Introduction
Cholangiocarcinoma (CCA) is the most common biliary
duct malignancy and the second most common primary liver cancer,
accounting for 10-20% of all primary hepatic malignancies (1). CCA arises from bile duct epithelial
cells, and there are three subtypes based on anatomic location:
Intrahepatic (iCCA), perihilar (pCCA), and distal (dCCA). The
etiology of CCA includes lithiasis (2), primary sclerosing cholangitis
(3), parasitic infection (4), congenital abnormalities (5), chronic liver disease (6), cirrhosis (6), metabolic abnormalities (7), and lifestyle (7). The incidence of iCCA appears to be
increasing (8); the rates of CCA
in North America, Japan, and Australia have been rising over the
past two decades (9). Although
surgery is the preferred treatment, the 5-year postoperative
survival rate is markedly low. Chemotherapy can be used for
inoperable cases; however, the highly desmoplastic nature, rich
tumor microenvironment, and profound genetic heterogeneity all
contribute to iCCA therapeutic resistance (9).
The nonsteroidal drug aspirin is an
anti-inflammatory and anticoagulation agent used to prevent and
reduce the risk of cardiovascular events. According to some
clinical analyses, long-term use is also associated with a
reduction in cancer risk, including colon (10), breast (11), and hepatocellular carcinoma
(12). Aspirin use also has a
significant inverse association with the development of all three
CCA subtypes and an approximately 3-fold reduction in CCA risk
(13). Numerous anticancer
mechanisms for aspirin have been identified, such as inhibition of
cyclooxygenase (14), activating
key molecular targets in the AMPK, mTOR, STAT3, and NF-κB pathways
(15), decreasing the levels of
reactive oxygen species and glucose consumption (16), inducing autophagy via
JNK/p-Bcl2/Beclin-1, AMPK/mTOR, and GSK-3 signaling (17), inducing apoptosis and mitochondrial
dysfunction by increasing oxidative stress (18), and changing the tumor
microenvironment by affecting platelets (19,20).
However, the effect of aspirin on CCA remains unknown. Elucidation
of the mechanism of aspirin in CCA could contribute to the
development of new therapeutic agents in the future.
The purpose of the present study was to determine
the antitumor effects of aspirin in CCA and identify the key
molecular targets and microRNAs (miRNAs) associated with this
effect.
Materials and methods
Chemicals
Aspirin was obtained from Wako Pure Chemical
Industries, Ltd. The prepared solution was diluted in cell culture
medium as per the requirement of cells and fresh pH 7.2 to 7.5,
within the range suitable for cell growth was used.
Cell lines and cell culture
Human CCA cell lines (HuCCT-1, RBE, and TKKK) were
obtained from the Japanese Research Resources Bank. HuCCT-1 and RBE
cells were grown in RPMI-1640 media (Gibco-Invitrogen; Thermo
Fisher Scientific, Inc.) supplemented with 10% fetal bovine serum
(FBS) (product no. 557-30355; FUJIFILM Wako Pure Chemical
Industries, Ltd.) and penicillin/streptomycin (100 mg/l;
Invitrogen; Thermo Fisher Scientific, Inc.). TKKK cells were
maintained in DMEM (Gibco-Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 10% FBS and penicillin/streptomycin. All
cell lines were grown in a humidified incubator at 5%
CO2 and 37°C.
Cell viability assay
Cell viability assays were performed using the Cell
Counting Kit-8 (Dojindo Molecular Technologies, Inc.) according to
the manufacturer's instructions. Briefly, cells (5,000 cells in 100
µl/well) were seeded into 96-well plates and allowed to
adhere, followed by treatment with 0, 2.5, 5, or 10 mmol/l aspirin
for 48 h. The medium was replaced with 100 µl of fresh
medium containing 10% of CCK-8 reagent, and cells were incubated at
37°C for 3 h. The absorbance was measured at 450 nm using a
multi-grating microplate reader SH-9000Lab (CORONA Electric Co.,
Ltd.). Experiments were carried out three times. Compared with RBE,
HuCCT-1 cells were more sensitive to aspirin treatment in the cell
viability assay. Moreover, considering the xenograft model, HuCCT-1
cells can be easily transplanted. Thus, HuCCT-1 cells were selected
for further study in vitro and in vivo.
Flow cytometric analysis of the cell
cycle
Flow cytometric analyses were performed using the
Cycle Phase Determination kit (Cayman Chemical Company). HuCCT-1
and TKKK cells (1.0×106 cells/100-mm dish) were treated
with 2.5 mmol/l aspirin or without for 24 to 48 h. Cells were
trypsinized and resuspended in phosphate-buffered saline (PBS) at a
density of 1×106 cells/ml. Approximately
1×106 cells were stained in 100 µl of PBS with 10
µl RNase A (250 µg/ml) and 10 µl propidium
iodide (PI) stain (100 µg/ml) and incubated at room
temperature in the dark for 30 min. Flow cytometry (FCM) was
performed to compare the proportion of aspirin-treated and control
cells in each phase of the cell cycle. FCM was performed using a
Cytomics FC 500 flow cytometer (Beckman Coulter, Inc.) with an
argon laser (488 nm), and the percentages of cells were analyzed
using the Kaluza software version v2.1 (Beckman Coulter, Inc.). The
experiments were repeated thrice.
Apoptosis analysis
Aspirin-mediated apoptosis was analyzed using FCM
and Annexin V-FITC Early Apoptosis Detection kit (Cell Signaling
Technology, Inc.). HuCCT-1 cells (1.0×106 cells/100-mm
dish) were treated with 2.5 mmol/l aspirin or without for 48 h.
Apoptotic and necrotic cells were analyzed by double staining with
FITC-conjugated Annexin V and PI per the manufacturer's
instructions. FCM was conducted using a Cytomics FC 500 flow
cytometer with an argon laser (488 nm) to compare the proportion of
apoptotic cells in the aspirin-treated and control groups, and data
were analyzed using the Kaluza software version v2.1. The
experiments were repeated thrice.
Apoptosis analysis by ELISA
ELISA was performed to analyze the levels of
caspase-cleaved cytokeratin 18 (cCK18) using the M30 Apoptosense
ELISA kit (cat. no. 10011; Peviva; Diapharma) according to the
manufacturer's instructions. HuCCT-1 cells (5,000 cells/well) were
seeded into 96-well plates and treated with 2.5 mmol/l aspirin for
48 h. Subsequently, the cells were lysed in polyoxyethylene octyl
phenyl ether (Pure Chemical Industries, Ltd.) and further analyzed
according to the manufacturer's instructions. The experiments were
repeated thrice.
Western blot analysis
HuCCT-1 and TKKK cells were seeded
(1.0×106 cells/100-mm dish) and treated with 2.5 mmol/l
aspirin for 24 or 48 h. The cells were lysed with PRO-PREP complete
protease inhibitor mixture (iNtRON Biotechnology, Korea).
Supernatants were collected, and the protein concentrations were
measured using a NanoDrop 2000 spectrofluorometer (Thermo Fisher
Scientific, Inc.). Protein aliquots (10 µg) were resuspended
in sample buffer and separated on 12% Tris-glycine gradient gels
via sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The
resolved proteins were transferred to a nitrocellulose membrane and
blocked with a blocking buffer containing 2% skimmed milk (GE
Healthcare) in TBST with 0.1% Tween 20 (cat. no. T9142; Takara Bio,
Inc.) for 30 min at room temperature. Subsequently, the membranes
were incubated overnight at 4°C with primary antibodies in 5% serum
(cat. no. 9048-46-8; FUJIFILM Wako Pure Chemical Industries, Ltd.)
followed by incubation with horseradish peroxidase (HRP)-conjugated
secondary antibodies in 2% skimmed milk. The following primary
antibodies were used: Cyclin D1 (SP4) (1:2,000 dilution; cat. no.
MA5-14512), retinoblastoma (Rb) (1:1,000 dilution; cat. no.
MA1-34070), and cyclin E (HE-12) (1:1,000 dilution; MS-870-P1) were
obtained from Thermo Fisher Scientific, Inc.; phosphorylated Rb
(pS780) (1:1,000 dilution; cat. no. 558554) was obtained from BD
Biosciences; Cdk2 (1: 5,000 dilution; cat. no. sc-163) was obtained
from Santa Cruz Biotechnology, Inc.; and anti-β-actin (1:5,000
dilution; product no. A5441) was purchased from Sigma-Aldrich;
Merck KGaA. The membranes were washed again with TBST and incubated
with HRP-conjugated anti-mouse (dilution 1:2,000; product no. 7076)
and anti-rabbit (dilution 1:2,000; product no. 7074) IgG secondary
antibodies obtained from Cell Signaling Technology, Inc. for 1 h at
room temperature. Finally, the signals were visualized using a
typically enhanced chemiluminescent (ECL) kit (cat. no. 45-000-999;
Cytiva), and blots were imaged using ImageQuant LAS 4010 (GE
Healthcare).
miRNA microarray
Cells were treated with 2.5 mmol/l aspirin for 48 h,
and total RNA was extracted using the miRNeasy Mini kit (Qiagen
GmbH) according to the manufacturer's instructions. After
confirming the purity and quantity of each sample using an Agilent
2100 Bioanalyzer and an RNA 6000 Nano kit (both from Agilent
Technologies), respectively, the samples were labeled using a
miRCURY Hy3 Power Labeling kit (Exiqon A/S) and hybridized to a
human miRNA Oligo Chip (v.21; Toray Industries, Inc.). Chips were
scanned using the 3D-Gene Scanner 3000 (Toray Industries, Inc.).
The 3D-Gene extraction software version 1.2 (Toray Industries,
Inc.) was used to calculate the raw signal intensity of the images.
The raw data were analyzed using the GeneSpring GX 10.0 software
(Agilent Technologies, Inc.) to assess the differences in miRNA
expression between the aspirin-treated and control samples. Global
normalization was performed on raw data obtained above the
background level. Differentially expressed miRNAs were determined
using Welch's t-test.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis of miRNAs
qPCR was used to validate the expression levels
obtained from the miRNA assay. Total RNA was extracted as
previously described above and diluted to 2.0 ng/µl. TaqMan
microRNA assays (Applied Biosystems; Thermo Fisher Scientific,
Inc.) were adopted to determine the expression of miRNAs using U6
small nuclear RNA (RNU6B) as an internal control. miRNAs
were reverse transcribed using the TaqMan microRNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.). Reverse transcription was performed in 20-µl reaction
volumes consisting of 5 µl of RNA, 3 µl of 5X RT
primer, and 12 µl of reverse transcription Master Mix. qPCR
was performed in the MicroAmp Fast Optical 96-Well Reaction Plate
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Each well
contained a 20-µl reaction consisting of 2 µl of
cDNA, 1 µl of 20X qPCR assay, 7 µl of nuclease-free
water, and 10 µl of TaqMan Fast Advanced Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.). PCR was performed
using the ViiA7 real-time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with the following reaction steps: Hold at
50°C for 2 min, denaturation at 95°C for 20 sec followed by 40
cycles of 1 sec at 95°C and 20 sec at 60°C. The relative expression
of miR-340-5p was calculated using the comparative Cq method
according to the following formula: 2−ΔΔCq
(ΔCq=miRCq-U6Cq, ΔΔCq=ΔCq-average control ΔCq) (21). The primer sequences are as follows:
miR-340-5p forward, 5′-GCG GTT ATA AAG CAA TGA GA-3′ and reverse,
5′-GTG CGT GTC GTG GAG TCG-3′; U6 forward, 5′-GCT TCG GCA GCA CAT
ATA CTA AAA T-3′ and reverse, 5′-CGC TTC ACG AAT TTG CGT GTC
AT-3′.
Bioinformatics analysis used for
prediction of the target genes of miR-340-5p and functional and
network analyses
For bioinformatics analysis, miRDB database
(www.mirdb.org) was used to predict the target
genes of miR-340-5p and the Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway analyses was used for upregulated miRNAs depending
on the Database for Annotation, Visualization, and Integrated
Discovery (DAVID database) (22).
miRNA regulatory network used mirPath v.3 (http://snf-515788.vm.okeanos.grnet.gr) (23); miRGator v3.0 (http://mirgator.kobic.re.kr) (24) and miTarBase (http://mirtarbase.cuhk.edu.cn/php/index.php)
(25) online software to reveal
the expression of miR-340-5p in related cancer and normal tissue
and the relationship with the underlying target gene. The cut-off
criterion was set as P<0.05.
Cell transfection
For transfection, 5×105 HuCCT-1cells were
seeded in 6-well plates with antibiotic-free medium 1 day before
transfection to reach a confluence of 90% at the time of
transfection. Cells were transfected with miR-340-5p mimic (50 nM)
(sense, 5′-UUA UAA AGC AAU GAG ACU GAU U-3′ and antisense, 5′-UCA
GUC UCA UUG CUU UAU AAT T-3′), inhibitor (50 nM) (5′-AAU CAG UCU
CAU UGC UUU AUA A-3′), or negative control miRNA (50 nM) (mimic
negative control 5′-UUG UAC UAC ACA AAA GUA CUG-3′; inhibitor
negative control 5′-CAG UAC UUU UGU GUA GUA CAA-3′) (Life
Technologies; Thermo Fisher Scientific, Inc.) using Lipofectamine
RNAiMAX Reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The
transfection complex was prepared according to the manufacturer's
instructions and added to the cells, and the plates were incubated
in a humidified atmosphere with 5% CO2 at 37°C. Cells
cultured with transfection reagent as untransfected control. The
medium was replaced 6 h post-transfection. Cell samples were
collected at 0 or 48 h after transfection for further analysis.
Colony formation assay
Cells were trypsinized for 5 min and resuspended at
a density of 1×103/ml. Five hundred microliters were
seeded into 6-well plates, and 1.5 ml RPMI-1640 medium containing
10% FBS was added to each well. The plates were incubated at 37°C
at 5% CO2, and the medium was changed every 3 days until
conspicuous colonies were observed. Colonies were stained with 0.1%
crystal violet for 5 min at room temperature and positive colony
formation (>50 cells/colony) was evaluated by counting the
number of colonies.
Xenograft model analysis
The animal study was conducted in accordance with
the guidelines set by the Committee on Experimental Animals of the
Kagawa University. All experimental protocols were approved
(approval no. 18674) by the Institutional Review Board of the
Department of Laboratory Animal Science of Kagawa University
(Kawaga, Japan). Thirty-five female athymic mice (BALB/c-nu/nu; 6
weeks old; 19-21 g) were purchased from Japan SLC (Shizuoka,
Japan). The mice were maintained at 20-25°C with 30-60% humidity
under a 12:12 h light/dark cycle, using a laminar airflow rack and
had continuous free access to sterilized (γ-irradiated) food (CL-2;
CLEA Japan, Inc.) and autoclaved water. Mice were subcutaneously
inoculated with 1.5×106 HuCCT-1 cells in the right
flank. When the xenografts were palpable with an approximate
diameter of 3 mm, we randomly assigned the animals to three groups
of 10 animals each. These groups were treated with 60 mg/kg
aspirin, 100 mg/kg aspirin, or vehicle (10% ethanol in PBS) by
intraperitoneal injection every day. The tumor volume
(mm3) was calculated as tumor length (mm) x tumor width
(mm)2/2. For humane endpoints, if difficulty in feeding
and/or intake of water; apparent poor physical condition; a rapid
and non-recoverable weight loss (over 20% body weight); and/or a
significant increase in tumor size was observed (tumor size more
than 10% of body weight and/or tumor diameter more than 20 mm), the
experiments were discontinued. Animals were euthanized through
CO2 euthanasia with 20% displacement of cage volume/min.
Sacrifice was confirmed by observation of unconsciousness, absence
of heartbeat and absence of breathing. All animals were sacrificed
on day 28 of treatment. The experiments were performed from
February 20, 2019 to March 28, 2019.
Immunohistochemistry
Immunohistochemistry was performed on tumor tissues
obtained from the xeno-grafted mice. We prepared 5-µm-thick
sections from formalin-fixed (10% formalin at room temperature for
24 h), paraffin-embedded tissue blocks which were deparaffinized,
rehydrated, and subjected to immunohistochemistry studies.
Following a blocking step at room temperature for 30 min using a
Vectastain Elite ABC kit (Vector Laboratories, Inc.), tissue
sections were incubated with primary anti-bodies [Cyclin D1 (EP12);
1:200 dilution; cat. no. 241R-4, Sigma-Aldrich; Merck KGaA].
Sections were washed and incubated with anti-rabbit IgG Antibody
(1:50 dilution; cat. no. BP-9100-50; Vector Laboratories) at room
temperature for 1 h and then with a streptavidin-peroxidase
solution. Color reactions involved the use of 3,3′-diaminobenzidine
(DAB) with Mayer's hematoxylin counterstaining at room temperature
for 10 sec. The specificity of immunostaining was evaluated using
non-immune mouse IgG (1:50 dilution; cat. no. BP-9200-50; Vector
Laboratories) at room tempera-ture for 1 h as a negative control
for the primary antibody. Sections were examined microscopically
for specific staining, and nuclei with a brown color regardless of
staining intensity were regarded as positive. Cyclin D1 positivity
was calculated at a magnification of ×40 by dividing the number of
positive cells by the total number of cells counted in five random
fields and expressed as a percentage. Images were captured using an
Olympus BX51 microscope and Olympus DP72 camera (magnification,
×40; Olympus Corporation).
Statistical analysis
GraphPad Prism software version 6.0 (GraphPad
Software, Inc.) was used for all analyses. A two-tailed unpaired
Student's t-test was used to determine statistical significance
between different groups. Two-way analysis of variance (ANOVA) or
mixed ANOVA was performed to test the comparisons and corrected by
Tukey's post hoc test. One-way analysis of variance before the
Tukey's post hoc test was performed to test the comparisons. A
P-value of <0.05 was considered to indicate a statistically
significant difference.
Results
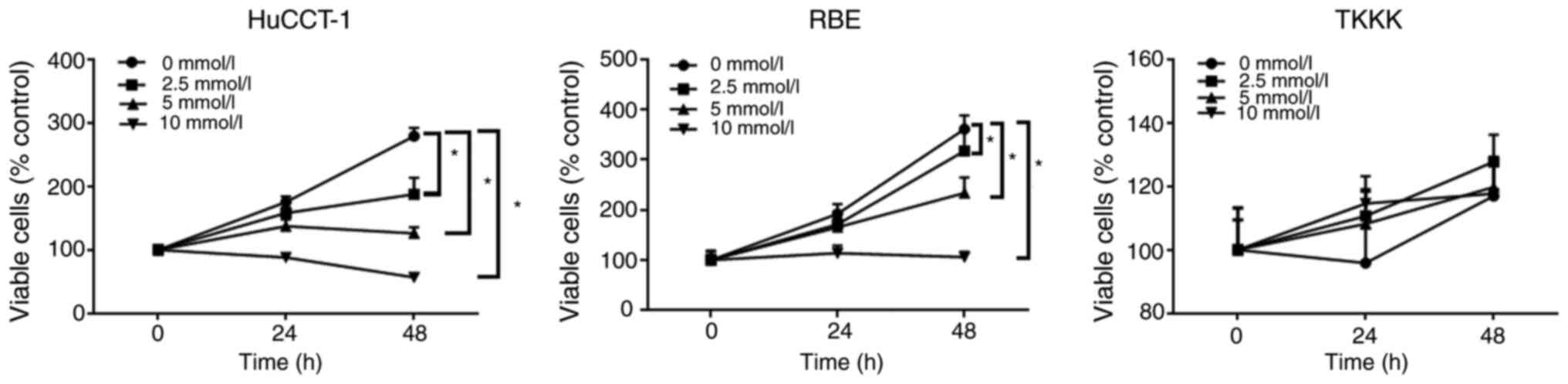
Aspirin inhibits the proliferation of
most human CCA cells
The anti-proliferative effects of aspirin on human
CCA cells were determined using the HuCCT-1, RBE, and TKKK CCA cell
lines. Cells were treated with 2.5, 5, or 10 mmol/l aspirin for 48
h, and the anti-proliferative effect of aspirin was assessed using
the cell viability assay. Untreated cells were used as controls.
Aspirin inhibited cell proliferation in CCA cells in a dose and
time-dependent manner, except in TKKK cells (Fig. 1).
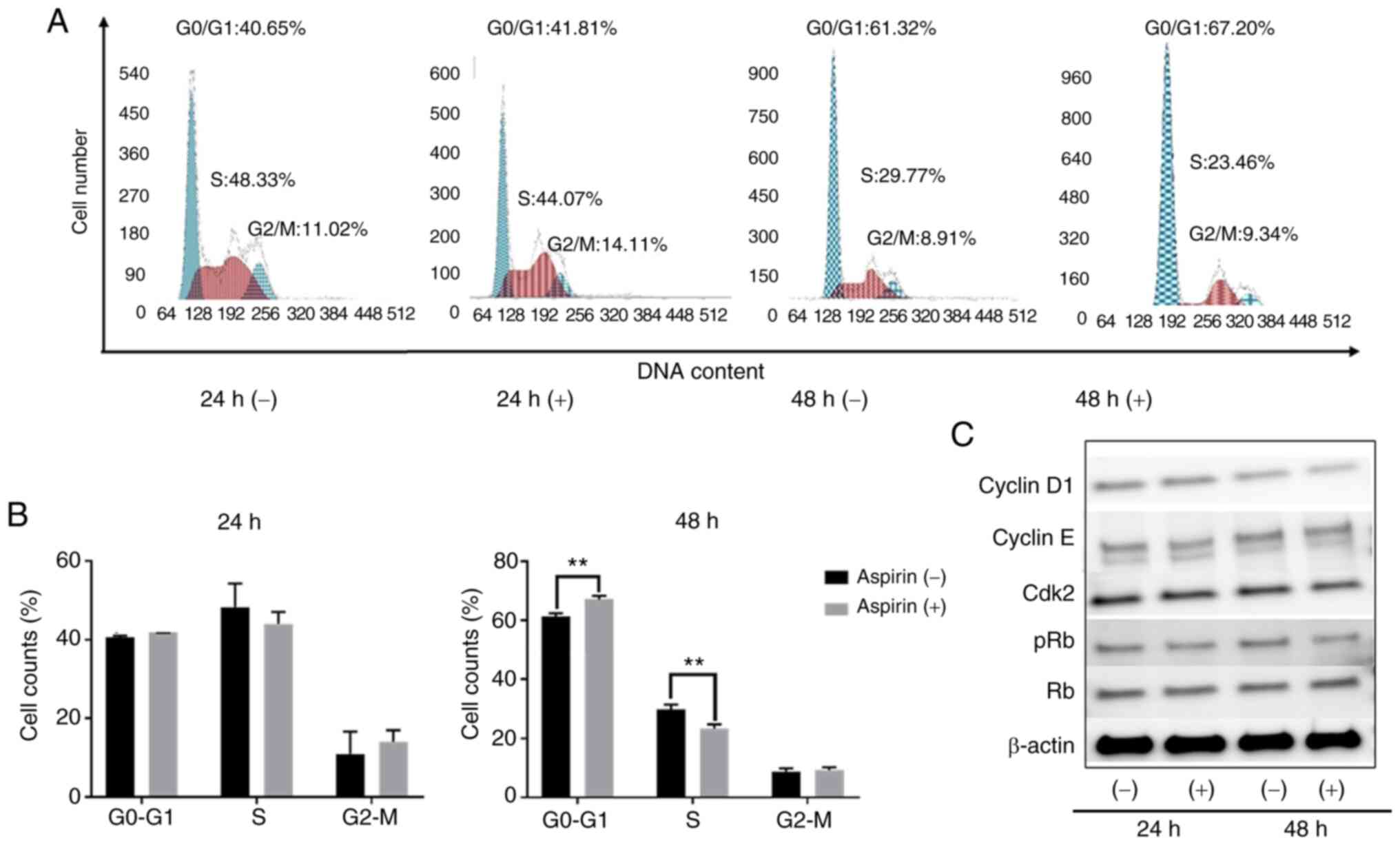
Aspirin induces cell cycle arrest in the
G0/G1 phase and regulates cell-cycle related
proteins in HuCCT-1 cells
To determine whether aspirin affected the cell cycle
in CCA cells, HuCCT-1 cells were treated with aspirin and FCM was
performed to examine cell cycle progression. Western blotting was
also performed to evaluate the expression of cell-cycle related
proteins. Cells were treated with 2.5 mmol/l aspirin for 24 or 48
h, and untreated cells were used as the control. Following aspirin
treatment, the population in the G0/G1 phase
significantly increased, whereas cells in the S phase decreased
(Fig. 2A and B). Western blot
results indicated that treatment with aspirin for 48 h
significantly modulated cyclin D1, a key protein expressed in the
early G1 phase. Cyclin D1 is a key regulator of the cell
cycle and is involved in the transition from the G1
phase to the S phase (26). The
levels of phosphorylated Rb decreased with aspirin treatment,
suggesting that the treated cells were in G1 arrest
(Fig. 2C).
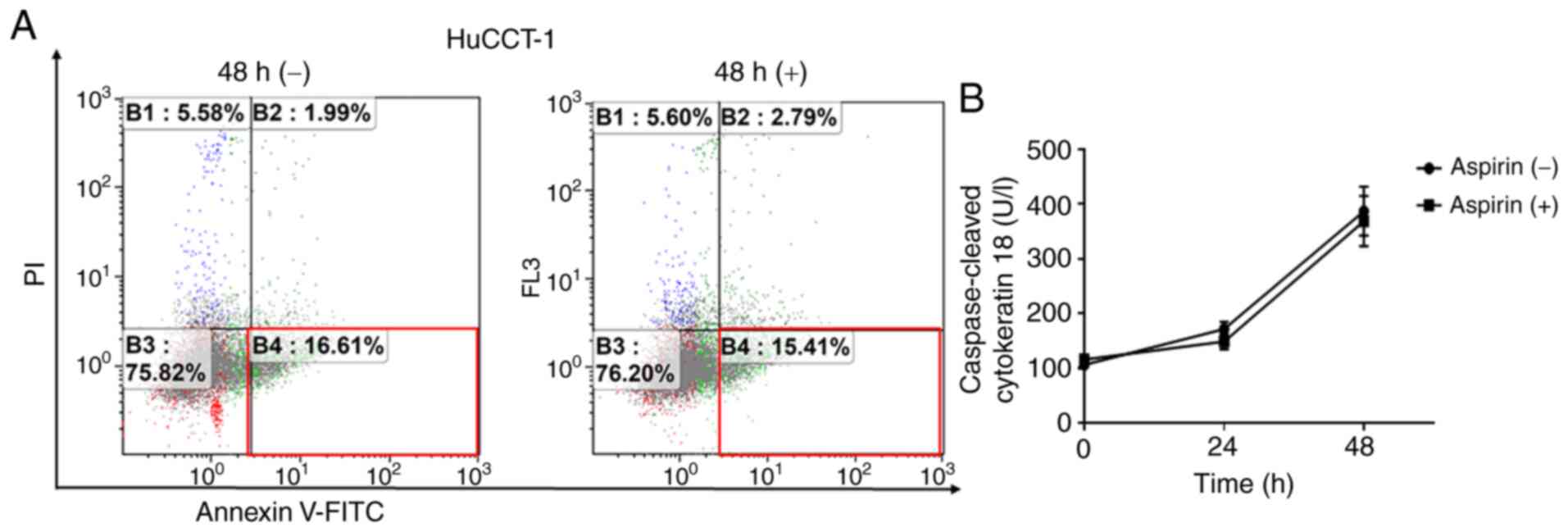
Aspirin does not induce cell apoptosis in
HuCCT-1 cells
To determine whether aspirin induced apoptosis in
HuCCT-1 cells, FCM was used to detect apoptotic cells after aspirin
treatment. The different quadrants represent living cells (lower
left quadrant), early apoptotic cells (lower right quadrant), and
late apoptotic cells (upper right quadrant). The proportion of
early apoptotic cells with aspirin treatment and without treatment
were similar after 48 h (Fig. 3A).
Additionally, there was no obvious difference in cCK-18 levels
between treated and untreated cells after 24- or 48-h treatments
(Fig. 3B).
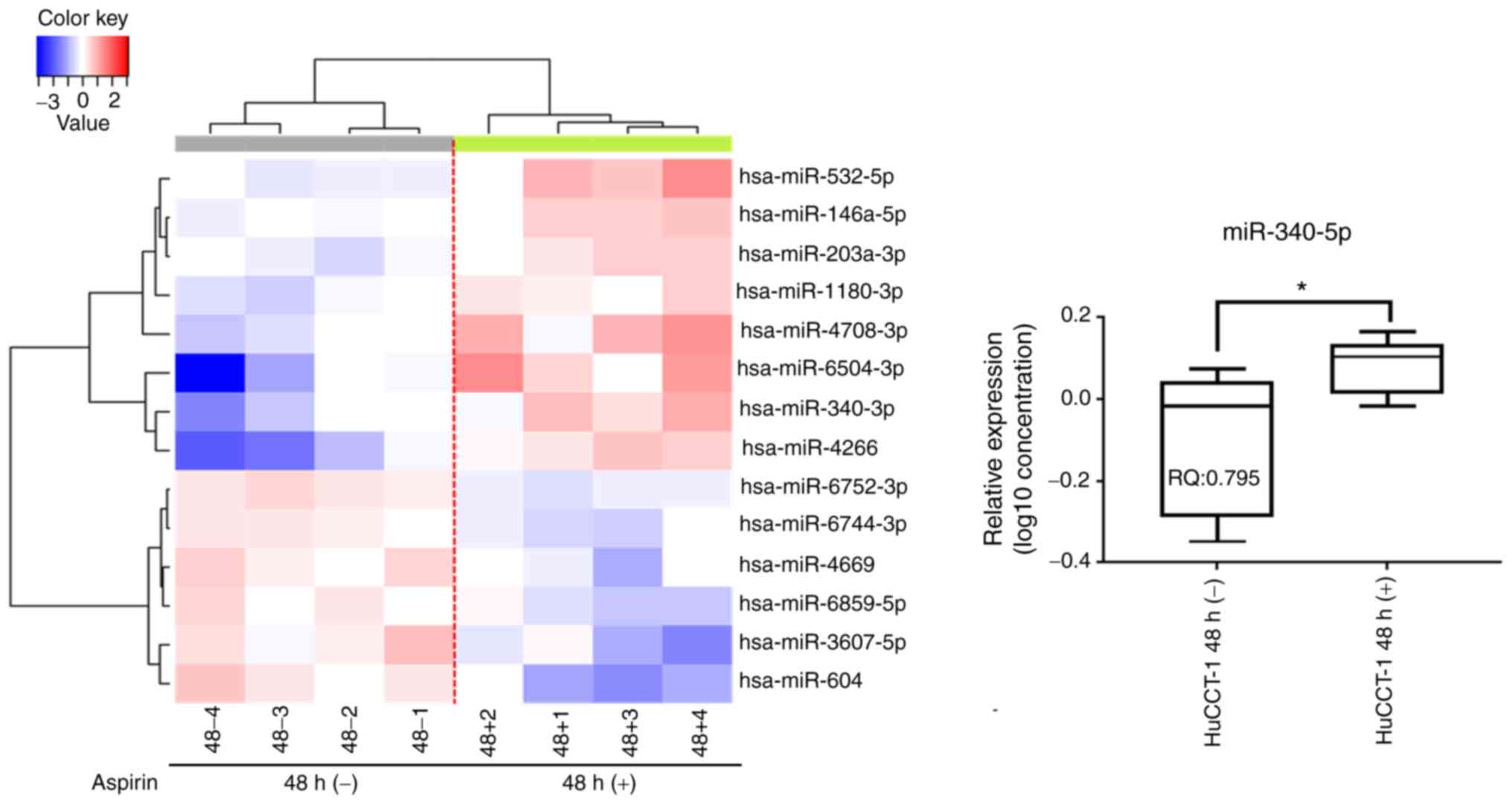
Aspirin affects miRNA expression in
HuCCT-1 cells
A customized microarray platform was used to analyze
the expression of 2,555 miRNAs in aspirin-treated and control
HuCCT-1 cells. Treatment with 2.5 mmol/l aspirin for 48 h induced
the overexpression of eight miRNAs, whereas the expression of six
miRNAs was decreased (Table I;
Fig. 4). Unsupervised hierarchical
clustering analysis was conducted by calculating Pearson's centered
correlation coefficient, and the results indicated that
differentially expressed miRNAs in aspirin-treated HuCCT-1 cells
clustered together. Previous studies have indicated that miR-340-5p
has tumor-suppressive properties (27,28),
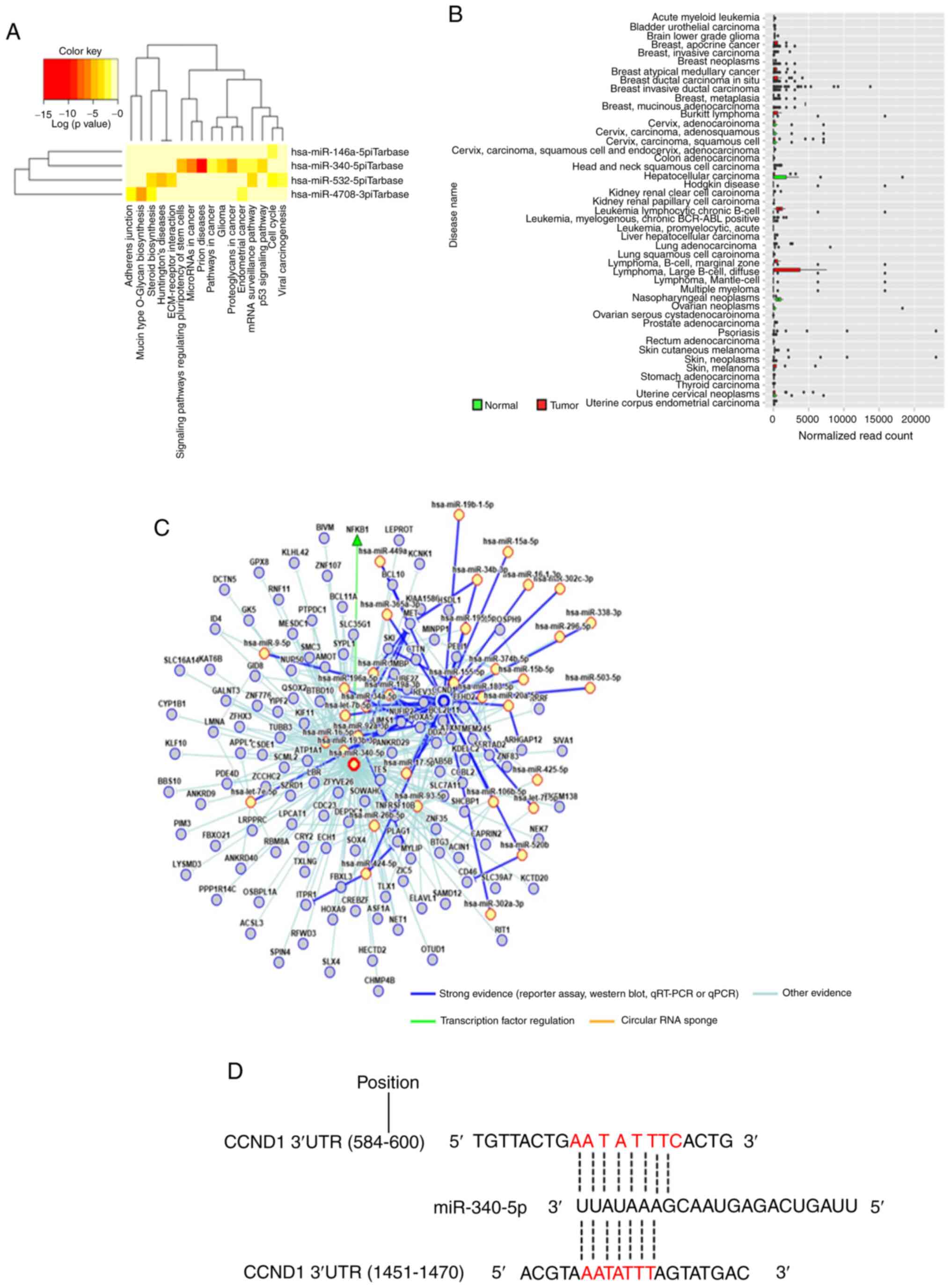
and miR-340-5p exhibited functions in different parts in the KEGG
pathway analyses of upregulated miRNAs, which included 'prion
diseases', 'microRNA in cancer', 'proteoglycans in cancer', and
'signaling pathways regulating pluripotency of stem cells' as well
as others (Fig. 5A). Although the
expression of miR-340-5p was not clearly revealed in CCA, it was
decreased in most types of cancer (Fig. 5B). The miRNA regulatory network and
miRDB database predicted that CCND1 may be a target of miR-340-5p
(Fig. 5C and D). Thus, it was
selected for further study, and RT-qPCR indicated that miR-340-5p
levels were significantly upregulated in the aspirin-treated cells
compared to the untreated cells (Fig.
4). Numerous other studies have revealed the effects of miRNAs
on CCA in recent years (Table II)
(29-47).
 | Table IStatistical results and chromosomal
locations of microRNAs in HuCCT-1 cells. |
Table I
Statistical results and chromosomal
locations of microRNAs in HuCCT-1 cells.
| miRNA | Fold change
(Treated/Untreated) | P-value | Chromosomal
location |
|---|
A, Upregulated
|
|
hsa-miR-6504-3p | 3.092 | 0.021 | 16 |
| hsa-miR-4266 | 2.701 | 0.005 | 2q13 |
|
hsa-miR-4708-3p | 2.158 | 0.028 | 14 |
| hsa-miR-532-5p | 2.055 | 0.039 | X |
| hsa-miR-340-5p | 2.029 | 0.039 | 5 |
|
hsa-miR-203a-3p | 1.552 | 0.018 | 14 |
|
hsa-miR-1180-3p | 1.547 | 0.009 | 17 |
|
hsa-miR-146a-5p | 1.522 | 0.022 | 5 |
|
B, Downregulated
|
|
hsa-miR-6744-3p | 0.654 | 0.003 | 11 |
|
hsa-miR-6752-3p | 0.652 | 0.000 | 11 |
|
hsa-miR-6859-5p | 0.640 | 0.041 | |
| hsa-miR-4669 | 0.636 | 0.030 | 9q34.2 |
|
hsa-miR-3607-5p | 0.526 | 0.045 | |
| hsa-miR-604 | 0.445 | 0.011 | 10p11.23 |
| Table IIPrevious studies examining microRNAs
in cholangiocarcinoma. |
Table II
Previous studies examining microRNAs
in cholangiocarcinoma.
| MicroRNA
(promoter) | References | MicroRNA
(inhibitor) | References |
|---|
| miR-191 | Li et al
(2017) (29) | miR-195 | Li et al
(2017) (30) |
| miR-205-5p | Kitdumrongthum
et al (2018) (31) | miR-410 | Palumbo et
al (2016) (32) |
| miR-21 | Lampis et al
(2018) (33) | miR-203 | Li et al
(2015) (34) |
| miR-181c | Wang et al
(2016) (35) | miR-15a | Utaijaratrasmi
et al (2018) (36) |
| miR-193-3p | Han et al
(2018) (37) | miR-34a | Han et al
(2016) (38) |
| miR-383 | Wan et al
(2018) (27) | miR-433 | Mansini et
al (2018) (28) |
| miR-22 | |
| miR-199a-3p | Li et al
(2017) (39) |
| miR-144 | Yang et al
(2014) (40) |
| miR-590-3p | Zu et al
(2017) (41) |
| miR-101 | Deng et al
(2015) (42) |
| miR-26b-5p | Fan et al
(2018) (43) |
| miR-24 | Ehrlich et
al (2017) (44) |
| miR-122 | Liu et al
(2015) (45) |
| miR-26a | Wang and Lv (2016)
(46) |
| miR-551b-3p | Chang et al
(2019) (47) |
miR-340-5p inhibits the proliferation of
HuCCT-1 cells and decreases the expression levels of cyclin D1
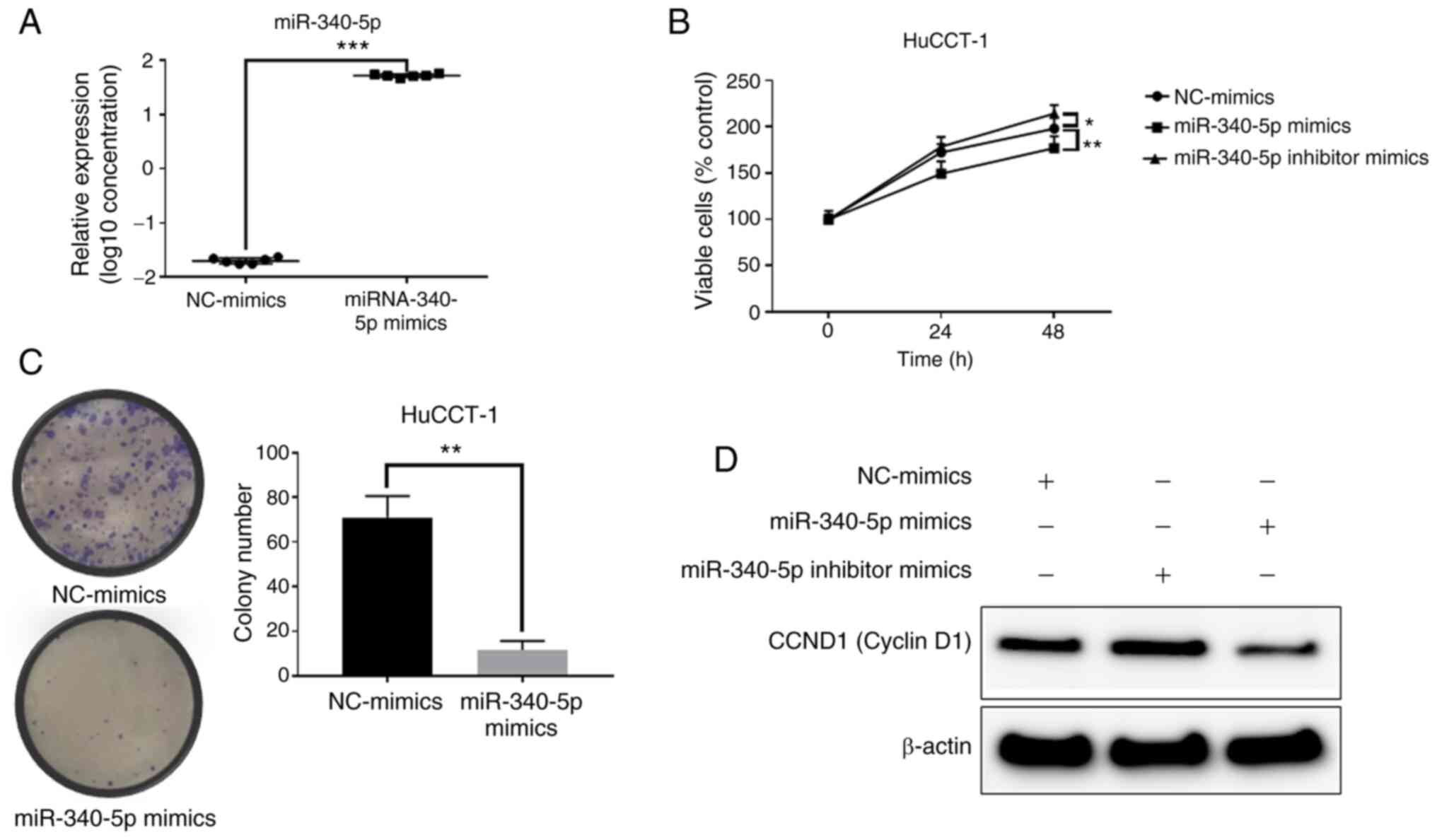
After transfection of miR-340-5p mimics, miR-340-5p
expression was significantly increased in the HuCCT-1 cells
(Fig. 6A). Transfection of
miR-340-5p mimics decreased proliferation in HuCCT-1 cells
(Fig. 6B). Moreover,
overexpression of miR-340-5p decreased the levels of cyclin D1,
whereas inhibition induced increased cyclin D1 levels (Fig. 6D). The colony formation assay
indicated that overexpression of miR-340-5p decreased the cell
proliferation ability of HuCCT-1 cells (Fig. 6C). Therefore, increasing the levels
of miR-340-5p inhibited cyclin D1 expression and decreased the cell
proliferation ability.
Aspirin-nonresponsive TKKK cells express
low levels of miR-340-5p
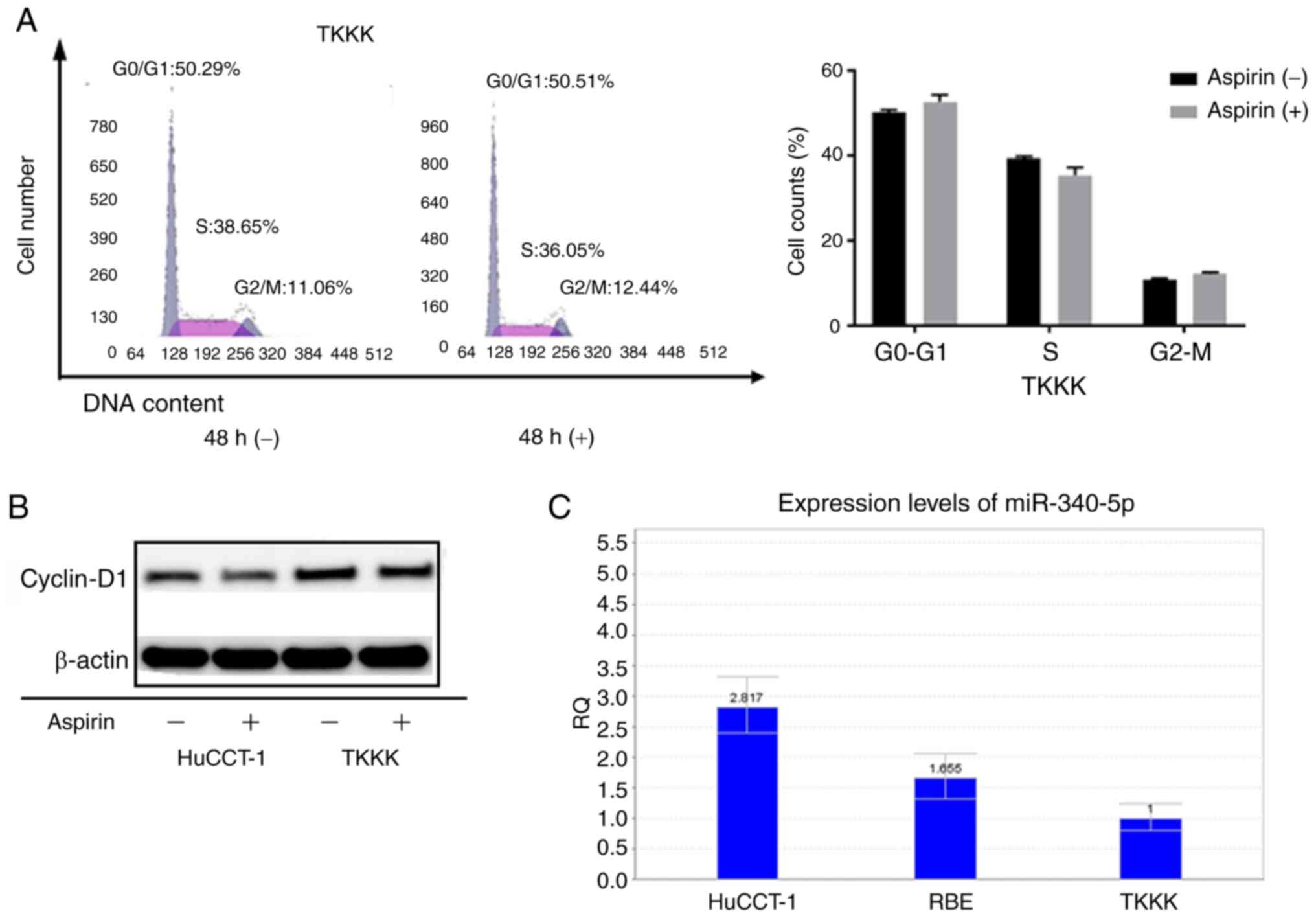
As the CCA cell line TKKK did not exhibit response
to aspirin, the cell cycle progression in TKKK cells as compared to
HuCCT-1 cells was assessed. Forty-eight hours of aspirin treatment
in TKKK cells revealed no obvious difference in the proportion of
cells in each phase of the cell cycle (Fig. 7A). In addition, the levels of
cyclin D1 were not significantly altered (Fig. 7B). Relative quantification of
miR-340-5p was assessed in all cell lines (Fig. 7C). Expression was lowest in TKKK
cells, which may indicate that cell lines with high expression of
miR-340-5p are more sensitive to cell cycle arrest with aspirin
treatment.
Aspirin inhibits tumor proliferation in
vivo
Based on the results obtained from in vitro
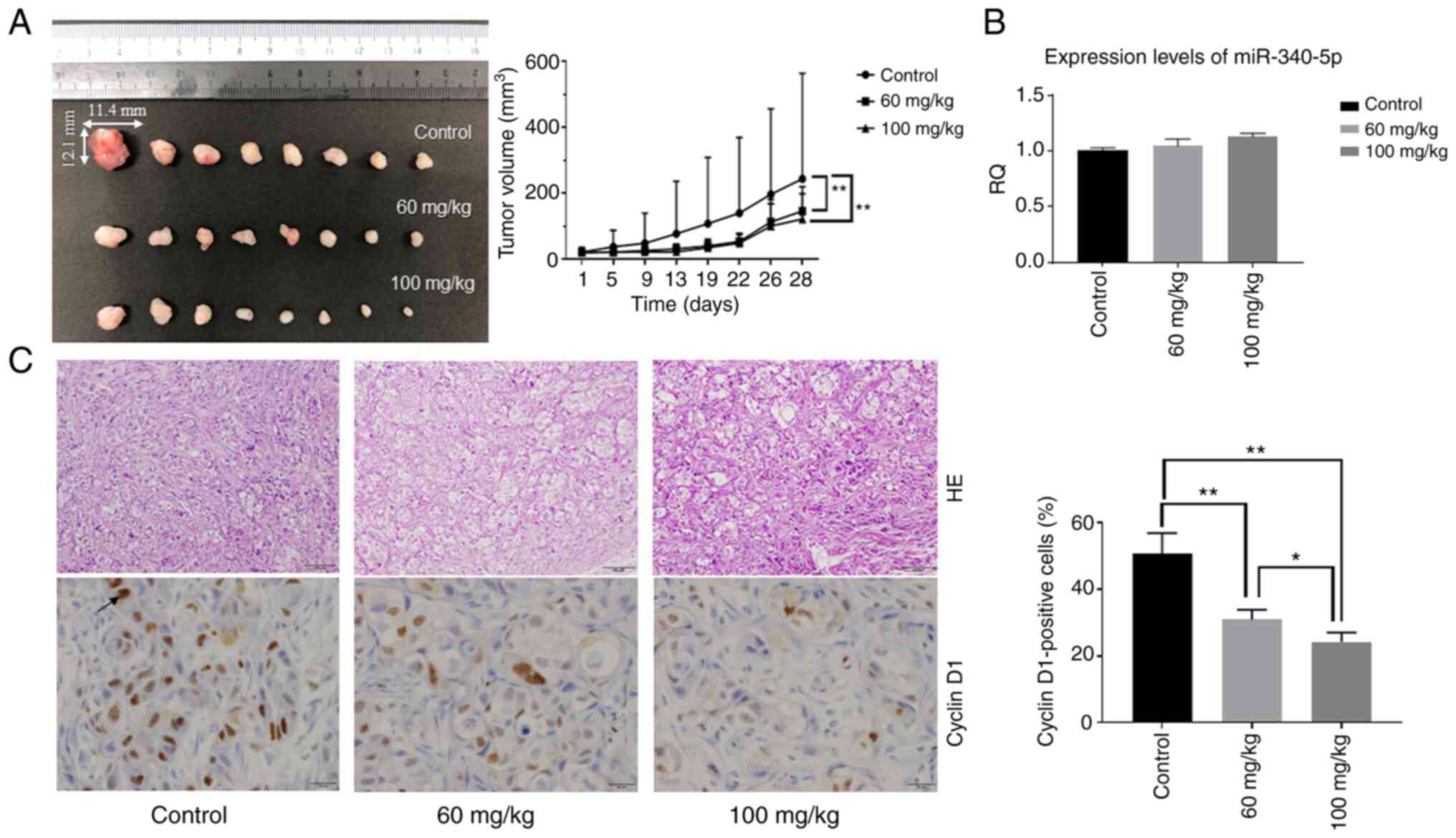
studies, the effect of aspirin in an in vivo model of CCA
was assessed. Nude mice were injected subcutaneously with HuCCT-1
cells followed by intraperitoneal injection of aspirin. The present
results revealed that tumor growth was significantly inhibited in
mice treated with aspirin compared to untreated mice (P<0.05)
(Fig. 8A). No mice succumbed
during the observation period. Expression levels of miR-340-5p in
tumor tissue were not significantly different, although the RQ was
slightly increased in the two aspirin-treated groups (Fig. 8B). H&E-stained images of the
xenografted tumor tissues revealed no significant
histo-pathological differences between the aspirin-treated and
control mice. Immunohistochemical staining of cyclin D1 indicated
that cyclin D1-positive cells in the aspirin-treated groups were
reduced in number compared with the control group (Fig. 8C).
Discussion
CCA is an aggressive cancer with high mortality and
poor prognosis that accounts for approximately 3% of
gastrointestinal malignancies (8).
The 5-year survival rate of CCA is only 10%, and the median
survival is 24 months (48).
Initial analysis revealed that aspirin use was associated with a
reduced iCCA risk in men (HR=0.64, 95% CI=0.42-0.98) (49). Subsequent analysis has revealed
that aspirin use has a significant inverse association with the
development of all three CCA subtypes and leads to an approximately
3-fold reduction in CCA risk (13). This is thought to be due to the
anti-inflammatory effect of aspirin. In the present study, we
further elucidated the mechanism of the effect of aspirin on CCA.
To the best of our knowledge, the present study is the first study
revealing that aspirin inhibits the proliferation of CCA cells
in vivo.
The anti-inflammatory dose of aspirin varies from
0.5-2.5 mM (50); therefore,
relying on data from the anti-proliferation assay, 2.5 mM was
selected as the concentration of aspirin, which does not have
off-target cytotoxicity. It was observed that aspirin inhibited the
proliferation of CCA cells (HuCCT-1 and RBE) and induced cell cycle
arrest (HuCCT-1) at the G0/G1 phase, which
was correlated with a marked decrease in the expression of cyclin
D1. Aspirin also decreased the phosphorylation of Rb. The
expression of cell cycle-related molecules is related to cancer
progression and prognosis (51),
and aspirin has been revealed to inhibit the expression of cyclin
D1 in other types of cancer, such as oral squamous cell carcinoma
(52) and glioblastoma multiforme
(53). To determine whether
aspirin induced apoptosis, HuCCT-1 cells were treated with 2.5 mM
aspirin and analyzed using FCM; the levels of cCK18 were also
measured. However, there was no evidence that aspirin induced
apoptosis in HuCCT-1 cells. These data indicated that aspirin
inhibited CCA cell proliferation mainly through cell cycle arrest.
However, aspirin has also been revealed to induce apoptosis in
numerous types of cancers (52-55);
this discrepancy could be due to differences in the properties of
different types of cancers.
miRNAs are short, noncoding, endogenous,
single-stranded RNA molecules 19-25 nucleotides in length that
regulate target gene expression (56). They are known to regulate the
development and progression of various cancers (57). A miRNA expression array was used to
identify the miRNAs associated with the antitumor effects of
aspirin. miR-340-5p that was significantly upregulated in response
to aspirin treatment in cells, was not significantly different in
tumor tissue, although the relative quantification (RQ) was
slightly increased in the two aspirin-treated groups. Recent
studies have indicated that miR-340-5p inhibited non-small cell
lung cancer cell growth and metastasis by targeting ZNF503
(58) and suppressed osteo-sarcoma
development via targeting STAT3 (59). In the present study, overexpression
of miR-340-5p inhibited proliferation in HuCCT-1 cells. In
addition, overexpression of miR-340-5p decreased the levels of
cyclin D1. The predicted sequence from the miRDB database and
bioinformatics analysis indicated that cyclin D1 may be an
miR-340-5p target, and miR-340-5p overexpression decreased cyclin
D1 levels in cells; immunohistochemical staining indicated that
cyclin D1-positive cells in aspirin-treated mice were reduced
compared with control mice. Therefore, it is theorized that aspirin
works partially through the miR-340-5p/cyclin D1 axis to inhibit
HuCCT-1 cell proliferation.
In the present study, aspirin treatment had no
effect on TKKK cells. To investigate the difference between TKKK
cells and HuCCT-1 cells, FCM was used to analyze changes in the
cell cycle with aspirin treatment. Aspirin did not induce
G0/G1 arrest in TKKK cells, and there was no
difference in the levels of cyclin D1 after 48 h of treatment. In
addition, the relative levels of miR-340-5p were assessed in all
cell lines, and TKKK cells had the lowest expression. Therefore,
aspirin may not suppress proliferation in TKKK cells because it
cannot utilize the miR-340-5p/cyclin D1 axis.
In the present in vivo model, aspirin
inhibited the growth of subcutaneous CCA tumors in athymic nude
mice. In accordance with the in vitro results and previous
studies (23,60), in the in vivo experiment,
the tumor volumes in both treatment groups (low-dosage and
high-dosage) were significantly decreased compared with the tumor
volume in the control group. However, there was no obvious
difference in tumor volume between the low-dosage and high-dosage
groups; long-term treatment may be required to see a difference
based on dosage.
In conclusion, the present study indicated that
aspirin inhibited cell proliferation and tumor growth in some CCA
cell lines by inducing G0/G1 phase cell cycle
arrest, and the underlying mechanism may partially be through the
miR-340-5p/cyclin D1 axis to induce cell cycle arrest.
Funding
No funding was received.
Availability of data and materials
All data supporting the conclusions of the present
study have been documented in this study.
Authors' contributions
TS, AM, HK and TM conceived and designed the
experiments. TS, HI, JG, NN, SL, MN, HY, TN, KO, YS and KF analyzed
and interpreted the data. TS performed the experiments. TS wrote,
reviewed and edited the manuscript. TM reviewed and edited the
manuscript for important intellectual content. All authors
reviewed, read and approved the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved (approval
no. 18674) by the Institutional Review Board of the Department of
Laboratory Animal Science of Kagawa University (Kawaga, Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
We thank Ms. Kayo Hirose, Ms. Keiko Fujikawa, Ms.
Miwako Watanabe, Ms. Megumi Okamura, and Ms. Fuyuko Kokado at the
Department of Gastroenterology and Neurology of Kagawa University
(Kagawa, Japan) for their assistance.
Abbreviations:
|
CCA
|
cholangiocarcinoma
|
|
NSAID
|
non-steroidal anti-inflammatory
drug
|
|
FCM
|
flow cytometer
|
|
AMPK
|
AMP-activated protein kinase
|
|
miRNA
|
microRNA
|
|
NF-κB
|
nuclear factor κ-light-chain-enhancer
of activated B cells
|
|
JNK
|
c-Jun-N-terminal kinase
|
|
GSK-3
|
glycogen synthase kinase-3
|
|
mTOR
|
mammalian target of rapamycin
|
|
STAT3
|
signal transducers and activators of
transcription 3
|
|
p-Bcl-2
|
phosphorylated B-cell lymphoma-2
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
Cdk
|
cyclin-dependent kinase
|
References
|
1
|
Patel T: Increasing incidence and
mortality of primary intrahepatic cholangiocarcinoma in the United
States. Hepatology. 33:1353–1357. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xiao J, Zhu J, Liu Z, Wan R, Li Y and Xiao
W: Role of surgical treatment for hepatolithiasis-associated
intrahepatic cholangio-carcinoma: A retrospective study in a single
institution. J Cancer Res Ther. 13:756–760. 2017. View Article : Google Scholar
|
|
3
|
Arbelaiz A, Azkargorta M, Krawczyk M,
Santos-Laso A, Lapitz A, Perugorria MJ, Erice O, Gonzalez E,
Jimenez-Agüero R, Lacasta A, et al: Serum extracellular vesicles
contain protein biomarkers for primary sclerosing cholangitis and
cholangiocar-cinoma. Hepatology. 66:1125–1143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim TS, Pak JH, Kim JB and Bahk YY:
Clonorchis sinensis, an oriental liver fluke, as a human biological
agent of cholangiocar-cinoma: A brief review. BMB Rep. 49:590–597.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang MH, Lee YJ and Kim H: Intrahepatic
cholangiocarcinoma arising in Caroli's disease. Clin Mol Hepatol.
20:402–405. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shi Y, Jiang Z, Yang Y, Zheng P, Wei H,
Lin Y, Lv G and Yang Q: Clonorchis sinensis infection and
co-infection with the hepatitis B virus are important factors
associated with cholan-giocarcinoma and hepatocellular carcinoma.
Parasitol Res. 116:2645–2649. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Blechacz B: Cholangiocarcinoma: Current
knowledge and new developments. Gut Liver. 11:13–26. 2017.
View Article : Google Scholar :
|
|
8
|
Razumilava N and Gores GJ:
Cholangiocarcinoma. Lancet. 383:2168–2179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang H, Yang T, Wu M and Shen F:
Intrahepatic cholangio-carcinoma: Epidemiology, risk factors,
diagnosis and surgical management. Cancer Lett. 379:198–205. 2016.
View Article : Google Scholar
|
|
10
|
Ng K, Meyerhardt JA, Chan AT, Sato K, Chan
JA, Niedzwiecki D, Saltz LB, Mayer RJ, Benson AB III, Schaefer PL,
et al: Aspirin and COX-2 inhibitor use in patients with stage III
colon cancer. J Natl Cancer Inst. 107:3452014.PubMed/NCBI
|
|
11
|
Barron TI, Flahavan EM, Sharp L, Bennett K
and Visvanathan K: Recent prediagnostic aspirin use, lymph node
involvement, and 5-year mortality in women with stage I-III breast
cancer: A nationwide population-based cohort study. Cancer Res.
74:4065–4077. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Simon TG, Ma Y, Ludvigsson JF, Chong DQ,
Giovannucci EL, Fuchs CS, Meyerhardt JA, Corey KE, Chung RT, Zhang
X and Chan AT: Association between aspirin use and risk of
hepatocellular carcinoma. JAMA Oncol. 4:1683–1690. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Choi J, Ghoz HM, Peeraphatdit T, Baichoo
E, Addissie BD, Harmsen WS, Therneau TM, Olson JE, Chaiteerakij R
and Roberts LR: Aspirin use and the risk of cholangiocarcinoma.
Hepatology. 64:785–796. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guenzle J, Garrelfs NWC, Goeldner JM and
Weyerbrock A: Cyclooxygenase (COX) inhibition by acetyl salicylic
acid (ASA) enhances antitumor effects of nitric oxide in
glioblastoma in vitro. Mol Neurobiol. 56:6046–6055. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Henry WS, Laszewski T, Tsang T, Beca F,
Beck AH, McAllister SS and Toker A: Aspirin suppresses growth in
PI3K-mutant breast cancer by activating AMPK and inhibiting mTORC1
signaling. Cancer Res. 77:790–801. 2017. View Article : Google Scholar :
|
|
16
|
Liu YX, Feng JY, Sun MM, Liu BW, Yang G,
Bu YN, Zhao M, Wang TJ, Zhang WY, Yuan HF and Zhang XD: Aspirin
inhibits the proliferation of hepatoma cells through controlling
GLUT1-mediated glucose metabolism. Acta Pharmacol Sin. 40:122–132.
2019. View Article : Google Scholar :
|
|
17
|
Huang Z, Fang W, Liu W, Wang L, Liu B and
Liu S and Liu S: Aspirin induces Beclin-1-dependent autophagy of
human hepatocellular carcinoma cell. Eur J Pharmacol. 823:58–64.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Raza H, John A and Benedict S:
Acetylsalicylic acid-induced oxidative stress, cell cycle arrest,
apoptosis and mitochondrial dysfunction in human hepatoma HepG2
cells. Eur J Pharmacol. 668:15–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pavlovic N, Rani B, Gerwins P and
Heindryckx F: Platelets as key factors in hepatocellular carcinoma.
Cancers (Basel). 11:10222019. View Article : Google Scholar
|
|
20
|
Malehmir M, Pfister D, Gallage S,
Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M,
Surewaard BGJ, Rath D, et al: Platelet GPIbα is a mediator and
potential interventional target for NASH and subsequent liver
cancer. Nat Med. 25:641–655. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar :
|
|
23
|
Vlachos IS, Zagganas K, Paraskevopoulou
MD, Georgakilas G, Karagkouni D, Vergoulis T, Dalamagas T and
Hatzigeorgiou AG: DIANA-miRPath v3.0: Deciphering microRNA function
with experimental support. Nucleic Acids Res. 43:W460–W466. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cho S, Jang I, Jun Y, Yoon S, Ko M, Kwon
Y, Choi I, Chang H, Ryu D, Lee B, et al: MiRGator v3.0: A microRNA
portal for deep sequencing, expression profiling and mRNA
targeting. Nucleic Acids Res. 41(Database issue): D252–D257. 2013.
View Article : Google Scholar :
|
|
25
|
Huang HY, Lin YC, Li J, Huang KY, Shrestha
S, Hong HC, Tang Y, Chen YG, Jin CN, Yu Y, et al: miRTarBase 2020:
Updates to the experimentally validated microRNA-target
inter-action database. Nucleic Acids Res. 48:D148–D154. 2020.
|
|
26
|
Pietenpol JA and Stewart ZA: Cell cycle
checkpoint signaling: Cell cycle arrest versus apoptosis.
Toxicology. 181-182:475–481. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wan P, Chi X, Du Q, Luo J, Cui X, Dong K,
Bing Y, Heres C and Geller DA: miR-383 promotes cholangiocarcinoma
cell proliferation, migration, and invasion through targeting IRF1.
J Cell Biochem. 119:9720–9729. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mansini AP, Lorenzo Pisarello MJ, Thelen
KM, Cruz-Reyes M, Peixoto E, Jin S, Howard BN, Trussoni CE, Gajdos
GB, LaRusso NF, et al: MicroRNA (miR)-433 and miR-22 dysregulations
induce histone-deacetylase-6 overexpression and ciliary loss in
cholangiocarcinoma. Hepatology. 68:561–573. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li H, Zhou ZQ, Yang ZR, Tong DN, Guan J,
Shi BJ, Nie J, Ding XT, Li B, Zhou GW and Zhang ZY: MicroRNA-191
acts as a tumor promoter by modulating the TET1-p53 pathway in
intrahepatic cholangiocarcinoma. Hepatology. 66:136–151. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li L, Piontek K, Ishida M, Fausther M,
Dranoff JA, Fu R, Mezey E, Gould SJ, Fordjour FK, Meltzer SJ, et
al: Extracellular vesicles carry microRNA-195 to intrahepatic
cholangiocarcinoma and improve survival in a rat model. Hepatology.
65:501–514. 2017. View Article : Google Scholar
|
|
31
|
Kitdumrongthum S, Metheetrairut C,
Charoensawan V, Ounjai P, Janpipatkul K, Panvongsa W,
Weerachayaphorn J, Piyachaturawat P and Chairoungdua A:
Dysregulated microRNA expression profiles in cholangiocarcinoma
cell-derived exosomes. Life Sci. 210:65–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Palumbo T, Poultsides GA, Kouraklis G,
Liakakos T, Drakaki A, Peros G, Hatziapostolou M and Iliopoulos D:
A functional microRNA library screen reveals miR-410 as a novel
anti-apoptotic regulator of cholangiocarcinoma. BMC Cancer.
16:3532016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lampis A, Carotenuto P, Vlachogiannis G,
Cascione L, Hedayat S, Burke R, Clarke P, Bosma E, Simbolo M,
Scarpa A, et al: MIR21 drives resistance to heat shock protein 90
inhibition in cholangio-carcinoma. Gastroenterology.
154:1066–1079.e5. 2018. View Article : Google Scholar
|
|
34
|
Li J, Gao B, Huang Z, Duan T, Li D, Zhang
S, Zhao Y, Liu L, Wang Q, Chen Z and Cheng K: Prognostic
significance of microRNA-203 in cholangiocarcinoma. Int J Clin Exp
Pathol. 8:9512–9516. 2015.PubMed/NCBI
|
|
35
|
Wang J, Xie C, Pan S, Liang Y, Han J, Lan
Y, Sun J, Li K, Sun B, Yang G, et al: N-myc downstream-regulated
gene 2 inhibits human cholangiocarcinoma progression and is
regulated by leukemia inhibitory factor/MicroRNA-181c negative
feedback pathway. Hepatology. 64:1606–1622. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Utaijaratrasmi P, Vaeteewoottacharn K,
Tsunematsu T, Jamjantra P, Wongkham S, Pairojkul C, Khuntikeo N,
Ishimaru N, Sirivatanauksorn Y, Pongpaibul A, et al: The
microRNA-15a-PAI-2 axis in cholangiocarcinoma-associated
fibroblasts promotes migration of cancer cells. Mol Cancer.
17:102018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han YL, Yin JJ and Cong JJ: Downregulation
of microRNA-193-3p inhibits the progression of intrahepatic
cholangiocarcinoma cells by upregulating TGFBR3. Exp Ther Med.
15:4508–4514. 2018.PubMed/NCBI
|
|
38
|
Han Y, Meng F, Venter J, Wu N, Wan Y,
Standeford H, Francis H, Meininger C, Greene J Jr, Trzeciakowski
JP, et al: miR-34a-dependent overexpression of Per1 decreases
cholangiocarcinoma growth. J Hepatol. 64:1295–1304. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li Q, Xia X, Ji J, Ma J, Tao L, Mo L and
Chen W: MiR-199a-3p enhances cisplatin sensitivity of
cholangiocarcinoma cells by inhibiting mTOR signaling pathway and
expression of MDR1. Oncotarget. 8:33621–33630. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang R, Chen Y, Tang C, Li H, Wang B, Yan
Q, Hu J and Zou S: MicroRNA-144 suppresses cholangiocarcinoma cell
proliferation and invasion through targeting platelet activating
factor acetylhydrolase isoform 1b. BMC Cancer. 14:9172014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zu C, Liu S, Cao W, Liu Z, Qiang H, Li Y,
Cheng C, Ji L and Li J and Li J: MiR-590-3p suppresses
epithelial-mesenchymal tran-sition in intrahepatic
cholangiocarcinoma by inhibiting SIP1 expression. Oncotarget.
8:34698–34708. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Deng G, Teng Y, Huang F, Nie W, Zhu L,
Huang W and Xu H: MicroRNA-101 inhibits the migration and invasion
of intrahe-patic cholangiocarcinoma cells via direct suppression of
vascular endothelial growth factor-C. Mol Med Rep. 12:7079–7085.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fan F, Lu J, Yu W, Zhang Y, Xu S, Pang L
and Zhu B: MicroRNA-26b-5p regulates cell proliferation, invasion
and metastasis in human intrahepatic cholangiocarcinoma by
targeting S100A7. Oncol Lett. 15:386–392. 2018.PubMed/NCBI
|
|
44
|
Ehrlich L, Hall C, Venter J, Dostal D,
Bernuzzi F, Invernizzi P, Meng F, Trzeciakowski JP, Zhou T,
Standeford H, et al: miR-24 inhibition increases menin expression
and decreases cholangio-carcinoma proliferation. Am J Pathol.
187:570–580. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu N, Jiang F, He TL, Zhang JK, Zhao J,
Wang C, Jiang GX, Cao LP, Kang PC, Zhong XY, et al: The roles of
MicroRNA-122 overexpression in inhibiting proliferation and
invasion and stimulating apoptosis of human cholangiocarcinoma
cells. miR-24 inhibition increases menin expression and decreases
cholangio-carcinoma proliferation. Sci Rep. 5:165662015. View Article : Google Scholar
|
|
46
|
Wang P and Lv L: miR-26a induced the
suppression of tumor growth of cholangiocarcinoma via KRT19
approach. Oncotarget. 7:81367–81376. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chang W, Wang Y, Li W, Shi L and Geng Z:
MicroRNA-551b-3p inhibits tumour growth of human cholangiocarcinoma
by targeting Cyclin D1. J Cell Mol Med. 23:4945–4954. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rizvi S and Gores GJ: Pathogenesis,
diagnosis, and management of cholangiocarcinoma. Gastroenterology.
145:1215–1229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Petrick JL, Sahasrabuddhe VV, Chan AT,
Alavanja MC, Beane-Freeman LE, Buring JE, Chen J, Chong DQ,
Freedman ND, Fuchs CS, et al: NSAID use and risk of hepatocellular
carcinoma and intrahepatic cholangiocarcinoma: The liver cancer
pooling project. Cancer Prev Res (Phila). 8:1156–1162. 2015.
View Article : Google Scholar
|
|
50
|
Dovizio M, Bruno A, Tacconelli S and
Patrignani P: Mode of action of aspirin as a chemopreventive agent.
Recent Results Cancer Res. 191:39–65. 2013. View Article : Google Scholar
|
|
51
|
Masaki T, Shiratori Y, Rengifo W, Igarashi
K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H,
Watanabe S, et al: Cyclins and cyclin-dependent kinases:
Comparative study of hepatocellular carcinoma versus cirrhosis.
Hepatology. 37:534–543. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang X, Feng H, Li Z, Guo J and Li M:
Aspirin is involved in the cell cycle arrest, apoptosis, cell
migration, and invasion of oral squamous cell carcinoma. Int J Mol
Sci. 19:20292018. View Article : Google Scholar :
|
|
53
|
Pozzoli G, Marei HE, Althani A, Boninsegna
A, Casalbore P, Marlier LNJL, Lanzilli G, Zonfrillo M, Petrucci G,
Rocca B, et al: Aspirin inhibits cancer stem cells properties and
growth of glio-blastoma multiforme through Rb1 pathway modulation.
J Cell Physiol. Jan 30–2019.Epub ahead of print. View Article : Google Scholar
|
|
54
|
Hossain MA, Kim DH, Jang JY, Kang YJ, Yoon
JH, Moon JO, Chung HY, Kim GY, Choi YH, Copple BL and Kim ND:
Aspirin induces apoptosis in vitro and inhibits tumor growth of
human hepatocellular carcinoma cells in a nude mouse xenograft
model. Int J Oncol. 40:1298–1304. 2012. View Article : Google Scholar
|
|
55
|
Choi BH, Chakraborty G, Baek K and Yoon
HS: Aspirin-induced Bcl-2 translocation and its phosphorylation in
the nucleus trigger apoptosis in breast cancer cells. Exp Mol Med.
45:e472013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lu TX and Rothenberg ME: MicroRNA. J
Allergy Clin Immunol. 141:1202–1207. 2018. View Article : Google Scholar :
|
|
57
|
Paliouras AR, Monteverde T and Garofalo M:
Oncogene-induced regulation of microRNA expression: Implications
for cancer initiation, progression and therapy. Cancer Lett.
421:152–160. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lu G and Zhang Y: MicroRNA-340-5p
suppresses non-small cell lung cancer cell growth and metastasis by
targeting ZNF503. Cell Mol Biol Lett. 24:342019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rongxin S, Pengfei L, Li S, Xiaochen J and
Yihe H: MicroRNA-340-5p suppresses osteosarcoma development by
down-regulating the Wnt/β-catenin signaling pathway via targeting
the STAT3 gene. Eur Rev Med Pharmacol Sci. 23:982–991.
2019.PubMed/NCBI
|
|
60
|
Yue W, Zheng X, Lin Y, Yang CS, Xu Q,
Carpizo D, Huang H, DiPaola RS and Tan XL: Metformin combined with
aspirin significantly inhibit pancreatic cancer cell growth in
vitro and in vivo by suppressing anti-apoptotic proteins Mcl-1 and
Bcl-2. Oncotarget. 6:21208–21224. 2015. View Article : Google Scholar : PubMed/NCBI
|