Introduction
Extracellular glucose is transferred into cells by
glucose transporters and the majority of the transferred glucose is
used for ATP synthesis through the glycolytic pathway and the
tricarboxylic acid cycle. Only 2-5% of the transferred glucose
enters the hexosamine biosynthetic pathway (HBP).
Glutamine-fructose-6-phosphate aminotransferase (GFAT) is the
rate-limiting enzyme of the HBP that catalyzes the conversion of
fructose 6-phosphate to glucosamine 6-phosphate (GlcN6P). This
reaction is the first step in the HBP. Subsequent steps metabolize
GlcN6P to the major end product uridine
5-diphospho-N-acetylglucosamine (UDP-GlcNAc), which is the
essential precursor of glycoproteins and glycolipids in the
endoplasmic reticulum and Golgi apparatus. Moreover, UDP-GlcNAc is
the direct donor for O-linked β-N-acetylglucosamine
(O-GlcNAc) modification on numerous proteins (1).
O-GlcNAc is a post-translational modification
of nuclear, cytoplasmic and mitochondrial proteins. Protein
O-GlcNAcylation is a dynamic and reversible process carried
out by two single enzymes: O-GlcNAc transferase (OGT) and
O-GlcNAcase (OGA). OGT transfers O-GlcNAc from
UDP-GlcNAc to the hydroxyl groups of serine or threonine residues
of target protein substrates, while OGA catalyzes its removal.
O-GlcNAcylation is emerging as a key regulator of diverse
cellular processes, such as signal transduction, transcriptional
regulation and proteasomal degradation (2). Aberrant O-GlcNAcylation in
cells is closely associated with a number of human diseases,
including cancer, metabolic disorders and cardiovascular disease
(3). Increased OGT and
O-GlcNAc levels have been observed in various types of
cancer and have been found to promote cancer growth and progression
(4).
Protein O-GlcNAcylation and the donor
substrate UDP-GlcNAc levels within the cell are modulated by the
availability of glucose, fatty acids, amino acids and nucleotides.
Therefore, O-GlcNAc is proposed as a nutrient sensor and
metabolic regulator (5).
Moreover, there is increasing evidence to suggest that
O-GlcNAc is a novel regulator of the cellular stress
response. In response to numerous forms of cellular stress or
injury, global O-GlcNAc levels are dynamically elevated in
both in vitro and in vivo models of heat stress,
oxidative stress, endoplasmic reticulum stress, hypoxia,
ischemia-reperfusion injury and trauma hemorrhage (6). However, it has been demonstrated
that O-GlcNAc levels of subproteome can decline (7). Alterations in the expression,
activity, localization and targeting of OGT and OGA, as well as tje
increased flux through the HBP, have been associated with
stress-induced changes in O-GlcNAcylation (8). However, it remains unclear whether
cells and tissues coordinate all of these mechanisms to affect
stress-induced changes in O-GlcNAcylation, or whether
different cells and tissues induce specific pathways depending on
the type of stress (6,9).
Cisplatin is the most widely agent for the treatment
of various types of solid malignancies. It generates intra- and
inter-strand purine crosslinks that interfere with DNA replication,
which leads to irreparable DNA damage, followed by apoptosis.
Cisplatin has also been shown to bind to mitochondrial DNA,
phospholipids and other molecules. However, resistance to cisplatin
can develop, which limits its effectiveness in clinical practice
(10). Recently, it was
demonstrated that decreased O-GlcNAcylation through the
inhibition of the HBP potentiates cisplatin cytotoxicity in
non-small cell lung cancer cells (11). Conversely, another study
demonstrated that suppressed O-GlcNAcylation via the
downregulation of OGT decreased the sensitivity of ovarian cancer
to cisplatin (12).
Global protein O-GlcNAcylation has been
reported to increase in response to various types of stress;
however, little is known with regards to the underlying mechanisms
through which chemotherapeutic agents affect the levels of
O-GlcNAc in cancer cells. In the present study, it was found
that cisplatin elevated O-GlcNAc levels in lung cancer cells
by altering the activity of OGT, OGA and AMP-activated protein
kinase (AMPK). In addition, the sensitivity of cancer cells to
cisplatin was not affected by the changes in O-GlcNAcylation
induced by the inhibition of OGT and OGA in vitro and in
vivo.
Materials and methods
Cell lines and cell culture
The lung cancer cell line NCI-H1299 (CRL-5803),
breast cancer cell line, MCF-7 (HTB-22) and the hepatoblastoma cell
line, Hep G2 (HB8065) were purchased from the Shanghai Institute of
Biochemistry and Cell Biology, Chinese Academy of Sciences. All
three cell lines were initially derived from ATCC. The
taxol-resistant cell line, H1299/Taxol, was a gift from Dr Hongying
Zhen (Department of Cell Biology, School of Basic Medical Sciences,
Peking University Health Science Center). All cell lines were
maintained in a humidified atmosphere containing 5% CO2
at 37°C in RPMI-1640 medium or DMEM supplemented with 10% FBS, 100
U/ml penicillin and 100 µg/ml streptomycin.
Reagents
Cisplatin, PUGNAc, alloxan,
6-diazo-5-oxo-L-nor-Leucine (DON), oligomycin and
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) were
obtained from Sigma-Aldrich; Merck KGaA. Adriamycin was obtained
from Shanghai Biochempartner Co., Ltd. Vincristine was obtained
from Guangrun Biotechnology.
Sulforhodamine B (SRB) assay
Cancer cells were incubated in 96-well plates for 24
h. Following the addition of 0, 2, 8 and 16 µg/ml of CDDP or
in combination with 10 µM of alloxan or 100 µM of
PUGNAc, the plates were incubated at 37°C for an additional 48 h in
a 5% CO2 incubator. The culture medium was then
discarded and the cells were fixed in situ by the gentle
addition of 100 µl of cold 10% (w/v) trichloroacetic acid
followed by incubation for 60 min at 4°C. The supernatant was
discarded and the plates were washed 5 times with tap water and
air-dried. SRB solution (100 µl) at 0.4% (w/v) in 1% acetic
acid was added and the plates were incubated for 20 min at room
temperature. After staining, the unbound dye was removed by washing
5 times with 1% acetic acid and the plates were air-dried. The
bound stain was subsequently solubilized with 10 mM Tris (pH 10.5)
and the absorbance was read at 515 nm on a Bio-Rad 550 ELISA
microplate reader (Bio-Rad Laboratories, Inc.). The optical density
(OD) was then analyzed with SPSS software 17.0 (IBM Corp.).
Western blot analysis
The cells were trypsinized, washed with PBS and then
lysed with buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl,
2 mM EDTA, 2 mM EGTA, 1 mM dithiothreitol, 1% Nonidet P-40, 0.1%
SDS, protease inhibitors (1 mM PMSF, 5 mg/ml aprotinin, 5 mg/ml
leupeptin and 5 mg/ml pepstatin) and phosphatase inhibitors (20 mM
β-glycerophosphate, 50 mM NaF, and 1 mM Na3VO4). The
tissues of nude mice (described below) were washed with PBS and
were then ground with the same lysis buffer. The lysates were
incubated at 4°C for 20 min and centrifuged at 12,000 × g for 15
min at 4°C. Equal amounts of the lysate (20 or 30 µg) were
resolved by a 10% sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to polyvinylidene
difluoride membranes (EMD Millipore). The membranes were blocked in
5% non-fat skim milk/TBST [20 mM Tris-HCl (pH7.4), 150 mM NaCl and
0.1% Tween-20] at room temperature for 2 h and detected with
primary anti-bodies at room temperature for 2 h. The membranes were
then blotted for 1 h at room temperature with an appropriate
horseradish peroxidase-linked secondary antibody (dilution 1:5,000,
ZB-2301, ZB-2305; ZSGB-Bio), followed by enhanced chemiluminescence
western blot detection reagents (Amersham Pharmacia Biotech).
Proteins were analyzed using Image Lab software (Bio-Rad
Laboratories, Inc.).
The primary antibodies, OGT (SAB2702273, dilution
1:1,000), OGA (HPA036141, dilution 1:1,000), O-GlcNAc
(MABS157, dilution 1:1,000), anti-AMPK α1 antibody (cat. no.
07-350, dilution 1:1,000), p-AMPK α (pThr172) (SAB4503754, dilution
1:1,000), p-serine (PSR-45) (P5747, dilution 1:1,000) were
purchased from Sigma-Aldrich; Merck KGaA. GFAT1 (D-9) (sc-377479,
dilution 1:1,000), glucose transporter member 1 (Glut1; sc-7903,
dilution 1:1,000), pyruvate kinase isozyme M2 (PKM2; sc-365684,
dilution 1:1,000), phosphofructokinase 1 (PFK1; sc-67028, dilution
1:1,000), HK2 (sc-374091, dilution 1:1,000) and β-actin (sc-47778,
dilution 1:1,000) were purchased from Santa Cruz Biotechnology,
Inc.
Immunoprecipitation assays
Cells were harvested in lysis buffer (50 mM Tris,
150 mM NaCl, 1% NP-40 and 0.5% sodium deoxycholate) and
supernatants incubated with rotation at 4°C with either anti-GFAT1
overnight. Protein G-agarose (Invitrogen; Thermo Fisher Scientific,
Inc.) was added the following day, and lysates were placed on the
rotator at 4°C for 4 h. Protein G-agarose beads were isolated by
centrifugation at 200 × g for 1 min at 4°C, washed 3 times with
lysis buffer and heated for 5 min at 100°C in loading buffer.
Samples were run on 10% SDS-PAGE and then probed by western blot
analysis for an antibody specific for the phosphorylation of
serine, as described above.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total mRNA was isolated from the H1299 cells using
TRIzol® reagent, and complementary DNA (cDNA) was
synthesized from 100 ng total RNA using the cDNA Synthesis kit
(Invitrogen; Thermo Fisher Scientific, Inc.). qPCR was performed
with SYBR-Green qPCR SuperMix (Invitrogen; Thermo Fisher
Scientific, Inc.). The sequences of the primers for OGT, OGA and
GAPDH amplification were as follows: OGT forward, 5′-TCC TGA TTT
GGT ACT GTG TTC GC-3′ and reverse, 5′-AAG CTA CTG CAA AGT TCG
GTT-3′; OGA forward, 5′-GAA GGA GAG TCA AGC GAC GTT-3′ and reverse,
5′-TCC ATA ACC CAA GGT CTT CCA T-3′; GAPDH forward, 5′-GGA GCG AGA
TCC CTC CAA AAT-3′ and reverse, 5′-GGC TGT TGT CAT ACT TCT CAT
GG-3′. The expression of OGA and OGT was normalized to that of
GAPDH and analyzed using the 2−ΔΔCq method (13).
Enzymatic activity assay of OGT and
OGA
OGT activity was measured in whole-cell lysates,
which were incubated with reaction buffer containing recombinant
glutathione S-transferase-tagged p62 and 100 µM UDP-GlcNAc
for 2 h, as previously described (14). Recombinant His-tagged p62 fragment
used as a substrate for OGT was extracted from Escherichia
coli (Tiangen Biotech Co., Ltd.) and purified on Ni-NTA His
Bind Resin (Merck KGaA). The reaction was terminated by the
addition of 2 mM glutathione, and supernatants were mixed with
SDS-PAGE sample buffer, subjected to 10% SDS-PAGE, and
immunoblotted using anti-O-GlcNAc antibodies.
OGA activity assays were performed as described in a
previous study (14). The
whole-cell lysates were incubated with assay buffer (50 mM sodium
cacodylate, pH6.5, 50 mM N-acetylgalactosamine, 2 mM
p-nitrophenyl-N-acetyl-D-glucosaminide) for 1 h at 37°C. The
reaction was terminated by the addition of 0.5 M sodium carbonate.
Hydrolyzed p-nitrophenol was measured at 400 nm using a
spectrophotometer (Agilent Technologies, Inc.).
Measurement of UDP-GlcNAc levels
The levels of UDP-GlcNAc were measured in cell
extracts as previously described with minor modifications (15). Cells were counted using a
hemocytometer (Sigma-Aldrich; Merck KGaA) and the cell extracts
were homogenized at room temperature for 10 min in 4 volumes of
perchloric acid (300 mM). The solution was adjusted to pH 7.0 with
1 M NaOH and then boiled for 10 min. The precipitates were
centrifuged at 13,800 × g for 10 min at room temperature. The lipid
was extracted from the supernatants with 2 volumes of
tri-n-octylamine:1,1,2-trichlorofluoroethane (1:4). The aqueous
phase was filtered through a 0.22-µM filter and then stored
at -80°C until analysis by HPLC. HPLC was performed on a YMC-Pack
Polyamine II HPLC Columns (250×4.6 mm, 5 µm), eluted with 10
mM potassium dihydrogen phosphate for 60 min at a flow rate of 0.5
ml/min. UDP-GlcNAc levels were quantified using a UV
spectrophotometer (Agilent Technologies, Inc.) at 254 nm, compared
with the standard curve.
Enzymatic activity assay of GFAT1
The enzymatic activity of GFAT was examined the
established glutamate dehydrogenase method (16). The generation of glutamate, one of
the products in the GFAT1 reaction, was assayed as the reduction of
acetylpyridine adenine dinucleotide (APAD) to APADH by the
glutamate dehydrogenase reaction with glutamate, which is
determined directly using a UV spectrophotometer (Agilent
Technologies, Inc.) at 370 nm. Briefly, the H1299 cells were
collected using GFAT buffer (50 mM Tris, 5 mM EDTA, 5 mM
glutathione, 5 mM D-Glucose 6-phosphate disodium salt hydrate and
50 mM KCl, pH 8.5). Cells were disrupted with an ultrasonic
cytometer and centrifuged at 13,800 × g for 10 min at 4°C. The
supernatants were collected for protein quantification and activity
assay. The aliquots of the cell lysate were incubated in 100
µl of the reaction mixture (10 mM fructose-6-phosphate, 6 mM
glutamine, 0.3 mM APAD, 50 mM KCl, 100 mM
KH2PO4 and 6 U of glutamate dehydrogenase) at
30°C for 2 h in a 96-well plate. The change in absorbance by the
reduction of APAD was monitored at 370 nm using a microplate
spectrometer (Model 680; Bio-Rad Laboratories, Inc.). The
absorbance values of the reaction mixture containing GFAT buffer
instead of the cell lysate were employed as a reference.
Intracellular glucose contents assay
The glucose oxidase method was used to measure the
glucose contents in the cells following the manufacturer's
instructions (Applygen Technologies, Inc.). Briefly, the H1299
cells were counted and the cell extracts were disrupted by
ultrasonic in buffer. The cell lysate was kept at 95°C for 10 min
and then centrifuged at 8,000 × g at 4°C. The supernatants were
used to determine the glucose contents.
Measurement of ATP and AMP/ATP
ratios
H1299 cells were treated in the presence or absence
of 8 µg/ml cisplatin or 2 µg/ml oligomycin for 24 h.
Endogenous levels of ATP and AMP in lysates of treated cells were
detected using an Enhanced ATP Assay kit following the
manufacturer's instructions (Shanghai Biyuntian Biological Co.,
Ltd.). The luciferase enzyme included in the assay was used to
catalyze the generation of light from ATP and luciferin. ADP was
measured by its conversion to ATP, which was detected using the
same reaction. The intracellular ATP and AMP contents were
calculated according to the standard curve made by ATP standards
and normalized to the cell number in each sample.
Inhibition of tumor growth in vivo
The research protocol was in accordance with the
institutional guidelines of the Animal Care and Use Committee of
Shandong University (no. 2016020, Jinan, China). The mice were
housed in pathogen-free ventilated cages and fed with standard
commercial diets and water in a temperature-controlled environment
(25±2°C) with a 12-h day/night cycle. The nude mouse experiment was
divided into the pre-experiment and formal experiment stage. The
purpose of the preliminary experiment was to determine the optimal
dosage of PUGNAc which can alter protein O-GlcNAcylation. In
the preliminary experiment, 40 female BALB/c (nu/nu) mice
(weighing, 20±2 g; 4-6 weeks old) were purchased from the Animal
Center of the China Academy of Medical Sciences (Beijing, China).
The H1299 lung cancer cells (5.0×106) suspended in 100
µl PBS, were subcutaneously inoculated into the lower right
flanks of the nude mice. When the tumors reached a volume of
100-150 mm3, 27 mice were used to determine the
appropriate dosage of PUGNAc. The mice were divided randomly into 9
groups (n=3 in each group) as follows: i) The control group (PBS);
ii) PUGNAc (1 mg/kg, 24 h) group; iii) PUGNAc (1 mg/kg, 48 h)
group; iv) PUGNAc (1 mg/kg, 72 h) group; v) PUGNAc (1 mg/kg, 96 h)
group; vi) PUGNAc (2 mg/kg, 24 h) group; vii) PUGNAc (2 mg/kg, 48
h) group; viii) PUGNAc (2 mg/kg, 72 h) group; and ix) PUGNAc (2
mg/kg, 96 h) group. PUGNAc (1 mg/kg or 2 mg/kg) was then injected
intravenously for each group, and the mice were sacrificed after
24, 48, 72 or 96 h, respectively. The tumor tissues were obtained
by dissection from mice that were subjected to cervical dislocation
following deep anesthesia by an intraperitoneal injection with
pentobarbital sodium injection (100 mg/kg). Protein O-GlcNAc
levels in tumors were measured by western blot analysis. In the
formal experiment, a total of 34 female BALB/c (nu/nu) mice
(weighing, 20±2 g; 4-6 weeks old) were purchased from the Animal
Center of the China Academy of Medical Sciences (Beijing, China).
The H1299 lung cancer cells (5.0×106) suspended in 100
µl PBS, were subcutaneously inoculated into the lower right
flanks of the nude mice. When the tumors reached a volume of
100-150 mm3, 24 mice were divided randomly into 4 groups
(n=6 in each group): Control group (PBS), CDDP group, PUGNAc group
and CDDP + PUGNAc group. The mice were injected intravenously 4
times with 100 µl of PBS, CDDP (6 mg/kg), PUGNAc (1 mg/kg),
or a combination of CDDP (6 mg/kg) and PUGNAc (1 mg/kg)
respectively during the 16 days of treatment. The doses of PUGNAc
were determined based on the preliminary experiment and a previous
study (17). The diameter of the
tumor was measured twice a week using a caliper. Tumor volume was
calculated with the following formula: v=ab2/2, where
'a' and 'b' are the long diameter and the perpendicular short
diameter of the tumor, respectively. The body weights were measured
twice a week. The mice with tumor volumes >2,000 mm3
or a weight loss of >20% were sacrificed in advance. On the 16th
day, all mice were injected with pentobarbital sodium injection
(100 mg/kg) intraperitoneally, and were then subjected to cervical
dislocation following deep anesthesia. Tumor, liver and lung
tissues from mice were obtained by dissection.
Statistical analysis
SPSS software version 17.0 (IBM Corp.) and GraphPad
Prism 6 (GraphPad Software, Inc.) were used for statistical
analysis. All data represent the means ± SD of 3 independent
experiments. Statistical tests included independent a samples
t-test and one-way ANOVA with post hoc Tukey's test. P-values
<0.05 were considered to indicate statistically significant
differences.
Results
Chemotherapeutic agents increase global
protein O-GlcNAcylation
To investigate whether chemotherapeutic agents lead
to increases in total protein O-GlcNAcylation in various
types of cancer cells, the H1299, HepG2 and MCF-7 cells were
treated with cisplatin, adriamycin and vincristine for 24 h,
respectively. The results of western blot analysis revealed that
all cancer cells exhibited an elevation in O-GlcNAc levels
upon treatment with the different cytotoxic drugs; vincristine
treatment led to a slight increase in O-GlcNAc levels, with
no significant difference (Fig.
1A). In addition, the effect of cisplatin on the
O-GlcNAc levels was examined in the multi-drug resistant
cells, H1299/Taxol. O-GlcNAcylation was enhanced upon
treatment of the H1299/Taxol cells with cisplatin at high
concentrations (Fig. 1B).
Furthermore, cisplatin increased the O-GlcNAc levels in both
a time- and concentration-dependent manner in the H1299 cells
(Fig. 1C and D). In the present
study, cisplatin at concentrations of 2, 8 and 16 µg/ml
inhibited cell growth by ~10, 45 and 67% after the H1299 cells were
treated for 48 h (Fig. S1). The
O-GlcNAc levels increased gradually and reached maximal
levels at 24 h and then declined to baseline levels at 72 h when
the H1299 cells were exposed to 8 µg/ml cisplatin for
various periods of time (Fig.
1C). These results suggested that the the global protein
O-GlcNAc levels were upregulated in cancer cells in response
to certain types of chemotherapeutic agents.
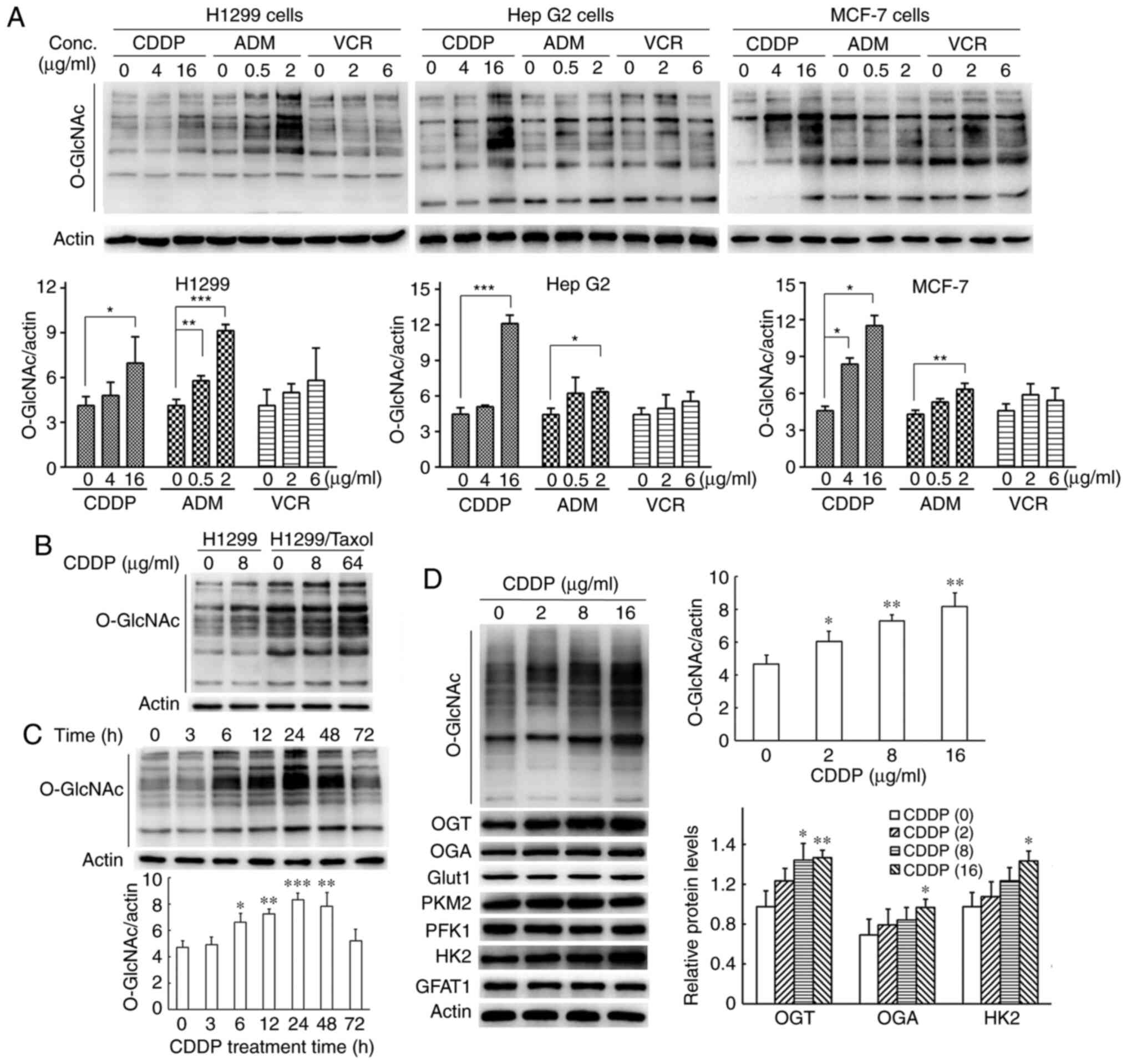 | Figure 1Chemotherapeutic agents increase
global protein O-GlcNAc levels in cancer cells. (A) The lung
cancer cell line, H1299, hepatoblastoma cell line, Hep G2, and the
breast cancer cell line, MCF-7, were treated with cisplatin,
adriamycin and vincristine at various concentrations for 24 h.
Total cell extracts were harvested for western blot analysis.
Protein O-GlcNAc levels were semi-quantified by densitometry
and normalized against those of β-actin. (B) The multi-drug
resistant cancer cells, H1299/Taxol, and parental cells, H1299,
were treated with cisplatin for 24 h. Total cell extracts were
harvested for western blot analysis. (C) H1299 cells were exposed
to 8 µg/ml cisplatin for the indicated times and then lysed
for western blot analysis. Protein O-GlcNAc levels were
semi-quantified by densitometry and normalized against that of
β-actin. (D) H1299 cells were exposed to cisplatin for 24 h at the
indicated concentrations and then lysed for western blot analysis.
Protein O-GlcNAcylation and other protein expression levels
were semi-quantified by densitometry and normalized against that of
β-actin. All data are shown as the means ± SD of 3 independent
experiments. *P<0.05, **P<0.01,
***P<0.001 vs. control. CDDP, cisplatin; AMD,
Adriamycin; VCR, vincristine; OGT, O-GlcNAc transferase;
OGA, O-GlcNAcase. |
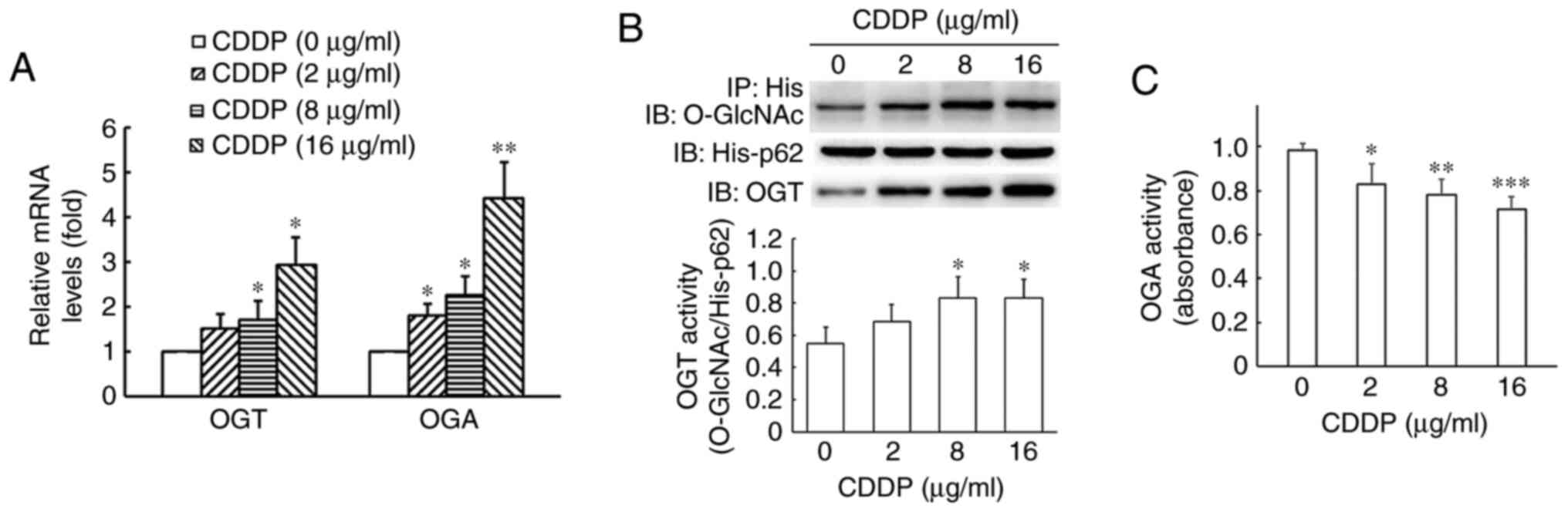
Cisplatin increases the enzymatic
activity of OGT and OGA
As shown in Fig.
1D, the results of western blot analysis revealed that
cisplatin increased both the OGT and OGA protein levels in the
H1299 cells. Moreover, the OGT and OGA mRNA levels were
significantly enhanced following treatment of the H1299 cells with
cisplatin (Fig. 2A). OGT and OGA
activity assays were then performed with total lysates from the
H1299 cells. OGT activity toward the p62 peptide substrate was
increased; however, OGA activity toward the PNP-GlcNAc substrate
gradually decreased and was 83.9, 78.1 and 71.5% of the control
following treatment of the H1299 cells with cisplatin at the
concentrations of 2, 8 and 16 µg/ml for 24 h (Fig. 2B and C).
Alloxan, an inhibitor of OGT, markedly downregulated
the global O-GlcNAc levels and reversed the
cisplatin-induced increase in O-GlcNAc levels in the H1299
cells (Fig. 3A). The results of
SRB assay revealed that the decrease in O-GlcNAc levels
induced by alloxan did not result in any changes in cisplatin
cytotoxicity (Figs. 3B and
S2A). In addition, the OGA
inhibitor, PUGNAc, was used to increase the global protein
O-GlcNAc levels. Although treatment with 100 µM
PUGNAc led to a 2.2-fold increase in the O-GlcNAc levels in
the H1299 cells compared with the control (Fig. 3C), PUGNAc alone did not affectd
cell survival (Fig. S2B). PUGNAc
enhanced the cisplatin-induced elevation in O-GlcNAc levels;
however, combined treatment with PUGNAc and cisplatin induced the
same cell growth rate as with cisplatin treatment alone (Fig. 3C and D). The aforementioned
results suggested that alteration of global O-GlcNAc levels via the
inhibition of OGT or OGA did not influence the sensitivity of H1299
cells to cisplatin.
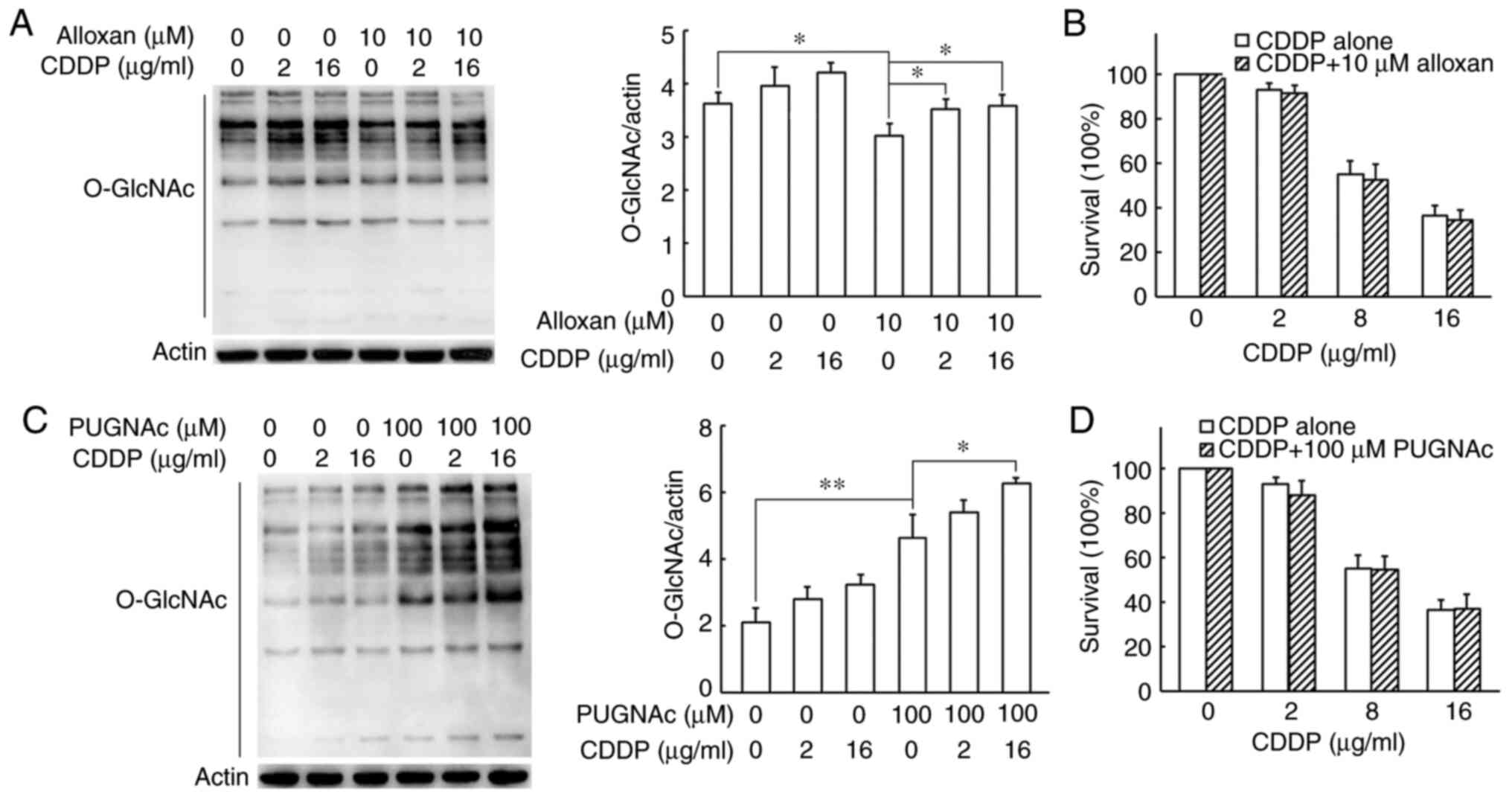 | Figure 3Effects of inhibition of OGT and OGA
on cisplatin-induced O-GlcNAcylation and cisplatin
cytotoxicity in H1299 cells. (A) H1299 cells were treated with
cisplatin alone or in combination with the OGT inhibitor, alloxan,
for 24 h. Cells were collected and lysed for western blot analysis.
Protein O-GlcNAc levels were semi-quantified by densitometry
and normalized against those of β-actin. (B) In SRB assay, H1299
cells were exposed to 0, 2, 8 or 16 µg/ml cisplatin or the
combination with 10 µM of alloxan for 48 h respectively. (C)
H1299 cells were treated with cisplatin alone or in combination
with the OGA inhibitor, PUGNAc for 24 h. Cells were collected and
lysed for western blot analysis. Protein O-GlcNAc levels
were semi-quantified by densitometry and normalized against those
of β-actin. (D) SRB assay was performed in H1299 cells. The cells
were exposed to 0, 2, 8 or 16 µg/ml cisplatin or the
combination with 100 µM of PUGNAc for 48 h respectively. All
data are shown as the means ± SD of 3 independent experiments.
*P<0.05, **P<0.01. CDDP, cisplatin;
OGT, O-GlcNAc transferase; OGA, O-GlcNAcase. |
To further explore the mechanisms underlying the
upregulation of the global O-GlcNAc levels induced by
cisplatin, the protein expression levels of enzymes related to
glycolysis and the HBP were examined. The results of western blot
analysis revealed that only HK2 expression was increased upon
cisplatin treatment and no changes were observed in the protein
levels of enzymes, such as Glut1, PFK1, PKM2 and GFAT1 (Fig. 1D).
Cisplatin increases UDP-GlcNAc via the
activation of GFAT1 and the inhibition of AMPK activity
It is commonly known that the activity of OGT is
sensitive to the intracellular concentration of UDP-GlcNAc.
Therefore, intracellular UDP-GlcNAc was measured using HPLC in
H1299 cells following cisplatin treatment. Compared with the
control cells, intracellular UDP-GlcNAc levels were enhanced 1.77-,
2.29- and 2.85-fold when the H1299 cells were exposed to cisplatin
at 2, 8 and 16 µg/ml for 24 h (Fig. 4A). The UDP-GlcNAc levels increased
as the duration of treatment increased and were 5.10-fold greater
at 72 h than at 0 h (Fig.
4B).
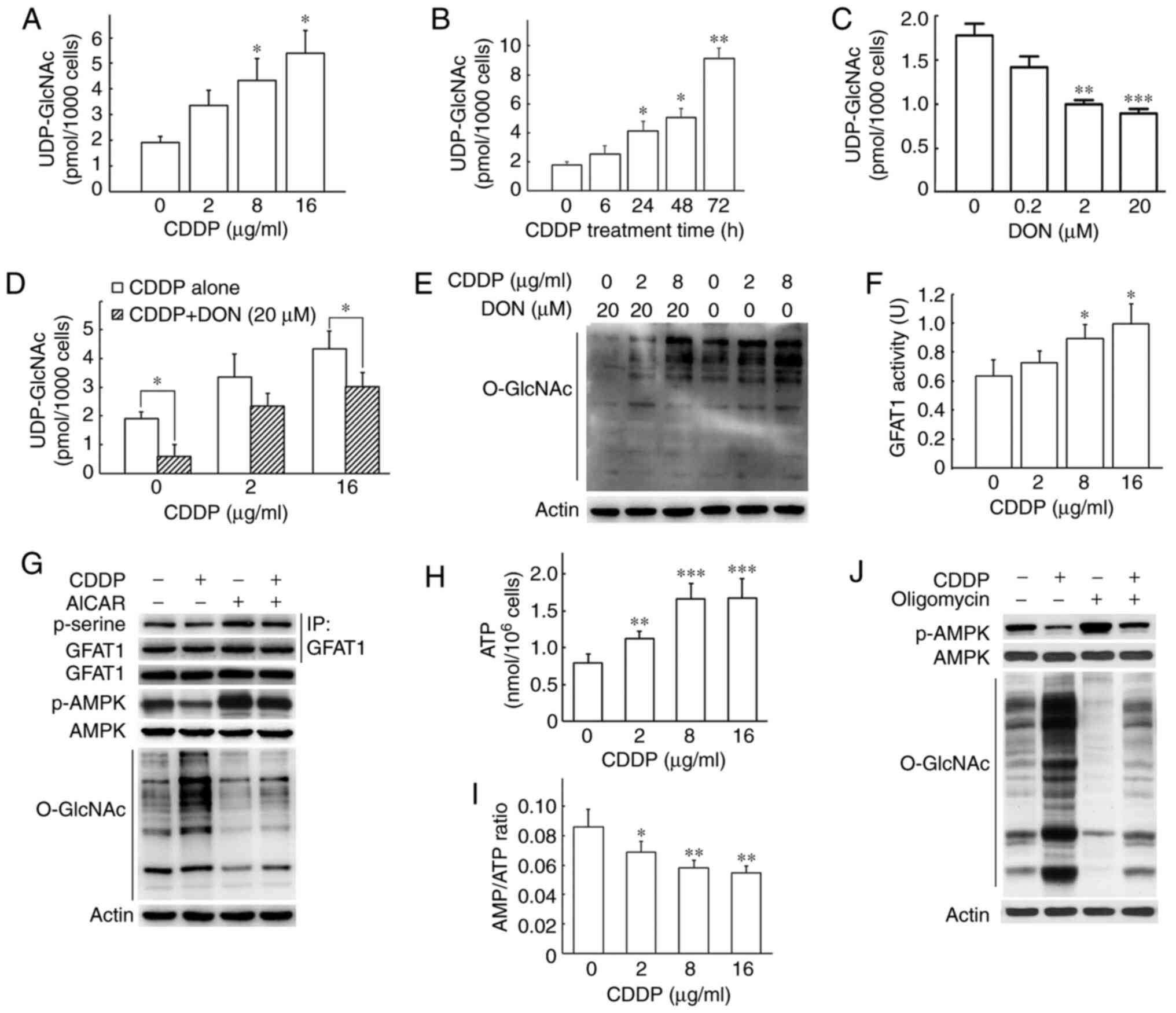 | Figure 4Cisplatin treatment enhances the
intracellular UDP-GlcNAc content by altering the activity of AMPK
and GFAT1 in H1299 cells. (A) Cisplatin increased intracellular
UDP-GlcNAc levels. UDP-GlcNAc was measured using HPLC after the
H1299 cells were treated with cisplatin for 24 h at the indicated
concentrations. (B) Intracellular UDP-GlcNAc was measured using
HPLC after the H1299 cells were treated with 8 µg/ml
cisplatin for the indicated periods of time. (C) The GFAT1
inhibitor, DON, inhibited the production of UDP-GlcNAc. H1299 cells
were treated with DON for 24 h and the cell lysate was used for
HPLC analysis. (D) DON prevented the cisplatin-induced increase in
UDP-GlcNAc levels. H1299 cells were treated with cisplatin in the
presence or absence of DON for 24 h and the cell lysate was used
for HPLC analysis. (E) DON prevented cisplatin-induced increase in
protein O-GlcNAcylation. H1299 cells were treated with
cisplatin in the presence or absence of DON for 24 h and the cell
lysate was used for western blot analysis. (F) Cisplatin enhanced
GFAT1 activity. GFAT1 activity was measured after the H1299 cells
were treated with cisplatin at the indicated concentrations. The
supernatants of cell lysate were mixed with reaction buffer and the
absorbance was monitored at 370 nm using a microplate spectrometer.
(G) Cisplatin counteracted the AMPK-induced phosphorylation of
GFAT1. H1299 cells were treated with cisplatin in the presence or
absence of the AMPK activator, AICAR, for 24 h. The cell lysate was
used for immunoprecipitation and western blot analysis. GFAT1 was
first enriched by immunoprecipitation, and then p-serine antibody,
a broad-spectrum phosphorylated serine antibody, was used to detect
the phosphorylation of GFAT1. (H and I) Cisplatin decreased the
AMP/ATP ratio. Intracellular ATP and AMP were detected after H1299
cells were treated with cisplatin for 24 h. H1299 cells cultured in
a 96-well plate were mixed with reaction buffer containing
luciferin and luciferase and light was measured using a
luminometer. (J) The AMP/ATP ratio affected cisplatin-induced AMPK
activation and protein O-GlcNAc levels. H1299 cells were
treated with cisplatin (8 µg/ml) in the presence or absence
of ATP synthase oligomycin (2 µg/ml) for 24 h. The cell
lysate was used for western blot analysis. All data are shown as
the means ± SD of 3 independent experiments. *P<0.05,
**P<0.01, ***P<0.001 vs. control. CDDP,
cisplatin; AMPK, AMP-activated protein kinase; GFAT1,
glutamine-fructose-6-phosphate aminotransferase (isomerizing) 1;
DON, 6-diazo-5-oxo-L-nor-Leucine; AICAR,
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside. |
DON is a glutamine analogue that selectively
inactivates glutamine-utilizing pathways to irreversibly inhibit
GFAT1 activity (18,19). As shown in Fig. 4C, DON significantly downregulated
the UDP-GlcNAc levels in a concentration-dependent manner in the
H1299 cells. Moreover, DON significantly reversed the
cisplatin-induced elevation in the UDP-GlcNAc levels, thereby
resulting in significant decreases in global O-GlcNAc levels
in the H1299 cells treated with DON and cisplatin (Fig. 4D and E). Taken together, these
findings indicated that cisplatin resulted in the elevation of
intracellular UDP-GlcNAc, which was involved in the regulation of
global protein O-GlcNAcylation.
GFAT1 is the first step of the HBP and the
rate-limiting enzyme for the synthesis of UDP-GlcNAc, the end
product of the HBP. The GFAT1 enzymatic activity assay using cell
lysates indicated that cisplatin treatment enhanced GFAT1 activity
in a concentration-dependent manner (Fig. 4F). AICAR, an activator of AMPK,
can reduce GFAT1 activity by enhancing GFAT1 phosphorylation
(20). In the present study,
GFAT1 was enriched in advance by immunoprecipitation, and the
phosphorylation of GFAT1 was then detected by a p-serine anti-body
to reflect its activation. Treatment with cisplatin not only
decreased GFAT1 phosphorylation, but also counteracted the
AICAR-induced increase in GFAT1 phosphorylation (Fig. 4G), suggesting that cisplatin
upregulated GFAT1 activity by inhibiting GFAT1 phosphorylation. As
shown in Fig. 4G, cisplatin
decreased AMPK phosphorylation. The phosphorylation of Thr172 is
the hallmark of AMPK activation. Furthermore, the AICAR-induced
AMPK activation decreased the O-GlcNAc levels, an effect
which was prevented by cisplatin treatment, indicating that the
cisplatin-induced inactivation of AMPK was involved in the
upregulation of O-GlcNAc levels in cells treated with
cisplatin (Fig. 4G). AMPK is
primarily involved in monitoring cellular energy status by sensing
the AMP/ATP and/or ADP/ATP ratios (21). Thus, the present study measured
the ATP and AMP levels in H1299 cells treated with cisplatin.
Cisplatin led to an increase in ATP levels and to a significant
decrease in the ratio of AMP/ATP (Fig. 4H and I). Intracellular ATP is
produced either through glycolysis or through oxidative
phosphorylation in the mitochondria. However, ATP is mainly
produced by ATP synthase in human cells (22). Oligomycin is a known inhibitor of
the membrane motor of the mitochondrial ATP synthase for >50
years (23). In the present
study, oligomycin treatment at a concentration of 2 µg/ml
led to a 14% decrease in cell viability, decreased the
intracellular ATP contents from 0.81 nmol/105 cells to
0.54 nmol/105 cells and increased the AMP/ATP ratio from
0.084 to 0.113, compared with the H1299 cells without oligomycin
treatment (Fig. S3). As
oligomycin decreased ATP production, oligomycin treatment alone
enhanced AMPK activation and reduced the global O-GlcNAc
levels (Fig. 4J). Furthermore,
oligomycin abrogated the cisplatin-induced enhancement in the
O-GlcNAc levels, suggesting that AMPK activation was
downregulated by the cisplatin-induced decrease in the AMP/ATP
ratio (Fig. 4J). Taken together,
these findings demonstrated that cisplatin enhanced the
intracellular UDP-GlcNAc level by modulating the AMP/AMPK/GFAT1
pathway.
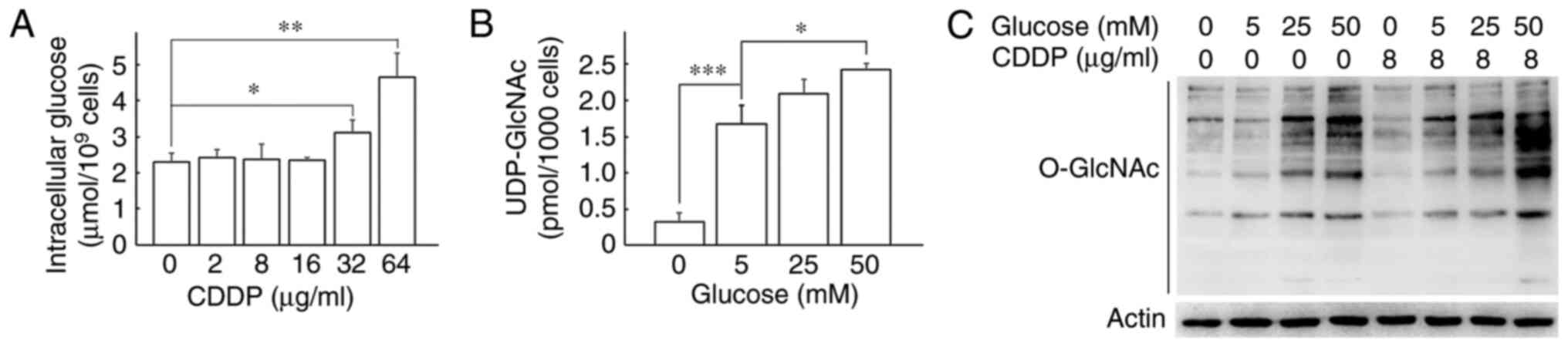
Glucose consumption is not involved in
the increase in cisplatin-induced O-GlcNAcylation
As GFAT1 is mostly activated by high glucose levels,
the present study then investigated whether cisplatin promotes
glucose uptake in H1299 cells. Intracellular glucose contents were
measured after the cells were treated with cisplatin for 24 h. As
shown in Fig. 5A, the glucose
contents were unaltered following cisplatin treatment at 2, 8 and
16 µg/ml; however, treatment with 32 and 64 µg/ml
cisplatin led to significant increases in the glucose contents. The
intracellular glucose level did not exhibit any marked changes
after the H1299 cells were exposed to 8 µg/ml CDDP for 0, 6,
12, 24, 48 and 72 h (Fig. S4).
Subsequently, the effects of extracellular glucose on the
intracellular UDP-GlcNAc level and protein O-GlcNAcylation
in the H1299 cells were examined. As shown in Fig. 5B and C, the intracellular
UDP-GlcNAc level and the protein O-GlcNAc levels were
gradually enhanced when the cells were cultured in medium with
increasing glucose concentrations. Cells cultured in 0 mM glucose
exhibited the lowest UDP-GlcNAc levels, whereas those cultured in
25 mM glucose demonstrated 6.27-fold greater UDP-GlcNAc levels than
the cells cultured in 0 mM glucose. The cells cultured in high
glucose exhibited increased cisplatin-induced O-GlcNAc
levels than the cells cultured in low glucose. The aforementioned
results demonstrated that the upregulation of both GFAT1 and
UDP-GlcNAc were not due to glucose consumption in H1299 cells.
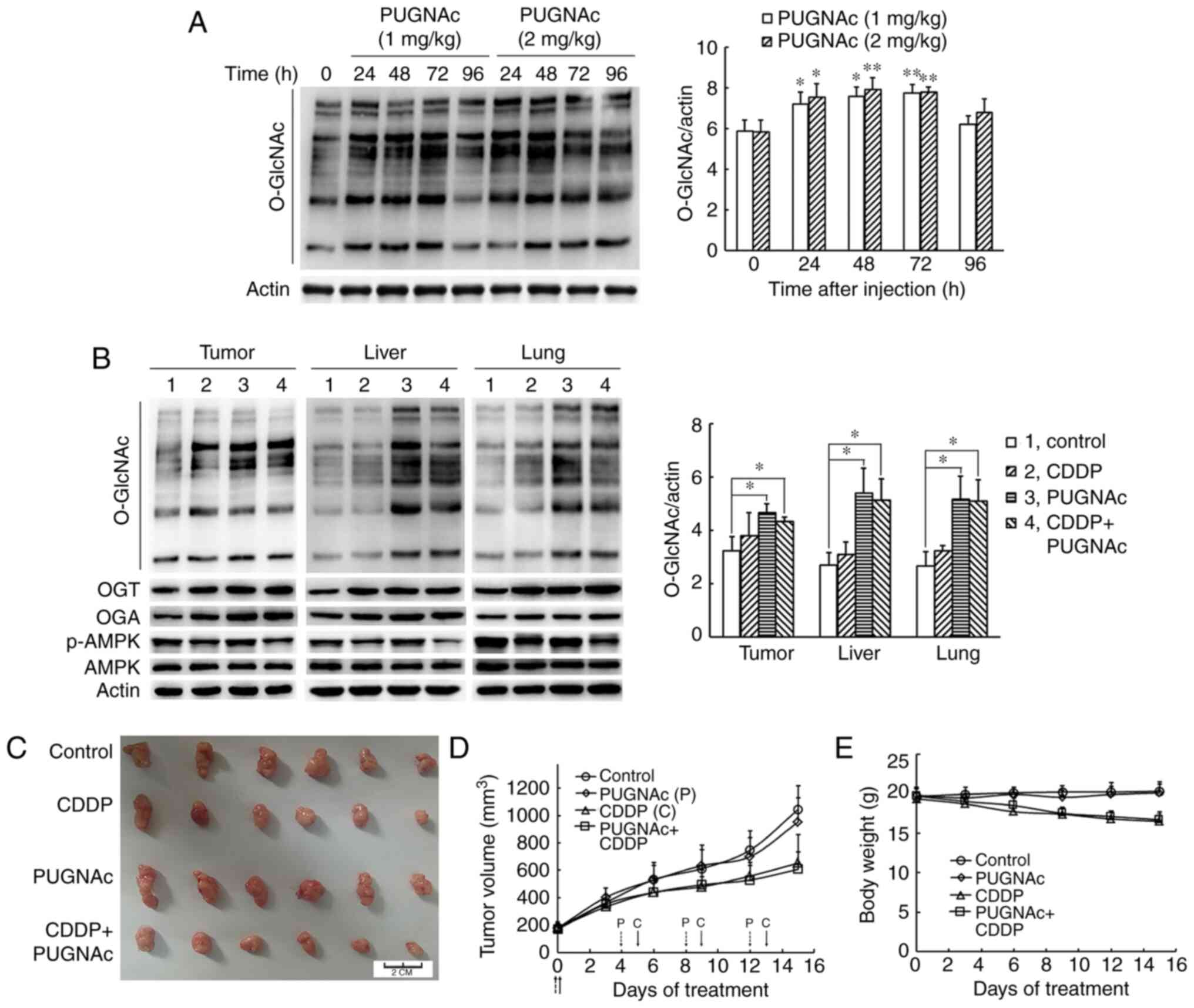
Cisplatin-induced elevation of protein
O-GlcNAc does not sensitize cancer cells to cisplatin in vivo
In order to examine whether protein
O-GlcNAcylation affects the antitumor effect of cisplatin
in vivo, the OGA inhibitor, PUGNAc, was selected to enhance
the global protein O-GlcNAc levels. First, the
concentrations and duration of PUGNAc administration were
determined. In the preliminary experiment using nude mice bearing
H1299 cells, the mice were sacrificed at 24, 48, 72 and 96 h
following the injection of 1 and 2 mg/kg PUGNAc via the tail vein,
and the O-GlcNAc levels in the tumors were measured. As shown in
Fig. 6A, treatment with both 1
and 2 mg/kg PUGNAc significantly increased the global
O-GlcNAc levels at 24 h, and these high levels were
maintained for 72 h in the tumor cells, suggesting that PUGNAc had
the potential to be used to enhance O-GlcNAc levels in mice.
Subsequently, 1 mg/kg PUGNAc was administered via the tail vein on
days 0, 4, 8 and 12. Cisplatin at a dose of 6 mg/kg body weight was
administered on days 0, 5, 9 and 13. All nude mice were sacrificed
on day 16. Consistent with the results observed in vitro,
cisplatin treatment notably enhanced the OGT and OGA protein
expression levels in the tissues of nude mice. Moreover, cisplatin
decreased the AMPK phosphorylation levels (Fig. 6B). As compared with the PBS
control group, the O-GlcNAc levels were significantly
increased in the tumor, liver and lung tissues of the
PUGNAc-treated group, as well as in the group treated with PUGNAc
and cisplatin (Fig. 6B). Although
PUGNAc promoted O-GlcNAcylation, PUGNAc alone did not affect
the tumor growth and bodyweight of the mice. The dosage of 6 mg/kg
cisplatin administered 4 times resulted in a 14.7% weight loss.
Cisplatin treatment alone inhibited tumor growth by 37.5% and
combined treatment with cisplatin and PUGNAc inhibited tumor growth
by 41.8%, indicating that the enhanced O-GlcNAcylation did
not affect the sensitivity of tumor cells to cisplatin in
vivo (Fig. 6C-E).
Discussion
Protein O-GlcNAc modification plays crucial
regulatory roles in cellular signaling. Global protein
O-GlcNAc levels are increased in response to numerous types
of cellular stresses, including drug treatment (6). However, less is known concerning the
underlying mechanisms through which chemotherapeutic agents affect
the O-GlcNAcylation in cancer cells. The present study
illustrated that cisplatin treatment elevated the global
O-GlcNAc levels in lung cancer cells in a
concentration-dependent manner by regulating the enzymatic activity
of OGT and OGA, and increased the intracellular UDP-GlcNAc levels
via the inhibition of AMPK activation. It was also demonstrated
that alterations in O-GlcNAc levels via the inhibition of
OGT and OGA did not affect the sensitivity of lung cancer cells to
cisplatin.
In the present study, the effect on the levels of
O-GlcNAc by CDDP was more evident in MCF-7 and HepG2 cell
lines, than in H1299. However, mouse tumor xenograft models using
mice injected with H1299 cells were constructed in advance; thus,
only the H1299 cells were selected for use in subsequent
experiments, which is a limitation of the present study. As another
potential limitation, it would have been prudent to compare the
O-GlcNAc level in cisplatin-sensitive cells and
cisplatin-resistant cells. However, due to the unsuccessful
development of cisplatin-resistant H1299 cells by the long-term
exposure of H1299 cells to an increasing concentration of CDDP, the
H1299/Taxol cell line was used in the experiments, which is
multi-drug resistant cell line. In the future, the authors aim to
focus on the effects on the O-GlcNAc levels between
cisplatin-sensitive and cisplatin-resistant cells.
In the present study, the global O-GlcNAc
levels were elevated upon the cisplatin treatment of H1299 lung
cancer cells in a concentration- and time-dependent manner.
Cisplatin enhanced the protein expression, mRNA levels and
enzymatic activity of OGT. Although >1,000 proteins have been
found to be modified with O-GlcNAc, only OGT and OGA are
involved in the process to add or remove the moiety of
O-GlcNAc. OGT is directly regulated by the concentration of
UDP-GlcNAc, with its activity increasing as UDP-GlcNAc levels
increase. Moreover, the substrate specificity of OGT changes at
different UDP-GlcNAc concentrations (24). In the present study, the amount of
intracellular UDP-GlcNAc was gradually upregulated after the H1299
cells were exposed to cisplatin for different periods of time.
These data demonstrated that the enhanced OGT activity was
principally regulated by the elevation of UDP-GlcNAc. It is also
important to note that the increase in intracellular UDP-GlcNAc
levels is likely to be cell-dependent. The levels of intracellular
UDP-GlcNAc are early markers of cisplatin treatment in brain tumor
cells, while levels remain unaltered in resistant cells (25). Duarte et al reported that
the treatment of lung cancer cells A549 with cisplatin for 48 h
resulted in a 2-fold elevation of UDP-GlcNAc levels; however, this
effect was not observed in osteosarcoma cells treated with a
comparable dose of cisplatin (26,27). The induction of UDP-GlcNAc by
other chemotherapeutic drugs remains to be demonstrated.
Notably, in the present study, cisplatin also
increased the OGA protein and mRNA levels, but significantly
decreased OGA activity. At present, little is known about the
regulation of OGA, although it can be cleaved by caspase-3 and is
O-GlcNAcylated, phosphorylated, ubiquitinated and
acetylated. However, the impact of these modifications on the
localization, substrate specificity or activity of OGA has not been
reported (6).
GFAT is the first step of the HBP and a
rate-limiting enzyme that plays a key role in the regulation of the
glucose through the HBP. Among the three identified human GFAT
isoforms, GFAT1 is the major form that is ubiquitously expressed.
GFAT1 is overexpressed in various types of cancer, and GFAT1
overexpression predicts a worse progression and pathological
outcomes in various types of cancer (28,29). The present study illustrated that
the elevation in the UDP-GlcNAc level induced by cisplatin was a
result of an enhanced GFAT1 activity, rather than changes in
intracellular glucose. Cisplatin dose-dependently upregulated GFAT1
activity; moreover, cisplatin inhibited the phosphorylation of
GFAT1. This result is consistent with the findings of previous
studies, which demonstrated that GFAT1 phosphorylation
downregulates its activity (16,20,30). Relatively, the regulation of GFAT1
is complex; it is known that it is phosphorylated by two kinases,
cAMP-dependent protein kinase at serine 205 and by AMPK at serine
243. AMPK is a conserved sensor of cellular energy changes and is
activated by increased AMP/ATP and/or ADP/ATP ratios. In the
present study, cisplatin treatment resulted in an enhanced ATP
content and a decreased AMP/ATP ratio, suggesting that decreased
AMP/ATP inhibited AMPK activation.
In the present study, the inhibitor of GFAT1, DON,
abrogated the cisplatin-induced elevation of UDP-GlcNAc; therefore,
DON significantly prevented the increase in global O-GlcNAc
levels induced by cisplatin in H1299 cells. A recent study
demonstrated that the inhibition of GFAT1 activity by DON
suppressed cell proliferation and exerted a synergic or additive
effect with cisplatin in inducing cancer cell death (11). Another recent study revealed that
treatment with DON sensitized pancreatic tumors cells to anti-PD1
therapy, resulting in tumor regression and prolonged survival
(31). The role of GFAT1 in
cancer has also drawn increasing attention in recent years. Recent
data indicated that GFAT1 inhibitors were effective in cancer
treatment, indicating that targeting GFAT1 may provide novel
adjuvant approaches for the clinical treatment of cancer (32).
The flux through the HBP and thus the synthesis of
UDP-GlcNAc is regulated mainly due to the metabolism of glucose
(33). The present study found
that cisplatin at concentrations of 2, 8 and 16 µg/ml did
not alter intracellular glucose consumption; however, the global
O-GlcNAc levels were increased in a concentration-dependent
manner upon cisplatin treatment at the same concentrations in H1299
cells. Compared with cells cultured in low glucose, the
cisplatin-induced O-GlcNAc levels were higher when the H1299
cells were cultured in medium containing a higher glucose
concentration. Moreover, a higher glucose concentration resulted in
both increased UDP-GlcNAc and protein O-GlcNAcylation than
the lower glucose concentration. The aforementioned results
confirmed that the cisplatin-induced increase in the protein
O-GlcNAc level was not related to glucose consumption. A
previous study demonstrated that treatment with 0.6 µg/ml
cisplatin for 48 h resulted in a 1.5-fold increase in glucose
uptake in cisplatin-sensitive cells and no changes in
cisplatin-resistant cell lines (34). However, another study reported
that cisplatin decreased glucose uptake and 5-fluorouracil
upregulated glucose metabolism in lung cancer A549 cells (35). Collectively, glucose uptake is
cell-specific in response to the treatment with chemotherapeutic
agents, including cisplatin. In the present study, glucose
consumption was not involved in the upregulation of
O-GlcNAcylation.
A number of studies have suggested that global
protein O-GlcNAc levels are transiently elevated in response
to moderate stress stimuli and the elevated O-GlcNAc levels
promote cell survival (24,36). By contrast, decreased
O-GlcNAc levels of sensitize cells and tissues to apoptosis
and necrosis (8). In the present
study, the data demonstrated that the changes in O-GlcNAc
levels via the inhibition of OGT or OGA did not affect the
sensitivity of H1299 cells to cisplatin in vitro and in
vivo. Alloxan, an inhibitor of OGT, significantly counteracted
the cisplatin-induced increase in O-GlcNAc levels; however,
the changes in O-GlcNAc levels did not affect the growth
inhibitory effect of cisplatin on H1299 cells. At the
concentrations tested in the present study, the OGA inhibitor,
PUGNAc, was non-toxic and had no effect on the growth rate of H1299
cells. It was also previously reported that PUGNAc did not affect
OGT activity and UDP-GlcNAc levels (37). Although PUGNAc markedly enhanced
the cisplatin-induced O-GlcNAc level in the present study,
combined treatment with cisplatin and PUGNAc inhibited cell growth
at the same rate as treatment with cisplatin alone in H1299 cells.
Similarly, in nude mice injected with H1299 cancer cells, the group
treated with both cisplatin and PUGNAc exhibited no changes in
tumor growth inhibition compared with the group treated with
cisplatin alone. The aforementioned results demonstrated that the
turnover of O-GlcNAc was not essential for tumor growth, and
the changes in O-GlcNAc levels did not affect the
sensitivity of H1299 cells to cisplatin. The study by Zhou et
al demonstrated that the downregulation of OGT increased
cisplatin resistance in ovarian cancer, but had no effect on the
efficacy of paclitaxel (12). By
contrast, another study revealed that reducing
hyper-O-GlcNAcylation by OGT knockdown facilitated the
chemosensitivity of bladder cancer cells to cisplatin (38). These results indicated that the
effects of O-GlcNAc on the sensitivity of cells to
chemotherapeutic agents need to be further explored in the
future.
Taken together, the present study demonstrated that
cisplatin augmented the global protein O-GlcNAc levels by
altering the enzymatic activity of OGT and OGA. Cisplatin-reduced
AMPK activation prevented GFAT1 phosphorylation and then promoted
the activity of GFAT1. Cisplatin-induced GFAT1 activation improved
production of the donor substrate UDP-GlcNAc through the HBP. The
alteration of O-GlcNAcylation did not affect the sensitivity
of lung cancer cells to cisplatin in vitro and in
vivo. These findings may prove to be useful in enhancing the
current understanding of the roles of O-GlcNAcylation in
chemotherapy.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
DiW and YS designed the study, planned the
experiments, analyzed the data and wrote the manuscript. DiW, JW,
DaW, XH and NZ performed the experiments. All authors have read and
approved the final manuscript. DiW and JW confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of the Department of Laboratory Animal
Science of Shandong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by Shandong Province Major
Science and Technology Innovation Project (grant no. 2018CXGC1402),
and Fundamental Research Projects of Shandong University (grant no.
2017JC022).
References
|
1
|
Wells L and Hart GW: O-GlcNAc turns
twenty: Functional implications for post-translational modification
of nuclear and cytosolic proteins with a sugar. FEBS Lett.
546:154–158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joiner CM, Li H, Jiang J and Walker S:
Structural characterization of the O-GlcNAc cycling enzymes:
Insights into substrate recognition and catalytic mechanisms. Curr
Opin Struct Biol. 56:97–106. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nie H and Yi W: O-GlcNAcylation, a sweet
link to the pathology of diseases. J Zhejiang Univ Sci B.
20:437–448. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanover JA, Chen W and Bond MR: O-GlcNAc
in cancer: An Oncometabolism-fueled vicious cycle. J Bioenerg
Biomembr. 50:155–173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hart GW: Nutrient regulation of signaling
and transcription. J Biol Chem. 294:2211–2231. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martinez MR, Dias TB, Natov PS and Zachara
NE: Stress-induced O-GlcNAcylation: an adaptive process of injured
cells. Biochem Soc Trans. 45:237–249. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee A, Miller D, Henry R, Paruchuri VD,
O'Meally RN, Boronina T, Cole RN and Zachara NE: Combined
anti-body/Lectin enrichment identifies extensive changes in the
O-GlcNAc sub-proteome upon oxidative stress. J Proteome Res.
15:4318–4336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Groves JA, Maduka AO, O'Meally RN, Cole RN
and Zachara NE: Fatty acid synthase inhibits the O-GlcNAcase during
oxidative stress. J Biol Chem. 292:6493–6511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zachara NE, Molina H, Wong KY, Pandey A
and Hart GW: The dynamic stress-induced 'O-GlcNAc-ome' highlights
functions for O-GlcNAc in regulating DNA damage/repair and other
cellular pathways. Amino Acids. 40:793–808. 2011. View Article : Google Scholar
|
|
10
|
Chen SH and Chang JY: New insights into
mechanisms of cisplatin resistance: From tumor cell to
microenvironment. Int J Mol Sci. 20:41362019. View Article : Google Scholar :
|
|
11
|
Chen W, Do KC, Saxton B, Leng S, Filipczak
P, Tessema M, Belinsky SA and Lin Y: Inhibition of the hexosamine
biosynthesis pathway potentiates cisplatin cytotoxicity by
decreasing BiP expression in non-small-cell lung cancer cells. Mol
Carcinog. 58:1046–1055. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou F, Yang X, Zhao H, Liu Y, Feng Y, An
R, Lv X, Li J and Chen B: Down-regulation of OGT promotes cisplatin
resistance by inducing autophagy in ovarian cancer. Theranostics.
8:5200–5212. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Kang JG, Park SY, Ji S, Jang I, Park S,
Kim HS, Kim SM, Yook JI, Park YI, Roth J and Cho JW: O-GlcNAc
protein modification in cancer cells increases in response to
glucose deprivation through glycogen degradation. J Biol Chem.
284:34777–34784. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taylor RP, Geisler TS, Chambers JH and
McClain DA: Up-regulation of O-GlcNAc transferase with glucose
deprivation in HepG2 cells is mediated by decreased hexosamine
pathway flux. J Biol Chem. 284:3425–3432. 2009. View Article : Google Scholar :
|
|
16
|
Eguchi S, Oshiro N, Miyamoto T, Yoshino K,
Okamoto S, Ono T, Kikkawa U and Yonezawa K: AMP-activated protein
kinase phosphorylates glutamine: Fructose-6-phosphate
amidotransferase 1 at Ser243 to modulate its enzymatic activity.
Genes Cells. 14:179–189. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nöt LG, Brocks CA, Vámhidy L, Marchase RB
and Chatham JC: Increased O-linked beta-N-acetylglucosamine levels
on proteins improves survival, reduces inflammation and organ
damage 24 h after trauma-hemorrhage in rats. Crit Care Med.
38:562–571. 2010. View Article : Google Scholar
|
|
18
|
Chen R, Lai LA, Sullivan Y, Wong M, Wang
L, Riddell J, Jung L, Pillarisetty VG, Brentnall TA and Pan S:
Disrupting glutamine metabolic pathways to sensitize
gemcitabine-resistant pancreatic cancer. Sci Rep. 7:79502017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Asthana A, Ramakrishnan P, Vicioso Y,
Zhang K and Parameswaran R: Hexosamine biosynthetic pathway
inhibition leads to AML cell differentiation and cell death. Mol
Cancer Ther. 17:2226–2237. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zibrova D, Vandermoere F, Göransson O,
Peggie M, Mariño KV, Knierim A, Spengler K, Weigert C, Viollet B,
Morrice NA, et al: GFAT1 phosphorylation by AMPK promotes
VEGF-induced angiogenesis. Biochem J. 474:983–1001. 2017.
View Article : Google Scholar
|
|
21
|
Lin SC and Hardie DG: AMPK: Sensing
glucose as well as cellular energy status. Cell Metab. 27:299–313.
2018. View Article : Google Scholar
|
|
22
|
Patel BA, D'Amico TL and Blagg BSJ:
Natural products and other inhibitors of F1FO
ATP synthase. Eur J Med Chem. 207:1127792020. View Article : Google Scholar
|
|
23
|
Zhou W and Faraldo-Gómez JD: Membrane
plasticity facilitates recognition of the inhibitor oligomycin by
the mitochondrial ATP synthase rotor. Biochim Biophys Acta
Bioenerg. 1859:789–796. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zachara NE, O'Donnell N, Cheung WD, Mercer
JJ, Marth JD and Hart GW: Dynamic O-GlcNAc modification of
nucleocytoplasmic proteins in response to stress. A survival
response of mammalian cells. J Biol Chem. 279:30133–30142. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pan X, Wilson M, Mirbahai L, McConville C,
Arvanitis TN, Griffin JL, Kauppinen RA and Peet AC: In vitro
metabonomic study detects increases in UDP-GlcNAc and UDP-GalNAc,
as early phase markers of cisplatin treatment response in brain
tumor cells. J Proteome Res. 10:3493–3500. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Duarte IF, Ladeirinha AF, Lamego I, Gil
AM, Carvalho L, Carreira IM and Melo JB: Potential markers of
cisplatin treatment response unveiled by NMR metabolomics of human
lung cells. Mol Pharm. 10:4242–4251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duarte IF, Lamego I, Marques J, Marques
MP, Blaise BJ and Gil AM: Nuclear magnetic resonance (NMR) study of
the effect of cisplatin on the metabolic profile of MG-63
osteosarcoma cells. J Proteome Res. 9:5877–5886. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang C, Peng P, Li L, Shao M, Zhao J, Wang
L, Duan F, Song S, Wu H, Zhang J, et al: High expression of GFAT1
predicts poor prognosis in patients with pancreatic cancer. Sci
Rep. 6:390442016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li L, Shao M, Peng P, Yang C, Song S, Duan
F, Jia D, Zhang M, Zhao J, Zhao R, et al: High expression of GFAT1
predicts unfavorable prognosis in patients with hepatocellular
carcinoma. Oncotarget. 8:19205–19217. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chang Q, Su K, Baker JR, Yang X, Paterson
AJ and Kudlow JE: Phosphorylation of human glutamine:
Fructose-6-phosphate amidotransferase by cAMP-dependent protein
kinase at serine 205 blocks the enzyme activity. J Biol Chem.
275:21981–21987. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sharma NS, Gupta VK, Garrido VT, Hadad R,
Durden BC, Kesh K, Giri B, Ferrantella A, Dudeja V, Saluja A and
Banerjee S: Targeting tumor-intrinsic hexosamine biosynthesis
sensitizes pancreatic cancer to anti-PD1 therapy. J Clin Invest.
130:451–465. 2020. View Article : Google Scholar :
|
|
32
|
Lemberg KM, Vornov JJ, Rais R and Slusher
BS: We're Not 'DON' Yet: Optimal dosing and prodrug delivery of
6-Diazo-5-oxo-L-norleucine. Mol Cancer Ther. 17:1824–1832. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Laczy B, Fülöp N, Onay-Besikci A, Des
Rosiers C and Chatham JC: Acute regulation of cardiac metabolism by
the hexosamine biosynthesis pathway and protein O-GlcNAcylation.
PLoS One. 6:e184172011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hudson CD, Savadelis A, Nagaraj AB, Joseph
P, Avril S, DiFeo A and Avril N: Altered glutamine metabolism in
platinum resistant ovarian cancer. Oncotarget. 7:41637–41649. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao JG, Ren KM and Tang J: Overcoming
5-Fu resistance in human non-small cell lung cancer cells by the
combination of 5-Fu and cisplatin through the inhibition of glucose
metabolism. Tumour Biol. 35:12305–12315. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X and Qian K: Protein
O-GlcNAcylation: Emerging mechanisms and functions. Nat Rev Mol
Cell Biol. 18:452–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Haltiwanger RS, Grove K and Philipsberg
GA: Modulation of O-linked N-acetylglucosamine levels on nuclear
and cytoplasmic proteins in vivo using the peptide
O-GlcNAc-beta-N-acetylglucosaminidase inhibitor
O-(2-acetamido-2-deoxy-D-glucopyranosylidene)
amino-N-phenylcarbamate. J Biol Chem. 273:3611–3617. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang L, Chen S, Zhang Z, Zhang J, Mao S,
Zheng J, Xuan Y, Liu M, Cai K, Zhang W, et al: Suppressed OGT
expression inhibits cell proliferation while inducing cell
apoptosis in bladder cancer. BMC Cancer. 18:11412018. View Article : Google Scholar : PubMed/NCBI
|