Zhong S, Yang C,
Wang R, Mao L and Chen Z and Chen Z: EphA2-to-YAP pathway drives
gastric cancer growth and therapy resistance. Int J Cancer.
146:1937–1949. 2020. View Article : Google Scholar
|
|
20
|
Kikuchi S, Kaibe N, Morimoto K, Fukui H,
Niwa H, Maeyama Y, Takemura M, Matsumoto M, Nakamori S, Miwa H, et
al: Overexpression of Ephrin A2 receptors in cancer stromal cells
is a prognostic factor for the relapse of gastric cancer. Gastric
Cancer. 18:485–494. 2015. View Article : Google Scholar
|
|
21
|
Takaoka RTC, Sertorio ND, Magalini LPJ,
Dos Santos LM, Souza HR, Iyomasalon MM, Possebon L, Costa SS and
Girol AP: Expression profiles of Annexin A1, formylated peptide
receptors and cyclooxigenase-2 in gastroesophageal inflammations
and neoplasias. Pathol Res Pract. 214:181–186. 2018. View Article : Google Scholar
|
|
22
|
Wang X, Zhi Q, Liu S, Xue SL, Shen C, Li
Y, Wu C, Tang Z, Chen W, Song JL, et al: Identification of specific
biomarkers for gastric adenocarcinoma by ITRAQ proteomic approach.
Sci Rep. 6:388712016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang ZQ, Li XJ, Liu GT, Xia Y, Zhang XY
and Wen H: Identification of Annexin A1 protein expression in human
gastric adenocarcinoma using proteomics and tissue microarray.
World J Gastroenterol. 19:7795–7803. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheng TY, Wu MS, Lin JT, Lin MT, Shun CT,
Huang HY, Hua KT and Kuo ML: Annexin A1 is associated with gastric
cancer survival and promotes gastric cancer cell invasiveness
through the formyl peptide receptor/extracellular signal-regulated
kinase/integrin beta-1-binding protein 1 pathway. Cancer.
118:5757–5767. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dunne PD, Dasgupta S, Blayney JK, McArt
DG, Redmond KL, Weir JA, Bradley CA, Sasazuki T, Shirasawa S, Wang
T, et al: EphA2 expression is a key driver of migration and
invasion and a poor prognostic marker in colorectal cancer. Clin
Cancer Res. 22:230–242. 2016. View Article : Google Scholar
|
|
26
|
Saito T, Masuda N, Miyazaki T, Kanoh K,
Suzuki H, Shimura T, Asao T and Kuwano H: Expression of EphA2 and
E-cadherin in colorectal cancer: Correlation with cancer
metastasis. Oncol Rep. 11:605–611. 2004.PubMed/NCBI
|
|
27
|
Sato Y, Kumamoto K, Saito K, Okayama H,
Hayase S, Kofunato Y, Miyamoto K, Nakamura I, Ohki S, Koyama Y and
Takenoshita S: Up-regulated Annexin A1 expression in
gastro-intestinal cancer is associated with cancer invasion and
lymph node metastasis. Exp Ther Med. 2:239–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Onozawa H, Saito M, Saito K, Kanke Y,
Watanabe Y, Hayase S, Sakamoto W, Ishigame T, Momma T, Ohki S and
Takenoshita S: Annexin A1 is involved in resistance to 5-FU in
colon cancer cells. Oncol Rep. 37:235–240. 2017. View Article : Google Scholar
|
|
29
|
Ydy LR, do Espirito Santo GF, de Menezes
I, Martins MS, Ignotti E and Damazo AS: Study of the Annexin A1 and
its associations with carcinoembryonic antigen and mismatch repair
proteins in colorectal cancer. J Gastrointest Cancer. 47:61–68.
2016. View Article : Google Scholar
|
|
30
|
Radke S, Austermann J, Russo-Marie F,
Gerke V and Rescher U: Specific association of annexin 1 with
plasma membrane-resident and internalized EGF receptors mediated
through the protein core domain. FEBS Lett. 578:95–98. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
White IJ, Bailey LM, Aghakhani MR, Moss SE
and Futter CE: EGF stimulates annexin 1-dependent inward
vesiculation in a multivesicular endosome subpopulation. EMBO J.
25:1–12. 2006. View Article : Google Scholar
|
|
32
|
Poeter M, Radke S, Koese M, Hessner F,
Hegemann A, Musiol A, Gerke V, Grewal T and Rescher U: Disruption
of the annexin A1/S100A11 complex increases the migration and
clonogenic growth by dysregulating epithelial growth factor (EGF)
signaling. Biochim Biophys Acta. 1833:1700–1711. 2013. View Article : Google Scholar
|
|
33
|
Akram ON, DeGraff DJ, Sheehan JH, Tilley
WD, Matusik RJ, Ahn JM and Raj GV: Tailoring peptidomimetics for
targeting protein-protein interactions. Mol Cancer Res. 12:967–978.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iyer VV: A review of stapled peptides and
small molecules to inhibit protein-protein interactions in cancer.
Curr Med Chem. 23:3025–3043. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ferreira LG, Oliva G and Andricopulo AD:
Protein-protein interaction inhibitors: Advances in anticancer drug
design. Expert Opin Drug Discov. 11:957–968. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jafary F, Ganjalikhany MR, Moradi A,
Hemati M and Jafari S: Novel peptide inhibitors for lactate
dehydrogenase A (LDHA): A survey to inhibit LDHA activity via
disruption of protein-protein interaction. Sci Rep. 9:46862019.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yeon M, Byun J, Kim H, Kim M, Jung HS,
Jeon D, Kim Y and Jeoung D: CAGE binds to beclin1, rregulates
autophagic flux and CAGE-derived peptide confers sensitivity to
anti-cancer drugs in non-small cell lung cancer cells. Front Oncol.
8:5992018. View Article : Google Scholar
|
|
38
|
Liang L, Wang H, Shi H, Li Z, Yao H, Bu Z,
Song N, Li C, Xiang D, Zhang Y, et al: A designed peptide targets
two types of modifications of p53 with anti-cancer activity. Cell
Chem Biol. 25:761–774 e765. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Henninot A, Collins JC and Nuss JM: The
current state of peptide drug discovery: Back to the future? J Med
Chem. 61:1382–1414. 2018. View Article : Google Scholar
|
|
40
|
Di L: Strategic approaches to optimizing
peptide ADME proper-ties. AAPS J. 17:134–143. 2015. View Article : Google Scholar
|
|
41
|
Li JY, Xiao T, Yi HM, Yi H, Feng J, Zhu
JF, Huang W, Lu SS, Zhou YH, Li XH and Xiao ZQ: S897
phosphorylation of EphA2 is indispensable for EphA2-dependent
nasopharyngeal carcinoma cell invasion, metastasis and stem
properties. Cancer Lett. 444:162–174. 2019. View Article : Google Scholar
|
|
42
|
Zhu JF, Huang W, Yi HM, Xiao T, Li JY,
Feng J, Yi H, Lu SS, Li XH, Lu RH, et al: Annexin A1-suppressed
autophagy promotes nasopharyngeal carcinoma cell invasion and
metastasis by PI3K/AKT signaling activation. Cell Death Dis.
9:11542018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mozayan A and Khaled A: Elucidation of
therapeutic peptide binding partners from isolated mitochondria.
Cureus. 10:e28982018.PubMed/NCBI
|
|
44
|
Yi HM, Yi H, Zhu JF, Xiao T, Lu SS, Guan
YJ and Xiao ZQ: A five-variable signature predicts radioresistance
and prognosis in nasopharyngeal carcinoma patients receiving
radical radio-therapy. Tumour Biol. 37:2941–2949. 2016. View Article : Google Scholar
|
|
45
|
Waterhouse A, Bertoni M, Bienert S, Studer
G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C,
Bordoli L, et al: SWISS-MODEL: Homology modelling of protein
structures and complexes. Nucleic Acids Res. 46:W296–W303. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kozakov D, Hall DR, Xia B, Porter KA,
Padhorny D, Yueh C, Beglov D and Vajda S: The ClusPro web server
for protein-protein docking. Nat Protoc. 12:255–278. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sastry GM, Adzhigirey M, Day T,
Annabhimoju R and Sherman W: Protein and ligand preparation:
Parameters, proto-cols, and influence on virtual screening
enrichments. J Comput Aided Mol Des. 27:221–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Maier JA, Martinez C, Kasavajhala K,
Wickstrom L, Hauser KE and Simmerling C: ff14SB: Improving the
accuracy of protein side chain and backbone parameters from ff99SB.
J Chem Theory Comput. 11:3696–3713. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tang CE, Guan YJ, Yi B, Li XH, Liang K,
Zou HY, Yi H, Li MY, Zhang PF, Li C, et al: Identification of the
amyloid β-protein precursor and cystatin C as novel epidermal
growth factor receptor regulated secretory proteins in
nasopharyngeal carci-noma by proteomics. J Proteome Res.
9:6101–6111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xiao T, Zhu W, Huang W, Lu SS, Li XH, Xiao
ZQ and Yi H: RACK1 promotes tumorigenicity of colon cancer by
inducing cell autophagy. Cell Death Dis. 9:11482018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Michiue H, Sakurai Y, Kondo N, Kitamatsu
M, Bin F, Nakajima K, Hirota Y, Kawabata S, Nishiki T, Ohmori I, et
al: The acceleration of boron neutron capture therapy using
multi-linked mercaptoundecahydrododecaborate (BSH) fused
cell-penetrating peptide. Biomaterials. 35:3396–3405. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pelay-Gimeno M, Glas A, Koch O and
Grossmann TN: Structure-based design of inhibitors of
protein-protein interactions: Mimicking peptide binding epitopes.
Angew Chem Int Ed Engl. 54:8896–8927. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Qvit N, Rubin SJS, Urban TJ, Mochly-Rosen
D and Gross ER: Peptidomimetic therapeutics: Scientific approaches
and opportunities. Drug Discov Today. 22:454–462. 2017. View Article : Google Scholar
|
|
54
|
Sabet O, Stockert R, Xouri G, Brüggemann
Y, Stanoev A and Bastiaens PIH: Ubiquitination switches EphA2
vesicular traffic from a continuous safeguard to a finite
signalling mode. Nat Commun. 6:80472015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Naudin C, Sirvent A, Leroy C, Larive R,
Simon V, Pannequin J, Bourgaux JF, Pierre J, Robert B, Hollande F
and Roche S: SLAP displays tumour suppressor functions in
colorectal cancer via destabilization of the SRC substrate EPHA2.
Nat Commun. 5:31592014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Annamalai B, Liu X, Gopal U and Isaacs JS:
Hsp90 is an essential regulator of EphA2 receptor stability and
signaling: Implications for cancer cell migration and metastasis.
Mol Cancer Res. 7:1021–1032. 2009. View Article : Google Scholar : PubMed/NCBI
|