1. Introduction
The drug riluzole [2-amino 6
(trifluoromethoxy)benzothiazole], due to its neuroprotective
qualities, was Food and Drug Administration-approved in 1995 for
the treatment and management of amyotrophic lateral sclerosis
(1). Riluzole was indicated to
block voltage-dependent sodium channels in a dose-dependent manner
(2,3). While the precise mechanism of action
of riluzole is still under investigation, numerous studies have
demonstrated that the effect of riluzole is derived from its
ability to block glutamate release and enhance glutamate reuptake
(4), thus leading to the
inhibition of glutamate-dependent signaling. Glutamate signaling is
hyperactive in numerous neurological diseases in which riluzole may
have beneficial effects (5). Of
note, cancers of numerous tissues also rely on glutamate signaling
to survive and proliferate (6).
Consequently, several investigations have explored the efficacy of
riluzole treatment in different cancers types, both in vitro
and in vivo (Table I). Due
to its efficacy, low toxicity and tolerability, riluzole became a
potential treatment for a number of cancer types (7-9).
Numerous studies have uncovered multiple mechanisms by which
riluzole targets each specific cell type. An understanding of the
molecular targets of riluzole and underlying mechanism in specific
cancer types may improve the current application of riluzole in
cancer treatment. The present review focused on all available data
pertaining to the effects of riluzole in cancers of various tissue
origins and its potential as a therapeutic agent, as represented in
Fig. 1.
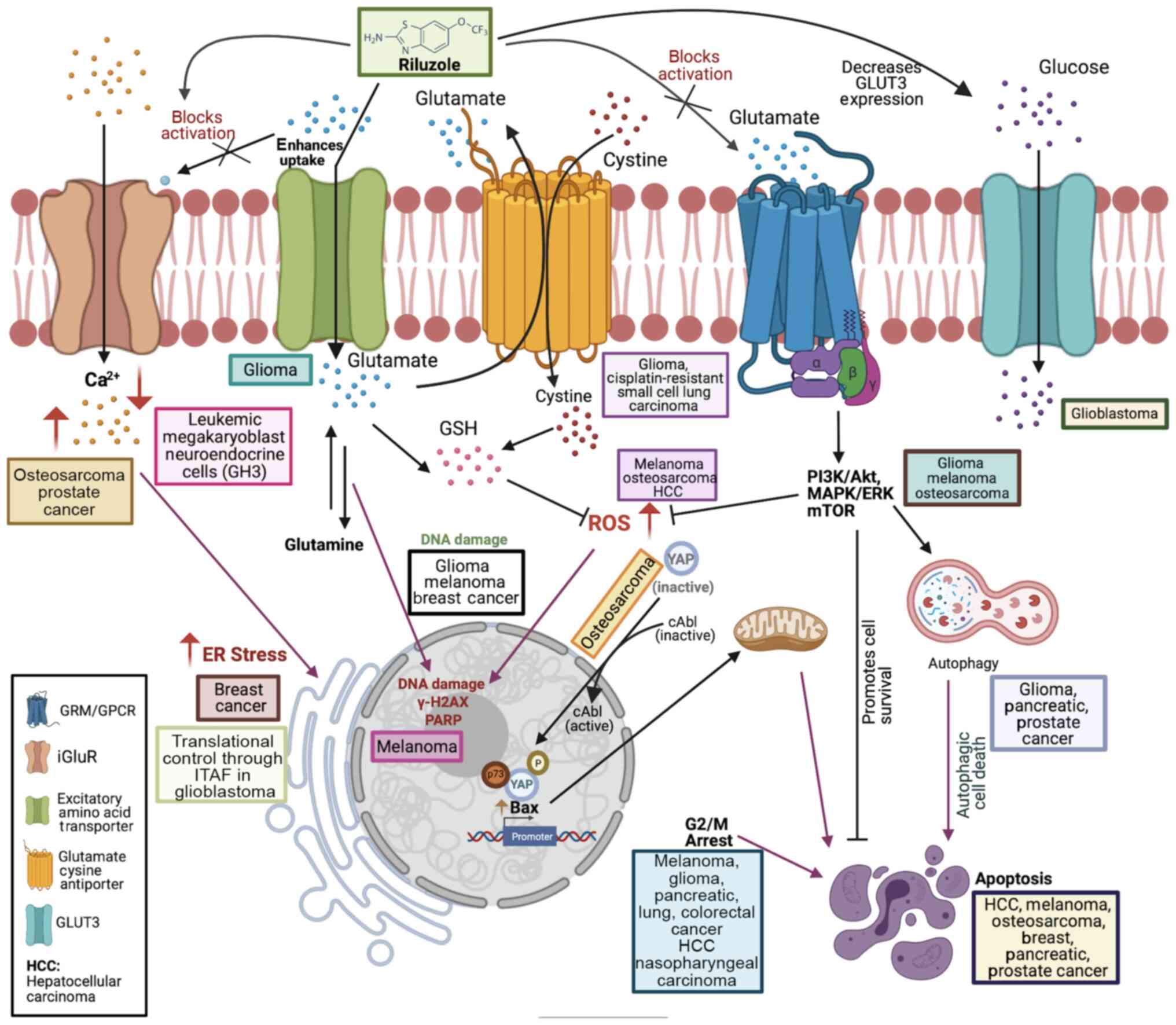 | Figure 1Schematic representation of receptors
and pathways targeted by riluzole. Riluzole was indicated to
increase Ca2+ levels in osteosarcoma and prostate
cancer, while decreasing Ca2+ levels in leukemic
megakaryoblast neuroendocrine cells (GH3). Increased
Ca2+ levels contribute to ER stress, as observed in
breast cancer. Riluzole is known to block protein translation in
glioblastoma through ITAF, IRES trans-acting factor. Riluzole is
thought to inhibit glutamate release by blocking the
voltage-dependent sodium channels (not shown) and enhances
glutamate uptake through excitatory amino acid transporter, which
regulates extracellular glutamate levels. Glioma cells lack a
functional glutamate uptake system, leading to excessive
extracellular glutamate. Riluzole blocks glutamate cystine
antiporter in glioma and cisplatin-resistant small cell lung
carcinoma. Inhibition of glutamate cystine antiporter by riluzole
reduces cystine import, thereby decreasing GSH synthesis, which in
turn leads to increase in ROS, as observed in melanoma,
osteosarcoma and HCC. Increases in ROS lead to DNA damage as
reported in glioma, melanoma and breast cancer. In melanoma cells,
riluzole elevates γ-H2AX levels and increases PARP cleavage. DNA
damage caused by riluzole leads to cell cycle arrest in G2/M phase,
as observed in melanoma, pancreatic cancer, HCC and nasopharyngeal
carcinoma. Increased ROS may contribute to phosphorylation of YAP
by cAbl kinase to promote apoptosis in osteosarcoma. Inhibition of
glutamate release by riluzole prevents activation of GRM and
signaling through these receptors, as reported in glioma, melanoma
and osteosarcoma. Blockage of these pathways by riluzole induces
autophagic death in glioma, pancreatic cancer and prostate cancer.
Riluzole induces apoptosis in breast cancer, melanoma, HCC,
prostate cancer, pancreatic cancer and osteosarcoma. Riluzole was
also indicated to decrease glucose transporter GLUT3 levels,
thereby decreasing glucose import in glioblastoma. Thus, riluzole
targets numerous types of receptors/transporters and associated
signaling pathways to cause cell death in various cancer types. The
figure was rendered using Biorender.com. ROS, reactive oxygen species; HCC,
hepatocellular carcinoma; ER, endoplasmic reticulum; GSH,
glutathione; GRM, metabotropic glutamate receptors; GPCR, G
protein-coupled receptor; YAP, YES-associated protein; PARP, poly
(adenosine diphosphate ribose) polymerase; iGluR, ionotropic
glutamate receptors; ITAF, IRES trans-acting factor. |
 | Table IEffects of riluzole on cancer
cells. |
Table I
Effects of riluzole on cancer
cells.
| Cancer type/cell
lines | Mechanism | (Refs.) |
|---|
| Pancreatic
cancer | | |
| PANC1, SW1990,
BXPC3, ASPC1 | Autophagy G2/M cell
cycle arrest Apoptosis | (67) |
| Colorectal
cancer | | |
| HCT116, H630,
HCT8, CACO2 and HT29 | Sensitizes cells to
cisplatin reduces cell viability in vitro and polyp
development in vivo G2/M arrest, apoptosis | (77,92) |
| Hepatocellular
carcinoma | | |
| SNU449, Huh-7 | G2/M cell cycle
arrest Apoptosis | (52) |
| Melanoma | | |
| SKMEL2, C8161,
UACC903 and 1205Lu | G2/M cell cycle
arrest MAPK/PI3K/AKT signaling DNA damage Apoptosis | (28,32,38,53) |
| Prostate
cancer | | |
|
LNCaP-androgen-dependent | ER stress | (44,71) |
|
C4-2-androgen-independent | Autophagy | |
| 22Rv1 | Apoptosis | |
| VCaP | | |
| CWR1-R1ca | | |
| Breast cancer | | |
| SUM149 | Apoptosis | (79,91) |
| SUM102 | ER stress | |
| SUM229 | | |
| Glioblastoma | | |
| LN229, T98G, short
term PDX patient-derived line | | |
| GBM6 | Translational
control | (57) |
| U87MG
glioblastoma | | |
| Neuroblastoma | | |
|
Neuron-neuroblastoma hybrid (NSC-34D),
IM32 neuroblastoma cells | Calcium levels | (41) |
| Lung cancer | | |
| A549 | G2/M arrest,
apoptosis | (63) |
| Glioma | | |
| U87MG glioma
cells, U118MG & LN229 | Cytotoxicity, tumor
suppression, DNA damage | (7,33) |
| Brain tumor
stem-like cell lines used: 11SP and 64SP | Autophagy
Sensitizes to radiation | |
| C6 cells | G2/M arrest,
apoptosis | (63) |
| Human
nasopharyngeal carcinoma | | |
| CNE1, CNE2 and
HNE1 | ATM/P53 G2/M arrest
Sensitizes to radiation | (64) |
| Osteosarcoma | Inhibits cell
proliferation | (35) |
| LM7 and OS482 | Apoptosis | |
| LM7 | cAbl kinase
activation YAP phosphorylation at Y357, binding to p73 and Bax
promoter activation | (55) |
2. Mechanisms of action of riluzole
Consequences of blocking glutamate
secretion with riluzole
Glutamate signaling is frequently mentioned in the
context of the central nervous system, where it acts as the major
excitatory neurotransmitter; however, glutamate signaling is
operational in numerous different cell types (10). For instance, there is a strong
link between glutamate signaling, cell survival and differentiation
of peripheral tissues, including bone (10-12). The key receptors in the glutamate
pathway are classified into two broad categories: Ionotropic
glutamate receptors and metabotropic-glutamate receptors
(mGluR/GRM) (5,13). While the glutamate receptors
largely differ in structure and mechanism of action, these
receptors frequently share similar functions and interact with each
other in a cooperative or non-cooperative manner (14). Ionotropic glutamate receptors are
involved in the regulation of ion intake, including certain
essential ions such as Ca2+, which is responsible for
maintaining cellular homeostasis. The metabotropic glutamate
receptors are G protein-coupled receptors, which mediate signal
transduction pathways involved in several cellular processes, such
as cell stress response, survival, growth and proliferation
(5).
Cystine-glutamate antiporter (xCT) is a glutamate
cystine antiporter that regulates the antioxidant system in cells
that contributes to growth, metastasis and invasion of cancer cells
(15). In this context, cancer
cell lines of multiple tissue origin have been indicated to secrete
glutamate via the xCT transporter (16,17). Of note, gliomas increase glutamate
levels in the extracellular space by mislocalized excitatory amino
acid transporter 1 (EAAT1) to the nucleus and decreasing the levels
of EAAT2, while simultaneously increasing glutamate secretion via
the xCT transporter (18).
Excessive secretion of glutamate by glioma causes neurotoxicity to
facilitate glioma growth (19).
The silencing of the xCT by small interfering RNA in glioma
decreased glutamate secretion, neurodegeneration and brain edema
(20). Of note, riluzole induced
cytotoxicity in GRM3-expressing glioma in vitro and reduced
tumor size in xenograft mice (7).
In melanoma, riluzole dose-dependently decreased cell proliferation
in vitro. It is noteworthy that downregulation of the xCT
was observed in xenograft-bearing animals that were treated with
riluzole, suggesting that xCT is a possible molecular target of
riluzole (21). Another
remarkable study suggested riluzole's growth inhibitory effect on
cisplatin-resistant small cell lung cancer cells in vitro
via the upregulation of both the xCT and CD44 variant, of which
CD44 is known to stabilize xCT (22). Although riluzole reduced tumor
size in vivo, the effects of riluzole were indicated to be
independent of glutamate signaling (22). Thus, blocking xCT and subsequent
inhibition of glutamate receptors may be one of the mechanisms by
which riluzole prevents cell growth.
Riluzole modulates glutamate-dependent
and glutamate-independent intracellular signaling pathways
In numerous cancer cell types, glutamate receptors
are overexpressed to enhance cancer cell survival and proliferation
(9). Tumorigenesis is linked to
several major intracellular signaling pathways, including the
PI3K/Akt/mTOR, Ras-MAP-ERK and MAPK/ERK pathways (23-26). A genomic study revealed a high
frequency of mutations across pathways, suggesting potential broad
cancer-targeting strategies (27). For instance, GRM1 is overexpressed
in melanoma cells, whereas the GRM3 receptor is expressed in
gliomas (7,28). Stimulation of GRM1 in melanomas
and GRM3 in glioma cells by agonists leads to the activation of
MAPK signaling, more specifically to the phosphorylation of ERK
(7,28). In malignant melanoma, GRM3 is
mutated, resulting in hypersensitivity of the MEK-MAPK pathway, and
overexpression of GRM5 increased the activation of the MAPK pathway
(29,30). In melanoma, treatment with
riluzole effectively suppressed MAPK/ERK and PI3K/AKT pathway
hyperactivity and related cellular processes, including cell
proliferation in vitro, as well as in a phase 0 clinical
trial (28,31). While the efficacy of riluzole was
observed in melanoma, it appears that the presence of mutations in
N-Ras, B-Raf or phosphatase and tensin homolog (PTEN) was able to
hinder the effect of the drug (31,32). Of note, riluzole in combination
with a drug for mTOR, a downstream effector of PI3K/AKT, improved
the effect of riluzole on melanoma cells with these mutations
(32).
In human brain tumor-like stem cells (BTSCs) derived
from glioblastoma, riluzole inhibited cell growth by decreasing
GLUT3 transporter expression. Decreased GLUT3 expression resulted
in lower uptake of glucose by the BTSCs, which heavily rely on
aerobic glycolysis. Consequently, riluzole-induced decrease in
GLUT3 was indicated to depend on the decrease in phosphorylated
(p-)AKT, leading to a decrease in the induction of
hypoxia-inducible factor (HIF)1α expression (33). HIF1α is a transcriptional
regulator of the SLC2A3 gene, which encodes for GLUT3 glucose
transporter. Therefore, riluzole modulates metabolic activity of
cells by altering phosphorylation of AKT. Of note, poor prognosis
of glioblastoma is dependent on the overexpression of DNA
(cytosine-5-)-methyltransferase 1 (DNMT1), which causes
hypermethylation of tumor suppressor genes (34). Riluzole was indicated to decrease
DNMT1 gene expression as a consequence of decreased GLUT3
expression and reduced tumor size in mice (33). Therefore, riluzole inhibited cell
growth by altering AKT phosphorylation to control glucose
metabolism in BTSCs and indirectly altering the expression of tumor
suppressor genes to inhibit growth in glioblastoma cells.
Furthermore, in osteosarcoma expressing GRM5, riluzole blocked cell
proliferation by altering phosphorylation of AKT (at both T308 and
S473) and p70 S6 kinase at threonine 389, a hallmark of mTOR
activation, suggesting the activation of the PI3K/AKT/mTOR pathway
in osteosarcoma growth. In addition, riluzole altered
phosphorylation at ERK1/2and JNK1/2 kinases in osteosarcoma
(35). Another key pathway linked
to oncogenesis is Wnt/β-catenin signaling, which regulates the
amount of the transcriptional co-activator β-catenin (36). In malignant melanoma,
downregulation of β-catenin has an important role in disease
progression and contributes to poor prognosis (37). Of note, in melanoma, riluzole
increased the levels of WNT3A protein involved in the stimulation
of the Wnt/β-catenin pathway and melanocyte differentiation,
subsequently leading to decreased cell proliferation both in
vivo and in vitro (38). Thus, riluzole targets multiple
signaling pathways to block cell proliferation in glioma,
osteosarcoma and melanoma.
Riluzole regulates intracellular
Ca2+ in both cancerous and non-cancerous cells
In the endoplasmic reticulum (ER), calcium levels
influence protein folding and trafficking, whereas in mitochondria,
calcium influences mitochondrial permeability, which contributes to
the modulation of intracellular reactive oxygen species (ROS)
levels and may have a direct effect on mitochondrial-mediated
apoptosis inside the cell (39).
Although riluzole is known to inhibit glutamate release and hence
block glutamate signaling, certain studies reported its potential
for targeting Ca2+ signaling in cells. In one of these
studies, riluzole inhibited spontaneous Ca2+ signaling
in the immortalized growth hormone-secreting pituitary cell line
GH3 (40). In another study using
the neuroblastoma-spinal motor neuron fusion cell line NSC-34D
(non-cancerous), riluzole counteracted the upregulation of
Ca2+ increase and cell death induced by thapsigargin, a
known inhibitor of sarcoplasmic calcium ATPase (41). While the exact mechanism for
Ca2+ inhibition by riluzole still remains to be
determined in cancer cells, riluzole was reported to block
glutamate release and glutamate regulated Ca2+ entry in
leukemic megakaryoblasts and promoted differentiation by blocking
cell proliferation (42).
Contrary to this effect, riluzole increased cytosolic
Ca2+ increase in prostate cancer cells that increased ER
stress, and increased Ca2+ levels in MG63 osteosarcoma
cells through an unidentified pathway (43,44). These results suggested that
intracellular Ca2+ regulation is one of the cellular
processes in cancer that may be affected by riluzole.
Riluzole increases oxidative stress
Typically, the cell is thought to be under oxidative
stress when the intracellular levels of reactive oxygen species
(ROS), such as hydrogen peroxide (H2O2) and
ion species (O2−, OH), exceed the levels of
antioxidants. Tumor cells have comparatively higher metabolic
requirements and altered metabolism; consequently, they generate
increased ROS, which are employed by the cells for survival, cell
motility and metastasis, tumor progression and angiogenesis
(45,46). Glutathione (GSH) is a well-known
antioxidant that reduces oxidative stress and ROS levels by
accepting an electron from a free radical on ROS (47). Of note, GSH synthesis is directly
linked to glutamate release, since glutamate serves as one of the
precursor molecules of GSH (48).
In normal cells, the overload of intracellular glutamate and
upregulation of Ca2+ leads to increases in ROS and ER
stress, resulting in both DNA damage and subsequent apoptosis
(49). On the other hand, cancer
cells have a number of mechanisms in place to resist apoptosis. The
antioxidant production pathways are typically upregulated, which
include GSH synthesis and salvage pathways (50,51). A key player in GSH production is
xCT, which imports cystine for GSH synthesis and is overexpressed
in cancer (15).
By inhibiting glutamate release in cancer cells,
riluzole has been indicated to decrease the overall GSH levels
(22). The reduction in GSH leads
to a marked reduction in anti-oxidants and increased ROS, which
induces cell cycle arrest in G2/M phase and eventual apoptosis
(7,22,52,53). In one of the most recent studies
on DNA damage repair inhibition in melanoma, riluzole promoted the
accumulation of ROS that triggered DNA double-strand breaks
(54). However, this effect was
observed in both GluR1-positive and -negative melanoma cells, which
further added to the complexity of the mechanisms of riluzole
causing oxidative stress (54).
Of note, in hepatocellular carcinoma (HCC), riluzole treatment was
indicated to cause an increase in cellular glutamate content, which
led to a decrease in GSH production as a consequence of a decrease
in cysteine uptake by the cells. This further resulted in the
accumulation of ROS, leading to apoptosis. In addition,
administration of riluzole inhibited growth in HCC xenografts, and
reduced GSH and increased ROS levels in the tumors (52). In osteosarcoma, riluzole increased
ROS production, leading to the activation of cAbl kinase, which
participates in events leading to apoptosis (55). Of note, the increased ROS in
cisplatin-resistant lung cancer cells were further enhanced by
riluzole to induce cell death, making it the only agent so far to
successfully kill cisplatin-resistant cells (22). It is noteworthy that in cancer
cells where glutamate secretion occurs via xCT, riluzole may
increase intracellular levels of glutamate to cause
glutamate-induced oxidative stress while simultaneously blocking
GSH synthesis due to deficiency of cystine by blocking xCT
(21). While the mechanism
remains to be fully elucidated, the available studies suggested
that in numerous cancer types, glutamate release inhibition by
riluzole contributes to ROS production.
Riluzole and protein translation
In glioblastoma, inhibitors of mTOR kinase have been
indicated to enhance IRES-dependent protein synthesis of key
regulators of the cell cycle, leading to resistance to the mTOR
inhibitors. Furthermore, IRES-dependent translation was reported to
have a key role in tumor growth and resistance. One of the
significant proteins involved in response to the accumulation of
misfolded protein is IRES trans-acting factor (ITAF) heterogenous
nuclear ribonucleoprotein A1 (hnRNP A1). hnRNA A1 has been
indicated to regulate IRES-dependent translation of c-myc and
cyclin D1 genes (56). In a
recent study using surface plasmon resonance imaging, riluzole was
indicated to directly bind to hnRNP A1 through its specific binding
site to prevent IRES RNA binding. This was the first report to
demonstrate the interaction of riluzole with a protein directly to
regulate its activity (57). In a
recent study, riluzole was indicated to downregulate ITAF hnRNP A1
activity to decrease translation of c-myc and cyclin D1 genes, and
as a result, to assist in offsetting the resistance to mTOR
inhibitor in glioblastoma (57).
Thus, riluzole may exert its effect through direct interactions
with proteins outside of the glutamate metabolic and signaling
pathway, such as ITAF hnRNP A1.
Riluzole induces DNA damage
A cell may be able to sustain DNA damage from
intracellular factors such as oxidative stress and replication
errors to extracellular factors such as ultraviolet radiation and
chemical carcinogens (58). There
are two prominent classifications of DNA damage: Single-strand
breaks (SSBs) and double-strand breaks (DSBs) (59). It is well known that cells have
adopted several strategies to repair DNA damage; for instance, SSBs
are repaired through a base excision repair pathway, whereas for
DSBs, the homologous recombination repair or the non-homologous
end-joining pathways are utilized. Damages that arise at a
particular nucleotide also have specialized repair mechanisms, such
as the nucleotide excision repair and mismatch repair pathways.
Cell cycle checkpoints serve as a protective mechanism against DNA
damage. Together, these pathways comprise the cellular DNA damage
response (DDR) (59,60). Dysregulation of DDR is a common
finding in various cancers and is typically associated with
mutations of specific proteins in a given pathway. For instance,
mutation in p53 allows the damaged cell to pass the cell cycle
checkpoint and proliferate despite not satisfying the checkpoint
requirements. Cells with DDR defects frequently become desensitized
to radiation and chemotherapy (59).
Multiple lines of evidence suggest that riluzole
causes damaged cells to accumulate at cell cycle checkpoints,
eventually triggering apoptosis. Specifically, riluzole treatment
in glioma and melanoma cells produce increased levels of DDR
proteins, such as poly(adenosine diphosphate ribose) polymerase
(PARP) and H2AX (7,53). These two biomarkers are frequently
used to assess drug efficacy, since both are frequently present
when DNA is damaged. The DSBs are known to induce the
phosphorylation of histone H2AX, while PARP cleavage is associated
with activation of SSB repair (59). Furthermore, the accumulation of
DNA damage is also associated with a reduction in glutamate release
and GSH levels, suggesting that the production of ROS stimulated by
riluzole treatment is the main contributor to the DNA damage in
these cancer cells (7,53). A similar increase in
phosphorylation of H2AX was also observed in a phase II clinical
trial for advanced melanoma (61). These studies strongly suggest that
riluzole may cause DNA damage, most likely due to induction of ROS
in cancer cells.
Riluzole causes G2/M cell cycle
arrest
In numerous studies, riluzole has been indicated to
cause G2/M cell cycle arrest in cancer cells. For instance, the HCC
cell lines SNU 449 and Huh-7 exhibited cell cycle arrest in G2/M
phase upon exposure to riluzole. Riluzole was indicated to elevate
the expression of cyclin B1 and depress the expression of p21 and
p-cdc2, leading to G2/M cell cycle inhibition in these cells
(35). Riluzole also induced G2/M
phase arrest in the pancreatic cancer cell lines PANC1 and ASPC1 in
a dose-dependent manner, while concomitantly decreasing the levels
of the regulatory protein cyclin-dependent kinase 1 (40). In an in vivo brain
metastasis study using GRM1-expressing human melanoma cells,
riluzole caused G2/M phase arrest and DNA damage. In addition,
Riluzole increased radiosensitivity, resulting in enhanced DNA
damage and reduced metastases in an animal model (62). Riluzole also induced cell cycle
arrest in A549 cells (lung cancer), as well as glioma and
colorectal adenocarcinoma cells (63). Furthermore, in studies involving
human nasopharyngeal carcinoma, Riluzole induced G2/M arrest and
apoptosis (64). In summary,
these studies provide evidence that riluzole causes cell cycle
arrest in G2/M phase in HCC, pancreatic cancer, melanoma and
nasopharyngeal carcinoma cells.
Autophagy
Autophagy, also known as 'self-digestion', is a
process of elimination of misfolded proteins and damaged
organelles. It is characterized by the autophagosome formation
around the components and the fusion of autophagosome with the
lysosome for degradation (65).
Of note, intracellular calcium and ROS levels are also implicated
in autophagy regulation. In normal cells, basal regulation of
autophagy contributes to homeostasis; however, in cancer, autophagy
dysregulation is linked to uncontrolled cell growth and
proliferation, making it a desirable target for cancer therapy
(65,66). As observed in pancreatic cell
lines, riluzole resulted in the upregulation of an autophagy
substrate, p62, as well as cell death in a dose-dependent manner
(67). In another study using a
castrate-resistant prostate cancer cell line expressing GRM1,
riluzole upregulated intracellular Ca2+ levels and
increased the expression of autophagy markers such as Beclin 1,
LC3AII (microtubule associated protein1 light chain 3AII) and p62,
leading to autophagy-mediated degradation of androgen receptor
(44). Furthermore, in glioma,
riluzole caused downregulation of PI3K/Akt signaling and GLUT3
expression, which led to autophagic cell death (33). Together, these studies indicated
that riluzole interferes with autophagic pathways to induce cell
death in various cancers types.
Riluzole induces apoptosis
Typically, upregulated PI3K/Akt and MAPK/ERK
pathways contribute to cancer survival via activated Akt, mTOR,
ERK, Ras and Braf proteins that downregulate pro-apoptotic proteins
while activating anti-apoptotic proteins. Apoptosis is induced via
the intrinsic apoptotic pathway or externally by ligand binding to
the cell receptor via the extrinsic apoptotic pathway (68). Both pathways include sequential
caspase cleavage, with slight differences in caspases involved in
the process. The intrinsic apoptotic pathway is generally triggered
in cells under oxidative stress or when DNA sustains serious
unrepairable damage. The process is regulated by pro-apoptotic
proteins such as Bax and Bak, and anti-apoptotic proteins like
Bcl-2 and Bcl-xl. When the apoptotic processes are initiated, the
apoptosome is formed, which is made up of Apaf-1 and cytochrome
c (released from mitochondria), as well as pro-caspase-9.
The formation of this complex then cleaves and activates caspase 9
to further activate downstream caspases (caspases3/6/7) to induce
apoptotic cell death (68). In
cancer cells, aberrant downregulation of apoptotic proteins and
inhibition of apoptosis is common.
In certain studies on melanoma, HCC, pancreatic
cancer, prostate cancer and breast cancer, riluzole treatment was
indicated to induce apoptotic cell death by alteration of different
cellular processes, ranging from oxidative stress induction,
autophagy inhibition, and downregulation of survival intracellular
signaling pathways (7,52,67,69). In a number of these studies,
apoptosis was assessed by detecting caspase-3 and caspase-9 levels,
which are common apoptotic markers. In HCC, cleaved caspase-3 and
-9, as well as PARP, were increased with riluzole treatment
(52). Similarly, pancreatic
cancer cells also exhibited an increase in caspase-3 (67). Furthermore, in melanomas, both
GRM1-positive and GRM1-negative, an increased amount of cleaved
PARP and caspase-3 was observed following treatment with riluzole
and radiation (70). In prostate
cancer cells lines, independent of their androgen-dependent status,
riluzole decreased cell viability by activation of caspase-3, -8
and -9 (71). In addition,
riluzole was also indicated to induce apoptosis following cell
cycle arrest via the activation of the ATM/p53 pathway in
nasopharyngeal carcinoma (64). A
recent study by our group suggested that in osteosarcoma, riluzole
activated c-Abl kinase, which is typically activated during DNA
damage response. Activated c-Abl kinase was indicated to
phosphorylate YES-associated protein (60), a transcription coactivator, to
facilitate its interaction with P73, a homolog of P53.
Riluzole-mediated YAP and P73 complex was reported to activate Bax
promoter to regulate pro-apoptotic activity (55). Previous studies by our group also
suggested that iron oxide nanoparticle-delivered riluzole induced
apoptosis in vitro in osteosarcoma cells and shrunk
osteosarcoma tumors in a xenograft mouse model (72,73). Since apoptosis ultimately leads to
the elimination of cancer cells, a better understanding of the
induction of riluzole-mediated cell death, the effects of riluzole
on different cellular processes and how they are related to
apoptosis may further improve the use of riluzole in different
cancer types.
3. Riluzole in cancer treatment
Mono- and combined therapy
A better understanding of cancer biology,
particularly the regulation of key factors and processes fueling
cell growth and metastasis, will bring advancements in cancer
treatment. Cancer cells are known to harbor multiple mutations
across different pathways to promote cell proliferation and
metastasis (74). While
monotherapy with single drugs is still in use, it is slowly being
replaced by newer and more efficient combinatorial treatments. In
addition to traditional radiation and chemotherapy, newer advanced
methods of cancer treatment are gaining momentum. Immunotherapy,
nanoparticles and gene therapy using the CRISPR/Cas9 (clustered
regularly interspaced short palindromic repeats and
CRISPR-associated protein 9) gene editing system are among the most
promising (75,76). According to studies by our group,
when delivered via iron oxide nanocage particles, riluzole caused
the highest reduction in osteosarcoma tumor size in nude mouse
xenografts (72,73).
In keeping with its promising potency and efficacy,
riluzole is being tested in combination with other drugs to enhance
its actions. Riluzole is frequently observed to have synergistic
effects with other drugs to decrease cell proliferation and
viability while also promoting apoptosis. Select drug combinations
may amplify these effects in different cancer cell lines. For
instance, riluzole in combination with paclitaxel in
triple-negative breast cancer, or cisplatin in colorectal cancer,
or sorafenib in melanoma, was demonstrated to have synergistic
effects (77-79). Combinatorial treatment with
riluzole and GRM3 antagonist LY341495 blocked the MAPK signaling
pathway in glioma, sensitized glioma to radiation and decreased
anchorage-independent colony growth (7). In an in vivo study in mice
with intra-cranially injected melanoma cells, combinatorial
treatment led to a significant decrease in tumor volume (62). In melanoma, a combination of
sorafenib with riluzole was tested in comparison to another potent
inhibitor, PLX4720, with riluzole. While both combinations had
synergistic effects, the inhibitory effects of sorafenib and
riluzole on cell proliferation were the strongest (78). In HCC, riluzole with sorafenib had
an additive effect in decreasing cell proliferation (52). Melanomas harboring PTEN and NRAS
mutations exhibit resistance to the action of riluzole after
prolonged treatment (19). Of
note, riluzole in combination with mTOR or AKT inhibitors had
promising results in vitro and in xenograft studies in
melanoma with these mutations, and in glioblastoma (32,57). Table II provides a summary of
noteworthy experiments that include riluzole in combination with
other drugs. While the majority of these studies are limited and
confined to animal experiments at the most, they may provide
information on mechanisms of action of riluzole or possibly lead to
clinical trials in the future.
| Table IICombination therapy with
riluzole. |
Table II
Combination therapy with
riluzole.
|
Subjects/samples | Cancer type | Therapeutic agents
combined with riluzole | Mechanism | Observed effects
with riluzole | (Refs.) |
|---|
| Primary HCC from 4
patients | HCC | Sorafenib | Multikinase
inhibitor targets angiogenesis (Raf-1, b-Raf) target proliferation
(VEGF, PDGFB receptors) | Additive effect on
cell growth inhibition | (52) |
| MDA-MB-231, SUM149,
SUM229 in vitro and in xenograft | Triple-negative
breast cancer | Paclitaxel | Inhibitor of
tubulin, inhibit mitotic spindle assembly involved in chromosome
segregation and cell division, induced apoptosis | Synergistic cell
growth inhibition, induced apoptosis | (79) |
| HCT116 with knocked
down hERG expression | Colorectal
cancer | Cisplatin | Binds to DNA and
inhibits replication, promotes DNA damage, inhibits mitosis | Synergistic effect
in reduced viability of cisplatin-resistant cells due to hERG1
overexpression | (77) |
|
TREK+/+/C7/BL6 mice | Not cancerous | Oxaliplatin | Inhibits DNA
synthesis, DNA replication and transcription, induces apoptosis.
Neurotoxic side effects (elevated glutamate release) | Reduced neurotoxic
side effects due to TREK-1 potassium channel | (92) |
| Melanoma cell lines
for In vitro C8161 (WT BRAF & NRAS), UACC903 UACC930,
HT144 (BRAF & PTEN mt) SKMEL2 (NRAS mt) For in vivo
C8161 UACC903 All cell lines are GRM1-positive except UACC930 | Melanoma | Rapamycin | mTOR inhibitor
Combination therapy was more effective than with individual
agent | Decreased
anchorage-independent growth and tumor growth in xenograft.
Combination therapy effective regardless of BRAF mutation and
PI | (32) |
| Melanoma cell line
expressing GRM1: UAC903, 1205Lu, C8161 with either B-RAF WT or mt,
in vitro and xenograft | Melanoma | Sorafenib | Multikinase
inhibitor targets angiogenesis (Raf-1, b-Raf) target proliferation
(VEGF, PDGFB receptors) | Synergistic effect
Reduced PI3K/Akt signaling, reduced cell proliferation on C8161,
additive effect on UAC903 and 1205Lu | (78) |
| Melanoma cell line
expressing GRM1: UAC903, 1205Lu, C8161 with either B-RAF WT or mt,
In vitro and xenograft | Melanoma | PLX4720 | Inhibit
B-RafV600E | Synergistic effect
but less efficacy compares to with sorafenib | (78) |
| Intracranially
injected melanoma C8161-luc+ | Melanoma | Radiation | Increased
apoptosis | Enhanced the effect
of radiation | (62) |
| GRM3 expressing
cell line U87, and T98G cell line, patients' primary samples with
detectable GRM3 expression | Glioma | Radiation | ROS, DNA damage,
apoptosis | Enhanced ROS
accumulation, reduced PI3K/Akt and MAPK/ERK signaling and DNA
damage and apoptosis induction | (7) |
| LN229 and T98G cell
lines | Glioblastoma | pp242 | mTOR inhibitor | Synergistic effect
on proliferation inhibition, enhanced cell cycle arrest and
apoptosis induction | (57) |
| T98G and UG87 | Glioblastoma | TMZ | TMZ-induced
O6-methylguanine DNA methyltransferase (MGMT
expression) | Synergistic effect
in T98G cells but not UG87 Suppressed intracranial tumor
growth | (93) |
| LM7 | Osteosarcoma | Iron oxide
nanocage | Apoptosis | Iron oxide
nanocage-delivered riluzole was most effective on inducing
apoptosis both in vitro and in vivo | (72,73) |
Riluzole as a radiosensitizer
Aside from using riluzole in combination with other
drugs, it may be used as a radio-sensitizer to produce favorable
effects against cancer. The use of riluzole and radiation therapy
revealed a synergistic effect in vitro and in vivo in
both melanoma and glioma cell lines (7,70).
Malignant melanomas, which typically metastasize into the lungs and
brain, are particularly dangerous. Despite several scientific
advancements, the prognosis for advanced-stage melanoma patients
remains bleak. According to the American Cancer Society, the 5-year
relative survival rate for metastatic melanoma patients is only 27%
and these tumors are frequently resistant to chemotherapy and
radiation treatment, which makes them even more dangerous and prone
to reoccurrence. In an in vivo study in mice injected
intracranially with melanoma cells, riluzole in combination with
radiation led to a significant decrease in tumor volume and
GRM1-expressing melanoma exhibited increased apoptosis (62). In another in vitro study on
BTSCs, a lower dose of riluzole in combination with radiation
therapy unexpectedly resulted in enhanced growth inhibition
compared to a higher dose of the drug (33). However, a higher dose of riluzole
with radiation therapy proved to be more effective in vivo
(33). In addition, riluzole
sensitized human nasopharyngeal carcinoma cells to radiation
through the ATM/P53 signaling pathway and cytotoxicity was enhanced
compared to groups treated with riluzole alone both in vivo
and in vitro (64).
Toxicity and adverse effects of
riluzole
Riluzole was observed to have low toxicity in
patients with amyotrophic lateral sclerosis (ALS) treated with a
daily oral dose of ~50 mg (80).
The area under the curve for the serum concentration at 24 h was
~2,000 ng/ml (81,82). Riluzole is well tolerated, with
the most common adverse effect observed being headache (83). Other significant side effects were
nausea, vertigo, somnolence and asthenia, which were indicated to
be dose-related (84,85). Elevation of alanine
aminotransferase was also commonly observed in patients (86). Rare and less frequently reported
side effects are acute hepatitis, leukopenia and methemoglobinemia
(87). Much rarer cases of
hypersensitivity pneumonitis and multi-organ autoimmune syndrome
were also reported (88). More
recently, interstitial pneumonia was observed in 21% of 92 patients
enrolled (89). In patients with
ALS, riluzole prolonged tracheostomy-free survival by 3-6 months;
however, the long-term toxicity of riluzole requires to be
determined for cancer patients considering their longer survival
expectancy. In a study performed in Europe and North America,
riluzole was reported to be well-tolerated for up to 7 years
(90). Further studies require to
be performed to assess the long-term tolerability and side effects
of riluzole.
Limitations of riluzole
In breast cancer cells, riluzole-induced DNA damage
is independent of mGluR1 expression or ER status (8,91).
Of note, riluzole-induced DNA damage was not observed in the breast
cancer cell line MCF-7, which expresses wild-type P53, whereas
other cell lines expressing mutant P53 exhibited riluzole-induced
DNA damage (8). On the contrary,
in melanoma, riluzole induced DNA damage in an mGluR-dependent
manner (53). However, it remains
to be determined whether cell line-specific differences in riluzole
sensitivity, as observed in breast cancer, are dependent on
replicative stress induced by other mutations in cell lines besides
mutations in P53 (8). In any
particular cancer type, it requires to be determined whether the
response to riluzole is dependent on the mutational profile of the
cell line, particularly those mutations that predispose the cells
to oxidative or replicative stress.
4. Conclusion
The current review presents up-to-date information
of the mechanisms of action of riluzole. Riluzole has been reported
to interfere with diverse cellular processes, such as
stress-related response, survival and apoptosis. Various noteworthy
studies have demonstrated the cytotoxic effects of riluzole in the
following scenarios: i) Cisplatin-resistant lung cancer cells; ii)
mTOR inhibitor-resistant glioblastoma; iii) triple-negative breast
cancer cells in combination with paclitaxel; iv) sensitizing effect
on melanoma, gliomas and nasopharyngeal carcinoma to radiation.
Riluzole thus holds great promise for cancers that are challenging
to treat effectively. Investigations into the efficacy of riluzole
in cancers exhibiting drug resistance may be an effective strategy
for circumventing drug resistance in cancer. A deeper insight into
the specific mechanisms of riluzole alone and in combination with
other drugs in a cell type-dependent manner may improve the
efficacy of riluzole and facilitate translation into clinical
use.
Availability of data and materials
Data sharing is not applicable to this article, as
no datasets were generated or analyzed during the current
study.
Authors' contributions
All authors (AB, SL, CT, IT, TT, SSM) performed
literature searches and wrote and edited the article. All authors
read and approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors have no competing interests to
declare.
Acknowledgments
The authors are grateful for the critical comments
provided by Dr Muktar Mahajan (Department of Medical Laboratory
Sciences, Hunter College, New York, USA) as well as the careful
reviewing of the manuscript by Ms Syeda Maryam Azeem (The Graduate
Center, City University of New York, New York, USA) and Ms Shraddha
ChandThakuri (Department of Medical Laboratory Sciences, Hunter
College, New York, USA). The authors also thank Ms Syeda Maryam
Azeem (The Graduate Center, City University of New York, New York,
USA) for rendering the schematic representation in Fig. 1 using Biorender.com.
References
|
1
|
Doble A: The pharmacology and mechanism of
action of riluzole. Neurology. 47(6 Suppl 4): S233–S241. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Urbani A and Belluzzi O: Riluzole inhibits
the persistent sodium current in mammalian CNS neurons. Eur J
Neurosci. 12:3567–3574. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zona C, Siniscalchi A, Mercuri NB and
Bernardi G: Riluzole interacts with voltage-activated sodium and
potassium currents in cultured rat cortical neurons. Neuroscience.
85:931–938. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheah BC, Vucic S, Krishnan AV and Kiernan
MC: Riluzole, neuroprotection and amyotrophic lateral sclerosis.
Curr Med Chem. 17:1942–1999. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Willard SS and Koochekpour S: Glutamate
signaling in benign and malignant disorders: Current status, future
perspectives, and therapeutic implications. Int J Biol Sci.
9:728–742. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu LJ, Wall BA, Wangari-Talbot J and Chen
S: Metabotropic glutamate receptors in cancer. Neuropharmacology.
115:193–202. 2017. View Article : Google Scholar
|
|
7
|
Khan AJ, LaCava S, Mehta M, Schiff D,
Thandoni A, Jhawar S, Danish S, Haffty BG and Chen S: The glutamate
release inhibitor riluzole increases DNA damage and enhances
cytotoxicity in human glioma cells, in vitro and in vivo.
Oncotarget. 10:2824–2834. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dolfi SC, Medina DJ, Kareddula A, Paratala
B, Rose A, Dhami J, Chen S, Ganesan S, Mackay G, Vazquez A and
Hirshfield KM: Riluzole exerts distinct antitumor effects from a
metabotropic glutamate receptor 1-specific inhibitor on breast
cancer cells. Oncotarget. 8:44639–44653. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Prickett TD and Samuels Y: Molecular
pathways: Dysregulated glutamatergic signaling pathways in cancer.
Clin Cancer Res. 18:4240–4246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Skerry TM and Genever PG: Glutamate
signalling in non-neuronal tissues. Trends Pharmacol Sci.
22:174–181. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hinoi E, Takarada T, Ueshima T,
Tsuchihashi Y and Yoneda Y: Glutamate signaling in peripheral
tissues. Eur J Biochem. 271:1–13. 2004. View Article : Google Scholar
|
|
12
|
Cowan RW, Seidlitz EP and Singh G:
Glutamate signaling in healthy and diseased bone. Front Endocrinol
(Lausanne). 3:892012. View Article : Google Scholar
|
|
13
|
Hollmann M and Heinemann S: Cloned
glutamate receptors. Annu Rev Neurosci. 17:31–108. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reiner A and Levitz J: Glutamatergic
signaling in the central nervous system: Ionotropic and
metabotropic receptors in concert. Neuron. 98:1080–1098. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin W, Wang C, Liu G, Bi C, Wang X, Zhou Q
and Jin H: SLC7A11/xCT in cancer: Biological functions and
therapeutic implications. Am J Cancer Res. 10:3106–3126.
2020.PubMed/NCBI
|
|
16
|
Muir A, Danai LV, Gui DY, Waingarten CY,
Lewis CA and Vander Heiden MG: Environmental cystine drives
glutamine anaplerosis and sensitizes cancer cells to glutaminase
inhibition. Elife. 6. pp. e277132017, View Article : Google Scholar
|
|
17
|
Sharma MK, Seidlitz EP and Singh G: Cancer
cells release glutamate via the cystine/glutamate antiporter.
Biochem Biophys Res Commun. 391:91–95. 2010. View Article : Google Scholar
|
|
18
|
Ye ZC, Rothstein JD and Sontheimer H:
Compromised glutamate transport in human glioma cells:
Reduction-mislocalization of sodium-dependent glutamate
transporters and enhanced activity of cystine-glutamate exchange. J
Neurosci. 19:10767–10777. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye ZC and Sontheimer H: Glioma cells
release excitotoxic concentrations of glutamate. Cancer Res.
59:4383–4391. 1999.PubMed/NCBI
|
|
20
|
Savaskan NE, Heckel A, Hahnen E, Engelhorn
T, Doerfler A, Ganslandt O, Nimsky C, Buchfelder M and Eyüpoglu IY:
Small interfering RNA-mediated xCT silencing in gliomas inhibits
neurodegeneration and alleviates brain edema. Nat Med. 14:629–632.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shin SS, Jeong BS, Wall BA, Li J, Shan NL,
Wen Y, Goydos JS and Chen S: Participation of xCT in melanoma cell
proliferation in vitro and tumorigenesis in vivo. Oncogenesis.
7:862018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wangpaichitr M, Wu C, Li YY, Nguyen DJM,
Kandemir H, Shah S, Chen S, Feun LG, Prince JS, Kuo MT and Savaraj
N: Exploiting ROS and metabolic differences to kill cisplatin
resistant lung cancer. Oncotarget. 8:49275–49292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martin GS: Cell signaling and cancer.
Cancer Cell. 4:167–174. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hemmings BA and Restuccia DF: PI3K-PKB/Akt
pathway. Cold Spring Harb Perspect Biol. 4:a0111892012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nicholson KM and Anderson NG: The protein
kinase B/Akt signalling pathway in human malignancy. Cell Signal.
14:381–395. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Schmelzle T and Hall MN: TOR, a central
controller of cell growth. Cell. 103:253–262. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sanchez-Vega F, Mina M, Armenia J, Chatila
WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia
S, et al: Oncogenic signaling pathways in the cancer genome atlas.
Cell. 173:321–337.e10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Namkoong J, Shin SS, Lee HJ, Marin YE,
Wall BA, Goydos JS and Chen S: Metabotropic glutamate receptor 1
and glutamate signaling in human melanoma. Cancer Res.
67:2298–2305. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi KY, Chang K, Pickel JM, Badger JD II
and Roche KW: Expression of the metabotropic glutamate receptor 5
(mGluR5) induces melanoma in transgenic mice. Proc Natl Acad Sci
USA. 108:15219–15224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Prickett TD, Wei X, Cardenas-Navia I, Teer
JK, Lin JC, Walia V, Gartner J, Jiang J, Cherukuri PF, Molinolo A,
et al: Exon capture analysis of G protein-coupled receptors
identifies activating mutations in GRM3 in melanoma. Nat Genet.
43:1119–1126. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yip D, Le MN, Chan JL, Lee JH, Mehnert JA,
Yudd A, Kempf J, Shih WJ, Chen S and Goydos JS: A phase 0 trial of
riluzole in patients with resectable stage III and IV melanoma.
Clin Cancer Res. 15:3896–3902. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rosenberg SA, Niglio SA, Salehomoum N,
Chan JL, Jeong BS, Wen Y, Li J, Fukui J, Chen S, Shin SS and Goydos
JS: Targeting glutamatergic signaling and the PI3 kinase pathway to
halt melanoma progression. Transl Oncol. 8:1–9. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sperling S, Aung T, Martin S, Rohde V and
Ninkovic M: Riluzole: A potential therapeutic intervention in human
brain tumor stem-like cells. Oncotarget. 8:96697–96709. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rajendran G, Shanmuganandam K, Bendre A,
Muzumdar D, Goel A and Shiras A: Epigenetic regulation of DNA
methyltransferases: DNMT1 and DNMT3B in gliomas. J Neurooncol.
104:483–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liao S, Ruiz Y, Gulzar H, Yelskaya Z, Ait
Taouit L, Houssou M, Jaikaran T, Schvarts Y, Kozlitina K, Basu-Roy
U, et al: Osteosarcoma cell proliferation and survival requires
mGluR5 receptor activity and is blocked by Riluzole. PLoS One.
12:e01712562017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nusse R and Clevers H: Wnt/β-catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kageshita T, Hamby CV, Ishihara T,
Matsumoto K, Saida T and Ono T: Loss of beta-catenin expression
associated with disease progression in malignant melanoma. Br J
Dermatol. 145:210–216. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Biechele TL, Camp ND, Fass DM, Kulikauskas
RM, Robin NC, White BD, Taraska CM, Moore EC, Muster J, Karmacharya
R, et al: Chemical-genetic screen identifies riluzole as an
enhancer of Wnt/β-catenin signaling in melanoma. Chem Biol.
17:1177–1182. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Duchen MR: Mitochondria and calcium: From
cell signalling to cell death. J Physiol. 529(Pt 1): 57–68. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Beltran-Parrazal L and Charles A: Riluzole
inhibits spontaneous Ca2+ signaling in neuroendocrine
cells by activation of K+ channels and inhibition of Na+
channels. Br J Pharmacol. 140:881–888. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hemendinger RA, Armstrong EJ III, Radio N
and Brooks BR: Neurotoxic injury pathways in differentiated mouse
motor neuron-neuroblastoma hybrid (NSC-34D) cells in vitro-limited
effect of riluzole on thapsigargin, but not staurosporine, hydrogen
peroxide and homocysteine neurotoxicity. Toxicol Appl Pharmacol.
258:208–215. 2012. View Article : Google Scholar
|
|
42
|
Kamal T, Green TN, Morel-Kopp MC, Ward CM,
McGregor AL, McGlashan SR, Bohlander SK, Browett PJ, Teague L,
During MJ, et al: Inhibition of glutamate regulated calcium entry
into leukemic megakaryoblasts reduces cell proliferation and
supports differentiation. Cell Signal. 27:1860–1872. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jan CR, Lu YC, Jiann BP, Chang HT and
Huang JK: Effect of riluzole on cytosolic Ca2+ increase
in human osteosarcoma cells. Pharmacology. 66:120–127. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wadosky KM, Shourideh M, Goodrich DW and
Koochekpour S: Riluzole induces AR degradation via endoplasmic
reticulum stress pathway in androgen-dependent and
castration-resistant prostate cancer cells. Prostate. 79:140–150.
2019. View Article : Google Scholar
|
|
45
|
Liou GY and Storz P: Reactive oxygen
species in cancer. Free Radic Res. 44:479–496. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pelicano H, Carney D and Huang P: ROS
stress in cancer cells and therapeutic implications. Drug Resist
Updat. 7:97–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chakravarthi S, Jessop CE and Bulleid NJ:
The role of glutathione in disulphide bond formation and
endoplasmic-reticulum-generated oxidative stress. EMBO Rep.
7:271–275. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Janáky R, Ogita K, Pasqualotto BA, Bains
JS, Oja SS, Yoneda Y and Shaw CA: Glutathione and signal
transduction in the mammalian CNS. J Neurochem. 73:889–902. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao SS and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress in cell fate decision and
human disease. Antioxid Redox Signal. 21:396–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hayes JD, Dinkova-Kostova AT and Tew KD:
Oxidative stress in cancer. Cancer Cell. 38:167–197. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kennedy L, Sandhu JK, Harper ME and
Cuperlovic-Culf M: Role of glutathione in cancer: From mechanisms
to therapies. Biomolecules. 10:14292020. View Article : Google Scholar :
|
|
52
|
Seol HS, Lee SE, Song JS, Lee HY, Park S,
Kim I, Singh SR, Chang S and Jang SJ: Glutamate release inhibitor,
Riluzole, inhibited proliferation of human hepatocellular carcinoma
cells by elevated ROS production. Cancer Lett. 382:157–165. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wall BA, Wangari-Talbot J, Shin SS, Schiff
D, Sierra J, Yu LJ, Khan A, Haffty B, Goydos JS and Chen S:
Disruption of GRM1-mediated signalling using riluzole results in
DNA damage in melanoma cells. Pigment Cell Melanoma Res.
27:263–274. 2014. View Article : Google Scholar :
|
|
54
|
Cerchio R Jr, Marinaro C, Foo TK, Xia B
and Chen S: Nonhomologous end-joining repair is likely involved in
the repair of double-stranded DNA breaks induced by riluzole in
melanoma cells. Melanoma Res. 30:303–308. 2020. View Article : Google Scholar
|
|
55
|
Raghubir M, Azeem SM, Hasnat R, Rahman CN,
Wong L, Yan S, Huang YQ, Zhagui R, Blyufer A, Tariq I, et al:
Riluzole-induced apoptosis in osteosarcoma is mediated through
Yes-associated protein upon phosphorylation by c-Abl Kinase. Sci
Rep. 11:209742021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jo OD, Martin J, Bernath A, Masri J,
Lichtenstein A and Gera J: Heterogeneous nuclear ribonucleoprotein
A1 regulates cyclin D1 and c-myc internal ribosome entry site
function through Akt signaling. J Biol Chem. 283:23274–23287. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Benavides-Serrato A, Saunders JT, Holmes
B, Nishimura RN, Lichtenstein A and Gera J: Repurposing potential
of Riluzole as an ITAF Inhibitor in mTOR therapy resistant
glioblastoma. Int J Mol Sci. 21:3442020. View Article : Google Scholar :
|
|
58
|
Basu AK: DNA Damage, mutagenesis and
cancer. Int J Mol Sci. 19:9702018. View Article : Google Scholar :
|
|
59
|
O'Connor MJ: Targeting the DNA damage
response in cancer. Mol Cell. 60:547–560. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Srinivas US, Tan BWQ, Vellayappan BA and
Jeyasekharan AD: ROS and the DNA damage response in cancer. Redox
Biol. 25:1010842019. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mehnert JM, Silk AW, Lee JH, Dudek L,
Jeong BS, Li J, Schenkel JM, Sadimin E, Kane M, Lin H, et al: A
phase II trial of riluzole, an antagonist of metabotropic glutamate
receptor 1 (GRM1) signaling, in patients with advanced melanoma.
Pigment Cell Melanoma Res. 31:534–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wall BA, Yu LJ, Khan A, Haffty B, Goydos
JS and Chen S: Riluzole is a radio-sensitizing agent in an in vivo
model of brain metastasis derived from GRM1 expressing human
melanoma cells. Pigment Cell Melanoma Res. 28:105–109. 2015.
View Article : Google Scholar
|
|
63
|
Lemieszek M, Stepulak A, Sawa-Wejksza K,
Czerwonka A, Ikonomidou C and Rzeski W: Riluzole inhibits
proliferation, migration and cell cycle progression and induces
apoptosis in tumor cells of various origins. Anticancer Agents Med
Chem. 18:565–572. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun L, Wu C, Ming J, Nie X, Guo E, Zhang W
and Hu G: Riluzole enhances the response of human nasopharyngeal
carcinoma cells to ionizing radiation via ATM/P53 signalling
pathway. J Cancer. 11:3089–3098. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yun CW and Lee SH: The roles of autophagy
in cancer. Int J Mol Sci. 19:34662018. View Article : Google Scholar :
|
|
66
|
Linder B and Kögel D: Autophagy in cancer
cell death. Biology (Basel). 8:822019.
|
|
67
|
Sun R, He X, Jiang X and Tao H: The new
role of riluzole in the treatment of pancreatic cancer through the
apoptosis and autophagy pathways. J Cell Biochem. Nov 11–2019.Epub
ahead of print.
|
|
68
|
Carneiro BA and El-Deiry WS: Targeting
apoptosis in cancer therapy. Nat Rev Clin Oncol. 17:395–417. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Le MN, Chan JL, Rosenberg SA, Nabatian AS,
Merrigan KT, Cohen-Solal KA and Goydos JS: The glutamate release
inhibitor Riluzole decreases migration, invasion, and proliferation
of melanoma cells. J Invest Dermatol. 130:2240–2249. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Khan AJ, Wall B, Ahlawat S, Green C,
Schiff D, Mehnert JM, Goydos JS, Chen S and Haffty BG: Riluzole
enhances ionizing radiation-induced cytotoxicity in human melanoma
cells that ectopically express metabotropic glutamate receptor 1 in
vitro and in vivo. Clin Cancer Res. 17:1807–1814. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Akamatsu K, Shibata MA, Ito Y, Sohma Y,
Azuma H and Otsuki Y: Riluzole induces apoptotic cell death in
human prostate cancer cells via endoplasmic reticulum stress.
Anticancer Res. 29:2195–2204. 2009.PubMed/NCBI
|
|
72
|
Raghubir M, Rahman CN, Fang J, Matsui H
and Mahajan SS: Osteosarcoma growth suppression by riluzole
delivery via iron oxide nanocage in nude mice. Oncol Rep.
43:169–176. 2020.
|
|
73
|
Rampersaud S, Fang J, Wei Z, Fabijanic K,
Silver S, Jaikaran T, Ruiz Y, Houssou M, Yin Z, Zheng S, et al: The
effect of cage shape on nanoparticle-based drug carriers:
Anticancer drug release and efficacy via receptor blockade using
dextran-coated iron oxide nanocages. Nano Lett. 16:7357–7363. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bayat Mokhtari R, Homayouni TS, Baluch N,
Morgatskaya E, Kumar S, Das B and Yeger H: Combination therapy in
combating cancer. Oncotarget. 8:38022–38043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Pucci C, Martinelli C and Ciofani G:
Innovative approaches for cancer treatment: Current perspectives
and new challenges. Ecancermedicalscience. 13:9612019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Falzone L, Salomone S and Libra M:
Evolution of cancer pharmacological treatments at the turn of the
Third Millennium. Front Pharmacol. 9:13002018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Fortunato A: The role of hERG1 ion
channels in epithelial-mesenchymal transition and the capacity of
riluzole to reduce cisplatin resistance in colorectal cancer cells.
Cell Oncol (Dordr). 40:367–378. 2017. View Article : Google Scholar
|
|
78
|
Lee HJ, Wall BA, Wangari-Talbot J, Shin
SS, Rosenberg S, Chan JL, Namkoong J, Goydos JS and Chen S:
Glutamatergic pathway targeting in melanoma: single-agent and
combinatorial therapies. Clin Cancer Res. 17:7080–7092. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Speyer CL, Bukhsh MA, Jafry WS, Sexton RE,
Bandyopadhyay S and Gorski DH: Riluzole synergizes with paclitaxel
to inhibit cell growth and induce apoptosis in triple-negative
breast cancer. Breast Cancer Res Treat. 166:407–419. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lacomblez L, Bensimon G, Leigh PN, Guillet
P and Meininger V: Dose-ranging study of riluzole in amyotrophic
lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study
Group II. Lancet. 347:1425–1431. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Groeneveld GJ, Van Kan HJ, Kalmijn S,
Veldink JH, Guchelaar HJ, Wokke JH and Van den Berg LH: Riluzole
serum concentrations in patients with ALS: Associations with side
effects and symptoms. Neurology. 61:1141–1143. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Groeneveld GJ, van Kan HJ, Lie-A-Huen L,
Guchelaar HJ and van den Berg LH: An association study of riluzole
serum concentration and survival and disease progression in
patients with ALS. Clin Pharmacol Ther. 83:718–722. 2008.
View Article : Google Scholar
|
|
83
|
Le Liboux A, Cachia JP, Kirkesseli S,
Gautier JY, Guimart C, Montay G, Peeters PA, Groen E, Jonkman JH
and Wemer J: A comparison of the pharmacokinetics and tolerability
of riluzole after repeat dose administration in healthy elderly and
young volunteers. J Clin Pharmacol. 39:480–486. 1999.PubMed/NCBI
|
|
84
|
Bellingham MC: A review of the neural
mechanisms of action and clinical efficiency of riluzole in
treating amyotrophic lateral sclerosis: What have we learned in the
last decade? CNS Neurosci Ther. 17:4–31. 2011. View Article : Google Scholar
|
|
85
|
Wokke J: Riluzole. Lancet. 348:795–799.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Miller RG, Mitchell JD, Lyon M and Moore
DH: Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron
disease (MND). Amyotroph Lateral Scler Other Motor Neuron Disord.
4:191–206. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Grant P, Song JY and Swedo SE: Review of
the use of the glutamate antagonist riluzole in psychiatric
disorders and a description of recent use in childhood
obsessive-compulsive disorder. J Child Adolesc Psychopharmacol.
20:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Sorenson EJ: An acute, life-threatening,
hypersensitivity reaction to riluzole. Neurology. 67:2260–2261.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Inoue-Shibui A, Kato M, Suzuki N,
Kobayashi J, Takai Y, Izumi R, Kawauchi Y, Kuroda H, Warita H and
Aoki M: Interstitial pneumonia and other adverse events in
riluzole-administered amyotrophic lateral sclerosis patients: A
retrospective observational study. BMC Neurol. 19:722019.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lacomblez L, Bensimon G, Leigh PN, Debove
C, Bejuit R and Truffinet P: Long-term safety of riluzole in
amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor
Neuron Disord. 3:23–29. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Speyer CL, Nassar MA, Hachem AH, Bukhsh
MA, Jafry WS, Khansa RM and Gorski DH: Riluzole mediates anti-tumor
properties in breast cancer cells independent of metabotropic
glutamate receptor-1. Breast Cancer Res Treat. 157:217–228. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Poupon L, Lamoine S, Pereira V, Barriere
DA, Lolignier S, Giraudet F, Aissouni Y, Meleine M, Prival L,
Richard D, et al: Targeting the TREK-1 potassium channel via
riluzole to eliminate the neuropathic and depressive-like effects
of oxaliplatin. Neuropharmacology. 140:43–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Yamada T, Tsuji S, Nakamura S, Egashira Y,
Shimazawa M, Nakayama N, Yano H, Iwama T and Hara H: Riluzole
enhances the antitumor effects of temozolomide via suppression of
MGMT expression in glioblastoma. J Neurosurg. 134:701–710. 2020.
View Article : Google Scholar : PubMed/NCBI
|














