Myelodysplastic syndrome (MDS) is a clonal
hematopoietic stem cell (HSC) disorder characterized by ineffective
erythropoiesis, dysplasia involving one or more cell lineages,
peripheral cytopenia and an increased risk of transformation to
acute myeloid leukemia (AML). In developed countries, the incidence
of MDS increases progressively with age and the annual incidence of
the disease is estimated to be 4 cases per 100,000 people, rising
to 30 cases per 100,000 people in those >70 years old (1,2).
Men have a higher incidence rate than women (2). Although the bone marrow (BM) of most
patients with MDS is normo- or hypercellular (NH-MDS), 10-20% of
patients with MDS have hypocellular BM (cellularity <20-30% in
the BM trephine biopsy) (2,3).
This subset is referred to as hypoplastic MDS (hMDS) in the World
Health Organization (WHO) classification of myeloid neoplasms
(4), but it is not currently
considered a separate entity. Most cases of hMDS are classified as
MDS with single- and multiple-lineage dysplasia in the WHO
classification system. Hypocellular BM is predominantly found in
low-risk MDS, but can also be observed in high-risk MDS (5,6).
hMDS shares some clinical manifestations with
NH-MDS, such as cytopenia, BM dyspoiesis, clonal chromosomal
changes and the possibility of transformation to AML. By contrast,
it shows distinctive features associated with decreased BM
cellularity, including more profound neutropenia and
thrombocytopenia, and a lower percentage of blasts (7). Furthermore, patients with hMDS have
less frequent abnormal karyotypes, a higher response rate to
immunosuppressive therapy (IST) and a more favourable prognosis
compared with patients with NH-MDS. Notably, patients with hMDS
tend to be younger (hMDS is the most common MDS type in pediatric
patients) (7). Since marrow
cellularity decreases with age, age-adjusted criteria of
hypocellularity have been proposed (e.g., <30% cellularity in
patients ≤70 years and <20% cellularity in patients >70
years) (8).
hMDS is initially treated as low-risk MDS, but
treatment may be tailored according to the degree of similarity to
aplastic anemia (AA) or MDS. AA-like treatment is based on IST with
anti-thymocyte globulin (ATG) and cyclosporine A, which suppresses
the activity of aberrant T cells and helps with BM recovery.
Approximately 50% of low-risk MDS patients show an objective IST
response, which is associated with hypocellular BM and increased
rates of transfusion independence (9). A high overall response rate (ORR)
(73%) was reported in a study that focused only on hMDS treated
with IST (10). Supportive care
in low-risk MDS includes red blood cell (RBC) transfusions,
antibiotics and erythropoietin for stimulation of RBC production.
Hypomethylating agents (HMAs), such as azacytidine or decitabine,
have recently been administered to high-risk patients, but these
agents are effective in only ~50% of MDS patients in the short
term, and a number of patients develop drug resistance and progress
to AML (11). Targeted therapy
using BCL2 and immune checkpoint inhibitors is being tested in
combination with HMAs. HMA therapy may be a reasonable option for
patients with hMDS who have high-risk cytogenetics and unfavourable
somatic mutations (12). HSC
transplantation (HSCT) is the only curative option for patients
with MDS; however, numerous patients are not eligible for HSCT due
to comorbidities usually associated with older age (13). Recently, Zhou et al
(14) evaluated the outcomes of
exclusively hMDS patients after allogenic HSCT; the patients had
favourable survival rate, and none of them relapsed within a
follow-up period of ~3 years.
AA is a rare BM failure (BMF) characterized by
hypoplastic or aplastic BM, a paucity of hematopoietic stem and
progenitor cells (HSPCs), and pancytopenia of the peripheral blood.
In North America and Europe, the incidence of AA is 2-3 cases per
million per year, but may be three-fold higher in Asian populations
(15). AA is a disease that
affects the young, typically within the first three decades of
life, with a median age of onset of ~20 years old. The second peak
occurs at ~60 years old (16). In
some cases, inherited conditions, such as Fanconi anemia,
Shwachman-Diamond syndrome and dyskeratosis congenital, can damage
stem cells and lead to AA (17).
Acquired AA is more frequent, and it may be caused by toxic
chemicals, radiation or idiosyncratic reactions to medications or
infections (18). However, in
>50% of cases, there is no identifiable cause and the condition
is then referred to as idiopathic AA (iAA). In iAA, a dysregulated
immune system destroys HSCs either directly by activation of
apoptosis or indirectly by overproduction of inflammatory
cytokines. Evolution to MDS or AML occurs in up to 20% of AA
patients, especially in those with an incomplete response to IST
(19).
Patients with mild or moderate AA generally do not
require immediate treatment, but patients with severe AA should be
treated as soon as possible after diagnosis. A crucial part of
patient care is supportive treatment that is focused on the
prevention of infections (antibiotics) and bleeding
(RBC/platelet/granulocyte transfusions). Immunosuppression with ATG
and cyclosporine A is frontline treatment in older patients with AA
and in patients for who matched BM donors are not available. A
total of 60-70% of patients with AA show long-term durable ORR
after IST (20) and may show
higher response rates for IST compared with those with hMDS
(21). Paroxysmal nocturnal
hemoglobinuria (PNH) clones have recently been shown to be a good
predictor of IST response in AA as well as MDS (22). Some patients with AA treated with
IST develop clonal hematopoiesis or somatic mutations and progress
to MDS or AML (23).
Corticosteroids, such as methylprednisolone, are often used with
immunosuppressants. Furthermore, AA therapy includes BM stimulants,
such as granulocyte monocyte colony-stimulating factor or platelet
growth factor (eltrombopag). Generally, HSCT is reserved for young
patients and those with severe AA (<50 years old) who are more
likely to have potentially fatal complications. Recently, Zhu et
al (24) performed a
meta-analysis of studies on HSCT and IST in AA, and observed longer
survival times in patients after first-line allo-HSCT compared with
times in those treated with first-line IST (24). However, the potential risks and
benefits of HSCT should be considered for each individual
patient.
Patients with hMDS and AA share overlapping clinical
and pathological features; thus, distinguishing between these
patients can be very difficult. An accurate diagnosis has important
clinical implications, as prognosis and treatment can be quite
different for these diseases. The differential diagnosis is mainly
based on the presence of dysgranulopoiesis,
dysmegakaryocytopoiesis, any ring sideroblasts, an increased
percentage of blasts and abnormal karyotype, all favouring the
diagnosis of hMDS (7). hMDS has a
greater risk of neoplastic progression and a shorter survival time
compared with AA (Table I)
(3,7). Clonal cytogenetic abnormalities are
considered typical of MDS, but they are usually found in only half
of all MDS patients, and cytogenetic analyses may be less reliable
when the BM is hypocellular (3).
An increased percentage of CD34+ cells and a tendency of
positive cells to form aggregates may be useful in distinguishing
hypoplastic myeloid neoplasms (hMDS and hypocellular AML) from AA
(25). Furthermore, elevated
levels of serum thrombopoietin have recently been reported in AA
compared with those in hMDS and may also help to discriminate
between these disorders (26).
MDS develops through a multistep process
encompassing an initial deleterious genetic event within a HSC and
successive genetic abnormalities, leading to clonal expansion and
malignant transformation (27).
In recent years, the understanding of the molecular pathogenesis of
MDS has been markedly improved by next-generation sequencing (NGS),
which has enabled the identification of a large spectrum of new
mutations across all MDS subtypes. There are >40 significantly
mutated genes in MDS, and these mutations account for nearly 90% of
patients with MDS (28).
Functionally, the mutations are grouped into several categories
based on their prevalence: RNA splicing factors [splicing factor 3B
subunit 1 (SF3B1), serine and arginine rich splicing factor
2 (SRSF2), zinc finger CCCH-type, RNA binding motif and
serine/arginine rich 2 (ZRSR2) and U2 small nuclear RNA
auxiliary factor 1/2 (U2AF1/2)], epigenetic regulators [Tet
methylcytosine dioxygenase 2 (TET2), DNA methyltransferase
3α (DNMT3A) and isocitrate dehydrogenase (NADP(+)) 1/2
(IDH1/2)], components of the cohesion complex (stromal
antigen 2, CCCTC-binding factor, structural maintenance of
chromosomes 1A and RAD21 cohesin complex component), chromatin
modifiers [ASXL transcriptional regulator 1 (ASXL1) and
enhancer of zeste 2 polycomb repressive complex 2 subunit
(EZH2)], transcription factors [tumor protein p53
(TP53), RUNX family transcription factor 1 (RUNX1),
ETS variant transcription factor 1 (ETV1) and GATA binding
protein 2 (GATA2)], signal transduction molecules [Fms
related receptor tyrosine kinase 3 (FLT3), Janus kinase 2
(JAK2), MPL proto-oncogene thrombopoietin receptor
(MPL), GNAS complex locus and KIT proto-oncogene receptor
tyrosine kinase], RAS pathway [KRAS proto-oncogene GTPase, NRAS
proto-oncogene GTPase (NRAS), Cbl proto-oncogene,
neurofibromin 1 and protein tyrosine phosphatase non-receptor type
11 (PTPN11)] and DNA repair [ATM serine/threonine kinase,
BRCA1/BRCA2-containing complex subunit 3, DNA cross-link repair 1C
and FA complementation group L]. Mutations in RNA splicing and DNA
methylation genes seem to occur early and are considered founder
mutations in >50% of patients with MDS (28). Mutations provide a wide range of
prognostic information, from benign to malignant and from good to
poor overall survival (OS) time. For example, TP53, EZH2,
ETS variant transcription factor 6 (ETV6), RUNX1,
ASXL1 and SRSF2 mutations predict shorter survival
time. The SF3B1 mutation is strongly associated with ring
sideroblasts and thus has been included as a diagnostic criterion
in MDS with ring sideroblasts (4).
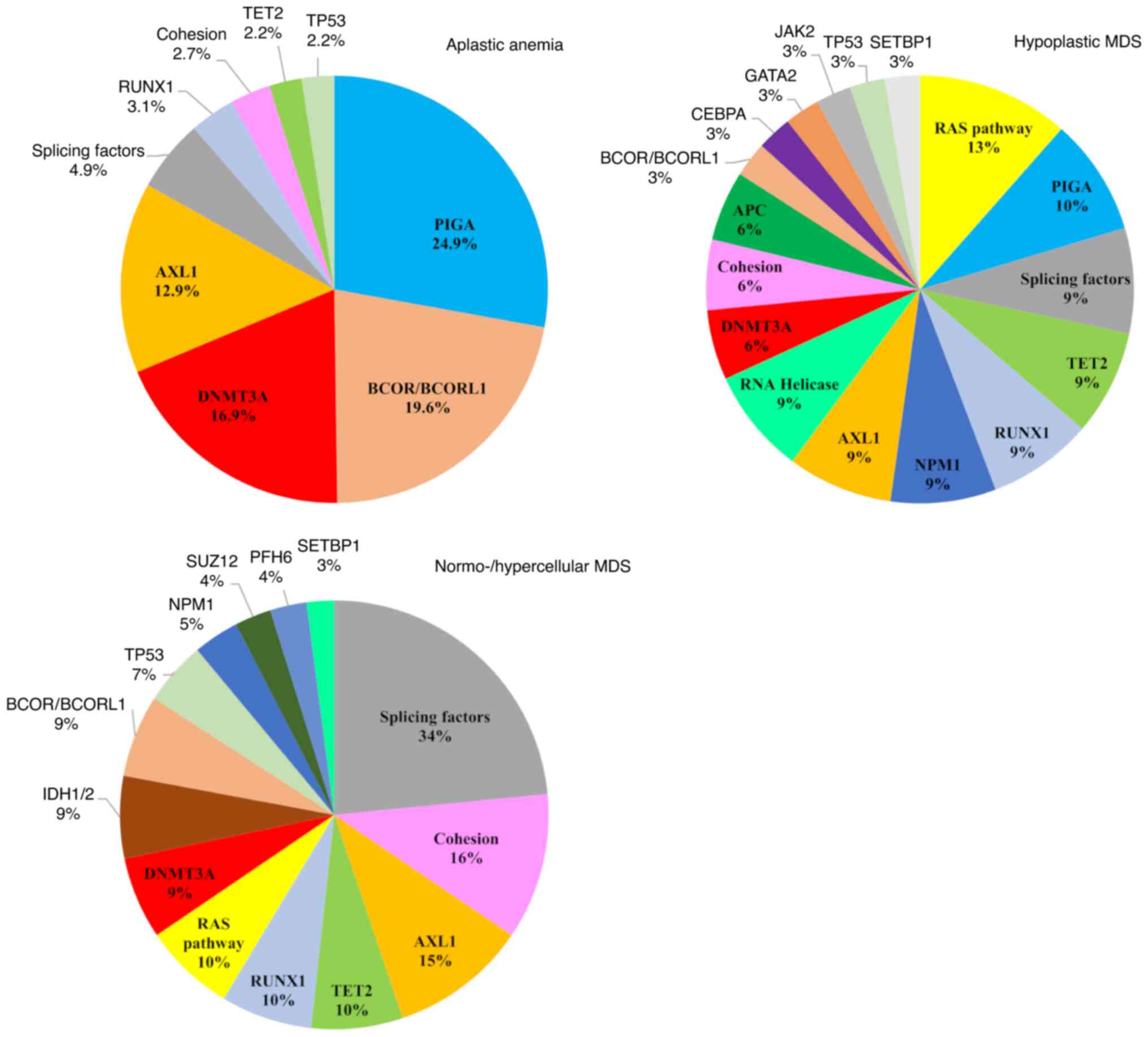
There are several reports concerning differences in
the mutational landscapes between hMDS and NH-MDS (Fig. 1). Nazha et al (29) compared the mutational profiles of
62 genes between patients with hMDS and NH-MDS. Patients with hMDS
acquired fewer somatic mutations and had smaller driver clones
compared with patients with NH-MDS. Splicing somatic mutations were
determined predominantly in patients with NH-MDS, as driver clones
were found exclusively in these patients. The study hypothesized
that the immune system in patients with hMDS may suppress the
driver clone by inhibiting its growth and genetic evolution, thus
limiting the acquisition of downstream somatic lesions. Notably,
some driver clones, such as SF3B1, SRSF2, TET2, ASXL1 and
BCL-6 coreceptor (BCOR), may overcome this inhibitory effect
(29). Yao et al (30) detected at least one gene mutation
(17 genes) in 35% of patients with hMDS, and the most common
mutation was an SF3B1 mutation. Patients with hMDS exhibited
significantly lower incidence rates of RUNX1, ASXL1, DNMT3A,
EZH2 and TP53 mutations, and a lower number of mutations
per subject compared with patients with NH-MDS; however, the number
was significantly higher in comparison with the number in patients
with AA. Schwartz et al (31) used a whole exome sequencing
approach to describe somatic and germline changes in pediatric MDS
and found prevalent Ras/MAPK pathway mutations compared with that
in adult MDS. Huang et al (6) did not find any difference in the
incidence of RAS, acute myeloid leukemia 1 protein, JAK2,
PTPN11 or FLT3/internal tandem duplication mutations
between hMDS and non-hMDS. Bono et al (5) reported mutational data from a
24-gene panel on a large cohort of hMDS patients (n=93) and
detected one or more somatic mutations in 38% of patients with
hMDS. In comparison to non-hMDS patients (n=239), the patients with
hMDS had a lower number of mutations per subject, but this number
was significantly higher than that found in the patients with AA.
The prevalence of splicing mutations (SF3B1 and
SRSF2) and comutation patterns of TET2, DNMT3A and
ASXL1 was lower in hMDS compared with that in non-hMDS. The
integration of mutational data into a scoring formula enabled the
separation hMDS patients with myeloid neoplasm-like profiles from
those with non-malignant profiles. It was suggested that hMDS more
likely represents a mixture of entities along a spectrum rather
than a homogeneous in-between category (5).
Taken together, these results suggest that the
mutational profile of hMDS overlaps with the profile of NH-MDS,
except for the lower incidence of mutations in splicing factors and
in ASXL1 and IDH1/2 genes. Patients with hMDS have
fewer somatic mutations, and overall, smaller driver clones.
In AA, the most frequently mutated genes are
phosphatidylinositol glycan anchor biosynthesis class A
(PIGA), BCOR/BCOR-like 1 (BCORL1),
DNMT3A and ASXL1, suggesting mechanisms of clonal
selection. Mutations in PIGA and BCOR/BCORL1 are more
specific to AA, while DNMT3A and ASXL1 mutations are
also found in MDS (Fig. 1).
PIGA somatic mutations are found in up to 40% of patients
with AA (16,32). PNH clones are detected in a higher
proportion of patients with AA (up to 60%) and have been shown to
escape T cell-mediated destruction. Blood cells with PIGA
mutations are likely less immunogenic and thus may acquire a
survival advantage (33). Somatic
mutations in JAK2/JAK3, RUNX1, TP53,
TET2, and CUB and sushi multiple domains 1 genes are less
common in AA, and SRSF2, U2AF1, MPL and Erb-B2
receptor tyrosine kinase 2 mutations are rare (<3% of acquired
AA cases) (34). Detected somatic
mutations in AA have mostly variant allelic frequencies of <10%
(23,34). Patients with AA and PIGA,
BCOR or BCORL1 mutations show a better response to
IST, as well as improved progression-free survival (PFS) and OS
rates, while DNMT3A, ASXL1, JAK2/JAK3
or RUNX1 mutations are associated with a worse IST response
and survival rate. Notably, mutations in DNMT3A or
ASXL1 increase the risk of developing MDS from AA (23,34,35). Keel et al (36) detected pathological mutations in
MPL and TP53 genes in young patients with AA and
MDS.
A high incidence of somatic mutations in MDS
suggests that mutational profiling of myelodysplasia-related genes
may help to distinguish AA from hMDS and may identify patients who
are at risk for progression. Kulasekararaj et al (23) used targeted high-throughput DNA
sequencing to determine somatic mutations in patients with acquired
AA. Somatic mutations (ASXL1, DNMT3A, BCOR) were
detected in 19% of patients with AA who had a longer disease
duration and a higher risk of MDS transformation than those without
mutations. Notably, the detection of ASXL1, DNMTA,
BCOR and TET2 mutations in the AA cohort coupled with
published expression data provides a role for the potential
association and cooperation between mutations in epigenetic
regulators and immune-mediated BMF. Similarly, Huang et al
(37) focused on a limited number
of genes and found mutations in epigenetic regulator genes,
including TET2 and ASXL1, in 17.4% of patients with
AA. By contrast, Heuser et al (38) identified somatic mutations in only
5.3% of patients with AA and suggested that mutations in myeloid
malignancy-related genes are rare in this disease.
In the last two decades, it has become increasingly
evident that ncRNAs are important regulators of biological
processes, including blood cell differentiation and immune
response. There are several categories of ncRNAs, such as microRNAs
(miRNAs), Piwi-interacting RNAs, small nucleolar RNAs and long
ncRNAs (lncRNAs) (39). miRNAs
are the most prolific class of ncRNAs and have been shown to play a
role in the pathogenesis of MDS (40). Comprehensive data are available on
expression miRNA profiles associated with MDS subtypes, disease
stages and treatment response, as well as on dysregulation of
specific miRNAs and their role in pathogenesis (Table II). As MDS originates in HSCs, a
number of studies have been performed on CD34+ cells.
Abundantly expressed miRNAs in CD34+ cells of patients
with MDS include, but are not limited to, let-7b,
miR-10a, miR-25, the miR-26 family,
miR-128a, miR-146, miR-155, miR-181a,
miR-222 and miR-223 (41). To date, no study has focused on
the differential expression of miRNAs between hMDS and NH-MDS. In
general, low-risk patients show distinctive expression profiles
compared with high-risk patients (42,43). Sokol et al (43) defined a unique signature of 10
miRNAs (miR-181a/b/c/d, miR-221, miR-376b,
miR-125b, miR-155, miR-130a and
miR-486-5p) that accurately differentiated low-risk patients
from high-risk patients. Notably, the 6-miRNA signature may
distinguish RA/refractory cytopenias with multilineage dysplasia
(RCMD) patients with a normal karyotype from those with trisomy 8,
who usually show a good response to IST.
lncRNAs represent another important class of ncRNAs
whose role in hematopoietic disorders is being explored. Studies on
lncRNAs in MDS display heterogeneity in experimental design (size
of patient cohort, MDS subtypes, technologies used and analytical
approaches), and thus far, no study has focused only on hMDS. The
very first study by Benetatos et al (58) revealed hypermethylation of the
maternally expressed 3 (MEG3) gene promoter in 34.9% of
patients with MDS, which may confer a worse overall prognosis.
Next, genome-wide studies defined the gene expression profiles of
lncRNAs in various specific groups of patients with MDS, such as
those with primary MDS (59,60), refractory anemia (RA) with excess
blasts type 2 (RAEB-2) MDS (61),
de novo MDS and MDS evolved from AA (62). Recently, Szikszai et al
(59) analyzed lncRNA expression
across all MDS subtypes and evaluated them in relation to disease
subtypes, cytogenetic and mutational aberrations, and risk of
progression. Comparative analysis between low- and high-risk
patients determined 16 deregulated lncRNAs [e.g., downregulated
RP11-897M7.1 and long intergenic non-protein coding RNA 539,
and upregulated T cell leukemia/lymphoma 6, long intergenic
non-protein coding RNA 1013, LEF1 antisense RNA 1 (LEF1-AS1)
and CTC-436K13.2 in low-risk patients] (59). Yao et al (60) attempted to use lncRNA expression
for the risk stratification of patients with MDS and integrated
four lncRNAs (TC07000551.hg.1, TC08000489.hg.1,
TC02004770.hg.1 and TC03000701.hg.1) whose expression
levels were associated with OS into a risk-scoring system. Higher
lncRNA scores were associated with high-risk MDS, complex
karyotype, and RUNX1, ASXL1, TP53,
SRSF2 and ZRSR2 mutations. In relation to the skewed
T cell repertoire in MDS, pathway analysis of differentially
expressed genes between patients with the highest and lowest lncRNA
risk scores determined T cell-related pathways [e.g., cytotoxic
T-lymphocyte-associated protein 4 signaling in cytotoxic T
lymphocytes and CD28 signaling in T helper (Th) cells] to be the
most significant (60). Hung
et al (63) recently
identified an association between higher KIAA0125 expression
(BM mononuclear cells) and high-risk MDS, ASXL1 and
NRAS mutations, and poorer OS and leukemia-free survival. A
recent study by Li et al (64) reported an association between a
higher expression level of LOC101928834 and a higher white
blood cell count, a higher blast percentage, RAEB subtype and a
shorter OS time in MDS. By contrast, LEF1-AS1 expression has
been shown to be significantly downregulated in patients with MDS
compared with that in healthy controls (65).
Although there are no studies describing
miRNA/lncRNA profiles exclusively in hMDS, there are reports
demonstrating that RA and RCMD categories (typical for the majority
of hMDS cases) show ncRNA expression patterns distinct from those
of other MDS subtypes. Moreover, levels of specific ncRNAs have
been successfully used for classification and stratification of
patients with MDS (42,43,60). It may be assumed that hMDS is also
associated with specific ncRNA profiles that differ from those of
AA and could be used for differentiation. The dysregulation of
ncRNAs detected in T cells derived from patients with AA and hMDS
indicates that these regulators may contribute to the
immunopathogenesis of these disorders.
The overlap of immunological features and the
responsiveness of a significant proportion of patients with hMDS/AA
to IST suggest that these distinctive clinical entities share an
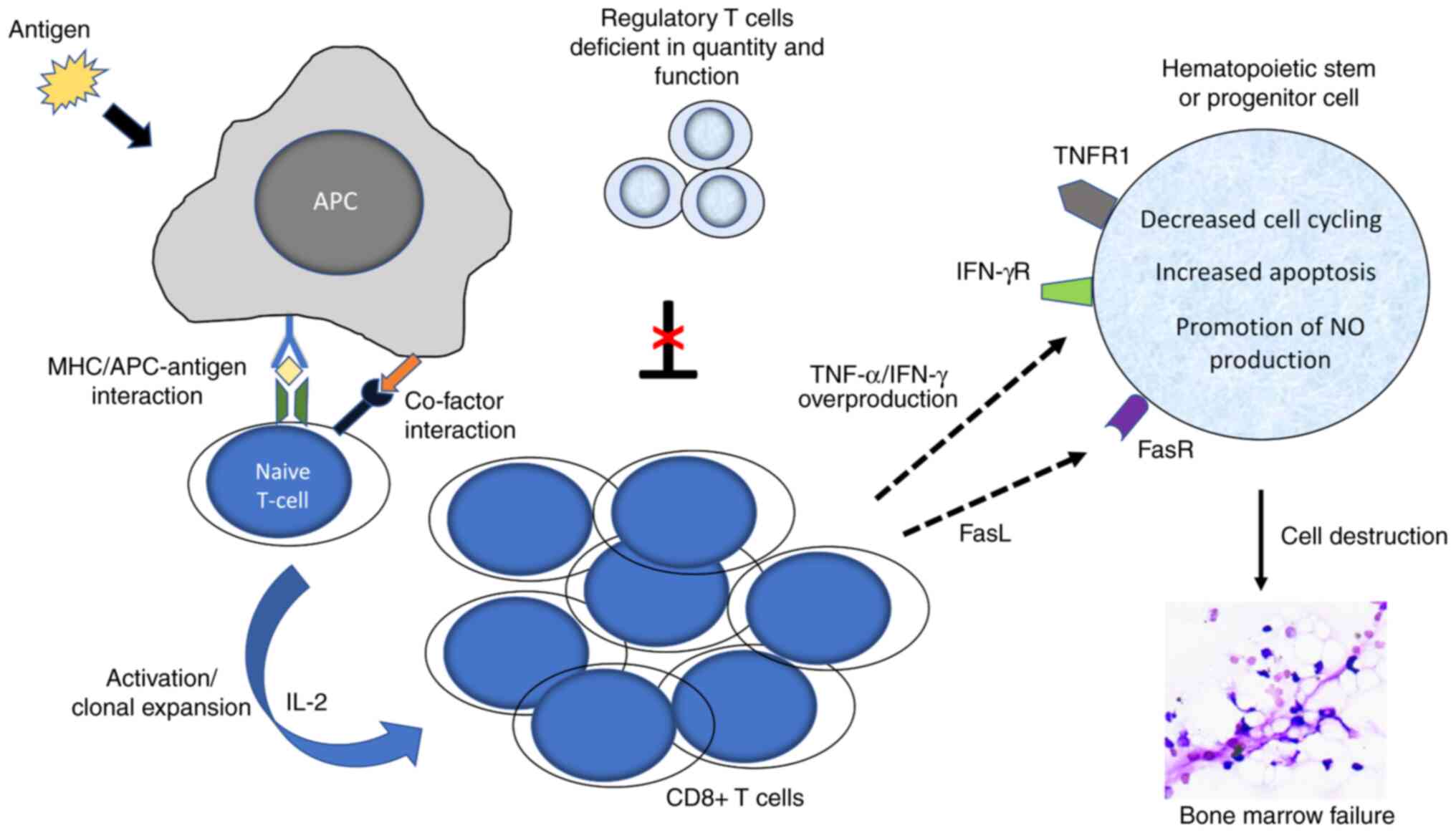
immune-mediated pathogenic mechanism. Clinical and experimental
studies have provided compelling evidence that HSPCs are damaged by
abnormally activated cytotoxic T cells (Fig. 2). Expanded CD4+ and
CD8+ T cell clones have been observed in the BM of both
patients with hMDS and those with AA (69,70). Melenhorst et al (71) analyzed the CD4+ and
CD8+ T cell repertoires in patients with MDS by flow
cytometry and PCR. Multiple T cell expansions (of both helper and
cytotoxic T (Tc) cells) were observed, as well as the functional
differentiation in vivo of T cells from memory to effector T
cells, in CD8+ cells. Similar findings were reported by
Fozza et al (72),
supporting the involvement of cytotoxic T cells either in antitumor
immune surveillance or in autoreactive aggression toward
hematopoietic precursors. Moreover, dominant T cell clones persist
in patients with MDS that is unresponsive to immunosuppression and
regress in responders (73).
Strong polarization of BM CD4+ cells toward Th1 and of
CD8+ cells toward Tc1 was observed in low-risk MDS
compared with that in AA, suggesting T cell stimulation from clones
of malignant hematopoietic cells (74). Regulatory T cells (Tregs) are
deficient in quantity and function in patients with early MDS
(75). The function of Tregs is
to suppress the autoreactivity of other T cell populations to
normal tissue; thus, their hypofunction may favour the autoimmune
destruction of HSPCs (76).
The antigens that trigger the immune response in
MDS are not known, but potential candidates [such as Wilms tumor
protein 1 (WT1)] have been suggested. As patients with MDS and
trisomy 8 often show a good response to IST, an immunological
mechanism underlying BMF has been proposed. Trisomy 8 cells express
high levels of WT1, and CD8+ T cells are able to
recognize WT1 peptides and induce IFN-γ expression in vitro,
suggesting that this antigen may contribute to the induction of an
immune response (77). Sloand
et al (78) further
demonstrated that marrow HSCs with trisomy 8 may escape T
cell-mediated destruction by overexpression of prosurvival protein
cyclin D1 and survivin. Other neoantigens or overexpressed
self-antigens [human leukocyte antigen (HLA)-A2-restricted
nonameric peptide] may also elicit an immune response (79).
Abnormal overproduction of proinflammatory
cytokines [such as TNF-α, IFN-γ and interleukin 17 (IL-17)] has
been reported in patients with MDS and contributes also to
ineffective hematopoiesis (80,81). In patients with low-risk MDS, the
G/A polymorphism in the TNF-α promoter is associated with
high levels of TNF-α produced by CD4+ and
CD8+ T lymphocytes (82), suggesting its role in anemia. In a
previous study, an increased frequency of CD4+ T cells
producing IFN-γ was detected in hMDS, and in vitro decrease
of interferon by cyclosporine led to improved hematopoiesis
(10). The production of IFN-γ
and TNF-α in low-risk MDS may be further enhanced by high levels of
IL-17 (83). Based on the
clinical/immunological/molecular features, Fattizzo et al
(84) recently defined two hMDS
phenotypes, namely, AA-like and MDS-like hMDS. The first is
characterized by prevailing inflammation and immune activation, and
a response to IST, and the second is characterized by genetic
lesions, clonal selection and an increased risk of leukemic
evolution.
Up to 80% of patients with AA show a response to T
cell-directed IST, supporting involvement of aberrant T cell
populations in the pathogenesis. As in hMDS, BM T cells are also
skewed toward oligo/polyclonal patterns in acquired AA (16,70,85). Giudice et al (85) observed oligoclonal characteristics
in CD8+CD57+ cells, as well as in total
CD8+ T cells from patients with AA. de Latour et
al (86) found an increased
population of CD3+CD4+IL-17-producing T cells
in patients with AA at presentation compared with that in controls,
and this correlated with disease activity. Abnormally activated T
cells destroy HSPCs through apoptosis [via Fas cell surface death
receptor (Fas)/Fas ligand, granzyme, perforin] and the
overproduction of proinflammatory cytokines. Extensive apoptosis of
BM HSPCs has been observed in patients with AA, indicating that
apoptosis is a major mechanism of cell destruction (87). BM CD34+ progenitor
cells and lymphocytes of patients with AA overexpress Fas, which is
involved in triggering the Fas-mediated apoptotic pathway (88). By contrast, normal expression of
Fas has been observed in patients with AA in remission (89). Overproduction of cytokines may
upregulate the expression of Fas (77).
As in hMDS, AA Tregs exhibit a decreased quantity
and ability to suppress the proliferation of autologous T cells.
Deep phenotyping of AA Tregs defined two specific Treg
subpopulations, Treg A and Treg B, that may predict the response to
IST. The Treg B subpopulation with a memory/activated phenotype was
overrepresented in IST responders, while the Treg A subpopulation
was significantly higher in non-responders. Furthermore, Tregs from
patients with AA were IL-2-sensitive and could be expanded in
vitro (90).
AA is strongly associated with PNH. PNH clones
deficient in glycosyl-phosphatidylinositol (GPI)-anchored proteins
appear to be spared by the immune attack mediated by T cells in BMF
syndromes. PNH clones are frequently found in acquired AA (≤60%)
and are also observed in MDS (10-20%, more common in low-risk
cases) (91). The mechanism of
this escape is not clear. It has been suggested that antigen
targets of T cell attack or coregulators are GPI-linked proteins.
Gargiulo et al (92)
demonstrated that CD1d-restricted, GPI-specific CD8+ T
cells are expanded in patients with PNH, suggesting that the GPI
may be targeted by autoreactive T cells and that these T cell
clones are responsible for the BMF in PNH. Hanaoka et al
(93) suggested that
immunoselection of PIGA mutant cells is due to a deficiency
in the stress-inducible GPI-linked membrane proteins UL16 binding
protein 1 and 2, which activate natural killer and T cells.
Furthermore, PIGA clones may acquire additional somatic
mutations (TET2, SUZ12 polycomb repressive complex 2
subunit, U2AF1 and JAK2), resulting in a
proliferative advantage (94).
Mechanisms and factors implicated in the immunopathogenesis of AA
and hMDS are summarized in Table
III.
In addition to the T cell-mediated immune response,
aging, which is associated with numerous changes in the immune
system, including chronic low-grade inflammation (known as
inflammaging), may be involved in the development of AA and hMDS in
elderly patients (95).
The genetic and molecular basis of an abnormal T
cell response is being studied. Several polymorphisms in cytokine
genes (e.g., IFN-γ, TNF-α and IL-6) have been linked to the high
production of proinflammatory cytokines in AA and MDS (82,96). Furthermore, specific HLA
haplotypes are associated with the AA phenotype and response to
IST, suggesting that cytotoxic T cells may target the autoantigens
presented on HSCs through these HLA class I molecules. HLA-DR15 (a
serological split of HLA-DR2) is overrepresented in AA patients and
MDS patients with RA compared with that in their healthy
counterparts (97). The presence
of this HLA allele is associated with a better response to IST in
AA (98). Notably, patients with
MDS bearing a PNH clone have a significantly higher HLA-DR15 allele
frequency (99). Katagiri et
al (100) demonstrated
frequent loss of HLA alleles associated with copy number-neutral
6pLOH in acquired AA. Notably, the missing HLA alleles in 6pLOH(+)
clones included HLA-A*02:01, A*02:06, A*31:01 and B*40:02, which
were overrepresented in the germline of patients with AA. Osumi
et al (101) suggested
that HLA-B*40:02 is one of the target antigens of T cells in
idiopathic AA and that mutations in this HLA allele contribute to
clonal escape. Babushok et al (102) screened patients with AA for
somatic HLA class 1 loss and detected it in 17% of cases.
Furthermore, the loss was correlated with a more severe disease
course and more frequent evolution to MDS. Mutations in
β2-microglobulin gene may represent another mechanism of MHC class
I loss leading to defective CD4-8+ cell-mediated
cytotoxicity (103).
Defective telomere homeostasis is also suggested to
play a role in the pathogenesis of AA and MDS. Approximately 35% of
patients with AA show telomere length shortening in peripheral
granulocytes and mononuclear cells. Patients with AA responsive to
IST do not possess telomeres that differ in length compared with
controls, while untreated patients and non-responders show marked
telomere shortening (104). The
degree of telomere erosion has been correlated with the severity of
AA, risk of relapse, overall survival rate and risk of clonal
evolution to MDS (105). In MDS,
telomere shortening is mostly linked to disease progression and
leukemic transformation into AML. A decrease in telomere length was
also observed in low-risk MDS patients with RA (42%) and patients
with low to intermediate-1 risk (43.1 and 30.8%, respectively),
according to the International Prognostic Scoring System, compared
with age-matched controls (106-108). Bouillon et al (109) found significantly shortened
age-adapted telomere length in both patients with hMDS and those
with AA, but patients with AA showed more accelerated telomere
shortening compared with patients with hMDS.
Mutations in telomerase complex genes (telomerase
RNA component and telomerase reverse transcriptase) have been
reported in AA and MDS (110,111); however, the mutations are
considered risk factors for BMF rather than genetic determinants
(112). Genetic variants of
other telomerase genes, i.e., telomeric repeat binding actor 1/2,
may be associated with risk for AA; however, they are rare
(113). Furthermore, the
presence of pathogenic regulator of telomere elongation helicase 1
variants, resulting in telomere erosion, has been associated with
AA and hMDS (114).
Potential implications of ncRNAs in the
immunopathogenesis of BMF were demonstrated the study by Hosokawa
et al (55), which
detected downregulation of miR-126-3p and miR-223-3p
in CD4+ T effector memory cells and downregulation of
miR-126-3p, miR-145-5p and miR-223-3p in
CD8+ T effector memory and terminal effector cells in
AA. miR-126-3p and miR-145-5p targeted MYC and
PIK3R2, which were upregulated in the CD4+ and
CD8+ T cells of the patients with AA. Notably,
successful IST was associated with the recovery of miRNA
levels.
Although acquired AA and hMDS represent distinct
clinical entities, they show considerable clinicopathological
similarities and are difficult to distinguish from each other. The
overlaps likely originate from a common pathogenic mechanism based
on cytotoxic T cell-mediated attack against certain antigens
located on stem or more lineage-restricted progenitor cells.
Despite the overlaps, these disorders differ in some
characteristics that are an important part of the differential
diagnosis. However, the cytological/morphological differences may
be subtle due to severe hypocellularity in some cases and need to
be evaluated carefully in the context of other findings.
Deep phenotyping has proposed that hMDS is a mixed
phenotypic entity comprising of two phenotypes, one resembling AA
(non-malignant BMF) and one closer to that of NH-MDS (BMF prone to
malignant transformation). A similar situation likely exists also
in AA, in which a small proportion of patients transform to MDS
and/or AML, even after successful IST in some cases. Identifying
patients at risk of disease progression is a crucial step for early
intervention and appropriate follow-up.
The NGS era has increased our knowledge of genetic
lesions in these disorders and improved the diagnostic specificity
of identifying malignant myelodysplasia; however, there are no
specific mutations that clearly separate AA from hMDS. Mutations in
BCOR/BCORL1, PIGA, DNMT3A and ASXL1 genes are
prevalent in AA, but DNMT3A and ASXL1 mutations are
also found in MDS. Clones with DNMT3A and ASXL1
mutations usually increase in size and predict a poorer response to
IST and progression to MDS/AML. By contrast, BCOR, BCORL1
and PIGA-mutated clones remain small or disappear and
predict a better response to IST and favourable outcomes of AA.
High diversity of mutational profiles, driver vs. passenger
mutations and infrequently mutated genes of unclear pathogenetic
relevance are challenging aspects of NGS testing. The role of other
molecular factors in BMF, such as ncRNAs, is also being explored.
With the development of RNA interference technology and
miRNA-inhibitory agents, these RNAs may provide novel therapeutic
approaches in autoimmune disorders.
In conclusion, the diagnostic criteria defining
boundaries between AA and hMDS remain the focus of debate and will
surely be refined by the incorporation of molecular features into
classification schemes.
Not applicable.
HV wrote the manuscript. MB critically revised the
manuscript. All authors read and approved the final manuscript.
Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This study was supported by Ministry of Health of the Czech
Republic (grant no. NU21-03-00565).
|
1
|
Germing U, Aul C, Niemeyer CM, Haas R and
Bennett JM: Epidemiology, classification and prognosis of adults
and children with myelodysplastic syndromes. Ann Hematol.
87:691–699. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Germing U, Strupp C, Kündgen A, Bowen D,
Aul C, Haas R and Gattermann N: No increase in age-specific
incidence of myelodysplastic syndromes. Haematologica. 89:905–910.
2004.
|
|
3
|
Durrani J and Maciejewski JP: Idiopathic
aplastic anemia vs hypocellular myelodysplastic syndrome.
Hematology Am Soc Hematol Educ Program. 2019:97–104. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arber DA, Orazi A, Hasserjian R, Thiele J,
Borowitz MJ, Le Beau MM, Bloomfield CD, Cazzola M and Vardiman JW:
The 2016 revision to the World Health Organization classification
of myeloid neoplasms and acute leukemia. Blood. 127:2391–2405.
2016. View Article : Google Scholar
|
|
5
|
Bono E, McLornan D, Travaglino E, Gandhi
S, Gallì A, Khan AA, Kulasekararaj AG, Boveri E, Raj K, Elena C, et
al: Clinical, histopathological and molecular characterization of
hypoplastic myelodysplastic syndrome. Leukemia. 33:2495–2505. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang TC, Ko BS, Tang JL, Hsu C, Chen CY,
Tsay W, Huang SY, Yao M, Chen YC, Shen MC, et al: Comparison of
hypoplastic myelodysplastic syndrome (MDS) with
normo-/hypercellular MDS by International prognostic scoring
system, cytogenetic and genetic studies. Leukemia. 22:544–550.
2008. View Article : Google Scholar
|
|
7
|
Marisavljevic D, Cemerikic V, Rolovic Z,
Boskovic D and Colovic M: Hypocellular myelodysplastic syndromes:
Clinical and biological significance. Med Oncol. 22:169–175. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yue G, Hao S, Fadare O, Baker S,
Pozdnyakova O, Galili N, Woda BA, Raza A and Wang SA:
Hypocellularity in myelodysplastic syndrome is an independent
factor which predicts a favorable outcome. Leuk Res. 32:553–558.
2008. View Article : Google Scholar
|
|
9
|
Stahl M, DeVeaux M, de Witte T, Neukirchen
J, Sekeres MA, Brunner AM, Roboz GJ, Steensma DP, Bhatt VR,
Platzbecker U, et al: The use of immunosuppressive therapy in MDS:
Clinical outcomes and their predictors in a large international
patient cohort. Blood Adv. 2:1765–1772. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Selleri C, Maciejewski JP, Catalano L,
Ricci P, Andretta C, Luciano L and Rotoli B: Effects of
cyclosporine on hematopoietic and immune functions in patients with
hypoplastic myelodysplasia: In vitro and in vivo studies. Cancer.
95:1911–1922. 2002. View Article : Google Scholar
|
|
11
|
Gil-Perez A and Montalban-Bravo G:
Management of myelodysplastic syndromes after failure of response
to hypomethylating agents. Ther Adv Hematol.
10:20406207198470592019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nazha A, Narkhede M, Radivoyevitch T,
Seastone DJ, Patel BJ, Gerds AT, Mukherjee S, Kalaycio M, Advani A,
Przychodzen B, et al: Incorporation of molecular data into the
revised international prognostic scoring system in treated patients
with myelodysplastic syndromes. Leukemia. 30:2214–2220. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bond DR, Lee HJ and Enjeti AK: Unravelling
the epigenome of myelodysplastic syndrome: Diagnosis, prognosis,
and response to therapy. Cancers (Basel). 12:–3128. 2020.
View Article : Google Scholar
|
|
14
|
Zhou M, Wu L, Zhang Y, Mo W, Li Y, Chen X,
Wang C, Pan S, Xu S, Zhou W, et al: Outcome of allogeneic
hematopoietic stem cell transplantation for hypoplastic
myelodysplastic syndrome. Int J Hematol. 112:825–834. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Issaragrisil S, Kaufman DW, Anderson T,
Chansung K, Leaverton PE, Shapiro S and Young NS: The epidemiology
of aplastic anemia in Thailand. Blood. 107:1299–1307. 2006.
View Article : Google Scholar
|
|
16
|
Shallis RM, Ahmad R and Zeidan AM:
Aplastic anemia: Etiology, molecular pathogenesis, and emerging
concepts. Eur J Haematol. 101:711–720. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Alter BP: Diagnosis, genetics, and
management of inherited bone marrow failure syndromes. Hematology
Am Soc Hematol Educ Program. 2007:29–39. 2007. View Article : Google Scholar
|
|
18
|
Young NS: Aplastic anemia. N Engl J Med.
379:1643–1656. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Afable MG II, Tiu RV and Maciejewski JP:
Clonal evolution in aplastic anemia. Hematology Am Soc Hematol Educ
Program. 2011:90–95. 2011. View Article : Google Scholar
|
|
20
|
Risitano AM: Immunosuppressive therapies
in the management of acquired immune-mediated marrow failures. Curr
Opin Hematol. 19:3–13. 2012. View Article : Google Scholar
|
|
21
|
Koh Y, Lee HR, Kim HK, Kim I, Park S, Park
MH, Kim BK, Yoon SS and Lee DS: Hypoplastic myelodysplastic
syndrome (h-MDS) is a distinctive clinical entity with poorer
prognosis and frequent karyotypic and FISH abnormalities compared
to aplastic anemia (AA). Leuk Res. 34:1344–1350. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fattizzo B, Dunlop A, Ireland R, Kassam S,
Yallop D, Mufti G, Marsh J and Kulasekararaj A: Prevalence of small
PNH clones and their prognostic significance in patients tested for
unusual indications: A single center experience. Br J Haematol.
185:1252019.
|
|
23
|
Kulasekararaj AG, Jiang J, Smith AE,
Mohamedali AM, Mian S, Gandhi S, Gaken J, Czepulkowski B, Marsh JC
and Mufti GJ: Somatic mutations identify a subgroup of aplastic
anemia patients who progress to myelodysplastic syndrome. Blood.
124:2698–2704. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu Y, Gao Q, Hu J, Liu X, Guan D and
Zhang F: Allo-HSCT compared with immunosuppressive therapy for
acquired aplastic anemia: A system review and meta-analysis. BMC
Immunol. 21:102020. View Article : Google Scholar :
|
|
25
|
Bennett JM and Orazi A: Diagnostic
criteria to distinguish hypocellular acute myeloid leukemia from
hypocellular myelodysplastic syndromes and aplastic anemia:
Recommendations for a standardized approach. Haematologica.
94:264–268. 2009. View Article : Google Scholar
|
|
26
|
Feng X, Scheinberg P, Wu CO, Samsel L,
Nunez O, Prince C, Ganetzky RD, McCoy JP Jr, Maciejewski JP and
Young NS: Cytokine signature profiles in acquired aplastic anemia
and myelodysplastic syndromes. Haematologica. 96:602–606. 2011.
View Article : Google Scholar :
|
|
27
|
Warlick ED and Smith BD: Myelodysplastic
syndromes: Review of pathophysiology and current novel treatment
approaches. Curr Cancer Drug Targets. 7:541–558. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ganguly BB and Kadam NN: Mutations of
myelodysplastic syndromes (MDS): An update. Mutat Res Rev Mutat
Res. 769:47–62. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nazha A, Seastone D, Radivoyevitch T,
Przychodzen B, Carraway HE, Patel BJ, Carew J, Makishima H, Sekeres
MA and Maciejewski JP: Genomic patterns associated with hypoplastic
compared to hyperplastic myelodysplastic syndromes. Haematologica.
100:e434–e437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao CY, Hou HA, Lin TY, Lin CC, Chou WC,
Tseng MH, Chiang YC, Liu MC, Liu CW, Kuo YY, et al: Distinct
mutation profile and prognostic relevance in patients with
hypoplastic myelodysplastic syndromes (h-MDS). Oncotarget.
7:63177–63188. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schwartz JR, Ma J, Lamprecht T, Walsh M,
Wang S, Bryant V, Song G, Wu G, Easton J, Kesserwan C, et al: The
genomic landscape of pediatric myelodysplastic syndromes. Nat
Commun. 8:15572017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mufti GJ and Marsh JCW: Somatic mutations
in aplastic anemia. Hematol Oncol Clin North Am. 32:595–607. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stanley N, Olson TS and Babushok DV:
Recent advances in understanding clonal haematopoiesis in aplastic
anaemia. Br J Haematol. 177:509–525. 2017. View Article : Google Scholar :
|
|
34
|
Yoshizato T, Dumitriu B, Hosokawa K,
Makishima H, Yoshida K, Townsley D, Sato-Otsubo A, Sato Y, Liu D,
Suzuki H, et al: Somatic mutations and clonal hematopoiesis in
aplastic anemia. N Engl J Med. 373:35–47. 2015. View Article : Google Scholar
|
|
35
|
Marsh JC and Kulasekararaj AG: Management
of the refractory aplastic anemia patient: What are the options?
Blood. 122:3561–3567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Keel SB, Scott A, Sanchez-Bonilla M, Ho
PA, Gulsuner S, Pritchard CC, Abkowitz JL, King MC, Walsh T and
Shimamura A: Genetic features of myelodysplastic syndrome and
aplastic anemia in pediatric and young adult patients.
Haematologica. 101:1343–1350. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang J, Ge M, Lu S, Shi J, Li X, Zhang J,
Wang M, Yu W, Shao Y, Huang Z, et al: Mutations of ASXL1 and TET2
in aplastic anemia. Haematologica. 100:e172–e175. 2015. View Article : Google Scholar :
|
|
38
|
Heuser M, Schlarmann C, Dobbernack V,
Panagiota V, Wiehlmann L, Walter C, Beier F, Ziegler P, Yun H, Kade
S, et al: Genetic characterization of acquired aplastic anemia by
targeted sequencing. Haematologica. 99:e165–e167. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lekka E and Hall J: Noncoding RNAs in
disease. FEBS Lett. 592:2884–2900. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kuang X, Chi J and Wang L: Deregulated
microRNA expression and its pathogenetic implications for
myelodysplastic syndromes. Hematology. 21:593–602. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rhyasen GW and Starczynowski DT:
Deregulation of microRNAs in myelodysplastic syndrome. Leukemia.
26:13–22. 2012. View Article : Google Scholar
|
|
42
|
Dostalova Merkerova M, Krejcik Z, Votavova
H, Belickova M, Vasikova A and Cermak J: Distinctive microRNA
expression profiles in CD34+ bone marrow cells from patients with
myelodysplastic syndrome. Eur J Hum Genet. 19:313–319. 2011.
View Article : Google Scholar :
|
|
43
|
Sokol L, Caceres G, Volinia S, Alder H,
Nuovo GJ, Liu CG, McGraw K, Clark JA, Sigua CA, Chen DT, et al:
Identification of a risk dependent microRNA expression signature in
myelodysplastic syndromes. Br J Haematol. 153:24–32. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Boultwood J, Fidler C, Strickson AJ,
Watkins F, Gama S, Kearney L, Tosi S, Kasprzyk A, Cheng JF, Jaju RJ
and Wainscoat JS: Narrowing and genomic annotation of the commonly
deleted region of the 5q-syndrome. Blood. 99:4638–4641. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pidíkova P, Reis R and Herichova I: miRNA
clusters with down-regulated expression in human colorectal cancer
and their regulation. Int J Mol Sci. 21:46332020. View Article : Google Scholar :
|
|
46
|
Takagi T, Iio A, Nakagawa Y, Naoe T,
Tanigawa N and Akao Y: Decreased expression of microRNA-143 and
-145 in human gastric cancers. Oncology. 77:12–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Starczynowski DT, Kuchenbauer F,
Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra
M, Wells RA, et al: Identification of miR-145 and miR-146a as
mediators of the 5q-syndrome phenotype. Nat Med. 16:49–58. 2010.
View Article : Google Scholar
|
|
48
|
Votavova H, Grmanova M, Dostalova
Merkerova M, Belickova M, Vasikova A, Neuwirtova R and Cermak J:
Differential expression of microRNAs in CD34+ cells of 5q-syndrome.
J Hematol Oncol. 4:12011. View Article : Google Scholar
|
|
49
|
Barreyro L, Chlon TM and Starczynowski DT:
Chronic immune response dysregulation in MDS pathogenesis. Blood.
132:1553–1560. 2018. View Article : Google Scholar :
|
|
50
|
Gañán-Gómez I, Wei Y, Yang H, Pierce S,
Bueso-Ramos C, Calin G, Boyano-Adánez Mdel C and García-Manero G:
Overexpression of miR-125a in myelodysplastic syndrome CD34+ cells
modulates NF-κB activation and enhances erythroid differentiation
arrest. PLoS One. 9:e934042014. View Article : Google Scholar
|
|
51
|
Srivastava J, Chaturvedi CP, Rahman K,
Gupta R, Sharma A, Chandra D, Singh MK, Gupta A, Yadav S and
Nityanand S: Differential expression of miRNAs and their target
genes: Exploring a new perspective of acquired aplastic anemia
pathogenesis. Int J Lab Hematol. 42:501–509. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lu S, Yadav AK and Qiao X: Identification
of potential miRNA-mRNA interaction network in bone marrow T cells
of acquired aplastic anemia. Hematology. 25:168–175. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Adhikari S and Mandal P: Integrated
analysis of global gene and microRNA expression profiling
associated with aplastic anaemia. Life Sci. 228:47–52. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hosokawa K, Kajigaya S, Feng X, Desierto
MJ, Fernandez Ibanez MD, Rios O, Weinstein B, Scheinberg P,
Townsley DM and Young NS: A plasma microRNA signature as a
biomarker for acquired aplastic anemia. Haematologica. 102:69–78.
2017. View Article : Google Scholar
|
|
55
|
Hosokawa K, Muranski P, Feng X, Keyvanfar
K, Townsley DM, Dumitriu B, Chen J, Kajigaya S, Taylor JG, Hourigan
CS, et al: Identification of novel microRNA signatures linked to
acquired aplastic anemia. Haematologica. 100:1534–1545. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sun YX, Li H, Feng Q, Li X, Yu YY, Zhou
LW, Gao Y, Li GS, Ren J, Ma CH, et al: Dysregulated
miR34a/diacylglycerol kinase ζ interaction enhances T-cell
activation in acquired aplastic anemia. Oncotarget. 8:6142–6154.
2017. View Article : Google Scholar
|
|
57
|
Giudice V, Banaszak LG,
Gutierrez-Rodrigues F, Kajigaya S, Panjwani R, Ibanez MDPF, Rios O,
Bleck CK, Stempinski ES, Raffo DQ, et al: Circulating exosomal
microRNAs in acquired aplastic anemia and myelodysplastic
syndromes. Haematologica. 103:1150–1159. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Benetatos L, Hatzimichael E, Dasoula A,
Dranitsaris G, Tsiara S, Syrrou M, Georgiou I and Bourantas KL: CpG
methylation analysis of the MEG3 and SNRPN imprinted genes in acute
myeloid leukemia and myelodysplastic syndromes. Leuk Res.
34:148–153. 2010. View Article : Google Scholar
|
|
59
|
Szikszai K, Krejcik Z, Klema J, Loudova N,
Hrustincova A, Belickova M, Hruba M, Vesela J, Stranecky V, Kundrat
D, et al: LncRNA profiling reveals that the deregulation of H19,
WT1-AS, TCL6, and LEF1-AS1 is associated with higher-risk
myelodysplastic syndrome. Cancers (Basel). 12:27262020. View Article : Google Scholar
|
|
60
|
Yao CY, Chen CH, Huang HH, Hou HA, Lin CC,
Tseng MH, Kao CJ, Lu TP, Chou WC and Tien HF: A 4-lncRNA scoring
system for prognostication of adult myelodysplastic syndromes.
Blood Adv. 1:1505–1516. 2017. View Article : Google Scholar
|
|
61
|
Liu K, Beck D, Thoms JAI, Liu L, Zhao W,
Pimanda JE and Zhou X: Annotating function to differentially
expressed LincRNAs in myelodysplastic syndrome using a
network-based method. Bioinformatics. 33:2622–2630. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wu Z, Gao S, Zhao X, Chen J, Keyvanfar K,
Feng X, Kajigaya S and Young NS: Long noncoding RNAs of single
hematopoietic stem and progenitor cells in healthy and dysplastic
human bone marrow. Haematologica. 104:894–906. 2019. View Article : Google Scholar :
|
|
63
|
Hung SY, Lin CC, Hsu CL, Yao CY, Wang YH,
Tsai CH, Hou HA, Chou WC and Tien HF: The expression levels of long
non-coding RNA KIAA0125 are associated with distinct clinical and
biological features in myelodysplastic syndromes. Br J Haematol.
192:589–598. 2021. View Article : Google Scholar
|
|
64
|
Li N, Ma Y, Wang W, Yin CC, Wu W, Sun R,
Zhao G, Li S and Wang X: LOC101928834, a novel lncRNA in
Wnt/β-catenin signaling pathway, promotes cell proliferation and
predicts poor clinical outcome in myelodysplastic syndromes. Clin
Sci (Lond). 134:1279–1293. 2020. View Article : Google Scholar
|
|
65
|
Congrains-Castillo A, Niemann FS, Santos
Duarte AS and Olalla-Saad ST: LEF1-AS1 long non-coding RNA,
inhibits proliferation in myeloid malignancy. J Cell Mol Med.
23:3021–3025. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang J, Liu X, Hao C, Lu Y, Duan X, Liang
R, Gao G and Zhang T: MEG3 modulates TIGIT expression and CD4 + T
cell activation through absorbing miR-23a. Mol Cell Biochem.
454:67–76. 2019. View Article : Google Scholar
|
|
67
|
Jiang S, Xia M, Yang J, Shao J, Liao X,
Zhu J and Jiang H: Novel insights into a treatment for aplastic
anemia based on the advanced proliferation of bone marrow-derived
mesenchymal stem cells induced by fibroblast growth factor 1. Mol
Med Rep. 12:7877–7882. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lu S, Song X, Chen J and Qiao X:
Identification of differentially expressed lncRNAs and mRNAs in
children with acquired aplastic anemia by RNA sequencing. Biomed
Res Int. 2020:89620902020. View Article : Google Scholar :
|
|
69
|
Risitano AM, Maciejewski JP, Green S,
Plasilova M, Zeng W and Young NS: In-vivo dominant immune responses
in aplastic anaemia: Molecular tracking of putatively pathogenetic
T-cell clones by TCR beta-CDR3 sequencing. Lancet. 364:355–364.
2004. View Article : Google Scholar
|
|
70
|
Risitano AM, Kook H, Zeng W, Chen G, Young
NS and Maciejewski JP: Oligoclonal and polyclonal CD4 and CD8
lymphocytes in aplastic anemia and paroxysmal nocturnal
hemoglobinuria measured by V beta CDR3 spectratyping and flow
cytometry. Blood. 100:178–183. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Melenhorst JJ, Eniafe R, Follmann D,
Nakamura R, Kirby M and Barrett AJ: Molecular and flow cytometric
characterization of the CD4 and CD8 T-cell repertoire in patients
with myelodys- plastic syndrome. Br J Haematol. 119:97–105. 2002.
View Article : Google Scholar
|
|
72
|
Fozza C, Contini S, Galleu A, Simula MP,
Virdis P, Bonfigli S and Longinotti M: Patients with
myelodysplastic syndromes display several T-cell expansions, which
are mostly polyclonal in the CD4(+) subset and oligoclonal in the
CD8(+) subset. Exp Hematol. 37:947–955. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Kochenderfer JN, Kobayashi S, Wieder ED,
Su C and Molldrem JJ: Loss of T-lymphocyte clonal dominance in
patients with myelodysplastic syndrome responsive to
immunosuppression. Blood. 100:3639–3645. 2002. View Article : Google Scholar
|
|
74
|
Li X, Xu F, He Q, Wu L, Zhang Z and Chang
C: Comparison of immunological abnormalities of lymphocytes in bone
marrow in myelodysplastic syndrome (MDS) and aplastic anemia (AA).
Intern Med. 49:1349–1355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Solomou EE, Rezvani K, Mielke S, Malide D,
Keyvanfar K, Visconte V, Kajigaya S, Barrett AJ and Young NS:
Deficient CD4+ CD25+ FOXP3+ T regulatory cells in acquired aplastic
anemia. Blood. 110:1603–1606. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bouchliou I, Miltiades P, Nakou E,
Spanoudakis E, Goutzouvelidis A, Vakalopoulou S, Garypidou V,
Kotoula V, Bourikas G, Tsatalas C and Kotsianidis I: Th17 and
Foxp3(+) T regulatory cell dynamics and distribution in
myelodysplastic syndromes. Clin Immunol. 139:350–359. 2011.
View Article : Google Scholar
|
|
77
|
Sloand EM and Barrett AJ:
Immunosuppression for myelodys- plastic syndrome: How bench to
bedside to bench research led to success. Hematol Oncol Clin North
Am. 24:331–341. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Sloand EM, Mainwaring L, Fuhrer M,
Ramkissoon S, Risitano AM, Keyvanafar K, Lu J, Basu A, Barrett AJ
and Young NS: Preferential suppression of trisomy 8 compared with
normal hematopoietic cell growth by autologous lymphocytes in
patients with trisomy 8 myelodysplastic syndrome. Blood.
106:841–851. 2005. View Article : Google Scholar
|
|
79
|
Sloand EM, Melenhorst JJ, Tucker ZC,
Pfannes L, Brenchley JM, Yong A, Visconte V, Wu C, Gostick E,
Scheinberg P, et al: T-cell immune responses to Wilms tumor 1
protein in myelodysplasia responsive to immunosuppressive therapy.
Blood. 117:2691–2699. 2011. View Article : Google Scholar :
|
|
80
|
Kitagawa M, Saito I, Kuwata T, Yoshida S,
Yamaguchi S, Takahashi M, Tanizawa T, Kamiyama R and Hirokawa K:
Overexpression of tumor necrosis factor (TNF)-alpha and interferon
(IFN)-gamma by bone marrow cells from patients with myelodysplastic
syndromes. Leukemia. 11:2049–2054. 1997. View Article : Google Scholar
|
|
81
|
Allampallam K, Shetty VT and Raza A:
Cytokines and MDS. Cancer Treat Res. 108:93–100. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Stifter G, Heiss S, Gastl G, Tzankov A and
Stauder R: Over-expression of tumor necrosis factor-alpha in bone
marrow biopsies from patients with myelodysplastic syndromes:
Relationship to anemia and prognosis. Eur J Haematol. 75:485–491.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang Z, Li X, Guo J, Xu F, He Q, Zhao Y,
Yang Y, Gu S, Zhang Y, Wu L and Chang C: Interleukin-17 enhances
the production of interferon-γ and tumour necrosis factor-α by bone
marrow T lymphocytes from patients with lower risk myelodysplastic
syndromes. Eur J Haematol. 90:375–384. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Fattizzo B, Serpenti F, Barcellini W and
Caprioli C: Hypoplastic myelodysplastic syndromes: Just an overlap
syndrome? Cancers (Basel). 13:1322021. View Article : Google Scholar
|
|
85
|
Giudice V, Feng X, Lin Z, Hu W, Zhang F,
Qiao W, Ibanez MDPF, Rios O and Young NS: Deep sequencing and flow
cytometric characterization of expanded effector memory
CD8+CD57+ T cells frequently reveals T-cell
receptor Vβ oligoclonality and CDR3 homology in acquired aplastic
anemia. Haematologica. 103:759–769. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
de Latour RP, Visconte V, Takaku T, Wu C,
Erie AJ, Sarcon AK, Desierto MJ, Scheinberg P, Keyvanfar K, Nunez
O, et al: Th17 immune responses contribute to the pathophysiology
of aplastic anemia. Blood. 116:4175–4184. 2010. View Article : Google Scholar :
|
|
87
|
Vibhuti, Tripathy NK and Nityanand S:
Massive apoptosis of bone marrow cells in aplastic anaemia. Br J
Haematol. 117:993–994. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Callera F and Falcão RP: Increased
apoptotic cells in bone marrow biopsies from patients with aplastic
anaemia. Br J Haematol. 98:18–20. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Callera F, Garcia AB and Falcão RP:
Fas-mediated apoptosis with normal expression of bcl-2 and p53 in
lymphocytes from aplastic anaemia. Br J Haematol. 100:698–703.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kordasti S, Costantini B, Seidl T, Perez
Abellan P, Martinez Llordella M, McLornan D, Diggins KE,
Kulasekararaj A, Benfatto C, Feng X, et al: Deep phenotyping of
Tregs identifies an immune signature for idiopathic aplastic anemia
and predicts response to treatment. Blood. 128:1193–1205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Young NS and Maciejewski JP: Genetic and
environmental effects in paroxysmal nocturnal hemoglobinuria: This
little PIG-A goes 'Why? Why? Why?'. J Clin Invest. 106:637–641.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gargiulo L, Papaioannou M, Sica M, Talini
G, Chaidos A, Richichi B, Nikolaev AV, Nativi C, Layton M, de la
Fuente J, et al: Glycosylphosphatidylinositol-specific,
CD1d-restricted T cells in paroxysmal nocturnal hemoglobinuria.
Blood. 121:2753–2761. 2013. View Article : Google Scholar
|
|
93
|
Hanaoka N, Kawaguchi T, Horikawa K,
Nagakura S, Mitsuya H and Nakakuma H: Immunoselection by natural
killer cells of PIGA mutant cells missing stress-inducible ULBP.
Blood. 107:1184–1191. 2006. View Article : Google Scholar
|
|
94
|
Shen W, Clemente MJ, Hosono N, Yoshida K,
Przychodzen B, Yoshizato T, Shiraishi Y, Miyano S, Ogawa S,
Maciejewski JP and Makishima H: Deep sequencing reveals stepwise
mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin
Invest. 124:4529–4538. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sadighi Akha AA: Aging and the immune
system: An overview. J Immunol Methods. 463:21–26. 2018. View Article : Google Scholar
|
|
96
|
Gidvani V, Ramkissoon S, Sloand EM and
Young NS: Cytokine gene polymorphisms in acquired bone marrow
failure. Am J Hematol. 82:721–724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Saunthararajah Y, Nakamura R, Nam JM,
Robyn J, Loberiza F, Maciejewski JP, Simonis T, Molldrem J, Young
NS and Barrett AJ: HLA-DR15 (DR2) is overrepresented in
myelodysplastic syndrome and aplastic anemia and predicts a
response to immunosuppression in myelodysplastic syndrome. Blood.
100:1570–1574. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Maciejewski JP, Follmann D, Nakamura R,
Saunthararajah Y, Rivera CE, Simonis T, Brown KE, Barrett JA and
Young NS: Increased frequency of HLA-DR2 in patients with
paroxysmal nocturnal hemoglobinuria and the PNH/aplastic anemia
syndrome. Blood. 98:3513–3519. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wang H, Chuhjo T, Yasue S, Omine M and
Nakao S: Clinical significance of a minor population of paroxysmal
nocturnal hemoglobinuria-type cells in bone marrow failure
syndrome. Blood. 100:3897–3902. 2002. View Article : Google Scholar
|
|
100
|
Katagiri T, Sato-Otsubo A, Kashiwase K,
Morishima S, Sato Y, Mori Y, Kato M, Sanada M, Morishima Y,
Hosokawa K, et al: Frequent loss of HLA alleles associated with
copy number-neutral 6p LOH in acquired aplastic anemia. Blood.
118:6601–6609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Osumi T, Miharu M, Saji H, Kusunoki Y,
Kojima H, Nakamura J and Shimada H: Nonsense mutation in
HLA-B*40-02 in a case with acquired aplastic anaemia: A possible
origin of clonal escape from autoimmune insult. Br J Haematol.
162:706–707. 2013. View Article : Google Scholar
|
|
102
|
Babushok DV, Duke JL, Xie HM, Stanley N,
Atienza J, Perdigones N, Nicholas P, Ferriola D, Li Y, Huang H, et
al: Somatic HLA mutations expose the role of class I-mediated
autoimmunity in aplastic anemia and its clonal complications. Blood
Adv. 1:1900–1910. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zijlstra M, Bix M, Simister NE, Loring JM,
Raulet DH and Jaenisch R: Beta 2-microglobulin deficient mice lack
CD4-8+ cytolytic T cells. Nature. 344:742–746. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Brümmendorf TH, Maciejewski JP, Mak J,
Young NS and Lansdorp PM: Telomere length in leukocyte
subpopulations of patients with aplastic anemia. Blood. 97:895–900.
2001. View Article : Google Scholar
|
|
105
|
Scheinberg P, Cooper JN, Sloand EM, Wu CO,
Calado RT and Young NS: Association of telomere length of
peripheral blood leukocytes with hematopoietic relapse, malignant
transformation, and survival in severe aplastic anemia. JAMA.
304:1358–1364. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Boultwood J, Fidler C, Kusec R, Rack K,
Elliott PJ, Atoyebi O, Chapman R, Oscier DG and Wainscoat JS:
Telomere length in myelodysplastic syndromes. Am J Hematol.
56:266–271. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Rollison DE, Epling-Burnette PK, Park JY,
Lee JH, Park H, Jonathan K, Cole AL, Painter JS, Guerrier M,
Meléndez-Santiago J, et al: Telomere length in myelodysplastic
syndromes. Leuk Lymphoma. 52:1528–1536. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sanz GF, Sanz MA and Greenberg PL:
Prognostic factors and scoring systems in myelodysplastic
syndromes. Haematologica. 83:358–368. 1998.PubMed/NCBI
|
|
109
|
Bouillon AS, Ferreira MS, Werner B, Hummel
S, Panse JP, Reinecke P, Schemenau J, Haas R, Traulsen A,
Bruemmendorf TH, et al: Comprehensive analysis of telomere biology
in patients with aplastic anemia and hypoplastic myelodysplastic
syndrome: Further evidence for a common mechanism. Blood.
126:28582015. View Article : Google Scholar
|
|
110
|
Yamaguchi H, Calado RT, Ly H, Kajigaya S,
Baerlocher GM, Chanock SJ, Lansdorp PM and Young NS: Mutations in
TERT, the gene for telomerase reverse transcriptase, in aplastic
anemia. N Engl J Med. 352:1413–1424. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ueda Y, Calado RT, Norberg A, Kajigaya S,
Roos G, Hellstrom-Lindberg E and Young NS: A mutation in the H/ACA
box of telomerase RNA component gene (TERC) in a young patient with
myelodysplastic syndrome. BMC Med Genet. 15:682014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Young NS: Current concepts in the
pathophysiology and treatment of aplastic anemia. Hematology Am Soc
Hematol Educ Program. 2013:76–81. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Savage SA, Calado RT, Xin ZT, Ly H, Young
NS and Chanock SJ: Genetic variation in telomeric repeat binding
factors 1 and 2 in aplastic anemia. Exp Hematol. 34:664–671. 2006.
View Article : Google Scholar
|
|
114
|
Marsh JCW, Gutierrez-Rodrigues F, Cooper
J, Jiang J, Gandhi S, Kajigaya S, Feng X, Ibanez MDPF, Donaires FS,
Lopes da Silva JP, et al: Heterozygous RTEL1 variants in bone
marrow failure and myeloid neoplasms. Blood Adv. 2:36–48. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Thol F, Friesen I, Damm F, Yun H,
Weissinger EM, Krauter J, Wagner K, Chaturvedi A, Sharma A,
Wichmann M, et al: Prognostic significance of ASXL1 mutations in
patients with myelodysplastic syndromes. J Clin Oncol.
29:2499–2506. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Jerez A, Clemente MJ, Makishima H, Rajala
H, Gómez-Seguí I, Olson T, McGraw K, Przychodzen B, Kulasekararaj
A, Afable M, et al: STAT3 mutations indicate the presence of
subclinical T-cell clones in a subset of aplastic anemia and myelo-
dysplastic syndrome patients. Blood. 122:2453–2459. 2013.
View Article : Google Scholar :
|
|
117
|
Kuehn HS, Ouyang W, Lo B, Deenick EK,
Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, et
al: Immune dysregulation in human subjects with heterozygous
germline mutations in CTLA4. Science. 345:1623–1627. 2014.
View Article : Google Scholar
|
|
118
|
Wlodarski MW, Collin M and Horwitz MS:
GATA2 deficiency and related myeloid neoplasms. Semin Hematol.
54:81–86. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Ogawa S: Clonal hematopoiesis in acquired
aplastic anemia. Blood. 128:337–347. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
West RR, Stafford DA, White AD, Bowen DT
and Padua RA: Cytogenetic abnormalities in the myelodysplastic
syndromes and occupational or environmental exposure. Blood.
95:2093–2097. 2000. View Article : Google Scholar
|
|
121
|
Negoro E, Nagata Y, Clemente MJ, Hosono N,
Shen W, Nazha A, Yoshizato T, Hirsch C, Przychodzen B, Mahfouz RZ,
et al: Origins of myelodysplastic syndromes after aplastic anemia.
Blood. 130:1953–1957. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Pons A, Nomdedeu B, Navarro A, Gaya A, Gel
B, Diaz T, Valera S, Rozman M, Belkaid M, Montserrat E and Monzo M:
Hematopoiesis-related microRNA expression in myelodysplastic
syndromes. Leuk Lymphoma. 50:1854–1859. 2009. View Article : Google Scholar
|
|
123
|
Krejčík Z, Beličková M, Hruštincová A,
Kléma J, Zemanová Z, Michalová K, Čermák J, Jonášová A and
Dostálová Merkerová M: Aberrant expression of the microRNA cluster
in 14q32 is associated with del(5q) myelodysplastic syndrome and
lenalidomide treatment. Cancer Genet. 208:156–161. 2015. View Article : Google Scholar
|
|
124
|
Starczynowski DT, Kuchenbauer F, Wegrzyn
J, Rouhi A, Petriv O, Hansen CL, Humphries RK and Karsan A:
MicroRNA-146a disrupts hematopoietic differentiation and survival.
Exp Hematol. 39:167–178.e4. 2011. View Article : Google Scholar
|
|
125
|
Chen Y, Zhao G, Li N, Luo Z, Wang X and Gu
J: Role of 4-aminobutyrate aminotransferase (ABAT) and the lncRNA
co-expression network in the development of myelodysplastic
syndrome. Oncol Rep. 42:509–520. 2019.PubMed/NCBI
|
|
126
|
Kordasti S, Marsh J, Al-Khan S, Jiang J,
Smith A, Mohamedali A, Abellan PP, Veen C, Costantini B,
Kulasekararaj AG, et al: Functional characterization of CD4+ T
cells in aplastic anemia. Blood. 119:2033–2043. 2012. View Article : Google Scholar
|
|
127
|
Serio B, Risitano A, Giudice V, Montuori N
and Selleri C: Immunological derangement in hypocellular
myelodysplastic syndromes. Transl Med UniSa. 8:31–42.
2014.PubMed/NCBI
|
|
128
|
Kordasti SY, Afzali B, Lim Z, Ingram W,
Hayden J, Barber L, Matthews K, Chelliah R, Guinn B, Lombardi G, et
al: IL-17-producing CD4(+) T cells, pro-inflammatory cytokines and
apoptosis are increased in low risk myelodysplastic syndrome. Br J
Haematol. 145:64–72. 2009. View Article : Google Scholar
|
|
129
|
Zhang HF, Huang ZD, Wu XR, Li Q and Yu ZF:
Comparison of T lymphocyte subsets in aplastic anemia and
hypoplastic myelodysplastic syndromes. Life Sci. 189:71–75. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Serio B, Selleri C and Maciejewski JP:
Impact of immunogenetic polymorphisms in bone marrow failure
syndromes. Mini Rev Med Chem. 11:544–552. 2011. View Article : Google Scholar : PubMed/NCBI
|