1. Introduction
Tumours are a series of diseases caused by
uncontrolled cell proliferation mainly due to a change in genes. A
tumour often forms in a local site and invades the surrounding
tissues so as to induce metastasis (1). As the most prevalent disease in the
world, it arises under the synergistic and sequential effects of
multiple oncogenic factors such as chemical carcinogens, ionising
radiation, viruses and bacteria, which can induce the
transformation of proto-oncogenes to oncogenes and the inactivation
of tumour-suppressor genes (2,3).
Under the influence of these factors, alterations in apoptosis
regulatory genes and DNA repair genes develop, accompanied by
abnormal expression levels of cellular proteins (4).
According to the cellular characteristics, tumour
morphology, treatment method and degree of harm to the body,
tumours can be divided into: i) solid tumours which can be detected
by clinical examination such as X-ray, CT scan, ultrasound, or
palpation (5,6), and ii) non-solid tumours which are
mainly present in the blood circulation and not visible to the
naked eye or on imaging (7). In
general, non-solid tumours have a wide distribution of tumour cells
in the blood and bone marrow, and thus cannot be removed
surgically, but only by chemotherapy (8). In contrast, the majority of solid
tumours can be treated with a wider range of strategies, such as
surgery, chemotherapy, radiotherapy, immunotherapy, tumour
biotherapy, oncolytic virotherapy, target treatment, hormone
therapy, minimally invasive interventional therapy, microwave
therapy, radiofrequency therapy and cryotherapy (9,10).
Among these treatments, immunotherapy, characterised as having high
specificity, precise targeting capability, powerful antitumour
effects and low side effects, relies on activation of the patient.s
own immune system to kill tumour cells which makes the target
different from other treatments (surgeries, chemotherapy,
radiotherapy and targeted therapies), and shows bright and
unparalleled prospects due to the unusual and miraculous effects
(11).
As the most prominent component of immunotherapy,
monoclonal antibodies (mAbs) are highly homogeneous antibodies
produced by a single B-cell clone and directed only against a
specific antigenic epitope (12).
While it has the advantages of high purity, high sensitivity, high
specificity, low cross-reactivity and low cost of preparation, some
disadvantages also exist, such as its production and preparation
requiring certain technology (13). However, with the optimisation of
preparation techniques and the production of numerous mAb drugs
(such as abciximab and rituximab) over the years, the scope of
application of mAbs has gradually broadened, and they have been
widely used in immune checkpoint therapy, targeted tumour therapy,
radioimmunotherapy and near-infrared photoimmunotherapy (NIR-PIT)
to date, specifically showing great development prospect in tumour
therapy (9,14,15). As mAb research can be applied to
many other areas of technical research, it not only drives the
research process of full human mAb preparation, but also perfectly
demonstrates its unparalleled value in tumour control and treatment
research, under the efforts of countless researchers.
In view of these factors, this review will focus on
the relatively mature techniques for the preparation of mAbs and
the application of mAbs to demonstrate the importance of mAb
research. This review reviewed 242 articles published mainly
between 2005 and 2021, including the PubMed, Excerpt Medica
Database, Medline, OVID and the Cochrane Library databases, by
searching the key word monoclonal antibody, immunotherapy or
tumour.
2. Immunotherapy and antibodies
Tumour immunotherapy is a therapy used to restore
the normal antitumour immune response of the body by restarting and
maintaining the tumour immune cycle for tumour control and
clearance, which has a major impact on the treatment of metastatic
tumours and has altered the standards of care for many types of
tumours (16). As its
indispensable components, antibodies are specific binding
immunoglobulins produced by plasma cells derived from B lymphocytes
or memory cells in response to antigen stimulation by the body.s
immune system (17,18). Its functions refer to combining
with antigens and effectively removing foreign bodies such as
invading microorganisms and parasites (19).
In general, antibodies can be divided into
polyclonal antibodies and mAbs. Polyclonal antibodies are produced
from multiple B cell clones after the body is stimulated by a
variety of antigenic determinants, which can be regarded as a
mixture of multiple mAbs (20).
In contrast, mAbs are the antibodies that can target the particular
antigen determining cluster, characterised as high specificity,
strong binding force, high purity, low cost and mass production
(18). As a kind of highly
specific and homologous antibody, the mAb was first produced by
Köhler and Milstein in 1975 with the use of the hybridoma
technique, which used the HAT culture medium to screen for
hybridoma cells that could grow steadily, recognise a particular
antigenic epitope and produce mAbs (21). In 1982, Levy of the Stanford
Medical Centre in the US prepared a unique mAb against B-cell
lymphoma; the patient.s condition was alleviated and the tumour
disappeared after treatment with this unique anti-body. This was
the first time that mAbs had been used in clinical treatment
(22), and showed promise for
application as targeted therapies for tumours, inflammation, and
cardiovascular, autoimmune and infectious diseases. Due to the
great contribution that hybridoma technology has made to the field
of life sciences, Milstein and Köhler were awarded the Nobel Prize
in Medicine and Physiology in 1984 (14), which indicated the people.s
recognition of mAbs and how optimistic people are about their
prospects to some extent. Soon after this, orthoclone was produced
by Ortho Biotech, which was also named muromomab-CD3. This was
approved by the food and drug administration (FDA) as the first mAb
drug in 1986 and was used to inhibit acute rejection of kidney
transplantation and treat human diseases (23), opening a new era of mAb
therapy.
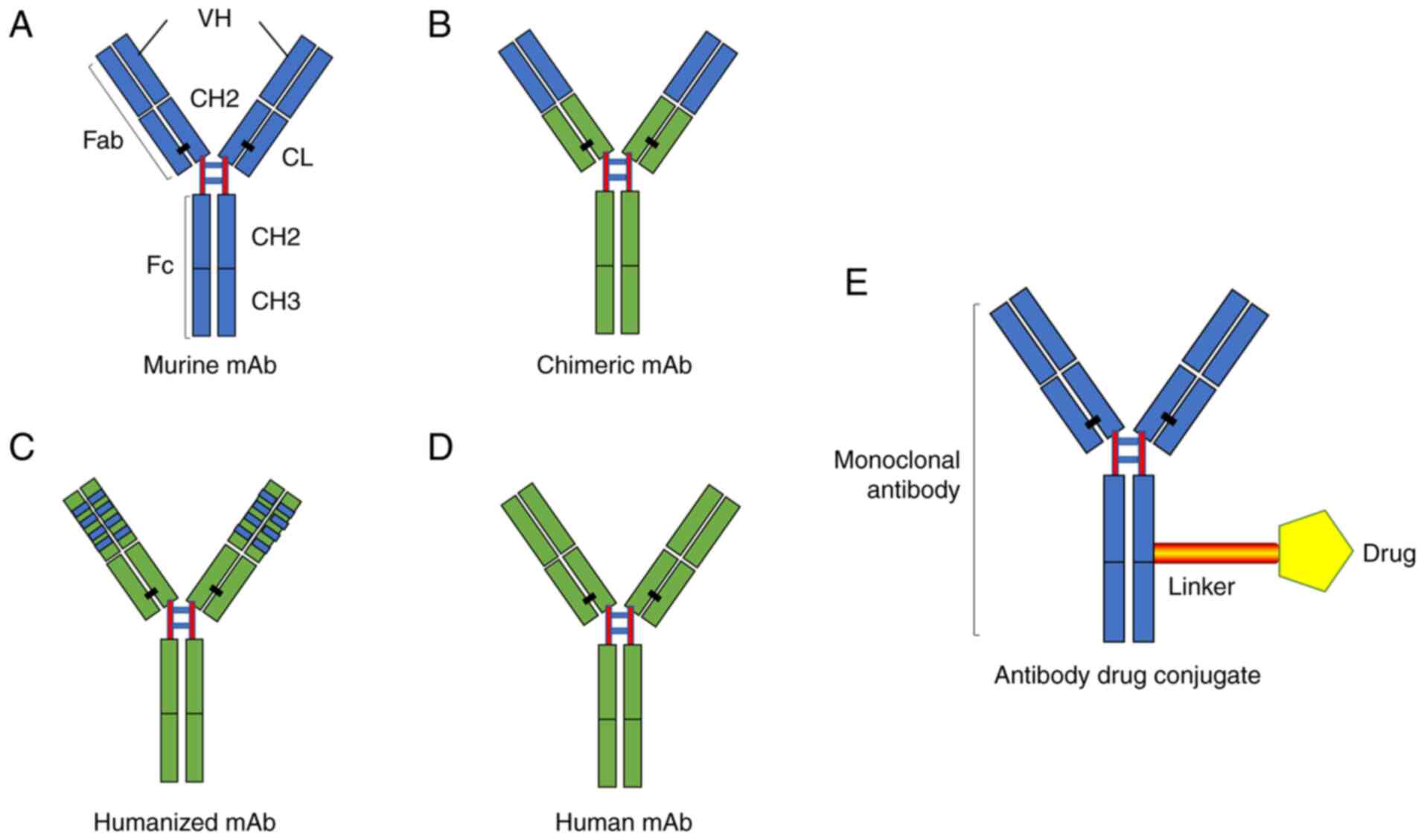
To date, mAbs have undergone different stages of
optimisation and development, including murine mAbs, humanised mAbs
and fully human mAbs (24). The
advent of hybridoma technology has made possible the implementation
of the large-scale preparation of uniform murine mAbs (25). Compared with the polyclonal
antibodies studied in the past, murine mAbs showed a huge
difference in terms of specificity and consistency, as even the
different batches of polyclonal antibodies prepared with the same
antigen cannot guarantee their consistency but perfectly consistent
murine mAbs can be produced continuously once the hybridoma is
successfully prepared (21,26). Nevertheless, the murine mAbs, as
the heterologous protein, may lead to an immune response and the
production of the human anti-murine antibody (HAMA) in vivo
which can in turn clear the murine mAbs, resulting in the emergence
of autoimmune diseases and an ultimate reduction in therapeutic
effectiveness (27,28).
The humanised mAb refers to the murine mAb
reconstructed by gene cloning and DNA recombination (29). The construction forms the constant
parts of mAb (the CH and CL regions) or all parts of the mAb
encoded by human antibody genes, leading to the basic preservation
of the affinity and specificity of the original murine mAb and a
reduction of its heterology (30). As the most widely used mAb, the
advantage of humanised mAbs is that they can overcome the human
anti-murine antibody reaction, preventing the rapid elimination of
mAbs as foreign proteins by the immune system, and improving the
biological activity of mAbs (31).
As the ideal antibody for treatment, fully human
mAbs have the humanised V and C regions (29). With the use of transgenes, the
transchromosome technique or some other technique, all of the genes
encoding human antibodies can be transferred into genetically
engineered animals with their antibody genes deleted, so that the
animals can express human antibodies and achieve the goal of full
humanisation (32). At present,
the human hybridoma technique, the EBV transformation of B
lymphocytes, phage display, the transgenic mouse technique and the
preparation of a single B cell antibody can all be used to produce
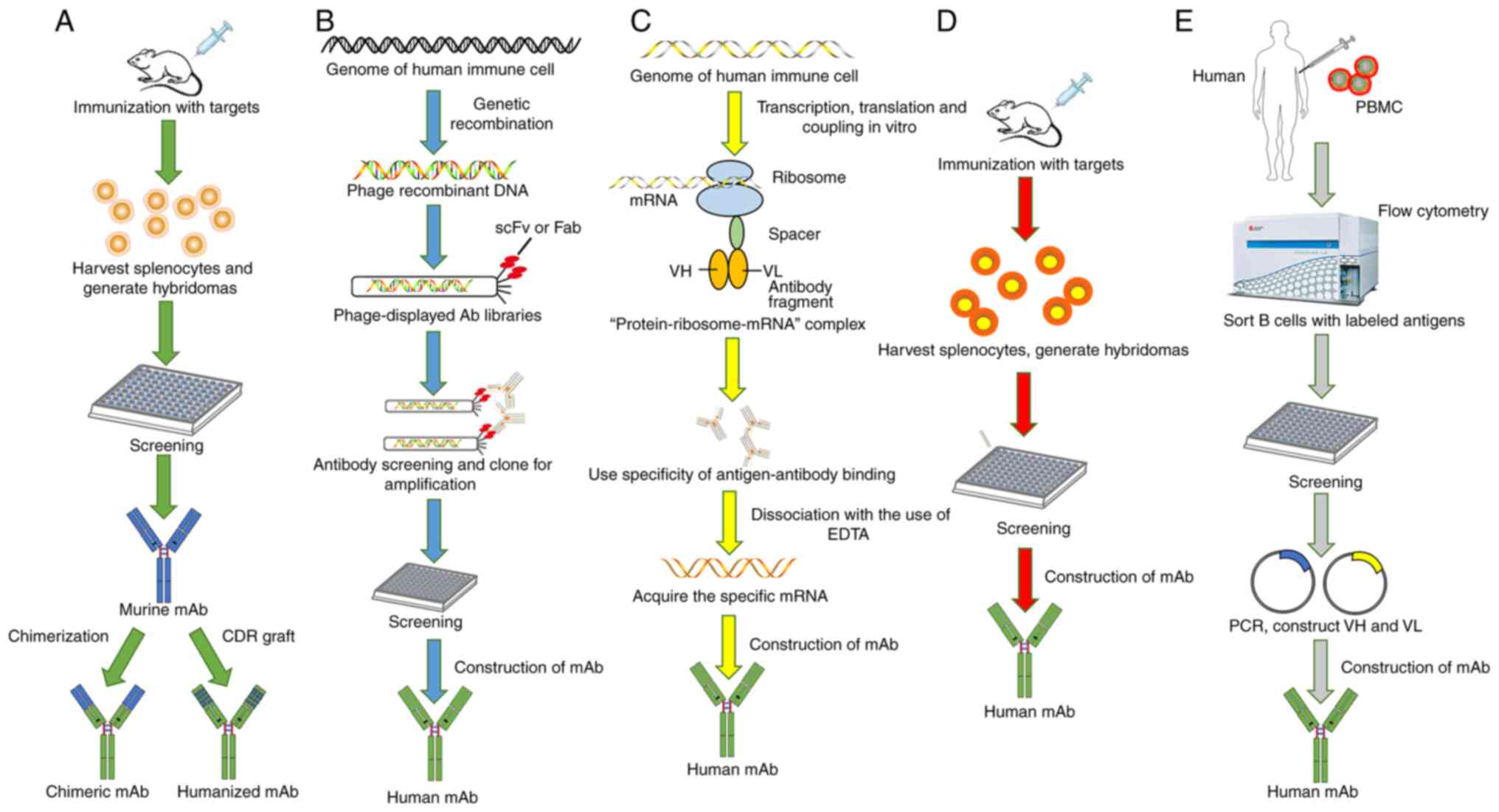
fully human mAbs (33-35) (Fig.
1). Among them, mAbs obtained by transgenic mouse technology
are relatively complete, and those obtained by phage display
technology are generally incomplete (35).
3. Monoclonal antibody preparation
techniques and their applications
To date, many relatively mature mAb preparation
technologies, e.g. the hybridoma technique, the phage display
technique, the transgenic mouse technique, the ribosome display
technique, and single B cell antibody preparation techniques, can
be selected depending on the characteristics of the desired
antibody (Fig. 2).
Hybridoma technique
Development history
As the earliest technology used to produce mAbs, the
hybridoma technique, also known as the lymphocytic hybridoma
technique, is developed from somatic cell fusion technology, which
enables the realisation of the large-scale preparation of the
uniform murine mAbs (26). It was
first invented by Köhler and Milstein in 1975 to produce hybridoma
cells through the fusion of mouse myeloma cells and immunised
animal spleen cells, which have the ability to reproduce endlessly
and secrete highly specific antibodies that can recognise specific
antigens-mAbs (21). As they did
not choose to patent the hybridoma technique, it is allowed to be
used in academia and the pharmaceutical industry, leading to
potential future treatments for a range of diseases including
tumours (36).
Advantages and disadvantages
Although antibodies produced by the hybridoma
technique possess attractive advantages, such as good specificity,
high purity and large-scale production (25), some defects of murine-derived
antibodies remain unavoidable. On the one hand, the low affinity of
murine mAbs to the Fc fragment on the immunocyte surface, can cause
light antibody-dependent cell-mediated cytotoxicity (ADCC),
resulting in a mild killing effect on tumour cells (37). In addition, the killing effect has
a short time in which to take effect because of the short half-life
of murine mAbs in the blood (38).
On the other hand, murine mAbs cause immunogenicity
(39) and can further produce
HAMA (27), which means that the
repeated use of murine mAbs can lead to decreased efficiency and
harm to humans due to allergic reactions (26). Additionally, it is not uncommon
for patients treated with mAbs to produce human anti-murine
immunoglobulin responses, possibly due to the immune deficiency
associated with certain types of tumours (40). Therefore, some early murine mAbs,
such as the E5 murine mAb, not only failed to achieve the desired
effect in the treatment, but increased the mortality of patients,
leading to a period of downturn in the development of mAb
preparation and mAb treatment (41).
Clinical therapeutic applications
Rituximab, as the first lymphoma mAb developed in
1982, was shown to alleviate the condition of tumour patients,
which raises great hope for the use of mAbs in tumour treatment
(42). In addition, the first mAb
drug, anti-cd3 mAb OKT3, was approved by the US FDA to enter the
market in 1986; this mAb can alleviate the anti-rejection reaction
during organ transplantation (23,34). Overall, the mAb drugs produced at
that time were murine mAbs and rabbit mAbs, which have some
clinical drawbacks (26).
Recently, with the advances in technology, human hybridoma
technology has been developed as a new mAb preparation technology
on the basis of mouse hybridoma technology and rabbit hybridoma
technology, which causes the fusion of immunised human B cells and
human myeloma cells to produce hybrid cells that can divide
indefinitely and secrete antibodies (26). However, the technology has
produced a very limited number of multiple myeloma cell lines, has
a low cell fusion success rate and easily causes the loss of
chromosomes (43).
DNA recombinant antibody technique
Development process
Advances in genetic engineering techniques
facilitate the development of chimeric (murine/human) mAbs. With
the use of DNA recombination technology, chimeric genes consisting
of a combination of the Variable (V) region gene of murine
antibodies and the Constant (C) region gene of human antibodies,
inserted into the expression vector containing the C region of
human antibodies, is used to express chimeric mAbs which possess a
humanised C region and heterogeneous V region. The resulting
antibody (44) causes reduced
immunogenicity of the allogenic antibody while retaining the
ability of the parental antibody to specifically bind to the
antigen. However, because there is still some residual
immunogenicity in the FR of V region, HAMA may be induced (45). In view of this, primate antibodies
produced by immunising macaques can be chosen as the heterologous
antibody to chimerism with the C region, because the V region of
primate antibodies show few differences to the V region of human
antigens, thus decreasing the immunogenicity.
Advantages and disadvantages
Compared with murine mAbs prepared by the hybridoma
technique which are limited in clinical application due to their
ability to cause HAMA reactions in vivo, the chimeric mAbs
prepared by DNA recombinant antibody technique can significantly
alleviate adverse reactions because of an approximately 70%
reduction in immunogenicity of the heterologous antibody, thus
improving the curative effect (46). For example, infliximab is capable
of preventing and reducing inflammation as the chimeric mAb to
tumour necrosis factor (TNF)-α in the treatment of rheumatoid
arthritis (RA) and Crohn.s disease (47). In addition, it also has the
effector functions of human antibodies because of the existence of
a humanised Fc fraction, which makes the chimeric mAb possess more
potent complement-dependent cytotoxicity (CDC) and ADCC (48). On the basis of the mechanism, the
rituximab chimeric anti-CD20 mAb was developed to treat relapsed
indolent lymphoma because the cell-surface antigen CD20 is
expressed on more than 90% of B-cell lymphomas and chronic
lymphocytic leukaemias (49).
Abciximab, a Fab fragment of a chimeric mAb, functions as a GP
IIb/IIIa receptor antagonist to significantly decrease the size of
coronary artery aneurysms in children with Kawasaki disease by
promoting vascular remodelling, and decrease the risk to go through
early stent thrombosis in diabetic patients with ST-segment
elevation myocardial infarction (50,51). In addition, basiliximab and
cetuximab, serving as chimeric mAb, can function to prevent early
acute or slow rejection reaction after organ or allogeneic
hematopoietic stem cell transplantation so as to treat acute
graft-vs. -host disease (52,53), and treat different cancers
including recurrent and metastatic head and neck squamous cell
carcinoma and metastatic colorectal cancer (54,55), respectively. However, chimeric
antibodies can partially solve the problem of heterologous protein
rejection, but they may still induce an HAMA reaction, interfere
with antibody efficacy and induce a hypersensitivity reaction due
to the fact that they also contain the murine V region which limits
their clinical application to some extent (56,57).
Phage display
Phage display techniques can clone the
peptide-coding or protein-coding gene fragment into the appropriate
position of the phage shell protein structure (58), so that the foreign
polypeptide/protein and shell protein are expressed in the form of
a fusion of each other and then displayed on the phage surface as
the progeny phage reassembles (59,60). The demonstrated polypeptide or
protein can maintain a relatively independent spatial structure and
biological activity, which is conducive to the recognition and
binding of target molecules, providing a way to screen for
single-chain antibodies with high specificity and binding ability
(61). In theory, as long as
enough of this type of peptide is expressed in the library, one or
more phage can bind to these targets.
Development process
Phage display was pioneered by Greg Winter and his
colleagues (59). In 1985, G.P.
Smith developed phage display technology (62) based on the research of phage
biology and molecular biology, which show unique advantages in
virus infection, including HIV infection and tumour diagnosis and
treatment. Later, in 1987, Geysen et al proposed that short
peptides containing key amino acid residues can mimic the antigenic
determinants of proteins and the interaction between proteins is
achieved by the interaction between local peptides (63). In 1988, Parmley and Smith proposed
the idea that the construction of a random peptide library could
provide insight into the antigen-determining cluster epitopes
recognised by antibodies (64).
Subsequently, Scott and Smith fused random short peptides to the
surface protein PIII of filamentous phage and displayed it on the
surface of the phage, creating the first phage random peptide
library (65). In the same year,
McCafferty et al used phage display technology to screen for
single-chain antibodies to lysozyme bacteria, propelling phage
display technology into an era of widespread application (66). Recently, because of the pioneering
work and application in the phage display of peptides and
antibodies, Professors George P. Smith and Gregory P. Winter both
won a quarter share of the 2018 Nobel Prize in Chemistry (59). Until now, the application scope of
phage display technology has been expanding, and the technology has
also been constantly improving and developing.
Advantages and disadvantages
The emergence of phage display technology has opened
up a simple and fast route for the production of genetically
engineered mAbs, which bypasses the technical difficulty of
hybridomas (35). It clones and
amplifies VH and VL gene fragments in human lymphocyte spectrum by
RT-PCR, and randomly combines gene fragments into expression
vectors, in order to construct a large-capacity human antibody
library (27). In addition, the
phage display can simplify the cloning process and acquire a large
amount of material to produce peptides or proteins because of the
small size of the phage genome and high efficiency of the phage
infection (67,68).
The phage display offers the direct physical link
between a protein and its genetic material, which helps people to
effectively screen the desired cloning again and again, and then
amplify it (67). In the process
of library screening, specific phage clones are enriched
continuously due to their specific affinity for ligands, and
relatively rare clones that can bind ligands can be quickly and
effectively screened out from a large library (58). Therefore, the biggest advantage of
phage display is that once the phage library is established,
specific antibodies against the target antigen can be directly
screened from the library according to the needs within 23 weeks,
which greatly reduces the preparation cycle of mAbs (61). In addition, by specific
construction, the filamentous phage may act as a vector, and
generate a peptide library of phage display that contains hundreds
of millions of unique peptides, which are conducive to their
application in antiviral research (69). However, due to the different
binding properties of antibodies in bacteria and eukaryotic cells,
the applicability of the technology is limited to a certain extent
(70). The processes of the phage
display refer to bacterial transformation, phage packaging, and
even transmembrane secretion processes, which limit the capacity of
the phage display library and their molecular diversity. At
present, the capacity of the phage display library is usually
1011 (27). Also,
limited by the expression system, the antibody library is not large
enough to support the acquisition of some rare antibodies and not
all sequences are well expressed in phages, because the realisation
of some protein functions acquire folding, transportation, membrane
insertion and complexation, resulting in the need for additional
selection pressure during in vivo screening (45). It is difficult to obtain
antibodies that inhibit the growth or function of phages or the
expression host, as the phage display system depends on the
expression of intracellular genes, which may make the diversity of
the library decrease rapidly (71). Also, a phage display library
cannot take on the effective mutation and recombination in
vitro, which in turn limits the genetic diversity of the
molecules in the library (72,73). Nevertheless, these temporary
shortcomings cannot obscure the great potential of its
applications.
Clinical therapeutic applications
Nowadays, with the establishment of more phage
display libraries, the construction of advanced genetic operating
system and the development of more efficient phage display systems,
phage display technology plays an important role in different
fields, especially in protein and antibody-related fields (34). Phage display technology has become
an advantageous tool for detecting the protein spatial structure,
exploring the binding sites between receptors and ligands, and
searching for ligands with high affinity and biological activity
(74). It has had a far-reaching
impact on research into the mutual recognition of protein
molecules, the preparation of phage-functionalised biosensors, the
development of new vaccines and tumour therapy (75,76). In addition, Humira, ramucirumab
and other mAbs developed by phage display technology have been
widely used in clinical practice, especially Humira, which is
widely applied in the treatment of rheumatoid arthritis (RA),
psoriatic arthritis, Crohn.s disease, ankylosing spondylitis and
uveitis (45).
Transgenic animal technology
Development process
Transgenic animal technology is mainly based on the
idea of 'why can.t mice be more like people.. It transfers human
antibody loci into animals including mice, chickens and cows, and
rearranges and re-expresses human antibody V region genes in their
lymphocytes, so that transgenic animals can produce B lymphocytes
which fully express human antibodies; this is essentially the
partial humanisation of animals (77-79). Under the stimulation of antigens,
these lymphocytes can be cloned and differentiated continuously to
form plasma cells that are capable of producing high-affinity human
antibodies (80). In addition,
transgenic animals carrying human DNA fragments have complete
functions, including effective homologous conversion and affinity
maturation, which can produce high affinity human antibodies after
the animals are immunised by any target antigen (37).
As early as 1985, the production of fully human
antibodies using transgenic mice was first proposed by Alt et
al (81). Later, many
difficulties, including the large size of the human Ig loci, were
followed but overcome one after another (82). In 1996 and 1997, Medarex and
Abgenix successfully established the HuMab-Mouse®
(Medarex), which significantly improved the efficiency of full
human mAb production (83) and
XenoMouse™ (Abgenix) (84,85).
Advantages, disadvantages and clinical
therapeutic applications
In the past three decades, transgenic animal
technology through genetic engineering has been envisaged to
improve food quality, animal production and the production of
biological products, to reduce or minimise the environmental impact
of animal production and to add value to animal products. Recently,
with the advance of the ability for targeted genome engineering via
genome editing methods such as TALENs, ZFN and the CRISPR/Cas9
system, this technique has been widely used to obtain a series of
human mAbs against the interleukin-6 (IL-6) receptor, TNF receptor
and epidermal growth factor receptor (EGFR), which play important
roles in the treatment of tumours and other diseases (86,87). mAbs against the human IL-6
receptor can show strong antitumour activity in vivo against
multiple myeloma cells by inhibiting IL-6 functions (87). H-R3, as a humanised anti-EGFR
antibody with antitumour, anti-proliferative, anti-angiogenic and
pro-apoptotic properties, can act as an effective EGFR antagonist
to inhibit signal transduction, in order to directly or indirectly
affect cell proliferation, cell survival and angiogenesis-inducing
capacity (88). As an important
milestone in validating XenoMouse strains as well as other human
immunoglobulin-producing mouse technologies, the first fully human
mAb, panitumumab, which was developed from XenoMouse technology and
approved by a regulatory agency, has a positive risk-benefit
profile in advanced, chemotherapy refractory colorectal tumours and
has the potential to increase treatment rates of this disease in
earlier lines of therapy (89).
Recently, the first transgenic rabbit strain for human antibody
production has been created with the discovery that the anti-body
diversification mechanism at work in rabbits can act on the
fragments of the human transgenic immunoglobulin gene (90), which further expands the
application of human mAbs in drug development and promises to lead
to new treatments for various diseases.
Ribosome display technology
Ribosome display technology is a powerful tool for
protein screening using functional protein interactions in
vitro (91). By associating
genotypes with protein phenotypes, it can use specific ligands of
target proteins to select target proteins and corresponding gene
sequences from the protein display library (92). It combines the correctly folded
protein and its mRNA on the ribosome at the same time to form
mRNA-ribosome-protein trimer, in order to screen some high-affinity
proteins with specific binding to target molecules, including
antibodies, peptides and enzymes (93,94). The preparative technique involves
different key processes, including specific processing and
modification of the DNA that encodes proteins, transcription and
translation, affinity screening in vitro, the separation of
mRNA and molecular orientation evolution in vitro (95).
Development process
The ribosomal display technology has undergone a
certain period of research from the time it was proposed to the
time it was developed and matured. In 1994, Mattheakis et al
of the Afflymax Institute in the US put the ideas of their
predecessors into practice for the first time and established the
prototype of ribosome display technology, which mainly used
'polypeptide-polyribosome-mRNA. complex to construct peptide
libraries on polyribosomes, thus screening the polypeptide ligands
of immobilized mAbs with an affinity constant of 109
(Nmol level) from a peptide library with a capacity of
1012 (96). Later,
Hanes and Plückthun improved the polyribosome display technology
and established a new technology, ribosome display technology, in
1997, for the screening of complete functional proteins such as
antibodies in vitro, on the basis of previous research
results (93).
Advantages and disadvantages
Traditional screening techniques have insurmountable
drawbacks, mainly related to cell transfection, phage packaging,
transmembrane secretion and protein degradation in library
construction and screening (97,98). The library capacity and molecular
diversity of phage or mRNA display technology are somewhat limited,
which reduces the efficiency of library screening (71). In contrast, a ribosome display is
a powerful way to screen large libraries and acquire molecular
evolution (99,100). It has the advantages of simple
library construction, large library capacity, strong molecular
diversity, simple screening methods and no need for selection
pressure, and can even improve the affinity of target proteins by
introducing mutation and recombination technology (93,94). As a system to produce and screen
folded proteins entirely in vitro, the ribosome display
technique is shown to greatly exploit replicability of mRNA,
allowing efficient enrichment of target genes, avoiding the step of
bacterial transformation and making the technique unconstrained by
the efficiency of cellular transformation (101). On the one hand, the technique
greatly increases library capacity and screening throughput, and
makes it easy to build a very large volume of antibody library
(101). On the other hand, while
the expressed proteins have the correct spatial fold conformation,
the technique can be combined with some special PCR techniques to
improve the protein expression diversity (28,102,103). It also can be used for the
screening and research of cytotoxic fractions (94).
However, there are still some technical problems
that need to be further advanced. Undoubtedly, maintaining mRNA
stability and preventing the degradation of mRNA is the first
problem in a ribosome display system (28). Facing the problem, Yamaguchi et
al reported a novel screening method-cDNA display, which
prevents the degradation of mRNA by promoting the binding of mRNA
to linkers and the reverse transcriptional synthesis of cDNA, thus
converting mRNA-protein fusions to cDNA-protein fusions and
avoiding problems due to the stability of mRNA (104). The use of modified nucleotides
as substrates for transcription reactions can also stabilise mRNA
(105). In addition, how to
construct the more stable 'mRNA-ribosome-protein. complex was one
of the problems (106), as it
only occurs in cases where the complex is complicated but its
stability is poor in practice due to ribosomal display. To solve
the problem, Roberts and Szostak developed a simpler and more
effective display system-mRNA display system, which allows mRNA to
bind to its encoded polypeptide in the presence of puromycin to
form a stable mRNA-peptide complex that screens for the target
peptide (107). The anti-small
stable RNA A (anti-SsrA) oligonucleotides were designed by Muranaka
et al to inhibit the function of SsrA and obviously promote
the form of the 'mRNA-ribosome-protein. complex (108). In addition, how to improve the
display of large molecular protein in the ribosome is also a
problem that needs to be solved.
Clinical therapeutic applications
At present, there are numerous reports on the
preparation of human mAbs by ribosomal display technology, and the
advantages of this technology represent the developmental direction
of mAbs (28). On the one hand,
it can be applied widely in antibody engineering, proteomics,
epitope mapping, and synthetic enzymes (93,103). On the other hand, it also opens
up a new way to screen new therapeutic antibodies and new drugs for
diagnosis and treatment in tumours, autoimmune diseases, infectious
diseases, and inflammatory disorders (94). Ribosome display technology, as a
new cloning display technology, will show a more extensive
application space in protein interaction research, new drug
development and proteomics (95,109).
A single B-cell monoclonal antibody
generation technology
Development process
As early as 2003, Wardemann et al prepared
autoreactive antibodies with the use of early human B-cell
precursors isolated from the bone marrow, to examine the structure,
development and silencing of autoreactive B cells (110). In 2004, Traggiai et al
immortalised the isolated human memory B cell with EBV and screened
35 mAbs that were well neutralised against influenza virus, which
makes it possible for memory B cells to produce mAbs (111). Since then, monoclonal B-cell
technology for generating mAbs has been gradually applied to
various experimental research and has made great contributions to
the development of many fields of life science as an important tool
for modern life science research.
Advantages and disadvantages
In recent years, single B-cell antibody preparation
techniques have begun to spring up and have gradually become widely
used, alongside the development of molecular cellular biology
(112). This is because mAbs
prepared by single B-cell antibody technology have the
characteristics of full human origin, high specificity and
uniformity, showing unique advantages and good application
prospects in the treatment of pathogenic microbial infections,
tumours, autoimmune diseases and organ transplantation (113,114).
Compared with other mAb preparation techniques,
monoclonal B-cell technology, is a technique for the cloning and
expression of B-cell antibodies with single antigenic specificity
in vitro, which preserves the natural pairing in the V
region of the light and heavy chain, and has the advantages of good
gene diversity, high efficiency, full humanisation and the small
number of cells required (115).
Studies have shown that human memory B cells can survive in humans
for more than 50 years, providing a historical record of the
specific antibodies produced during most of the host.s lifetime
(116). In contrast, antibodies
in the body fluids used in most traditional methods usually decay
after macroglobulin clearance, which means that people lose their
protective antibody within a few years (117). In addition, it is proposed that
memory B cells in the blood of virus-infected patients may store
records of early infection with the virus in patients-the genes,
which provide a new direction for the research and development of
mAbs (118).
Therapeutic application in clinical
Currently, the preparation of memory B-cell
antibodies has become a popular method used to prepare the
humanised antibody, which also promotes immunological research
including antibody affinity maturation, the defence mechanism
against vaccine immunity, vaccine development, and the treatment of
tumours and autoimmune diseases at the same time (1,119). With the maturity and improvement
of B-cell sorting technology, subsequent PCR gene amplification
methods and the high-throughput analysis and identification of
antibody genes, memory B-cell antibody preparation technology will
play an unprecedented important role in the diagnosis,
pharmacodynamic and clinical application in the future, leading to
a new era of therapeutic antibody research (120-122). As this approach has also been
successful in widely isolating neutralizing antibodies against
viruses including SARS and H5N1 influenza, it can provide not only
neutralizing antibodies for passive serum therapy, but also
information for vaccine design, and is expected to accelerate the
development of therapeutics in the field of infectious diseases
(119,123). Additionally, Wrammert et
al produced anti-H1N1 antibodies in 2009 by isolating plasma
cells from peripheral blood, to analyse the characteristics of
antibodies in detail generated from plasma blasts induced by
pandemic H1N1 infection (124).
4. Application of monoclonal antibodies in
tumour therapy
Mechanisms of monoclonal antibodies as
antitumour drugs
Traditionally, mAbs produce cytotoxic effects in
tumour cells though ADCC, CDC, changing signal transduction,
elimination of the cell-surface antigen, and targeted conveying
payloads (125).
ADCC
In general, ADCC is achieved by the specific binding
of mAbs and the targeted antigen of tumour cells (126). Namely, the Fc fraction of mAbs
can bind to the receptor of immune effector cells (NK cells,
macrophages, neutrophils, granulocytes), and achieve activation of
intracellular signals in the next moment, resulting in ADCC
(126,127). NK cells activated by antibodies
can release cellular cytotoxic granules (perforin and granzyme) to
achieve cell apoptosis on the one hand, while they can release
cytokines and chemokines to inhibit cell proliferation and
angiogenesis on the other (128).
CDC
CDC refers to the cytotoxic effect involving
complement; that is, after the binding of specific mAbs and the
corresponding antigens on the surface of the cell membrane, the
complex activates the classical pathway of complement and forms an
attack membrane complex to induce a lysis effect on target cells
(127,129). It is worth noting that, although
CDC does not directly preside over the antitumour effects of most
mAbs, it produces a variety of factors that enhance ADCC (130,131).
Changing signal transduction
Almost every clinically effective unconjugated mAb,
directly or indirectly, interferes with the signal transduction
that influences the proliferation and survival of targeted cell
populations (132). Growth
factor receptors are some of the most commonly targeted
tumour-associated antigens whose activation under normal conditions
induces mitotic reactions and promotes cell survival, are
overexpressed in numerous malignancies, leading to promotion of
tumour cell growth and insensitivity to chemotherapy drugs
(133-135). Therefore, the use of mAbs is
likely to normalise the cell growth rate and restore the
sensitivity of cells to cytotoxic drugs by reducing the signal that
passes through these receptors (136). For example, the pertuzumab block
shows receptor heterodimerisation (the dimerisation of HER2 with
HER3 and other HER family receptors) that is required for signal
transduction to play an antitumour role (130,137).
Application of monoclonal antibodies in
molecular-targeted therapy
Targeted antitumour drugs provide a new concept
concerning tumour therapy, referring to several tumour-associated
signaling pathways and targets. For instance, the most common
target antigens in solid tumours refer to epithelial cell adhesion
molecule (Ep-CAM; also known as epithelial glycoprotein-2, EGP-2/GA
733-2), carcinoembryonic antigen (CEA), EGFR family including EGFR
(also known as c-erbB-1), HER2/neu (c-erbB-2), HER3 (c-erbB-3) and
HER4 (c-erbB-4) (135,138,139). Compared with them, the mAbs
applied in lymphoma usually target CD52, CD20, CD30, CD22, CD37 and
CD79 (135,136), with the easier achievement of a
better effect because it is simpler to manage tumour penetration.
In contrast to the above, tumour stroma and tumour vasculature
offer some unique targets for antibody-based interaction because
the new generation of tissue and vasculature show some components
that differ from those in the normal situation, leading to the
situation whereby fibroblast activation protein (FAP) (140) and tenascin-C (TNC) (141) are regarded as targets in the
tumour stroma, Fibronectin ED-B (142) and prostate-specific membrane
antigen (PSMA) (143) are
regarded as targets in the tumour vasculature. In addition, ligands
including vascular endothelial growth factor (VEGF) are thought to
target cell-surface receptors expressed on tumour cells or their
supporting tissue (140,144).
Application of monoclonal antibodies in
immune checkpoint therapy (ICI)
The fight between the immune system and tumour
cells is a long-term dynamic process, which has both positive and
mutual influences (145). In the
process whereby a healthy individual.s immune cells detect and kill
tumour cells via the antitumour immune response, activated T cells
cause upregulated expression of several surface receptors, which
can bind with relevant ligands expressed highly on the surface of
tumour cells, resulting in inhibition of the immune response and
downregulation of potent immune response (146). These surface receptors, namely
the suppressive regulatory molecules, are essential for maintaining
self-tolerance, preventing autoimmune response and minimizing
tissue damage by regulating the time and intensity of the immune
response; this is called the immune checkpoint (146,147). The immune checkpoint results in
the inhibition of cellular function, meaning that the body cannot
produce an effective antitumour immune response, ultimately leading
to immune escape of the tumour (148).
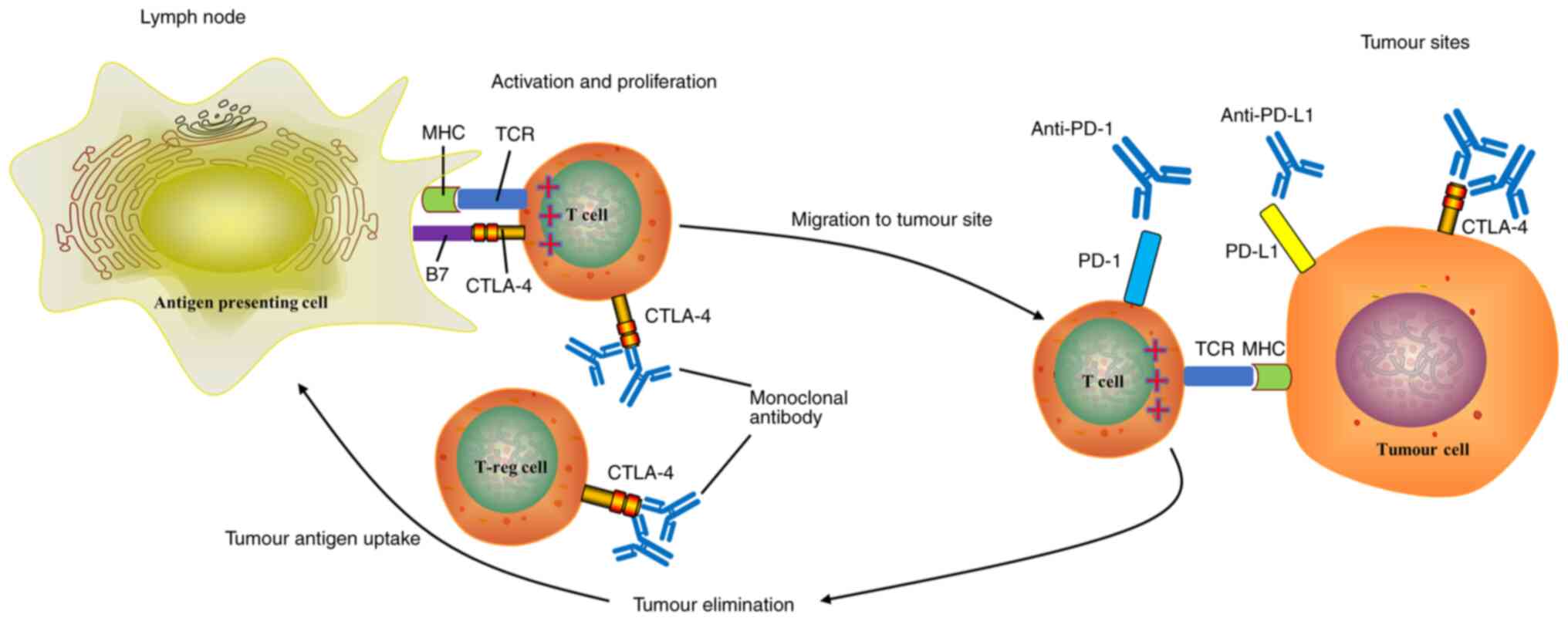
On the theoretical basis of the immune checkpoint,
some mAbs have been developed as immune checkpoint inhibitors to
block the interaction between tumour cells expressing immune
checkpoints and immune cells, in order to block the inhibitory
effect of tumour cells on immune cells (148,149). During the occurrence and
development of tumours, immune checkpoint inhibitors can enhance
the immune function of the body and restore the recognition ability
of T cells, in order to eliminate tumours or slow down the
development of tumours (150,151) (Fig. 3). Recently, tumour-related immune
checkpoint molecules mainly include programmed death-1 (PD-1),
cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), T-cell
immunoglobulin mucin 3 (TIM3) and lymphocyte activation gene-3
(LAG3) (145,146,150).
Anti-CTLA-4
CTLA-4 (also known as CD152) is a transmembrane
protein expressed on the surface of activated CD4+ T
cells, activated CD8+ T cells, and regulatory T cells
(Treg cells), which can bind to the CD86(B7-2) and CD80(B7-1)
ligand to negatively regulate T-cell activation (152). In addition, there is an
intracellular pool of CTLA-4 in recycling endosomes of Treg and
memory T cells which can be rapidly cycled to the cell surface upon
activation (153). Therefore,
the anti-CTLA-4 mAb can decrease Treg cells and activate T cell
immune response by terminating the activity of CTLA-4 (145). Ipilimumab is an mAb that blocks
CTLA-4, which was approved by the FDA to treat melanoma in 2011
(150,154). The blocking effect of ipilimumab
on CTLA-4 is, that Ipilimumab binds to CTLA-4 to further impede the
interaction between CTLA-4 receptor and the B7 ligand, thus
increasing the activation and proliferation of T cells (145). This means that the antitumour
effect of ipilimumab on melanoma is indirect and the action
mechanism may be to help the body.s immune system recognize, target
and attack melanoma cells (155). Hence, it is possible for
ipilimumab to be used as a carrier of imaging agent with good
targeting properties (156). In
addition, it is reported that patients with human CTLA-4
insufficiency develop immune dysregulation, lymphadenopathy and
hepatosplenomegaly (153,157,158).
Therefore, anti-CTLA-4 mAbs should be able to inhibit the
inhibitory signalling pathway and maintain the killing function of
T cells, thereby killing tumour cells (146).
Although some anti-CTLA-4 mAbs do not cause an
adverse reaction similar to cytotoxic drugs, including
myelosuppression and alopecia, due to the different actions of
cytotoxic drugs, they cause pathological damage to the body while
producing an antitumour reaction, namely immune-related adverse
events (IRAEs) (146,149). Different from others, the
toxicity profile of ipilimumab mainly manifests as symptoms
associated with infusion, including rigor, pruritus, fatigue,
nausea, dizziness, colitis, less frequently hypophysitis, hepatitis
pneumonitis, and even hypotension, angioneurotic oedema, and
dyspnoea (159-161). When facing the problems that
occur in anti-CTLA-4 immunotherapy, combination therapy is put
forward which may thereby provide greater antitumour activity than
either agent alone by enhancing antitumour immune responses, and
presenting a miraculous, unprecedented therapeutic effect (159). For example, in the treatment of
progressive melanoma, the combined blocking of PD-1/PD-L1 and
CTLA-4 can further improve efficacy in patients compared with the
single blocking of PD-1/PD-L1 or CTLA-4 (162). Based on this, a trial involving
patients with advanced melanoma was carried out and revealed that
nivolumab plus ipilimumab provides the longer progression-free and
overall survival, and better health-related quality of life than
ipilimumab alone (159,163). From another perspective, CTLA-4
inhibition can synergize with local chemotherapy, improving
applicability and sensitivity to immune-checkpoint inhibition
(164).
At present, not only ipilimumab, but also some
other anti-CTLA-4 mAbs, have been optimised and developed, and have
now attracted more and more attention from the public (148). Another anti-CTLA-4 mAb,
tremelimumab, which was exploited by Pfizer, is being investigated
in clinical trials (165).
However, the research into tremelimumab has made little progress,
meaning that ipilimumab is still regarded as the most promising
anti-CTLA-4 mAb applied in tumour treatment (148).
Anti-PD-1 and anti-PD-L1
PD-1 (also known as CD279), as a member of the
immunoglobulin superfamily which is mainly expressed on the surface
of activated T cells, can be used as an immunosuppressive molecule
to regulate the immune system and promote self-tolerance by
downregulating the response of the immune system to human cells and
suppressing the inflammatory activity of T cells, which may prevent
the immune system from killing tumour cells (166,167). PD-1 has at least two ligands,
PD-L1 (also known as CD274 or B7-H1) and PD-L2 (CD273 or B7-DC)
(146). In general, as one of
the means by which human tissue protects them, PD-1 can bind with
specific ligands on the immunocyte surface to prevent immune cells
from activating and killing normal cells (166,168). Additionally, the binding of PD-1
with ligands promotes the programmed death of T cells and reduces
the apoptosis of regulatory T cells by suppressing the T cell
activation signal primed by the interaction between MHC and TCR, in
order to make the tumour cells acquire immune escape (146). Certain types of malignant
tumours express a mass of PD-1 on the cell surface; therefore, they
evade the attack of immune cells by powerfully suppressing the
activation of immune cells (129). PD-1 is also expressed on the
surface of activated B cells and macrophages, indicating that PD-1
negatively regulates the immune response more widely than CTLA-4
(145). Therefore, immune
regulation targeting of PD-1 plays an important role in antitumour,
anti-infection, anti-autoimmune diseases and organ transplantation
survival (169). Facing the
situation, anti-PD-1 mAbs are manufactured as PD-1 inhibitors to
activate the immune system to attack tumours and treat some types
(145).
Compared with anti-CTLA-4 mAb therapy,
immunotherapy with anti-PD-1 mAbs has a broader antitumour effect
and fewer overall side effects (170,171). The biggest difference between
immune drugs and others is that it shows more persistent efficacy
which could lead to long-term survival or even a clinical cure for
patients with advanced disease (146). Currently, there are various
drugs approved by the FDA, including nivolumab (also known as
Opdivo) and pembrolizumab (also known as Keytruda) (172). The indications of nivolumab
include melanoma, non-small cell lung cancer (NSCLC), renal cell
carcinoma (RCC), classical Hodgkin.s lymphoma (CHL), squamous cell
carcinoma of the head and neck (SCCHN), and urothelial carcinoma
(148). In contrast,
pembrolizumab is mainly applied in melanoma, NSCLC, and SCCHN
(167,172).
At present, the anti-PD-1 mAbs are mostly used in
combination therapies, because traditional therapy using anti-PD-1
mAbs alone can lead to various IRAEs, including fatigue, skin rash,
colitis, hypophysitis, pneumonitis, myasthenia gravis and
interstitial nephritis (171).
For tumours that are highly dependent on the immunosuppressive
mechanism of PD-1/PD-L1, such as malignant melanoma, Hodgkin.s
lymphoma, certain types of lung tumour, and colon tumours, it has
shown remarkable efficacy (147), but for the vast majority of
unselected solid tumours, the efficacy of PD-1 inhibitor alone is
not high. In comparison, combination therapy improves the treatment
effect and transforms patients who are not suitable for PD-1
inhibitor treatment into those who can benefit from it (173). First, the anti-PD-1 mAb can
combine with another immunotherapy drug (174). A combination of the PD-1 and
CTLA-4 mAbs has been shown to be more effective in treating several
tumours, including malignant melanoma, than either antibody alone
(162). In addition, PD-1 can
combine with anti-CTLA-4 or VEGF tyrosine kinase inhibitors (TKIs)
to adjust first-line therapy for metastatic kidney carcinoma
(175). Secondly, PD-1 in
combination with chemotherapy is considered a promising treatment
strategy (176). Chemotherapy
has a profound impact on the antitumour immune by directly
regulating immune cellular subsets or indirectly stimulating the
immune system through the induction of immunogenic cell death,
revealing the existence of synergy between cytotoxic chemotherapy
and immune checkpoint inhibition (176). This combination therapy is also
approved for the first-line treatment of advanced NSCLC (176). Nevertheless, there are some
patients with chemotherapy-refractory metastatic solid tumours; for
those patients, PD-1 inhibitor combined with radiotherapy is
regarded as a salvage treatment (177,178). This means that the anti-PD-1 may
combine with radiotherapy to improve the overall survival (179). In addition, anti-PD-1 can
combine with targeted drugs including acitinib, and levatinib
(180,181). Recent studies have shown that
the association of acitinib with pembrolizumab provides improved
clinical benefit in patients with previously untreated advanced
renal cell carcinoma, which is well tolerated (182). In addition, the strategy of
using anti-PD-1 mAbs in combination with oncolytic virus (OV) to
enhance antitumour immunity and therapy has been developed
(183). To validate this,
research into the effect of IL-15-armed OV in combination with PD-1
inhibitors in mice with colon or ovarian carcinoma processes, has
shown some results including tumour regression and the prolongation
of overall survival (184).
Moreover, a personalised mutanome vaccine can be used in
combination therapy with anti-PD-1 mAb as it enhances the
persistence of anti-PD-1-mediated effect and extends anti-PD-1
therapies to patients with no preexisting T cell response (185). Finally, the anti-PD-1 mAb can
combine with novel tumour-specific immune cells, such as the
chimeric antigen receptor T-cell (CAR-T), to produce better
therapeutic effects (186). Some
research has found that PD-1 blocks CAR-T cell therapy within solid
tumours, therefore the anti-PD-1 mAb which prevents the
PD-1-related inhibition of CAR-T cell response can increase the
levels of cytolysis and cytokine secretion and enhance the in
vivo antitumour function of CAR-T cells (187,188).
PD-L1, expressed on the surface of tumour cells,
can bind with PD-1 on the surface of activated T cells and B cells
to conduct inhibitory signals and reduce T cell proliferation
(146,189). As promising new agents, there
are some anti-PD-L1 mAbs approved by the FDA in clinic, including
atezolizumab (also known as Tecentriq) which is used to treat
locally advanced or metastatic urothelial carcinoma, durvalumab
(also known as Imfinzi) which is used to treat locally advanced or
metastatic urothelial carcinoma and NSCLC, and avelumab (also known
as Bavencio) which is used to treat meningioma, metastatic Merkel
cell carcinoma and carcinoma of the urinary bladder (128,167,190). In theory, compared with
anti-PD-1 mAbs which bind to PD-L2, anti-PD-L1 mAbs have specific
effects and demonstrate a certain superiority (146). The anti-PD-L1 mAbs can block the
co-suppression of B7-1 and PD-1, which is conducive to fully
activate the function of T cells and produce cytokines (191,192). Therefore, anti-PD-L1 mAbs may
more fully activate the immune system to kill tumours (191). Furthermore, it has been shown
that durvalumab as a third-line or later treatment can
significantly benefit advanced NSCLC patients with EGFR mutations
or ALK rearrangements (EGFR+/ALK+) with ≥25%
of tumour cells expressing PD-L1, although it is unsuitable for
patients with EGFR+/ALK+ to use the immune
checkpoint inhibitor because of the low curative effect and
subsequent severe adverse reactions (193). In summary, anti-PD-1 mAbs and
anti-PD-L1 mAbs each have their own indications and application
scope, and the combined utilisation can achieve mutual
complementarity in the interest of our common development (146).
Others
In addition, there are various other useful
checkpoints, such as the lymphocyte activation gene-3 (LAG-3),
B7-H3, B7-H4, T cell immunoglobulin-3 (TIM-3), T cell
immunoglobulin and ITIM domain protein (TIGIT), and V-domain
immunoglobulin-containing suppressor of T cell activation (VISTA)
(194-197), which are either entering the
clinic or under active development.
Potential combined therapeutic
strategies
Radioimmunotherapy,
chemo-immunoconjugate and immunotoxin
In tumour-guided therapy, mAbs against tumour
antigens are used in the guidance of chemotherapy drugs or
radiotherapy drugs to the target organ, thereby directly killing
the tumour cells or producing antibody-directed enzyme prodrug
therapy (ADEPT) by specifically activating prodrugs within the
tumour (198).
Radioimmunoconjugates
Radiation can act directly on DNA molecules and
cause their damage, by ionising water molecules in living organisms
to produce free radicals which break macromolecules and lead to
cell damage (199). Based on
this theory, mAbs with specific affinity for tumours can be
utilised as a carrier of highly active radiopharmaceutical agents,
thus forming radioimmunoconjugates that target tumour tissue to
kill tumour cells or inhibit their growth while reducing radiation
damage to normal tissue, using the ionising radiation effects of
radioisotopes (11).
Chemo-immunoconjugate
The cytotoxic agent can conjugate with antitumour
mAbs to form chemo-immunoconjugate, which can bind to the surface
of antigen-positive tumour cells through the guidance of mAbs,
inducing the internalisation of the conjugates (200). After that, these chemical drugs
play their cytotoxic effects by binding to DNA molecules, thus
killing tumour cells by inhibiting cell DNA and protein synthesis,
interfering with cell nucleic acid or protein function, and
inhibiting mitosis (201,202).
Common conjugates include cisplatin, cyclophosphamide, etoposide,
adriamycin, paclitaxel, methotrexate, and vinblastine (203,204).
Immunotoxin
Immunotoxin has a specific affinity for tumour cell
surface antigens and can release bacterial or plant protein toxins
to tumour cells without harming normal cells (205). Once the toxin enters the cell,
it kills the tumour cell by inhibiting protein synthesis and
altering signalling transmission (206). The main toxins currently used in
reagents are diphtheria toxin, abrin, ricin, gelonin, and
Pseudomonas aeruginosa endotoxin (198,207,208).
Antibody-drug conjugates (ADCs)
Antibody-based immunotherapy has been a major and
rational therapeutic strategy in the clinical management of
oncology (209,210). In clinical practice, therapeutic
mAbs have a limited effectiveness in the treatment of solid tumours
due to their large molecular weight, but a high degree of targeting
(198). With a few exceptions
such as mAbs to HER2, EGFR and CD20, most mAbs can bind with
effector molecules by using specific linkers to produce
antibody-drug conjugates (ADCs), which expand the scope of medical
treatment while possessing highly targeted selection, achieving the
complementary advantages of the two therapeutic drugs, which have
little antitumour effects after binding the target antigen
(15,198). In contrast, small molecule
chemicals are highly effective against tumour cells, in spite of
the fact that they are less selective and may cause serious side
effects, accidentally injuring normal cells due to off-target
toxicity (211). Therefore, mAbs
can bind with effector molecules by using specific linkers to
produce ADCs, which expand the scope of medical treatment while
owning highly targeted selection, achieving the complementary
advantages of the two therapeutic drugs (17,135). The mAbs can bind with effector
molecules by using specific linkers to produce ADCs, which expand
the scope of medical treatment while possessing highly targeted
selection, achieving the complementary advantages of the two
therapeutic drugs that can usually target tumour-associated
antigens or specific receptors on the surface of tumour cells and
show a selective directing effect on tumour cells (135). The effector molecules that act
as payloads which produce a killing effect on tumours include
radiopharmaceutical agents, cytotoxic agents and bacterial or plant
protein toxins, conjugating respectively with mAbs to form
radioimmunoconjugates, chemo-immunoconjugates and immunotoxins used
in tumour-guided therapy (198).
Currently, ADCs that have been approved by the FDA include
brentuximab vedotin, trastuzumab emtansine, gemtuzumab ozogamicin,
Inotuzumab ozogamicin, and polatuzumab vedotin (211,212).
Action mechanism
Generally, ADCs are injected intravenously into the
blood system to prevent the hydrolysis of mAbs by gastric acid and
protease and are distributed into the tumour tissue by exosmosis of
the endothelial pores and the endocytosis of endothelial cells
(198). After the mAbs
specifically direct drugs to the surface of tumour cells expressing
tumour-specific antigens, ADCs come into tumour cells by
internalisation (211,213). As a general rule, there are
three distinguished pathways to internalise, including
clathrin-mediated endocytosis, caveolae-mediated endocytosis and
pinocytosis (211). Later, with
the influence of the acidic environment of the cytoplasm, some ADCs
with cleavable linkers release effector molecules which can damage
tumour cells, while other ADCs undergo the enzymatic fracture of
linkers or mAb degradation with the influence of lysosomal protease
(214). Finally, payload and
degradation products are released into the cytoplasm of tumour
cells, disturbing their cellular action mechanism, affecting the
tumour microenvironment and inducing the death of cells (213). As ADCs are formed from
maytansinoid drugs which are derivatives of maytansine and huC242,
humanised mAbs which bind to the CanAg antigen expressed on
colorectal tumours, pancreatic tumours and certain NSCLCs,
huC242-maytansinoid conjugates can be disintegrated with the
influence of lysosomal acidic conditions in order to release the
maytansinoid, which contributes greatly to the antitumour effect of
conjugates and the bystander effect (214). If the released payload is
permeable, a bystander effect is produced, which means that the
internalised payload enters and kills adjacent tumour cells,
showing the effect of apoptotic tumour cells on bystander tumour
cells (215). Although ADCs
cannot directly kill the adjacent antigen-negative tumour cells,
they can kill the antigen-positive tumour cells in order to
indirectly kill the adjacent tumour cells by the bystander effect
(216). Not only do ADCs damage
the growing tumour, they can also disrupt the structure supporting
tumour growth, such as tumour stromal cells and tumour vessels, in
order to enhance the antitumour effect (217). More importantly, it was proposed
that the activation of bystander effects by apoptotic tumour cells
may be crucial to achieving the permanent eradication of tumours
(215).
Basic strategies for selecting monoclonal
antibodies
As an important part of ADCs, mAbs used in
tumour-guided therapy must have some special characteristics.
First, the ideal mAb must effectively bind to antigens on target
cells so that the cytotoxic drugs are concentrated at the site of
the tumour (212). Secondly, the
mAbs should bind selectively to tumour cells and have little
cross-reactivity with normal cells (212). If the antibody selectivity is
poor or the selected antigen is present in normal tissues,
cytotoxic drugs will be delivered to normal cells, resulting in
targeted toxicity including allergic reactions, rashes and alopecia
(218,219). In addition, the Fc fractions of
some mAbs should have the affinity to bind with the Fc receptor of
immune cells, thereby activating the killer effect of immunocytes
(211). Thirdly, mAbs must own
the ability to induce the internalisation of tumour cells,
resulting in the release of the payload in the cytoplasm (220). In this way, the cytotoxic
molecule can be released to extend the extent of damage to tumour
cells, while mAbs play a certain antitumour role (212). Fourth, mAbs should be optimised
to significantly reduce the non-specific binding of ADC drugs and
prolong the half-life of ADCs in the blood (221). The immune interaction of the
constant Fc fragment of an ADC is one of the major determinants of
its cyclic half-life (222). As
a consequence, humanised mAbs and fully human mAbs should be
selected and the Fc fraction should be modified to decrease a part
of immunogenicity and immunotoxicity and increase the cyclic
half-life (223). Fifth, the
molecular weight of mAbs should be appropriate. If the molecular
weight is too large, ADCs will have difficulty penetrating the
capillary endodermis and extracellular spaces (217). If the molecular weight is too
small, the half-life will be influenced (224). Finally, the mAbs of ADCs should
have some of the function of mAbs, including ADCC and CDC (217), which means that the mAb alone
can be seen as an effective drug.
Nowadays, all ADCs in clinical trials use IgG
because the biomolecule not only contains multiple natural sites
for conjugation, but can also be modified to produce other
conjugate sites (225). Due to
their high affinity to target antigens and long circulating
half-life in the blood, IgG can accumulate in the tumour region
(226). Also, compared with
others, IgG1 is most often chosen as the antibody part of ADCs
(211). Generally, different IgG
subtypes have different immune functions including ADCC and CDC
(217). Compared with IgG4 and
IgG2 subtypes, human IgG1 and IgG3 have stronger ADCC and CDC
(217,227). Furthermore, IgG3 antibodies have
a short half-life and rapid clearance compared to IgG1, IgG2 and
IgG4, making them impossible to use in ADC synthesis (223). As a result, IgG1 is being used
more selectively for ADC development.
In the research of tumour treatments, the
development of fully human mAbs is very important, as murine mAbs
and chimeric mAbs induce the immunogenicity of allogenic antibodies
which can cause allergic reactions in humans (212). In early studies, the use of
murine mAbs often triggered a severe immune response in humans, and
patients produced human anti-murine antibodies which greatly
reduced the therapeutic effect (228). Therefore, it is necessary to
develop an mAb preparation technique, in order to make it possible
to use better humanised or fully human mAbs as an essential
component in an ADC in the future.
Near-infrared photoimmunotherapy
(NIR-PIT)
Traditional tumour treatments, such as surgery,
chemotherapy, radiation therapy and photodynamic therapy (PDT),
often damage the function of normal cells while killing diseased
tissue; this can break the delicate balance between the pathogen
tissue and the surrounding healthy cells (229). For example, after the
photosensitiser is administered and enriched in the tumour, the PDT
uses certain wavelengths of visible light to activate the
photosensitiser, generating singlet oxygen to kill the tumour
cells, in order to achieve a therapeutic effect (230). In the process, it is inevitable
to damage normal tissues or organs because of the accumulation of
photosensitisers in normal cells (231). In contrast, NIR-PIT, as a
molecularly targeted phototherapy with the selective killing of
diseased tissue developed on the basis of photodynamic therapy and
immunotherapy, uses mAbs to direct the near-infrared,
water-soluble, silicon-phthalocyanine derivative,
IRdye700DX(IR700), to tumour sites, solving the problem of the low
selectivity of photodynamic therapy (229,232).
As early as the beginning of the 1980s, Mew et
al started studying PIT in vitro and in vivo,
indicating that photo-immunoconjugates show higher selectivity to
tumour tissues than photosensitisers or mAbs alone (233,234). In subsequent years, although
various photosensitisers, cross-linking methods and mAbs were
developed and applied to photoimmunotherapy (PIT), the application
of photo-immunoconjugate was still limited in vivo due to
the hydrophobic nature of the photosensitisers (235). Later, a new type of PIT was
developed in 2011 by Mitsunaga et al, NIR-PIT, which uses a
target-specific photosensitiser based on NIR phthalocyanine dye,
IR700, in combination with mAb targeting EGFR (236). In this treatment, mAb-IR700
conjugate binds to tumour cells that overexpress antibody targets
(235). When irradiated with
near-infrared light, IR700 in the conjugate is activated and
rapidly destroys the hydrophobic tumour cell membrane, resulting in
the death of the cancer cells (229,237). In addition, the conjugate in
vivo can indirectly activate cytotoxic T cells to kill tumours
and inhibit tumour metastasis and recurrence by targeting CD44,
CD133 and other tumour stem cell biomarkers (229,232).
In short, mAbs, as a specialised tool for
identifying specific proteins on the surface of cancer cells, can
act as a carrier to selectively deliver the photosensitisers which
have a poor targeting ability to the tumour site, helping
photosensitisers to locate and attach to cancer cells (238). More importantly,
photoimmunotherapy could be applied to a range of cancers simply by
altering mAbs in photo-immunoconjugates which have different
targets, such as EGFR, HER2, PSMA, CD25, CEA, mesothelin, GPC3,
CD47, CD20 and PD-L1 (139,229,238). Therefore, it is necessary to
design and manufacture mAbs with better properties so that the PIT
has a greater prospect.
5. Monoclonal antibody drugs and the market
value
To date, biotechnology medicines have been
developing rapidly, and half of the pharmaceuticals are synthesised
by biotechnology companies around the world, especially those drugs
with complex molecular structures including multi-specific drugs;
this is because biotechnology is simpler than chemical synthesis
and can produce higher economic efficiency (239). Therefore, as important
components of biotechnology medicines, mAb drugs produced by
lymphocyte hybridoma technology or genetic engineering technology,
among others, have been widely used in the medical and biological
fields as diagnostic and treatment agents in the last 30 years
(24,240). From the perspective of the
ingredients of mAbs, these drugs can be divided into four
generations: a) Murine-derived mAb drugs (-momab); b) human-murine
chimeric mAb drugs (-ximab); c) humanised mAb drugs (-zumab); and
d) fully human mAb drugs (-mumab) (29).
According to the investigative report of the
American Pharmaceutical Research and Producers Association, the
antibody drugs currently under development and already on the
market are summarised as follows. To date, the FDA has approved 108
mAb drugs, which have made a breakthrough in tumour immunotherapy
and greatly improved the survival of patients with certain types of
tumours and other diseases (24,39) (Table
I). From the perspective of diseases, tumours, and auto-immune,
infectious, endocrine, cardiovascular and neurological diseases are
the six sectors with the largest market size, all worth billions of
dollars (24,58) (Table II). In summary, mAbs have become
a new force that cannot be ignored in biological drugs at present,
and it will be the main force in the field of biomedicine in the
future (239).
 | Table IMonoclonal antibody drugs approved by
FDA from 1986 to November 2021. |
Table I
Monoclonal antibody drugs approved by
FDA from 1986 to November 2021.
| Trade name (generic
name) | Company | Disease target | Antibody
target | FDA approval
date | Clinical
trials |
|---|
| Othoclone
(muromomab) | Ortho Biotech |
Allotransplantation | CD3 | 1986 | NCT01932554 |
| Reopro
(abciximab) | Centocor | Cardiovascular
disease (CVD) | Glycoprotein
IIβ/IIIα | 12/22/1994 | NCT01932554 |
| Panorex
(edrecolomab) | Centocor | Tumour | Glycoprotein
17-1A | 1995 | NCT00002968 |
| Rituxan
(rituximab) | IDEC/Genentech | Non-Hodgkin.s
lymphoma | CD20 | 11/26/1997 | NCT02433522 |
| Zenapax
(daclizumab) | PDL | Renal
transplantation | CD25 | 12/10/1997 | NCT01051349 |
| Simulect
(basiliximab) | Novartis | Renal
transplantation | CD25 | 5/12/1998 | NCT00724022 |
| Synagis
(palivizumab) | MedImmune | Respiratory tract
infection (RTI) | F protein of
RSV | 6/19/1998 | NCT02442427 |
| Remicade
(infliximab) | Centocor | Rheumatoid
arthritis (RA) | TNF-α | 8/24/1998 | NCT02096861 |
| Herceptin
(trastuzumab) | Genentech | Breast
carcinoma | HER-2 | 9/25/1998 | NCT00045032 |
| ENBREL
(etanercept) | Amgen/Wyeth | RA and
psoriasis | TNFR-Fc | 11/2/1998 | NCT02376790 |
| Mylotarg
(gemtuzumab) | Celltech/Wyeth | Leukaemia | CD33 | 5/17/2000 | NCT00927498 |
| Campath
(alemtuzumab) | Ilex/Millennium
Pharmaceuticals/Berlex | Lymphoma | CD52 | 5/7/2001 | NCT00530348 |
| Zevalin
(ibritumomab) | IDEC | Lymphoma | CD20-Y90 | 2/19/2002 | NCT00220285 |
| Humira
(adalimumab) | CAT/Abbott | RA | TNF-α | 12/31/2002 | NCT02745080 |
| Raptiva
(efalizumab) | Xoma/Genentech | Psoriasis | CD11a | 10/27/2003 | NCT00256139 |
| Bexxar
(tositumomab) | Corixa Corp. and
GlaxoSmithKline | Non-Hodgkin.s
lymphoma | CD20 | 6/27/2003 | NCT00022945 |
| Xolair
(omalizumab) |
Genentech/Tanox/Novartis | Moderate to severe
allergic asthma | IgE | 6/20/2003 | NCT01157117 |
| Avastin
(bevacizumab) | Genentech | Colorectal
carcinoma | VEGF | 2/26/2004 | NCT00976911 |
| Erbitux
(cetuximab) | ImClone/BMS | Colorectal
carcinoma | EGFR | 2/12/2004 | NCT01228734 |
| Tysabri
(natalizumab) | Biogen Idec | Multiple
sclerosis | α4-integrin | 11/23/2004 | NCT02730455 |
| Lucentis
(ranibizumab) | Genentech | Neovascular
age-related macular degeneration | VEGF-A | 6/30/2006 | NCT01489189 |
| Vectibix
(panitumumab) | Amgen | Colorectal
carcinoma | EGFR | 9/27/2006 | NCT01328171 |
| Soliris
(eculizumab) | Alexion
Pharmaceuticals | Paroxysmal
nocturnal haemoglobinuria (PNH) | Complement protein
5a | 3/16/2007 | NCT00122330 |
| Cimzia
(certolizumab) | UCB | Moderate to severe
RA in adults | TNF-α | 4/22/2008 | NCT01087788 |
| Ilaris
(canakunumab) | Novartis
Pharmaceutical Corp. |
Cryopyrin-associated periodic
syndrome | IL-1β | 6/17/2009 | NCT01327846 |
| Stelara
(ustekinumab) | Centocor Orth
Biotech Inc. | Psoriasis | IL-12/23 | 9/25/2009 | NCT01369355 |
| Arzerra
(ofatumumab) | Genmab and GSK | Chronic
granulocytic leukaemia | CD20 | 10/26/2009 | NCT01457924 |
| Actemra
(tocilizumab) | Genentech Inc. | Rare childhood
arthritis | IL-6 receptor | 1/8/2010 | NCT01331837 |
| Prolia
(denosumab) | Amgen Inc. | Osteoporosis in
postmenopausal women | IgG-2 | 6/1/2010 | NCT00523341 |
| Benlysta
(belimumab) | HGS and GSK | Systemic lupus
erythematosus in adult patients | BLyS | 3/9/2011 | NCT01639339 |
| Yervoy
(ipilimumab) | Bristol Myers
Squibb Co. | Metastatic
melanoma | CTLA-4 | 3/25/2011 | NCT02899299 |
| Adcetris
(brentuximab) | Seattle Genetics,
Inc. | Hodgkin lymphoma
(HL) and recurrent anaplastic large cell lymphoma | CD30 | 8/19/2011 | NCT01100502 |
| Perjeta
(pertuzumab) | Genentech Inc. | End-stage breast
carcinoma | HER-2 | 6/9/2012 | NCT00545688 |
| Abthrax
(raxibacumab) | Human Genome
Science Inc. | Inhalational
anthrax | Anthracis
Toxin | 12/17/2012 | NCT02339155 |
| Kadcyla
(ado-trastuzumab emtansine) | Genentech,
Inc. | Breast
carcinoma | HER2 | 2/22/2013 | NCT02675829 |
| Simponi Aria
(golimumab) | Janssen Biotech,
Inc. | RA | TNF | 7/23/2013 | NCT02846545 |
| Gazyva
(obinutuzumab) | Genentech | Chronic lymphocytic
leukaemia (CLL) | CD20 | 11/5/2013 | NCT02242942 |
| Sylvany
(siltuximab) | Janssen
Biotech | Multicentre
Castleman disease | IL-6 | 4/23/2014 | NCT01024036 |
| Entyvio
(vedolizumab) | Takeda
Pharmaceuticals USA | Ulcerative colitis
and Crohn.s disease | α4β7 integrin | 5/20/2014 | NCT00783718 |
| Keytruda
(permbrolizumab) | Merck Sharp &
Dohme | Non-small cell lung
carcinoma and head-neck tumours | PD-1 | 9/4/2014 | NCT02775435 |
| Cyramza
(ramucirumab) | Eli Lilly and
Co. | Advanced stomach
cancer, adenocarcinoma of gastroesophageal junction | VEGFR2 | 11/7/2014 | NCT01170663 |
| Cosentyx
(ecukinumab) | Novartis | Plaque
psoriasis | IL-17a | 1/21/2015 | NCT02745080 |
| Unituxin
(dinutuximab) | United Therapeutics
Corp. | Neuroblastoma in
children | PD-L1 | 3/10/2015 | NCT01767194 |
| Praluent
(alirocumab) | Sanofi | Decrease LDL-C | PCSK9
inhibitor | 7/24/2015 | NCT01663402 |
| Repatha
(evolocumab) | Amgen | Decrease LDL-C | PCSK9
inhibitor | 8/27/2015 | NCT02392559 |
| Praxbind
(idarucizumab) | Boehringer
Ingelheim | Anti-coagulating
effect therapy | Dabigatran | 10/16/2015 | NCT02104947 |
| Nucala
(mepolizumab) |
GlaxoSmithKline | Severe asthma | IL-5 | 11/4/2015 | NCT01000506 |
| Darzalex
(daratumumab) | Johnson and
Johnson | Multiple
myeloma | CD-38 | 11/16/2015 | NCT03277105 |
| Portrazza
(necitumumab) | Eli Lilly and
Co. | Squamous non-small
cell lung cancer | EGFR | 11/24/2015 | NCT00981058 |
| Empliciti
(elotuzumab) | Bristol-Myers
Squibb | Multiple
myeloma | SLAMF7 protein
targeted | 11/30/2015 | NCT01239797 |
| Anthim
(obiltoxaximab) | Elusys Therapeutics
Inc. | Inhalational
anthrax | / | 3/18/2016 | NCT01932242 |
| Taltz
(ixekizumab) | Eli Lilly and
Co. | Moderate-to-severe
plaque psoriasis. | IL-17A | 3/22/2016 | NCT02757352 |
| Cinqair
(reslizumab) | Teva Respiratory
LLC | Asthma | IL-5 | 3/23/2016 | NCT0250162 |
| Tecentriq
(atezolizumab) | Genentech | Non-small cell lung
cancer and urothelial carcinoma | PD-L1 | 5/18/2016 | NCT02425891 |
| Zinbryta
(daclizumab) | Biogen | Multiple
sclerosis | IL-2 | 5/27/2016 | NCT01797965 |
| Lartruvo
(olaratumab) | Eli Lilly and
Co. | Soft-tissue
sarcoma | PDGFR-α | 10/19/2016 | NCT01185964 |
| Zinplava
(bezlotoxumab) | Merck Sharp
Dohme | Clostridium
difficile infection | Clostridium
difficile toxin A and B | 10/21/2016 | NCT01513239 |
| Siliq
(brodalumab) | Valeant | Plaque
psoriasis | IL-17R | 2/15/2017 | NCT01708629 |
| Bavencio
(avelumab) | Pfizer and Merck
& Co | Ovarian and gastric
cancer | PD-1 | 3/23/2017 | NCT02603432 |
| Dupixent
(dupilumab) | Sanofi and
Regeneron Pharmaceuticals | Atopic
dermatitis | IL-4Rα | 3/28/2017 | NCT02414854 |
| Ocrevus
(ocrelizumab) | Roche Holdings | Multiple
sclerosis | CD20 | 3/28/2017 | NCT02545868 |
| Imfinzi
(durvalumab) | Astrazeneca | Metastatic
urothelial carcinoma | PD-L1 | 5/1/2017 | NCT03043872 |
| Kevzara
(sarilumab) | Sanofi and
Regeneron Pharmaceuticals | RA | IL-6R | 5/22/2017 | NCT01768572 |
| Tremfya
(guselkumab) | Johnson and
Johnson | Plaque
psoriasis | IL-23 | 7/13/2017 | NCT03162796 |
| Besponsa
(inotuzumab ozogamicin) | Pfizer | Acute lymphocytic
leukaemia | CD22 | 8/17/2017 | NCT01535989 |
| Fasenra
(benralizumab) | Astrazeneca AB | Severe asthma | IL-5 | 11/14/2017 | NCT03557307 |
| Hemlibra
(emicizumab) | Roche Group | Type A von
Willebrand disease | FIXa-FX | 11/16/2017 | NCT03020160 |
| Trogarzo
(ibalizumab-uiyk) | TaiMed
Biologics | HIV | CD4+T cell
receptor | 3/6/2018 | NCT02475629 |
| Ilumya
(tildrakizumab) | SUN Pharma | Psoriasis | IL-23 | 3/20/2018 | NCT02980692 |
| Crysvita
(burosumab-twza) | Ultragenyx
Pharmaceutical | Rhachitis | FGF23 | 4/17/2018 | NCT02915705 |
| Aimovig
(erenumab-aooe) | Amgen Inc. | Migraine | CGRP | 5/17/2018 | NCT02066415 |
| Poteligeo
(mogamulizumab-kpkc) | Kyowa Kirin | Granuloma fungoides
and Sézary syndrome | CCR4 (cellular
chemokine receptor type 4) | 8/8/2018 | NCT01728805 |
| Takhzyro
(lanadelumab) | DYAX Corp. | Types I and II
hereditary angioedema | Kallikrein | 8/23/2018 | NCT02586805 |
| Lumoxiti
(moxetumomab pasudotox-tdfk) | AstraZeneca | Hairy cell
leukaemia | CD-22 | 9/13/2018 | NCT01829711 |
| Ajovy
(fremanezumab) | Teva | Episodic
migraine | Calcitonin
gene-related peptide (CGRP) | 9/14/2018 | NCT02621931 |
| Emgality
(galcanezumab) | Eli Lilly and
Co. | Episodic
migraine | CGRP | 9/27/2018 | NCT02614261 |
| Libtayo
(cemiplimab-rwlc) | Sanofi and
Regeneron | Cutaneous squamous
cell carcinoma | PD-L1 | 9/28/2018 | NCT03836105 |
| Gamifant
(emapalumab-lzsg) | Swedish Orphan
Biovitrum AB | Haemophagocytic
lymphohistiocytosis | IFNγ | 11/20/2018 | NCT01818492 |
| Ultomiris
(ravulizumab) | Alexion Pharm | PNH | Complement (C5
protein) | 12/21/2018 | NCT02946463 |
| Cablivi
(caplacizumab-yhdp) | Ablynx | Acquired thrombotic
thrombocytopenic purpura | von Willebrand
factor | 2/6/2019 | NCT02553317 |
| Evenity
(romosozumab-aqqg) | Amgen | Osteoporosis | Slerostin | 4/9/2019 | NCT01631214 |
| Skyrizi
(risankizumab-rzaa) | AbbVie | Plaque
psoriasis | IL-23 | 6/10/2019 | NCT04433442 |
| Polivy (polatuzumab
vedotin-piiq) | Genentech and Roche
Group | Diffuse large
B-cell lymphoma | CD79b | 6/10/2019 | NCT05006534 |
| Beovu
(brolucizumab-dbll) | Novartis | Wet type
age-related macular degeneration | VEGF | 10/7/2019 | NCT04690062 |
| Adakveo
(crizanlizumab-tmca) | Novartis | Sickle cell
anaemia | P-selectin | 11/15/2019 | NCT05020873 |
| Padcev (enfortumab
vedotin-ejfv) | Astellas | Refractory bladder
cancer | Nectin-4 | 12/18/2019 | NCT05014139 |
| Enhertu
(fam-trastuzumab deruxtecan-nxki) | Daiichi Sankyo
Pharmaceutical | Metastatic
HER2-positive breast cancer | HER2 | 12/20/2019 | NCT05113251 |
| Tepezza
(teprotumumab-trbw) | Horizon Pharma | Thyroid eye
disease | IGF-1R | 1/21/2020 | NCT05002998 |
| Vyepti
(eptinezumab-jjmr) | Lundbeck | Migraine in
adults | CGRP | 2/21/2020 | NCT04921384 |
| Sarclisa
(isatuximab) | Sanofi | Multiple
myeloma | CD38 | 3/2/2020 | NCT04802031 |
| Trodelvy
(sacituzumab govitecan-hziy) | Gilead | Metastatic
triple-negative breast cancer | Trop-2 | 4/22/2020 | NCT04559230 |
| Uplizna
(inebilizumab-cdon) | Viela Bio | Neuromyelitis
optica spectrum disorder | CD19 | 6/11/2020 | NCT02200770 |
| Monjuvi
(tafasitamab-cxix) | MorphoSys and
Incyte | Relapsed or
refractory diffuse large B-cell lymphoma | CD19 | 7/31/2020 | NCT04680052 |
| Blenrep (belantamab
mafodotin-blmf) |
GlaxoSmithKline | Multiple
myeloma | BCMA | 8/5/2020 | NCT05002816 |
| Enspryng
(satralizumab-mwge) | Roche | Neuromyelitis
optica spectrum disorder | IL-6R | 8/14/2020 | NCT04660539 |
| Inmazeb
(atoltivimab, maftivimab, and odesivimab-ebgn) | Regeneron
Pharmaceuticals | Ebola virus | / | 10/14/2020 | / |
| Danyelza
(naxitamab-gqgk) | Y-Mabs
Therapeutics | High-risk
refractory or relapsed neuroblastoma | Gangliosides
GD2 | 11/25/2020 | NCT04909515 |
| Margenza
(margetuximab) | Macrogenics | HER2-positive
breast cancer | HER2 | 12/16/2020 | NCT04425018 |
| Ebanga
(ansuvimab-zykl) | Ridgeback
Biotherapeutics | Ebola virus | Glycoprotein | 12/21/2020 | NCT05067166 |
| Evkeeza
(evinacumab-dgnb) | Regeneron
Pharmaceuticals | Homozygous familial
hypercholesterolemia | ANGPTL3 | 2/11/2021 | NCT04863014 |
| Jemperli
(dostarlimab-gxly) |
Glaxosmithkline | Endometrial
cancer | PD-1 | 4/22/2021 | NCT04581824 |
| Zynlonta
(loncastuximab tesirine-lpyl) | Adc Therapeutics
Sa | Relapsed or
refractory large B-cell lymphoma | CD19 | 4/23/2021 | NCT03589469 |
| Aduhelm
(amivantamab-vmjw) | Janssen
Biotech | Non-small cell lung
cancer | EGFR/MET | 5/21/2021 | NCT04606381 |
| Zynlonta
(aducanumab-avwa) | Biogen Inc | Alzheimer.s
disease | Amyloid
beta-protein | 6/7/2021 | NCT02484547 |
| Saphnelo
(anifrolumab-fnia) | AstraZeneca | Moderate-to severe
systemic lupus erythematousus | IFN receptor | 7/30/2021 | NCT04877691 |
| Tivdak (tisotumab
vedotin-tftv) | Seagen | Recurrent or
metastatic cervical cancer with disease progression on or after
chemotherapy | Tissue factor | 9/20/2021 | NCT03657043 |
| Table IIMonoclonal antibody drug of global
drug sales TOP100 in 2020. |
Table II
Monoclonal antibody drug of global
drug sales TOP100 in 2020.
| Ranking | Drug | Sale (billion
dollars) | Manufacturer | Adaptation
disease |
|---|
| 1 | Humria®
(adalimumab) | 19.832 | Abbvie | Autoimmune
disease |
| 2 |
Keytruda® (pablizumab) | 14.38 | MRK | Melanoma and
non-small lung cancer (NSCLC) |
| 7 | Opdivo®
(nivolumab) | 7.81 | Bristol-Myers
Squibb | Melanoma and
NSCLC |
| 8 | Stelara®
(ustekinumab) | 7.707 | Johnson &
Johnson (J&J) | Psoriasis |
| 14 | Avastin®
(bevacizumab) | 5.321 | Roche | Cancers including
colon cancer |
| 16 | Ocrevus®
(ocrelizumab) | 4.611 | Roche | Multiple sclerosis
(MS) |
| 18 |
Darzalex® (daratumumab) | 4.19 | J&J | Multiple
myeloma |
| 19 | Perjeta®
(pertuzumab) | 4.139 | Roche | Breast
carcinoma |
| 20 |
Remicade® (infliximab) | 4.077 | J&J/MSD | Autoimmune
disease |
| 21 | Soliris®
(eculizumab) | 4.064 | Alexion | Paroxysmal
nocturnal haemoglobinuria (PNH) |
| 22 |
Dupixent® (dupilumab) | 4.045 | Sanofi | Atopic
dermatitis |
| 23 |
Cosentyx® (secukinumab) | 3.995 | Novartis | Psoriasis |
| 25 |
Herceptin® (trastuzumab) | 3.978 | Roche | Cancers including
breast carcinoma |
| 31 |
Lucentis® (ranibizumab) | 3.473 | Roche/Novartis | Age-related macular
degeneration (ARMD) |
| 32 | Rituxan®
(rituximab) | 3.418 | Roche | Leukaemia |
| 35 | Xolain®
(omalizumab) | 3.281 | Roche/Novartis | Asthma |
| 37 | Entyvio®
(vedolizumab) | 3.252 | Takeda | Ulcerative
enteritis and Crohn.s disease |
| 42 | Actemra®
(tocilizumab) | 3.05 | Roche | Autoimmune
disease |
| 46 |
Tecentriq® (atezolizumab) | 2.919 | Roche | Metastatic
urothelial carcinoma |
| 50 |
Prolia®/Xgeva®
(denosumab) | 2.763 | Amgen/Daiichi
Sankyo | Osteoporosis |
| 61 |
Hemlibra® (emicizumab) | 2.335 | Roche | Haemophilia |
| 65 | Simponi®
(golimumab) | 2.243 | J&J | Autoimmune
disease |
| 69 | Cimzia®
(certolizumab) | 1.887 | UCB | Autoimmune
disease |
| 72 | Imfinzi®
(durvalumab) | 2.042 | AstraZeneca | Lung cancer |
| 78 | Tysabri®
(natalizumab) | 1.946 | Biogen | MS |
| 79 | Xgeva®
(denosumab) | 1.935 | Amgen | Giant cell
tumour |
| 82 | Kadcyla®
(ado-trastuzumab emtansine) | 1.445 | Roche | Her2-positive
metastatic breast cancer |
| 84 | Taltz®
(ixekizumab) | 1.788 | Eli Lilly and Co.
(LLY) | Plaque
psoriasis |
| 90 | Yervoy®
(ipilimumab) | 1.682 | Bristol-Myers
Squibb | Melanoma and
NSCLC |
| 98 | Erbitux®
(cetuximab) | 1.552 | LLY/Merck | Colorectal
tumour |
6. Prospects
In the past 30 years, from murine-derived mAbs to
fully human mAbs, the preparation of mAbs has made great progress.
The research into human antibodies has made a breakthrough in the
last 10 years, which has played an important role in medicine.
Nowadays, mAbs are mainly used in the treatment of tumours, organ
transplant rejection, autoimmune diseases and other diseases.
Because of their good targeting, mAbs have a quick effect, small
side effects and good effects. They are taking an increasing share
in the sales of biotechnology drugs and have broad development
prospects. However, the mature affinity of antibodies, the
stability of antibodies secreted by human hybrid tumour cells and
the mass production of antibodies still need to be solved.
With the development of the human genome project,
the high specificity of mAbs has meant that they will play an
irreplaceable role in the in-depth study of the subtle structure of
proteins. Additionally, the emergence of phage display technology
and ribosome display technology has greatly shortened the
preparation cycle of mAbs and reduced their production costs. In
addition, the safety of fully human mAbs has greatly promoted their
wide application in the clinical treatment of infectious diseases,
tumours, organ transplantation, haematological diseases, toxic
diseases, allergic diseases, autoimmune diseases and other aspects.
It is believed that with the development of molecular biology
technology, especially the murine antibody humanisation technology,
antibody library technology and transgenic technology, the clinical
application of mAbs will become more extensive.
Availability of data and materials
Not applicable.
Authors. contributions
TC and ZL provided the concepts of this review and
designed its framework. JL and JD conducted the research, selected
the literature findings and wrote the manuscript. All authors
edited the manuscript. All authors read and approved the final
manuscript for publication.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by grants from the National Natural
Science Foundation of China (no. 82060638) and 'Double 10-Thousand
Plan. of Jiangxi Province (Innovation and Technology Professionals
as the High-End Talent).
References
|
1
|
Patel A: Benign vs malignant tumors. JAMA
Oncol. 6:14882020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Murray PG and Young LS: An etiological
role for the Epstein-Barr virus in the pathogenesis of classical
Hodgkin lymphoma. Blood. 134:591–596. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keum N and Giovannucci E: Global burden of
colorectal cancer: Emerging trends, risk factors and prevention
strategies. Nat Rev Gastroenterol Hepatol. 16:713–732. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hasanpourghadi M, Pandurangan AK and
Mustafa MR: Modulation of oncogenic transcription factors by
bioactive natural products in breast cancer. Pharmacol Res.
128:376–388. 2018. View Article : Google Scholar
|
|
5
|
Stark A, Donahue TR, Reber HA and Hines
OJ: Pancreatic cyst disease: A review. JAMA. 315:1882–1893. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Travis WD, Asamura H, Bankier AA, Beasley
MB, Detterbeck F, Flieder DB, Goo JM, MacMahon H, Naidich D,
Nicholson AG, et al: The IASLC lung cancer staging project:
Proposals for coding T categories for subsolid nodules and
assessment of tumor size in part-solid tumors in the forthcoming
eighth edition of the TNM classification of lung cancer. J Thorac
Oncol. 11:1204–1223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clara-Trujillo S, Gallego Ferrer G and
Gómez Ribelles JL: In vitro modeling of non-solid tumors: How far
can tissue engineering go? Int J Mol Sci. 21:57472020. View Article : Google Scholar :
|
|
8
|
Shimada A: Hematological malignancies and
molecular targeting therapy. Eur J Pharmacol. 862:1726412019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dunn-Pirio AM and Vlahovic G:
Immunotherapy approaches in the treatment of malignant brain
tumors. Cancer. 123:734–750. 2017. View Article : Google Scholar
|
|
10
|
Cassetta L and Pollard JW: Targeting
macrophages: Therapeutic approaches in cancer. Nat Rev Drug Discov.
17:887–904. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schweizer C, Schubert P, Rutzner S,
Eckstein M, Haderlein M, Lettmaier S, Semrau S, Gostian AO, Frey B,
Gaipl US, et al: Prospective evaluation of the prognostic value of
immune-related adverse events in patients with non-melanoma solid
tumour treated with PD-1/PD-L1 inhibitors alone and in combination
with radiotherapy. Eur J Cancer. 140:55–62. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ovacik M and Lin K: Tutorial on monoclonal
antibody pharmacokinetics and its considerations in early
development. Clin Transl Sci. 11:540–552. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cymer F, Beck H, Rohde A and Reusch D:
Therapeutic monoclonal antibody N-glycosylation-structure, function
and therapeutic potential. Biologicals. 52:1–11. 2018. View Article : Google Scholar
|
|
14
|
Alkan SS: Legends of allergy/immunology:
Georges Köhler and the discovery of MONOCLONAL antibodies. Allergy.
74:1412–1414. 2019.PubMed/NCBI
|
|
15
|
Seaman S, Zhu Z, Saha S, Zhang XM, Yang
MY, Hilton MB, Morris K, Szot C, Morris H, Swing DA, et al:
Eradication of tumors through simultaneous ablation of
CD276/B7H3-positive tumor cells and tumor vasculature. Cancer Cell.
31:501–515.e8. 2017. View Article : Google Scholar
|
|
16
|
Fay EK and Graff JN: Immunotherapy in
prostate cancer. Cancers (Basel). 12:17522020. View Article : Google Scholar
|
|
17
|
Arlotta KJ and Owen SC: Antibody and
antibody derivatives as cancer therapeutics. Wiley Interdiscip Rev
Nanomed Nanobiotechnol. 11:e15562019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Starr CG and Tessier PM: Selecting and
engineering monoclonal antibodies with drug-like specificity. Curr
Opin Biotechnol. 60:119–127. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiu ML and Gilliland GL: Engineering
antibody therapeutics. Curr Opin Struct Biol. 38:163–173. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wootla B, Denic A and Rodriguez M:
Polyclonal and monoclonal antibodies in clinic. Methods Mol Biol.
1060:79–110. 2014. View Article : Google Scholar
|
|
21
|
Köhler G and Milstein C: Continuous
cultures of fused cells secreting antibody of predefined
specificity. Nature. 256:495–497. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Miller RA, Maloney DG, Warnke R and Levy
R: Treatment of B-cell lymphoma with monoclonal anti-idiotype
antibody. N Engl J Med. 306:517–522. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
An Z: Monoclonal antibodies-a proven and
rapidly expanding therapeutic modality for human diseases. Protein
Cell. 1:319–330. 2010. View Article : Google Scholar
|
|
24
|
Grilo AL and Mantalaris A: The
increasingly human and profitable monoclonal antibody market.
Trends Biotechnol. 37:9–16. 2019. View Article : Google Scholar
|
|
25
|
Garrard LJ and Zhukovsky EA: Antibody
expression in bacteriophage systems: The future of monoclonal
antibodies? Curr Opin Biotechnol. 3:474–480. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Parray HA, Shukla S, Samal S, Shrivastava
T, Ahmed S, Sharma C and Kumar R: Hybridoma technology a versatile
method for isolation of monoclonal antibodies, its applicability
across species, limitations, advancement and future perspectives.
Int Immunopharmacol. 85:1066392020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shim H: Antibody phage display. Adv Exp
Med Biol. 1053:21–34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Groves MA and Osbourn JK: Applications of
ribosome display to antibody drug discovery. Expert Opin Biol Ther.
5:125–135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ,
Li HJ and Wu HC: Development of therapeutic antibodies for the
treatment of diseases. J Biomed Sci. 27:12020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schmid AS and Neri D: Advances in antibody
engineering for rheumatic diseases. Nat Rev Rheumatol. 15:197–207.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuramochi T, Igawa T, Tsunoda H and
Hattori K: Humanization and simultaneous optimization of monoclonal
antibody. Methods Mol Biol. 1904:213–230. 2019. View Article : Google Scholar
|
|
32
|
Goydel RS, Weber J, Peng H, Qi J, Soden J,
Freeth J, Park H and Rader C: Affinity maturation, humanization,
and co-crystallization of a rabbit anti-human ROR2 monoclonal
antibody for therapeutic applications. J Biol Chem. 295:5995–6006.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu Y, Li C, Xia S, Tian X, Kong Y, Wang Z,
Gu C, Zhang R, Tu C, Xie Y, et al: Identification of human
single-domain antibodies against SARS-CoV-2. Cell Host Microbe.
27:891–898.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Frenzel A, Schirrmann T and Hust M: Phage
display-derived human antibodies in clinical development and
therapy. MAbs. 8:1177–1194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pucca MB, Cerni FA, Janke R,
Bermúdez-Méndez E, Ledsgaard L, Barbosa JE and Laustsen AH: History
of envenoming therapy and current perspectives. Front Immunol.
10:15982019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ribatti D: From the discovery of
monoclonal antibodies to their therapeutic application: An
historical reappraisal. Immunol Lett. 161:96–99. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Elgundi Z, Reslan M, Cruz E, Sifniotis V
and Kayser V: The state-of-play and future of antibody
therapeutics. Adv Drug Deliv Rev. 122:2–19. 2017. View Article : Google Scholar
|
|
38
|
Paci A, Desnoyer A, Delahousse J, Blondel
L, Maritaz C, Chaput N, Mir O and Broutin S:
Pharmacokinetic/pharmacodynamic relationship of therapeutic
monoclonal antibodies used in oncology: Part 1, monoclonal
antibodies, antibody-drug conjugates and bispecific T-cell
engagers. Eur J Cancer. 128:107–118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zaroff S and Tan G: Hybridoma technology:
The preferred method for monoclonal antibody generation for in vivo
applications. Biotechniques. 67:90–92. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Schroff RW, Foon KA, Beatty SM, Oldham RK
and Morgan AC Jr: Human anti-murine immunoglobulin responses in
patients receiving monoclonal antibody therapy. Cancer Res.
45:879–885. 1985.PubMed/NCBI
|
|
41
|
Angus DC, Birmingham MC, Balk RA, Scannon
PJ, Collins D, Kruse JA, Graham DR, Dedhia HV, Homann S and
MacIntyre N: E5 murine monoclonal antiendotoxin antibody in
gram-negative sepsis: A randomized controlled trial. E5 study
investigators JAMA. 283:1723–1730. 2000.
|
|
42
|
Karmali R, Kimby E, Ghielmini M, Flinn IW,
Gordon LI and Zucca E: Rituximab: A benchmark in the development of
chemotherapy-free treatment strategies for follicular lymphomas.
Ann Oncol. 29:332–340. 2018. View Article : Google Scholar
|
|
43
|
Crowe JE Jr: Recent advances in the study
of human antibody responses to influenza virus using optimized
human hybridoma approaches. Vaccine. 27(Suppl 6): G47–G51. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gonzales NR, De Pascalis R, Schlom J and
Kashmiri SV: Minimizing the immunogenicity of antibodies for
clinical application. Tumour Biol. 26:31–43. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Alfaleh MA, Alsaab HO, Mahmoud AB,
Alkayyal AA, Jones ML, Mahler SM and Hashem AM: Phage display
derived monoclonal antibodies: From bench to bedside. Front
Immunol. 11:19862020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
LoBuglio AF, Wheeler RH, Trang J, Haynes
A, Rogers K, Harvey EB, Sun L, Ghrayeb J and Khazaeli MB:
Mouse/human chimeric monoclonal antibody in man: Kinetics and
immune response. Proc Natl Acad Sci USA. 86:4220–4224. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Targan SR, Hanauer SB, van Deventer SJ,
Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF and
Rutgeerts PJ: A short-term study of chimeric monoclonal antibody
cA2 to tumor necrosis factor alpha for Crohn.s disease. Crohn.s
disease cA2 study group. N Engl J Med. 337:1029–1035. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu AY, Robinson RR, Murray ED Jr,
Ledbetter JA, Hellström I and Hellström KE: Production of a
mouse-human chimeric monoclonal antibody to CD20 with potent
Fc-dependent biologic activity. J Immunol. 139:3521–3526.
1987.PubMed/NCBI
|
|
49
|
McLaughlin P, Grillo-López AJ, Link BK,
Levy R, Czuczman MS, Williams ME, Heyman MR, Bence-Bruckler I,
White CA, Cabanillas F, et al: Rituximab chimeric anti-CD20
monoclonal antibody therapy for relapsed indolent lymphoma: Half of
patients respond to a four-dose treatment program. J Clin Oncol.
16:2825–2833. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Piccolo R, Eitel I, Galasso G,
Dominguez-Rodriguez A, Iversen AZ, Abreu-Gonzalez P, Windecker S,
Thiele H and Piscione F: 1-Year outcomes with intracoronary
abciximab in diabetic patients undergoing primary percutaneous
coronary intervention. J Am Coll Cardiol. 68:727–738. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bachlava E, Loukopoulou S, Karanasios E,
Chrousos G and Michos A: Management of coronary artery aneurysms
using abciximab in children with Kawasaki disease. Int J Cardiol.
220:65–69. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu SN, Zhang XH, Xu LP, Wang Y, Yan CH,
Chen H, Chen YH, Han W, Wang FR, Wang JZ, et al: Prognostic factors
and long-term follow-up of basiliximab for steroid-refractory acute
graft-versus-host disease: Updated experience from a large-scale
study. Am J Hematol. 95:927–936. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Furuya Y, Jayarajan SN, Taghavi S, Cordova
FC, Patel N, Shiose A, Leotta E, Criner GJ, Guy TS, Wheatley GH, et
al: The impact of alemtuzumab and basiliximab induction on patient
survival and time to bronchiolitis obliterans syndrome in double
lung transplantation recipients. Am J Transplant. 16:2334–2341.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aranda E, García-Alfonso P, Benavides M,
Sánchez Ruiz A, Guillén-Ponce C, Safont MJ, Alcaide J, Gómez A,
López R, Manzano JL, et al: First-line mFOLFOX plus cetuximab
followed by mFOLFOX plus cetuximab or single-agent cetuximab as
maintenance therapy in patients with metastatic colorectal cancer:
Phase II randomised MACRO2 TTD study. Eur J Cancer. 101:263–272.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Strohbehn GW and Vokes EE: Palbociclib: A
new partner for cetuximab? Lancet Oncol. 20:1195–1196. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Strohl WR: Current progress in innovative
engineered antibodies. Protein Cell. 9:86–120. 2018. View Article : Google Scholar :
|
|
57
|
Presta LG: Engineering of therapeutic
antibodies to minimize immunogenicity and optimize function. Adv
Drug Deliv Rev. 58:640–656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kumar R, Parray HA, Shrivastava T, Sinha S
and Luthra K: Phage display antibody libraries: A robust approach
for generation of recombinant human monoclonal antibodies. Int J
Biol Macromol. 135:907–918. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Saw PE and Song EW: Phage display
screening of therapeutic peptide for cancer targeting and therapy.
Protein Cell. 10:787–807. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ledsgaard L, Kilstrup M, Karatt-Vellatt A,
McCafferty J and Laustsen AH: Basics of antibody phage display
technology. Toxins (Basel). 10:2362018. View Article : Google Scholar
|
|
61
|
Greenwood J, Willis AE and Perham RN:
Multiple display of foreign peptides on a filamentous
bacteriophage. Peptides from plasmodium falciparum circumsporozoite
protein as antigens. J Mol Biol. 220:821–827. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Smith GP: Filamentous fusion phage: Novel
expression vectors that display cloned antigens on the virion
surface. Science. 228:1315–1317. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Geysen HM, Tainer JA, Rodda SJ, Mason TJ,
Alexander H, Getzoff ED and Lerner RA: Chemistry of antibody
binding to a protein. Science. 235:1184–1190. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Parmley SF and Smith GP:
Antibody-selectable filamentous fd phage vectors: Affinity
purification of target genes. Gene. 73:305–318. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Scott JK and Smith GP: Searching for
peptide ligands with an epitope library. Science. 249:386–390.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
McCafferty J, Griffiths AD, Winter G and
Chiswell DJ: Phage antibodies: Filamentous phage displaying
antibody variable domains. Nature. 348:552–554. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tan Y, Tian T, Liu W, Zhu Z and C JY:
Advance in phage display technology for bioanalysis. Biotechnol J.
11:732–745. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Deng X, Wang L, You X, Dai P and Zeng Y:
Advances in the T7 phage display system (Review). Mol Med Rep.
17:714–720. 2018.
|
|
69
|
Burritt JB, Bond CW, Doss KW and Jesaitis
AJ: Filamentous phage display of oligopeptide libraries. Anal
Biochem. 238:1–13. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Huang J, Doria-Rose NA, Longo NS, Laub L,
Lin CL, Turk E, Kang BH, Migueles SA, Bailer RT, Mascola JR and
Connors M: Isolation of human monoclonal antibodies from peripheral
blood B cells. Nat Protoc. 8:1907–1915. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wang Z, Li Y, Hou B, Pronobis MI, Wang M,
Wang Y, Cheng G, Weng W, Wang Y, Tang Y, et al: An array of 60,000
antibodies for proteome-scale antibody generation and target
discovery. Sci Adv. 6:eaax22712020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Galán A, Comor L, Horvatić A, Kuleš J,
Guillemin N, Mrljak V and Bhide M: Library-based display
technologies: Where do we stand? Mol Biosyst. 12:2342–2358. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lim CC, Choong YS and Lim TS: Cognizance
of molecular methods for the generation of mutagenic phage display
antibody libraries for affinity maturation. Int J Mol Sci.
20:18612019. View Article : Google Scholar :
|
|
74
|
Goracci M, Pignochino Y and Marchiò S:
Phage display-based nanotechnology applications in cancer
immunotherapy. Molecules. 25:8432020. View Article : Google Scholar :
|
|
75
|
Rahbarnia L, Farajnia S, Babaei H, Majidi
J, Veisi K, Ahmadzadeh V and Akbari B: Evolution of phage display
technology: From discovery to application. J Drug Target.
25:216–224. 2017. View Article : Google Scholar
|
|
76
|
Petrenko VA: Landscape phage: Evolution
from phage display to nanobiotechnology. Viruses. 10:3112018.
View Article : Google Scholar :
|
|
77
|
Brüggemann M and Neuberger MS: Strategies
for expressing human antibody repertoires in transgenic mice.
Immunol Today. 17:391–397. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Brüggemann M, Osborn MJ, Ma B, Hayre J,
Avis S, Lundstrom B and Buelow R: Human antibody production in
transgenic animals. Arch Immunol Ther Exp (Warsz). 63:101–108.
2015. View Article : Google Scholar
|
|
79
|
Laffleur B, Pascal V, Sirac C and Cogné M:
Production of human or humanized antibodies in mice. Methods Mol
Biol. 901:149–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chen WC and Murawsky CM: Strategies for
generating diverse antibody repertoires using transgenic animals
expressing human antibodies. Front Immunol. 9:4602018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Alt FW, Blackwell TK and Yancopoulos GD:
Immunoglobulin genes in transgenic mice. Trends Genet. 1:231–236.
1985. View Article : Google Scholar
|
|
82
|
Frippiat JP, Williams SC, Tomlinson IM,
Cook GP, Cherif D, Le Paslier D, Collins JE, Dunham I, Winter G and
Lefranc MP: Organization of the human immunoglobulin lambda
light-chain locus on chromosome 22q11.2. Hum Mol Genet. 4:983–991.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Fishwild DM, O'Donnell SL, Bengoechea T,
Hudson DV, Harding F, Bernhard SL, Jones D, Kay RM, Higgins KM,
Schramm SR and Lonberg N: High-avidity human IgG kappa monoclonal
antibodies from a novel strain of minilocus transgenic mice. Nat
Biotechnol. 14:845–851. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mendez MJ, Green LL, Corvalan JR, Jia XC,
Maynard-Currie CE, Yang XD, Gallo ML, Louie DM, Lee DV, Erickson
KL, et al: Functional transplant of megabase human immunoglobulin
loci recapitulates human antibody response in mice. Nat Genet.
15:146–156. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lonberg N: Human antibodies from
transgenic animals. Nat Biotechnol. 23:1117–1125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Siegel SA, Shealy DJ, Nakada MT, Le J,
Woulfe DS, Probert L, Kollias G, Ghrayeb J, Vilcek J and Daddona
PE: The mouse/human chimeric monoclonal antibody cA2 neutralizes
TNF in vitro and protects transgenic mice from cachexia and TNF
lethality in vivo. Cytokine. 7:15–25. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Tsujinaka T, Fujita J, Ebisui C, Yano M,
Kominami E, Suzuki K, Tanaka K, Katsume A, Ohsugi Y, Shiozaki H and
Monden M: Interleukin 6 receptor antibody inhibits muscle atrophy
and modulates proteolytic systems in interleukin 6 transgenic mice.
J Clin Invest. 97:244–249. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Crombet-Ramos T, Rak J, Pérez R and
Viloria-Petit A: Antiproliferative, antiangiogenic and proapoptotic
activity of h-R3: A humanized anti-EGFR antibody. Int J Cancer.
101:567–575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Jakobovits A, Amado RG, Yang X, Roskos L
and Schwab G: From XenoMouse technology to panitumumab, the first
fully human antibody product from transgenic mice. Nat Biotechnol.
25:1134–1143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ros F, Offner S, Klostermann S, Thorey I,
Niersbach H, Breuer S, Zarnt G, Lorenz S, Puels J, Siewe B, et al:
Rabbits transgenic for human IgG genes recapitulating rabbit B-cell
biology to generate human antibodies of high specificity and
affinity. MAbs. 12:18469002020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
He M and Taussig MJ: Eukaryotic ribosome
display with in situ DNA recovery. Nat Methods. 4:281–288. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Thom G and Groves M: Ribosome display.
Methods Mol Biol. 901:101–116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Plückthun A: Ribosome display: A
perspective. Methods Mol Biol. 805:3–28. 2012. View Article : Google Scholar
|
|
94
|
Rothe A, Hosse RJ and Power BE: Ribosome
display for improved biotherapeutic molecules. Expert Opin Biol
Ther. 6:177–187. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ministro J, Manuel AM and Goncalves J:
Therapeutic antibody engineering and selection strategies. Adv
Biochem Eng Biotechnol. 171:55–86. 2020.
|
|
96
|
Mattheakis LC, Bhatt RR and Dower WJ: An
in vitro polysome display system for identifying ligands from very
large peptide libraries. Proc Natl Acad Sci USA. 91:9022–9026.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hammers CM and Stanley JR: Antibody phage
display: Technique and applications. J Invest Dermatol. 134:1–5.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Loh B, Kuhn A and Leptihn S: The
fascinating biology behind phage display: Filamentous phage
assembly. Mol Microbiol. 111:1132–1138. 2019. View Article : Google Scholar
|
|
99
|
Zahnd C, Amstutz P and Plückthun A:
Ribosome display: Selecting and evolving proteins in vitro that
specifically bind to a target. Nat Methods. 4:269–279. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hammerling MJ, Fritz BR, Yoesep DJ, Kim
DS, Carlson ED and Jewett MC: In vitro ribosome synthesis and
evolution through ribosome display. Nat Commun. 11:11082020.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
He M and Khan F: Ribosome display:
Next-generation display technologies for production of antibodies
in vitro. Expert Rev Proteomics. 2:421–430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lagoutte P, Lugari A, Elie C, Potisopon S,
Donnat S, Mignon C, Mariano N, Troesch A, Werle B and Stadthagen G:
Combination of ribosome display and next generation sequencing as a
powerful method for identification of affibody binders against
β-lactamase CTX-M15. N Biotechnol. 50:60–69. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Rouet R, Jackson KJL, Langley DB and
Christ D: Next-generation sequencing of antibody display
repertoires. Front Immunol. 9:1182018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Yamaguchi J, Naimuddin M, Biyani M, Sasaki
T, Machida M, Kubo T, Funatsu T, Husimi Y and Nemoto N: cDNA
display: A novel screening method for functional disulfide-rich
peptides by solid-phase synthesis and stabilization of mRNA-protein
fusions. Nucleic Acids Res. 37:e1082009. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lipovsek D and Plückthun A: In-vitro
protein evolution by ribosome display and mRNA display. J Immunol
Methods. 290:51–67. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ueda T, Kanamori T and Ohashi H: Ribosome
display with the PURE technology. Methods Mol Biol. 607:219–225.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Roberts RW and Szostak JW: RNA-peptide
fusions for the in vitro selection of peptides and proteins. Proc
Natl Acad Sci USA. 94:12297–12302. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Muranaka N, Hohsaka T and Sisido M:
Four-base codon mediated mRNA display to construct peptide
libraries that contain multiple nonnatural amino acids. Nucleic
Acids Res. 34:e72006. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Dufner P, Jermutus L and Minter RR:
Harnessing phage and ribosome display for antibody optimisation.
Trends Biotechnol. 24:523–529. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Wardemann H, Yurasov S, Schaefer A, Young
JW, Meffre E and Nussenzweig MC: Predominant autoantibody
production by early human B cell precursors. Science.
301:1374–1377. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Traggiai E, Becker S, Subbarao K,
Kolesnikova L, Uematsu Y, Gismondo MR, Murphy BR, Rappuoli R and
Lanzavecchia A: An efficient method to make human monoclonal
antibodies from memory B cells: Potent neutralization of SARS
coronavirus. Nat Med. 10:871–875. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Bushey RT, Moody MA, Nicely NL, Haynes BF,
Alam SM, Keir ST, Bentley RC, Roy Choudhury K, Gottlin EB, Campa
MJ, et al: A Therapeutic antibody for cancer, derived from single
human B cells. Cell Rep. 15:1505–1513. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Tiller T: Single B cell antibody
technologies. N Biotechnol. 28:453–457. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Rudkin FM, Raziunaite I, Workman H, Essono
S, Belmonte R, MacCallum DM, Johnson EM, Silva LM, Palma AS, Feizi
T, et al: Single human B cell-derived monoclonal anti-Candida
antibodies enhance phagocytosis and protect against disseminated
candidiasis. Nat Commun. 9:52882018. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Rajan S, Kierny MR, Mercer A, Wu J,
Tovchigrechko A, Wu H, Dall Acqua WF, Xiao X and Chowdhury PS:
Recombinant human B cell repertoires enable screening for rare,
specific, and natively paired antibodies. Commun Biol. 1:52018.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Buisman AM, de Rond CG, Oztürk K, Ten
Hulscher HI and van Binnendijk RS: Long-term presence of memory
B-cells specific for different vaccine components. Vaccine.
28:179–186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Jilg W, Schmidt M and Deinhardt F: Decline
of anti-HBs after hepatitis B vaccination and timing of
revaccination. Lancet. 335:173–174. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Inoue T, Moran I, Shinnakasu R, Phan TG
and Kurosaki T: Generation of memory B cells and their
reactivation. Immunol Rev. 283:138–149. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
von Bredow B, Arias JF, Heyer LN, Moldt B,
Le K, Robinson JE, Zolla-Pazner S, Burton DR and Evans DT:
Comparison of antibody-dependent cell-mediated cytotoxicity and
virus neutralization by HIV-1 Env-specific monoclonal antibodies. J
Virol. 90:6127–6139. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
von Boehmer L, Liu C, Ackerman S, Gitlin
AD, Wang Q, Gazumyan A and Nussenzweig MC: Sequencing and cloning
of antigen-specific antibodies from mouse memory B cells. Nat
Protoc. 11:1908–1923. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Lei L, Tran K, Wang Y, Steinhardt JJ, Xiao
Y, Chiang CI, Wyatt RT and Li Y: Antigen-specific single B cell
sorting and monoclonal antibody cloning in guinea pigs. Front
Microbiol. 10:6722019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Cao Y, Su B, Guo X, Sun W, Deng Y, Bao L,
Zhu Q, Zhang X, Zheng Y, Geng C, et al: Potent neutralizing
antibodies against SARS-CoV-2 identified by high-throughput
single-cell sequencing of convalescent patients. B cells. Cell.
182:73–84.e16. 2020. View Article : Google Scholar
|
|
123
|
Lanzavecchia A, Corti D and Sallusto F:
Human monoclonal antibodies by immortalization of memory B cells.
Curr Opin Biotechnol. 18:523–528. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Wrammert J, Koutsonanos D, Li GM,
Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig
M, Lipkin WI, et al: Broadly cross-reactive antibodies dominate the
human B cell response against 2009 pandemic H1N1 influenza virus
infection. J Exp Med. 208:181–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Hafeez U, Gan HK and Scott AM: Monoclonal
antibodies as immunomodulatory therapy against cancer and
autoimmune diseases. Curr Opin Pharmacol. 41:114–121. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Walcheck B and Wu J: iNK-CD64/16A cells: A
promising approach for ADCC? Expert Opin Biol Ther. 19:1229–1232.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Decaup E, Rossi C, Gravelle P, Laurent C,
Bordenave J, Tosolini M, Tourette A, Perrial E, Dumontet C, Poupot
M, et al: A tridimensional model for NK cell-mediated ADCC of
follicular lymphoma. Front Immunol. 10:19432019. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Giles AJ, Hao S, Padget M, Song H, Zhang
W, Lynes J, Sanchez V, Liu Y, Jung J, Cao X, et al: Efficient ADCC
killing of meningioma by avelumab and a high-affinity natural
killer cell line, haNK. JCI Insight. 4:e1306882019. View Article : Google Scholar :
|
|
129
|
Pockley AG, Vaupel P and Multhoff G: NK
cell-based therapeutics for lung cancer. Expert Opin Biol Ther.
20:23–33. 2020. View Article : Google Scholar
|
|
130
|
Adams GP and Weiner LM: Monoclonal
antibody therapy of cancer. Nat Biotechnol. 23:1147–1157. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Seguin-Devaux C, Plesseria JM, Verschueren
C, Masquelier C, Iserentant G, Fullana M, Józsi M, Cohen JHM and
Dervillez X: FHR4-based immunoconjugates direct
complement-dependent cytotoxicity and phagocytosis towards
HER2-positive cancer cells. Mol Oncol. 13:2531–2553. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wyant T, Fedyk E and Abhyankar B: An
overview of the mechanism of action of the monoclonal antibody
vedolizumab. J Crohns Colitis. 10:1437–1444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Czyz M: Fibroblast growth factor receptor
signaling in skin cancers. Cells. 8:5402019. View Article : Google Scholar :
|
|
134
|
Jimenez-Pascual A and Siebzehnrubl FA:
Fibroblast growth factor receptor functions in glioblastoma. Cells.
8:7152019. View Article : Google Scholar :
|
|
135
|
Lee YT, Tan YJ and Oon CE: Molecular
targeted therapy: Treating cancer with specificity. Eur J
Pharmacol. 834:188–196. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Weiner GJ: Building better monoclonal
antibody-based therapeutics. Nat Rev Cancer. 15:361–370. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Howie LJ, Scher NS, Amiri-Kordestani L,
Zhang L, King-Kallimanis BL, Choudhry Y, Schroeder J, Goldberg KB,
Kluetz PG, Ibrahim A, et al: FDA approval summary: Pertuzumab for
adjuvant treatment of HER2-positive early breast cancer. Clin
Cancer Res. 25:2949–2955. 2019. View Article : Google Scholar
|
|
138
|
Touat M, Idbaih A, Sanson M and Ligon KL:
Glioblastoma targeted therapy: Updated approaches from recent
biological insights. Ann Oncol. 28:1457–1472. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Xu MJ, Johnson DE and Grandis JR:
EGFR-targeted therapies in the post-genomic era. Cancer Metastasis
Rev. 36:463–473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Frezzetti D, Gallo M, Maiello MR,
D'Alessio A, Esposito C, Chicchinelli N, Normanno N and De Luca A:
VEGF as a potential target in lung cancer. Expert Opin Ther
Targets. 21:959–966. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Yalcin F, Dzaye O and Xia S: Tenascin-C
function in glioma: Immunomodulation and beyond. Adv Exp Med Biol.
1272:149–172. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Lieverse RIY, Van Limbergen EJ, Oberije
CJG, Troost EGC, Hadrup SR, Dingemans AC, Hendriks LEL, Eckert F,
Hiley C, Dooms C, et al: Stereotactic ablative body radiotherapy
(SABR) combined with immunotherapy (L19-IL2) versus standard of
care in stage IV NSCLC patients, ImmunoSABR: A multicentre,
randomised controlled open-label phase II trial. BMC Cancer.
20:5572020. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wester HJ and Schottelius M: PSMA-targeted
radiopharmaceuticals for imaging and therapy. Semin Nucl Med.
49:302–312. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Apte RS, Chen DS and Ferrara N: VEGF in
signaling and disease: Beyond discovery and development. Cell.
176:1248–1264. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Topalian SL, Taube JM, Anders RA and
Pardoll DM: Mechanism-driven biomarkers to guide immune checkpoint
blockade in cancer therapy. Nat Rev Cancer. 16:275–287. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhang JC, Chen WD, Alvarez JB, Jia K, Shi
L, Wang Q, Zou N, He K and Zhu H: Cancer immune checkpoint blockade
therapy and its associated autoimmune cardiotoxicity. Acta
Pharmacol Sin. 39:1693–1698. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Chamoto K, Al-Habsi M and Honjo T: Role of
PD-1 in immunity and diseases. Curr Top Microbiol Immunol.
410:75–97. 2017.PubMed/NCBI
|
|
148
|
Darvin P, Toor SM, Sasidharan Nair V and
Elkord E: Immune checkpoint inhibitors: Recent progress and
potential biomarkers. Exp Mol Med. 50:1–11. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Postow MA, Sidlow R and Hellmann MD:
Immune-related adverse events associated with immune checkpoint
blockade. N Engl J Med. 378:158–168. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Gotwals P, Cameron S, Cipolletta D,
Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S,
Sabatos-Peyton C, Petruzzelli L, et al: Prospects for combining
targeted and conventional cancer therapy with immunotherapy. Nat
Rev Cancer. 17:286–301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Abril-Rodriguez G and Ribas A: SnapShot:
Immune checkpoint inhibitors. Cancer Cell. 31:848–848.e1. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Rowshanravan B, Halliday N and Sansom DM:
CTLA-4: A moving target in immunotherapy. Blood. 131:58–67. 2018.
View Article : Google Scholar
|
|
153
|
Lo B and Abdel-Motal UM: Lessons from
CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol.
49:14–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Duperret EK, Trautz A, Stoltz R, Patel A,
Wise MC, Perales-Puchalt A, Smith T, Broderick KE, Masteller E, Kim
JJ, et al: Synthetic DNA-encoded monoclonal antibody delivery of
anti-CTLA-4 antibodies induces tumor shrinkage in vivo. Cancer Res.
78:6363–6370. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Specenier P: Ipilimumab in melanoma.
Expert Rev Anticancer Ther. 16:811–826. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Carter BW, Bhosale PR and Yang WT:
Immunotherapy and the role of imaging. Cancer. 124:2906–2922. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Kuehn HS, Ouyang W, Lo B, Deenick EK,
Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, et
al: Immune dysregulation in human subjects with heterozygous
germline mutations in CTLA4. Science. 345:1623–1627. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Schubert D, Bode C, Kenefeck R, Hou TZ,
Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schäffer AA,
Grüning BA, et al: Autosomal dominant immune dysregulation syndrome
in humans with CTLA4 mutations. Nat Med. 20:1410–1416. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Chesney J, Puzanov I, Collichio F, Singh
P, Milhem MM, Glaspy J, Hamid O, Ross M, Friedlander P, Garbe C, et
al: Randomized, open-label phase II study evaluating the efficacy
and safety of talimogene laherparepvec in combination with
ipilimumab versus ipilimumab alone in patients with advanced,
unresectable melanoma. J Clin Oncol. 36:1658–1667. 2018. View Article : Google Scholar :
|
|
160
|
Soularue E, Lepage P, Colombel JF, Coutzac
C, Faleck D, Marthey L, Collins M, Chaput N, Robert C and Carbonnel
F: Enterocolitis due to immune checkpoint inhibitors: A systematic
review. Gut. 67:2056–2067. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Spain L, Diem S and Larkin J: Management
of toxicities of immune checkpoint inhibitors. Cancer Treat Rev.
44:51–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Rotte A: Combination of CTLA-4 and PD-1
blockers for treatment of cancer. J Exp Clin Cancer Res.
38:2552019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Larkin J, Chiarion-Sileni V, Gonzalez R,
Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J,
Dummer R, et al: Five-year survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 381:1535–1546. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Sidaway P: Immunotherapy: Local
chemotherapy synergizes with CTLA-4 inhibition. Nat Rev Clin Oncol.
15:2022018.PubMed/NCBI
|
|
165
|
Weber JS, Kähler KC and Hauschild A:
Management of immune-related adverse events and kinetics of
response with ipilimumab. J Clin Oncol. 30:2691–2697. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Han Y, Liu D and Li L: PD-1/PD-L1 pathway:
Current researches in cancer. Am J Cancer Res. 10:727–742.
2020.PubMed/NCBI
|
|
167
|
Alsaab HO, Sau S, Alzhrani R, Tatiparti K,
Bhise K, Kashaw SK and Iyer AK: PD-1 and PD-L1 checkpoint signaling
inhibition for cancer immunotherapy: Mechanism, combinations, and
clinical outcome. Front Pharmacol. 8:5612017. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Xu-Monette ZY, Zhou J and Young KH: PD-1
expression and clinical PD-1 blockade in B-cell lymphomas. Blood.
131:68–83. 2018. View Article : Google Scholar :
|
|
169
|
Du S, McCall N, Park K, Guan Q, Fontina P,
Ertel A, Zhan T, Dicker AP and Lu B: Blockade of tumor-expressed
PD-1 promotes lung cancer growth. Oncoimmunology. 7:e14087472018.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X
and Wu K: Biomarkers for predicting efficacy of PD-1/PD-L1
inhibitors. Mol Cancer. 17:1292018. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Naidoo J, Page DB, Li BT, Connell LC,
Schindler K, Lacouture ME, Postow MA and Wolchok JD: Toxicities of
the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann
Oncol. 26:2375–2391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Sunshine J and Taube JM: PD-1/PD-L1
inhibitors. Curr Opin Pharmacol. 23:32–38. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Patel SA and Minn AJ: Combination cancer
therapy with immune checkpoint blockade: Mechanisms and strategies.
Immunity. 48:417–433. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Hayashi H and Nakagawa K: Combination
therapy with PD-1 or PD-L1 inhibitors for cancer. Int J Clin Oncol.
25:818–830. 2020. View Article : Google Scholar
|
|
175
|
Aggen DH, Drake CG and Rini BI: Targeting
PD-1 or PD-L1 in metastatic kidney cancer: Combination therapy in
the first-line setting. Clin Cancer Res. 26:2087–2095. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Mathew M, Enzler T, Shu CA and Rizvi NA:
Combining chemotherapy with PD-1 blockade in NSCLC. Pharmacol Ther.
186:130–137. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Kong Y, Zhao X, Zou L, Xing P, Ma Y, Tian
Y and Zhang L: PD-1 inhibitor combined with radiotherapy and GM-CSF
as salvage therapy in patients with chemotherapy-refractory
metastatic solid tumors. J Clin Oncol. 38(Suppl 15): e151732020.
View Article : Google Scholar
|
|
178
|
Kordbacheh T, Honeychurch J, Blackhall F,
Faivre-Finn C and Illidge T: Radiotherapy and anti-PD-1/PD-L1
combinations in lung cancer: Building better translational research
platforms. Ann Oncol. 29:301–310. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Walshaw RC, Honeychurch J, Illidge TM and
Choudhury A: The anti-PD-1 era-an opportunity to enhance
radiotherapy for patients with bladder cancer. Nat Rev Urol.
15:251–259. 2018. View Article : Google Scholar
|
|
180
|
Sheng X, Yan X, Chi Z, Si L, Cui C, Tang
B, Li S, Mao L, Lian B, Wang X, et al: Axitinib in combination with
toripalimab, a humanized immunoglobulin G4 monoclonal
antibody against programmed cell death-1, in patients with
metastatic mucosal melanoma: An open-label phase IB trial. J Clin
Oncol. 37:2987–2999. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Kato Y, Tabata K, Kimura T,
Yachie-Kinoshita A, Ozawa Y, Yamada K, Ito J, Tachino S, Hori Y,
Matsuki M, et al: Lenvatinib plus anti-PD-1 antibody combination
treatment activates CD8+ T cells through reduction of
tumor-associated macrophage and activation of the interferon
pathway. PLoS One. 14:e02125132019. View Article : Google Scholar
|
|
182
|
Atkins MB, Plimack ER, Puzanov I, Fishman
MN, McDermott DF, Cho DC, Vaishampayan U, George S, Olencki TE,
Tarazi JC, et al: Axitinib in combination with pembrolizumab in
patients with advanced renal cell cancer: A non-randomised,
open-label, dose-finding, and dose-expansion phase 1b trial. Lancet
Oncol. 19:405–415. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Kohlhapp FJ and Kaufman HL: Molecular
pathways: Mechanism of action for talimogene laherparepvec, a new
oncolytic virus immunotherapy. Clin Cancer Res. 22:1048–1054. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma
C, Ravindranathan R, Dai E, Roy EJ, Guo ZS and Bartlett DL:
Superagonist IL-15-armed oncolytic virus elicits potent antitumor
immunity and therapy that are enhanced with PD-1 blockade. Mol
Ther. 26:2476–2486. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Sahin U and Türeci Ö: Personalized
vaccines for cancer immunotherapy. Science. 359:1355–1360. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Sui H, Ma N, Wang Y, Li H, Liu X, Su Y and
Yang J: Anti-PD-1/PD-L1 therapy for non-small-cell lung cancer:
Toward personalized medicine and combination strategies. J Immunol
Res. 2018:69849482018. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Rafiq S, Yeku OO, Jackson HJ, Purdon TJ,
van Leeuwen DG, Drakes DJ, Song M, Miele MM, Li Z, Wang P, et al:
Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances
anti-tumor efficacy in vivo. Nat Biotechnol. 36:847–856. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Shi X, Zhang D, Li F, Zhang Z, Wang S,
Xuan Y, Ping Y and Zhang Y: Targeting glycosylation of PD-1 to
enhance CAR-T cell cytotoxicity. J Hematol Oncol. 12:1272019.
View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Xu J, Sun HH, Fletcher CD, Hornick JL,
Morgan EA, Freeman GJ, Hodi FS, Pinkus GS and Rodig SJ: Expression
of programmed cell death 1 ligands (PD-L1 and PD-L2) in histiocytic
and dendritic cell disorders. Am J Surg Pathol. 40:443–453. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Xia L, Liu Y and Wang Y: PD-1/PD-L1
blockade therapy in advanced non-small-cell lung cancer: Current
status and future directions. Oncologist. 24(Suppl 1): S31–S41.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B
and Freeman GJ: PD-L1 binds to B7-1 only in Cis on the same cell
surface. Cancer Immunol Res. 6:921–929. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Inman BA, Longo TA, Ramalingam S and
Harrison MR: Atezolizumab: A PD-L1-blocking antibody for bladder
cancer. Clin Cancer Res. 23:1886–1890. 2017. View Article : Google Scholar
|
|
193
|
Garassino MC, Cho BC, Kim JH, Mazières J,
Vansteenkiste J, Lena H, Corral Jaime J, Gray JE, Powderly J,
Chouaid C, et al: Durvalumab as third-line or later treatment for
advanced non-small-cell lung cancer (ATLANTIC): An open-label,
single-arm, phase 2 study. Lancet Oncol. 19:521–536. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Lui Y and Davis SJ: LAG-3: A very singular
immune checkpoint. Nat Immunol. 19:1278–1279. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Maruhashi T, Sugiura D, Okazaki IM and
Okazaki T: LAG-3: From molecular functions to clinical
applications. J Immunother Cancer. 8:e0010142020. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Anderson AC, Joller N and Kuchroo VK:
Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized
functions in immune regulation. Immunity. 44:989–1004. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Das M, Zhu C and Kuchroo VK: Tim-3 and its
role in regulating anti-tumor immunity. Immunol Rev. 276:97–111.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Thomas A, Teicher BA and Hassan R:
Antibody-drug conjugates for cancer therapy. Lancet Oncol.
17:e254–e262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Sharabi AB, Lim M, DeWeese TL and Drake
CG: Radiation and checkpoint blockade immunotherapy:
Radiosensitisation and potential mechanisms of synergy. Lancet
Oncol. 16:e498–e509. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Yaghoubi S, Karimi MH, Lotfinia M, Gharibi
T, Mahi-Birjand M, Kavi E, Hosseini F, Sineh Sepehr K, Khatami M,
Bagheri N, et al: Potential drugs used in the antibody-drug
conjugate (ADC) architecture for cancer therapy. J Cell Physiol.
235:31–64. 2020. View Article : Google Scholar
|
|
201
|
Qin SY, Cheng YJ, Lei Q, Zhang AQ and
Zhang XZ: Combinational strategy for high-performance cancer
chemotherapy. Biomaterials. 171:178–197. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Yu WD, Sun G, Li J, Xu J and Wang X:
Mechanisms and therapeutic potentials of cancer immunotherapy in
combination with radiotherapy and/or chemotherapy. Cancer Lett.
452:66–70. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Pérez-Herrero E and Fernández-Medarde A:
Advanced targeted therapies in cancer: Drug nanocarriers, the
future of chemotherapy. Eur J Pharm Biopharm. 93:52–79. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Nadal R and Bellmunt J: Management of
metastatic bladder cancer. Cancer Treat Rev. 76:10–21. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Akbari B, Farajnia S, Ahdi Khosroshahi S,
Safari F, Yousefi M, Dariushnejad H and Rahbarnia L: Immunotoxins
in cancer therapy: Review and update. Int Rev Immunol. 36:207–219.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Alewine C, Hassan R and Pastan I: Advances
in anticancer immunotoxin therapy. Oncologist. 20:176–185. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Polito L, Djemil A and Bortolotti M: Plant
toxin-based immunotoxins for cancer therapy: A short overview.
Biomedicines. 4:122016. View Article : Google Scholar
|
|
208
|
Madhumathi J, Devilakshmi S, Sridevi S and
Verma RS: Immunotoxin therapy for hematologic malignancies: Where
are we heading? Drug Discov Today. 21:325–332. 2016. View Article : Google Scholar
|
|
209
|
Kumar M, Thangavel C, Becker RC and
Sadayappan S: Monoclonal antibody-based immunotherapy and its role
in the development of cardiac toxicity. Cancers (Basel). 13:862020.
View Article : Google Scholar
|
|
210
|
Tse BW, Collins A, Oehler MK, Zippelius A
and Heinzelmann-Schwarz VA: Antibody-based immunotherapy for
ovarian cancer: Where are we at? Ann Oncol. 25:322–331. 2014.
View Article : Google Scholar
|
|
211
|
Chau CH, Steeg PS and Figg WD:
Antibody-drug conjugates for cancer. Lancet. 394:793–804. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Khongorzul P, Ling CJ, Khan FU, Ihsan AU
and Zhang J: Antibody-drug conjugates: A comprehensive review. Mol
Cancer Res. 18:3–19. 2020. View Article : Google Scholar
|
|
213
|
Tsuchikama K and An Z: Antibody-drug
conjugates: Recent advances in conjugation and linker chemistries.
Protein Cell. 9:33–46. 2018. View Article : Google Scholar :
|
|
214
|
Erickson HK, Park PU, Widdison WC, Kovtun
YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS and Blättler WA:
Antibody-maytansinoid conjugates are activated in targeted cancer
cells by lysosomal degradation and linker-dependent intracellular
processing. Cancer Res. 66:4426–4433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Huang Y, Lee C, Borgström P and Gjerset
RA: Macrophage-mediated bystander effect triggered by tumor cell
apoptosis. Mol Ther. 15:524–533. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Staudacher AH and Brown MP: Antibody drug
conjugates and bystander killing: Is antigen-dependent
internalisation required? Br J Cancer. 117:1736–1742. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Beck A, Goetsch L, Dumontet C and Corvaïa
N: Strategies and challenges for the next generation of
antibody-drug conjugates. Nat Rev Drug Discov. 16:315–337. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Birrer MJ, Moore KN, Betella I and Bates
RC: Antibody-drug conjugate-based therapeutics: State of the
science. J Natl Cancer Inst. 111:538–549. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Mahalingaiah PK, Ciurlionis R, Durbin KR,
Yeager RL, Philip BK, Bawa B, Mantena SR, Enright BP, Liguori MJ
and Van Vleet TR: Potential mechanisms of target-independent uptake
and toxicity of antibody-drug conjugates. Pharmacol Ther.
200:110–125. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Lambert JM and Berkenblit A: Antibody-drug
conjugates for cancer treatment. Annu Rev Med. 69:191–207. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Duerr C and Friess W: Antibody-drug
conjugates-stability and formulation. Eur J Pharm Biopharm.
139:168–176. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Theocharopoulos C, Lialios PP, Gogas H and
Ziogas DC: An overview of antibody-drug conjugates in oncological
practice. Ther Adv Med Oncol. Oct 4–2020.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Abdollahpour-Alitappeh M, Lotfinia M,
Gharibi T, Mardaneh J, Farhadihosseinabadi B, Larki P, Faghfourian
B, Sepehr KS, Abbaszadeh-Goudarzi K, Abbaszadeh-Goudarzi G, et al:
Antibody-drug conjugates (ADCs) for cancer therapy: Strategies,
challenges, and successes. J Cell Physiol. 234:5628–5642. 2019.
View Article : Google Scholar
|
|
224
|
Liu L: Pharmacokinetics of monoclonal
antibodies and Fc-fusion proteins. Protein Cell. 9:15–32. 2018.
View Article : Google Scholar :
|
|
225
|
Yu B and Liu D: Antibody-drug conjugates
in clinical trials for lymphoid malignancies and multiple myeloma.
J Hematol Oncol. 12:942019. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Gébleux R and Casi G: Antibody-drug
conjugates: Current status and future perspectives. Pharmacol Ther.
167:48–59. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Tiller KE and Tessier PM: Advances in
antibody design. Annu Rev Biomed Eng. 17:191–216. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Ponziani S, Di Vittorio G, Pitari G,
Cimini AM, Ardini M, Gentile R, Iacobelli S, Sala G, Capone E,
Flavell DJ, et al: Antibody-drug conjugates: The new frontier of
chemotherapy. Int J Mol Sci. 21:55102020. View Article : Google Scholar :
|
|
229
|
Kobayashi H and Choyke PL: Near-infrared
photoimmunotherapy of cancer. Acc Chem Res. 52:2332–2339. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Ozog DM, Rkein AM, Fabi SG, Gold MH,
Goldman MP, Lowe NJ, Martin GM and Munavalli GS: Photodynamic
therapy: A clinical consensus guide. Dermatol Surg. 42:804–827.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Larue L, Myrzakhmetov B, Ben-Mihoub A,
Moussaron A, Thomas N, Arnoux P, Baros F, Vanderesse R, Acherar S
and Frochot C: Fighting hypoxia to improve PDT. Pharmaceuticals
(Basel). 12:1632019. View Article : Google Scholar
|
|
232
|
Nagaya T, Nakamura Y, Okuyama S, Ogata F,
Maruoka Y, Choyke PL, Allen C and Kobayashi H: Syngeneic mouse
models of oral cancer are effectively targeted by anti-CD44-Based
NIR-PIT. Mol Cancer Res. 15:1667–1677. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Mew D, Lum V, Wat CK, Towers GH, Sun CH,
Walter RJ, Wright W, Berns MW and Levy JG: Ability of specific
monoclonal antibodies and conventional antisera conjugated to
hematoporphyrin to label and kill selected cell lines subsequent to
light activation. Cancer Res. 45:4380–4386. 1985.PubMed/NCBI
|
|
234
|
Mew D, Wat CK, Towers GH and Levy JG:
Photoimmunotherapy: Treatment of animal tumors with tumor-specific
monoclonal antibody-hematoporphyrin conjugates. J Immunol.
130:1473–1477. 1983.PubMed/NCBI
|
|
235
|
Wang M, Rao J, Wang M, Li X, Liu K, Naylor
MF, Nordquist RE, Chen WR and Zhou F: Cancer photo-immunotherapy:
From bench to bedside. Theranostics. 11:2218–2231. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Mitsunaga M, Ogawa M, Kosaka N, Rosenblum
LT, Choyke PL and Kobayashi H: Cancer cell-selective in vivo near
infrared photoimmunotherapy targeting specific membrane molecules.
Nat Med. 17:1685–1691. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Isobe Y, Sato K, Nishinaga Y, Takahashi K,
Taki S, Yasui H, Shimizu M, Endo R, Koike C, Kuramoto N, et al:
Near infrared photoimmunotherapy targeting DLL3 for small cell lung
cancer. EBioMedicine. 52:1026322020. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Nishimura T, Mitsunaga M, Ito K, Kobayashi
H and Saruta M: Cancer neovasculature-targeted near-infrared
photoimmunotherapy (NIR-PIT) for gastric cancer: Different
mechanisms of phototoxicity compared to cell membrane-targeted
NIR-PIT. Gastric Cancer. 23:82–94. 2020. View Article : Google Scholar
|
|
239
|
Deshaies RJ: Multispecific drugs herald a
new era of biopharmaceutical innovation. Nature. 580:329–338. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Kaplon H, Muralidharan M, Schneider Z and
Reichert JM: Antibodies to watch in 2020. MAbs. 12:17035312020.
View Article : Google Scholar :
|