Introduction
Under aerobic conditions, the majority of
proliferating cancer cells preferentially metabolize glucose via
glycolysis, which is a primary metabolic hallmark of cancer cells
(1-3). Pyruvate dehydrogenase kinases (PDKs)
are key molecules in the mitochondria, which via decarboxylation,
regulate the oxidative metabolism of pyruvate into acetyl-CoA. The
physiological role of the PDK family is to phosphorylate the
pyruvate dehydrogenase (PDH) complex (4). The inhibition of PDKs can
restimulate the mitochondria metabolism of pyruvate in cancer cells
(5), which subsequently inhibits
tumor growth (6). Studies have
revealed that the expression of PDK4, one of four PDK isoforms
(PDK1-4), is frequently upregulated in various cancer types; thus,
its regulation is critical for metabolic changes in cancer cells
(7,8). Trinidad et al (9) reported that PDK4-knockdown using
specific small interfering (si)RNAs suppressed lung and colorectal
cancer cell proliferation, which suggests that PDK4 is an
attractive target for cancer therapy. Despite reports demonstrating
the therapeutic inhibition of PDK activity by dichloroacetate (DCA)
(10-12), the half maximal inhibitory
concentration necessary for PDK4 inhibition is in the range of
57.8-500 µM (7,13), and DCA exhibited reversible
peripheral neuropathy in a number of clinical trials (10,14). Previous studies have attempted to
identify novel compounds that can suppress PDK4 activity at
sub-micromolar concentrations (13,14). Our previous study demonstrated
that cryptotanshinone (CPT), which is obtained from the traditional
Chinese herb Salvia miltiorrhiza Bunge (danshen),
effectively inhibited PDK4 activity at low concentrations
(micromolar-order) to suppress malignant phenotypes, including
three-dimensional (3D)-spheroid formation, anchorage-independent
growth, cellular proliferation and in vivo tumor growth of
human pancreatic and colorectal cancer (15).
Invasiveness and metastasis are key defining
characteristics of cancer cell malignancy, and metastasis is a
major cause of cancer-associated death (16,17). Although invasiveness and
metastasis are considered critical targets in the development of
new therapeutic strategies, effective reagents that target these
processes have yet to be identified. Previously, changes in energy
metabolism have been shown to be closely associated with the
invasive and metastatic abilities of cancer cells (18,19). Changes in cancer cell metabolism
increase acid production in the cancer cells involved (20), which leads to normal cell death
(21), extracellular matrix
degradation by proteolytic enzymes (22), and enhanced migration and invasion
capacity. Therefore, targeting cancer cell metabolism may provide a
novel approach for inhibiting cancer cell invasiveness and
metastasis, and result in more favorable treatment outcomes.
Bladder cancer is the 10th most common cancer type
worldwide, with ~550,000 new cases diagnosed in 2018 (23). Bladder cancer is a complex disease
associated with high morbidity and mortality rates. The treatment
outcomes of radical cystectomy for muscle invasive bladder cancer
(MIBC) combined with chemotherapy are insufficient (24), and the five-year survival rate is
only ~5% in patients with metastatic bladder cancers (25). As there have been no significant
improvements in the treatment outcome over the last three decades
(26), new treatment strategies
for MIBC and metastatic bladder cancers are necessary. PDK4
expression has been shown to be markedly higher in high-grade than
in low-grade bladder cancers without the overexpression of PDK1, 2
or 3 (27). Therefore, the aim of
the present study was to identify novel compounds to treat
intractable bladder cancer by investigating the effects of CPT on
the invasive and metastatic capacity of human bladder cancer
cells.
Materials and methods
Cell culture
The human bladder cancer cell lines (T24 and J82)
were cultured with RPMI-1640 (Nacalai Tesque, Inc.) supplemented
with 10% fetal calf serum (FCS; Sigma-Aldrich; Merck KGaA),
penicillin (100 U/ml; Meiji Seika Pharma, Co., Ltd.) and
streptomycin (100 µg/ml; Meiji Seika Pharma, Co., Ltd.), at
37°C (5% CO2) in a humidified atmosphere. The SUIT-2
human pancreatic cancer cell line was cultured in Dulbecco's
modified Eagle's medium (DMEM, Nacalai Tesque, Inc.). Additionally,
a nonadherent culture was created by coating the culture dishes
with poly-(2-hydroxyethyl methacrylate) reagent (poly-HEMA;
Sigma-Aldrich; Merck KGaA). T24 cells were purchased from the
American Type Culture Collection in 2000, and maintained in our
laboratory. J82 and SUIT-2 cells were purchased from the American
Type Culture Collection in 2019 and the Japanese Collection of
Research Bioresources Cell Bank (Osaka, Japan) in 2018,
respectively. The T24 cell line was authenticated by short tandem
repeat analysis in 2018, as previously described (28).
Reagents
CPT was purchased from Cosmo Bio Co., Ltd., and was
dissolved in dimethyl sulfoxide (DMSO) to prepare 10-mM stock
solutions for the in vitro experiments.
IL-6-hydroxymethyl-chiro-inositol2(R)-2-O-methyl-3-O-octadecylcarbonate
(Akt inhibitor, Calbiochem; Merck KGaA), temsirolimus (Tokyo
Chemical Industry Co., Ltd.) and PD98059 (Cell Signaling
Technology, Inc.) were dissolved in DMSO to prepare the 20-mM stock
solutions for the in vitro experiments. The same volume of
DMSO was added to the control samples in all experiments.
3D-spheroid formation assay
The 3D-spheroid formation assay was performed using
96-well V-bottom plates (PrimeSurface®; Sumitomo
Bakelite Co., Ltd.) as previously described (28). Briefly, 1×103 cells
were seeded in triplicate into each well in complete culture
medium. After three days of incubation at 37°C (5% CO2)
in a humidified atmosphere, images of spheroid formation were
captured using phase contrast microscopy (magnification, ×4;
Olympus Corporation). To determine the number of viable cells in
the 3D-spheroid, the ATP content was quantified using the
luminescence-based CellTiter-Glo® 3D cell viability
assay (Promega Corporation) according to the manufacturer's
protocol. The Caspase-Glo® 3/7 assay (Promega
Corporation) was used to measure caspase-3 and -7 activities
according to the manufacturer's protocol.
In vitro invasion assay
The in vitro invasive potential of cancer
cells was determined using a Matrigel™ Basement Membrane Matrix
Invasion Chamber (chamber size, 6.4 mm; membrane surface area, 0.3
cm2; pore size, 8 µm; BD Biosciences), according
to the manufacturer's instructions (29). A total of 500 µl cell
suspension (2×104 T24 and 4×104 J82 cells/ml)
was added to each chamber, and incubated for two or three days,
respectively, at 37°C in a humidified 5% CO2 atmosphere.
Culture medium without FCS was used in upper chambers, and 750
µl culture medium with 10% FCS was added to the lower
chambers. Non-invasive cells were removed from the upper surface of
the membrane using a cotton swab. The invasive cells on the
underside of the membrane were harvested using Diff-Quik™ (Kokusai
Shiyaku Co., Ltd.) and counted under a light microscope
(magnification, ×100). The duration of each Diff-Quik™ staining
step was 3 min at room temperature. Each sample was then analyzed
in triplicate.
Cell cycle analysis
Cell cycle distribution was evaluated using the
Cycletest Plus DNA Reagent kit with a FACSCalibur flow cytometer
system and ModFit 3.2 software (both Becton, Dickinson and
Company). Briefly, the cells were collected after treatment with
CPT (0, 10 and 20 µM at 37°C for 24 h) and fixed in 70%
ethanol at -20°C for 18 h. The cells were washed twice with
phosphate-buffered saline and then pelleted by centrifugation at
400 × g at room temperature for 5 min. The cell pellets were
incubated with 250 µl solution A (trypsin in a spermine
tetrahydrochloride detergent buffer) at room temperature for 10
min, 200 µl solution B (trypsin inhibitor and ribonuclease A
in citrate stabilizing buffer with spermine tetrahydrochloride) at
room temperature for 10 min, and then 200 µl solution C
(propidium iodide and spermine tetrahydrochloride in citrate
stabilizing buffer) in the dark at 4°C for 10 min. The cells were
then flow cytometrically.
Small interfering RNA (siRNA)
transfection
For the siRNA experiment, cells were transfected
with 50 nM Silencer® Select PDK4 siRNA (s10262; Thermo
Fisher Scientific, Inc.) and β-catenin siRNA (sense; 5′-CAG GGG GUU
GUG GUU AAG CUC UU-3′, antisense; 5′-AAG AGC UUA ACC ACA ACC CCC
UG-3′) (30) using
Lipofectamine® RNAiMax (Invitrogen; Thermo Fisher
Scientific, Inc.) for 24 h according to the manufacturer's
protocol. A non-specific siRNA duplex (GeneDesign, Inc.) served as
the control.
Western blotting
The 60-mm culture dishes were precoated with
poly-HEMA at room temperature for 24 h. A total of 5×105
cells were seeded into a poly-HEMA-coated dish, and cultured with
CPT for 48 h (37°C) under nonadherent culture conditions. Cell
lysate preparation and western blotting were conducted as
previously described (31).
Amersham ECL western blotting detection kit (Cytiva) was used for
visualization according to the manufacturer's protocol. The
experiments were repeated at least three times. Mouse monoclonal
antibodies for α-tubulin (1:1,000; DM1A, cat. no. T9026) were
purchased from Sigma-Aldrich; Merck KGaA. Mouse monoclonal
antibodies for E-cadherin (1:500; 36/E, cat. no. 610181) were
purchased from BD Biosciences. Anti-PDH mouse monoclonal antibodies
(1:500; cat. no. ab110330), anti-phospho-PDH rabbit polyclonal
antibodies (1:500; S293; cat. no. ab92696), and anti-PDK4 rabbit
polyclonal antibodies (1:500; cat. no. ab63157) were purchased from
Abcam. Rabbit monoclonal antibodies for 4E-BP1 (1:500; 53H11, cat.
no. #9644), phospho-4E-BP1 (1:500; S65, 174A9, cat. no. #9456),
phospho-Akt (1:500; S473, D9E, cat. no. #4060), Erk1/2 (1:500;
137F5, cat. no. #4695), phospho-Erk1/2 (1:500; T202/Y204, 20G11,
cat. no. #4376), MEK1/2 (1:500; 47E6, cat. no. #9126),
phospho-MEK1/2 (1:500; S221, 166F8, cat. no. #2338), S6K (1:500;
49D7, cat. no. #2708), phospho-S6K (1:500; T421/S424, #9204),
β-catenin (1:500; D10A8, #8480), phospho-β-catenin (1:500; S552,
D8E11, cat. no. #5651), N-cadherin (1:500; D4R1H, cat. no. #13116),
and caspase-3 (1:500; 8G10, cat. no. #9665); rabbit polyclonal
antibodies for Akt (1:500; cat. no. #9272) and phospho-β-catenin
(1:500; S33/37/T41, #9561); and mouse monoclonal antibodies for
CD44 (1:1,000; 156-3C11, cat. no. #3570) and epithelial cell
adhesion molecule (EpCAM) (1:500; VU1D9, cat. no. #2929) were
purchased from Cell Signaling Technology, Inc. Rabbit polyclonal
antibodies for lamin A/C (1:500; cat. no. #10298-1-AP) were
purchased from ProteinTech Group, Inc. The horse radish
peroxidase-conjugated anti-mouse IgG (1:10,000, cat. no. NA931,
from sheep) and anti-rabbit IgG (1:10,000, cat. no. NA934, from
donkey) secondary antibodies were purchased from Cytiva. Caspase-3
control cell extracts (cat. no. #9663) for the negative and
positive controls for apoptosis analysis were purchased from Cell
Signaling Technology, Inc.
Subcellular fractionation
Cytoplasmic and nuclear protein fractions were
extracted using NE-PER® cytoplasmic and nuclear
extraction regents (Pierce; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Western blot analysis was
used to analyze the extracted protein samples. α-tubulin and lamin
A/C were used as cytoplasmic and nuclear endogenous controls,
respectively.
Laboratory animals
A total of 10 female, five-week-old
BALB/c-nu/nu nude mice (5 control and 5 CPT-treated mice,
weight 16-18 g) were purchased from CLEA Japan and housed in a
specific pathogen-free room with controlled temperature (20-22°C)
and humidity (50-60%), and a preset light-dark cycle (12:12 h). All
mice were allowed ad libitum access to food (CE-2; CLEA
Japan, Inc.) and water. The use of animals in the experimental
protocols was reviewed and approved by the Management Committee of
the Research Center for Animal Life Science at Shiga University of
Medical Science (Shiga, Japan).
Orthotopic pancreatic cancer model of
nude mice
A total of 10 5-week-old mice were anesthetized by
intraperitoneal injection of pentobarbital (50 mg/kg) and
maintained by inhalation of 1% isoflurane. The abdominal cavity was
opened using a 1.5-cm wide longitudinal laparotomy pointing
slightly to the left. Then, 2×105 SUIT-2 cells in 100
µl DMEM were injected into the tail of the pancreas with a
27-gauge needle. Both the operation and injection were performed by
the same person (CJK and YT, respectively). The pancreas was placed
back into the abdominal cavity, which was then closed using
interrupted suturing with 4-0 Nylon, and penicillin G (200 U/20 g)
was injected intramuscularly to prevent postoperative infection.
After seven days, five mice were intraperitoneally administered 40
mg/kg of CPT (suspended in 8% DMSO and 2% Solutol) every two days
for two weeks. The control mice were administered a vehicle (8%
DMSO and 2% Solutol) every two days for two weeks. Finally, the
mice were sacrificed by cervical dislocation one day after the last
administration. The pancreas, heart, lungs, liver and kidneys were
resected and fixed with 10% buffered formalin at 4°C for 24 h. The
intestine with the mesentery were also resected, and the tumor
nodules of the mesentery were counted macroscopically. The volume
of the largest tumor nodule in one mouse was calculated using the
following formula: 3.14xAxB2/6, where A and B represent
the long and short diameters, respectively. A and B were measured
using the histological preparations of hematoxylin and eosin
staining.
Histopathological analyses
Serial 3-µm sections of formalin-fixed (10%,
4°C for 24 h), paraffin-embedded tissues were histologically
evaluated by hematoxylin and eosin staining and light microscope.
For the immunohistochemical analyses, dewaxed sections were
assessed using β-catenin antibodies via the
streptavidin-biotin-peroxidase method [Histofine® MAX-PO
(MULTI); Nichirei Biosciences, Inc.], according to the
manufacturer's instructions.
Statistical analyses
All quantitative data are presented as the mean ±
standard deviation. One-way ANOVA following by Tukey's test for
multiple comparisons, Wilcoxon's rank sum test, and Welch's t-test
were used for the statistical analyses. All analyses were performed
using the R statistical software package, version 2.6.2 (https://www.r-project.org/), and P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of CPT on bladder cancer cell
invasiveness in vitro
To assess the suppressive activity of CPT on the
invasiveness of human bladder cancer, its effects on the
3D-spheroid formation and invasiveness of two human bladder cancer
cell lines (T24 and J82) were investigated. The effect of CPT on
PDK4 activity was evaluated by monitoring the phosphorylation PDH,
the target protein of PDK4. Under nonadherent culture conditions,
10-20 µM CPT effectively suppressed PDH phosphorylation at
serine 293, a hallmark of PDH activity (32), in a dose-dependent manner
(Fig. 1A). The expression levels
of PDH and α-tubulin (the internal control protein) remained
unaffected by CPT treatment, while PDK4 expression was suppressed
by 20 µM CPT. The antioncogenic effect of CPT was assessed
by evaluating T24 and J82 cell 3D-spheroid formation, a method is
frequently used in cancer research to more closely mimic the tumor
environment (33). 3D-spheroid
formation was morphologically altered in these cell lines by CPT
treatment (3-10 µM; Fig.
S1). The edge of the spheroid became less distinct following
CPT treatment. Moreover, CPT treatment (3-20 µM)
significantly decreased the ATP concentration in these cell lines
in a dose-dependent manner, which is closely associated with the
number of viable cancer cells in the spheroids (Fig. 1B). Furthermore, the invasiveness
of both cell lines was significantly suppressed by CPT treatment
(Fig. 1C-D). The induction of
apoptosis was also assessed by monitoring the cleavage of
caspase-3. Caspase-3 activation (cleaved, lower band) was not
detected in T24 and J82 cells (Fig.
1E). Furthermore, the Caspase-Glo® 3/7 assay was
used to measure caspase-3 and -7 activities. The number of viable
cells in 3D-spheroids was assessed using ATP activity in T24 and
J82 cells. There were significantly fewer viable T24 and J82 cells
following CPT treatment (Fig.
S2A). Similarly, the caspase-3/7 activities decreased in these
cells, and the caspase-3/7/ATP activity ratio was not increased
following CPT treatment (Fig.
S2B-C). This result was most likely due to the decrease in
caspase-3/7 activities reflecting the decrease in the viable number
of T24 and J82 spheroids after CPT treatment. Furthermore, flow
cytometry was used to assess the pro-apoptotic effect of CPT. As
shown in Figs. 1F and S3, CPT treatment increased the
sub-G1 fraction at a concentration of 20 µM in
both cell lines. These results indicate that CPT suppressed
invasiveness and 3D-spheroid formation in bladder cancer cells, and
that apoptosis may play a partial role in these anti-oncogenic
activities.
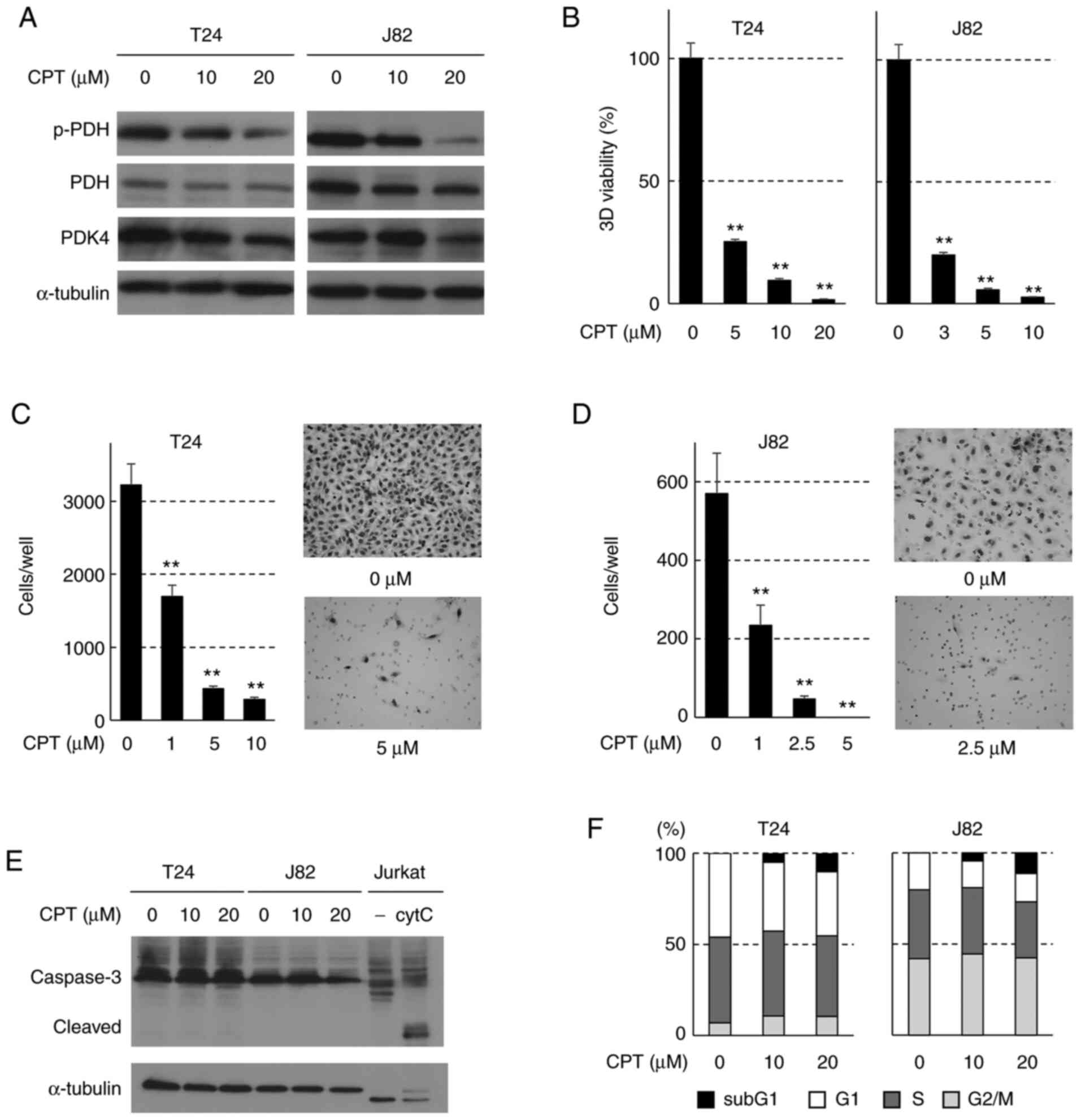 | Figure 1Effects of CPT on the invasiveness of
human bladder cancer cells in vitro. (A) Western blot
detection of PDH and PDK4 in T24 and J82 cells treated with CPT for
48 h. α-tubulin was used as the internal control. (B) Effects of
CPT on the viability of spheroids formed from T24 and J82 cells.
Bars indicate the mean ± SD. Adenosine triphosphate content was
assessed using CellTiter-Glo® 3D assays.
**P<0.001 compared with untreated cells using Tukey's
test. Effects of CPT on the invasiveness of (C) T24 and (D) J82
cells. Each sample was assayed in triplicate, and the bars
represent the mean ± SD. Images of T24 cells invading the Matrigel
membrane. Images were captured at ×100 magnification.
**P<0.001 compared with untreated cells using Tukey's
test. (E) Western blot analysis of caspase-3 cleavage in T24 and
J82 cells treated with CPT. Untreated (−) or cytC-treated Jurkat
cell extracts (caspase-3 control cell extracts, Cell Signaling
Technology, Inc.) were used as negative or positive controls,
respectively. α-tubulin was used as the internal control. (F)
Apoptotic cytotoxicity of CPT in T24 and J82 cells. Stacked bars
indicate the cell cycle profiles after treatment with CPT for 24 h;
the sub-G1 fraction, presented as the black area on the top of each
bar, indicates the proportion of the apoptotic cells. CPT,
cryptotanshinone; PDH, pyruvate dehydrogenase; PDK4, pyruvate
dehydrogenase kinase 4; p-, phosphorylated; cytC, cytochrome C. |
Alterations in invasion-related proteins
following CPT treatment
To clarify the molecular mechanisms by which CPT
suppresses cellular invasiveness, changes in the expression of
proteins associated with epithelial-mesenchymal transition (EMT)
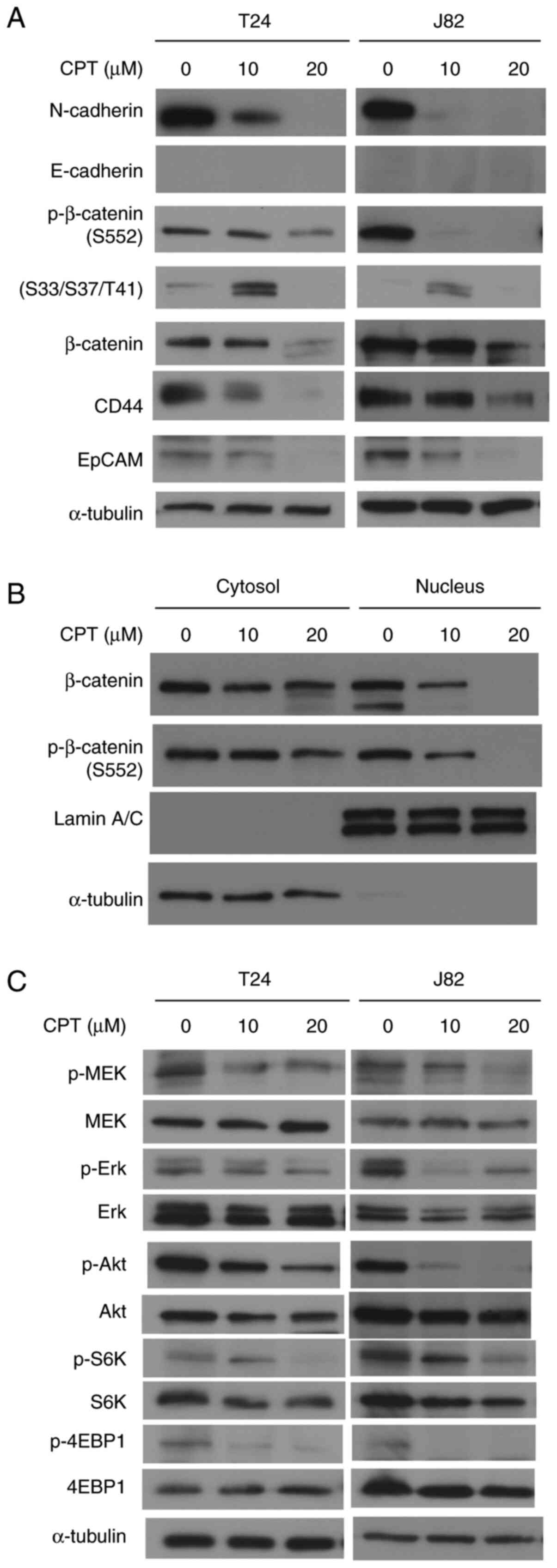
were evaluated in T24 and J82 cells. Figs. 2A and S4 show that CPT treatment significantly
decreased N-cadherin expression in a dose-dependent manner.
However, E-cadherin expression was not detected in these cells. A
CPT concentration of 10-20 µM also suppressed β-catenin
phosphorylation at S552 in a dose-dependent manner. β-catenin
phosphorylation at S552 induces β-catenin accumulation in the
nucleus and increases its transcriptional activity (34). However, 10 µM CPT enhanced
β-catenin phosphorylation at S33/S37/T41, which has been shown to
promote its ubiquitylation and proteasomal degradation (35,36). In the present study, CPT treatment
at 20 µM suppressed both total β-catenin expression and
β-catenin phosphorylation at S33/S37/T41 (Figs. 2A and S4). This dual function of CPT for
β-catenin phosphorylation is thought to be responsible for the
suppression of cell invasiveness via β-catenin suppression. A
subcellular fractionation assay was then performed to detect
changes in the subcellular localization of β-catenin in T24 cells
(Fig. 2B). CPT treatment
significantly suppressed the phosphorylation of β-catenin at S552
and its accumulation in the nucleus. By contrast, the levels of
β-catenin and phosphorylated β-catenin at S552 in the cytoplasm
were weakly decreased by CPT, compared with the levels in the
nucleus. The β-catenin signaling pathway has been reported to play
a crucial role in the EMT process via the nuclear translocation of
β-catenin (37). Therefore, these
results indicate that CPT can effectively suppress the accumulation
of β-catenin in the nucleus.
Since 3D-spheroid formation is considered to be a
phenotype of cancer stem cells (15), and because these spheroids are
associated with invasiveness (28), alterations in the expression of
CD44 and EpCAM, which are stem cell markers of human bladder
cancer, were investigated in T24 and J82 cells. Changes in
cancer-related signaling pathway proteins, such as those of the
MEK/Erk, Akt and mTOR cascades, were also investigated. As shown in
Figs. 2A and S4, CPT suppressed the cancer stem cell
phenotypes of these cells. Additionally, CPT suppressed MEK and Erk
phosphorylation (Fig. 2C).
Furthermore, CPT treatment decreased Akt phosphorylation at S473
and suppressed the phosphorylation of S6K (Fig. S4) and 4E-BP1, the target proteins of mTOR
kinase (Fig. 2C). These results
indicate that CPT suppressed the MEK/Erk, Akt and mTOR signaling
pathways in these bladder cancer cell lines.
The role of β-catenin in the CPT-induced
suppression of N-cadherin expression and cellular invasiveness
To confirm whether the alterations in EMT-associated
protein levels, growth-associated signaling pathways, and cancer
stemness markers induced by CPT were the result of inhibited PDK4
activity, PDK4 expression was knocked down in T24 cells, which
effectively suppressed the expression of PDK4 and PDH
phosphorylation (Fig. 3A).
PDK4-knockdown significantly decreased ATP concentration and
suppressed cellular invasiveness, as well as altering the
morphology of the spheroid (Figs. 3B
and C, and S5). As shown in
Fig. 3D, it also suppressed the
phosphorylation of β-catenin at S552, N-cadherin expression, the
MEK/Erk, Akt and mTOR signaling pathways, and CD44 expression.
However, PDK4-siRNA did not alter β-catenin phosphorylation at
S33/S37/T41. Taken together, these results support the finding that
the suppressive effects caused by CPT were the result of PDK4
inactivation.
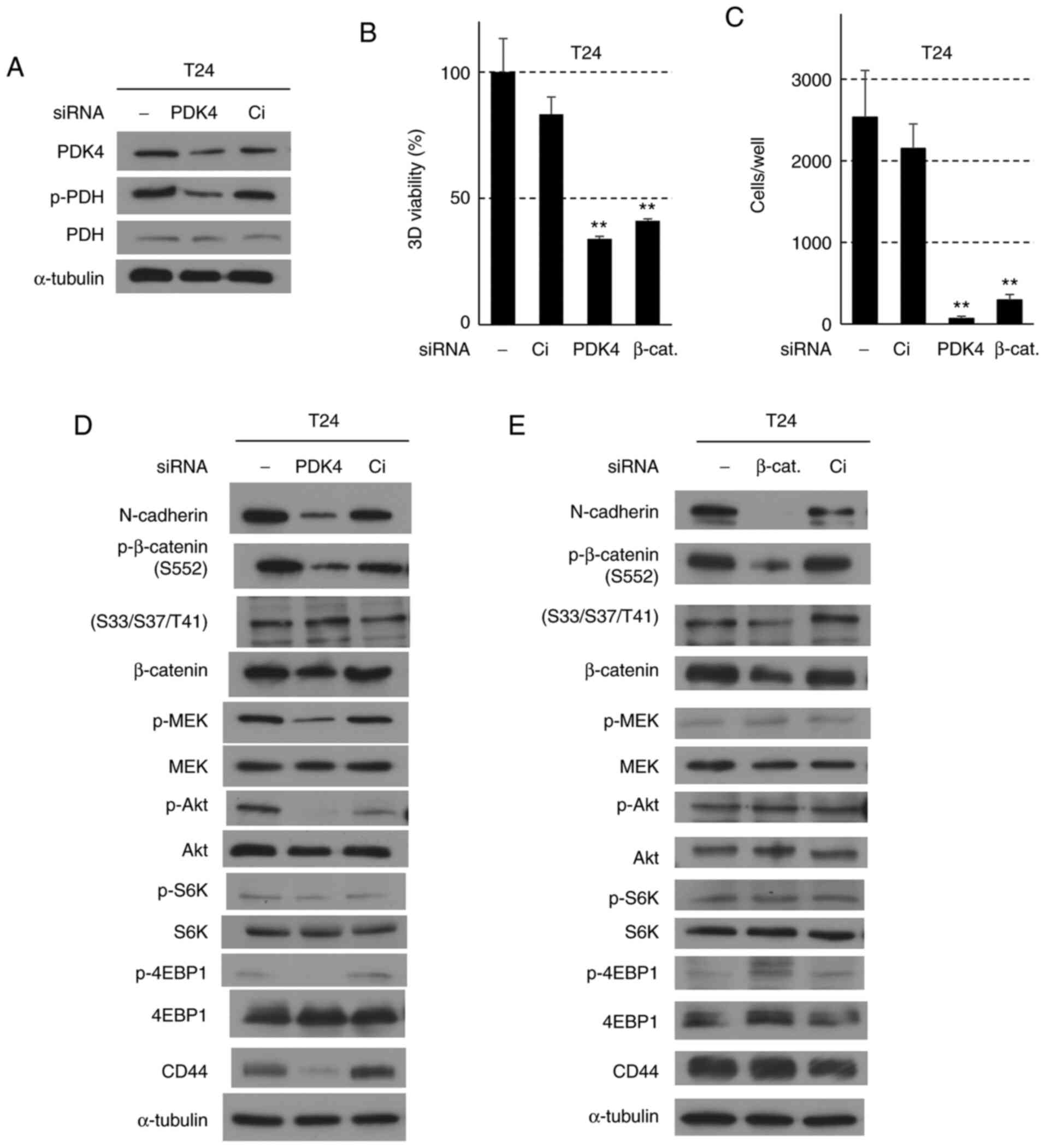 | Figure 3Effects of PDK4 and β-catenin
siRNA-knockdown in T24 cells. Cells were treated with siRNA for 24
h, followed by nonadherent culture for 48 h, or 3D-spheroid
formation for 72 h. (A) Western blot analysis for PDK4 and PDH
proteins in T24 cells treated with Ci (siRNA control) and PDK4
siRNAs. α-tubulin was used as the internal control. (B) Effects of
PDK4- and β-catenin-knockdown on the viability in T24 cells. Bars
indicate the mean ± SD. Adenosine triphosphate content was assessed
using the CellTiter-Glo® 3D assay.
**P<0.001 compared with Ci control using Tukey's
test. (C) Effects of siRNA for PDK4 and β-catenin on the
invasiveness in T24 cells. Each sample was assayed in triplicate,
and the bars represent the mean ± SD.**P<0.001 using
Turkey's test compared with Ci control cells. (D and E) Western
blot analysis for epithelial-mesenchymal transition, MEK, Akt, and
mTOR pathway proteins in T24 cells treated with Ci, PDK4 and
β-catenin siRNAs, respectively. α-tubulin was used as the internal
control. CPT, cryptotanshinone; PDH, pyruvate dehydrogenase; PDK4,
pyruvate dehydrogenase kinase 4; p-, phosphorylated; 3D,
three-dimensional; Ci, siRNA control; 4EBP1, eukaryotic translation
initiation factor 4E-binding protein 1. |
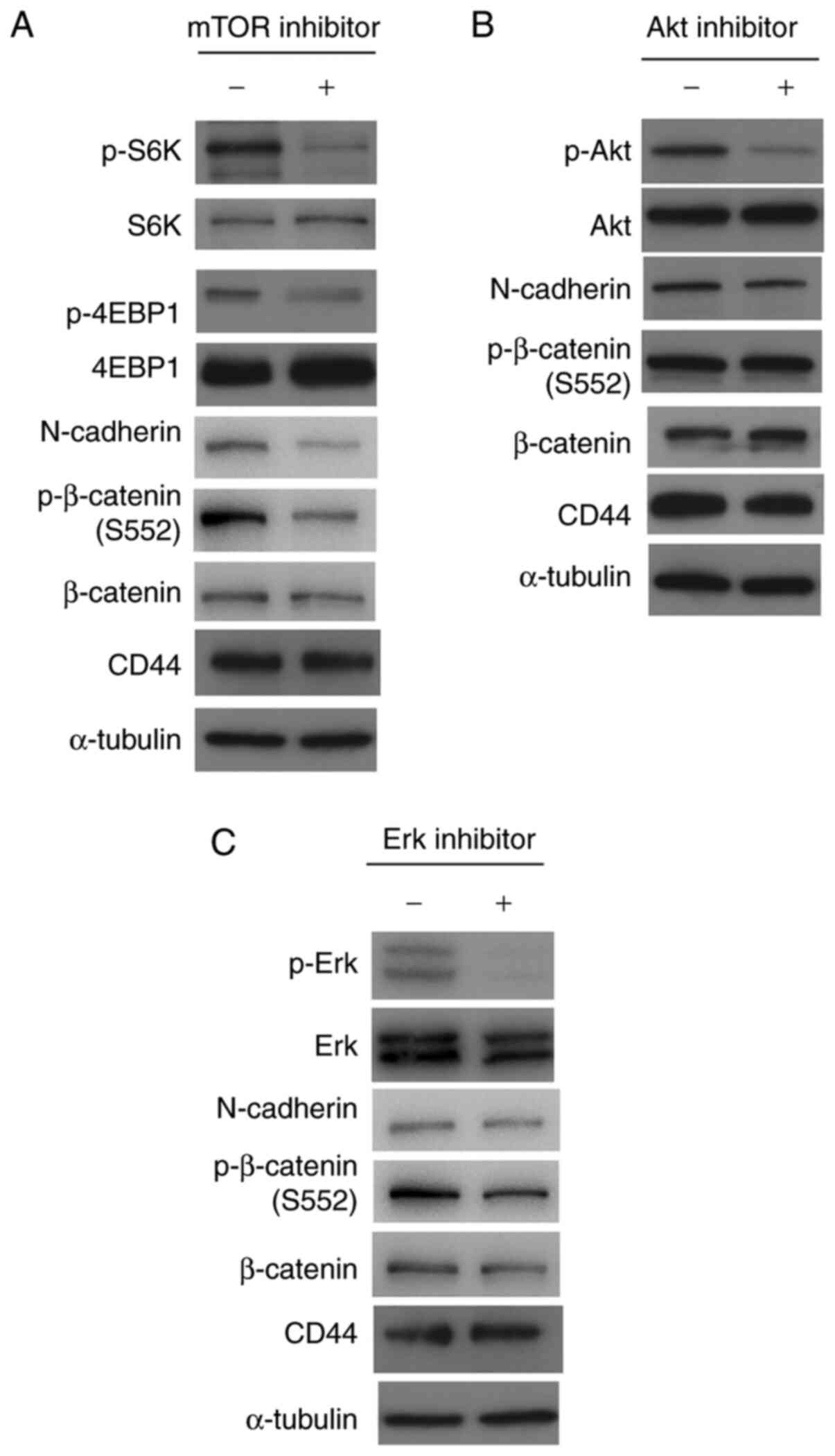
To clarify the roles of N-cadherin and β-catenin in
the CPT-induced suppression of invasiveness, β-catenin was knocked
down in T24 cells. β-catenin-siRNA significantly decreased the ATP
concentration and suppressed cellular invasiveness, 3D-spheroid
formation, and β-catenin expression (Figs. 3B, C and E, and S5). Phosphorylation at S552 and
N-cadherin expression were also suppressed (Fig. 3E). The MEK/Erk, Akt and mTOR
signaling pathways, as well as CD44 expression, were not affected
by the β-catenin-knockdown. These results demonstrate that the
inactivation of β-catenin plays a crucial role in suppressing
cellular invasiveness; moreover, N-cadherin acts downstream of
β-catenin to suppress the invasiveness induced by CPT. To assess
the roles of Erk, Akt and mTOR in the suppression of β-catenin and
N-cadherin, T24 cells were treated with inhibitors of these
kinases. As shown in Fig. 4, the
inhibitors (40 µM) acted on their target kinases to suppress
the mTOR targets, and the phosphorylation of S6K and 4EBP1
(Fig. 4A), Akt (Fig. 4B) and Erk (Fig. 4C). Although Akt and Erk inhibitors
could not suppress β-catenin phosphorylation at S552 and N-cadherin
expression, the mTOR inhibitor, temsirolimus, suppressed β-catenin
phosphorylation at S552 and N-cadherin expression (Fig. 4D). These results indicate that the
mTOR pathway acts upstream of β-catenin/N-cadherin in T24 cells.
Additionally, Akt, mTOR, and Erk inhibitors did not suppress CD44
expression (Fig. 4D), indicating
that the downregulation of CD44 by CPT occurs independently of
these signaling pathways.
Antioncogenic effects of CPT in an in
vivo orthotopic pancreatic cancer model of nude mice
To investigate the effects of CPT on in vivo
metastasis, the aim was to establish a subcutaneous cancer cell
xenograft model of lung metastasis via injection of J82 cells into
the tail vein, and a liver metastasis model via injection into the
spleens of nude mice. However, J82 metastatic tumors could not be
detected in these experimental models. Therefore, an orthotopic
pancreatic cancer model was created, which closely mimics
pancreatic tumor formation using the highly metastatic pancreatic
cancer cell line, SUIT-2 (38).
The effects of CPT treatment in SUIT-2 cells were similar to those
in bladder cancer cells; specifically, multiple oncogenic signaling
pathways components were suppressed, including the phosphorylation
of PDH and β-catenin at S552, and the expression of N-cadherin and
CD44 (Fig. 5A). SUIT-2 cells were
injected into the pancreases of nude mice, and after one week, CPT
was repeatedly administered (40 mg/kg, intraperitoneally) every two
days for two weeks. At day 1 post-injection, SUIT-2 cells formed a
pancreatic cancer-like tumor and disseminated nodules of the
mesentery. Pancreatic tumor formation was moderately suppressed by
CPT treatment in the pancreatic orthotopic cancer model (Fig. 5B-D). The volume of the largest
tumor nodule in one mouse was 0.011±0.008 and 0.061±0.028 ml in
CPT-treated and control groups, respectively. CPT treatment
significantly decreased tumor growth in the pancreatic orthotopic
cancer model (P=0.005). Furthermore, histopathological analyses
revealed that tumor morphology in the CPT-treated mice was similar
to that in the control mice. However, β-catenin was diffusely
expressed in the cytoplasm of pancreatic cancer cells, while this
was observed at the cytoplasmic membrane in normal pancreatic cells
(Fig. 5E). β-catenin expression
was markedly suppressed in the CPT-treated pancreatic tumors
compared with the control tumors (Fig. 5F), and there were significantly
fewer tumor nodules in the mesenterium after CPT treatment compared
with the control tumors (Fig.
5G-I). Although histopathological examinations of the heart,
lungs, liver and kidneys were also performed, no side effects from
CPT treatment were observed under these experimental conditions.
These results indicate that CPT suppressed in vivo tumor
growth and peritoneal dissemination in an orthotopic pancreatic
cancer model of nude mice.
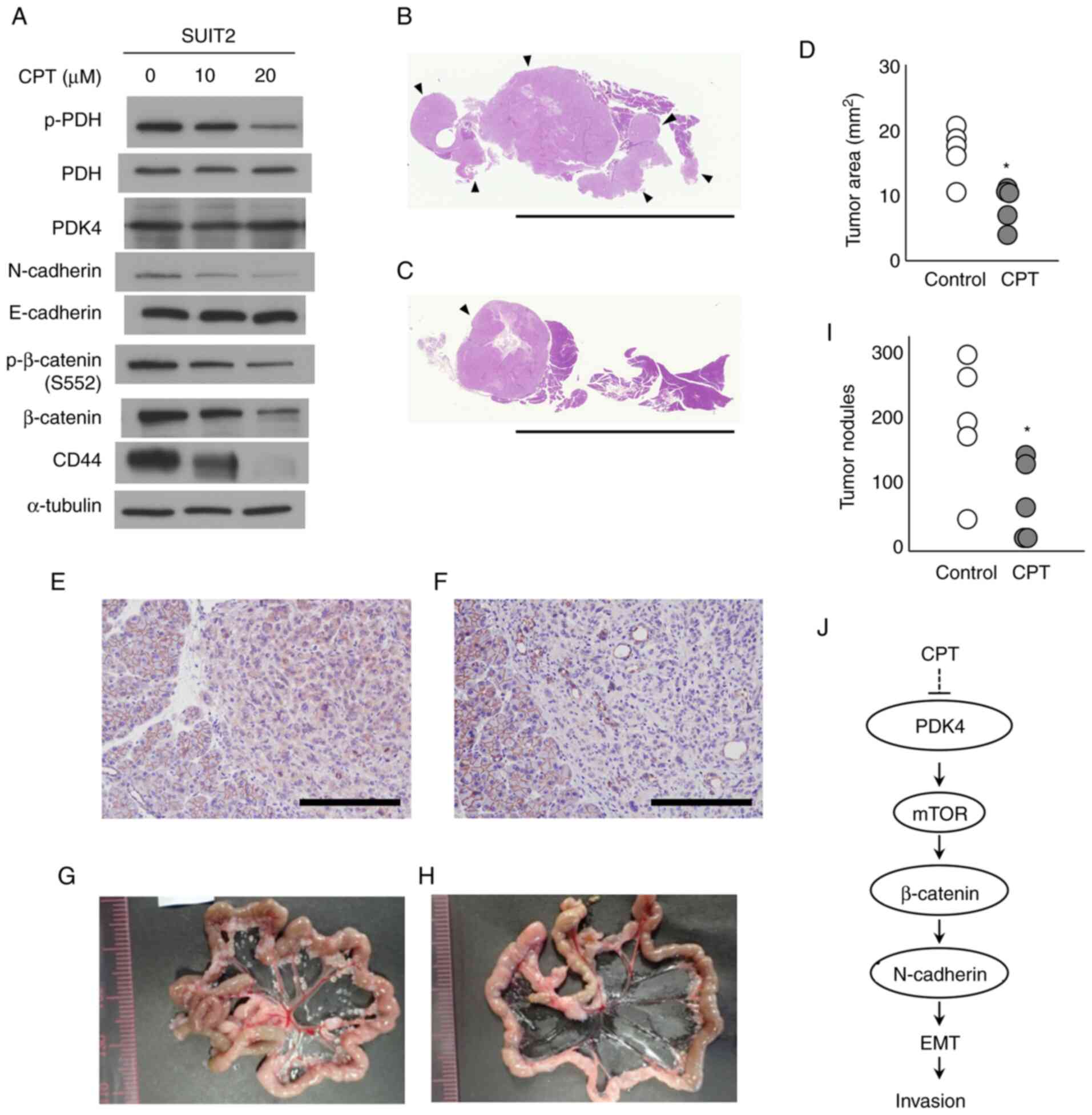 | Figure 5Effects of CPT in an in vivo
orthotopic pancreatic cancer model of nude mice. (A) Western blot
analysis for PDK4, PDH and proteins regulating EMT and cancer
stemness in SUIT-2 cells, following treatment with CPT for 48 h.
α-tubulin was used as the internal control. Low-magnification
histological sections of the pancreas from the (B) control and (C)
CPT-injected mice. Scale bar, 10 mm. Hematoxylin and eosin
staining; arrows represent the tumor areas. (D) Tumor area size of
the pancreas of SUIT-2 cells from the control and CPT-injected
mice. *P<0.05 according to the Wilcoxon's rank sum
test. Immunostaining for β-catenin of the pancreas from the (E)
control and (F) CPT-injected mice. Scale bar, 200 µm;
magnification, ×200. Peritoneal metastatic nodules of SUIT-2 cells
from the (G) control and (H) CPT-injected mice. (I) Number of
peritoneal SUIT-2 cell metastatic nodules from the control and
CPT-injected mice. *P<0.05 according to Wilcoxon's
rank sum test. (J) Schematic illustration of the signaling pathway
responsible for the suppressive effect of CPT on the invasiveness
of bladder cancer cells. CPT, cryptotanshinone; PDH, pyruvate
dehydrogenase; PDK4, pyruvate dehydrogenase kinase 4; p-,
phosphorylated; EMT, epithelial-mesenchymal transition. |
Discussion
In the present study, CPT, a novel PDK4 inhibitor,
was shown to suppress the invasiveness of bladder cancer cells
in vitro. Furthermore, CPT inhibited in vivo tumor
growth and peritoneal dissemination in an orthotopic pancreatic
cancer model of nude mice. Although multiple molecular changes were
observed in human bladder cancer cells following CPT treatment,
including those of the β-catenin/N-cadherin, Erk, Akt and mTOR
signaling pathways, as well as cancer stemness, siRNA-knockdown
experiments highlighted the critical role of the
β-catenin/N-cadherin axis in the suppression of cellular
invasiveness downstream of PDK4 (Fig.
5J). Inhibitor experiments also indicated that CPT suppressed
the β-catenin/N-cadherin axis via the mTOR pathway. As illustrated
in Fig. 5J, CPT-associated
inhibition of PDK4 leads to the suppression of mTOR. Downstream of
mTOR, β-catenin was suppressed, causing the downregulation of
N-cadherin expression, a master regulator of EMT in bladder cancer
cells.
In the present study, CPT was revealed to exert a
dual function in β-catenin phosphorylation. CPT (10 µM)
enhanced β-catenin phosphorylation at S33/S37/T41 and suppressed
phosphorylation at S552 in human bladder cancer cells.
Phosphorylation of β-catenin at S33/S37/T41 has been shown to
promote its ubiquitylation and proteasomal degradation (35,36). The suppression of β-catenin
phosphorylation at S33/S37/T41 induced its cytoplasmic
accumulation, and some of the free β-catenin translocated to the
nucleus, resulting in deregulated cell cycle progression (35,36). Conversely, β-catenin
phosphorylation at S552 is known to promote its transcriptional
activity and cancer cell invasiveness (39). Therefore, CPT is considered to
suppress the invasion of bladder cancer cells by both promoting the
ubiquitylation and degradation of β-catenin, as well as suppressing
its transcriptional activity. In the present study, 20 µM
CPT markedly suppressed β-catenin phosphorylation at S33/S37/T41 in
bladder cancer cells, which appeared to be attributed to the
decrease in total β-catenin expression due to its ubiquitylation
and degradation. The enhanced nuclear accumulation of β-catenin has
been observed in bladder cancer tissues, resulting in enhanced
transcription of genes such as Snail and Twist (40). In the present study, β-catenin
nuclear localization was decreased by CPT treatment in bladder
cancer cells, which suggests that the alteration in β-catenin
function is closely associated with CPT-induced suppression of
tumor growth and cellular invasiveness.
CPT also suppressed the expression of cancer stem
cell markers, CD44 and EpCAM, in bladder cancer cells, reflecting
the inhibitory effect of CPT on cancer stem cell proliferation. In
fact, CPT inhibited the representative phenotype of cancer stem
cells and 3D-spheroid formation in bladder cancer cells.
Furthermore, CPT significantly suppressed in vivo tumor
growth and peritoneal dissemination in an orthotopic mouse model of
SUIT-2 pancreatic cancer. CD44 is a cancer stem cell marker and a
major surface receptor for hyaluronate, that has been shown to play
a role in intracellular adhesion, cell-matrix adhesion, migration
and tumor cell invasiveness and metastasis (41-43). Van Grevenstein et al
(44) suggested that interference
with CD44 may decrease the incidence of peritoneal tumor recurrence
after curative resection of pancreatic cancer. In bladder cancer,
CD44, Oct4 and EpCAM have been shown to be highly expressed in, and
associated with the progression of, bladder cancer (45,46). The present study also investigated
the expression of Oct4 and retinal dehydrogenase 1. However, the
expression of these proteins was low and not affected by CPT
treatment in T24 and J82 cells (data not shown).
The mTOR signaling pathway has been reported to be a
master regulator for the maintenance of cancer cell stemness
(45,47,48). Previous research has demonstrated
that cancer stem cells undergo metabolic alterations, including
high glycolytic activity and low mitochondrial respiration
(49). The results of the present
study also suggest that the metabolic alterations caused by
inhibiting PDK4 activity can suppress the stemness of bladder and
pancreatic cancer cells. PDK4-knockdonwn analysis indicated that
the inactivation of PDK4 contributed to decreased CD44 expression,
although its suppression was independent of that of the
mTOR/β-catenin/N-cadherin axis. N-cadherin has been shown to play a
central role in the induction of EMT, which primarily contributes
to the invasion and metastasis of malignant cancers (50). The dual suppression of CPT-induced
EMT and cancer cell stemness via PDK4 inactivation may present a
novel therapeutic approach for intractable malignant cancers.
The results of the present study also revealed that
mTOR was a critical downstream target molecule of PDK4 for
suppressing cellular invasiveness by CPT via β-catenin and
N-cadherin. The mTOR (mTORC1) pathway is a central signaling
pathway that controls metabolic processes (51,52). Studies using specific inhibitors
have elucidated the role of mTOR in the control of cell
proliferation and metabolism (53). The Wnt/β-catenin and Akt/mTOR
pathways have been shown to be critically involved in colorectal
cancer development (54) and are
primarily regulated by feedback mechanisms. They are connected at
multiple levels involving common upstream and downstream effectors.
In the present study, mTOR acted upstream of β-catenin to suppress
EMT and invasiveness following CPT administration. mTOR
orchestrates metabolic reprogramming by regulating nutrient uptake
and flux. Although various clinical trials have used rapamycin and
its analogs, their therapeutic effects are limited by modest
antitumor activity and increased toxicity. The results of the
present study demonstrated that CPT was able to suppress the mTOR
pathway, as well as other signaling cascades, at low
concentrations. CPT also suppressed metastasis and tumor growth in
an in vivo mouse orthotopic model without causing severe
side effects. We also hypothesize that CPT and conventional mTOR
inhibitors may synergistically suppress tumor growth, invasion and
metastasis at low concentrations by altering cellular metabolism;
however, the detailed mechanism by which PDK4 inactivation
suppresses mTOR activity requires further investigation. Although
our previous study investigated the inhibitory effect of CPT on
PDK4 by in vitro, whether CPT can directly bind to and
inhibit PDK4 in vivo remains to be elucidated.
Our previous study reported that CPT significantly
inhibited the 3D-spheroid formation of pancreatic cancer cells at
micromolar-order concentrations, without inducing apoptosis
(15). In the current study, the
induction of apoptosis was detected at low levels in T24 and J82
bladder cancer cells. Liu et al (55) reported that CPT promoted the
apoptosis of bladder cancer cells in a dose-dependent manner. In
their report, apoptosis was potently induced with 40-80 µM
CPT, a higher concentration than that used in the present study
(10-20 µM), suggesting that a difference in CPT
concentration might contribute to the induction of apoptosis in
bladder cancer cells. Furthermore, a low concentration of CPT was
sufficient enough to suppress invasion and tumor growth, suggesting
its potential as a clinical cancer therapy without considerable
side effects. Cisplatin-based chemotherapy remains the standard
first-line treatment for advanced bladder cancer (24), though patient outcomes remain
insufficient. Thus, novel strategies for the treatment of advanced
bladder cancer are required. The PDH-PDK axis has been considered a
crucial therapeutic target in cancer (7,10,11) because PDK4 plays a pivotal role in
the altered metabolism of malignant cancers (7,9).
This therapeutic strategy warrants small molecules that effectively
inhibit PDK4 activity without exhibiting toxicity. However, the
current findings on CPT will contribute to targeting or suppressing
the invasion and metastasis of intractable malignant cancers.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
CJK and HI designed the study. CJK, TT, YT and HI
performed and analyzed the experiments. KIM designed and performed
the immunohistochemical analyses. SK and AK were involved in
conceiving the project and provided several important suggestions
to the research plan. CJK, TT, YT and HI wrote the paper. CJK, TT
and HI confirmed the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consult to
participate
All animal experiments were performed in accordance
with the care and use guidelines of Shiga University of Medical
Science and approved by the Management Committee of the Research
Center for Animal Life Science at Shiga University of Medical
Science.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Ms. Akiyo Ushio
(Shiga University of Medical Science) for their technical
assistance.
Funding
The present study was supported by the Japan Society for the
Promotion of Scientific KAKENHI (Grants-in-Aid for Scientific
Research from the Japan Society for the Promotion of Science),
grant nos. 19K09687, 19K07480 and 19K07663.
Abbreviations:
|
ATP
|
adenosine triphosphate
|
|
CPT
|
cryptotanshinone
|
|
DMSO
|
dimethyl sulfoxide
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
DCA
|
dichloroacetate
|
|
EMT
|
epithelial-mesenchymal transition
|
|
MIBC
|
muscle invasive bladder cancer
|
|
PDH
|
pyruvate dehydrogenase
|
|
PDK
|
pyruvate dehydrogenase kinase
|
|
poly-HEMA
|
poly-(2-hydroxyethyl methacrylate)
|
|
siRNA
|
small interfering RNA
|
|
3D
|
three-dimensional
|
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hsu PP and Sabatini DM: Cancer cell
metabolism: Warburg and beyond. Cell. 134:703–707. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sugden MC and Holness MJ: Mechanisms
underlying regulation of the expression and activities of the
mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem.
112:139–149. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kinnaird A, Dromparis P, Saleme B, Gurtu
V, Watson K, Paulin R, Zervopoulos S, Stenson T, Sutendra G, Pink
DB, et al: Metabolic modulation of clear-cell renal cell carcinoma
with dichloroacetate, an inhibitor of pyruvate dehydrogenase
kinase. Eur Urol. 69:734–744. 2016. View Article : Google Scholar
|
|
6
|
Bonnet S, Archer SL, Allalunis-Turner J,
Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta
L, Bonnet S, et al: A mitochondria-K+ channel axis is
suppressed in cancer and its normalization promotes apoptosis and
inhibits cancer growth. Cancer Cell. 11:37–51. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saunier E, Benelli C and Bortoli S: The
pyruvate dehydrogenase complex in cancer: An old metabolic
gatekeeper regulated by new pathways and pharmacological agents.
Int J Cancer. 138:809–817. 2016. View Article : Google Scholar
|
|
8
|
Leclerc D, Pham DN, Lévesque N, Truongcao
M, Foulkes WD, Sapienza C and Rozen R: Oncogenic role of PDK4 in
human colon cancer cells. Br J Cancer. 116:930–936. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Trinidad AG, Whalley N, Rowlinson R,
Delpuech O, Dudley P, Rooney C and Critchlow SE: Pyruvate
dehydrogenase kinase 4 exhibits a novel role in the activation of
mutant KRAS, regulating cell growth in lung and colorectal tumour
cells. Oncogene. 36:6164–6176. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stacpoole PW: Therapeutic targeting of the
pyruvate dehydrogenase complex/pyruvate dehydrogenase kinase
(PDC/PDK) axis in cancer. J Natl Cancer Inst. 109:2017 View Article : Google Scholar
|
|
11
|
Sutendra G and Michelakis ED: Pyruvate
dehydrogenase kinase as a novel therapeutic target in oncology.
Front Oncol. 3:382013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kankotia S and Stacpoole PW:
Dichloroacetate and cancer: New home for an orphan drug? Biochim
Biophys Acta. 1846:617–629. 2014.PubMed/NCBI
|
|
13
|
Yamane K, Indalao IL, Chida J, Yamamoto Y,
Hanawa M and Kido H: Diisopropylamine dichloroacetate, a novel
pyruvate dehydrogenase kinase 4 inhibitor, as a potential
therapeutic agent for metabolic disorders and multiorgan failure in
severe influenza. PLoS One. 9:e980322014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chu QS, Sangha R, Spratlin J, Vos LJ,
Mackey JR, McEwan AJ, Venner P and Michelakis ED: A phase I
open-labeled, single-arm, dose-escalation, study of dichloroacetate
(DCA) in patients with advanced solid tumors. Invest New Drugs.
33:603–610. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tambe Y, Terado T, Kim CJ, Mukaisho K,
Yoshida S, Sugihara H, Tanaka H, Chida J, Kido H, Yamaji K, et al:
Antitumor activity of potent pyruvate dehydrogenase kinase 4
inhibitors from plants in pancreatic cancer. Mol Carcinog.
58:1726–1737. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fares J, Fares MY, Khachfe HH, Salhab HA
and Fares Y: Molecular principles of metastasis: A hallmark of
cancer revisited. Signal Transduct Target Ther. 5:282020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dillekås H, Rogers MS and Straume O: Are
90% of deaths from cancer caused by metastases? Cancer Med.
8:5574–5576. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kamarajugadda S, Stemboroski L, Cai Q,
Simpson NE, Nayak S, Tan M and Lu J: Glucose oxidation modulates
anoikis and tumor metastasis. Mol Cell Biol. 32:1893–1907. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Han T, Kang D, Ji D, Wang X, Zhan W, Fu M,
Xin HB and Wang JB: How does cancer cell metabolism affect tumor
migration and invasion? Cell Adh Migr. 7:395–403. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gatenby RA and Gawlinski ET: The
glycolytic phenotype in carcinogenesis and tumor invasion: Insights
through mathematical models. Cancer Res. 63:3847–3854.
2003.PubMed/NCBI
|
|
21
|
Williams AC, Collard TJ and Paraskeva C:
An acidic environment leads to p53 dependent induction of apoptosis
in human adenoma and carcinoma cell lines: Implications for clonal
selection during colorectal carcinogenesis. Oncogene. 18:3199–3204.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lardner A: The effects of extracellular pH
on immune function. J Leukoc Biol. 69:522–530. 2001.PubMed/NCBI
|
|
23
|
GLOBCAN 2018 Cancer fact sheet: Bladder.
http://gco.iarc.fr/today/data/factsheets/cancers/30-Bladder-fact-sheet.pdf.
Accessed December 22, 2020.
|
|
24
|
von der Maase H, Sengelov L, Roberts JT,
Ricci S, Dogliotti L, Oliver T, Moore MJ, Zimmermann A and Arning
M: Long-term survival results of a randomized trial comparing
gemcitabine plus cisplatin, with methotrexate, vinblastine,
doxorubicin, plus cisplatin in patients with bladder cancer. J Clin
Oncol. 23:4602–4608. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wahafu W, Liu S, Xu W, Wang M, He Q, Song
L, Wang M, Yang F, Hua L, Niu Y and Xing N: The long-term efficacy
of one-shot neoadjuvant intra-arterial chemotherapy combined with
radical cystectomy versus radical cystectomy alone for bladder
cancer: A propensity-score matching study. BMC Urol. 19:1172019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Goto T and Miyamoto H: Why has the
prognosis for muscle-invasive bladder cancer not significantly
improved after decades of therapeutic advancements? Expert Rev
Anticancer Ther. 20:229–231. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Woolbright BL, Choudhary D, Mikhalyuk A,
Trammel C, Shanmugam S, Abbott E, Pilbeam CC and Taylor JA III: The
role of pyruvate dehydrogenase Kinase-4 (PDK4) in bladder cancer
and chemoresistance. Mol Cancer Ther. 17:2004–2012. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim CJ, Terado T, Tambe Y, Mukaisho K,
Sugihara H, Kawauchi A and Inoue H: Anti-oncogenic activities of
cyclin D1b siRNA on human bladder cancer cells via induction of
apoptosis and suppression of cancer cell stemness and invasiveness.
Int J Oncol. 52:231–240. 2018.
|
|
29
|
Kim CJ, Nishi K, Isono T, Okuyama Y, Tambe
Y, Okada Y and Inoue H: Cyclin D1b variant promotes cell
invasiveness independent of binding to CDK4 in human bladder cancer
cells. Mol Carcinog. 48:953–964. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang Q, Miao S, Han X, Li C, Zhang M, Cui
K, Xiong T, Chen Z, Wang C and Xu H: MicroRNA-3619-5p suppresses
bladder carcinoma progression by directly targeting β-catenin and
CDK2 and activating p21. Cell Death Dis. 9:9602018. View Article : Google Scholar
|
|
31
|
Tambe Y, Hasebe M, Kim CJ, Yamamoto A and
Inoue H: The drs tumor suppressor regulates glucose metabolism via
lactate dehydrogenase-B. Mol Carcinog. 55:52–63. 2016. View Article : Google Scholar
|
|
32
|
Zhang W, Zhang SL, Hu X and Tam KY:
Targeting tumor metabolism for cancer treatment: Is pyruvate
dehydrogenase kinases (PDKs) a viable anticancer target? Int J Biol
Sci. 11:1390–1400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weiswald LB, Bellet D and Dangles-Marie V:
Spherical cancer models in tumor biology. Neoplasia. 17:1–15. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fang D, Hawke D, Zheng Y, Xia Y,
Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T and Lu Z:
Phosphorylation of beta-catenin by Akt promotes beta-catenin
transcriptional activity. J Biol Chem. 282:11221–11229. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mulholland DJ, Dedhar S, Coetzee GA and
Nelson CC: Interaction of nuclear receptors with the
Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr
Rev. 26:898–915. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Daugherty RL and Gottardi CJ:
Phospho-regulation of Beta-catenin adhesion and signaling
functions. Physiology (Bethesda). 22:303–309. 2007.
|
|
37
|
Han J, Xie C, Pei T, Wang J, Lan Y, Huang
K, Cui Y, Wang F, Zhang J, Pan S, et al: Deregulated
AJAP1/β-catenin/ZEB1 signaling promotes hepatocellular carcinoma
carcinogenesis and metastasis. Cell Death Dis. 8:e27362017.
View Article : Google Scholar
|
|
38
|
Higuchi T, Yokobori T, Naito T, Kakinuma
C, Hagiwara S, Nishiyama M and Asao T: Investigation into
metastatic processes and the therapeutic effects of gemcitabine on
human pancreatic cancer using an orthotopic SUIT-2 pancreatic
cancer mouse model. Oncol Lett. 15:3091–3099. 2018.PubMed/NCBI
|
|
39
|
Lu Z, Ghosh S, Wang Z and Hunter T:
Downregulation of caveolin-1 function by EGF leads to the loss of
E-cadherin, increased transcriptional activity of beta-catenin, and
enhanced tumor cell invasion. Cancer Cell. 4:499–515. 2003.
View Article : Google Scholar
|
|
40
|
Jing Y, Cui D, Guo W, Jiang J, Jiang B, Lu
Y, Zhao W, Wang X, Jiang Q, Han B and Xia S: Activated androgen
receptor promotes bladder cancer metastasis via Slug mediated
epithelial-mesenchymal transition. Cancer Lett. 348:135–145. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gunthert U, Hofmann M, Rudy W, Reber S,
Zöller M, Haussmann I, Matzku S, Wenzel A, Ponta H and Herrlich P:
A new variant of glycoprotein CD44 confers metastatic potential to
rat carcinoma cells. Cell. 65:12–24. 1991. View Article : Google Scholar
|
|
42
|
Nagano O and Saya H: Mechanism and
biological significance of CD44 cleavage. Cancer Sci. 95:930–935.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ponta H, Sherman L and Herrlich PA: CD44:
From adhesion molecules to signalling receptors. Nat Rev Mol Cell
Biol. 4:33–45. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
van Grevenstein WM, Hofland LJ, Jeekel J
and van Eijck CH: The expression of adhesion molecules and the
influence of inflammatory cytokines on the adhesion of human
pancreatic carcinoma cells to mesothelial monolayers. Pancreas.
32:396–402. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Abugomaa A, Elbadawy M, Yamawaki H, Usui T
and Sakaki K: Emerging roles of cancer stem cells in bladder cancer
progression, tumorigenesis, and resistance to chemotherapy: A
potential therapeutic target for bladder cancer. Cells. 9:2352020.
View Article : Google Scholar :
|
|
46
|
Brunner A, Prelog M, Verdorfer I, Tzankov
A, Mikuz G and Ensinger C: EpCAM is predominantly expressed in high
grade and advanced stage urothelial carcinoma of the bladder. J
Clin Pathol. 61:307–310. 2008. View Article : Google Scholar
|
|
47
|
Xia P and Xu X: PI3K/Akt/mTOR signaling
pathway in cancer stem cells: From basic research to clinical
application. Am J Cancer. 5:1602–1609. 2015.
|
|
48
|
Matsubara S, Ding Q, Miyazaki Y, Kuwahara
T, Tsukasa K and Takao S: mTOR plays critical roles in pancreatic
cancer stem cells through specific and stemness-related functions.
Sci Rep. 3:32302013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Deshmukh A, Deshpande K, Arfuso F,
Newsholme P and Dharmarajan A: Cancer stem cell metabolism: A
potential target for cancer therapy. Mol Cancer. 15:692016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cao ZQ, Wang Z and Leng P: Aberrant
N-cadherin expression in cancer. Biomed Phamacother.
118:1093202019. View Article : Google Scholar
|
|
51
|
Magaway C, Kim E and Jacinto E: Targeting
mTOR and metabolism in cancer: Lessons and innovations. Cells.
8:15842019. View Article : Google Scholar
|
|
52
|
Valvezan AJ and Manning BD: Molecular
logic of mTORC1 signalling as a metabolic rheostat. Nat Metab.
1:321–333. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zou Z, Tao T, Li H and Zhu X: mTOR
signaling pathway and mTOR inhibitors in cancer: Progress and
challenges. Cell Biosci. 10:312020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Prossomariti A, Piazzi G, Alquati C and
Ricciardiello L: Are Wnt/β-catenin and PI3K/AKT/MTORC1 distinct
pathways in colorectal cancer. Cell Mol Gastroenterol Hepatol.
10:491–506. 2020. View Article : Google Scholar :
|
|
55
|
Liu Y, Lin F, Chen Y, Wang R, Liu J, Jin J
and An R: Cryptotanshinone inhibits bladder cancer cell
proliferation and promotes apoptosis via the PTEN/PI3K/AKT pathway.
J Cancer. 11:488–499. 2020. View Article : Google Scholar :
|