Introduction
Breast cancer (BC) has one of the highest incidences
of all malignant tumors worldwide and has the highest mortality
rate in females (1).
Specifically, in China, 270,000 patients were diagnosed with BC and
70,000 BC patients died from the disease in 2015 (2). BC has been reported as the most
common type of malignancy that is likely to metastasize to bone,
with a high prevalence of 70 to 90% in patients with known primary
BC in a study of postmortem cadavers (3,4).
Moreover, the prolonged survival of BC patients due to advances in
diagnosis and treatment has led to an increased rate of metastasis
(5). Approximately 65% of
advanced BC patients develop bone metastasis (6). Among them, 80% of BC bone metastases
are located in the spine; 70 to 80% of BC is the estrogen
receptor-positive (ER+) subtype, and luminal subtypes
(ER+) of BC are reported to be highly associated with
bone metastasis (7,8). Therefore, spinal metastasis (SM) of
ER+ BC is common among bone metastatic tumors, which is
consistent with our clinical experience. As the late stage of
ER+ BC, SM is highly malignant and always resistant to
chemotherapies (7,8). However, few studies have focused on
the SM of BC, and the potential mechanism underlying the
progression and SM of ER+ BC remains unclear.
We screened relevant genes involved in the SM of
ER+ BC and identified lysosomal protein transmembrane 5
(LAPTM5) as an SM-related gene. As a protein located in the
lysosomal membrane, LAPTM5 is thought to be preferentially
expressed in immune cells (9-11).
However, recent studies have reported that LAPTM5 expression is
downregulated in various types of cancers, including lung cancer,
esophageal squamous cell carcinoma, and glioblastoma, indicating
that the tumor-suppressive function of LAPTM5 and downregulated
expression of this gene may play a role in tumor progression
(12,13). However, the exact role of LAPTM5
in human BC remains unknown.
Cancer cells are characterized by their reprogrammed
nutrient metabolism (14,15). Recently, glutamine was reported to
be one of the key nutrients of BC, and glutamine metabolism
facilitates the proliferation, progression and chemoresistance of
BC (16,17). During the process of glutamine
metabolism, sodium-dependent neutral amino acid transporter type 2
(SLC1A5) and glutaminase 1 (GLS1) are two critical molecules. The
former mediates uptake of neutral amino acids including glutamine,
and the latter catalyzes glutamine into glutamate (18,19). Gene set enrichment analysis has
indicated that LAPTM5 expression is related to the nutrient
metabolism of ER+ BC. Moreover, some studies have shown
that glutamine metabolism can activate mammalian target of
rapamycin (mTOR), which plays an important role in regulating the
fundamental physiological functions of cancer cells, including
protein synthesis, proliferation, migration, and autophagy
(20,21). Therefore, we speculated that
glutamine-dependent activation of mTOR signaling may mediate the
biological function of LAPTM5.
C-X3-C motif chemokine ligand 1 (CX3CL1) has been
regarded as an essential mediator in tumor metastasis via binding
to its receptor CX3CR1-expressing cells to endothelial cells
(22). Our previous research and
other works confirmed that CX3CL1/CX3CR1 interaction is involved in
the survival, adhesion, and migration of breast cancer cells
(23,24). Furthermore, a higher level of
CX3CL1/CX3CR1 was found in vertebrae than in limb bone, which may
account for SM (23,25). However, the exact molecular
mechanism of CX3CL1/CX3CR1-mediated SM of breast cancer still
remains unclear.
Collectively, we hypothesized that downregulation of
LAPTM5 expression could enhance the progression of ER+
BC by activating glutamine-dependent mTOR signaling. To examine our
hypothesis, we detected the impact of LAPTM5 expression on
glutamine metabolism and activation of downstream mTOR signaling in
ER+ BC both in vitro and in vivo.
Subsequently, we explored the role of LAPTM5 in the SM of
ER+ BC using a mouse model of SM. The aims of this study
were to ascertain the role of LAPTM5 in tumorigenesis and the SM of
ER+ BC and reveal the underlying mechanism.
Materials and methods
ER+ BC specimens
A total of 19 clinical specimens were obtained from
the Department of Orthopedic Surgery, Zhongshan Hospital, Fudan
University and the Department of General Surgery, Xuhui-Zhongshan
Hospital, Fudan University. The specimens included 7 SM samples of
ER+ BC (median age, 53 years; range, 43-60 years), 6
samples of primary ER+ BC (median age, 49.5 years;
range, 45-60 years), and 6 corresponding tumor-adjacent normal
tissues of primary ER+ BC. The clinicopathological data
of these patients are shown in Table
SI. The acquisition of patient specimens and the research
procedures were approved (approval no. Y2019-085, 2019.02.27) by
the Ethics Committee of Zhongshan Hospital, Fudan University. The
relevant rules and regulations of the Ethics Committee of Zhongshan
Hospital, Fudan University were strictly followed, and informed
consent was provided by all patients.
Bioinformatics analysis
Gene Ontology (GO; http://geneontology.org/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; https://www.kegg.jp/kegg/) analyses were used to
identify the biological functions of LAPTM5 expression in
ER+ BC. The biological processes and enriched pathways
of the proteins encoded by the candidate genes were analyzed using
The Database for Annotation, Visualization and Integrated Discovery
(DAVID 6.8, https://david.ncifcrf.gov/home.jsp) (26,27). P<0.05 was set as the cut-off
criterion. The GEO2R web tool (http://ncbi.nlm.nih.gov/geo/geo2r/) was used to
analyze the gene expression profile of LAPTM5 in ER+ BC
tissues from patients with primary disease or SM (GSE14661)
(28). Gene Set Enrichment
Analysis (GSEA; https://www.gsea-msigdb.org/gsea/index.jsp) was
performed to screen the GO terms and KEGG pathways that may be
associated with LAPTM5 in the database.
Cell lines
293T cells (SCSP-502) and the human ER+
BC cell lines MCF-7 (SCSP-531) and T47D (KG115) were purchased from
The Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences. The above cells were maintained in Dulbecco's modified
Eagle's medium (DMEM, Gibco/Thermo Fisher Scientific, Inc.)
containing 10% fetal bovine serum (FBS, 10091-148, Gibco/Thermo
Fisher Scientific, Inc.) with incubation in humidified air
containing 5% CO2 at 37°C.
Reagents
The glutaminase inhibitor BPTES (S7753), the CX3CR1
inhibitor JMS-17-2 (S0135), the AKT inhibitor MK-2206 (S1078), and
docetaxel (S1148) were purchased from Selleck Chemicals (China).
Lipofectamine® 3000 was purchased from Invitrogen
(L3000001; Thermo Fisher Scientific, Inc.). Recombinant human
fractalkine/CX3CL1 was purchased from MedChemExpress (HY-P7355,
China).
Plasmids and lentivirus
The third-generation lentivirus packing system
including hU6-MCS-ubiquitin-EGFP-RES-puromycin, psPAX2, and pMD2.G
was purchased from GeneChem (China). Three RNAi sequences targeting
human LAPTM5 were designed (LAPTM5 shRNA1, gcG GTG
CTA CAG ATT GAT CAA; shRNA2, tcA TAA CCA GTT CAT CAA GAT; and
shRNA3, gcT CCA GGA AAT AAC AGT TAT). Then, the three shRNAs
targeting human LAPTM5 were designed, annealed, and inserted
into the lentivirus. For ER+ BC cell lines that stably
overexpressed LAPTM5, the lentivirus plasmid GV657-LAPTM5 was
constructed by subcloning the LAPTM5 coding sequence (Gene ID:
7805) into the modified GV657-puro backbone (GeneChem, China). The
abovementioned plasmids (7 µg psPAX2, 3 µg pMD2.G, 1
µg shRNA) were added to 293T cells at approximately 30%
density with 20 µl of Lipofectamine 3000 (L3000-015;
Invitrogen/Thermo Fisher Scientific, Inc.). After incubation for 6
to 12 h, the culture medium was changed to fresh DMEM containing
10% FBS. Culture medium containing the lentivirus was collected at
2 and 3 days, filtered through a 0.45-µm cellulose acetate
filter (Millipore) and stored in an ultra-low temperature
refrigerator. MCF-7 and T47D cells were infected by the lentivirus
with 6 µg/ml polybrene (TR-1003; Sigma-Aldrich/Merck KGaA)
for 72 h at 37°C. Finally, the successfully transfected cells were
screened out using puromycin (A1113802; Thermo Fisher Scientific,
Inc.) for 1 week.
Colony formation assay
ER+ BC cells in a logarithmic growth
state were digested with 0.25% trypsin and blown into single cells.
The cells were suspended in the culture medium and inoculated in a
culture dish at 500 cells/well. The cells were incubated at 37°C
with 5% CO2 and saturated humidity for 2 to 3 weeks.
When obvious clones appeared in the culture dish, the experiment
was terminated. The supernatant was discarded, and the cells were
washed twice with PBS buffer. Five milliliters of 4%
paraformaldehyde was added to fix the cells for 15 min. Then, the
fixing solution was removed, and 1% crystal violet was added to dye
for 10 min. After the dyeing solution was washed away with running
water, the clones could be counted with the naked eye and
photographed (E-M10 MarkIV; Olympus).
Wound healing assay
ER+ BC cells were seeded in 6-well plates
at a density of 1×105 cells per well and cultured to
approximately 90% confluence. A horizontal wound was created and
photographed immediately. The cells were then stimulated with 2
µM docetaxel or control PBS buffer. Finally, the images at
24 and 48 h were also captured (CKX53; Olympus), and the wound
areas after healing were measured and calculated.
Cell inhibition assay
ER+ BC cells were seeded in 96-well
culture plates (3,000 cells/well) and cultured at 37°C with 5%
CO2. For growth inhibition assays, after incubation for
12 h for cell adherence to culture plates, a concentration gradient
of docetaxel or control PBS buffer was added for 72 h of incubation
at 37°C. Then, 10 µl/well of Cell Counting Kit (CCK-8, CK04;
Dojindo, Japan) reagent was added after an additional 2 h
incubation at 37°C. Finally, the optical density (OD)450 value was
detected by a microplate reader, and the half maximal inhibitory
concentration (IC50) was calculated by GraphPad Prism 8
software (GraphPad Software, Inc.).
Western blot analysis
Protein samples were extracted from tissues or cells
by RIPA lysis buffer (P0013B; Beyotime Institute of Biotechnology)
containing phenylmethanesulfonyl fluoride (ST505, Beyotime
Institute of Biotechnology) and phosphorylase inhibitor (78445;
Thermo Fisher Scientific, Inc.). The protein concentration was
determined with a Pierce™ BCA protein quantification kit (23225;
Thermo Fisher Scientific, Inc.). Twenty micrograms of protein
sample was separated by 10% SDS-PAGE, transferred to a PVDF
membrane (ISEQ00010; Millipore), and blocked using 5% fat-free milk
for 1 h at 25°C. Then, the membranes were incubated with primary
antibodies against LAPTM5 (PA5-23585; Thermo Fisher Scientific,
Inc., 1:1,000), SLC1A5 [8057; Cell Signaling Technology, Inc.
(CST), 1:1,000], GLS1 (56750, CST, 1:1,000), glutaminase 1 (GLS2)
(ab150474; Abcam, 1:1,000), Raptor (48648, CST, 1:1,000), ribosomal
protein S6 kinase 1 (S6K1) (14130, CST, 1:1,000), phosphorylated
(p-) S6K1 (9209, CST, 1:1,000), eukaryotic translation initiation
factor 4E (eIF4E)-binding protein 1 (4EBP1) (9644, CST, 1:1,000),
p-4EBP1 (2855, CST, 1:1,000), matrix metallopeptidase 9 (MMP9)
(13667, CST, 1:1,000), cyclin D1 (55506, CST, 1:1,000), NFκB p65
(8242, CST, 1:1,000), NFκB p-p65 (3033, CST, 1:1,000), Histone H3
(4499, CST, 1:1,000), PI3K (4249, CST, 1:1,000), p-PI3K (13857,
CST, 1:1,000), AKT (4691, CST, 1:1,000), p-AKT Ser473 (4060, CST,
1:1,000), p-AKT Thr308 (13038, CST, 1:1,000), and β-actin (3700,
CST, 1:1,000) at 4°C overnight and incubated with anti-mouse
(43593, CST, 1:3,000) or anti-rabbit secondary antibodies (7074,
CST, 1:3,000) for 2 h at 25°C. Proteins were visualized using
Pierce™ ECL western substrate (32209; Thermo Fisher Scientific,
Inc.). Quantitative analysis of relative protein expression was
performed with ImageJ software (1.52 V; National Institutes of
Health).
Real time-quantitative PCR (qPCR)
Total RNA of tissues or cultured cells was extracted
by TRIzol reagent (15596026; Invitrogen/Thermo Fisher Scientific,
Inc.). Then, the PrimeScript™ RT reagent Kit (RR037Q; Takara) was
applied for reverse transcription. Real-time PCR was conducted
using TB Green® Fast qPCR Mix according to the
manufacturer's instructions (RR430S; Takara) using a thermocycler
(Thermo-ABI 7500; Thermo Fisher Scientific, Inc.). The primers used
in this experiment were purchased from Sangon Biotech (China) and
were as follows: LAPTM5: forward, 5′-GCG TCT TGT TGT TCA TCG
AGC-3′; reverse, 5′-CGA TCC TGA GGT AGC CCA T-3′; GAPDH: forward,
5′-GGA GCG AGA TCC CTC CAA AAT-3′; reverse, 5′-GGC TGT TGT CAT ACT
TCT CAT GG-3′. The following thermocycling conditions were used:
initial denaturation at 95°C for 30 sec followed by 35 cycles at
95°C for 5 sec and 60°C for 30 sec. The 2−∆∆Cq method
was used for relative quantification of the genes, and GAPDH was
used as the internal reference (29).
Animal experiments
Male nude mice aged 4-6 weeks (weight, 15-17 g) were
obtained from Vital River Laboratory Animal Technology (China) and
randomly assigned to experimental groups. The animals were housed
under controlled environmental conditions (12 h dark/light cycle;
20-22°C; humidity, 55±5%), and allowed free access to normal food
and water. A total of 15 mice (five in each group) were
anesthetized by intraperitoneal injection of 0.3% sodium
pentobarbital (30-60 mg/kg). For the in vivo subcutaneous
tumorigenesis assay, 2×106 blank, LAPTM5-sh3, and
LAPTM5-OE MCF-7 cells were injected into the subcutaneous right
forelimb armpit of the anesthetized mice. Then, 30 mg/kg docetaxel
was injected via the tail vein once every other day. Tumor size was
measured twice a week using a caliper to measure two perpendicular
tumor diameters. Tumor volume (mm3) was calculated as
follows: Volume=0.5 × length × width2. One month later,
the mice were sacrificed by intraperitoneal injection of sodium
pentobarbital (200 mg/kg, death was verified by respiratory and
cardiac arrest, and pupil dilation), and the tumors were
photographed and subjected to hematoxylin and eosin (H&E)
staining and immunohistochemistry (IHC) staining as previously
described (25).
For the SM assay, 1×106 blank or
LAPTM5-sh3 MCF-7 cells were injected into the left cardiac
ventricle of the anesthetized mice (five in each group) (25). Docetaxel (30 mg/kg) was
intravenously injected once every other day; BPTES (12.5 mg/kg) was
intraperitoneally injected every 3 days (30), and JMS-17-2 (10 nM) was added to
the cell suspension 30 min before inoculation (31). The development of SM was monitored
by bioluminescence imaging (BLI) once a week using a Xenogen IVIS
200 Imaging System (Xenogen, USA) for 6-8 weeks. Finally, the mice
were sacrificed when obvious SM could be observed via BLI in the
control group, and the tumors were scanned by micro-computed X-ray
tomography (micro-CT) and subjected to H&E and IHC staining.
Micro-CT is a non-destructive 3D X-ray imaging technique used to
study micro-changes of bone structure. Spinal metastasis of
ER+ BC mainly manifests as osteolytic destruction
pathologically; therefore, micro-CT could show cortical defect and
bone tissue destruction in the metastatic lesions of the spine. For
the convenience of readers, we marked cortical defects and bone
tissue destructions with red arrows in Fig. 7B. The animal experimental
procedures were approved by the Animal Ethics Committee of
Zhongshan Hospital, Fudan University (accession number: Y2021-322),
and their care was in accordance with institutional guidelines. No
mice died unexpectedly during the experiment.
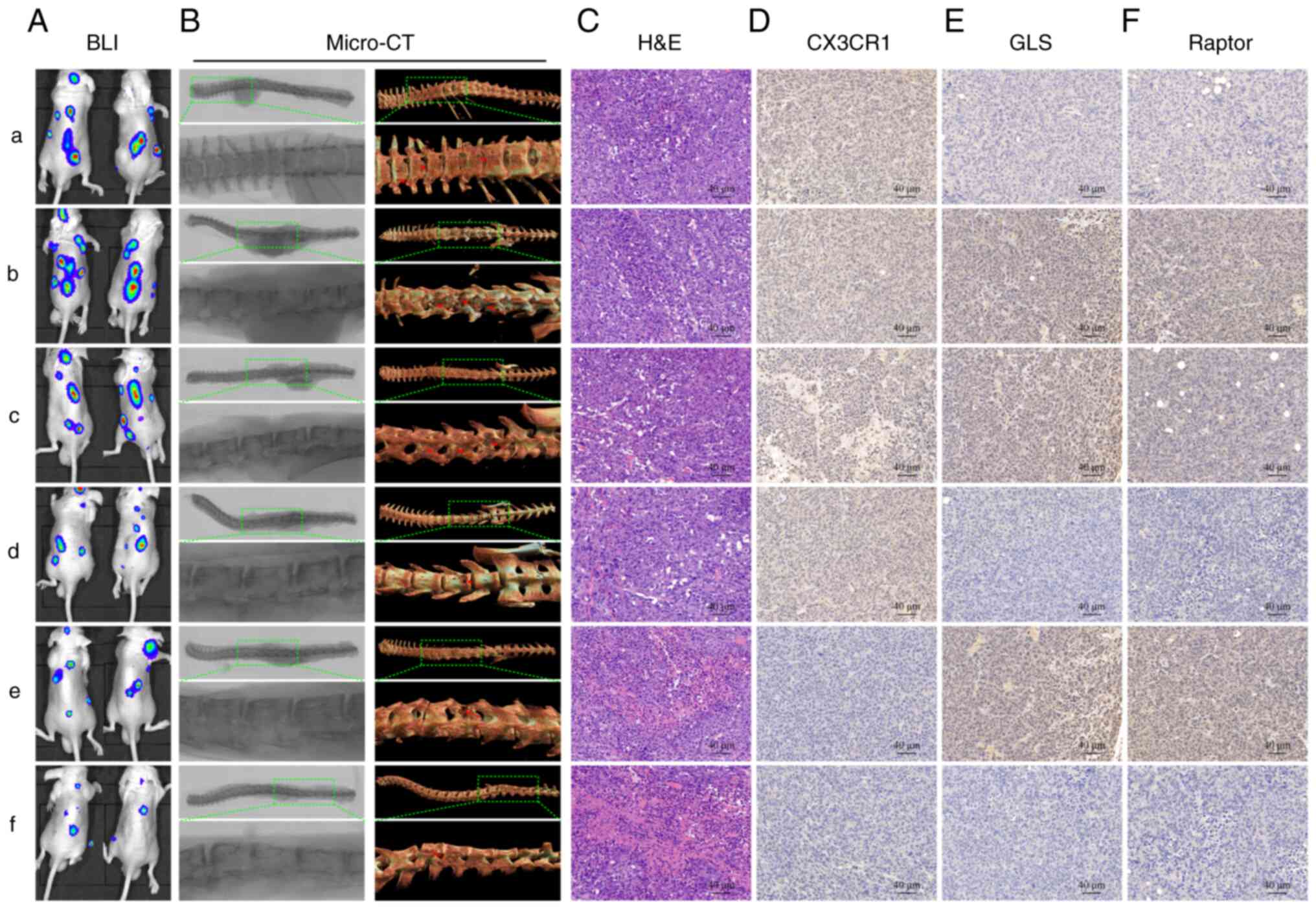 | Figure 7Blockade of CX3CL1/CX3CR1 and
glutaminase inhibits SM and enhances the chemosensitivity of
ER+ BC cells. (A) Representative bioluminescence imaging
(BLI) imaging of each mouse group. (B) Representative micro-CT
images of each group. (C) Hematoxylin and eosin (H&E) staining
of each group. (D-F) Immunohistochemistry (IHC) staining for
CX3CR1, GLS1, and Raptor in each group. Blank MCF-7 cells were used
as the control group (a). The other groups were established with
LAPTM5-sh3 (knockdown) cells, and groups (b-f) were injected with
saline (b), docetaxel (c), BPTES+docetaxel (d), JMS-17-2+docetaxel
(e), and BPTES+JMS-17-2+docetaxel (f). CX3CL1, C-X3-C motif
chemokine ligand 1; CX3CR1, C-X3-C motif chemokine receptor 1;
ER+ BC, estrogen receptor-positive breast cancer; SM,
spinal metastasis; LAPTM5, lysosomal protein transmembrane 5; GLS1,
glutaminase 1. |
Statistical analysis
All data are presented as the mean ± SD of at least
three repeated experiments. One-way analysis of variance (ANOVA) or
two-tailed Student's t-test using GraphPad Prism 8 software
(GraphPad Software, Inc.) was applied to analyze differences among
groups. A P-value of <0.05 was considered statistically
significant (*P<0.05, **P<0.01, and
***P<0.001, as denoted in the figures/legends).
Results
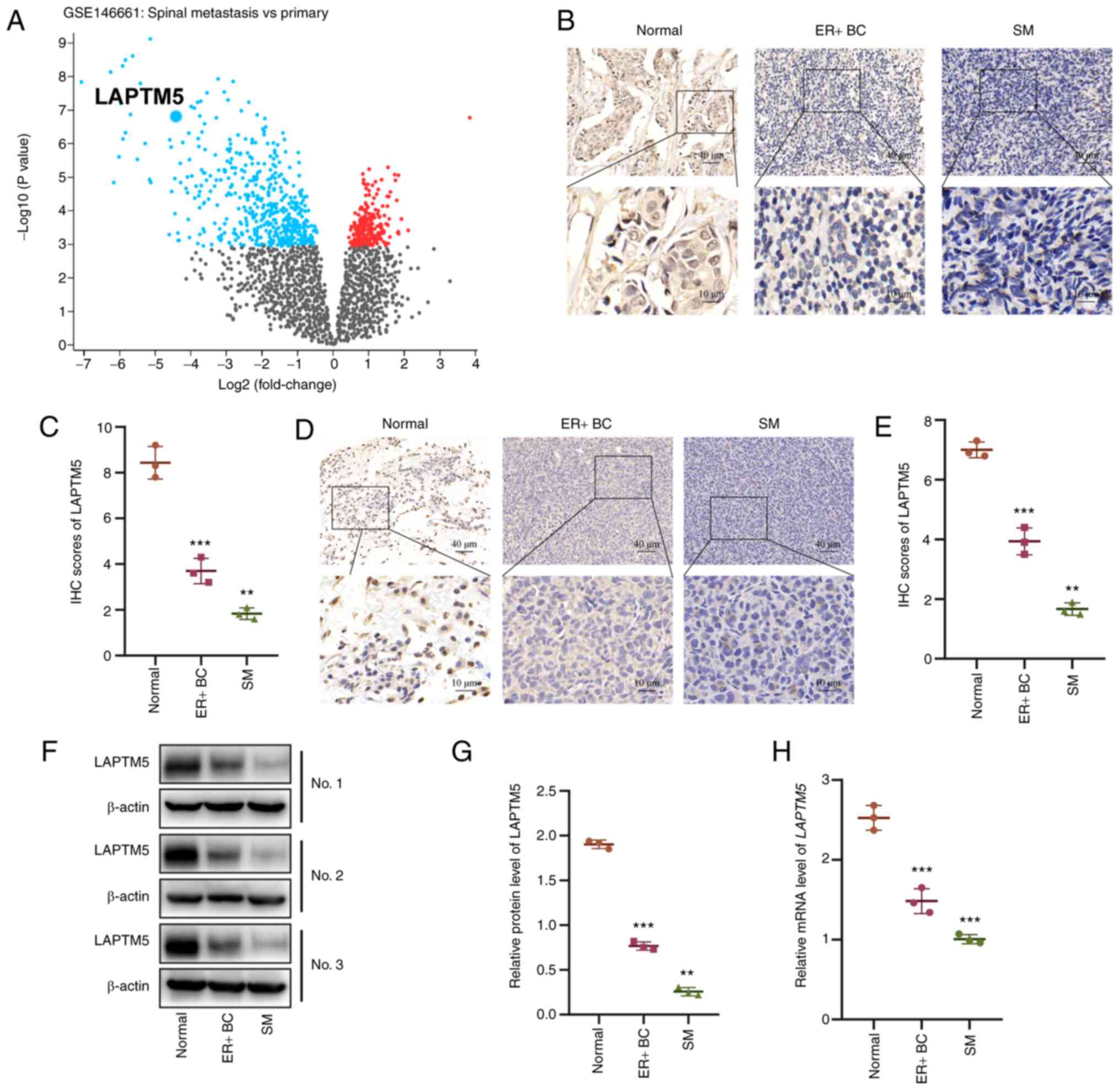
The LAPTM5 level is substantially
decreased in SM samples of ER+ BC
LAPTM5 has been reported as a tumor suppressor in
many types of cancer, but its role in ER+ BC is still
unclear. First, we analyzed the gene expression profile of
ER+ BC tissues from patients with primary disease or SM
using the GEO2R web tool (32).
The results showed that LAPTM5 was one of the top genes with
downregulated expression in SM samples compared with primary
tumors, indicating that it was negatively related to tumor
progression and involved in SM in ER+ BC (Figs. 1A and S1). To verify the above sequencing
results, we collected primary ER+ BC samples and SM
samples from patients undergoing surgery in our hospital. As shown
in Fig. 1B and C, IHC staining
demonstrated decreased expression of LAPTM5 in ER+ BC
both in the primary site and SM compared with the tumor-adjacent
normal tissues, but the lowest level was observed in the SM
samples. Furthermore, we established orthotopic ER+ BC
and SM mouse models to strengthen our findings. The IHC (Fig. 1D and E), western blot analysis
(Fig. 1F and G), and qPCR
(Fig. 1H) results revealed that
LAPTM5 expression was reduced sequentially in tumor-adjacent normal
tissues, primary ER+ BC, and SM. Consistent with the
above findings, the differences between each group were
statistically significant. Collectively, these results demonstrated
that downregulation of LAPTM5 expression is correlated with the
spinal metastasis of ER+ BC.
LAPTM5 expression is correlated with the
proliferation, migration, chemoresistance, and tumorigenesis of
ER+ BC
To further examine the role of LAPTM5 in
ER+ BC, we established LAPTM5-knockdown (sh) and
-overexpressing (OE) MCF-7 and T47D cell lines. As shown in
Figs. 2A-C and S2A and B, the LAPTM5-sh3 groups had the
lowest expression level of LAPTM5 and were chosen for the following
experiments. We first observed the impact of LAPTM5 expression on
the proliferation and migration of ER+ BC cells with or
without treatment with the chemotherapeutic drug docetaxel. The
colony formation results showed an obviously enhanced growth rate
of the LAPTM5-sh3 cells and an impaired proliferative capacity of
the LAPTM5-OE cells without docetaxel treatment (Figs. 2D and E, S2C and D). After treatment with
docetaxel, the growth rate of the cells in all three groups was
reduced, but the LAPTM5-sh3 cells exhibited significant resistance
to docetaxel, as they still formed many clones. Similarly, in the
wound healing assay, the LAPTM5-sh3 cells showed the highest
healing rate among the cells in all groups, while the LAPTM5-OE
cells almost lost their migratory ability (Figs. 2F and G, S2E and F). Moreover, although docetaxel
treatment for 48 h caused varying degrees of inhibition of the
healing rate, the LAPTM5-sh3 cells still exhibited stronger
migratory ability than other groups. The above cell experiments
demonstrated that LAPTM5 inhibits the in vitro proliferation
and migration of ER+ BC cells but promote their
chemosensitivity.
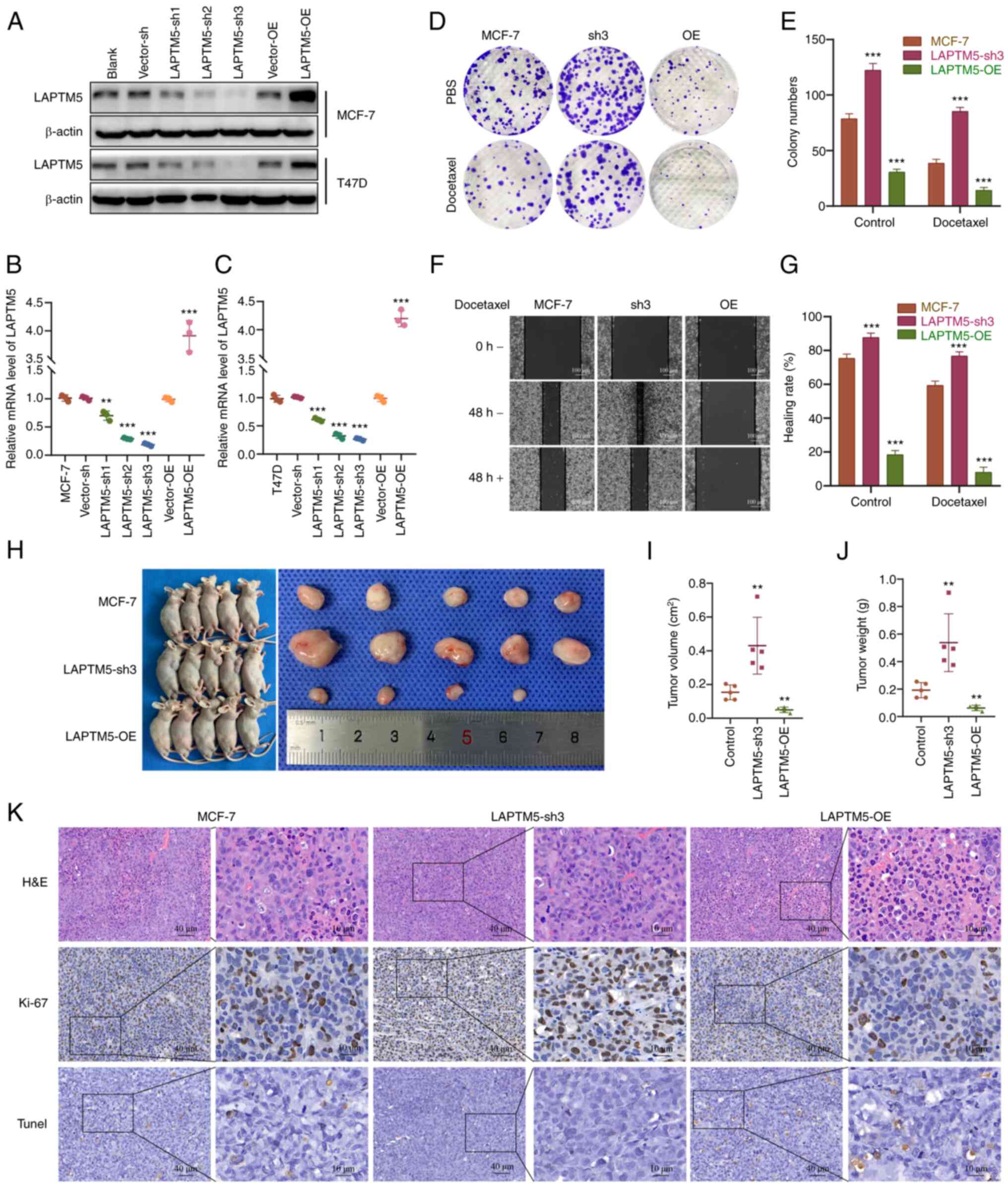 | Figure 2LAPTM5 is negatively associated with
the proliferation and migration in vitro and tumorigenesis
in vivo of ER+ BC cells under docetaxel
treatment. (A) The establishment of LAPTM5-knockdown (sh1-3) or
-overexpressing (OE) MCF-7 and T47D cell lines. The relative mRNA
levels of LAPTM5 in MCF-7 (B) and T47D (C) cells.
**P<0.01 and ***P<0.001, compared with
the blank MCF-7 and T47D cells. (D) Colony formation results of
blank, LAPTM5-sh3 and LAPTM5-OE MCF-7 cells with or without
docetaxel treatment. (E) Statistical analysis of the colony
formation assay. ***P<0.001, compared with the blank
MCF-7 cells. (F) The wound healing results of blank, LAPTM5-sh3 and
LAPTM5-OE MCF-7 cells with or without docetaxel treatment. (G)
Statistical analysis of the wound healing assay.
***P<0.001, compared with the blank MCF-7 cells. (H)
Subcutaneous tumorigenesis of blank, LAPTM5-sh3 and LAPTM5-OE MCF-7
cells under docetaxel treatment. (I and J) Statistical analyses of
tumor volume and weight. **P<0.01, compared with the
mouse group inoculated with MCF-7 blank cells. (K) H&E, Ki-67
and TUNEL staining results of the above three groups. LAPTM5,
lysosomal protein transmembrane 5; ER+ BC, estrogen
receptor-positive breast cancer. |
Then, an in vivo subcutaneous tumorigenesis
assay was conducted. Fig. 2H-J
showed stronger tumorigenic ability of the LAPTM5-sh3
ER+ BC cells compared with the blank or LAPTM5-OE MCF-7
cells when mice were injected with docetaxel. The results of the
histopathological examinations are shown in Figs. 2K, S2G and H. H&E and Ki-67 staining
indicated the state of cell proliferation of the LAPTM5-sh3 group,
while H&E and TUNEL staining demonstrated obvious necrosis of
the LAPTM5-OE group under docetaxel treatment. Taken together,
these data demonstrated that LAPTM5 was negatively associated with
the proliferation and migration in vitro and tumorigenesis
in vivo of the ER+ breast cancer cells under
docetaxel treatment.
LAPTM5 regulates the malignant
progression of ER+ BC through glutamine-mediated mTOR
signaling
Before investigating the molecular mechanism
underlying the regulatory effect of LAPTM5 on ER+ BC, we
detected the IC50 of docetaxel in blank, LAPTM5-sh3, and
LAPTM5-OE MCF-7 cells. As shown in Fig. 3A-D, the IC50 value of
the LAPTM5-sh3 group was significantly higher than the values of
the other groups, while LAPTM5 overexpression promoted the
chemosensitivity of cancer cells. Given that the gene set
enrichment analysis indicated that LAPTM5 may regulate cell
metabolism and that glutamine is one of the key nutrient sources
and is crucial for the biological function of BC (Fig. S3), glutamine metabolism was
tested to verify our speculation. As shown in Fig. 3E-G, the glutamine transporters
SLC1A5 and GLS1 showed significant upregulated expression when
LAPTM5 was inhibited, illustrating the effect of LAPTM5 on
glutamine metabolism. In addition, no significant changes were
found in the glutaminase 2 (GLS2) expression between
LAPTM5-silenced and control ER+ BC cells (Fig. S4). In addition as shown in
Fig. 3E-G, the mTOR complex 1
(mTORC1) component Raptor and its downstream factors ribosomal
protein S6 kinase B1 (S6K1) and eukaryotic translation initiation
factor 4E binding protein 1 (4EBP1) were found to be significantly
activated in the LAPTM5-sh3 cells. After phosphorylation of S6K1
(p-S6K1) and 4EBP1 (p-4EBP1), matrix metallopeptidase 9 (MMP9),
cyclin D1, and intranuclear and phosphorylated NFκB p65 (NFκB
p-p65) were subsequently activated, explaining the enhanced
migration, proliferation and chemoresistance of LAPTM5-sh3 cells.
In contrast, the opposite results were observed in LAPTM5-OE cells.
Above all, these results indicated that LAPTM5 enhanced the
progression and resistance to docetaxel in ER+ BC cells
via activation of glutamine-mediated mTOR signaling.
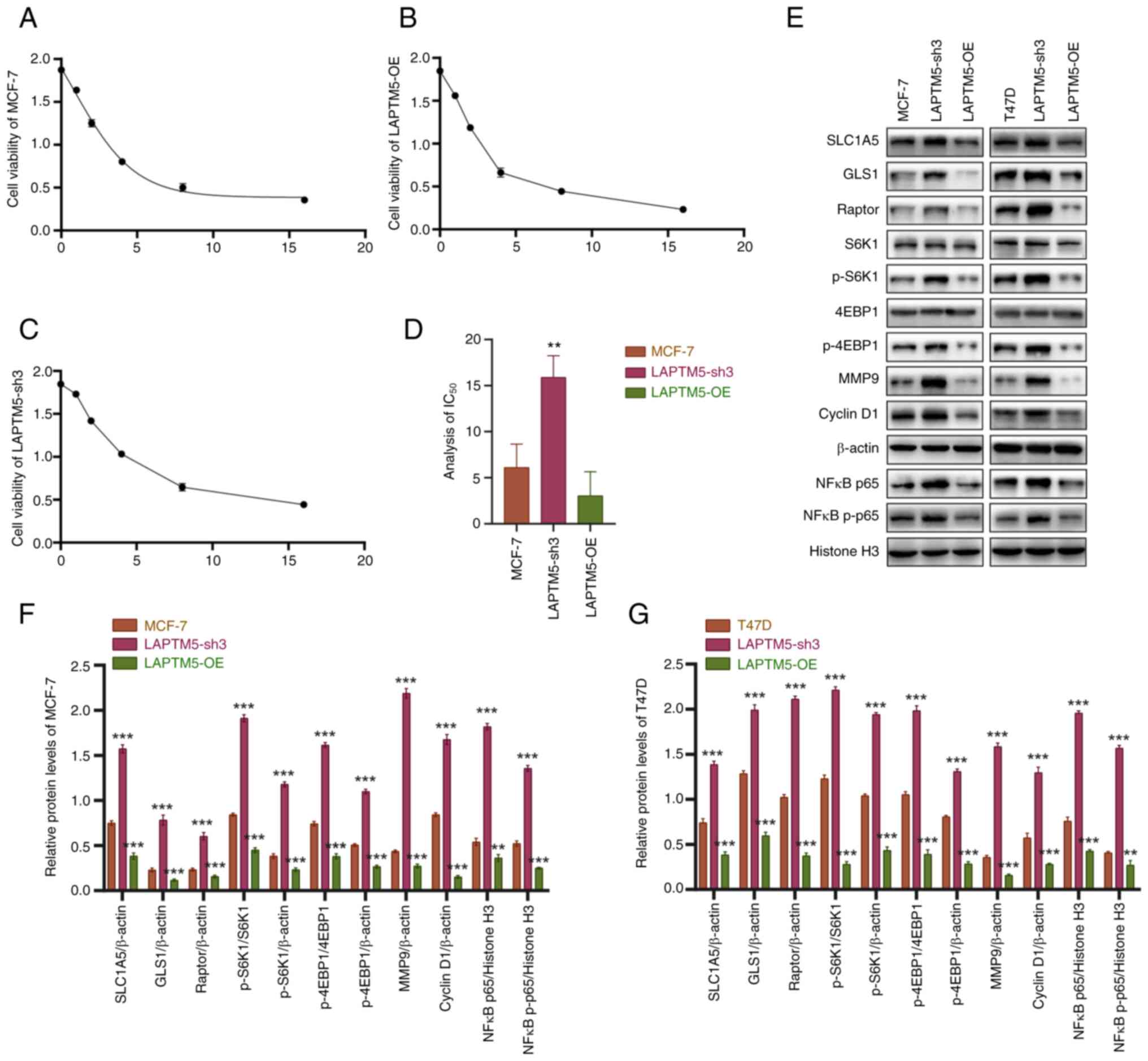 | Figure 3LAPTM5 enhances the resistance to
docetaxel in ER+ BC cells via the activation of glutamine-mediated
mTOR signaling. (A-C) Cell viability curves of blank (A),
LAPTM5-sh3 (knockdown) (C) and LAPTM5-OE (overexpressing) MCF-7 (B)
cells treated with 0, 1, 2, 4, 8, or 16 µM docetaxel for 24
h. (D) Statistical analysis of the half maximal inhibitory
concentration (IC50) values of the above three cell
lines. **P<0.01, compared to the blank MCF-7 cells.
(E) Protein expression and densitometric analysis results of key
molecules of glutamine metabolism and mTOR signaling in blank,
LAPTM5-sh3 and LAPTM5-OE MCF-7 and T47D cells. (F and G) Relative
protein levels of LAPTM5 in the different MCF-7 (F) and T47D (G)
cells. **P<0.01 and ***P<0.001,
compared with the blank MCF-7 and T47D cells. LAPTM5, lysosomal
protein transmembrane 5; ER+ BC, estrogen
receptor-positive breast cancer; SLC1A5, sodium-dependent neutral
amino acid transporter type 2; GLS1, glutaminase 1; S6K1, ribosomal
protein S6 kinase 1; 4EBP1, eukaryotic translation initiation
factor 4E (eIF4E)-binding protein 1; MMP9, matrix metallopeptidase
9; nuclear factor κB, NFκB; p-, phosphorylated. |
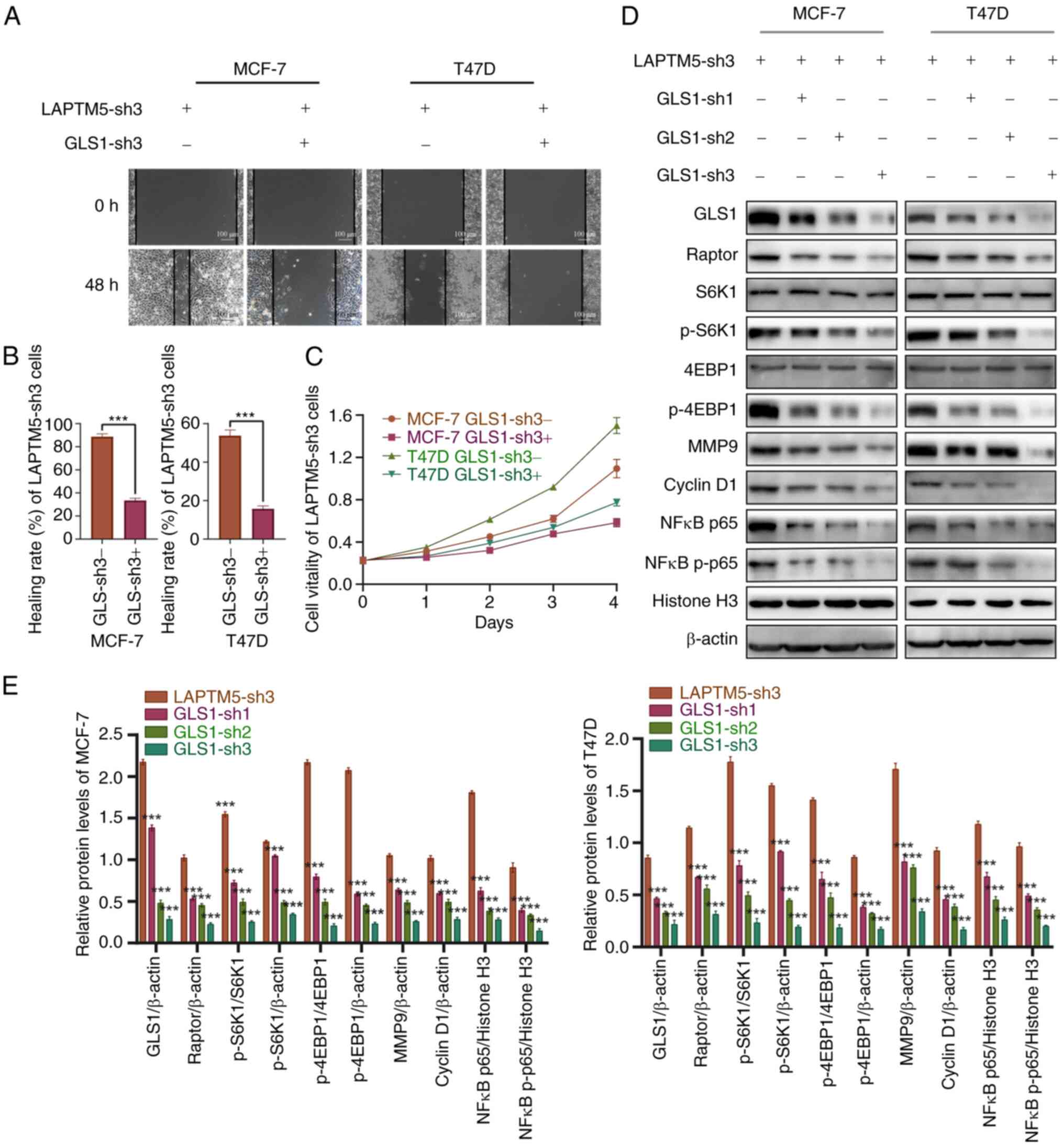
Glutaminase inhibitor BPTES blocks the
effect of LAPTM5 inhibition
To further elucidate the molecular mechanism of
LAPTM5 in regulating ER+ BC, we treated blank or
LAPTM5-sh3 cells with docetaxel, the glutaminase inhibitor BPTES,
or both docetaxel and BPTES. As shown in Fig. 4A, after treatment with docetaxel
alone, the activation of mTORC1 and the phosphorylation of its
downstream factors S6K1 and 4EBP1 was suppressed in the blank
ER+ BC cells by the chemotherapeutic drug. However, in
LAPTM5-sh3 cells, Raptor, p-S6K1 and p-4EBP1 were even more highly
activated than in cells without docetaxel treatment, indicating
that downregulation of LAPTM5 expression decreased the
chemosensitivity of ER+ BC cells. In contrast, the
opposite results were observed in the LAPTM5-OE cells (Fig. 4L). Fig. 4B-K, M and N shows the quantitative
analysis of the protein levels of Raptor, p-S6K1, and p-4EBP1 in
the MCF-7 and T47D cell lines. In addition, BPTES treatment
inhibited the activation of mTORC1 signaling in both blank and
LAPTM5-sh3 cells with or without docetaxel treatment, demonstrating
that glutamine-dependent mTORC1 signaling mediates the biological
function of LAPTM5 in ER+ BC. Furthermore, GLS1
silencing (GLS1 sh1-3) significantly reversed the increased
migration and proliferation of LAPTM5-sh3 ER+ BC cells (Fig. 5A-C). In addition, the activation
of mTORC1 signaling after LAPTM5 silencing was inhibited by
downregulation of GLS1 expression (Fig. 5D and E). These results further
confirmed that glutamine-induced mTOR signaling mediates the
biological function of LAPTM5 in ER+ BC. Therefore,
these data demonstrated that glutamine metabolism could be a
therapeutic target for ER+ BC at the late stage.
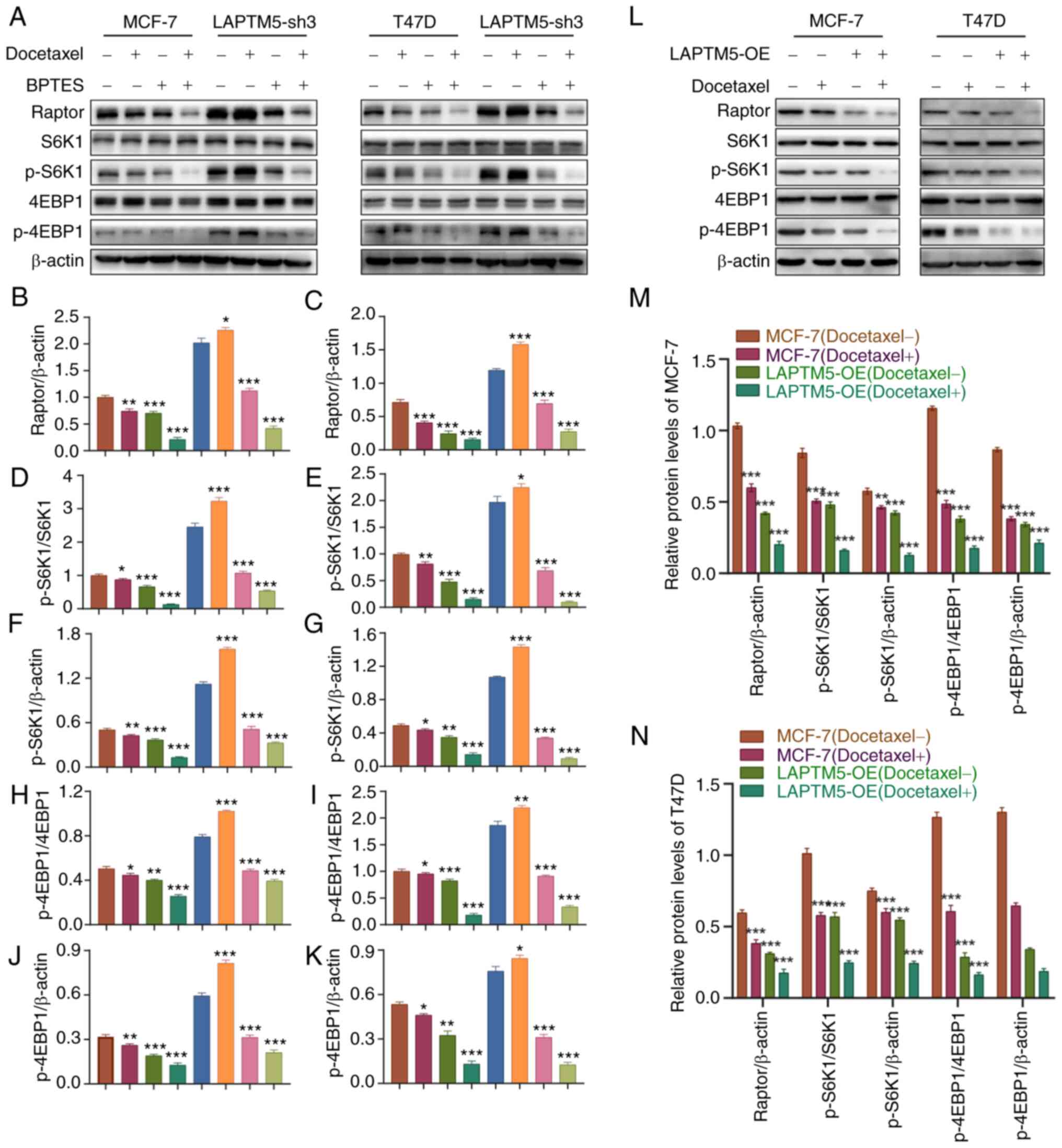 | Figure 4The glutaminase inhibitor BPTES
reverses activation of mTOR signaling in LAPTM5-knockdown
ER+ BC cells. (A) Protein expression of key molecules of
glutamine metabolism and mTOR signaling in the blank and LAPTM5-sh3
(knockdown) MCF-7 and T47D cells when treated with docetaxel or the
SLC1A5 inhibitor BPTES. (B-K) The relative protein expression of
Raptor, p-S6K1, and p-4EBP1 in MCF-7 and T47D cells. (L) Protein
expression of key molecules of glutamine metabolism and mTOR
signaling in the blank and LAPTM5-OE (overexpessing) MCF-7 and T47D
cells when treated with docetaxel. The relative protein expression
of Raptor, p-S6K1, and p-4EBP1 in MCF-7 (M) and T47D (N) cells.
*P<0.05, **P<0.01, and
***P<0.001, compared with the blank MCF-7 and T47D
cells. LAPTM5, lysosomal protein transmembrane 5; ER+
BC, estrogen receptor-positive breast cancer; S6K1, ribosomal
protein S6 kinase 1; 4EBP1, eukaryotic translation initiation
factor 4E (eIF4E)-binding protein 1; p-, phosphorylated. |
CX3CL1/CX3CR1 interaction mediates
vertebrae-specific inhibition of LAPTM5
In our previous studies, CX3CL1/CX3CR1 was shown to
be highly expressed in vertebrae and mediated SM in several types
of cancer (25,33-35). However, the role of CX3CL1/CX3CR1
in the SM of ER+ BC has not been fully elucidated.
First, we demonstrated the high expression levels of CX3CR1 in the
SM samples of ER+ BC in both human patients and mouse
models, while CX3CR1 expression was higher in the primary
ER+ BC tissue than that in the tumor-adjacent normal
tissue (Fig. 6A-C). Preliminary
experiments were conducted to determine the relationship between
CX3CL1 and LAPTM5. When stimulated by CX3CL1, LAPTM5 was
significantly inhibited in the ER+ BC cells, while
LAPTM5 inhibition was reversed after coculture with the
CX3CR1-specific inhibitors JMS-17-2 and CX3CL1, indicating a
negative regulatory effect of CX3CL1 on LAPTM5 expression (Fig. 6D). Then, blank and LAPTM5-OE cells
were used to further confirm the relationship between CX3CL1/CX3CR1
and LAPTM5. As shown in Fig. 6E,
CX3CL1 treatment significantly decreased LAPTM5 expression levels
in both the blank and LAPTM5-OE cells, while JMS-17-2 blocked the
inhibitory effect of CX3CL1 on LAPTM5. These results demonstrated
that the CX3CL1/CX3CR1 interaction could reduce LAPTM5 expression,
which may explain the lower expression of LAPTM5 in SM samples of
ER+ BC than that in primary tumor or normal tissues.
Furthermore, CX3CL1/CX3CR1-related signaling pathways were tested
to reveal the mechanism by which high spinal CX3CL1/CX3CR1 levels
influence the expression of LAPTM5. Among the various signaling
pathways, PI3K/AKT signaling was most obviously involved, as both
JMS-17-2 and the AKT-specific inhibitor MK-2206 reversed the
activation of PI3K/AKT signaling and the subsequent inhibition of
LAPTM5 (Fig. 6F). Collectively,
these results demonstrated that a high expression level of
CX3CL1/CX3CR1 in the spine facilitated SM in ER+ BC by
inhibiting LAPTM5 expression to promote the growth, migration, and
chemoresistance of cancer cells.
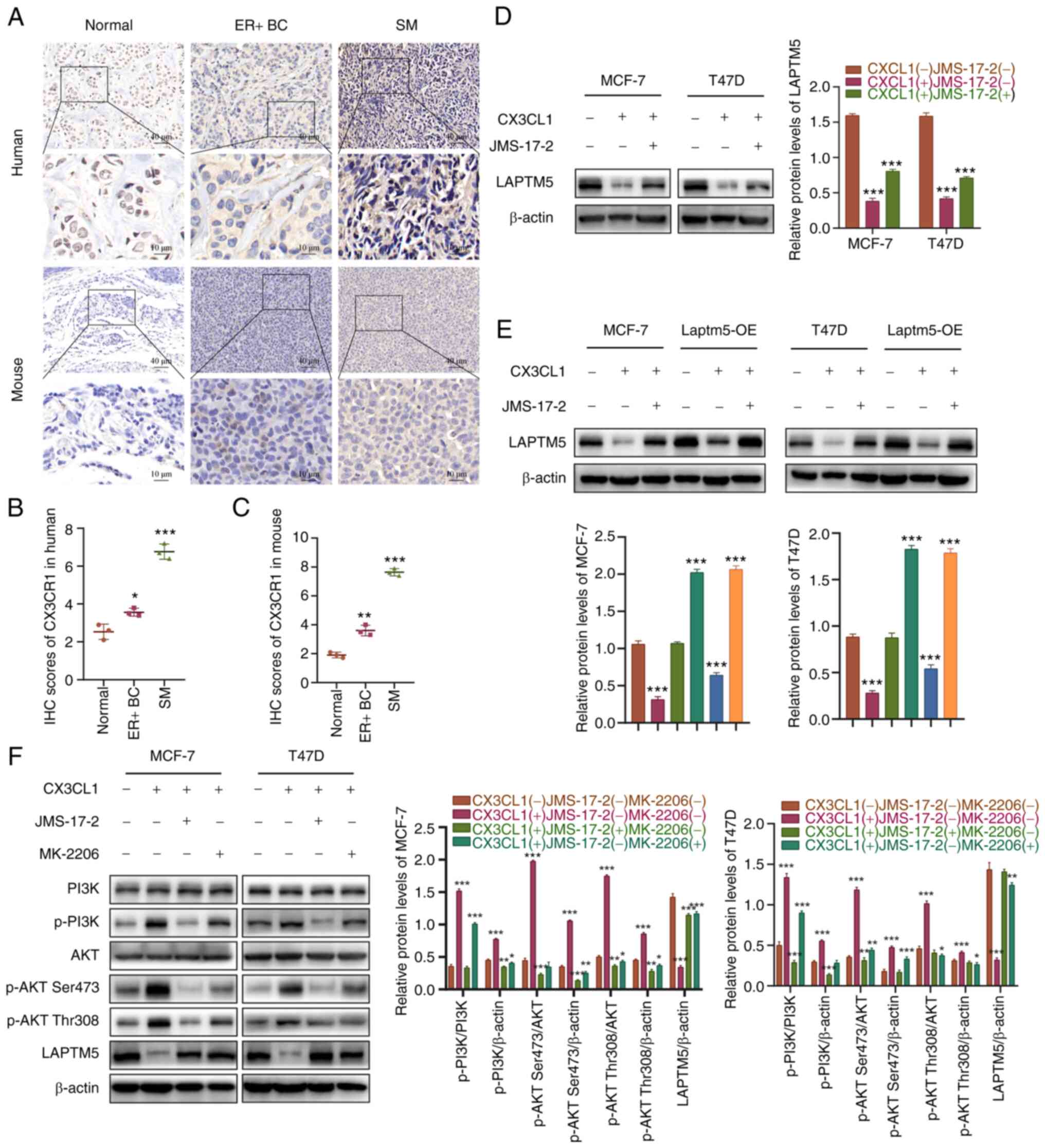 | Figure 6CX3CL1/CX3CR1 interaction mediates SM
of ER+ BC cells by activating PI3K/AKT signaling to
downregulate LAPTM5 expression. (A) IHC staining of CX3CR1 in ER+
BC tissue, tumor-adjacent normal tissue and SM tissue from patients
and mouse models. (B and C) Quantitative analysis of the IHC
results of CX3CR1. *P<0.05, **P<0.01,
and ***P<0.001, compared to the normal tissues. (D)
The protein level of LAPTM5 under CX3CL1 and/or the CX3CR1
inhibitor JMS-17-2 treatment. ***P<0.001, compared to
untreated cells. (E) The protein level of LAPTM5 in blank and
LAPTM5-OE (overexpressing) cells treated with CX3CL1 and/or the
CX3CR1 inhibitor JMS-17-2. ***P<0.001, compared to
untreated cells. (F) Activation of PI3K/AKT signaling and LAPTM5
expression under PBS, CX3CL1, CX3CL1+JMS-17-2, or CX3CL1+MK-2206
(AKT inhibitor) treatment. *P<0.05,
**P<0.01, and ***P<0.001, compared with
the untreated cells. LAPTM5, lysosomal protein transmembrane 5;
ER+ BC, estrogen receptor-positive breast cancer;
CX3CL1, C-X3-C motif chemokine ligand 1; CX3CR1, C-X3-C motif
chemokine receptor 1; SM, spinal metastasis; p-,
phosphorylated. |
Blockade of CX3CL1/CX3CR1 or glutamine
metabolism inhibits SM and enhances the chemosensitivity of
ER+ BC
Finally, we detected the role of the
CX3CL1/CX3CR1/LAPTM5/glutamine metabolic axis in the SM of
ER+ BC and tested the potential therapeutic targets with
an SM mouse model. As shown in Fig.
7A, the mouse model established with blank MCF-7 cells was set
as the control group (a). The other groups were established with
LAPTM5-sh3 cells and groups b-f were injected with saline (b),
docetaxel (c), BPTES+docetaxel (d), JMS-17-2+docetaxel (e), and
BPTES+JMS-17-2+docetaxel (f). One month after inoculation with
cancer cells and injection of drugs, BLI and ex vivo tumor
images visually showed the SM of ER+ BC, as more SM was
observed in group b, while docetaxel only slightly inhibited the SM
of cancer cells (Figs. 7A and
S5A). Moreover, both BPTES and
JMS-17-2 improved the therapeutic effect of docetaxel, while
co-injection of BPTES and JMS-17-2 improved chemosensitivity to the
greatest extent. As shown in Fig.
S5B and C, BPTES and JMS-17-2 treatment significantly delayed
the occurrence of SM and prolonged the survival of the mice.
Consistent with the above results, micro-CT was applied to observe
bone destruction by cancer cells, and the results exhibited obvious
lesions of the spine in groups a-c, while almost no lesions were
found in group f (Fig. 7B).
H&E staining showed similar results (Fig. 7C). IHC staining for CX3CR1, GLS1
and Raptor further confirmed that the inhibition of LAPTM5 promoted
SM of ER+ BC and that blockade of glutamine metabolism
and CX3CL1/CX3CR1 reversed the chemoresistance of LAPTM5-sh3 cells
(Fig. 7A and D-F).
Discussion
The exact role of lysosomal protein transmembrane 5
(LAPTM5) in human estrogen receptor-positive (ER+)
breast cancer (BC) has not been reported and is still unclear.
Given that LAPTM5 was reported to suppress several types of solid
tumors (12,13), our preliminary analysis indicated
that LAPTM5 expression was significantly downregulated in spinal
metastasis (SM) specimens of ER+ BC compared with its
primary lesions as determined from the GEO dataset. The
tumor-suppressive role of LAPTM5 in ER+ BC was verified
by detecting LAPTM5 expression in ER+ BC tissue and its
tumor-adjacent normal tissue and SM samples from patients and mouse
models. Moreover, these data indicated that decreased LAPTM5
expression was related to the SM of ER+ BC. In addition,
we also observed SM in mice injected with LAPTM5-overexpressing
(OE) MCF-7 cells, but no obvious SM could be found via
bioluminescence imaging (BLI) and micro-CT (data not shown). This
study mainly discussed the downregulation of LAPTM5 and its
involvement in the promotion of SM of ER+ BC and how to
prevent spinal metastasis via blocking downstream molecules of
LAPTM5; therefore, only results of LAPTM5-sh3 knockdown groups were
exhibited in Fig. 7. At the late
stage of ER+ BC, the degree of malignancy of cancer
cells in SM is much higher than that in the primary site, which is
characterized by faster growth and migration and enhanced
resistance to chemotherapies (7,8).
These results prompted us to determine the exact role and mechanism
of LAPTM5 in ER+ BC and how LAPTM5 mediates the
development of SM.
As a tumor suppressor, LAPTM5 mainly has a negative
regulatory effect on receptor-mediated signaling in T cells or B
cells (36,37). Although the direct impact of
LAPTM5 on cancer cells has attracted much attention in recent
years, data concerning ER+ BC have not been reported. In
our preliminary experiments, we investigated the tumorigenic
ability of LAPTM5-sh3, LAPTM5-OE, and blank MCF-7 cells. The
results showed that LAPTM5 silencing significantly enhanced the
in vivo tumorigenesis of ER+ BC in the absence of
docetaxel (data not shown). Interestingly, our results demonstrated
that decreased LAPTM5 expression strongly enhanced the growth and
migration rates, as well as resistance to chemotherapeutic drugs,
of ER+ BC. After we demonstrated the role of LAPTM5 in
ER+ BC, the underlying mechanism was also investigated.
As the gene set enrichment analysis indicated that nutrient
metabolism is closely related to LAPTM5 expression, we detected the
degree of glutamine metabolism of the blank ER+ BC cells
compared with the LAPTM5-sh or LAPTM5-OE cells. Here, for the first
time, we demonstrated that glutamine metabolism was regulated by
LAPTM5 and mediated the role of LAPTM5 in ER+ BC.
Glutamine promotes the growth and migration of
cancer cells, as it provides a nitrogen source for synthesizing
nucleotides and nonessential amino acids and a carbon source for
the tricarboxylic acid cycle and fatty acid synthesis (38,39). Recently, glutamine transporters
were found to be overexpressed in ER+ BC, indicating the
critical role and potential mechanism of glutamine metabolism in
ER+ BC (40).
Sodium-dependent neutral amino acid transporter type 2 (SLC1A5) and
glutaminase 1 (GLS1) are two major upstream regulators in the
glutamine metabolic process (18,41). SLC1A5, also known as ASCT2, is a
neutral amino acid transporter located on the cell surface that
mediates the uptake of glutamine (18). GLS1 can convert glutamine to
glutamate and then fuel the tricarboxylic acid cycle (41). The expression of both SLC1A5 and
GLS1 was upregulated when LAPTM5 was inhibited, suggesting that
LAPTM5 inhibition activates glutamine metabolism. Glutamine, as the
primary source of carbon and nitrogen for maintaining vital
activities of cancer cells, has been reported to facilitate poor
progression of BC (17).
Therefore, oncogenic alteration of LAPTM5 could reprogram glutamine
metabolism to regulate cancer cells.
Many signaling pathways are activated by
reprogrammed glutamine metabolism. Among them, mTORC1 was reported
to be one of the main downstream pathways of glutamine metabolism
(42,43). Studies have shown that molecules
that influence amino acid transportation or glutamine consumption
are involved in controlling mTOR recruitment and activation
(42). mTORC1, the complex
composed of mTOR, Raptor, and mLST8, is activated by amino acids,
including glutamine, and energy metabolism, which regulates
proliferation, migration, chemoresistance, and other
mTORC1-mediated processes (43,44). After the formation of mTORC1,
activated mTORC1 phosphorylates S6K1 and 4EBP1 to promote the
expression of metalloproteinase (MMP)9 and cyclin D1 and activate
NFκB signaling, explaining the enhanced migration, proliferation,
and chemo-resistance of ER+ BC (45). MMP9 is involved in degrading the
extracellular matrix in the tissue remodeling process to enhance
the migration and invasion of cancer cells, while cyclin D1 plays
an important role in driving cell proliferation and promoting
chemoresistance in BC (46,47). Moreover, NFκB renders BC cells
resistant to chemotherapies, and its inhibitors are considered
promising anti-BC drugs (48). As
mentioned earlier, we demonstrated that LAPTM5 inhibition could
activate SLC1A5 and GLS1 to enhance glutamine metabolism, which
resulted in activation of mTORC1. In contrast, the SLC1A5 inhibitor
BPTES reversed the activation of mTORC1 by downregulating LAPTM5
expression, further confirming that glutamine-dependent mTOR
activation mediates the role of LAPTM5 in ER+ BC.
Emerging evidence has shown that the interaction
and activation of CX3CL1/CX3CR1 facilitate the SM of circulating
tumor cells (25,33-35). Here, for the first time, we
demonstrated that CX3CL1/CX3CR1 interaction downregulated LAPTM5
expression, and the inhibition of the latter was proven to be
closely related to the enhanced proliferation, migration, and
chemoresistance of ER+ BC. We inferred that when
circulating ER+ BC cells reaches the spine, the abundant
CX3CL1 interacts with CX3CR1 on the surface of cancer cells to
downregulate LAPTM5 expression and then promotes the viability of
cancer cells even under chemotherapy treatment, facilitating the
invasion of ER+ BC in the spine. Furthermore, we found
that CX3CL1 stimulation downregulated LAPTM5 expression via
PI3K/AKT signaling, which was reported to be downstream of the
CX3CL1/CX3CR1 interaction (Fig.
8). Therefore, we believe that a high concentration of CX3CL1
mediates spine-specific inhibition of LAPTM5 to facilitate SM of
ER+ BC. Finally, with an SM mouse model, we verified the
tumor-suppressive role of LAPTM5 in ER+ BC and confirmed
the SM-specific targeting effect of the
CX3CL1/CX3CR1/LAPTM5/glutamine axis.
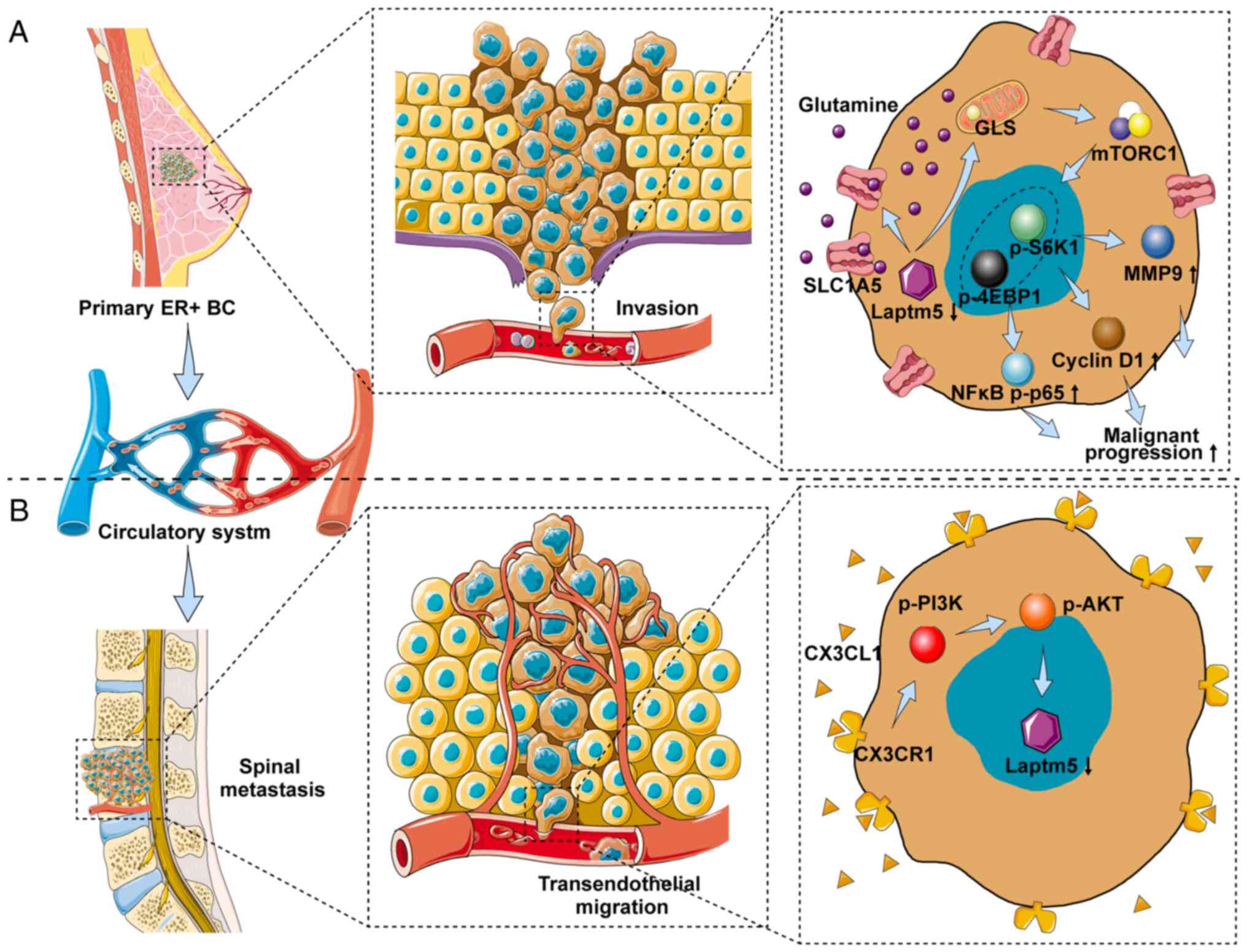 | Figure 8Schematic depicting the role of
LAPTM5 in regulating the malignant progression and SM of
ER+ BC. (A) LAPTM5 downregulation promotes the
proliferation, migration, and chemoresistance of ER+ BC
cells by activating glutamine-mediated mTOR signaling. (B)
CX3CL1/CX3CR1 interaction mediates the vertebrae-specific
inhibition of LAPTM5 and the SM of ER+ BC via PI3K/AKT
signaling. LAPTM5, lysosomal protein transmembrane 5;
ER+ BC, estrogen receptor-positive breast cancer;
CX3CL1, C-X3-C motif chemokine ligand 1; CX3CR1, C-X3-C motif
chemokine receptor 1; SM, spinal metastasis; GLS, glutaminase;
S6K1, ribosomal protein S6 kinase 1; 4EBP1, eukaryotic translation
initiation factor 4E (eIF4E)-binding protein 1; MMP9, matrix
metallopeptidase 9; nuclear factor κB, NFκB. |
In summary, we demonstrated that downregulation of
LAPTM5 expression could regulate glutamine metabolism through
SLC1A5 and GLS1 to activate mTOR signaling and promote the
proliferation, migration, and chemoresistance of ER+ BC.
In addition, a high level of CX3CL1 in the spine inhibited LAPTM5
expression through interaction with CX3CR1, indicating that the
CX3CL1/CX3CR1/LAPTM5/glutamine axis mediates the SM of
ER+ BC. Therefore, the CX3CL1/CX3CR1/LAPTM5/glutamine
metabolic axis may be a prospective therapeutic target for
ER+ BC and its SM, suggesting that upregulation of
LAPTM5 expression or blockade of its downstream signaling could
alleviate the malignancy of ER+ BC.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
QM and JD designed the experiments. LZ, LJ and HLia
collected the samples. QM, LZ, HLia, AH and HZ conducted the
experiments and acquired the data. JZ, XZ, HLin, LJ and XL analyzed
the data and confirmed the integrity of the data. QM and JD wrote
the manuscript. All authors read and approved the manuscript and
agree to be accountable for all aspects of the research in ensuring
that the accuracy or integrity of any part of the work are
appropriately investigated and resolved.
Ethics approval and consent to
participate
The animal experiments were approved (approval no.
2020-032, 2020.04.06) by the Animal Ethics Committee of Zhongshan
Hospital, Fudan University (Shanghai, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (nos. 82172738, 81972508 and 81772855).
References
|
1
|
Akram M, Iqbal M, Daniyal M and Khan AU:
Awareness and current knowledge of breast cancer. Biol Res.
50:332017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liang Y, Zhang H, Song X and Yang Q:
Metastatic heterogeneity of breast cancer: Molecular mechanism and
potential therapeutic targets. Semin Cancer Biol. 60:14–27. 2020.
View Article : Google Scholar
|
|
4
|
Scully OJ, Bay BH, Yip G and Yu Y: Breast
cancer metastasis. Cancer Genomics Proteomics. 9:311–320.
2012.PubMed/NCBI
|
|
5
|
Turner NC, Neven P, Loibl S and Andre F:
Advances in the treatment of advanced oestrogen-receptor-positive
breast cancer. Lancet. 389:2403–2414. 2017. View Article : Google Scholar
|
|
6
|
Chen WZ, Shen JF, Zhou Y, Chen XY, Liu JM
and Liu ZL: Clinical characteristics and risk factors for
developing bone metastases in patients with breast cancer. Sci Rep.
7:113252017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ma RY, Zhang H, Li XF, Zhang CB, Selli C,
Tagliavini G, Lam AD, Prost S, Sims AH, Hu HY, et al:
Monocyte-derived macrophages promote breast cancer bone metastasis
outgrowth. J Exp Med. 217:e201918202020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tahara RK, Brewer TM, Theriault RL and
Ueno NT: Bone metastasis of breast cancer. Adv Exp Med Biol.
1152:105–129. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Glowacka WK, Alberts P, Ouchida R, Wang JY
and Rotin D: LAPTM5 protein is a positive regulator of
proinflammatory signaling pathways in macrophages. J Biol Chem.
287:27691–27702. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu X, Shang X, Li J and Zhang S: The
prognosis and immune checkpoint blockade efficacy prediction of
tumor-infiltrating immune cells in lung cancer. Front Cell Dev
Biol. 9:7071432021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L, Zhang M, Chen X, He Y, Chen R,
Zhang J, Huang J, Ouyang C and Shi G: Identification of the
tubulointerstitial infiltrating immune cell landscape and immune
marker related molecular patterns in lupus nephritis using
bioinformatics analysis. Ann Transl Med. 8:15962020. View Article : Google Scholar
|
|
12
|
Berberich A, Bartels F, Tang Z, Knoll M,
Pusch S, Hucke N, Kessler T, Dong Z, Wiestler B, Winkler F, et al:
LAPTM5-CD40 crosstalk in glioblastoma invasion and temozolomide
resistance. Front Oncol. 10:7472020. View Article : Google Scholar
|
|
13
|
Nuylan M, Kawano T, Inazawa J and Inoue J:
Down-regulation of LAPTM5 in human cancer cells. Oncotarget.
7:28320–28328. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mossmann D, Park S and Hall MN: mTOR
signalling and cellular metabolism are mutual determinants in
cancer. Nat Rev Cancer. 18:744–757. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vander Heiden MG and DeBerardinis RJ:
Understanding the intersections between metabolism and cancer
biology. Cell. 168:657–669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reinfeld BI, Madden MZ, Wolf MM, Chytil A,
Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A and Do BT: et al
Cell-programmed nutrient partitioning in the tumour
microenvironment. Nature. 593:282–288. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cha YJ, Kim ES and Koo JS: Amino acid
transporters and glutamine metabolism in breast cancer. Int J Mol
Sci. 19:9072018. View Article : Google Scholar :
|
|
18
|
van Geldermalsen M, Wang Q, Nagarajah R,
Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG and Deng
N: et al ASCT2/SLC1A5 controls glutamine uptake and tumour growth
in triple-negative basal-like breast cancer. Oncogene.
35:3201–3208. 2016. View Article : Google Scholar :
|
|
19
|
Kodama M, Oshikawa K, Shimizu H, Yoshioka
S, Takahashi M, Izumi Y, Bamba T, Tateishi C, Tomonaga T, Matsumoto
M and Nakayama KI: A shift in glutamine nitrogen metabolism
contributes to the malignant progression of cancer. Nat Commun.
11:13202020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feng M, Xiong G, Cao Z, Yang G, Zheng S,
Qiu J, You L, Zheng L, Zhang T and Zhao Y: LAT2 regulates
glutamine-dependent mTOR activation to promote glycolysis and
chemoresistance in pancreatic cancer. J Exp Clin Cancer Res.
37:2742018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ramapriyan R, Caetano MS, Barsoumian HB,
Mafra ACP, Zambalde EP, Menon H, Tsouko E, Welsh JW and Cortez MA:
Altered cancer metabolism in mechanisms of immunotherapy
resistance. Pharmacol Ther. 195:162–171. 2019. View Article : Google Scholar
|
|
22
|
Korbecki J, Simińska D, Kojder K, Grochans
S, Gutowska I, Chlubek D and Baranowska-Bosiacka I:
Fractalkine/CX3CL1 in neoplastic processes. Int J Mol Sci.
21:37232020. View Article : Google Scholar :
|
|
23
|
Liang Y, Yi L, Liu P, Jiang L, Wang H, Hu
A, Sun C and Dong J: CX3CL1 involves in breast cancer metastasizing
to the spine via the Src/FAK signaling pathway. J Cancer.
9:3603–3612. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jiang G, Wang H, Huang D, Wu Y, Ding W,
Zhou Q, Ding Q, Zhang N, Na R and Xu K: The clinical implications
and molecular mechanism of CX3CL1 expression in urothelial bladder
cancer. Front Oncol. 11:7528602021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang K, Jiang L, Hu A, Sun C, Zhou L,
Huang Y, Chen Q, Dong J, Zhou X and Zhang F: Vertebral-specific
activation of the CX3CL1/ICAM-1 signaling network mediates
non-small-cell lung cancer spinal metastasis by engaging tumor
cell-vertebral bone marrow endothelial cell interactions.
Theranostics. 11:4770–4789. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang da W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1–13. 2009. View Article : Google Scholar
|
|
28
|
Lichter JG, Carruth E, Mitchell C, Barth
AS, Aiba T, Kass DA, Tomaselli GF, Bridge JH and Sachse FB:
Remodeling of the sarcomeric cytoskeleton in cardiac ventricular
myocytes during heart failure and after cardiac resynchronization
therapy. J Mol Cell Cardiol. 72:186–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
30
|
Xiang Y, Stine ZE, Xia J, Lu Y, O'Connor
RS, Altman BJ, Hsieh AL, Gouw AM, Thomas AG and Gao P: et al
Targeted inhibition of tumor-specific glutaminase diminishes
cell-autonomous tumorigenesis. J Clin Invest. 125:2293–2306. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shen F, Zhang Y, Jernigan DL, Feng X, Yan
J, Garcia FU, Meucci O, Salvino JM and Fatatis A: Novel
small-molecule CX3CR1 antagonist impairs metastatic seeding and
colonization of breast cancer cells. Mol Cancer Res. 14:518–527.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Montaudon E, Nikitorowicz-Buniak J, Sourd
L, Morisset L, El Botty R, Huguet L, Dahmani A, Painsec P, Nemati F
and Vacher S: et al PLK1 inhibition exhibits strong anti-tumoral
activity in CCND1-driven breast cancer metastases with acquired
palbociclib resistance. Nat Commun. 11:40532020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu W, Jiang L, Bian C, Liang Y, Xing R,
Yishakea M and Dong J: Role of CX3CL1 in diseases. Arch Immunol
Ther Exp (Warsz). 64:371–383. 2016. View Article : Google Scholar
|
|
34
|
Sun C, Hu A, Wang S, Tian B, Jiang L,
Liang Y, Wang H and Dong J: ADAM17-regulated CX3CL1 expression
produced by bone marrow endothelial cells promotes spinal
metastasis from hepatocellular carcinoma. Int J Oncol. 57:249–263.
2020.PubMed/NCBI
|
|
35
|
Yi L, Liang Y, Zhao Q, Wang H and Dong J:
CX3CL1 induces vertebral microvascular barrier dysfunction via the
Src/P115-RhoGEF/ROCK signaling pathway. Front Cell Neurosci.
14:962020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tu J, Kuang Z, Xie X, Wu S, Wu T and Chen
S: Prognostic and predictive value of a mRNA signature in
peripheral T-cell lymphomas: A mRNA expression analysis. J Cell Mol
Med. 25:84–95. 2021. View Article : Google Scholar
|
|
37
|
Zouali M: Transcriptional and metabolic
pre-B cell receptor-mediated checkpoints: Implications for
autoimmune diseases. Mol Immunol. 62:315–320. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cory JG and Cory AH: Critical roles of
glutamine as nitrogen donors in purine and pyrimidine nucleotide
synthesis: Asparaginase treatment in childhood acute lymphoblastic
leukemia. In Vivo. 20:587–589. 2006.PubMed/NCBI
|
|
39
|
Metallo CM, Gameiro PA, Bell EL, Mattaini
KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ and Guarente
L: et al Reductive glutamine metabolism by IDH1 mediates
lipogenesis under hypoxia. Nature. 481:380–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Karunakaran S, Ramachandran S,
Coothankandaswamy V, Elangovan S, Babu E, Periyasamy-Thandavan S,
Gurav A, Gnanaprakasam JP, Singh N and Schoenlein PV: et al SLC6A14
(ATB0,+) protein, a highly concentrative and broad specific amino
acid transporter, is a novel and effective drug target for
treatment of estrogen receptor-positive breast cancer. J Biol Chem.
286:31830–31838. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matés JM, Campos-Sandoval JA,
Santos-Jiménez JL and Márquez J: Dysregulation of glutaminase and
glutamine synthetase in cancer. Cancer Lett. 467:29–39. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jewell JL, Kim YC, Russell RC, Yu FX, Park
HW, Plouffe SW, Tagliabracci VS and Guan KL: Metabolism.
Differential regulation of mTORC1 by leucine and glutamine.
Science. 347:194–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng D, Yang Q, Wang H, Melick CH, Navlani
R, Frank AR and Jewell JL: Glutamine and asparagine activate mTORC1
independently of Rag GTPases. J Biol Chem. 295:2890–2899. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Csibi A, Fendt SM, Li C, Poulogiannis G,
Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A and Morrison
T: et al The mTORC1 pathway stimulates glutamine metabolism and
cell proliferation by repressing SIRT4. Cell. 153:840–854. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hua H, Kong Q, Zhang H, Wang J, Luo T and
Jiang Y: Targeting mTOR for cancer therapy. J Hematol Oncol.
12:712019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhen Y, Liu J, Huang Y, Wang Y, Li W and
Wu J: miR-133b inhibits cell growth, migration, and invasion by
targeting MMP9 in non-small cell lung cancer. Oncol Res.
25:1109–1116. 2017. View Article : Google Scholar
|
|
47
|
Shi Q and Li Y, Li S, Jin L, Lai H, Wu Y,
Cai Z, Zhu M, Li Q and Li Y: et al LncRNA DILA1 inhibits cyclin D1
degradation and contributes to tamoxifen resistance in breast
cancer. Nat Commun. 11:55132020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ling J and Kumar R: Crosstalk between NFkB
and glucocorticoid signaling: A potential target of breast cancer
therapy. Cancer Lett. 322:119–126. 2012. View Article : Google Scholar : PubMed/NCBI
|