Introduction
While breast cancer is a preeminent threat to the
healthcare of women worldwide, triple-negative breast cancer (TNBC)
is the most notable subtype due to its incomparable aggressiveness.
Currently, treatment for TNBC primarily relies only on systemic
cytotoxic chemotherapy, with a rather unsatisfactory prognostic
outcome (1). Therefore, effective
therapy for TNBC is urgently required.
Apoptosis, programmed cell death, is an intrinsic
phenomenon for normal embryonic development and tissue homeostasis.
Dysregulation of apoptosis is a critical step in cancer
pathogenesis and is one of the major barriers to effective cancer
treatment (2,3). The B-cell lymphoma 2 (BCL-2) protein
family regulates the apoptosis pathway through mitochondrial outer
membrane permeabilization, the cytosolic release of mitochondrial
protein, cytochrome c, and caspase activation (4,5).
Cancer cells resist apoptosis through various strategies, including
increased expression of anti-apoptotic BCL-2 family proteins. The
BCL-2 family includes anti-apoptotic members such as BCL-2, BCL-XL,
BCL-W, as well as myeloid cell leukemia sequence 1 (MCL-1), and
pro-apoptotic members such as BAX, BAK, and BH3-only (6).
MCL-1 has unique features among the BCL-2 family,
including structure, function, and regulation (4). MCL-1 has a high affinity for
pro-apoptotic BH3 peptides of BAX, BAK, BID, PUMA and NOXA compared
with other BCL-2 anti-apoptotic family proteins (7). MCL-1 expression is quickly and
readily induced by environmental stimuli including cytokines and
growth factors unlike other proteins in the family such as BCL-2
and BCL-XL (8). MCL-1 is
overexpressed in various human hematologic and solid cancers
(7), and MCL-1 gene amplification
has been observed in diverse cancers (9). MCL-1 is responsible for resistance
to chemotherapeutic agents, and its high expression is associated
with poor prognosis (10,11). The resistance to navitoclax
(ABT-263) and venetoclax (ABT-199) has been demonstrated to be
linked to high expression levels of MCL-1. In numerous instances,
this resistance could be overcome by treatment with agents that
downregulate, destabilize, or inactivate MCL-1 (12-14). Thus, MCL-1 has recently been
recognized as an emerging therapeutic target in cancer.
Acriflavine (ACF), a mixture of
3,6-diamino-10-methylacridinium chloride (trypaflavin) and
3,6-diaminoacridine (proflavine) has been used as an antimicrobial
agent a century ago (15).
Recently, its potential as an anticancer agent has been
highlighted. The antitumor activity of ACF has been reported in
various cancers, including colorectal, pancreas, and prostate
(16-21). Its mechanism is still under
investigation; however, HIF-1 inhibition is considered as the main
anticancer mechanism.
In the present study, the anticancer activity of ACF
on TNBC cells and its cell death mechanism were evaluated.
Furthermore, the interaction of MCL-1 protein in TNBC cells during
ACF-induced cancer apoptosis was revealed. The potential of MCL-1
downregulation by ACF as a breakthrough strategy against BCL-2
inhibitor resistance was evaluated. Additionally, the therapeutic
potential of ACF against non-small cell lung cancer (NSCLC) and
glioblastoma multiforme (GBM) was demonstrated.
Materials and methods
Reagents and antibodies
ACF was purchased from Sigma-Aldrich; Merck KGaA.
APC Annexin V Apoptosis Detection Kit was purchased from BioLegend,
Inc (cat. no. 640930). APC anti-human IgG was purchased from
BioLegend, Inc. (cat. no. 366906). ABT-263 was purchased from Santa
Cruz Biotechnology, Inc. (cat. no. sc-207241). Monoclonal
antibodies to detect MCL-1 (product no. 5453), GSK-3β (product no.
9315), phosphorylated (p)-GSK-3β (product no. 9323), GAPDH (product
no. 2118) and polyclonal antibodies to detect p-MCL1 (product no.
4579), BCL-XL (product no. 2762), BCL-2 (product no. 2872),
β-catenin (product no. 9562), cleaved PARP (product no. 9541),
caspase-8 (product no. 9746), caspase-9 (product no. 9502),
ubiquitin (product no. 3933), XAF1 (product no. 13805S) and XIAP
(product no. 2042) were purchased from Cell Signaling Technology,
Inc. Monoclonal antibody against β-actin (cat. no. A5316) was
obtained from Sigma-Aldrich; Merck KGaA. Monoclonal antibodies to
evaluate caspase-3 activation were purchased from Abcam (product
code ab136812). MG-132 (product no. M7449), and cycloheximide (CHX;
product no. C4859) were purchased from Sigma-Aldrich; Merck KGaA.
Protein A/G PLUS-Agarose (cat. no. sc-2003) was purchased from
Santa Cruz Biotechnology, Inc.
Cell line culture
The human cancer cell lines MDA-MB-231 (HTB-26),
HS578T (HTB-126), HCC-70 (CRL-2315), A549 (CCL-185), and NCI-H69
(HTB-119) cells were purchased from the American Type Culture
Collection (ATCC). U87 (glioblastoma of unknown origin), U343 and
U251 cells were kindly received as a gift from Professor Jongsun
Park (Department of Pharmacology, College of Medicine, Chungnam
National University, South Korea). STR profiling was performed on
all cell lines. All cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) or RPMI-1640 medium with 10% fetal bovine
serum (FBS) (all from Cytiva) and 1% penicillin, and cultured at
37°C with 5% CO2. Cells were routinely controlled to
exclude mycoplasma contamination.
Determination of cell viability
The In Vitro Toxicology Assay Kit (product
no. TOX6; Sigma-Aldrich; Merck KGaA) was used to determine cellular
viability. Briefly, cells (0.5-2×104 cells/well) were
seeded and attached to 96-well culture plates. The indicated doses
(0, 0.5, 1, 2, 10, 50, 100 µM) of ACF for different
experiments were administered for 6-48 h. Cells were fixed in 10%
trichloroacetic acid for 1 h at 4°C and washed five times with
water. Fixed cells were stained with 0.4% sulforhodamine B for 15
min at room temperature and washed five times with 1% acetic acid.
The incorporated dye was solubilized with 10 mM Tris Base, pH 8.8.
The absorbance was spectrophotometrically measured at 565 nm using
an EL800 microplate reader (BioTek Instruments, Inc.).
Cell death assay
Cell death was assayed using the Muse Annexin V
& Dead Cell kit (cat. no. MCH100105; Luminex Corporation)
according to the manufacturer's protocol. Briefly, 5×105
cells were collected, washed using cold phosphatebuffered saline
(PBS), and resuspended in a medium containing 1% FBS. Following
staining with the Muse Annexin V and Dead Cell reagent at room
temperature for 20 min, cells were analyzed using the Muse Cell
Analyzer (Luminex Corporation). Cell death was detected as the
percentage of Annexin V- and/or 7-amino-actinomycin D
(7-AAD)-positive cells.
Caspase-3/7 activity assay
Caspase-3/7 activity was analyzed using the Muse
Caspase-3/7 kit (cat. no. MCH100108; Luminex Corporation). Briefly,
5×105 cells were collected and washed using cold PBS.
Cells were resuspended in 1X assay buffer BA and mixed with Muse
Caspase-3/7 reagent. Following 30 min of incubation at 37°C, Muse
Caspase 7-AAD working solution was added to the cells. The cells
were detected using the Muse Cell Analyzer and results were
analyzed using Muse Cell Analyzer software version 1.6 (both from
Luminex Corporation). The results were reported as the percentages
of live cells (lower left panel, caspase-3/7-negative and
7-AAD-negative), apoptotic cells exhibiting caspase-3/7 activity
(lower right panel, caspase-3/7 positive and 7-AAD-negative), late
apoptotic/dead cells (upper right panel, caspase-3/7-positive and
7-AAD-positive), and necrotic cells (upper left panel,
caspase-3/7-negative and 7-AAD-positive).
Western blot analysis
Cells were lysed using lysis buffer (cat. no. 9803S;
Cell Signaling Technology, Inc.) supplemented with a protease
inhibitor cocktail (cat. no. 11697498001; Roche Diagnostics GmbH)
for 30 min at 4°C. Protein concentration was measured by
bicinchoninic acid (BCA) assay. A total of 10-50 µg of
protein per lane were separated using 8-14% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to nitrocellulose membranes (GE Healthcare Life
Sciences; Cytiva). The membranes were blocked using 2% skim milk
(product no. 70166; Sigma-Aldrich; Merck KGaA) for 1 h at room
temperature followed by immunoblotting with specific primary
antibodies (BCL-XL, BCL-2, GAPDH, β-actin, MCL-1, p-MCL1, XIAP,
XAF1, cleaved PARP, GSK-3β, and p-GSK-3β, ubiquitin, caspase-8 and
caspase-9) at a dilution of 1:2,000 overnight at 4°C. Horseradish
peroxidase-conjugated anti-mouse (product no. 7076P2) and
anti-rabbit (product no. 7074P2; both from Cell Signaling
Technology, Inc.) secondary antibodies were incubated at a dilution
of 1:1,000 at room temperature for 1 h. Following three washes with
Tris-buffered saline 0.1% Tween-20, immunoreactive bands were
visualized using an enhanced chemiluminescence detection system
(Cyanagen Srl).
Annexin V and 7-AAD staining
Cells (4-5×105 cells/well) were seeded
and attached to 6-well culture plates. The indicated doses (0, 1, 2
and 10 µM) of ACF were administered for 6-48 h. Cells were
resuspended in binding buffer or PBS and stained using Annexin
V-APC and 7-AAD for 15 min at room temperature. Flow cytometry was
performed using a FACSCanto II instrument (BD Biosciences) and
FlowJo v10.8.0 software (Tree Star, Inc.).
RNA extraction, cDNA synthesis, and
reverse transcription-quantitative (RT-q) PCR
The mRNA was isolated using TRIzol (Thermo Fisher
Scientific, Inc.). Extracted RNA was subjected to complementary
cDNA synthesis using TOPscript RT DryMIX (Enzynomics, Inc.)
according to the manufacturer's protocol. qPCR was performed using
SYBR Green TOPreal qPCR 2X PreMIX (Thermo Fisher Scientific, Inc.).
Samples were normalized according to GAPDH (forward, 5′-GGA GCG AGA
TCC CTC CAA AAT-3′ and reverse, 5′-GGC TGT TGT CAT ACT TCT CAT
GG-3′) and compared with assigned expression of untreated cells
using the MCL-1 primers (forward, 5′-CGA CGG CGT AAC AAA CT-3′ and
reverse, 5′-GGA AGA ACT CCA CAA ACC C-3′). Amplification was
conducted with the CFX96 Touch Real-Time PCR detection system, a
PCR machine (Bio-Rad Laboratories, Inc.). The thermocycling
conditions consisted of initial denaturation at 95°C for 30 sec,
followed by 40 cycles at 95°C for 5 sec, annealing at 60°C for 30
sec and extension at 72°C for 30 sec. All reactions were examined
in technical triplicate. The 2−ΔΔCq method (22) was used to calculate the relative
gene expression.
Mitochondrial membrane potential and
reactive oxygen species (ROS) measurement
Mitochondrial membrane potential and ROS generation
were evaluated using specific fluorescence probe staining including
tetramethylrhodamine-ethyl ester-perchlorate (TMRE; Thermo Fisher
Scientific, Inc.), and
6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate acetyl
ester (CM-H2DCFDA; Thermo Fisher Scientific, Inc.), respectively,
as previously reported (23).
Briefly, cells exposed to ACF (2 µM) and ABT-263 (1
µM) for 2, 6 or 24 h were incubated in 200 nM TMRE and 10
µM CM-H2DCFDA probe for 30 min at 37°C. Following washing
twice using PBS, the fluorescence signals of TMRE and DCF-DA were
detected and analyzed using a confocal microscope (LSM 700; Carl
Zeiss AG).
Post-translational regulation and
immunoprecipitation
For post-translational regulation of MCL-1,
MDA-MB-231 and HS578T, cells were treated with cycloheximide (CHX;
10 µg/ml) and/or ACF (10 µM) and after 0, 1 and 2 h
the expression of MCL-1 was determined using western blot analysis.
For the proteasomal regulation study, MDA-MB-231 and HS578T cells
were treated with MG-132 (10 µM) and/or ACF (10 µM)
and after 6 h the expression of MCL-1 was determined using western
blot analysis. For ubiquitination study, HS578T cells were treated
with MG-132 (10 µM) and/or ACF (10 µM) and after 6 h
the cells were harvested and washed twice with PBS. Cells were then
resuspended in Pierce® IP Lysis Buffer (25 mM Tris-HCl
pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol) mixed with
protease inhibitor and EDTA. A total of 100 µg of protein
was incubated with anti-MCL-1 antibody for 2 h at 4°C.
Subsequently, 20 µl of resuspended Protein A/G PLUS-Agarose
(cat. no. sc-2003; Santa Cruz Biotechnology, Inc.) was added and
incubated overnight at 4°C. Immunoprecipitates were collected and
washed 4 times with PBS by centrifugation at 1,000 × g for 5 min at
4°C. The supernatant was discarded and the beads were resuspended
with 40 µl of electrophoresis buffer and analyzed by
SDS-PAGE and western blotting.
Combination index (CI) analyses
Using the CompuSyn software (CompuSyn v1.0; CompuSyn
Inc.), the CI values were calculated according to the cell
viability at corresponding concentrations. The CI value, CI<1
indicates a synergistic effect, CI=1 indicates an additive effect
and CI>1 indicates and antagonistic effect (24,25).
Statistical analyses
Differences between groups were analyzed using
one-way ANOVA and subsequent Tukey's multiple comparison post hoc
test. Error bars represent ± standard deviation from three
independent experiments. All statistical analyses were conducted
using Graph-Pad Prism Version 4.0 (GraphPad Software, Inc.).
P<0.05 was considered to indicate a statistically significant
difference.
Results
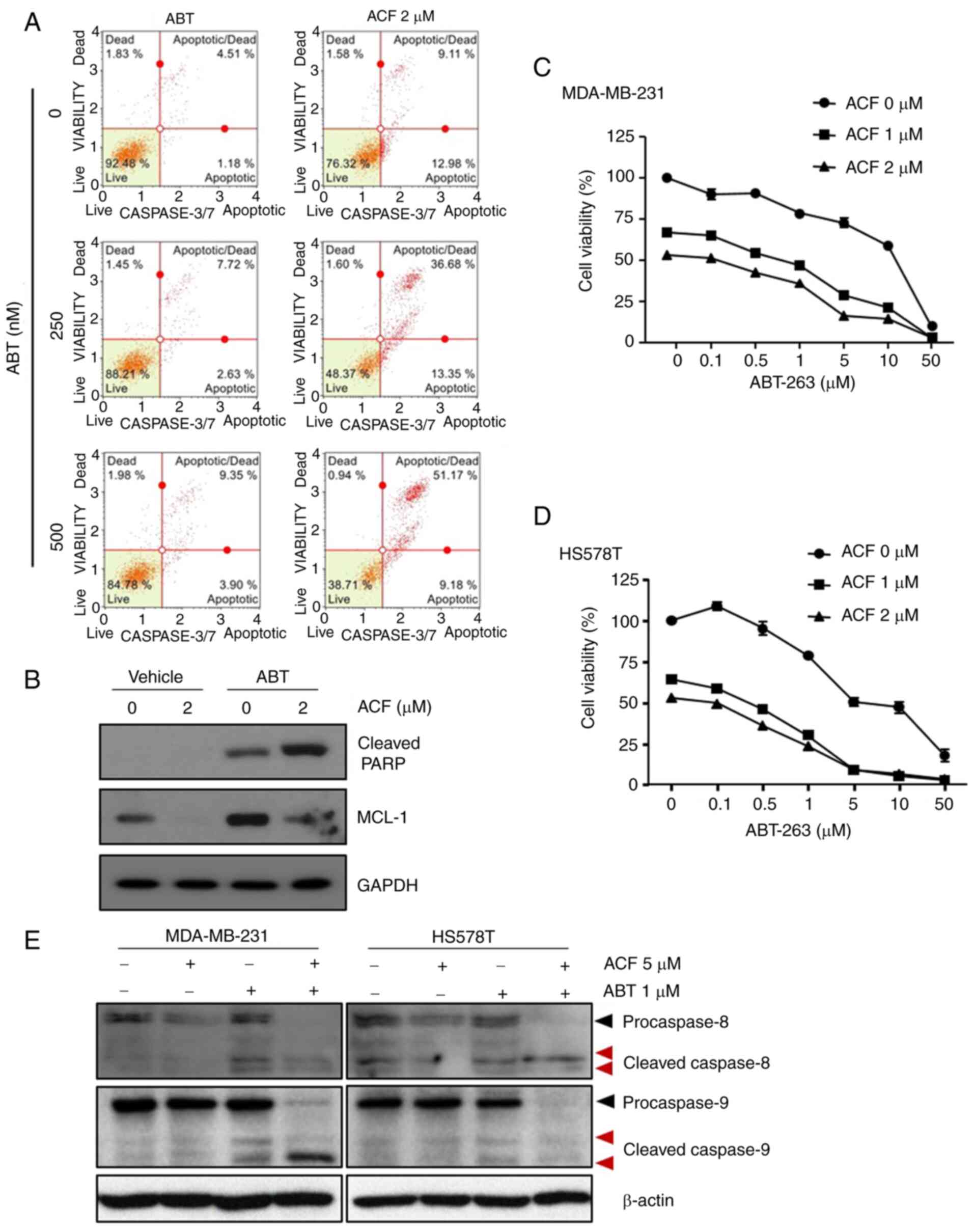
ACF induces apoptosis in TNBC cells in
time and dose-dependent manners
To assess whether ACF has anticancer activity on
TNBC cells under normoxic conditions, the effect of ACF on survival
and apoptosis in MDA-MB-231 and HS578T cells was examined. ACF (5
µM for 72 h) induced typical apoptosis morphologies in TNBCs
characterized by shrinkage of cells and fragmentation into
ultra-structured apoptotic bodies (Fig. 1A). The cell viability assay
revealed that the anticancer activity of ACF was dose- and
time-dependent (Fig. 1B). Flow
cytometric analysis revealed that ACF-induced cell death was
apoptosis via activation of caspase-3/7 (Fig. 1C and D). In addition, ACF-treated
cells exhibited increased levels of cleaved PARP, which is a known
cellular substrate of caspases (Fig.
1E). Then, the anticancer effect of ACF on different GBM (U87,
U251, and U343) and lung cancer (A549, NCI-H69) cell lines was
explored. Cytotoxicity and FACS data (Fig. S1) also revealed the anticancer
activity of ACF on GBM and lung cancer cell lines.
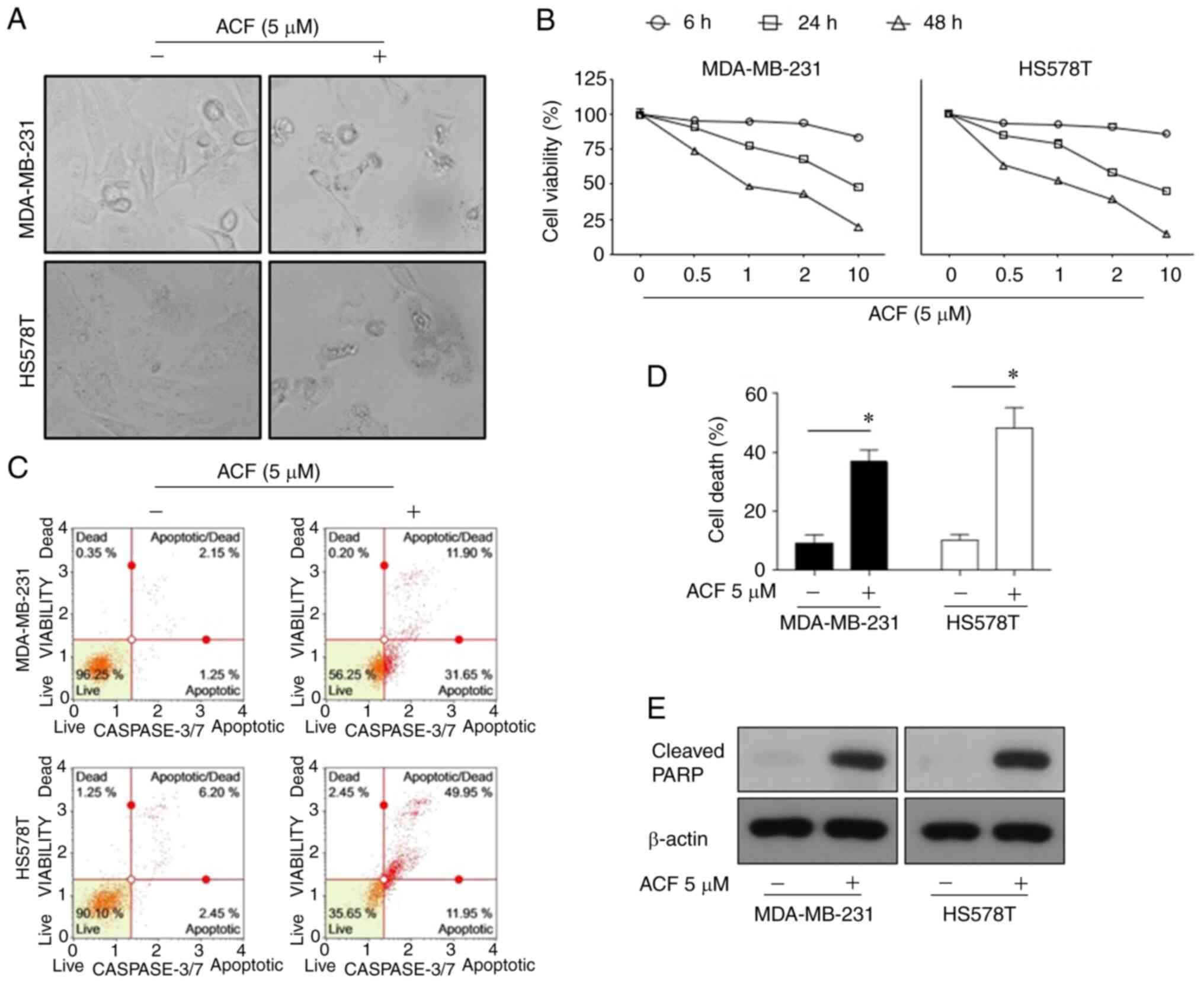 | Figure 1Apoptosis is promoted by (ACF) in
triple-negative breast cancer cell lines in a time and
dose-dependent manner. (A) Microscopic images (magnification, ×100)
of MDA-MB-231 and HS578T cells. (B) Effects of ACF on the viability
of MDA-MB-231 and HS578T cells. The cells were treated with various
concentrations (0, 0.5, 1, 2 and 10 µM) of ACF for 6, 24 and
48 h, and cell viability was determined using the sulforhodamine B
assay. (C) MDA-MB-231 and HS578T cells were treated with 0 or 5
µM ACF for 24 h. Caspase-3/7 activity was analyzed using
Muse Caspase-3/7 kit, as described in the Materials and methods. A
total of 4 populations of cells were distinguished: Live
[caspase-3/7(-)/7-AAD(-)], apoptotic [caspase-3/7(+)/7-AAD(-)],
apoptotic/dead cells [caspase-3/7(+)/7-AAD(+)], and necrotic
[caspase-3/7(-)/7-AAD(+)]. (D) MDA-MB-231 and HS578T cells were
treated with 0 or 5 µM ACF for 24 h. Cell death was detected
as the percentage of Annexin V and/or 7-AAD-positive cells. The
results are expressed as the percentage of surviving cells over
control cells. Each value is reported as the mean ± standard
deviation and is representative of results obtained from three
independent experiments. *P<0.05 compared with
non-treated cells. (E) MDA-MB-231 and HS578T cells were treated
with 0 or 5 µM ACF for 24 h and then western blot analysis
for cleaved PARP expression was performed. β-actin served as the
loading control. ACF, acriflavine; 7-AAD, 7-amino-actinomycin
D. |
ACF causes apoptosis via the intrinsic
pathway
To determine whether apoptosis signaling occurs via
the mitochondria-mediated intrinsic or death receptor-mediated
extrinsic apoptotic signaling pathway, the accumulation of ROS and
the level of mitochondrial membrane potential was assessed using
TMRE. ROS levels and mitochondrial membrane potential were not
altered following treatment with ABT-263, a BCL-2 protein
inhibitor. However, ACF markedly increased the ROS levels within 2
h and reduced the mitochondrial membrane potentials at 6 h
following treatment (Fig. 2).
From a molecular point of view, the expression of caspase-8 and
caspase-9 in ACF-treated (5 µM) cells was analyzed via
western blot analysis. A total of 24 h following ACF treatment, the
expression of caspase-9 was decreased while the expression of
caspase-8 remained unchanged and the cleaved form of caspase-9 was
not detected (Fig. S2).
Collectively, these data suggested that ACF induces apoptosis via
the intrinsic pathway.
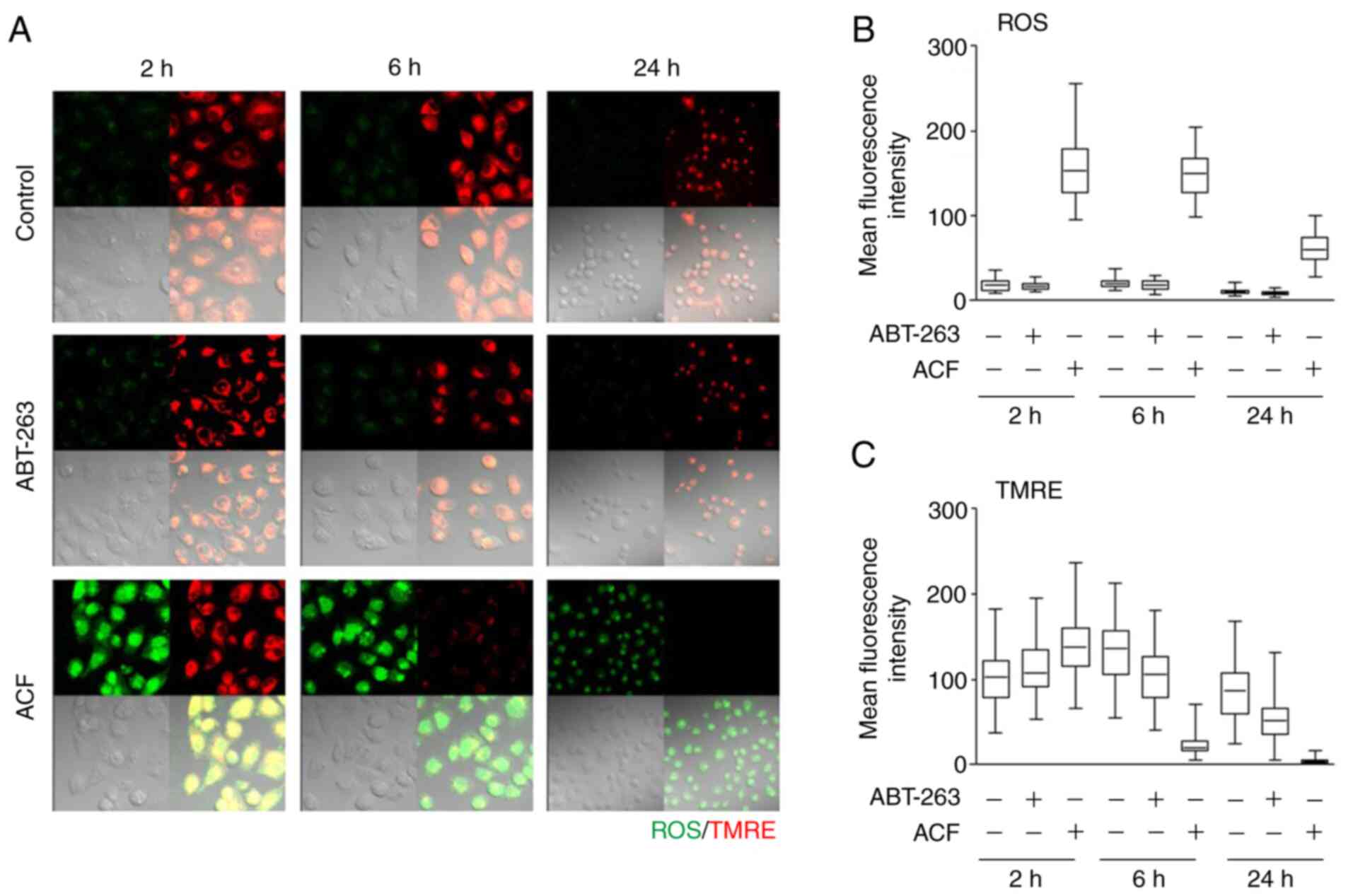 | Figure 2ACF induces mitochondrial damage and
ROS. (A) HS578T cells were treated with ABT-263 (1 µM) and
ACF (2 µM), and mitochondria membrane potential and ROS
generation were evaluated using specific fluorescence probe
staining including tetramethylrhodamine-ethyl ester-perchlorate,
and 6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate,
acetyl ester, respectively. Fluorescence images (magnification,
×200) were captured after treating cells with each drug for 2, 6
and 24 h. (B and C) Mean fluorescence intensity of control cells
and cells treated with ABT-263 and ACF was evaluated. ACF,
acriflavine; ROS, reactive oxygen species; TMRE,
tetramethylrhodamine-ethyl ester-perchlorate. |
Antitumor effect of ACF is mediated by
the downregulation of MCL-1 protein
To evaluate the mechanism of ACF-induced intrinsic
apoptosis, the protein levels of anti-apoptotic BCL-2 family
proteins were determined using western blotting. ACF downregulated
the protein levels of MCL-1, but not of other BCL-2 family proteins
(Fig. 3A). The time kinetic study
revealed that the MCL-1 protein level rapidly decreased and
completely disappeared at 3-6 h following ACF treatment, while the
majority of the cleaved PARP was formed 24 h following treatment
(Fig. 3B). These results
suggested the presence of other molecules between MCL-1 and cleaved
PARP during ACF-mediated apoptosis. To determine which molecules
linked MCL-1 downregulation and PARP cleavage, the XAF1 and XIAP
protein levels were evaluated. Expression of XAF1 protein was
increased and expression of XIAP was decreased at 24 h following
the ACF treatment (Fig. 3C).
Then, XAF1 and XIAP expression was evaluated at serial time-points.
The results revealed the increased expression of XAF1 at 1, 3, 10
and 24 h following ACF treatment and decreased expression at 6 h.
Expression of XIAP was decreased at 1, 3, 10 and 24 h but increased
at 6 h following ACF treatment in the MDA-MB-231 cell line. In the
HS578T cells, XAF1 increased at 3, 6, 10 and 24 h but decreased at
1 h and XIAP decreased at 1, 10 and 24 h and increased at 3 and 6 h
following ACF treatment (Fig.
3D). In addition, MCL-1 downregulation was also observed both
in hypoxic and normal conditions (Fig. S3). The aforementioned experiments
entailed that ACF treatment induces an antitumor effect by
downregulating MCL-1 not HIF-1 and it may also be associated with
the expression of XAF1 and XIAP in TNBC cells.
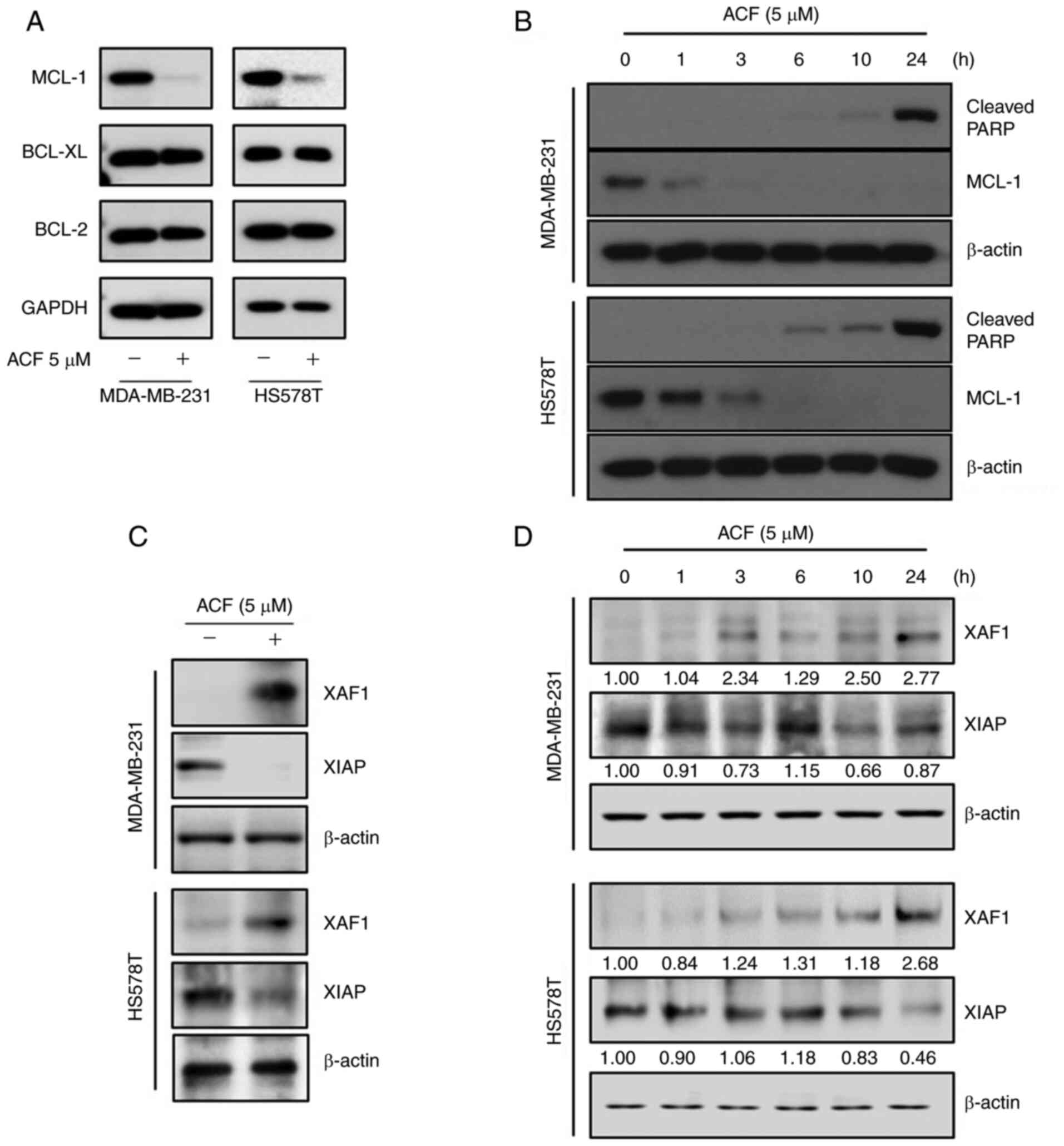 | Figure 3ACF suppresses anti-apoptotic
proteins MCL-1 and XIAP. (A) MDA-MB-231 and HS578T cells were
treated with 0 or 5 µM ACF for 6 h and then western blot
analysis for MCL-1, XIAP, BCL-2 and BCL-XL expression was
performed. (B) MDA-MB-231 and HS578T cells were treated with 5
µM ACF and expression of cleaved PARP was assessed after 0,
1, 3, 6, 10 and 24 h using western blot analysis. (C) MDA-MB-231
and HS578T cells were treated using 0 or 5 µM ACF for 24 h
and expression of XAF1 and XIAP was assessed using western blot
analysis. (D) MDA-MB-231 and HS578T cells were treated using 5
µM ACF for 0, 1, 3, 6, 10 and 24 h and the expression of
XAF1 and XIAP was assessed using western blot analysis. In all
experiments, GAPDH or β-actin served as the loading control. ACF,
acriflavine; MCL-1, myeloid cell leukemia sequence 1; BCL-2, B-cell
lymphoma 2. |
ACF induces downregulation of MCL-1 at a
transcriptional- and post-translational-dependent manner
MCL-1 protein expression is regulated in multiple
steps: transcription and post-translation. First, the mRNA levels
of MCL-1 were evaluated using RT-qPCR. ACF decreased the levels of
MCL-1 mRNA in MDA-MB-231 and HS578T cells (Fig. 4A). Secondly, it was evaluated if
MCL-1 degradation occurs under CHX (10 µg/ml), an inhibitor
of protein biosynthesis. ACF (10 µM) decreased MCL-1
expression even following inhibition of translation (Fig. 4B). This indicated that MCL-1 is
also downregulated following translation. Next, it was evaluated
whether ACF increases proteasomal degradation of MCL-1 using a
proteasome inhibitor, MG-132 (10 µM). The decrease in MCL-1
by ACF was blocked using MG-132 (Fig.
4C). Interestingly, cells treated with MG-132 alone
demonstrated markedly higher MCL-1 protein levels than those
treated with both MG-132 and ACF. Lastly, the ubiquitination of
MCL-1 protein was evaluated. Western blot data followed by
immunoprecipitation also revealed increased ubiquitination of MCL-1
protein by ACF treatment (Fig.
4D). A higher ubiquitination level in cells treated with MG-132
was also consistent with the previous experiment (Fig. 4C). However, the level of GSK3β
remained unchanged when treated with ACF (Fig. S4). Collectively, these data
indicated that MCL-1 downregulation by ACF occurs at
transcriptional and post-translational levels.
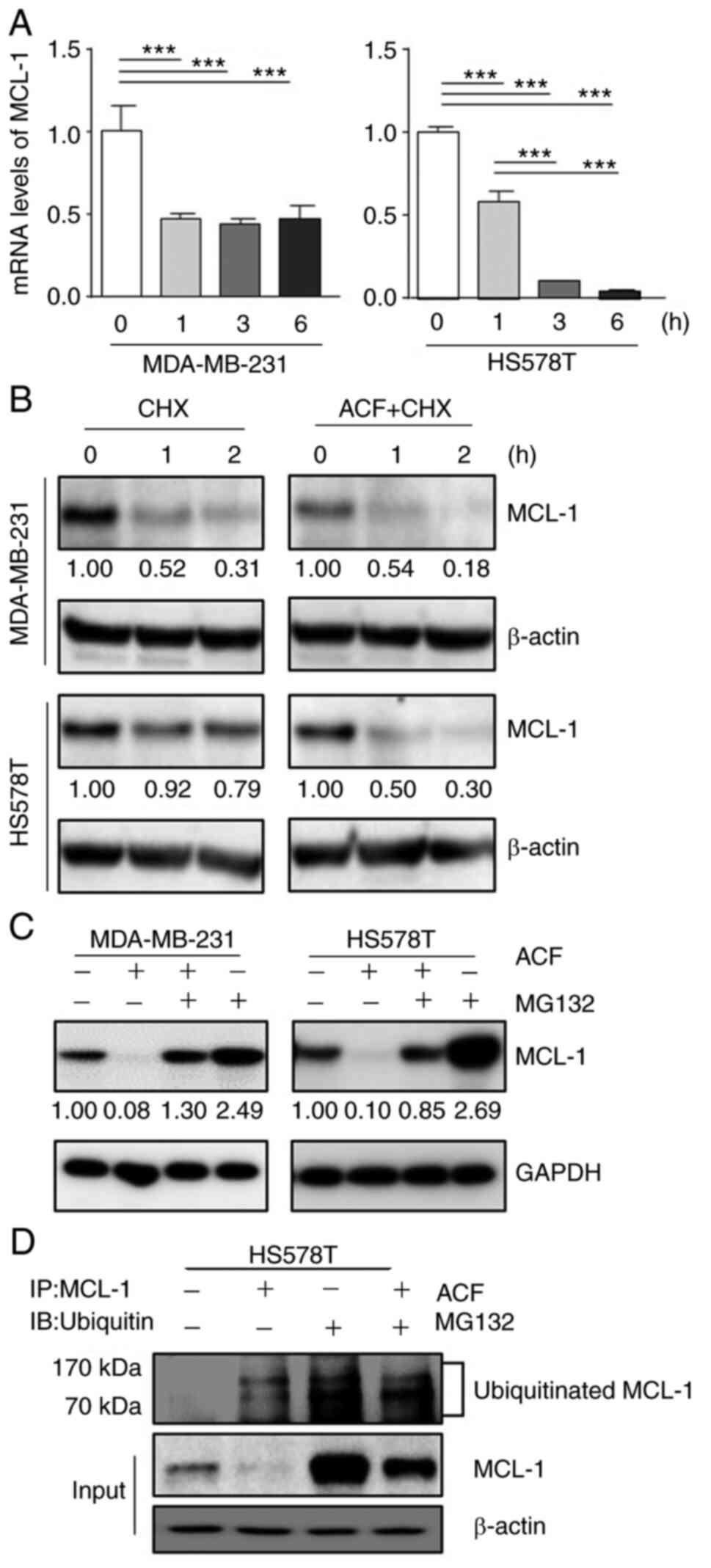 | Figure 4MCL-1 downregulation occurs at
transcriptional and post-translational levels. (A) MDA-MB-231 and
HS578T cells were treated using 5 µM ACF and the expression
of MCL-1 mRNA was evaluated after 0, 1, 3, and 6 h using reverse
transcription-quantitative PCR. Each value is reported as the mean
± standard deviation and is representative of results obtained from
three independent experiments. (B) MDA-MB-231 and HS578T cells were
incubated with cycloheximide (10 µg/ml), and/or ACF (10
µM) and after 0, 1 and 2 h the expression of MCL-1 was
determined using western blot analysis. (C) MDA-MB-231 and HS578T
cells were incubated with ACF and proteasome inhibitor, MG-132, and
expression of MCL-1 was determined using western blot analysis. (D)
HS578T cells were treated with 10 µM ACF and and/or MG-132,
and after 6 h MCL-1 was pulled down and the ubiquitination level
was analyzed using western blotting. ***P<0.001
compared with non-treated cells. ACF, acriflavine; MCL-1, myeloid
cell leukemia sequence 1. |
ACF and ABT-263 work synergistically by
overcoming the resistance to ABT-263
To determine whether ACF may be used as a treatment
strategy, HS578T cells were treated with ABT-263, a BCL-2 protein
inhibitor, with or without ACF. While ABT-263 or ACF alone was not
so effective in inducing apoptosis, when combined with 2 µM
ACF, apoptosis was induced in more than 30% of cells. Importantly,
apoptosis induced by the combination of ABT-263 and ACF was five
times greater than that by ABT-263 or ACF alone (Fig. 5A). As previously reported
(26), ABT-263 induced MCL-1
expression in TNBC cells, and ACF treatment prevented MCL-1
induction (Fig. 5B). The
combination treatment of ACF and ABT-263 revealed synergy in both
MDA-MB-231 and HS578T cells (Figs. 5C
and D and S3). To determine
the molecular pathways involved, the expression of caspase-8 and
caspase-9 was analyzed. Although the expression of active caspase-9
was detected after 24 h, caspase-8 remained unaltered when treated
with ACF (Fig. S2). However, the
expression of both caspases was not detected 6 h following ACF
treatment. Interestingly, combination of ACF and ABT-263 activated
both caspases suggesting the involvement of both an intrinsic and
extrinsic apoptotic pathway in the combination treatment (Fig. 5E). To determine whether the
synergistic effect of ACF and ABT-263 may be observed in other
cancer types, GBM (U87, U343, U251) and NSCLC (A549) cells were
treated with ACF and ABT-263. Immunoblotting data revealed that the
ABT-263 treatment upregulated the expression of MCL-1. However,
when treated with 2 µM ACF, the expression of MCL-1 was
downregulated again (Fig. 6A).
Addition of ACF to ABT-263 also increased apoptosis in GBM (U87,
U251 and U343) and NSCLC (A549) cell lines (Fig. 6B and C) compared with ABT-263
single therapy. Analysis of CI revealed synergistic apoptosis in
U251, U343 and A549 cell lines (Fig.
S5). Cell viability data also suggested that the combination
treatment could eliminate cancer cells in a dose-dependent manner
(Fig. 6D). These data suggested
that ACF could act synergistically with ABT-263 by preventing MCL-1
upregulation.
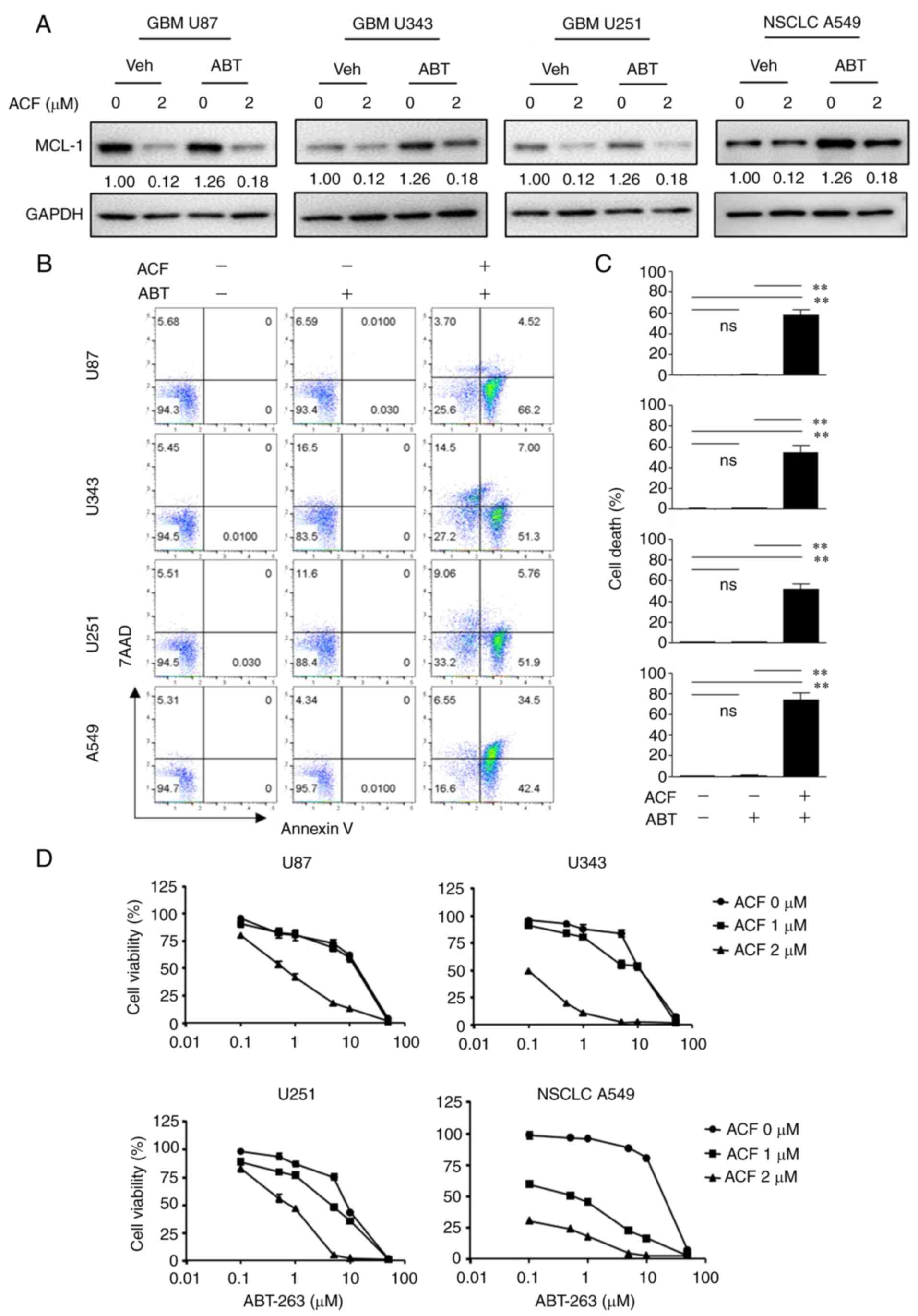 | Figure 6ACF overcomes the therapeutic
resistance of ABT-263. (A) U87, U343, U251 and A549 cells were
treated using ABT-263 and were incubated with 0 or 2 µM ACF
for 24 h, and the expression of MCL-1 was detected using western
blotting. (B) U87, U343, U251 and A549 cells were treated with ACF
(2 µM) and/or ABT-263 (0.5 µM) for 24 h and apoptosis
was evaluated using Annexin V and 7-AAD staining followed by FACS.
(C) Quantitative analysis of the percentage of cell death (Annexin
V+/7-AAD−). Data are presented as the means ±
standard deviation from two independent experiments. (D) U87, U343,
U251 and A549 cells were treated with indicated concentrations of
ACF and ABT-263 and cell viability was determined using the
sulforhodamine B assay. **P<0.01 compared with
non-treated and ABT alone treated cells. ACF, acriflavine; 7-AAD,
7-amino-actinomycin D; MCL-1, myeloid cell leukemia sequence 1. |
Discussion
The BCL-2 family proteins modulate the intrinsic
(mitochondrial) apoptotic pathway via the balance of pro- and
anti-apoptotic proteins. The BCL-2 family consists of three main
subclasses depending on the location of BCL-2 homology (BH) and
function: multidomain anti-apoptotic (BCL-2, MCL-1 and BCL-XL),
multidomain pro-apoptotic (BAX and BAK), and BH3-only pro-apoptotic
(BIM, PUMA and NOXA) (2).
Anti-apoptotic BCL-2 family members regulate apoptosis by isolating
the stimulants from interacting with BAX and BAK (27,28). BH3 proteins (BAD and NOXA) are
considered to cause apoptosis by engaging with anti-apoptotic
proteins, freeing activators to stimulate BAX and BAK (29). Furthermore, anti-apoptotic BCL-2
family proteins seldom directly inhibit BAX and BAK, both inducing
cell survival directly and indirectly, by sequestering BH3-only
proteins (27,28). One of the most well-known and
clinically advanced BCL-2 family target therapies is ABT-737 and
its clinical analog ABT-263 (navitoclax). Their molecular structure
mimics BAD BH3 and binds to the BH3 binding groove of BCL-2,
BCL-XL, and BCL-W (30,31). However, the potency of
BCL-2/BCL-XL inhibitors has been underwhelming (32). Their major limitation is the
unexpected upregulation of MCL-1. In addition, the most frequently
amplified gene in inhibitions of BCL-XL and BCL-2 is MCL-1, gaining
significant interest in anticancer therapeutics.
MCL-1 is overexpressed in a wide range of cancers,
including both solid and hematologic malignancies such as liver,
ovarian, prostate, hematologic, and breast cancers (33-38). Previous studies revealed that
MCL-1 is a crucial anti-apoptotic factor in TNBC (39,40). For instance, MCL-1-knockdown TNBC
cell lines revealed markedly reduced viability, while BCL-XL
silencing had a modest effect (41). Furthermore, Balko et al
revealed an overexpression of the MCL-1 gene in 54% of TNBC
patients who underwent neoadjuvant chemotherapy (42). They suggested that MCL-1 protects
TNBC cells against apoptosis induced by cytotoxic chemotherapy
(37). Moreover, the
overexpression of MCL-1 has been proposed as a marker of poor
prognosis in TNBC (39,43). Overall, MCL-1 may be one of the
prospective therapeutic targets for TNBC. Upregulation of
anti-apoptotic BCL-2 family proteins hallmarks numerous cancers and
occurs via various pathways. A prototype of this mechanism is MCL-1
upregulation, which contributes to protein stabilization by genetic
inactivation of the ubiquitin ligase complex protein F-box and WD
repeat domain-containing 7 (FBW7) (9,44,45). Given the importance of inducing
apoptosis in managing cancer treatment, agents inhibiting
anti-apoptotic proteins have garnered interest as a therapeutic
option against cancer. To overcome this MCL-1 upregulation by BCL-2
family inhibitors, trials were conducted to identify MCL-1
inhibitors in various cancers, including breast, hematologic, and
lung cancers. They combined the pre-existing BCL-2 family
inhibitors with novel MCL-1 inhibitors to overcome the potential
resistance to BCL-2 family inhibitors and improve therapeutic
outcomes (46,47).
In the present study, it was revealed that ACF could
regulate MCL-1 at the transcriptional and post-translational levels
in breast cancer cells. The anticancer mechanism of ACF with MCL-1
downregulation was also revealed to be effective in lung and GBM
cells. The limitation of the present study on the exact mechanism
of MCL-1 decrease in lung cancer and GBM cells should be explored
in further studies. Our study revealed that ACF promoted cell death
of TNBC cells (MDA-MB-231 and HS578T) through the intrinsic
apoptotic pathway in normoxia regardless of the HIF-1 pathway. Both
ubiquitin-dependent and independent degradation of MCL-1 was
reported. E3 ubiquitin-ligases confer a high degree of specificity
to ubiquitination by recognizing target proteins. Ubiquitination of
MCL-1 is mediated by five E3 ubiquitin ligases, including MCL-1
ubiquitin ligase E3 (MULE), glycogen synthase kinase 3β (GSK3β),
β-transducin repeats-containing protein (β-TrCP), FBW7, and
tripartite motif-containing 17 (TRIM17) (48). However, different E3
ubiquitin-ligases appear to regulate MCL-1 protein expression in
different cell types exposed to different stimuli. In the present
study, the exact mechanism in the post-translational regulation of
MCL-1 by ACF was not defined; however, ubiquitination was detected
and GSK3β was not involved. Furthermore, the combination of ACF and
ABT-263 generated apoptosis synergistically. This synergistic
effect may be due to their independent inhibitory mechanism.
ABT-263 stimulates TNBC apoptosis by inhibiting BCL-2, BCL-XL, and
BCL-W. By contrast, our study revealed that ACF hinders MCL-1 and
promotes TNBC apoptosis. It was assumed that as ABT and ACF work by
regulating different BCL-2 family molecules, they compensated each
other to enhance the antitumor effect. ACF is a mixture of two
acridine analogs that are mostly utilized as antimicrobial agents
(15). Goldie et al first
reported the anticancer effect of ACF (49). Its potential as an anticancer
agent has also been identified with the activity inhibiting the
topo-isomerase and hypoxia-inducing factor (HIF) pathway by
preventing the dimerization of HIF-1α and HIF-1β (16,50). Previously, it has also been
reported that ACF inhibited the epithelial-mesenchymal transition
in pancreatic cancer cells induced by TGF-β or cobalt
chloride-induced hypoxia via reduction of the activating
transcription factor 4 (20). ACF
has also been revealed to be active in the prostate, pancreas, and
colorectal tumor xenograft models in rodents when administered
locally or systemically (16,49,51). ACF revealed anticancer effects in
hepatocellular carcinoma and lung adenocarcinoma under normoxic
conditions (18,19). The present study demonstrated
cancer cell death by apoptosis caused by ACF in the normoxic state.
Changes were primarily observed in mitochondrial membrane
potentiality, which indicates an alteration in the function of the
intrinsic apoptotic pathway. This indicates the importance of
membranous stability, and only MCL-1 downregulation was observed
without changes in various factors controlling membrane stability,
including BCL-2. Considering these results, it is theorized that
cancer cell death by ACF is due to MCL-1 inhibition, and not HIF-1
inhibition.
In the present study, the potential of ACF in
treating TNBC cell lines in vitro, was investigated. To the
best of our knowledge, this is the first study to suggest a novel
MCL-1 inhibitory function of ACF. In addition, the present study
revealed a synergistic antitumor effect with ABT-263. Furthermore,
ACF demonstrated its therapeutic potency not only in TNBC but also
in lung cancer and GBM cells.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
AL, HOJ, KSK, and SGP conceptualized the study and
reviewed the writing of the manuscript. HYK and MMH wrote the
original draft of the manuscript and analyzed the data. HOJ, HJ,
JHP, IK, JYS, HKY, MMH and ICP investigated the anticancer effect
of ACF on cancer cells and analyzed and curated the data. MMH, HJ,
ICP, KSK, and SGP investigated the apoptotic signaling pathway and
MCL-1 regulation mechanisms and analyzed the data. HKK and JH
performed and analyzed the mitochondrial function assay. KSK and
SGP were involved in supervision, funding acquisition,
investigation, and project administration. KSK and SGP confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Korea Institute of
Radiological and Medical Sciences (grant no. 50531-2021), which is
funded by the Ministry of Science and Information and
Communications Technology of the Korean government (IWK) and the
Bio & Medical Technology Development Program of the National
Research Foundation (NRF) of Korea Funded by the Korean government
(MSIT) under grant no. 2019M3A9H1103607.
References
|
1
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adams JM and Cory S: The BCL-2 arbiters of
apoptosis and their growing role as cancer targets. Cell Death
Differ. 25:27–36. 2018. View Article : Google Scholar
|
|
5
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35(Suppl): S78–S103. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang MH and Reynolds CP: Bcl-2 inhibitors:
Targeting mitochondrial apoptotic pathways in cancer therapy. Clin
Cancer Res. 15:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ertel F, Nguyen M, Roulston A and Shore
GC: Programming cancer cells for high expression levels of Mcl1.
EMBO Rep. 14:328–336. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Craig RW: MCL1 provides a window on the
role of the BCL2 family in cell proliferation, differentiation and
tumorigenesis. Leukemia. 16:444–454. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Beroukhim R, Mermel CH, Porter D, Wei G,
Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J,
Urashima M, et al: The landscape of somatic copy-number alteration
across human cancers. Nature. 463:899–905. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ding Q, He X, Hsu JM, Xia W, Chen CT, Li
LY, Lee DF, Liu JC, Zhong Q, Wang X and Hung MC: Degradation of
Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced
tumor suppression and chemosensitization. Mol Cell Biol.
27:4006–4017. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Young AI, Timpson P, Gallego-Ortega D,
Ormandy CJ and Oakes SR: Myeloid cell leukemia 1 (MCL-1), an
unexpected modulator of protein kinase signaling during invasion.
Cell Adh Migr. 12:513–523. 2018. View Article : Google Scholar
|
|
12
|
Woo SM, Min KJ, Seo BR, Seo YH, Jeong YJ
and Kwon TK: YM155 enhances ABT-737-mediated apoptosis through
Mcl-1 downregulation in Mcl-1-overexpressed cancer cells. Mol Cell
Biochem. 429:91–102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Caenepeel S, Brown SP, Belmontes B, Moody
G, Keegan KS, Chui D, Whittington DA, Huang X, Poppe L, Cheng AC,
et al: AMG 176, a selective MCL1 inhibitor, is effective in
hematologic cancer models alone and in combination with established
therapies. Cancer Discov. 8:1582–1597. 2018.PubMed/NCBI
|
|
14
|
Ramsey HE, Fischer MA, Lee T, Gorska AE,
Arrate MP, Fuller L, Boyd KL, Strickland SA, Sensintaffar J, Hogdal
LJ, et al: A novel MCL1 inhibitor combined with venetoclax rescues
venetoclaxresistant acute myelogenous leukemia. Cancer Discov.
8:1566–1581. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wainwright M: Acridine-a neglected
antibacterial chromophore. J Antimicrob Chemother. 47:1–13. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee K, Zhang H, Qian DZ, Rey S, Liu JO and
Semenza GL: Acriflavine inhibits HIF-1 dimerization, tumor growth,
and vascularization. Proc Natl Acad Sci USA. 106:17910–17915. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Huang G, Li X, Zhang Y, Jiang Y,
Shen J, Liu J, Wang Q, Zhu J, Feng X, et al: Hypoxia induces
epithelial-mesenchymal transition via activation of SNAI1 by
hypoxia-inducible factor -1α in hepatocellular carcinoma. BMC
Cancer. 13:1082013. View Article : Google Scholar
|
|
18
|
Lee CJ, Yue CH, Lin YJ, Lin YY, Kao SH,
Liu JY and Chen YH: Antitumor activity of acriflavine in lung
adenocarcinoma cell line A549. Anticancer Res. 34:6467–6472.
2014.PubMed/NCBI
|
|
19
|
Lee CJ, Yue CH, Lin YY, Wu JC and Liu JY:
Antitumor activity of acriflavine in human hepatocellular carcinoma
cells. Anticancer Res. 34:3549–3556. 2014.PubMed/NCBI
|
|
20
|
Dekervel J, Bulle A, Windmolders P,
Lambrechts D, Van Cutsem E, Verslype C and van Pelt J: Acriflavine
inhibits acquired drug resistance by blocking the
epithelial-to-mesenchymal transition and the unfolded protein
response. Transl Oncol. 10:59–69. 2017. View Article : Google Scholar :
|
|
21
|
Mangraviti A, Raghavan T, Volpin F, Skuli
N, Gullotti D, Zhou J, Asnaghi L, Sankey E, Liu A, Wang Y, et al:
HIF-1α-targeting acriflavine provides long term survival and
radiological tumor response in brain cancer therapy. Sci Rep.
7:149782017. View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-(Delta Delta C(T) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Jeong SH, Kim HK, Song IS, Lee SJ, Ko KS,
Rhee BD, Kim N, Mishchenko NP, Fedoryev SA, Stonik VA and Han J:
Echinochrome A protects mitochondrial function in cardiomyocytes
against cardiotoxic drugs. Mar Drugs. 12:2922–2936. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ashton JC: Drug combination studies and
their synergy quantification using the Chou-Talalay method-letter.
Cancer Res. 75:24002015. View Article : Google Scholar
|
|
26
|
Williams MM, Lee L, Hicks DJ, Joly MM,
Elion D, Rahman B, McKernan C, Sanchez V, Balko JM, Stricker T, et
al: Key survival factor, Mcl-1, correlates with sensitivity to
combined Bcl-2/Bcl-xL blockade. Mol Cancer Res. 15:259–268. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Certo M, Del Gaizo Moore V, Nishino M, Wei
G, Korsmeyer S, Armstrong SA and Letai A: Mitochondria primed by
death signals determine cellular addiction to antiapoptotic BCL-2
family members. Cancer Cell. 9:351–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lovell JF, Billen LP, Bindner S,
Shamas-Din A, Fradin C, Leber B and Andrews DW: Membrane binding by
tBid initiates an ordered series of events culminating in membrane
permeabilization by Bax. Cell. 135:1074–1084. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kuwana T, Bouchier-Hayes L, Chipuk JE,
Bonzon C, Sullivan BA, Green DR and Newmeyer DD: BH3 domains of
BH3-only proteins differentially regulate Bax-mediated
mitochondrial membrane permeabilization both directly and
indirectly. Mol Cell. 17:525–535. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Willis SN, Chen L, Dewson G, Wei A, Naik
E, Fletcher JI, Adams JM and Huang DC: Proapoptotic bak is
sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by
BH3-only proteins. Genes Dev. 19:1294–1305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Edlich F, Banerjee S, Suzuki M, Cleland
MM, Arnoult D, Wang C, Neutzner A, Tjandra N and Youle RJ: Bcl-x(L)
retrotranslocates bax from the mitochondria into the cytosol. Cell.
145:104–116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oltersdorf T, Elmore SW, Shoemaker AR,
Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges
J, Hajduk PJ, et al: An inhibitor of Bcl-2 family proteins induces
regression of solid tumours. Nature. 435:677–681. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Souers AJ, Leverson JD, Boghaert ER,
Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH,
Fairbrother WJ, et al: ABT-199, a potent and selective BCL-2
inhibitor, achieves anti-tumor activity while sparing platelets.
Nat Med. 19:202–208. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bartlett NL: Highlights in lymphoma from
the 2013 American society of hematology annual meeting and
exposition: Commentary. Clin Adv Hematol Oncol. 12(Suppl 6): 18–23.
2014.PubMed/NCBI
|
|
35
|
Sieghart W, Losert D, Strommer S, Cejka D,
Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia
BP, Peck-Radosavljevic M and Wacheck V: Mcl-1 overexpression in
hepatocellular carcinoma: A potential target for antisense therapy.
J Hepatol. 44:151–157. 2006. View Article : Google Scholar
|
|
36
|
LaBelle JL, Katz SG, Bird GH, Gavathiotis
E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL
and Walensky LD: A stapled BIM peptide overcomes apoptotic
resistance in hematologic cancers. J Clin Invest. 122:2018–2031.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Reiner T, de Las Pozas A, Parrondo R,
Palenzuela D, Cayuso W, Rai P and Perez-Stable C: Mcl-1 protects
prostate cancer cells from cell death mediated by
chemotherapy-induced DNA damage. Oncoscience. 2:703–715. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Young AI, Law AM, Castillo L, Chong S,
Cullen HD, Koehler M, Herzog S, Brummer T, Lee EF, Fairlie WD, et
al: MCL-1 inhibition provides a new way to suppress breast cancer
metastasis and increase sensitivity to dasatinib. Breast Cancer
Res. 18:1252016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zervantonakis IK, Iavarone C, Chen HY,
Selfors LM, Palakurthi S, Liu JF, Drapkin R, Matulonis U, Leverson
JD, Sampath D, et al: Systems analysis of apoptotic priming in
ovarian cancer identifies vulnerabilities and predictors of drug
response. Nat Commun. 8:3652017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Campbell KJ, Dhayade S, Ferrari N, Sims
AH, Johnson E, Mason SM, Dickson A, Ryan KM, Kalna G, Edwards J, et
al: MCL-1 is a prognostic indicator and drug target in breast
cancer. Cell Death Dis. 9:192018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Goodwin CM, Rossanese OW, Olejniczak ET
and Fesik SW: Myeloid cell leukemia-1 is an important apoptotic
survival factor in triple-negative breast cancer. Cell Death
Differ. 22:2098–2106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Balko JM, Giltnane JM, Wang K, Schwarz LJ,
Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sánchez V, et al:
Molecular profiling of the residual disease of triple-negative
breast cancers after neoadjuvant chemotherapy identifies actionable
therapeutic targets. Cancer Discov. 4:232–245. 2014. View Article : Google Scholar :
|
|
43
|
Ozretic P, Alvir I, Sarcevic B, Vujaskovic
Z, Rendic-Miocevic Z, Roguljic A and Beketic-Oreskovic L: Apoptosis
regulator Bcl-2 is an independent prognostic marker for worse
overall survival in triple-negative breast cancer patients. Int J
Biol Markers. 33:109–115. 2018. View Article : Google Scholar
|
|
44
|
Inuzuka H, Shaik S, Onoyama I, Gao D,
Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al:
SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for
ubiquitylation and destruction. Nature. 471:104–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wertz IE, Kusam S, Lam C, Okamoto T,
Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, et al:
Sensitivity to antitubulin chemotherapeutics is regulated by MCL1
and FBW7. Nature. 471:110–114. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leverson JD, Zhang H, Chen J, Tahir SK,
Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, et al: Potent
and selective small-molecule MCL-1 inhibitors demonstrate on-target
cancer cell killing activity as single agents and in combination
with ABT-263 (navitoclax). Cell Death Dis. 6:e15902015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pan R, Ruvolo VR, Wei J, Konopleva M, Reed
JC, Pellecchia M, Andreeff M and Ruvolo PP: Inhibition of Mcl-1
with the pan-Bcl-2 family inhibitor (-)BI97D6 overcomes ABT-737
resistance in acute myeloid leukemia. Blood. 126:363–372. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Mojsa B, Lassot I and Desagher S: Mcl-1
ubiquitination: Unique regulation of an essential survival protein.
Cells. 3:418–437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Goldie H, Walker M, Graham T and Williams
F: Topical effect of acriflavine compounds on growth and spread of
malignant cells. J Natl Cancer Inst. 23:841–855. 1959.PubMed/NCBI
|
|
50
|
Hassan S, Laryea D, Mahteme H, Felth J,
Fryknas M, Fayad W, Linder S, Rickardson L, Gullbo J, Graf W, et
al: Novel activity of acriflavine against colorectal cancer tumor
cells. Cancer Sci. 102:2206–2213. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim SG, Kim CW, Ahn ET, Lee KY, Hong EK,
Yoo BI and Han YB: Enhanced anti-tumour effects of acriflavine in
combination with guanosine in mice. J Pharm Pharmacol. 49:216–222.
1997. View Article : Google Scholar : PubMed/NCBI
|