Introduction
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common cancer in the world with approximately 800,000
cases being diagnosed every year (1). Advanced HNSCC shows poor prognosis
and resistance to standard therapy consisting of cisplatin. Because
60-80% of HNSCC cases carry a TP53 mutation (2-4)
and human papillomavirus (HPV)-positive HNSCC shows impaired p53
function (5), therapeutic
targeting of the TP53 mutation has become a focus of
research. Additionally, as p53 is functional in normal somatic
cells, targeted therapy against TP53 mutations can relieve
adverse effects. Thus, owing to its selective cytotoxicity against
TP53-mutated carcinoma cells, adavosertib (Adv), a WEE1 G2
checkpoint kinase (WEE1) inhibitor, has been the recent focus of
study (6,7).
WEE1 is a kinase that regulates replication stress
and cell cycle arrest at the G2/M checkpoint. DNA damage during the
S-phase activates WEE1, which in turn phosphorylates
cyclin-dependent kinase 1 (CDK1) at Tyr15 to inactivate its kinase
activity (8). Because p53
regulates the G1/S checkpoint (9), TP53-mutated cells rely on the
G2/M checkpoint to arrest the cell cycle to repair their DNA
damage. Therefore, Adv treatment of TP53-mutated cells leads
to the cells entering M-phase without DNA repair, which in turn
causes cell death named mitotic catastrophe. Mitotic catastrophe is
a type of cell death characterized by DNA damage, abnormalities in
the mitotic apparatus, dysfunction of the mitotic checkpoint, and
failure in the occurrence of normal mitosis (10,11), but there is no clear definition.
As Adv abrogates cell cycle arrest, it was suggested that the
co-administration of DNA-damaging drugs with Adv enhances
cytotoxicity (7,12,13). In the present study, we sought to
identify drug combinations to enhance Adv-induced cytotoxicity
without losing selective cell death in TP53-mutated
cells.
It has been reported that histone deacetylase (HDAC)
inhibitors induce growth arrest, differentiation, and apoptosis
in vitro, and suppress tumor growth in xenograft mouse
models with various cancer cell lines including HNSCC (14-16). Additionally, HDAC inhibitors have
been shown to enhance cytotoxicity in combination with DNA-damaging
anticancer drugs (17,18) or Adv (19-21). We recently reported that
ricolinostat (RCS), a selective histone deacetylase 6 (HDAC6)
inhibitor, exhibits potent cell growth inhibition in HNSCC cell
lines in vitro in combination with the proteasome inhibitor
bortezomib (22). Of note, HDAC6
differs from other HDACs by deacetylating cytoplasmic proteins,
including α-tubulin. HDAC6 has also been reported to be involved in
the cell cycle via deacetylation of α-tubulin (23). Additionally, inhibition of HDAC6
has been reported to sensitize cancer cells to DNA-damaging drugs
(24-26). As the disruption of the cell cycle
or DNA damage enhances mitotic catastrophe, in the present study,
we assessed the effect of the combined treatment of Adv and RCS on
HNSCC cell lines with TP53 mutation (CAL27, SAS, HSC-3,
Detroit562, and OSC-19) or impaired p53 function by HPV-infection
(UPCI-SCC154).
Materials and methods
Reagents
Adavosertib (AZD1775, MK-1775, hereafter referred to
as Adv) was purchased from MedChemExpress. Ricolinostat (ACY-1215,
hereafter referred to as RCS) was purchased from Selleck Chemicals.
Adv and RCS were dissolved in dimethyl sulfoxide (DMSO) (Wako Pure
Chemical Industries) to make the stock solution at a concentration
of 10 mM for Adv and 5 mM for RCS. Doxorubicin (DOX) was purchased
from Cayman Chemical Co. z-VAD-fmk was purchased from Peptide
Institute (Japan).
Cell lines and culture conditions
The human oral squamous cell carcinoma cell line
CAL27, the human pharyngeal squamous carcinoma cell line
Detroit562, human tongue squamous carcinoma cell line UPCI-SCC154,
the human breast mammary gland adenocarcinoma cell lines MCF7 and
MDA-MB-231, and the human lung adenocarcinoma cell line A549 were
purchased from the American Type Culture Collection (ATCC). The
human tongue squamous carcinoma cell lines SAS, HSC-3, and OSC-19
cells were obtained from the JCRB Cell Bank (Osaka, Japan). A549
and MCF7 cells possess wild-type TP53. MDA-MB-231, CAL27,
HSC-3, SAS, Detroit562, and OSC-19 cells carry mutant TP53.
UPCI-SCC-154 cells are HPV-positive and show impaired p53 function.
CAL27, Detroit562, MCF7, MDA-MB-231, and A549 cells were cultured
in Dulbecco's modified Eagle medium (DMEM) supplemented with 10%
fetal bovine serum (FBS; Biosera) and 1% penicillin/streptomycin
solution (Wako Pure Chemical Industries). UPCI-SCC154 cells were
cultured in MEM supplemented with 10% FBS and 1%
penicillin/streptomycin solution. SAS and OSC-19 cells were
cultured in DMEM/F12 medium supplemented with 10% FBS and 1%
penicillin/streptomycin solution. HSC-3 cells were cultured in
Eagle's minimum essential medium (EMEM) supplemented with 10% FBS
and 1% penicillin/streptomycin solution. All cell lines were
cultured at 37°C in a humidified incubator containing 5%
CO2 and 95% air. In all experiments, as a control, cells
were treated with control medium containing less than 1% DMSO to
match the amount of solvent brought in by the drug. All cell line
experiments were conducted within 10 passages after thawing.
Mycoplasma contamination was tested routinely using the e-Myco™
Mycoplasma PCR Detection kit ver.2.0 (iNtRON Biotechnology,
Inc.).
Establishment of TP53-KO A549 and TP53-KO
MCF7 cells
TP53-KO A549 cells were established as previously
described (27). Briefly, A549
cells were transfected with pSpCas9 (BB)-2A-Puro (PX459) V2.0
plasmid vector (a gift from Dr Feng Zhang; plasmid cat. no. 48139;
Addgene) with the following sequence (5′-CAC CGT CCA TTG CTT GGG
ACG GCA A-3′), selected with puromycin for 2 days, and grown
without puromycin to select single colonies. TP53-KO MCF7 cells
were also established in the same way.
Establishment of
CAL27/H2B-mCherry/AcGFP-α-tubulin cells
To establish CAL27 cells expressing H2B-mCherry and
AcGFP-α-tubulin, CAL27 cells were infected with lentiviruses and
positive clones were selected using puromycin and blasticidin.
Lentiviruses were produced in 293T cells (ATCC) by transfection of
the following plasmids: pMD2.G (gift from Dr Didier Trono: Addgene
#12259), psPAX2 (gift from Dr Didier Trono: Addgene #12260),
pLenti6-H2B-mCherry (gift from Dr Torsten Wittmann: Addgene plasmid
#89766) (28), and
pLenti-AcGFP-α-tubulin. For pLenti-AcGFP-α-tubulin construction,
TUBA1A cDNA was cloned into pAcGFP1-Hyg-C1 vector (Takara Bio
Inc.), and then, the AcGFP-α-tubulin fused gene was inserted into
the pLentiN vector (gift from Dr Karl Munger: Addgene #37444)
(29).
RNA interference
For the gene silencing of HDAC6 in CAL 27 cells,
HDAC6 siRNA and control siRNA were synthesized as follows (Japan
Bio Services Co., Ltd.): siHDAC6#2: sense GCU UAU UUA AGU GUU AAU
AdT dT and antisense UAU UAA CAC UUA AAU AAG CdA dC; siHDAC6#3:
sense GGU UUU UGC UUU UUC AAC UdT dT and antisense AGU UGA AAA AGC
AAA AAC CdG dC; siHDAC6#4: sense GCA UAU GUA AUA AAG UAC AdT dT and
antisense UGU ACU UUA UUA CAU AUG CdA dA; control siLuc: sense CUU
ACG CUG AGU ACU UCG AdT dT and antisense UCG AAG UAC UCA GCG UAA
GdT dT. siRNAs were diluted to 200 nM in Opti-MEM I (Thermo Fisher
Scientific, Inc.). Transfection was performed using Lipofectamine
RNAiMAX transfection reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Knockdown (KD)
efficiency was assessed using western blotting.
Assessment of cell death
The dead cell counts were assessed by staining with
propidium iodide (PI) (FUJIFILM Wako Chemical Corp.), and the
number of red fluorescent signals was counted using the IncuCyte
ZOOM (Sartorius) automated live cell imaging system. The cells were
treated with Adv with or without RCS in the presence of PI (2.5
µg/ml) for up to 48 h in 96-well plates in tetraplicate
(30).
Morphological assessment
The cells were spread on glass slides using a
Cytospin 4 Centrifuge (Thermo Fisher Scientific, Inc.) to prepare
glass slides, stained with May-Grünwald-Giemsa and examined under a
digital microscope (BZ-X800; KEYENCE Co.).
Western blotting
The cells were lysed using RIPA lysis buffer
(Nacalai Tesque) added together with a protease and phosphatase
inhibitor cocktail (Nacalai Tesque). Equal amounts of proteins (25
µg) were loaded onto the gels (7.5, 10 and 15% gels were
used), separated by SDS-PAGE, and then transferred onto Immobilon-P
membranes (Millipore Corp.). These membranes were probed with
primary antibodies, such as anti-p53 antibody (Ab) (sc-126,
1/1,000), anti-β-actin Ab (sc-47778, 1/1,000), anti-HDAC6 Ab
(sc-11420, 1/1,000), anti-acetylated α-tubulin Ab (sc-23950,
1/1,000), and anti-α-tubulin Ab (sc-5286, 1/1,000) were purchased
from Santa Cruz Biotechnology, Inc. Anti-PARP Ab (#9542S, 1/1,000),
anti-caspase3 Ab (#9665S, 1/1,000), anti-phospho-p53 Ab (#9286,
1/1,000), anti-p21 Ab (#2947S, 1/1,000), anti-phospho-ATR Ab
(#9947, 1/1,000), anti-ATR Ab (#2790, 1/1,000), anti-phospho-Chk1
(Ser345) Ab (#2348, 1/1,000), anti-Chk1 Ab (#2360, 1/1,000),
anti-phospho-Chk2 (Thr68) Ab (#2197, 1/1,000), anti-Chk2 Ab (#3440,
1/1,000), anti-phospho-Cdc2 (Tyr15) Ab (#4539, 1/1,000), anti-Cdc2
Ab (#9116, 1/1,000), anti-H2A.X Ab (#7631, 1/1,000), and
anti-phospho-histone H2A.X (Ser139) Ab (#9718, 1/1,000) were
purchased from Cell Signaling Technology, Inc.
Assessment of mitotic catastrophes
CAL27/H2B-mCherry/AcGFP-α-tubulin cells were seeded
on collagen-coated glass-bottom dishes (CELLview #627870, Greiner).
The next day, the cells were treated with control medium, Adv, RCS,
or Adv + RCS, and time-lapse images were obtained every 10 min
using a confocal microscope LSM 700 equipped with CO2,
temperature, and humidity controller (Carl Zeiss) or fluorescent
microscope BZ-X800 equipped with a time-lapse module (BZ-H4XT)
(KEYENCE). The cells were maintained at 37°C and 5% CO2,
under humidified conditions. Time-lapse images from fluorescent
microscopy were used to analyze the cell fate.
Statistical analysis
All quantitative data are expressed as mean ±
standard deviation (SD). Statistical analyses for cell death
inhibition with z-VAD-fmk treatment, and cell death in combination
with Adv treatment and HDAC6 KD were performed using two-way ANOVA
followed by Bonferroni's multiple comparison test. For the analysis
of the M-phase duration, the Kruskal-Wallis test followed by Dunn's
multiple comparison test was used. Statistical significance was set
at P<0.05. All analyses were performed using GraphPad Prism 5
software (GraphPad Software, Inc.). The synergistic effect was
assessed as follows. Based on nesting all elapsed hours for each
experiment, mixed-effect linear regression analyses were performed
by setting the number of cell deaths as the dependent variables and
the concentration of the two anticancer drugs as independent
variables. To assess the possible synergistic effects of the two
drugs, the interaction term was added as one of the independent
variables in the model to calculate p-for-interaction. All
statistical analyses were performed using Stata version 17.0 (Stata
Corp.). When the mono treatment was enough to kill almost all
cells, the analysis indicated an 'antagonistic effect', because the
dead cell count in the combination treatment group was smaller than
the dead cell count in each mono treatment group.
Results
Combined treatment of Adv and RCS
synergistically induces cell death in TP53-mutated HNSCC cells
To assess the effect of the combined treatment of
Adv and RCS on TP53-mutated HNSCC cells, namely CAL27,
HSC-3, SAS, Detroit562, and OSC-19 cells, and HPV-positive
UPCI-SCC-154 cells, which showed impaired p53, these cells were
treated with Adv and RCS for 48 h and PI-positive dead cell number
was measured using a live cell imaging system. In HNSCC cells,
treatment with Adv and/or RCS induced cell death in a dose- and
time-dependent manner. Notably, co-administration of Adv and RCS
synergistically enhanced cell death in four out of five
TP53-mutated HNSCC cell lines (except Detroit562 cells) as
well as in the HPV-positive UPCI-SCC-154 cell line (Figs. 1 and S1). To address whether this synergistic
effect was ubiquitous, breast cancer cell lines (MDA-MB-231 and
MCF7) and a lung cancer cell line (A549) were also treated with Adv
and/or RCS for 48 h (Figs. 1 and
S1). Although co-administration
of RCS with Adv led to enhanced cell death in MDA-MB-231 cells with
TP53 mutation, little or no enhanced cell death was observed
in MCF7 and A549 cells, both carrying wild-type TP53.
Because Adv has been reported to induce mitotic catastrophe in
TP53-mutated cells, we hypothesized that RCS enhanced
Adv-induced mitotic catastrophe in TP53-mutated cells but
not in wild-type TP53 cells. To address this issue, we next
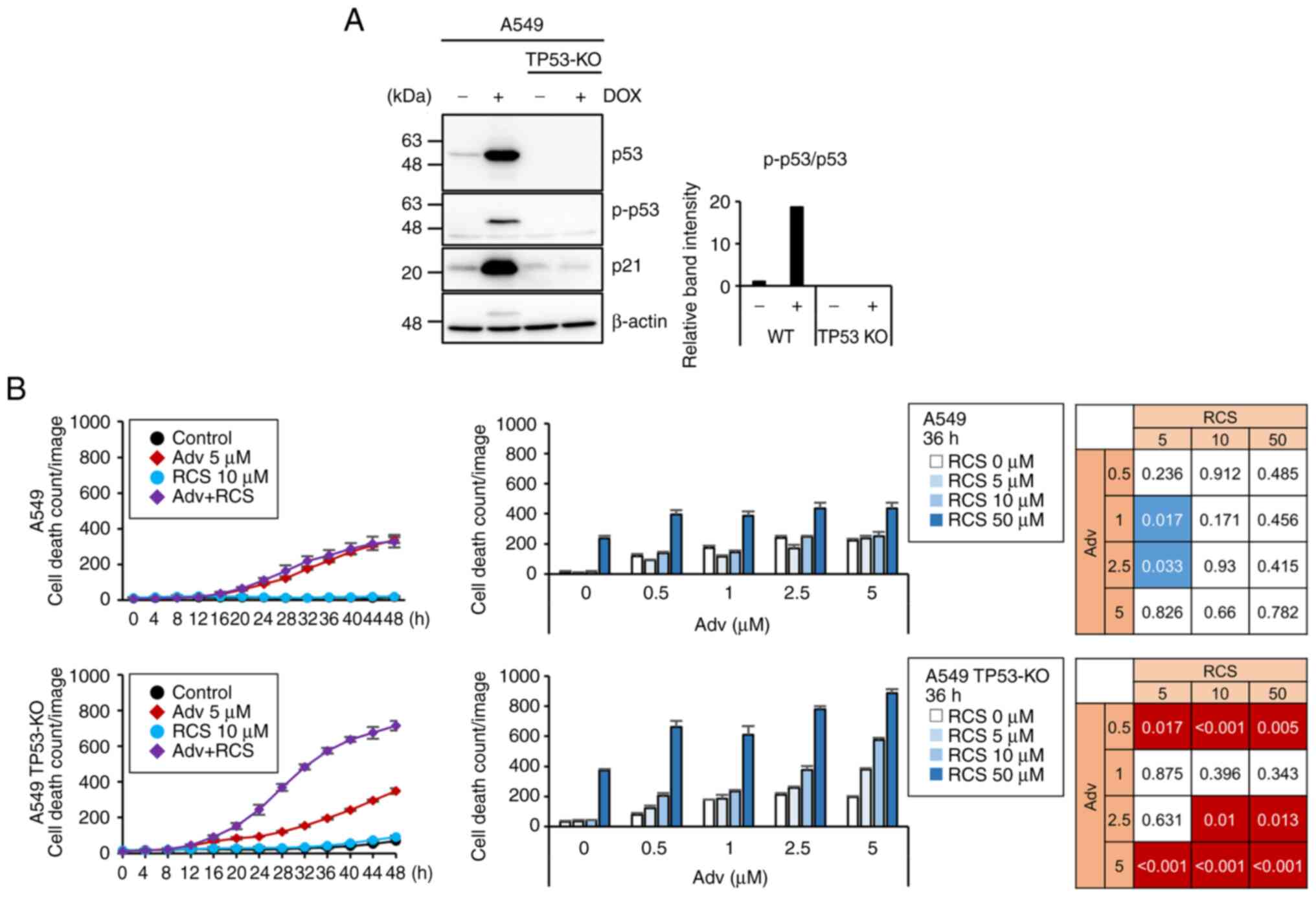
compared the induction of cell death between TP53 wild-type
and TP53 knockout (KO) A549 cells, which were established
using the CRISPR-Cas9 system (Fig.
2A). Although TP53-KO A549 cells did not show altered
sensitivity to Adv as compared to wild-type A549 cells,
co-administration of RCS significantly enhanced cell death in
TP53-KO but not TP53-WT A549 cells (Fig. 2B). To further confirm this
observation, we established TP53-KO MCF7 cells using the
CRISPR-Cas9 system (Fig. S2A).
Although wild-type MCF7 cells showed slight synergistic cell death
in the combined treatment of Adv and RCS, TP53-KO MCF7 cells showed
increased synergistic cell death by the combined treatment as shown
in A549 cells (Fig. S2B). This
indicated that the combination treatment of Adv and RCS can
synergistically induce cell death in cells lacking p53
function.
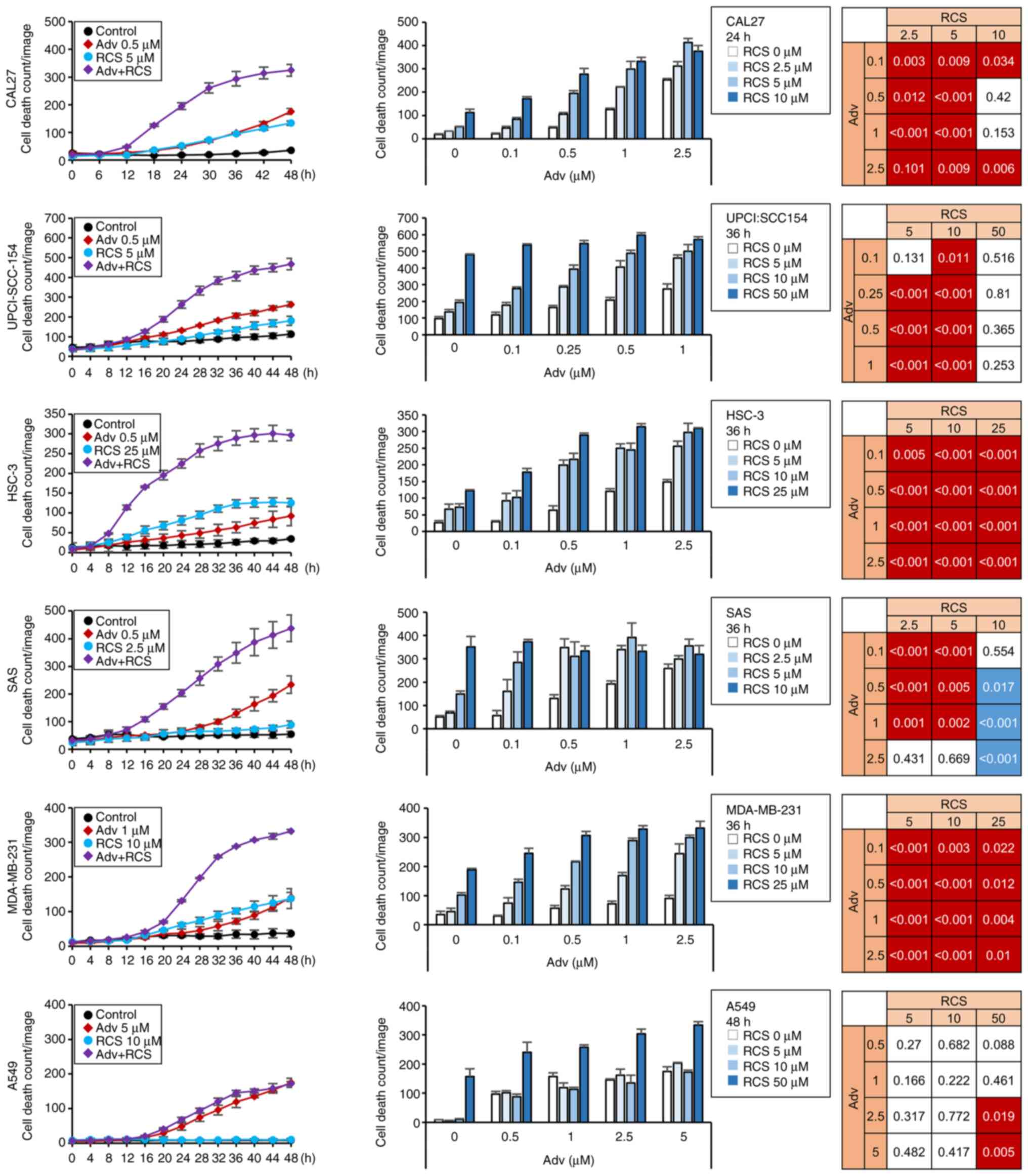 | Figure 1Ricolinostat enhances
adavosertib-induced cytotoxicity in HNSCC cell lines. CAL27,
UPCI-SCC-154, HSC-3, SAS, MDA-MB-231, and A549 cells were treated
with Adv in combination with RCS for up to 48 h. Cells were
monitored using IncuCyte live cell imaging system, and dead cell
number was assessed using PI staining. Time-dependent and
dose-dependent cell death numbers are shown in the left and the
middle panels, respectively. Representative data of three
independent experiments are shown. n=3, bar, mean ± SD.
Synergistically enhanced cell death in the combination treatment
was analyzed as described in Materials and methods and summarized
in the right panels. A significant synergistic effect with both Adv
and RCS treatment with P<0.05 is shown in red. An antagonistic
effect with P<0.05 is shown in blue. HNSCC, head and neck
squamous cell carcinoma; Adv, adavosertib; RCS, ricolinostat. |
Combined treatment of Adv and RCS in
cancer cells leads to mitotic catastrophe
To identify the mechanism by which the combined
treatment of Adv and RCS leads to enhanced cell death, we treated
CAL27 cells with Adv and RCS for 24 h and observed their cell
morphology. In response to drug treatment, CAL27 cells showed
nuclear fragmentation and chromatin condensation (Fig. 3A), which are characteristic
features of cells undergoing apoptosis. Western blotting revealed
that Adv or RCS treatment induced the cleavage of caspase-3, and
co-administration of the two drugs further increased the cleavage
of caspase-3 in CAL 27 cells (Fig.
3B). Additionally, Adv-and RCS-induced cell death was abolished
in the presence of a pan-caspase inhibitor, z-VAD-fmk (Fig. 3C). These data demonstrated that
co-administration of Adv and RCS induced apoptosis. Adv is known to
induce mitotic catastrophe, which subsequently leads to apoptotic
or non-apoptotic cell death (31). To investigate whether the
co-administration of Adv and RCS enhanced mitotic catastrophe, we
established CAL27 cells stably expressing AcGFP-α-tubulin and
histone-H2B-mCherry to monitor mitosis. Time-lapse imaging showed
that most cells treated with Adv could not complete mitosis and
underwent cell death, which indicated mitotic catastrophe (Fig. 3D-F). Additionally, Adv treatment
resulted in a prolonged duration of mitosis (Fig. 3G). On the contrary, although RCS
treatment induced cell death, most of the cell death occurred
during interphase, and most RCS-treated cells did not enter
metaphase (Fig. 3E and F). This
indicated that, unlike Adv, RCS hardly induced mitotic catastrophe.
Of note, when CAL27 cells were simultaneously exposed to Adv and
RCS, dead cell counts were increased and most of the cell death
occurred during mitosis (Fig.
3D-F). However, the duration of the mitotic phase until cell
death was not further extended as compared to the cells treated
with Adv alone (Fig. 3G). These
data showed that RCS enhanced the Adv-induced mitotic
catastrophe.
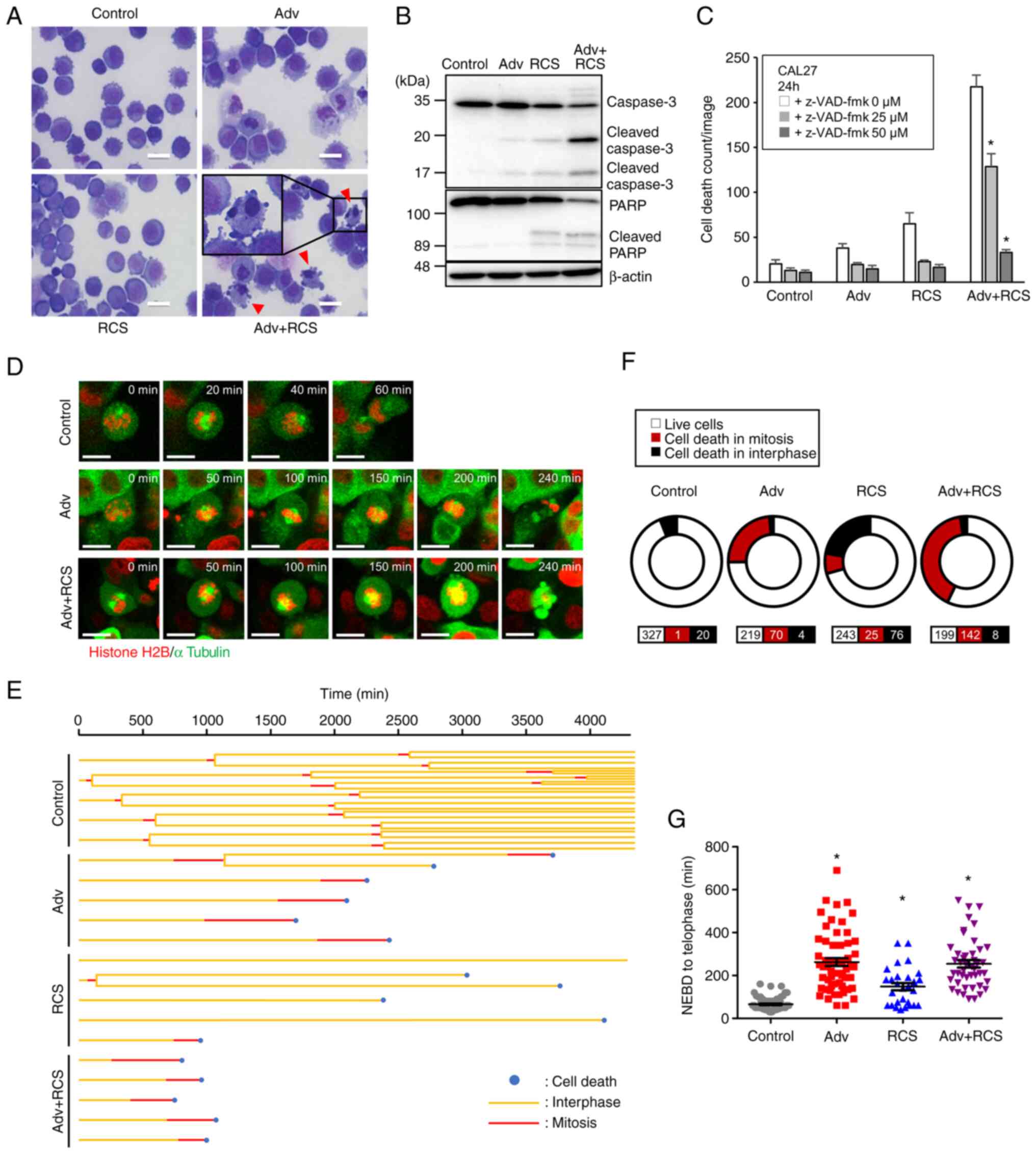 | Figure 3Co-administration of ricolinostat
enhances adavosertib-induced mitotic catastrophe in CAL27 cells.
(A) CAL27 cells were treated with control, Adv (0.5 µM), RCS
(5 µM), or Adv+RCS for 24 h and then stained with
May-Grünwald-Giemsa stain. Scale bar, 25 µm. Arrowheads
indicate nuclear chromatin condensation. (B) CAL27 cells were
treated with control, Adv (0.5 µM), RCS (5 µM), and
Adv + RCS for 24 h, and then, cleavage of PARP and caspase-3 were
assessed by western blotting. (C) CAL27 cells were treated with
control, Adv (0.5 µM), RCS (5 µM), or Adv + RCS in
the presence of z-VAD-fmk (0, 25, and 50 µM) for 24 h, and
dead cell number was monitored using IncuCyte live cell imaging
system by PI staining. n=7, bar, mean ± SD; *P<0.05
vs. 0 µM z-VAD-fmk. (D) Live cell imaging of CAL27 cells
expressing AcGFP-α-tubulin and Histone H2B-mCherry. Cells were
treated with control, Adv (0.5 µM), or Adv + RCS (5
µM) and monitored using confocal microscopy. Representative
images of cells after the nuclear envelope breakdown (NEBD) are
shown. Time after the NEBD is shown in the upper right corner of
each image. Scale bar, 20 µm. (E) Five representative cell
fates in each treated cell with control, Adv (0.5 µM), RCS
(5 µM), or Adv + RCS are shown in a tree diagram. (F) The
ratio of cell death in mitosis or interphase in each treated cell
is summarized in the pie chart. n for each condition is shown at
the bottom. Data from three independent experiments are summarized.
(G) Time from NEBD to telophase or cell death was assessed and
summarized. n=75, 59, 29, 46. Data from three independent
experiments are summarized. *P<0.05 vs. the control.
PARP, poly(ADP-ribose) polymerase; Adv, adavosertib; RCS,
ricolinostat. |
RCS promotes Adv-induced mitotic entry
along with the increment of γ-H2A.X expression
Next, to address how RCS enhances Adv-induced
mitotic catastrophe, we assessed the G2/M checkpoint and DNA damage
response (DDR)-related proteins by western blotting in CAL27 cells
(Fig. 4A). WEE1, a Ser/Thr
protein kinase family member, phosphorylates CDK1 at Tyr15,
inhibits its activity, and acts as a negative regulator for entry
into mitosis (G2 to M transition) (32). Thus, Adv treatment decreased CDK1
(Tyr15) phosphorylation (Fig.
4A). This indicated that CDK1 was activated, leading to
premature mitotic entry. At the same time, we observed increased
phosphorylation of Chk1 (Ser345), which indirectly inactivates
CDK1. This increased p-Chk1 presumably compensated for the
increased CDK1 activity. In contrast, RCS treatment suppressed Chk1
phosphorylation. Thus, co-administration of RCS with Adv suppressed
p-Chk1 and p-CDK1. This appeared to further promote forced mitotic
entry to induce mitotic catastrophe. In support of these findings,
co-administration of the two drugs resulted in further increase in
γ-H2A.X expression, a marker of DNA double-strand breaks. These
data suggest that RCS enhanced Adv-induced mitotic catastrophe by
suppressing p-Chk1 and p-CDK1 (Fig.
4A).
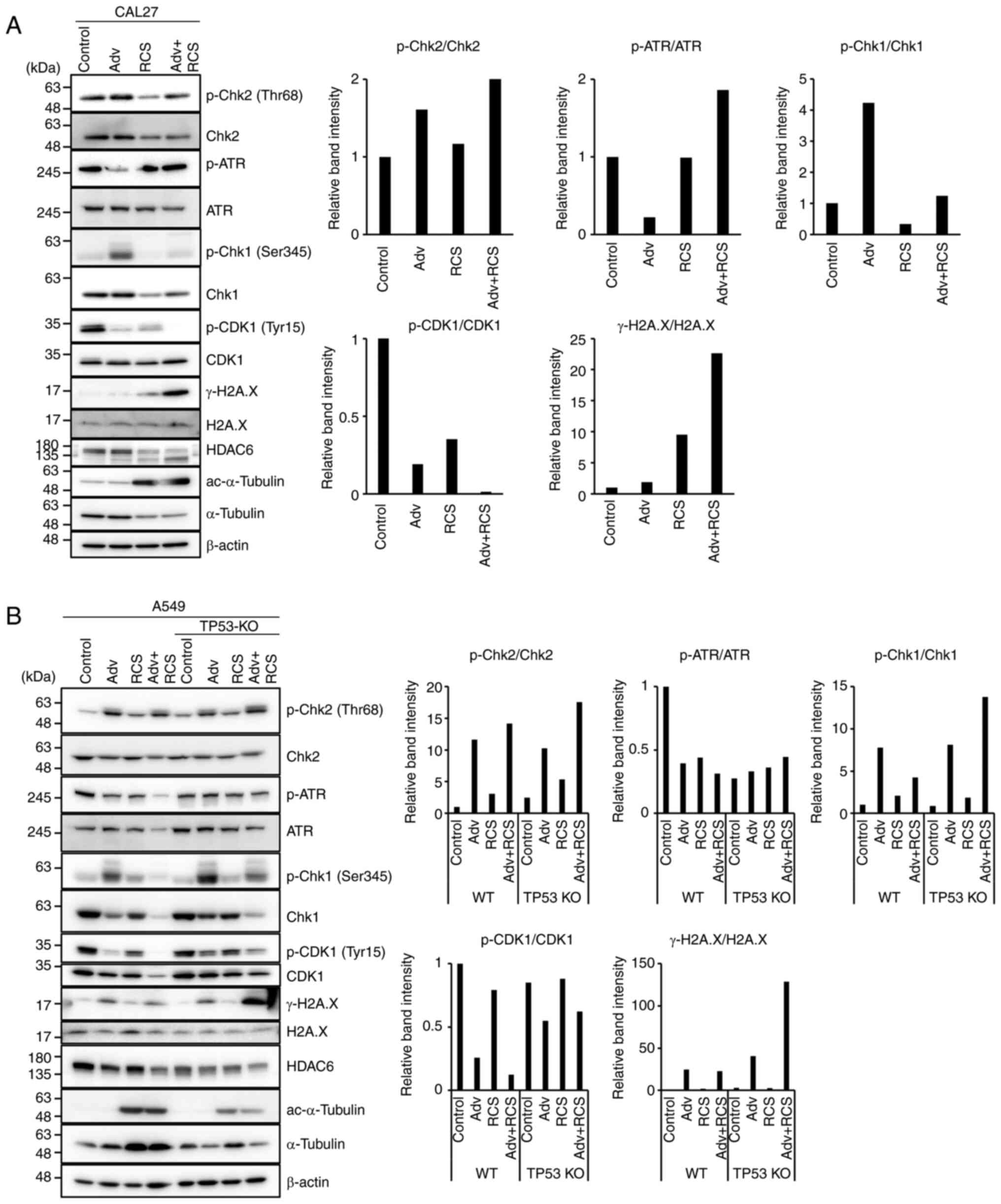 | Figure 4Ricolinostat suppresses
phosphorylation of Chk1 and further suppresses p-CDK1 when
co-administered with adavosertib. (A) CAL27 cells and (B) TP53-WT
and TP53-KO A549 cells were treated with Adv (0.5 µM), RCS
(5 µM), and Adv+RCS for 24 h (CAL27 cells) or 48 h (A549
cells), and then the expression of DNA damage response-related
proteins (p-Chk2, Chk2, p-ATR, ATR, p-Chk1, Chk1, p-CDK1, CDK1, and
γ-H2A.X) was assessed by western blotting. To assess the inhibitory
effect of HDAC6 by RCS, the level of acetylated (ac)-α-tubulin was
monitored. Expression of β-actin was assessed as the loading
control. The relative band intensity of each phosphorylated protein
was calculated and summarized at the right. Representative data of
three independent experiments are shown. Adv, adavosertib; RCS,
ricolinostat; WT, wild-type; Chk, checkpoint kinase; ATR, ATR
serine/threonine kinase; CDK1, cyclin-dependent kinase 1. |
As shown in Fig.
1, the pronounced cell death induction by combined treatment of
Adv and RCS appeared to be dependent on TP53 mutations. We
examined whether the co-administration of RCS suppressed p-Chk1 in
both WT and TP53-KO A549 cells (Fig.
4B). In both cells, Adv treatment increased p-Chk1 and
suppressed p-CDK1, similar to that observed in CAL27 cells.
Although co-administration of RCS with Adv suppressed p-CDK1 and
p-Chk1 in both cell lines, γ-H2A.X expression was increased only in
TP53-KO A549 cells. This may reflect the dependency of
TP53-mutated cells on the G2/M checkpoint. These data also
suggest that RCS enhances premature mitotic entry by suppressing
p-Chk1 in A549 cells. Additionally, we assessed the DDR-related
protein expression in Detroit562 cells which carry
mutated-TP53 but did not show pronounced cell death by
combined treatment of Adv and RCS. Detroit562 cells showed almost
the same result as WT A549 cells and failed to further upregulate
γ-H2A.X expression as compared to treatment with Adv alone
(Fig. S3). This suggests that
some of the TP53-mutated cell lines may have gained a
compensatory mechanism to p53 and are insensitive to
co-administration of RCS with Adv.
RCS enhanced cell death induction via
inhibition of HDAC6
It has been reported that although RCS selectively
inhibits HDAC6 (IC50=4.7 nM), it also inhibits HDAC1, 2,
and 3 (IC50=58, 48, and 51 nM, respectively) at higher
concentrations (33). To clarify
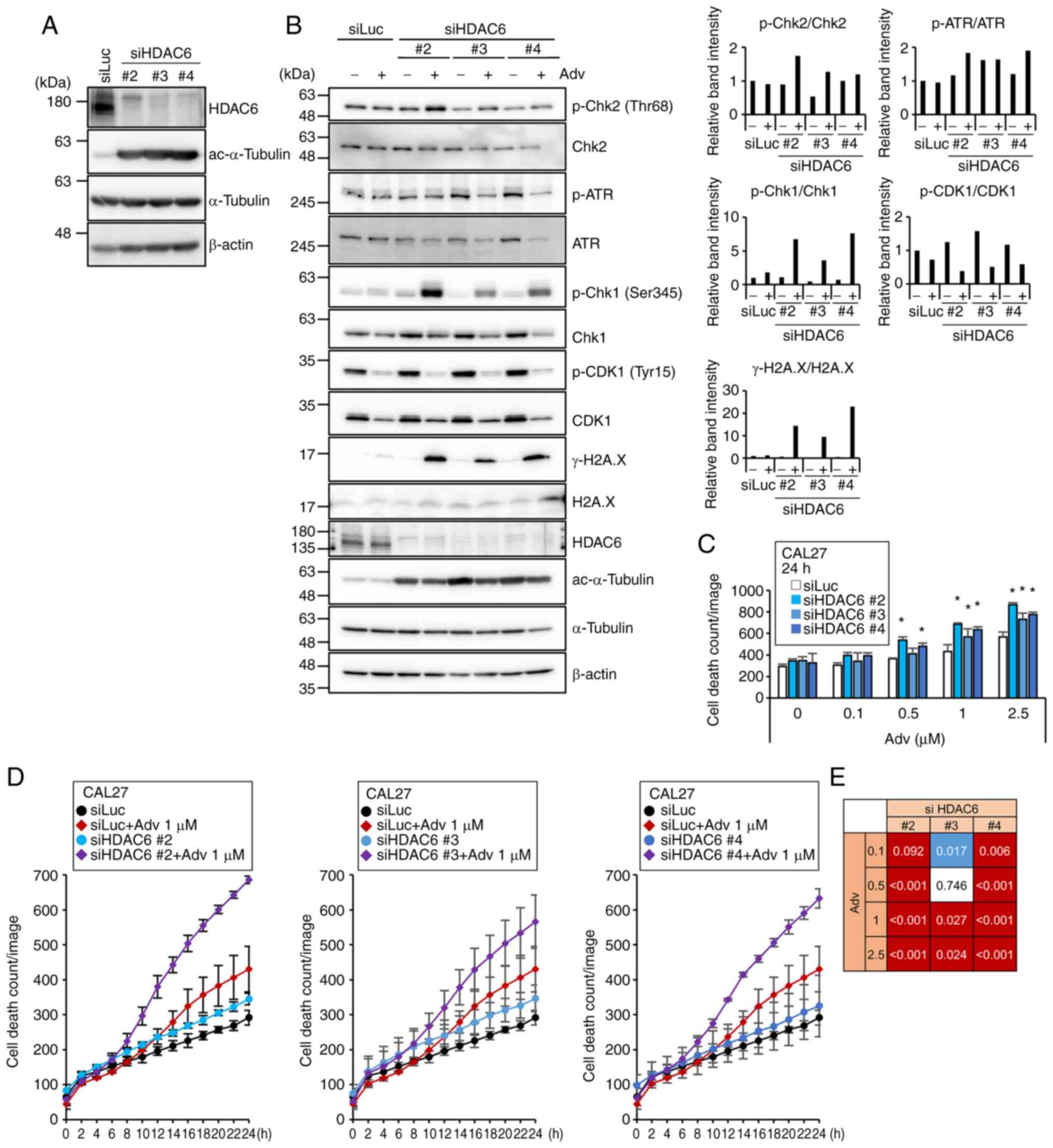
whether the enhanced cell death mediated by RCS was due to the
inhibition of HDAC6, we knocked down HDAC6 using three different
siRNAs (siHDAC6 #2, #3, and #4). All three siRNAs efficiently
knocked down HDAC6 and increased acetylated α-tubulin, which is a
substrate for HDAC6 (Fig. 5A).
Although KD of HDAC6 by itself did not change p-Chk1 levels or
γ-H2A.X expression levels, Adv treatment in HDAC6-KD cells resulted
in increased γ-H2A.X expression, as in the case of CAL27 cells
treated with RCS and Adv (Fig.
5B). However, we did not observe the suppression of p-Chk1 or
p-CDK1 in Adv-treated HDAC6-KD cells. In terms of cell death
induction, treatment with Adv in HDAC6-KD cells significantly and
synergistically increased cell death, as in the case of CAL27 cells
concomitantly treated with RCS and Adv (Fig. 5C-E). This suggested that increased
DNA damage and enhanced cell death by co-administration of RCS with
Adv appeared to have been caused by HDAC6 inhibition, but the
enhanced mitotic catastrophe caused by RCS addition to Adv
treatment may be independent of HDAC6 inhibition.
Discussion
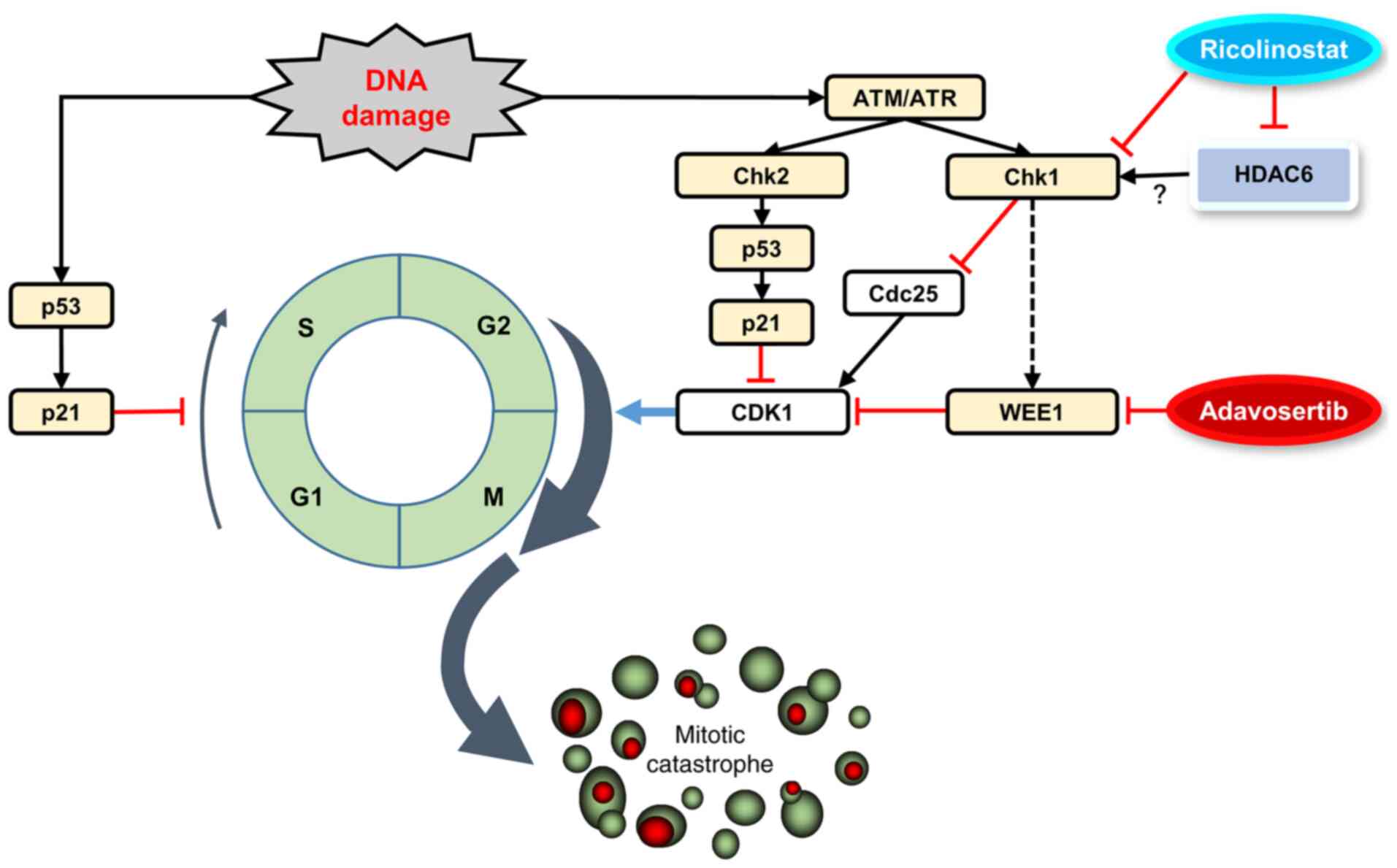
In the present study, the combined WEE1 G2
checkpoint kinase (WEE1) inhibitor adavosertib (Adv) and the
histone deacetylase 6 (HDAC6)-selective inhibitor ricolinostat
(RCS) treatment exhibited synergistic cytotoxicity in
TP53-mutated, impaired p53 function by HPV-infection, or
TP53-KO HNSCC, lung cancer, and breast cancer cell lines likely via
enhanced induction of Adv-induced mitotic catastrophe (Fig. 6). WEE1 phosphorylates the amino
acids Tyr15 and Thr14 of CDK1, which keeps the kinase activity of
CDK1 low and prevents entry into mitosis (32). As p53 regulates the G1/S
checkpoint, TP53-mutated cells rely on the G2/M checkpoint
to arrest the cell cycle to repair their DNA damage. Thus,
TP53-mutated cells with increased replication stress
appeared to exhibit greater sensitivity to WEE1 inhibitors
(34,35). The synergistic effect of the
two-drug combination shown in Fig.
1 appeared to be mediated through forced entry of the cell into
M phase by CDK1 activation. Indeed, the two-drug combination
resulted in enhanced dephosphorylation of CDK1, which is an active
state of CDK1, as compared to that in the treatment of Adv alone.
This was accompanied by pronounced γ-H2A.X expression, which
indicated an increase in DNA double-strand breaks in
TP53-mutated cells (Fig. 4A
and B). In Adv-treated cells, a compensatory mechanism appeared
to be induced, leading to an increased expression of p-Chk1.
However, combination treatment with RCS suppressed p-Chk1
upregulation and further activated CDK1 to promote premature
mitotic entry (Figs. 4 and
6).
Adv is an orally available, first-in-class,
reversible WEE1 inhibitor (36).
Most clinical trials using WEE1 inhibitors have been conducted as a
combination with chemotherapy or radiotherapy (37-40). In the case of WEE1 inhibitor
monotherapy against recurrent uterine serous carcinoma, which
frequently possesses TP53 mutation, the treatment showed
significant elongation of progression-free survival (PFS) and a
high response rate (41).
Consistent with our results (Figs.
1 and 2), several previous
studies have reported that sensitivity to Adv is dependent on
TP53 mutation status (6,7,42),
whereas a few other reports have shown that TP53 status does
not alter sensitivity to Adv (43). This may suggest that TP53
single gene mutation by itself is not sufficient to determine
sensitivity to WEE1 inhibitor monotherapy.
HDAC6 is a unique member of the HDAC subfamily
possessing two catalytic domains, namely DAC1 with E3-ligase
activity and DAC2 with deacetylase activity (44). HDAC6 deacetylates histones;
however, cytoplasmic proteins, namely α-tubulin, cortactin, and
heat shock protein 90 (HSP90), are also deacetylated by HDAC6
(45-47). The roles of HDAC6 in tumorigenesis
and cell-cycle progression have been reported (23,48,49). The overexpression of HDAC6 has
been reported in acute myeloid leukemia, oral squamous cell
carcinoma, and ovarian cancer (50-52), and higher HDAC6 expression appears
to correlate with tumor progression and malignancy in
hepatocellular carcinoma and esophageal squamous cell carcinoma
(53,54). In our study, HDAC6 inhibition by
RCS suppressed cell cycle progression (Fig. 3), which was consistent with
previous reports showing that RCS or other HDAC inhibitors caused
G2/M phase arrest (53,55). In contrast, in the presence of Adv
+ RCS, the cells underwent a pronounced mitotic catastrophe,
indicating increased entry into the M-phase. The molecular
mechanisms underlying this contradictory phenomenon remain
unclear.
Our study demonstrated that HDAC6 inhibition by RCS
suppressed p-Chk1 expression (Fig. 4A
and B), whereas HDAC6 KD slightly increased Chk1 (Fig. 5B). Thus, suppression of p-Chk1 by
RCS seemed to be HDAC6-independent. However, as described above, in
addition to deacetylase activity, HDAC6 possesses E3-ligase
activity, which ubiquitinates and degrades Chk1 and functions in
DDR. Therefore, it was reported that HDAC6 deletion resulted in an
increase in Chk1, which enhanced radiation-induced cell cycle
arrest in non-small cell lung cancer cells (44). In our system, although HDAC6 KD
suppressed both activities, RCS interacted only with DAC2 and
E3-ligase activity localized in DAC1 appeared to be intact. If the
increase in Chk1 due to DAC1 inhibition overcomes the inhibition of
p-Chk1 caused by DAC2 inhibition, the HDAC6 KD experiment alone is
not sufficient to determine whether RCS suppresses p-Chk1 via
HDAC6. To address this, we need to further evaluate the specific
disruption/deletion of the DAC2 domain in HDAC6.
In terms of the Chk1-CDK1 axis, Adv treatment of
HDAC6-KD cells resulted in increased γ-H2A.X and cell death despite
no decrease in p-Chk1 or p-CDK1 (Fig.
5). This indicates that the enhanced cell death by siHDAC6 or
RCS is not limited to be mediated by Chk1-CDK1. As previously
reported, we also showed that RCS treatment and HDAC6 KD resulted
in α-tubulin acetylation (Figs. 4
and 5) (45). It is well-known that
post-translational modifications of tubulins contribute to
microtubule dynamics, which are crucial for proper spindle
organization and cell cycle progression (56). CYLD, a tumor suppressor gene
product, has been reported to bind and inactivate HDAC6 along with
increased acetylation of tubulin, which negatively regulates
cell-cycle progression (23). In
addition, tubastatin A, an HDAC6 inhibitor, has been reported to
disrupt maturational progression and meiotic apparatus assembly in
mouse oocytes (57). Therefore,
it is still possible that HDAC6 inhibitors including RCS cause
abnormal mitosis by constitutively enhancing the acetylation of
tubulins, leading to disruption of microtubule dynamics. This may
also play a role in promoting the Adv-induced mitotic
catastrophe.
To the best of our knowledge, this is the first
report showing that RCS with a higher selectivity against HDAC6
suppresses p-Chk1. Although suppression of p-Chk1 by RCS may be
independent of HDAC6, enhanced DNA damage and cell death were
observed in both RCS-treated and HDAC6-KD cells in combination with
Adv treatment. As prominent cell death in Adv + RCS-treated cells
was only observed in TP53-mutated and TP53-KO cells, this
drug combination appears to be much less toxic to normal cells with
intact TP53 than in TP53-mutated cancer cells.
Applying this drug combination in the experiment with the tumor
xenograft model appears to have potential for future research.
Although in vivo studies remain to be carried out, this drug
combination with high specificity for cells lacking p53 function
could be a good candidate for the treatment of HNSCC patients
carrying TP53 mutations as well as human papillomavirus
(HPV)-positive HNSCC patients lacking p53 function.
Supplementary Data
Availability of data and materials
The datasets used during the present study are
available from the corresponding authors upon reasonable
request.
Authors' contributions
KM (Miyazawa) and NT conceived and designed the
present study. KM (Miyake), NT and HK (Kazama) performed the
experiments. KM (Miyake), NT and HK (Kikuchi) analyzed,
interpretated the data. KM (Miyake), NT and KM (Miyazawa) confirmed
the integrity of the data. KM (Miyazawa), NT, MH, KT and KM
(Miyake) wrote, reviewed and/or revised the manuscript. All authors
read and approved the manuscript and agree to be accountable for
all aspects of the research in ensuring that the accuracy or
integrity of any part of the work are appropriately investigated
and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
This study was supported by JSPS KAKENHI Grant Number 20K07298
(to NT) and the Cancer Research Grant afforded by the Tokyo Medical
University Cancer Research Foundation (to KM).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Agrawal N, Frederick MJ, Pickering CR,
Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et
al: Exome sequencing of head and neck squamous cell carcinoma
reveals inactivating mutations in NOTCH1. Science. 333:1154–1157.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stransky N, Egloff AM, Tward AD, Kostic
AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C,
McKenna A, et al: The mutational landscape of head and neck
squamous cell carcinoma. Science. 333:1157–1160. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang X and Sun Q: TP53 mutations,
expression and interaction networks in human cancers. Oncotarget.
8:624–643. 2017. View Article : Google Scholar :
|
|
5
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Indovina P and Giordano A: Targeting the
checkpoint kinase WEE1: Selective sensitization of cancer cells to
DNA-damaging drugs. Cancer Biol Ther. 9:523–525. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirai H, Arai T, Okada M, Nishibata T,
Kobayashi M, Sakai N, Imagaki K, Ohtani J, Sakai T, Yoshizumi T, et
al: MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor
efficacy of various DNA-damaging agents, including 5-fluorouracil.
Cancer Biol Ther. 9:514–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Den Haese GJ, Walworth N, Carr AM and
Gould KL: The Wee1 protein kinase regulates T14 phosphorylation of
fission yeast Cdc2. Mol Biol Cell. 6:371–385. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Castedo M, Perfettini JL, Roumier T,
Andreau K, Medema R and Kroemer G: Cell death by mitotic
catastrophe: A molecular definition. Oncogene. 23:2825–2837. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vitale I, Galluzzi L, Castedo M and
Kroemer G: Mitotic catastrophe: A mechanism for avoiding genomic
instability. Nat Rev Mol Cell Biol. 12:385–392. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meng X, Bi J, Li Y, Yang S, Zhang Y, Li M,
Liu H, Li Y, McDonald ME, Thiel KW, et al: AZD1775 increases
sensitivity to olaparib and gemcitabine in cancer cells with p53
mutations. Cancers (Basel). 10:1492018. View Article : Google Scholar
|
|
13
|
Lin X, Chen D, Zhang C, Zhang X, Li Z,
Dong B, Gao J and Shen L: Augmented antitumor activity by olaparib
plus AZD1775 in gastric cancer through disrupting DNA damage repair
pathways and DNA damage checkpoint. J Exp Clin Cancer Res.
37:1292018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Prystowsky MB, Adomako A, Smith RV,
Kawachi N, McKimpson W, Atadja P, Chen Q, Schlecht NF, Parish JL,
Childs G, et al: The histone deacetylase inhibitor LBH589 inhibits
expression of mitotic genes causing G2/M arrest and cell death in
head and neck squamous cell carcinoma cell lines. J Pathol.
218:467–477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Iglesias-Linares A, Yañez-Vico RM and
González-Moles MA: Potential role of HDAC inhibitors in cancer
therapy: Insights into oral squamous cell carcinoma. Oral Oncol.
46:323–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu YS, Kashida Y, Kulp SK, Wang YC, Wang
D, Hung JH, Tang M, Lin ZZ, Chen TJ, Cheng AL, et al: Efficacy of a
novel histone deacetylase inhibitor in murine models of
hepatocellular carcinoma. Hepatology. 46:1119–1130. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ozaki K, Kishikawa F, Tanaka M, Sakamoto
T, Tanimura S and Kohno M: Histone deacetylase inhibitors enhance
the chemosensitivity of tumor cells with cross-resistance to a wide
range of DNA-damaging drugs. Cancer Sci. 99:376–384. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ji M, Li Z, Lin Z and Chen L: Antitumor
activity of the novel HDAC inhibitor CUDC-101 combined with
gemcitabine in pancreatic cancer. Am J Cancer Res. 8:2402–2418.
2018.
|
|
19
|
Zhou L, Zhang Y, Chen S, Kmieciak M, Leng
Y, Lin H, Rizzo KA, Dumur CI, Ferreira-Gonzalez A, Dai Y and Grant
S: A regimen combining the Wee1 inhibitor AZD1775 with HDAC
inhibitors targets human acute myeloid leukemia cells harboring
various genetic mutations. Leukemia. 29:807–818. 2015. View Article : Google Scholar :
|
|
20
|
Qi W, Zhang W, Edwards H, Chu R,
Madlambayan GJ, Taub JW, Wang Z, Wang Y, Li C, Lin H and Ge Y:
Synergistic anti-leukemic interactions between panobinostat and
MK-1775 in acute myeloid leukemia ex vivo. Cancer Biol Ther.
16:1784–1793. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tanaka N, Patel AA, Tang L, Silver NL,
Lindemann A, Takahashi H, Jaksik R, Rao X, Kalu NN, Chen TC, et al:
Replication stress leading to apoptosis within the S-phase
contributes to synergism between vorinostat and AZD1775 in HNSCC
harboring high-risk TP53 mutation. Clin Cancer Res. 23:6541–6554.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hattori K, Takano N, Kazama H, Moriya S,
Miyake K, Hiramoto M, Tsukahara K and Miyazawa K: Synergistic
non-apoptotic cell death induction by simultaneously targeting
proteasome with bortezomib and HDAC6 with ricolinostat in head and
neck tumor cells. Oncol Lett. 22:6802021. View Article : Google Scholar
|
|
23
|
Wickström SA, Masoumi KC, Khochbin S,
Fässler R and Massoumi R: CYLD negatively regulates cell-cycle
progression by inactivating HDAC6 and increasing the levels of
acetylated tubulin. Embo J. 29:131–144. 2010. View Article : Google Scholar
|
|
24
|
Kim IA, No M, Lee JM, Shin JH, Oh JS, Choi
EJ, Kim IH, Atadja P and Bernhard EJ: Epigenetic modulation of
radiation response in human cancer cells with activated EGFR or
HER-2 signaling: Potential role of histone deacetylase 6. Radiother
Oncol. 92:125–132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Namdar M, Perez G, Ngo L and Marks PA:
Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA
damage and sensitizes transformed cells to anticancer agents. Proc
Natl Acad Sci USA. 107:20003–20008. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang L, Xiang S, Williams KA, Dong H, Bai
W, Nicosia SV, Khochbin S, Bepler G and Zhang X: Depletion of HDAC6
enhances cisplatin-induced DNA damage and apoptosis in non-small
cell lung cancer cells. PLoS One. 7:e442652012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Toriyama K, Takano N, Kokuba H, Kazama H,
Moriya S, Hiramoto M, Abe S and Miyazawa K: Azithromycin enhances
the cytotoxicity of DNA-damaging drugs via lysosomal membrane
permeabilization in lung cancer cells. Cancer Sci. 112:3324–3337.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pemble H, Kumar P, van Haren J and
Wittmann T: GSK3-mediated CLASP2 phosphorylation modulates
kinetochore dynamics. J Cell Sci. 130:1404–1412. 2017.PubMed/NCBI
|
|
29
|
Spangle JM, Ghosh-Choudhury N and Munger
K: Activation of cap-dependent translation by mucosal human
papillomavirus E6 proteins is dependent on the integrity of the
LXXLL binding motif. J Virol. 86:7466–7472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Szalai P and Engedal N: An image-based
assay for high-throughput analysis of cell proliferation and cell
death of adherent cells. Bio-protocol. 8:e28352018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vakifahmetoglu H, Olsson M and Zhivotovsky
B: Death through a tragedy: Mitotic catastrophe. Cell Death Differ.
15:1153–1162. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghelli Luserna di Rorà A, Cerchione C,
Martinelli G and Simonetti G: A WEE1 family business: Regulation of
mitosis, cancer progression, and therapeutic target. J Hematol
Oncol. 13:1262020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Santo L, Hideshima T, Kung AL, Tseng JC,
Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, et
al: Preclinical activity, pharmacodynamic, and pharmacokinetic
properties of a selective HDAC6 inhibitor, ACY-1215, in combination
with bortezomib in multiple myeloma. Blood. 119:2579–2589. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen X, Low KH, Alexander A, Jiang Y,
Karakas C, Hess KR, Carey JP, Bui TN, Vijayaraghavan S, Evans KW,
et al: Cyclin E overexpression sensitizes triple-negative breast
cancer to Wee1 kinase inhibition. Clin Cancer Res. 24:6594–6610.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Young LA, O'Connor LO, de Renty C,
Veldman-Jones MH, Dorval T, Wilson Z, Jones DR, Lawson D, Odedra R,
Maya-Mendoza A, et al: Differential activity of ATR and WEE1
inhibitors in a highly sensitive subpopulation of DLBCL linked to
replication stress. Cancer Res. 79:3762–3775. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Do K, Doroshow JH and Kummar S: Wee1
kinase as a target for cancer therapy. Cell Cycle. 12:3159–3164.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cuneo KC, Morgan MA, Sahai V, Schipper MJ,
Parsels LA, Parsels JD, Devasia T, Al-Hawaray M, Cho CS, Nathan H,
et al: Dose escalation trial of the Wee1 inhibitor Adavosertib
(AZD1775) in combination with gemcitabine and radiation for
patients with locally advanced pancreatic cancer. J Clin Oncol.
37:2643–2650. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leijen S, van Geel RM, Sonke GS, de Jong
D, Rosenberg EH, Marchetti S, Pluim D, van Werkhoven E, Rose S, Lee
MA, et al: Phase II study of WEE1 inhibitor AZD1775 plus
carboplatin in patients with TP53-mutated ovarian cancer refractory
or resistant to first-line therapy within 3 months. J Clin Oncol.
34:4354–4361. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lheureux S, Cabanero M, Cristea MC,
Mantia-Smaldone G, Olawaiye A, Ellard S, Weberpals JI, Hendrickson
AEW, Fleming GF, Welch S, et al: A randomized double-blind
placebo-controlled phase II trial comparing gemcitabine monotherapy
to gemcitabine in combination with adavosertib in women with
recurrent, platinum resistant epithelial ovarian cancer: A trial of
the Princess Margaret, California, Chicago and Mayo Phase II
Consortia. J Clin Oncol. 37:5518. 2019. View Article : Google Scholar
|
|
40
|
Oza AM, Estevez-Diz M, Grischke EM, Hall
M, Marmé F, Provencher D, Uyar D, Weberpals JI, Wenham RM, Laing N,
et al: A Biomarker-enriched, randomized phase II trial of
adavosertib (AZD1775) plus paclitaxel and carboplatin for women
with platinum-sensitive TP53-mutant ovarian cancer. Clin Cancer
Res. 26:4767–4776. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu JF, Xiong N, Campos SM, Wright AA,
Krasner C, Schumer S, Horowitz N, Veneris J, Tayob N, Morrissey S,
et al: Phase II study of the WEE1 inhibitor adavosertib in
recurrent uterine serous carcinoma. J Clin Oncol. 39:1531–1539.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ku BM, Bae YH, Koh J, Sun JM, Lee SH, Ahn
JS, Park K and Ahn MJ: Mutational status of TP53 defines the
efficacy of Wee1 inhibitor AZD1775 in KRAS-mutant non-small cell
lung cancer. Oncotarget. 8:67526–67537. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kreahling JM, Foroutan P, Reed D, Martinez
G, Razabdouski T, Bui MM, Raghavan M, Letson D, Gillies RJ and
Altiok S: Wee1 inhibition by MK-1775 leads to tumor inhibition and
enhances efficacy of gemcitabine in human sarcomas. PLoS One.
8:e575232013.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Moses N, Zhang M, Wu JY, Hu C, Xiang S,
Geng X, Chen Y, Bai W, Zhang YW, Bepler G and Zhang XM: HDAC6
regulates radiosensitivity of non-small cell lung cancer by
promoting degradation of Chk1. Cells. 9:22372020. View Article : Google Scholar :
|
|
45
|
Hubbert C, Guardiola A, Shao R, Kawaguchi
Y, Ito A, Nixon A, Yoshida M, Wang XF and Yao TP: HDAC6 is a
microtubule-associated deacetylase. Nature. 417:455–458. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kovacs JJ, Murphy PJ, Gaillard S, Zhao X,
Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB and Yao TP: HDAC6
regulates Hsp90 acetylation and chaperone-dependent activation of
glucocorticoid receptor. Mol Cell. 18:601–607. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang X, Yuan Z, Zhang Y, Yong S,
Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR,
et al: HDAC6 modulates cell motility by altering the acetylation
level of cortactin. Mol Cell. 27:197–213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y,
Barrientos T, Ordentlich P, Wang XF, Counter CM and Yao TP: The
cytoplasmic deacetylase HDAC6 is required for efficient oncogenic
tumorigenesis. Cancer Res. 68:7561–7569. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lernoux M, Schnekenburger M, Dicato M and
Diederich M: Anti-cancer effects of naturally derived compounds
targeting histone deacetylase 6-related pathways. Pharmacol Res.
129:337–356. 2018. View Article : Google Scholar
|
|
50
|
Bradbury CA, Khanim FL, Hayden R, Bunce
CM, White DA, Drayson MT, Craddock C and Turner BM: Histone
deacetylases in acute myeloid leukaemia show a distinctive pattern
of expression that changes selectively in response to deacetylase
inhibitors. Leukemia. 19:1751–1759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sakuma T, Uzawa K, Onda T, Shiiba M, Yokoe
H, Shibahara T and Tanzawa H: Aberrant expression of histone
deacetylase 6 in oral squamous cell carcinoma. Int J Oncol.
29:117–124. 2006.PubMed/NCBI
|
|
52
|
Bazzaro M, Lin Z, Santillan A, Lee MK,
Wang MC, Chan KC, Bristow RE, Mazitschek R, Bradner J and Roden RB:
Ubiquitin proteasome system stress underlies synergistic killing of
ovarian cancer cells by bortezomib and a novel HDAC6 inhibitor.
Clin Cancer Res. 14:7340–7347. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cao J, Lv W, Wang L, Xu J, Yuan P, Huang
S, He Z and Hu J: Ricolinostat (ACY-1215) suppresses proliferation
and promotes apoptosis in esophageal squamous cell carcinoma via
miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 9:8172018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kanno K, Kanno S, Nitta H, Uesugi N, Sugai
T, Masuda T, Wakabayashi G and Maesawa C: Overexpression of histone
deacetylase 6 contributes to accelerated migration and invasion
activity of hepatocellular carcinoma cells. Oncol Rep. 28:867–873.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chuang MJ, Wu ST, Tang SH, Lai XM, Lai HC,
Hsu KH, Sun KH, Sun GH, Chang SY, Yu DS, et al: The HDAC inhibitor
LBH589 induces ERK-dependent prometaphase arrest in prostate cancer
via HDAC6 inactivation and down-regulation. PLoS One. 8:e734012013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Vicente JJ and Wordeman L: The
quantification and regulation of microtubule dynamics in the
mitotic spindle. Curr Opin Cell Biol. 60:36–43. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ling L, Hu F, Ying X, Ge J and Wang Q:
HDAC6 inhibition disrupts maturational progression and meiotic
apparatus assembly in mouse oocytes. Cell Cycle. 17:550–556. 2018.
View Article : Google Scholar :
|