Introduction
Triple-negative breast cancer (TNBC) is one of the
most aggressive subtypes of breast cancer, due to its high
invasiveness, high recurrence and poor prognosis (1). At present, there is a lack of
effective therapeutic targets and drugs for the clinical treatment
of TNBC. Different proteins are expressed in various types of
breast cancer cells, including epidermal growth factor receptor
(EGFR), which is overexpressed in MDA-MB-231 cells, but lowly
expressed in MCF-7 cells (2).
Therefore, identifying key molecular targets and developing
specific targeted therapeutic drugs is important for the
development of novel therapeutic strategies for TNBC.
EGFR, a member of the EGFR family, is involved in
tumor cell proliferation, angiogenesis, tumor invasion and
metastasis (3). EGFR is
abnormally expressed in several solid tumors, including TNBC
(4). Among all TNBC cases, 50-75%
are associated with EGFR activation or overexpression (5). Due to the multidimensional role of
EGFR in the progression of cancer, targeting EGFR has emerged as a
potential anticancer therapeutic strategy.
Rac1 is involved in the regulation of various
cellular processes, including adhesion, migration, proliferation,
transcription, vesicle formation and cell apoptosis (6). Increasing evidence has demonstrated
that Rac1 is abnormally expressed and aberrant Rho family signaling
contributes to angiogenesis, invasion and metastasis. Patients with
high Rac1 expression levels were more susceptible to early tumor
recurrence and displayed poor prognosis (7). Our previous study demonstrated that
RP-4, a novel anthraquinone derivative, enhanced the sensitivity of
nasopharyngeal carcinoma cells to radiotherapy by targeting Rac1
(8). Rhein can inhibit the
production of matrix metallopeptpidase (MMP) by regulating the
Rac1/reactive oxygen species/MAPK/activator protein 1 signaling
pathway in human ovarian carcinoma cells (9). Several quinazoline derivatives were
used as first- and second-generation EGFR tyrosine kinase
inhibitors, including gefitinib and afatinib (10). Inspired by anthraquinone and
quinazoline derivatives, a series of novel
anthraqui-none-quinazoline hybrids, which were expected to be more
potent antitumor agents by inhibiting the activity of EGFR and
Rac1, were designed and synthesized in our previous study (11). Among these hybrids, hybrid 7B
displayed antiproliferative activity on several tumor cells,
especially on human MDA-MB-231 TNBC cells. However, the effect of
hybrid 7B on tumor metastasis and invasion is not completely
understood. The present study investigated whether hybrid 7B
suppressed breast cancer cell invasion and epithelial-mesenchymal
transition (EMT) by downregulating the expression of EGFR and Rac1.
The results indicated that targeting both EGFR and Rac1 might serve
as a novel and effective therapeutic strategy for TNBC.
Materials and methods
Drugs and reagents
4,5-Bis(benzyloxy)-9,10-dioxo-N-(4-(3-methy-lphenylamino)quinazolin-6-yl)anthracene-2-carboxamide
[hybrid 7B; purity, >98%] had previously been synthesized by our
team (11). Hybrid 7B was
dissolved in DMSO to make a 5,000 µM stock solution, which
was diluted according to the experimental requirements. Rhein was
purchased from Nanjing Langze Pharmaceutical Technology Co., Ltd.
Gefitinib is purchased from Qilu Pharmaceutical Co., Ltd. DMEM and
FBS were purchased from Gibco (Thermo Fisher Scientific, Inc.). EGF
was purchased from R&D Systems China Co., Ltd. MTT was
purchased from Merck KGaA. Crystal violet dye solution was obtained
from Beyotime Institute of Biotechnology. Transwell chambers,
Matrigel and the Annexin V-allophycocyanin (APC)/7-aminoactinomycin
D (7-AAD) kit were purchased from BD Biosciences.
Cell culture
MDA-MB-231, MCF-7 and MCF-10A cells were purchased
from China Center for Type Culture Collection. Cells were cultured
in DMEM supplemented with 10% FBS at 37°C with 5%
CO2.
Cell viability
Cell viability was determined by performing MTT
assays. Cells were inoculated (5x103 cells/well) into a
96-well plate for 24 h. Cells were incubated with or without
different concentrations of hybrid 7B [0 (control), 0.5, 1, 2, 4, 8
or 16 µM], rhein [0 (control), 16, 32, 64, 128 or 256
µM] and gefitinib [0 (control), 4, 8, 16, 32 or 64
µM) at 37°C for 48 h. Subsequently, cells were incubated
with 20 µl MTT (5 mg/ml) for 4 h. The cell medium was
discarded and 100 µl DMSO was added to each well for 15 min
with gentle agitation. Absorbance was measured at a wavelength of
490 nm using a microplate reader (BioTek Instruments, Inc.).
Relative cell viability was calculated according to the following
formula: Cell viability (%) = (mean absorbance of the test
wells/mean absorbance of the control wells) x100. The
IC30, IC50 and IC70 values were
calculated as the concentrations of hybrid 7B that inhibited cell
viability by 30, 50 and 70%, respectively.
Apoptosis detection
Cell apoptosis was detected using an Annexin
V-APC/7-AAD kit. Cells were seeded (3x105 cells/well)
into a 6-well plate. MDA-MB-231 cells were treated with different
concentrations of hybrid 7B (0, 1, 2 or 4 µM) for 48 h.
MCF-7 cells were treated with hybrid 7B (0, 4, 8 or 16 µM)
for 48 h. Following washing twice with PBS, cells were digested
with trypsin and centrifuged at room temperature for 5 min at 132 ×
g. Cells were collected and the supernatant was discarded. Cells
were resuspended in 100 µl binding buffer. Subsequently,
cells were incubated with 5 µl 7-AAD and 5 µl Annexin
V-APC in the dark at room temperature for 30 min. Apoptotic cells
(early and late apoptosis) were analyzed using a FACSCalibur flow
cytometer (Becton, Dickinson and Company) and Cell Quest Pro
software (version 6.0; Becton, Dickinson and Company).
Cell cycle
Cells were seeded (3×105 cells/well) into
a 6-well plate. MDA-MB-231 cells were treated with different
concentrations of hybrid 7B (0, 1, 2 or 4 µM) for 48 h.
MCF-7 cells were treated with different concentrations of hybrid 7B
(0, 4, 8 or 16 µM) for 48 h. Following washing with PBS,
cells were digested with trypsin and centrifuged at room
temperature for 5 min at 132 × g. Cells were washed twice with
precooled PBS and fixed with 70% precooled ethanol at 4°C
overnight. Subsequently, cells were centrifuged (132 × g for 5 min
at 25°C), the supernatant was discarded and 5 ml PBS was added to
cells, gently mixed and incubated for 15 min. Following
centrifugation (132 × g for 5 min at 25°C) and discarding the
supernatant, 1 ml DNA staining solution was added [Hangzhou
Multisciences (Lianke) Biotech Co., Ltd.]. Cells were oscillated
for 10 sec and incubated for 30 min at room temperature in the
dark. Cell cycle distribution was analyzed using a FACSCalibur flow
cytometer (Becton, Dickinson and Company) and ModFit LT software
(version 3.0; Verity Software House, Inc.).
Transmission electron microscopy
(TEM)
MDA-MB-231 and MCF-F cells (2.5x105
cells/well; 6-well plate) were treated with 2 or 8 µM hybrid
7B for 48 h, respectively. Cells were harvested by centrifugation
(132 × g at 5 min at 25°C) and fixed with 3% glutaraldehyde (1 ml)
at 4°C for 2 h. Subsequently, 1% osmium tetroxide buffer solution
was added to fix the sample at 4°C for 1 h. Cells were dehydrated
using an ethanol gradient (30, 50, 70, 90 and 100%; 10 min per
step; 25°C). Subsequently, cells were washed three times with 100%
acetone (10 min per wash; 25°C). Acetone and resin were permeated
for 2 h at 25°C at ratios of 3:1, 1:1 and 1:3. The last ratio was
permeated overnight, and then infiltrated with complete resin for
24 h at 25°C. Samples were embedded in resin and placed into the
oven (40°C for 15 h, 48°C for 13 h, 60°C for 24 h). Ultrathin
sections were stained with uranyl acetate for 2 h at 25°C, followed
by lead citrate for 15 min at 25°C. Cell ultrastructure was
observed and photographed by TEM using a H7650 transmission
electron microscope (Hitachi, Ltd.).
Mitochondrial membrane potential
detection
MDA-MB-231 cells (2.5x105 cells/well;
6-well plate) treated with hybrid 7B (0, 1, 2 or 4 µM) for
48 h. MCF-7 cells (2.5x105 cells/well; 6-well plate)
were treated with hybrid 7B (4, 8 or 16 µM) for 48 h.
Subsequently, cells were digested, collected, centrifuged (132 × g
for 5 min at 25°C) and resuspended in 0.5 ml medium. Cells were
incubated with 0.5 ml JC-1 working solution at 37°C for 20 min.
Subsequently, centrifugation was performed at 600 × g for 3-4 min
at 4°C to precipitate cells. The supernatant was discarded and
cells were washed twice with ice-cold JC-1 staining buffer.
Following centrifugation (132 × g for 5 min at 25°C), cells were
resuspended with JC-1 staining buffer, passed through a 400-mesh
nylon screen and detected via flow cytometry using a FACSCalibur
flow cytometer (Becton, Dickinson and Company) and CellQuest Pro
software (version 6.0; Becton, Dickinson and Company).
Cell invasion assay
Cell invasion was evaluated using a 24-well
Transwell chamber (diameter, 6.5 mm; pore size, 8 µm;
Corning, Inc.). Each chamber was coated with Matrigel (diluted 1:8
in serum-free medium) at 37°C for 3 h. To compare the invasive
ability between MCF-7 and MDA-MB-231 cells, MDA-MB-231 or MCF-7
cells were inoculated (4x104 cells/well) into the upper
chamber. To assess the effect of hybrid 7B on cell invasion,
MDA-MB-231 cells were inoculated in serum-free medium with
different concentrations of hybrid 7B (0, 0.25, 0.5 or 1 µM)
in the upper chamber. In the lower chamber, 500 µl medium
supplemented with 20% FBS was plated in the lower chamber.
Following incubation for 48 h at 37°C, the upper chamber was
removed and cells on the surface of the membrane were gently wiped
off using cotton swabs, The membrane was fixed with 4%
paraformaldehyde at 25°C for 15 min, then stained with 0.2% crystal
purple at 25°C for 2 min. Invading cells were visualized using a
light microscope and analyzed using ImageJ software (version 1.8.0;
National Institutes of Health).
Wound-healing assay
MDA-MB-231 cells were incubated in a 6-well plate.
Once a fusion monolayer had formed, a plastic pipette tip was used
to wound the monolayer. Cell medium was discarded and replaced with
medium containing 2% FBS and different concentrations of hybrid 7B
(0.25, 0.5 or 1 µM) for 48 h. The wounds were observed using
an light microscope in five randomly selected fields of view. The
following formula was used to calculate wound closure: Wound
closure (%) = [(the width of wound at 0 h-the width of wound at 48
h)/the width of wound at 0 h] x100.
Western blotting
Following treatment with different concentrations of
hybrid 7B (MDA-MB-231 cells, 1 or 2 µM; MCF-7 cells, 4 or 8
µM) or gefitinib (MDA-MB-231, 2 µM; MCF-7, 8
µM) for 48 h at 37°C, total protein was isolated from cells
using cold RIPA buffer containing 1% PMSF (Beyotime Institute of
Biotechnology). The lysate was collected by centrifugation at
12,000 × g for 10 min at 4°C. Protein concentrations were
determined by performing a bicinchoninic acid assay. Proteins (30
µg) were separated by 10% SDS-PAGE and transferred onto PVDF
membranes (Merck KGaA) following electrotransfer at 100 V constant
pressure for 90 min. Following blocking with 5% skimmed milk for 1
h at 25°C, the membranes were incubated at 4°C for 12 h with
primary antibodies targeted against: EGFR (cat. no. 2085),
phosphorylated (p)-EGFR (Tyr1068; cat. no. 3777), MMP-2 (cat. no.
40994), MMP-7 (cat. no. 3801), MMP-9 (cat. no. 13667), β-catenin
(cat. no. 8480), Vimentin (cat. no. 5741), snail family
transcriptional repressor 1 (Snail; cat. no. 3879), E-cadherin
(cat. no. 3195), GAPDH (cat. no. 5174), β-actin (cat. no. 4970) and
Rac1 (cat. no. 10485-2-AP). All antibodies were rabbit monoclonal
antibodies that were used at a dilution of 1:1,000. All antibodies
were purchased from Cell Signaling Technology, Inc., except for the
Rac1 primary antibody, which was purchased from ProteinTech Group,
Inc. Following washing with PBST (0.05% Tween-20) three times (10
min per wash), the membranes were incubated with a HRP-conjugated
secondary antibody (cat. no. ab6721; 1:30,000; Abcam) for 1 h at
room temperature. Following washing three times with PBST, protein
bands were visualized using an Odyssey scanner (LI-COR
Biosciences). Protein expression was semi-quantified using ImageJ
software (version 1.8.0; National Institutes of Health) with
β-actin as the loading control.
For EGF stimulation, cells were incubated with
different concentrations of hybrid 7B or gefitinib for 48 h,
followed by incubation with EGF (50 ng/ml; cat. no. 236-EG-200) for
20 min at 37°C.
Molecular docking
To investigate the interaction between compound
molecules and EGFR or Rac1 proteins, molecular docking research was
conducted using Molecular Operating Environment (MOE; version
10.2008) software provided by Chemical Computing Group ULC. London
dG scoring was used to evaluate the binding affinity of each
compound to EGFR or Rac1. The lower the score, the more hydrogen
bonds, and the tighter the interaction of amino acid residues,
which indicated a more stable and stronger interaction between the
compound and protein. The protein structures of EGFR [Protein Data
Bank (PDB) ID: 3W2S] and Rac1 (PDB ID: 2rmk) were obtained from the
protein database Research Collaboratory for Structural
Bioinformatics PDB (www.rcsb.org).
The chemical structure of Rhein, gefitinib and hybrid 7B were drawn
by ChemBio Draw Ultra software (version 12.0; PerkinElmer, Inc.)
and saved in PDB format.
Statistical analysis
Comparisons between two groups were analyzed using
the unpaired Student's t-test. Comparisons among multiple groups
were analyzed using one-way ANOVA followed by Tukey's post hoc
test. Data are presented as the mean ± SD of at least three
independent experiments. Statistical analyses were performed using
SPSS (version 20.0; IBM Corp.) and GraphPad Prism (version 8;
GraphPad Software, Inc.) software. P<0.05 was considered to
indicate a statistically significant difference.
Results
Inhibitory effect of hybrid 7B, Rhein and gefitinib
on MDA-MB-231, MCF-7 and MCF-10A cell viability. The MTT assay was
performed to assess the effects of hybrid 7B, Rhein and gefitinib
on MDA-MB-231 TNBC, MCF-7 breast cancer and MCF-10A normal breast
epithelial cell viability. The results indicated that hybrid 7B
inhibited MDA-MB-231 and MCF-7 cell viability, displaying
significantly lower IC50 values compared with Rhein and
gefitinib. In normal breast epithelial cells, the IC50
value of hybrid 7B was approximately twice that of the
IC50 value in MDA-MB-231 cells, suggesting that hybrid
7B displayed a certain level of cell selectivity.
In addition, the IC30, IC50
and IC70 values of MDA-MB-231 were close to 1, 2 and 4
µM, respectively. Moreover, the IC30,
IC50 and IC70 of MCF-7 cells were close to 4,
8 and 16 µM, respectively (Table I). Therefore, 1, 2 and 4 µM
were selected for MDA-MB-231 cells and 4, 8 and 16 µM were
selected for MCF-7 cells for subsequent experiments.
 | Table IInhibitory concentrations of hybrid
7B, Rhein and gefitinib in breast cancer cells. |
Table I
Inhibitory concentrations of hybrid
7B, Rhein and gefitinib in breast cancer cells.
A, MDA-MB-231 cells
|
|---|
| Compound |
IC30 |
IC50 |
IC70 |
| Hybrid 7B | 1.02±0.23 | 2.31±0.42a,b | 4.15±0.35 |
| Rhein | - | 163.96±33.36 | - |
| Gefitinib | - | 31.73±2.98 | - |
|
B, MCF-7 cells
|
| Compound |
IC30 |
IC50 |
IC70 |
|
| Hybrid 7B | 3.9±0.36 | 9.02±0.71a,b | 16.45±1.23 |
| Rhein | - | 120.19±10.98 | - |
| Gefitinib | - | 29.76±2.04 | - |
|
C, MCF-10A cells
|
| Compound |
IC30 |
IC50 |
IC70 |
|
| Hybrid 7B | - | 5.19±0.81a,b | - |
| Rhein | - | >100 | - |
| Gefitinib | - | 0.55±0.12 | - |
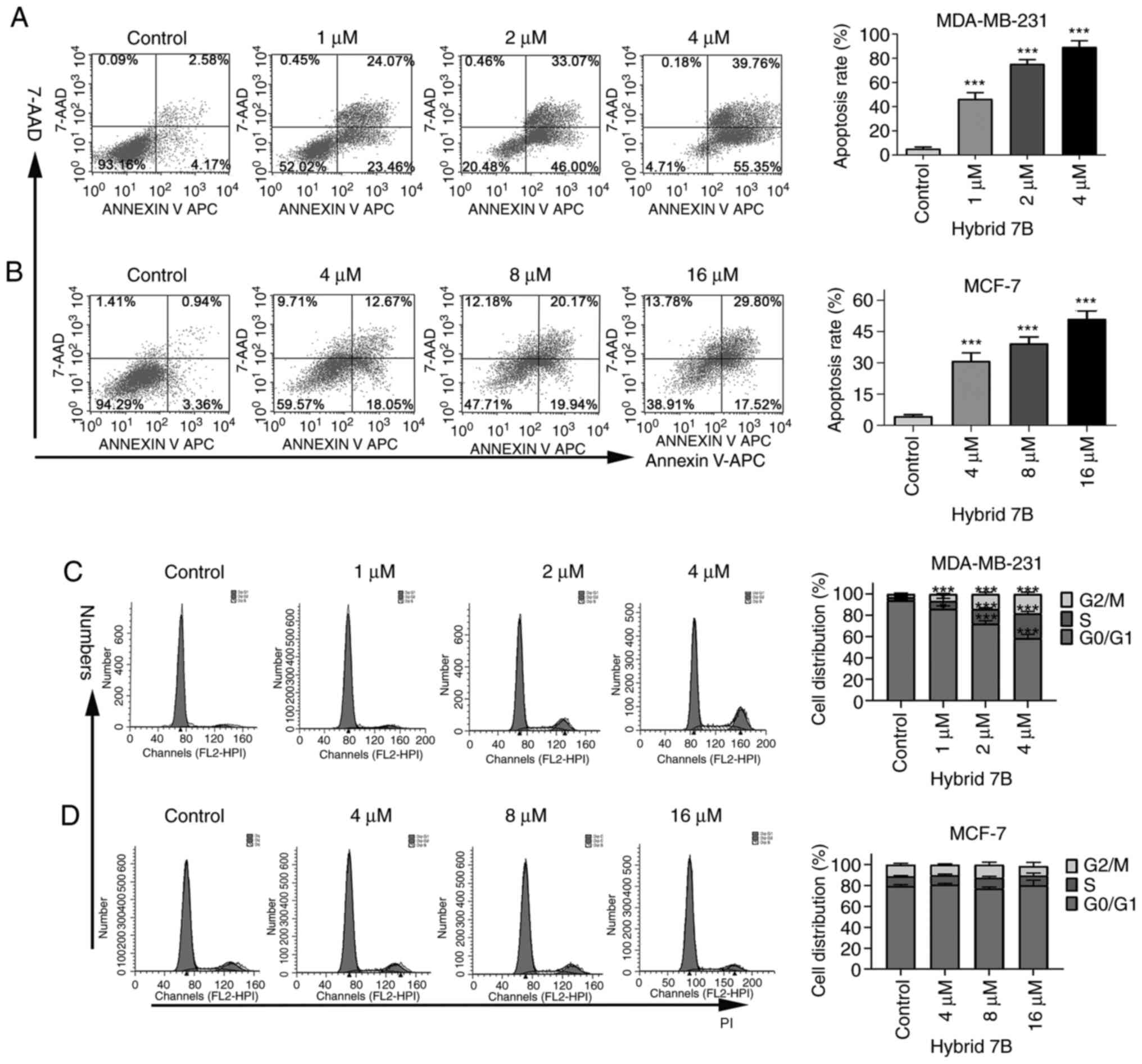
Hybrid 7B induces MDA-MB-231 and MCF-7
cell apoptosis
To determine the effect of hybrid 7B on breast
cancer cell apoptosis, flow cytometry was performed to assess
alterations in apoptotic rates following hybrid 7B treatment for 48
h. The apoptotic rates of MDA-MB-231 cells were 5.23±1.44,
46.35±5.34, 77.37±3.55 and 89.37±5.19% following treatment with 0,
1, 2 and 4 µM hybrid 7B, respectively (Fig. 1A). The apoptotic rates of MCF-7
cells were 4.43±0.71, 30.91±4.01, 39.37±3.07 and 51.10±3.84%
following treatment with 0, 4, 8 and 16 µM hybrid 7B,
respectively (Fig. 1B).
Increasing concentrations of hybrid 7B results in increased rates
of apoptosis in MDA-MB-231 and MCF-7 cells. The results suggested
that hybrid 7B significantly promoted MDA-MB-231 and MCF-7 cell
apoptosis compared with the control group. At the same
concentration (4 µM), the apoptotic rate of MDA-MB-231 cells
was notably higher compared with MCF-7 cells.
MDA-MB-231 cell cycle progression is
blocked by hybrid 7B at the G2/M phase
The cell cycle distribution following hybrid 7B
treatment for 48 h was analyzed via flow cytometry. Compared with
the control group, the proportion of MDA-MB-231 cells at the S and
G2/M phases was significantly increased by hybrid 7B
treatment in a concentration-dependent manner (Fig. 1C), indicating that hybrid 7B
blocked MDA-MB-231 cell cycle progression at the S and
G2/M phases. However, no significant differences in the
proportion of cells at different cell cycle phases was observed
among the control and hybrid 7B groups in MCF-7 cells (Fig. 1D).
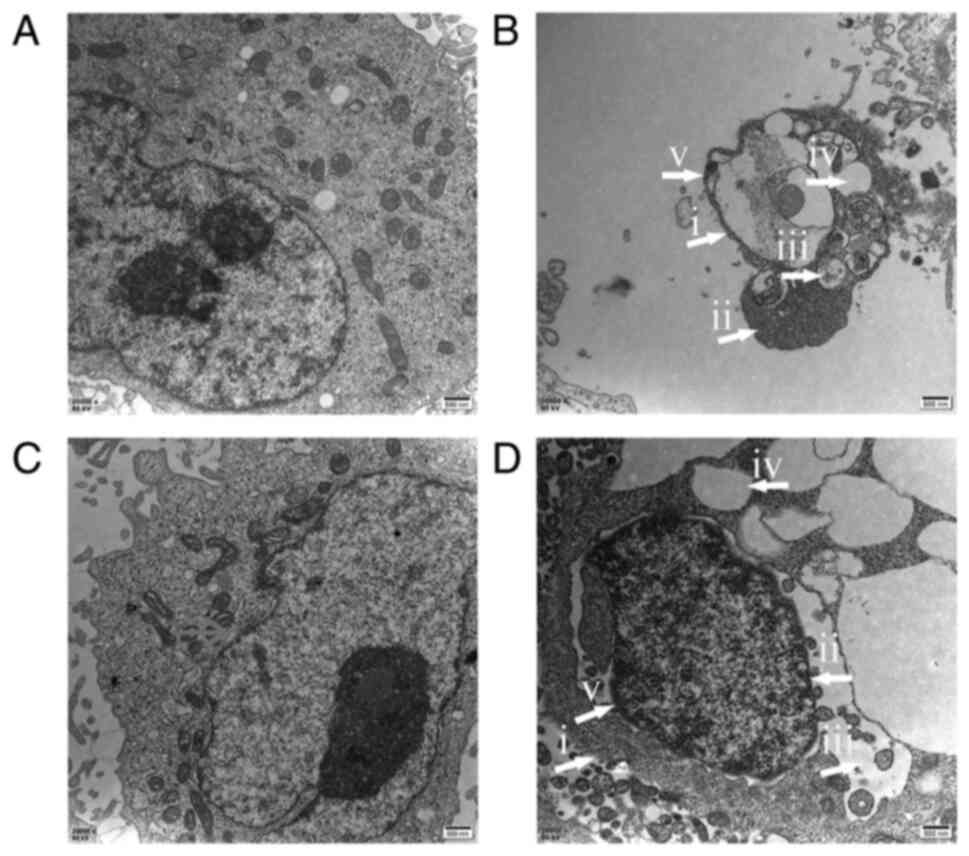
MDA-MB-231 and MCF-7 cells display
apoptotic morphological alterations following hybrid 7B
treatment
TEM is the gold standard for identifying various
modes of cell death (12). In the
control groups, cells and the nuclear membranes were intact and
smooth, the nuclear chromatin displayed a normal distribution, and
organelles, including the endoplasmic reticulum, mitochondria and
lysosomes, were well developed (Fig.
2A and C). In hybrid 7B-treated MDA-MB-231 (Fig. 2B) and MCF-7 (Fig. 2D) cells, typical morphological
alterations of apoptotic cells, including cell shrinkage, irregular
nuclei and chromatin concentrated at the edge of the nucleus,
swelling of mitochondria, disappearance of cristae, a large number
of vacuoles and apoptotic bodies, were observed via TEM.
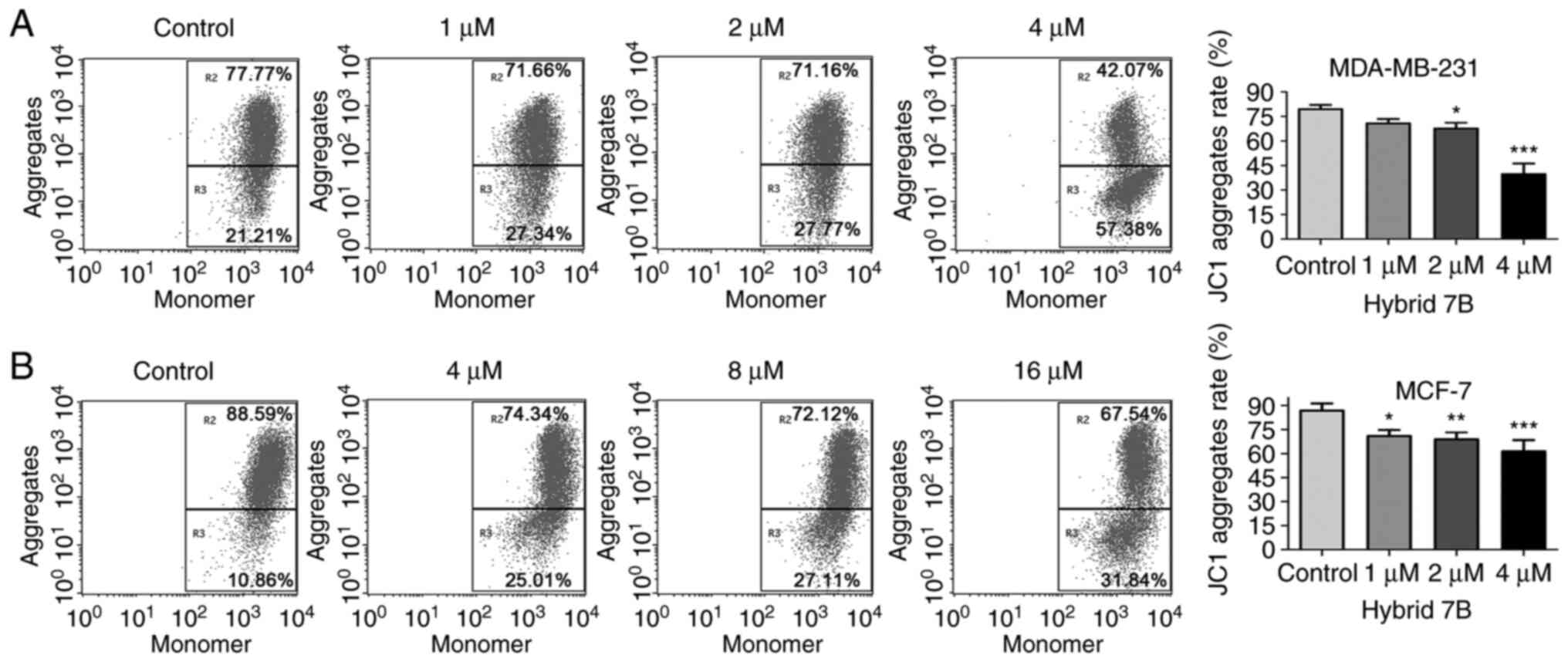
Hybrid 7B modulates mitochondrial
membrane potential in MDA-MB-231 and MCF-7 cells
To verify mitochondrial alterations, a JC-1
fluorescent probe was used to further detect the mitochondrial
membrane potential of cells. When the mitochondrial membrane
potential is high, JC-1 aggregates to form a polymer in the
mitochondrial matrix, resulting in red fluorescence. When the
mitochondrial membrane potential is low, JC-1 is a monomer and
produces green fluorescence. The percentage of MDA-MB-231 cells
with red fluorescence was 79.94±2.14, 71.29±2.13, 68.19±3.03 and
40.23±6.00% following treatment with 0, 1, 2 or 4 µM hybrid
7B, respectively (Fig. 3). The
percentage of MCF-7 cells with red fluorescence was 87.39±3.88,
71.52±3.22, 69.44±3.87 and 62.02±6.40% following treatment with 0,
4, 8 and 16 µM hybrid 7B, respectively. The results
demonstrated that the mitochondrial function of MCF-7 cells was
significantly impaired following treatment with hybrid 7B compared
with the control group, and the mitochondrial function of
MDA-MB-231 cells was significantly impaired at higher concentration
of hybrid 7B (2.4 µM).
Hybrid 7B decreases MDA-MB-231 cell
migration and invasion by inhibiting EMT
Under the same conditions, the invasive ability of
MDA-MB-231 cells was significantly higher compared with MCF-7 cells
(Fig. 4A). Therefore, MDA-MB-231
cells were selected for subsequent experiments. To avoid the effect
of hybrid 7B on cell viability, non-cytotoxic concentrations of
hybrid 7B (0.25, 0.5 and 1 µM), which were determined based
on the MTT assay results, were selected for the invasion assay. The
number of invading MDA-MB-231 cells was 60.00±12.35, 17.33±10.97
and 8.00±3.56 following treatment with 0.25, 0.5 and 1 µM
hybrid 7B, respectively, which was significantly reduced compared
with the control group (141.25±19.82; Fig. 4B). Similarly, the wound-healing
assay results demonstrated that the wound healing percentage of
untreated MDA-MB-231 cells was 95.28±3.04%, whereas following
treatment with 0.25, 0.5 and 1 µM hybrid 7B, the wound
healing percentages were 85.63±2.28, 70.00±5.21 and 41.27±4.06%,
respectively, which were significantly reduced compared with the
control group (Fig. 4C). The
results indicated that hybrid 7B significantly reduced MDA-MB-231
cell migration and invasion compared with the control group.
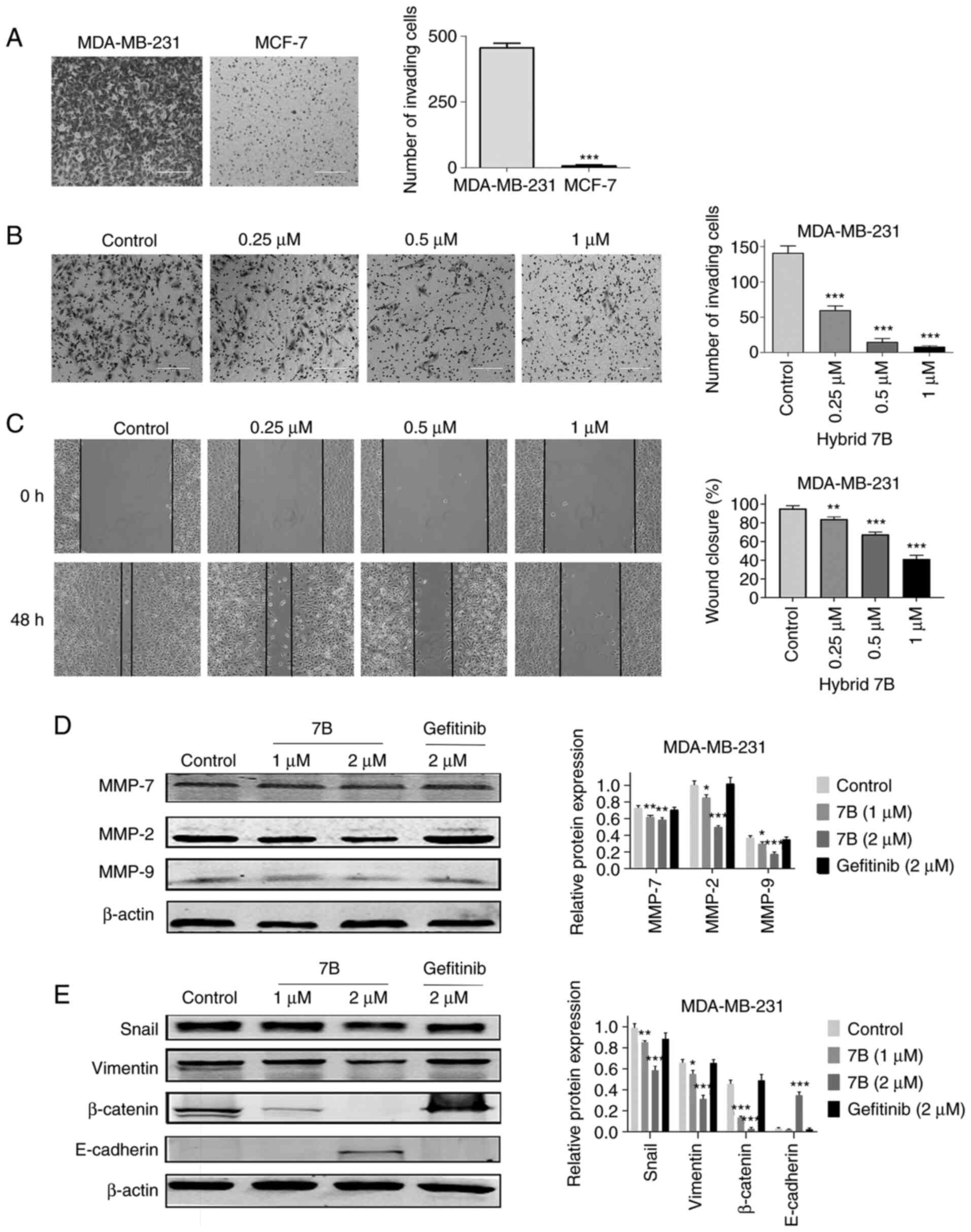 | Figure 4Effect of hybrid 7B on MDA-MB-231
cell invasion, migration and EMT. (A) Comparison between MDA-MB-231
cell invasion and MCF-7 cell invasion (scale bar, 200 µm).
(B) Effect of hybrid 7B on MDA-MB-231 cell invasion (scale bar, 200
µm). MDA-MB-231 cells were treated with hybrid 7B (0, 0.25,
0.5 or 1 µM) for 48 h (scale bar, 200 µm). (C) Effect
of hybrid 7B on MDA-MB-231 cell migration (magnification, x100).
MDA-MB-231 cells were treated with hybrid 7B (0, 0.25, 0.5 or 1
µM) for 48 h. Effect of hybrid 7B on (D) MMP and (E)
EMT-related protein expression in MDA-MB-231 cells. Cells were
treated with different doses of hybrid 7B or gefitinib for 48 h.
*P<0.05, **P<0.01 and
***P<0.001 vs. control. EMT, epithelial-mesenchymal
transition; MMP, matrix metallopeptidase; Snail, snail family
transcriptional repressor 1; 7B, hybrid 7B. |
Compared with the control group, hybrid 7B
significantly reduced the expression levels of MMP-2, MMP-7 and
MMP-9 in MDA-MB-231 cells in a dose-dependent manner (Fig. 4D). In addition, hybrid 7B
significantly decreased the expression levels of EMT-related
proteins, including β-catenin, vimentin and Snail, and
significantly upregulated E-cadherin expression levels compared
with the control group. The results indicated that hybrid 7B
reversed the EMT process of MDA-MB-231 cells, whereas the positive
control gefitinib did not display the same effect.
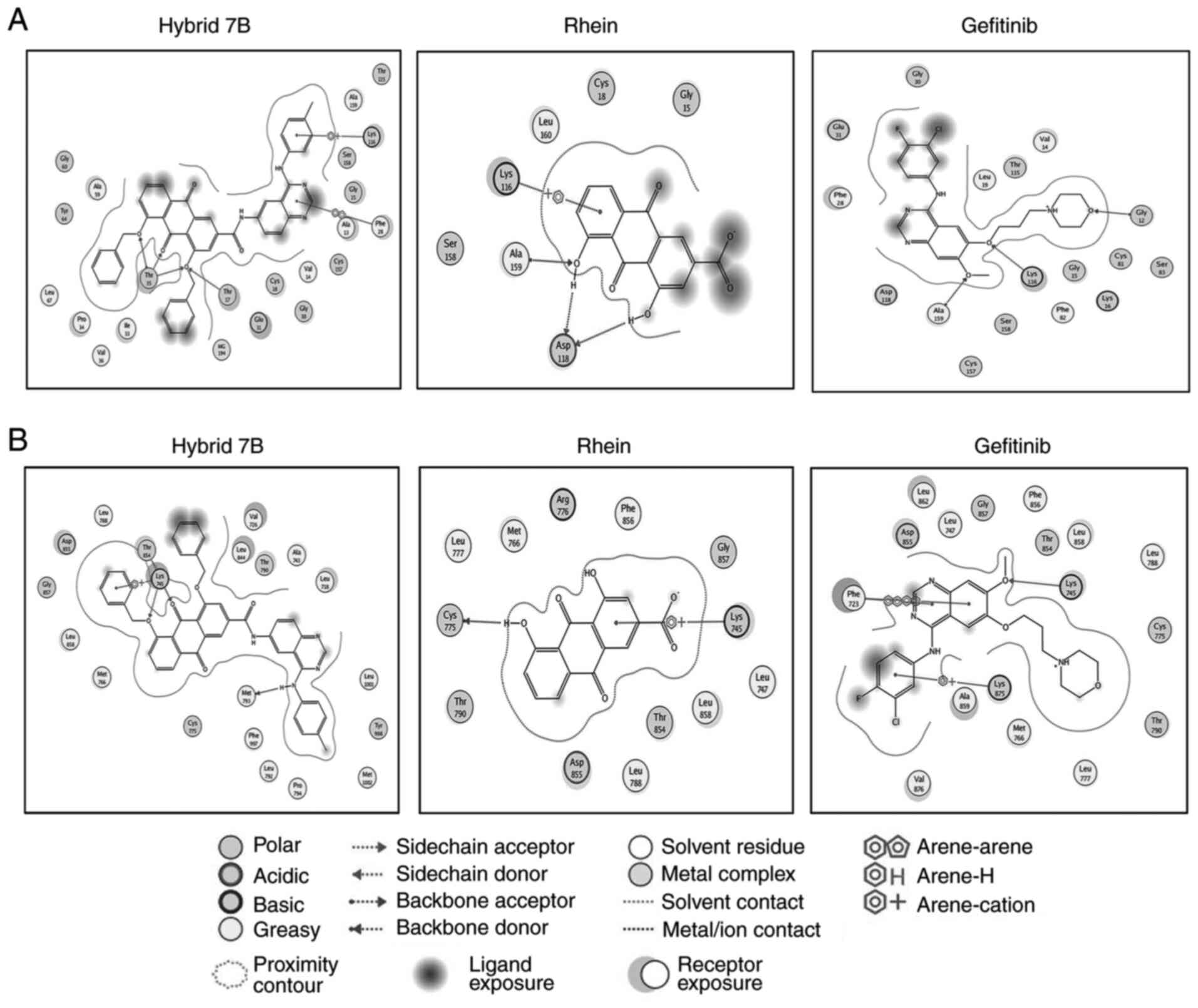
Docking results of hybrid 7B, Rhein,
gefitinib and Rac1
Molecular docking studies of hybrid 7B, Rhein and
gefitinib were performed using MOE docking software. The binding
modes of hybrid 7B, Rhein, gefitinib and Rac1 are presented in
Table II and Fig. 5A. Hybrid 7B formed four hydrogen
bonds with amino acid residues Thr17 and Thr35, Rhein formed three
hydrogen bonds with amino acid residues Asp118 and Ala159, and
gefitinib formed three hydrogen bonds with amino acid residues
Gly12, Lys116 and Ala159. The London dG scores of hybrid 7B-Rac1,
Rhein-Rac1 and gefitinib-Rac1 complexes were -31.215, -12.759 and
-25.953 kcal/mol, respectively (Table II). The higher the negative
value, the stronger the binding affinity between the compounds and
Rac1; therefore, it was hypothesized that hybrid 7B displayed a
stronger ability of targeting and regulating Rac1 compared with
Rhein and gefitinib.
| Table IIBinding affinity and interactions of
Rac1-ligand complexes. |
Table II
Binding affinity and interactions of
Rac1-ligand complexes.
| Compound | London dG
(kcal/mol) | Amino acid involved
in H-bonds | Distance of
hydrogen bond (Å) | Hydrophobic
interactions |
|---|
| Hybrid 7B | -31.215 | Thr17 | 2.90 | Ala159, Ala13 and
Phe28 |
| | Thr35(3) | 2.55/2.60/2.53 | Val14, Ala59,
Leu67, Pro34, Ile33 and Val36 |
| Rhein | -12.759 | Asp118(2) | 1.72/2.26 | Leu160 and
Ala159 |
| | Ala159 | 2.83 | |
| Gefitinib | -25.953 | Gly12 | 2.96 | Phe28, Leu19,V
al14, Ala159 and Phe 82 |
| | Lys116 | 3.06 | |
| | Ala159 | 2.96 | |
Docking results of hybrid 7B, Rhein,
gefitinib and EGFR
The London dG scores of 7B-EGFR, Rhein-EGFR and
gefitinib-EGFR complexes were -35.325, -16.330 and -27.211
kcal/mol, respectively (Table
III and Fig. 5B). The
interaction energies of hybrid 7B with EGFR were superior compared
with Rhein with EGFR. Hydrogen bonding interactions serve an
important role in the stability of a complex. The more hydrogen
bonds formed in the complex, the closer the hydrogen bond between
the receptor and the ligand, and the more conducive to the
stability of the complex (13).
The results demonstrated that three hydrogen bonds were observed in
hybrid 7B, whereas one hydrogen bond was identified in Rhein. The
number of amino acid residues involved in hybrid 7B-EGFR
interactions was higher compared with those involved in Rhein-EGFR
and gefitinib-EGFR interactions. The results indicated that hybrid
7B bound more tightly and strongly with EGFR compared with Rhein
and gefitinib.
| Table IIIBinding affinity and interactions of
EGFR-ligand complexes. |
Table III
Binding affinity and interactions of
EGFR-ligand complexes.
| Compound | London dG
(kcal/mol) | Amino acid involved
in H-bonds | Distance of
hydrogen bond (Å) | Hydrophobic
interactions |
|---|
| Hybrid 7B | -35.325 | Met 793 | 1.56 | Leu788, Val26,
Leu844, Ala743, Leu718, Leu858, Met766, |
| | Lys 745(2) | 2.66/2.26 | Met793, Leu1001,
Phe997, Leu792, Pro794 and Met 1002 |
| | Thr 854 | 2.62 | |
| Rhein | -16.330 | Cys 775 | 1.99 | Leu777, Met766,
Phe856 Leu747, Leu858, Leu 788 |
| Gefitinib | -27.211 | Lys 745 | 3.40 | Leu862, Leu747,
Phe856, Leu858, Leu788, Phe723, Ala859, |
| | | | Met766, Leu777 and
Val 876 |
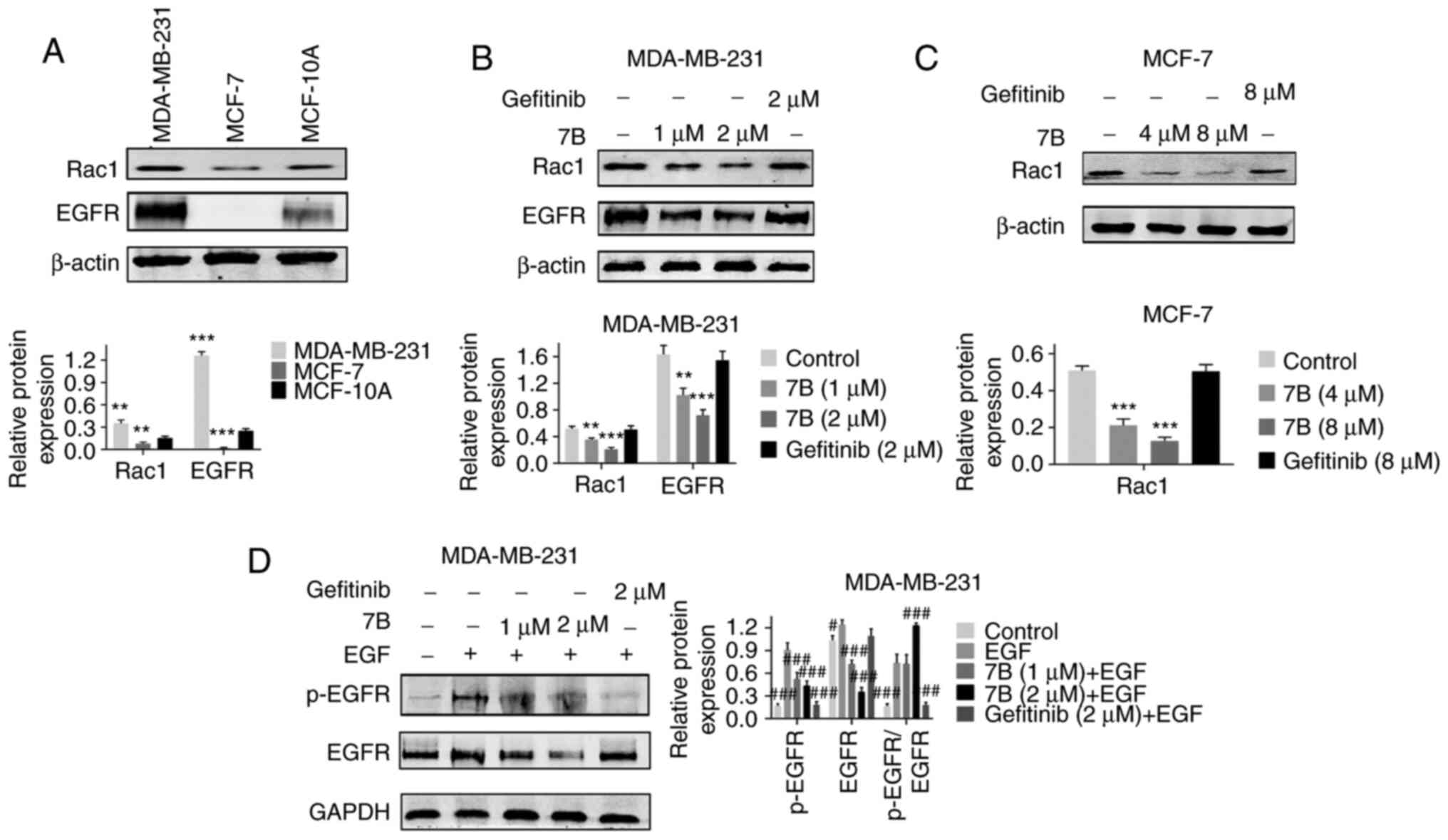
Hybrid 7B downregulates the expression
levels of Rac1, EGFR and p-EGFR
To further verify whether hybrid 7B displayed
targeted regulation of EGFR and Rac1 proteins in breast cancer
cells, western blotting was performed to detect their expression
level in breast cancer cells treated with hybrid 7B or gefitinib
(positive control). The results demonstrated that EGFR and Rac1
expression levels in MDA-MB-231 cells were significantly higher
compared with MCF-7 cells (Fig.
6A). EGFR protein was expressed at low levels in MCF-7 cells;
therefore, the effect of hybrid 7B on EGFR protein in MCF-7 cells
was not investigated.
Compared with the control group, hybrid 7B not only
significantly downregulated the expression levels of EGFR and Rac1
in MDA-MB-231 cells in a dose-dependent manner, but also partially
reversed the regulatory effect of EGF on p-EGFR in MDA-MB-231
cells, confirming the targeting effect of hybrid 7B on EGFR
(particularly p-EGFR) and Rac1 (Fig.
6B-D).
Discussion
The invasion and metastasis of malignant tumors is a
complex, multi-step process that involves the regulation of
multiple genes (14). Previous
studies have indicated that EGFR is closely associated with tumor
cell adhesion, migration, EMT and extracellular matrix (ECM)
degradation, and serves an important role in the process of
invasion and metastasis (4,15).
It has been reported that >50% of patients with TNBC display
EGFR overexpression (15), and
these patients can undergo EGFR-tyrosine kinase inhibitor (TKI)
therapy (16). However, no clear
clinical benefit regarding the use of EGFR-targeted TKIs, including
gefitinib, erlotinib and cetuximab, has been reported (17,18). One reason for treatment failure
may be that the progression of TNBC does not rely solely on the
EGFR signaling pathway, but is regulated by several proteins. When
EGFR is inhibited, there is an alternative response mechanism
dominated by other proteins in patients with breast cancer
(19). For instance, it has been
reported that Rac1 protein, which is also highly expressed in TNBC,
is a component of the signaling pathway and may serve as a
therapeutic target for tumor angiogenesis and metastasis (20). Another potential reason for
treatment failure may be that the binding mode of EGFR-TKIs and
EGFR also affects the curative effect of EGFR-TKIs. Gefitinib, the
first generation of oral EGFR-TKIs, has been reported to compete
with Mg-ATP binding sites in the EGFR-TK catalytic region,
preventing EGFR-induced signal activation. If there is a mutation
in the binding site or other compounds competitively bind to the
binding site, the antitumor effect of EGFR-TKI is reduced (21). Therefore, the synthesis of novel
multi-target EGFR-TKIs, which can inhibit the malignant
proliferation and metastasis of TNBC by acting on multiple targets,
may serve as a more effective strategy. Inspired by the results of
a lung cancer study (22),
Rac1-mediated signaling pathways are considered to be independent
of other downstream signaling pathways mediated by EGFR, including
PI3K/Akt or MEK1/2/ERK1/2 signaling pathways. Rac1 specific
inhibitor NSC23766 can inhibit gefitinib-resistant non-small cell
lung cancer cell viability and migration, even in the presence of
MEK and PI3K inhibitors (22). In
addition, it has been reported that Rac1 inhibitor NSC23766 and
HER1/EGFR-targeting drug erlotinib display a synergistic
antiproliferative effect in glioblastoma (23). Our previous study demonstrated
that compounds containing an anthraquinone structure regulated Rac1
expression (9). Simultaneously
targeting several signaling pathways is critical; therefore,
developing novel combinations targeting both EGFR and Rac1 proteins
may serve as an advantageous therapeutic approach. Based on the
pharmacophore combination principle, quinazoline and anthraquinone
scaffolds were organically fused, and hybrid 7B was synthesized
(11).
The results of molecular docking demonstrated that
the binding affinities of hybrid 7B to EGFR were stronger compared
with gefitinib, suggesting that the binding mode of hybrid 7B to
EGFR may not be consistent with that of gefitinib. The western
blotting results also indicated that hybrid 7B not only
significantly decreased the expression of p-EGFR (Fig. 6D), but also downregulated the
expression of EGFR compared with gefitinib (Fig. 6B), which was consistent with the
results obtained for the second-generation EGFR-TKI alphatinib
(24).
A decrease in the mitochondria membrane potential is
a hallmark of apoptosis initiation (25). The JC-1 fluorescent probe assay
was used in the present study to assess alterations in the
mitochondrial membrane potential following hybrid 7B treatment.
Numerous typical morphological features of apoptosis induced by
hybrid 7B treatment were verified by TEM, including cell shrinkage,
irregular nuclei, swelling of mitochondria, disappearance of
cristae, and vacuoles and apoptotic bodies in the cytoplasm. The
results indicated that hybrid 7B caused damage to the mitochondrial
membrane and decreased the membrane potential compared with the
control group.
Increasing evidence has demonstrated that TNBC
metastasis is associated with abnormal activation of EMT (26,27). EMT is the process of acquisition
of molecular alterations by which epithelial cancer cells lose
their epithelial features (E-cadherin and cytokeratin) and gain
mesenchymal features (vimentin, N-cadherin, Snail, Slug and Twist)
(28). Triple-negative breast
cancer cells are transformed by EMT, escape from the primary tumor
site, invade the stromal tissues and establish a distant secondary
tumor. Epithelial marker E-cadherin protein expression loss and
increased mesenchymal marker expression, including β-catenin and
vimentin, are features of the EMT phenotype (29). A previous study reported that
EGFR-TKI afatinib regulates EMT by inhibiting EGFR, and can
significantly reduce MMP-9 protein expression levels (30). Rac1 expression is also associated
with EMT, thus inhibiting Rac1 expression in gastric adenocarcinoma
cells blocks EMT, invasion and metastasis (31). Therefore, regulating the
expression of EGFR and Rac1 proteins may reverse the EMT process of
tumor cells. The results of the present study demonstrated that
hybrid 7B successfully reversed the EMT of MDA-MB-231 by
significantly downregulating the expression levels of β-catenin and
Vimentin, and significantly upregulating the expression levels of
E-cadherin compared with the control group. At the same
concentration (2 µM), the ability of hybrid 7B to reverse
EMT was notably superior compared with gefitinib, which may be
associated with synergistic inhibition of the expression of
multiple proteins. Therefore, it was hypothesized that hybrid 7B
may serve as a potential multi-target antitumor activity
compound.
MMPs are a group of calcium-dependent extracellular
proteases that facilitate ECM degradation, resulting in tumor
invasion and metastasis promotion. MMP expression is regulated by
EMT-related signal transduction pathways, including the TGF-β
signaling pathway (32). In
HT1080 fibrosarcoma cells cultured in three-dimensional collagen
gel, Rac1 mediated MMP-2 activation and MMP14 expression/processing
during the encounter between invading tumor cells and type I
collagen-rich stroma, thereby facilitating collagenolysis and cell
invasion (33). Using specific
inhibitors, Binker et al (34) revealed that not only is Rac1
activated following the lipopolysaccharide stimulation of NCI-H292
cells (human airway cells), but also that PI3K was the signaling
molecule downstream of EGFR that regulated Rac1 activity. In
addition, a study has reported that EGFR overexpression in tumor
cells is involved in MMP-9 upregulation (35). Therefore, the aforementioned
studies indicated that the occurrence of EMT, tumor invasion and
metastasis is closely associated with the expression of EGFR, Rac1
and MMPs. The results of the present study demonstrated that
following treatment with hybrid 7B, the protein expression levels
of MMP-2, MMP-7, MMP-9, EGFR and Rac1 were significantly decreased
in MDA-MB-231 cells compared with the control group. In addition,
MDA-MB-231 cell invasion and metastasis were significantly
inhibited by hybrid 7B treatment compared with the control group.
The aforementioned results further verified the multi-target
antitumor activity of hybrid 7B.
In the present study, the results demonstrated that
MDA-MB-231 cell cycle progression was blocked at the S and
G2/M phases, but there was no alteration in MCF-7 cell
cycle progression following hybrid 7B treatment. The aforementioned
result was similar to the findings reported by Elkhalifa et
al (36). In the
aforementioned study, compound 14 induced apoptosis in MDA-MB-231
and MCF-7 cells, and MDA-MB-231 cells displayed cell cycle arrest
at the G2/M phase, whereas the MCF-7 cell cycle was not
altered. However, in the present study, the
sub-G0/G1 peak was not identified by flow
cytometry in these two cell lines. Since any fractional DNA
content, not only that caused by apoptosis but also that caused by
other reasons, can result in multiple nuclear fragments, it has
been suggested that the sub-G1 peak is not a good
indicator of cell apoptosis and cannot fully represent the number
of apoptotic cells. Therefore, the apoptosis peak was not modeled
using ModFitLT software to analyze the cell cycle data in the
present study, thus a hypodiploid peak (sub-G1 peak) was not
presented in the present study.
Moreover, the results of the present study
demonstrated that compared with MDA-MB-231 cells, EGFR protein
expression in MCF-7 cells was notably lower, indicating that MCF-7
cells could not invade the matrix. This result was consistent with
the conclusions of two previous studies (2,37)
that reported that EGFR+ cancer cell lines were more
invasive compared with EGFR– cells. The aforementioned
result also provided a potential explanation for why the
IC50 value of hybrid 7B was markedly higher in MCF-7
compared with MDA-MB-231 cells due to the lack of an EGFR target in
MCF-7.
In the present study, the inhibitory effect of
hybrid 7B on TNBC cell viability and metastasis was only studied
in vitro. However, the possible signaling pathway underlying
hybrid 7B-induced suppression of cell migration, invasion and EMT
in MDA-MB-231 cells requires further investigation, including in
vivo studies.
In conclusion, the present study demonstrated that
hybrid 7B induced breast cancer cell apoptosis, arrested MDA-MB-231
cell cycle progression at the G2/M phase, and
significantly inhibited TNBC invasion and metastasis compared with
the control group. The mechanism underlying hybrid 7B was
associated with downregulation of EGFR and Rac1 protein expression
levels and reversal of TNBC EMT. Therefore, the results of the
present study indicated that hybrid 7B may serve as a potential
dual-target inhibitor of EGFR and Rac1.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JK, HH and DL conceived and designed the
experiments. JK, WT, JL, XL and LZ performed the experiments. JK
and YZ analyzed the data. WT, JL and LZ contributed reagents,
materials and analysis tools. JK, YZ and DL drafted the manuscript.
JK and DL confirm the authenticity of all the raw data. All authors
read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Guangxi Natural Science
Foundation (grant no. 2018GXNSFAA281064), the Innovation Project of
Guangxi Graduate Education (grant no. YCSW2019107) and the Program
of Key Laboratory of High-Incidence Tumor Prevention and Treatment
(Guangxi Medical University) and the Ministry of Education (China;
grant no. GK2019-22).
Abbreviations:
|
EMT
|
epithelial-mesenchymal transition
|
|
MMP
|
mitochondrial membrane potential
|
|
MOE
|
Molecular Operating Environment
|
|
TEM
|
transmission electron microscopy
|
|
TNBC
|
triple negative breast cancer
|
|
TKI
|
tyrosine kinase inhibitor
|
References
|
1
|
Yin L, Duan JJ, Bian XW and Yu SC:
Triple-negative breast cancer molecular subtyping and treatment
progress. Breast Cancer Res. 22:612020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McGovern UB, Francis RE, Peck B, Guest SK,
Wang J, Myatt SS, Krol J, Kwok JM, Polychronis A, Coombes RC and
Lam EW: Gefitinib (Iressa) represses FOXM1 expression via FOXO3a in
breast cancer. Mol Cancer Ther. 8:582–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lehmann BD, Bauer JA, Chen X, Sanders ME,
Chakravarthy AB, Shyr Y and Pietenpol JA: Identification of human
triple-negative breast cancer subtypes and preclinical models for
selection of targeted therapies. J Clin Invest. 121:2750–2767.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsuda N, Lim B, Wang X and Ueno NT:
Early clinical development of epidermal growth factor receptor
targeted therapy in breast cancer. Expert Opin Investig Drugs.
26:463–479. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peng B, He R, Xu Q, Yang Y, Hu Q, Hou H,
Liu X and Li J: Ginsenoside 20(S)-protopanaxadiol inhibits
triple-negative breast cancer metastasis in vivo by targeting
EGFR-mediated MAPK pathway. Pharmacol Res. 142:1–13. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gonzalez-Conchas GA, Rodriguez-Romo L,
Hernandez-Barajas D, Gonzalez-Guerrero JF, Rodriguez-Fernandez IA,
Verdines-Perez A, Templeton AJ, Ocana A, Seruga B, Tannock IF, et
al: Epidermal growth factor receptor overexpression and outcomes in
early breast cancer: A systematic review and a meta-analysis.
Cancer Treat Rev. 62:1–8. 2018. View Article : Google Scholar
|
|
7
|
Payapilly A and Malliri A:
Compartmentalisation of RAC1 signalling. Curr Opin Cell Biol.
54:50–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su Z, Li Z, Wang C, Tian W, Lan F, Liang
D, Li J, Li D and Hou H: A novel Rhein derivative: Activation of
Rac1/NADPH pathway enhances sensitivity of nasopharyngeal carcinoma
cells to radiotherapy. Cell Signal. 54:35–45. 2019. View Article : Google Scholar
|
|
9
|
Zhou G, Peng F, Zhong Y, Chen Y, Tang M
and Li D: Rhein suppresses matrix metalloproteinase production by
regulating the Rac1/ROS/MAPK/AP-1 pathway in human ovarian
carcinoma cells. Int J Oncol. 50:933–941. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roskoski R Jr: ErbB/HER protein-tyrosine
kinases: Structures and small molecule inhibitors. Pharmacol Res.
87:42–59. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang D, Su Z, Tian W, Li J, Li Z, Wang C,
Li D and Hou H: Synthesis and screening of novel
anthraquinone-quinazoline multitarget hybrids as promising
anticancer candidates. Future Med Chem. 12:111–126. 2020.
View Article : Google Scholar
|
|
12
|
Klöditz K and Fadeel B: Three cell deaths
and a funeral: Macrophage clearance of cells undergoing distinct
modes of cell death. Cell Death Discov. 5:652019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Daddam JR, Dowlathabad MR, Panthangi S and
Jasti P: Molecular docking and P-glycoprotein inhibitory activity
of flavonoids. Interdiscip Sci. 6:167–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stivarou T and Patsavoudi E: Extracellular
molecules involved in cancer cell invasion. Cancers (Basel).
7:238–265. 2015. View Article : Google Scholar
|
|
15
|
Williams CB, Soloff AC, Ethier SP and Yeh
ES: Perspectives on epidermal growth factor receptor regulation in
triple-negative breast cancer: Ligand-mediated mechanisms of
receptor regulation and potential for clinical targeting. Adv
Cancer Res. 127:253–281. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sporikova Z, Koudelakova V, Trojanec R and
Hajduch M: Genetic markers in triple-negative breast cancer. Clin
Breast Cancer. 18:e841–e850. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al-Mahmood S, Sapiezynski J, Garbuzenko OB
and Minko T: Metastatic and triple-negative breast cancer:
Challenges and treatment options. Drug Deliv Transl Res.
8:1483–1507. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baselga J, Albanell J, Ruiz A, Lluch A,
Gascón P, Guillém V, González S, Sauleda S, Marimón I, Tabernero
JM, et al: Phase II and tumor pharmacodynamic study of gefitinib in
patients with advanced breast cancer. J Clin Oncol. 23:5323–5333.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
El Guerrab A, Bamdad M, Bignon YJ,
Penault-Llorca F and Aubel C: Co-targeting EGFR and mTOR with
gefitinib and everolimus in triple-negative breast cancer cells.
Sci Rep. 10:63672020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bid HK, Roberts RD, Manchanda PK and
Houghton PJ: RAC1: An emerging therapeutic option for targeting
cancer angiogenesis and metastasis. Mol Cancer Ther. 12:1925–1934.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi Y, Su C, Cui W, Li H, Liu L, Feng B,
Liu M, Su R and Zhao L: Gefitinib loaded folate decorated bovine
serum albumin conjugated carboxymethyl-beta-cyclodextrin
nanoparticles enhance drug delivery and attenuate autophagy in
folate receptor-positive cancer cells. J Nanobiotechnology.
12:432014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kaneto N, Yokoyama S, Hayakawa Y, Kato S,
Sakurai H and Saiki I: RAC1 inhibition as a therapeutic target for
gefitinib-resistant non-small-cell lung cancer. Cancer Sci.
105:788–794. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim H, Samuel SL, Zhai G, Rana S, Taylor
M, Umphrey HR, Oelschlager DK, Buchsbaum DJ and Zinn KR:
Combination therapy with anti-DR5 antibody and tamoxifen for triple
negative breast cancer. Cancer Biol Ther. 15:1053–1060. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen Y, Chen X, Ding X and Wang Y:
Afatinib, an EGFR inhibitor, decreases EMT and tumorigenesis of
Huh-7 cells by regulating the ERK-VEGF/MMP9 signaling pathway. Mol
Med Rep. 20:3317–3325. 2019.PubMed/NCBI
|
|
25
|
Kim TI, Kim H, Lee DJ, Choi SI, Kang SW
and Kim EK: Altered mitochondrial function in type 2 granular
corneal dystrophy. Am J Pathol. 179:684–692. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Neelakantan D, Zhou H, Oliphant MUJ, Zhang
X, Simon LM, Henke DM, Shaw CA, Wu MF, Hilsenbeck SG, White LD, et
al: EMT cells increase breast cancer metastasis via paracrine GLI
activation in neighbouring tumour cells. Nat Commun. 8:157732017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim S and Lee JW: Membrane proteins
involved in epithelial-mesenchymal transition and tumor invasion:
Studies on TMPRSS4 and TM4SF5. Genomics Inform. 12:12–20. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jang MH, Kim HJ, Kim EJ, Chung YR and Park
SY: Expression of epithelial-mesenchymal transition-related markers
in triple-negative breast cancer: ZEB1 as a potential biomarker for
poor clinical outcome. Hum Pathol. 46:1267–1274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tien Y, Tsai CL, Hou WH, Chiang Y, Hsu FM,
Tsai YC and Cheng JC: Targeting human epidermal growth factor
receptor 2 enhances radiosensitivity and reduces the metastatic
potential of Lewis lung carcinoma cells. Radiat Oncol. 15:582020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoon C, Cho SJ, Chang KK, Park DJ, Ryeom
SW and Yoon SS: Role of Rac1 pathway in epithelial-to-mesenchymal
transition and cancer stem-like cell phenotypes in gastric
adenocarcinoma. Mol Cancer Res. 15:1106–1116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Santibanez JF, Obradović H, Kukolj T and
Krstić J: Transforming growth factor-β, matrix metalloproteinases,
and urokinase-type plasminogen activator interaction in the cancer
epithelial to mesenchymal transition. Dev Dyn. 247:382–395. 2018.
View Article : Google Scholar
|
|
33
|
Zhuge Y and Xu J: Rac1 mediates type I
collagen-dependent MMP-2 activation. Role in cell invasion across
collagen barrier. J Biol Chem. 276:16248–16256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Binker MG, Binker-Cosen AA, Richards D,
Oliver B and Cosen-Binker LI: LPS-stimulated MUC5AC production
involves Rac1-dependent MMP-9 secretion and activation in NCI-H292
cells. Biochem Biophys Res Commun. 386:124–129. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cox G, Jones JL and O'Byrne KJ: Matrix
metalloproteinase 9 and the epidermal growth factor signal pathway
in operable non-small cell lung cancer. Clin Cancer Res.
6:2349–2355. 2000.PubMed/NCBI
|
|
36
|
Elkhalifa D, Siddique AB, Qusa M, Cyprian
FS, El Sayed K, Alali F, Al Moustafa AE and Khalil A: Design,
synthesis, and validation of novel nitrogen-based chalcone analogs
against triple negative breast cancer. Eur J Med Chem.
187:1119542020. View Article : Google Scholar
|
|
37
|
Martin JL, de Silva HC, Lin MZ, Scott CD
and Baxter RC: Inhibition of insulin-like growth factor-binding
protein-3 signaling through sphingosine kinase-1 sensitizes
triple-negative breast cancer cells to EGF receptor blockade. Mol
Cancer Ther. 13:316–328. 2014. View Article : Google Scholar
|