Introduction
The overexpression of chromosome segregation 1-like
(CSE1L), also known as cellular apoptosis susceptibility protein,
has been previously reported to correlate positively with the
progression of various malignacies, such as gastric cancer and
colorectal cancer (CRC) (1–5).
However, this CSE1L-induced risk of CRC tumorigenisis can be
suppressed by the activation of wild-type p53 expression (6). It has been also reported that
different chemotherapeutic agents, including 5-fluorouracil,
cisplatin, and paclitaxel, can mediate differential apoptotic
effects in CSE1L-overexpressing CRC cancer cells (7,8).
The therapeutic efficacy of drugs against CRC can be reduced
through the suppression of paclitaxel-induced apoptosis in
CSE1L-expressing CRC cells (8,9).
Therefore, CSE1L knockdown may improve CRC treatment (8).
Gut microbes can regulate the gene expression
profile in colonic cells, which may in turn alter the course of CRC
(10,11). Short-chain fatty acids (SCFAs)
derived from microbial metabolism in the gut, including acetate,
propionate and butyrate, are key for the maintenance of intestinal
homeostasis (12,13). SCFAs can induce cell apoptosis and
cell cycle arrest to reduce cancer risk (14), rendering them to be potential
chemotherapeutic agents (15).
Butyrate-producing microorganisms in the gut have been reported to
prevent necrotic enteritis and reduce pathogen abundance (16–18). Therefore, dysbiosis caused by the
overpopulation of detrimental microbes and underpopulation of
beneficial butyrate-producing microbes in the gut may confer
clinical significance in CRC. However, the effects of butyrate in
CRC and the molecular mechanism underlying such effects remain
unclear (19). Butyrate has been
previously documented to downregulate the expression of a number of
genes, including placenta specific 8 protein, toll-like receptor 4
and glucose 6-phosphatase (10,20–22). It can attenuate the
lipopolysaccharide-induced inflammation of intestinal epithelial
cells whilst exerting antioncogenic potential in LS1034 or WiDr
human CRC cells by promoting p53 gene expression (23–25). In particular, patients with
inflammatory bowel disease or CRC were found to have lower
concentrations of the butyrate-producing microbe butyricicoccus
pullicaecorum in their stools (10,26). In addition, the culture
supernatant of B. pullicaecorum is rich in butyrates and can
strengthen intestinal barrier function (17,26), which supports the pharmabiotic
potential B. pullicaecorum for clinical application
(10,20,27,28). However, the possible association
between CSE1L and/or the butyrate-producing B. pullicaecorum
in the development of CRC remain poorly understood.
A previous study has indicated that suppression of
CSE1L expression in CRC cells may improve CRC treatment (8). Butyrate-producing microorganisms in
the gut have been reported to confer potentially anti-CRC effects
(17). Accordingly, a possible
strategy to alter CRC physiology would be to decrease CSE1L
expression in CRC cells by either B. pullicaecorum
administration or butyrate supplementation. However, information
concerning the potential effects remain unavailable. Therefore, the
aim of the present study was to explore the potential role of
butyrate in the molecular events mediated by CSE1L in CRC cell
lines with different p53 genotypes. Knockdown of CSE1L in HCT116
p53−/− cells, knockdown of p53 in HCT116
p53+/+ and CRC cell lines with very distinct differences
in the p53 status were applied to evaluate the effects of CSE1L on
the expression levels of wild-type p53. To examine the molecular
significance of butyrate supplementation on CSE1L expression and
CRC alleviation, cellular physiology of buyrate-treated HCT116
p53−/− cells in vitro and the colon tumors from
mice after B. pullicaecorum administration in vivo
were used. In this manner, the potential role of B.
pullicaecorum and CSE1L in CRC were investigated and
clarified.
Materials and methods
Induction of CRC in mice
A total of 17 male BALB/cByJNarl mice aged 4–6
weeks, weighing 22.7±0.6 g, were provided by the National
Laboratory Animal Center (National Applied Research Laboratories,
Taipei, Taiwan). All animal experiments were conducted in
compliance with ARRIVE (Animal Research: Reporting of In Vivo
Experiments) guidelines (29).
The protocols followed the principles of Reduction, Refinement and
Replacement and were approved by the Institutional Animal Care and
Use Committees of Cathay General Hospital (approval no. 107-008;
Taipei, Taiwan). Mice (at n=3–5 per plastic cage) were housed in an
individually ventilated cage rack system (Tecniplast Group) and had
free access to food and drinking water under the following
conditions: 50±10% humidity, 12-h light/dark cycle and 23±2°C
temperature. All efforts were made to minimize the number of mice
and their suffering. The mice were classified into the following
groups as previously described (17): i) Control group (n=5), consisting
of mice that did not receive any chemical or B.
pullicaecorum administration; ii) 1,2-dimethylhydrazine (DMH;
cat. no. D0741; Tokyo Chemical Industry Co., Ltd.)/dextran sulphate
sodium (DSS; cat. no. D5144; Tokyo Chemical Industry Co., Ltd.)
group (n=6), consisting of mice that received DMH (20 mg/kg) once
at the beginning of the experiment through intraperitoneal
injection, followed by 1 week of normal water and 1 week of DSS (30
g/l) in the drinking water, with two cycles of additional DSS
treatment (2 weeks of normal water + 1 week of DSS (30 g/l) in
drinking water); and iii) DMH/DSS/B. pullicaecorum group (n=6),
consisting of mice that received DMH/DSS in the same manner as
DMH/DSS group, but were treated with B. pullicaecorum every
7 days during the experiment. The body weight of each mouse was
monitored once a week. All mice were euthanized with CO2
in a cage when they showed weakness and rapid weight loss of 15–20%
or at the end of the experiment. The duration of this animal
experiment was 2–3 months. The CO2 flow rate was set to
displace 30% of the cage volume per minute. Immobility for >2
min and lack of spontaneous breathing were used to confirm animal
death before the colon samples were collected.
B. pullicaecorum administration by oral
gavage
The molecular effects of B. pullicaecorum on
colon tumor formation was evaluated. B. pullicaecorum
(3.125×107 colony-forming units in 100 µl of
modified peptone yeast extract broth) was administered by oral
gavage. B. pullicaecorum (cat. no. BCRC-81109; https://catalog.bcrc.firdi.org.tw/BcrcContent?bid=81109)
and the growth medium (modified peptone yeast extract broth; cat.
no. 967; Bioresource Collection and Research Center) were purchased
from the Bioresource Collection and Research Center (Hsinchu,
Taiwan) and cultured for 3 days under anaerobic conditions (10%
CO2 and 90% N2) at 37°C as described
previously (17).
Cell lines and reagents
In total, two human colon cell lines, CCD-18Co [cat.
no. CRL-1459; American Type Culture Collection (ATCC)], which
harbors wild-type p53 and FHC (cat. no. CRL-1831; ATCC), which
expresses the R273H p53 mutant (30), were acquired as non-transformed
colon cells (30,31). In addition, three human CRC cell
lines (LS 174T, cat. no. CL-188; T84, cat. no. CCL-248; HCT116
p53+/+, cat. no. CCL-247; all from ATCC) expressing
wild-type p53, two human CRC cell lines (SW480, cat. no. CCL-228;
SW620, cat. no. CCL-227; both from ATCC) carrying the R273H and
P309S double p53 mutation (32),
in addition to two p53-null cell lines [Caco-2, cat. no. HTB-37,
ATCC; HCT116 p53−/−, a gift from Professor Bert
Vogelstein (School of Medicine, Johns Hopkins University,
Baltimore, USA)] (33,34), were used as tumorigenic cancer
cells (35–38). These cell lines were expanded in
complete media [medium suggested for each cell line by the ATCC,
10% FBS (Asia Bioscience Co., Ltd.) and 1X antibiotic/antimycotic
solution (cat. no. 15240-062; Thermo Fisher Scientific, Inc.)] in a
humidified chamber with 95% air and 5% CO2 at 37°C with
some exceptions. Briefly, four cell lines (FHC, T84, HCT116
p53+/+ and HCT116 p53−/−) were cultured with
DMEM (cat. no. 12800-017; Thermo Fisher Scientific, Inc.) whereas
three cell lines (CCD-18Co, LS 174T and Caco-2) were cultured with
MEM (cat. no. 41500-034; Thermo Fisher Scientific). In addition,
SW480 and SW620 cell lines were cultured with Leibovitz's L-15
medium (cat. no. 11415-064; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS and 1X antibiotic/antimycotic solution,
but maintained under 100% atmospheric air (without CO2)
in a humidified incubator at 37°C.
To measure the expression of CSE1L in cells after
treatment with 5-fluouracil (5-FU; cat. no. F6627; Merck KGaA) or
sodium buyrate (NaB; cat. no. B5887; Merck KGaA), a total of
5×105 cells were cultured for 24 h at 37°C and before
chemicals were added as follows: HCT116 p53+/+ cells
with 5-FU (40 µM) for 24 h at 37°C, whereas HCT116
p53−/−, SW480 and SW620 cells with NaB (5 mM) for 24 or
48 h at 37°C.
To differentiate Caco-2 cells into a polarized
enterocyte-like monolayer, cells were seeded at 8×105
cells per well and cultured to confluence for 21 days at 37°C, with
changes of fresh MEM supplemented with 20% FBS and 1%
antibiotic/antimycotic solution every 1–2 days (39,40).
Lentiviral knockdown of p53 and
CSE1L
All lentiviral particles were obtained from the RNA
Technology Platform and Gene Manipulation Core (https://rnai.genmed.sinica.edu.tw/index.html).
Briefly, lentiviral particles were packaged in 293T cells (cat. no.
CRL-3216; ATCC) using the 2nd generation system, with the combined
ratio of lentiviral construct, packaging plasmid and envelope
plasmid at 1 µg: 900 ng:100 ng. The 293T cells were then
cultured in DMEM supplemented with 10% FBS and 0.1X
antibiotic/antimycotic solution in a humidified chamber with 95%
air and 5% CO2 at 37°C for 40 h. The cultured media were
spun (300 x g for 5 min) to remove any packaging cells and
supernatant containing viral paricles were collected. In total, two
lentiviral constructs, namely pLKO.1_ p53 (clone ID:
TRCN0000003753) encoding a short hairpin RNA (shRNA) targeting p53
(shp53) and pLKO.1_CSE1L (clone ID: TRCN0000061789) targeting CSE1L
(shCSE1L), were used for p53 knockdown in HCT116 p53+/+
cells and CSE1L knockdown in HCT116 p53−/− cells. The
control pLKO.1-luciferase (Luc; clone ID: TRCN0000072249) vector
targeting Luc was used as the negative control (shLuc-HCT116
p53+/+ for shp53 or shLuc-HCT116 p53−/− for
shCSE1L). A total of 1.25×105 cells/well were cultured
in six-well plates for 24 h at 37°C, before subsequent lentiviral
infections (multiplicity of infection, 3) were performed to knock
down the expression of target genes in the cells. Subsequently,
medium containing 2 mg/ml puromycin (Thermo Fisher Scientific,
Inc.) was used to select and maintain the stable clones. After a
48-h incubation at 37°C, transfection efficiency was determined
using reverse transcription-quantitative PCR (RT-qPCR).
RNA isolation, cDNA synthesis and gene
quantitation
Total RNA was extracted from the parental CRC cell
lines (CCD-18Co, FHC, LS 174T, T84, Caco-2, HCT116
p53+/+, HCT116 p53−/−, SW480 and SW620) and
their derived cells, using the Easy Pure Total RNA Spin kit (cat.
no. RT050; Bioman Scientific, Co., Ltd.) according to the
manufacturer's protocol. Single-stranded cDNA was generated from 1
µg total RNA in the presence of an oligo (dT)12
primer using the High-Capacity cDNA Reverse Transcription kit (cat.
no. 4368813; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. mRNA expression levels were quantified
through qPCR using the LightCycler® TaqMan Master mix
(cat. no. 04535286001; Roche Diagnostics GmbH) with a specific
thermocycling profile (95°C for 10 min, followed by 50 cycles at
95°C for 10 sec and 60°C for 20 sec) as described in a previous
study (10,17). Primer sequences and probe numbers
were CSE1L (Universal Probe: #27): forward,
5′-GTTGTCTACCGCCTGTCCA-3′ and reverse,
5′-AAATGCAGTTTAAAGCAGTGTCA-3′; c-Myc (Universal Probe: #34):
forward, 5′-CACCAGCAGCGACTCTGA-3′ and reverse,
5′-ACTCTGACCTTTTGCCAGGA-3′; p53 (Universal Probe: #12): forward,
5′-AGGCCTTGGAACTCAAGGAT-3′ and reverse, 5′-CCCTTTTTGGACTTCAGGTG-3′
and GAPDH (Universal Probe: #60): forward,
5′-CTCTGCTCCTCCTGTTCGAC-3′ and reverse 5′-ACGACCAAATCCGTTGACTC-3′.
Expression levels were quantified using the 2−ΔΔCq
method and normalized to the expression level of GAPDH (41). The human reference cDNA (HRC; cat.
no. 636692; Takara Bio, Inc.) was used as an expression control.
Gene expression data were obtained after performing ≥ three
independent experiments with similar results.
Preparation of whole cell extracts and
nuclear/cytosol fractions for western blotting
Whole-cell extracts from shLuc-HCT116
p53−/− and shCSE1L-HCT116 p53−/− cells were
prepared using PRO-PREP Protein Extraction Solution (Intron
Biotechnology, Inc.) in the presence of a protease inhibitor (cat.
no. P8340; Merck KGaA) according to the manufacturer's protocols.
Cell fractionation was performed using NE-PER Nuclear and
Cytoplasmic Extraction Reagents kit (cat. no. 78833; Thermo Fisher
Scientific, Inc.) to isolate the different protein fractions from
the cytoplasm and nuclei, according to the manufacturer's
protocols. The purity of non-nuclear and nuclear fractions was
determined using specific protein markers, namely Tubulin and
TATA-box binding protein (TBP), respectively. Each protein
concentration was then determined using a Bio-Rad Protein Assay
reagent (cat. no. 500-0006; Bio-Rad Laboratories, Inc.). Next, 30
µg of protein per lane was denatured at 95°C for 10 min,
separated using 12% SDS-PAGE in 1X NuPAGE LDS Sample Buffer (Thermo
Fisher Scientific, Inc.) and then transferred onto 0.2-µm
PolyScreen 2 PVDF Transfer membranes (PerkinElmer, Inc.). The
membranes were blocked with 3% bovine serum albumin (cat. no.
ALB001.100; BioShop Canada, Inc.) for 1 h at room temperature and
incubated with the following primary antibodies for 1 h at room
temperature: Anti-CSE1L (1:1,000; cat. no. 22219-1-AP; Proteintech
Group, Inc.), anti-p53 (1:500; cat. no. NCL-L-p53-DO7; Leica
Biosystems, Inc.), anti-cyclin A2 (CCNA2; 1:2,000; cat. no. 4656P;
Cell Signaling Technology, Inc.), anti-cyclin B2 (CCNB2; 1:2,000;
cat. no. ab185622; Abcam), anti-cyclin D1 (CCND1; 1:1,000; cat. no.
2978; Cell Signaling Technology, Inc.), anti-tubulin (1:1,000; cat.
no. sc-5286, Santa Cruz Biotechnology, Inc.), anti-TBP (1:1,000;
cat. no. 22006-1-AP, Proteintech Group, Inc.) and anti-GAPDH
(1:5,000; cat. no. 60004-1-Ig; Proteintech Group, Inc.). Expression
of GAPDH was used as the endogenous control gene. After incubation
of the primary antibodies, the membranes were incubated with a
HRP-conjugated anti-mouse IgG (H&L) secondary antibody
(1:5,000; cat. no. ab6808; Abcam) HRP-conjugated antirabbit IgG
secondary antibody (1:5,000; cat. no. L3012; Signalway Antibody
LLC) for 60 min at room temperature. Protein bands were visualized
using Western Lightning Chemiluminescence Reagent Plus
(PerkinElmer, Inc.) and an AlphaView software (version 3.2.2) of
the FluorChem FC2 Imaging System (Cell Biosciences, Inc.) according
to the manufacturers' protocols (Alpha Innotech FluorChem FC2
Imaging System; Cell Biosciences, Inc.).
Cell cycle analysis and immunofluorescent
staining
shLuc-HCT116 p53−/− cells and shCSE1L
HCT116 p53−/− cells were starved under low-serum
conditions (0.5%) for 24 h at 37°C and then cultured in complete
medium for 24 h at 37°C. They were then fixed in 70% prechilled
ethanol for >1 h at −20°C, washed twice with PBS, incubated with
1 µg/ml RNase A for 1 h at 37°C and stained with 5
µg/ml propidium iodide for 1 h at room temperature. Light
emission at 585 nm from propidium iodide-stained nuclei was
detected using a BD FACScan flow cytometer (BD Bioscienes). The
percentages of cells (from 1×104 cells) at different
phases of cell cycle were determined using FlowJo software (v. 8.7;
FlowJo LLC).
A total of 1.5×104 HCT116
p53−/− cells and HCT116 p53+/+ cells for
immunofluorescence staining were cultured on 12-mm cover slips (SPL
Life Sciences). The cells were probed with a diluted anti-CSE1L
antibody (1:50; cat. no. 22219-1-AP; Proteintech Group, Inc.) or
anti-Tubulin antibody (1:50; cat. no. sc-5286; Santa Cruz
Biotechnology, Inc.) for 16 h at 4°C after fixation with 4%
paraformaldehyde (Merck KGaA) in PBS for 10 min at room
temperature, permeabilization with 0.1% Triton X-100 (Merck KGaA)
in PBS for 35 min at room temperature, and blocking with 1.5%
normal horse serum blocking solution (cat. no. S-2000-20; Vector
Laboratories, Inc.; Maravai LifeSciences) in 10 ml PBS for 30 min
at room temperature. Next, the FITC-conjugated secondary antibody
(1:200; cat. no. AP132F; Merck KGaA) for CSE1L and the
Cy3-conjugated secondary antibody (1:200; cat. no. AP124C; Merck
KGaA) for α-tubulin were incubated for 1 h at room temperature.
Nuclear DNA was stained with 1 µg/ml DAPI (cat. no.
71-03-01; Kirkegaard & Perry Laboratories Inc.) for 15 min at
room temperature. The stained samples were dehydrated through an
ascending ethanol series and air-dried for 10 min at room
temperature before being mounted in VECTASHIELD®
HardSet™ Antifade Mounting Medium (cat. no. H-1400; Vector
Laboratories, Inc.; Maravai LifeSciences). They were then observed
using a Nikon Eclipse 80i fluorescence microscope at ×200
magnification (Nikon Corporation) before fluorescence was
quantified from > five fields of views (10 views for HCT116
p53+/+ cells and five views for HCT116 p53−/−
cells) using Adobe Photoshop (version CS6; Adobe Systems,
Inc.).
Immunohistochemical staining for mouse
tissues
Mouse colorectal samples were fixed with 4%
paraformaldehyde in PBS for 10 min at room temperture and embedded
in paraffin. Paraffin sections (3–5 µm thickness) were
obtained and then processed using the
avidin-biotin-immunoperox-idase method to measure the expression of
CSE1L and p53. Immunohistochemical staining was performed on an
auto-mated BenchMark GX slide stainer (Roche Diagnostics) in a
closed and fixed program, which included deparaffinization with EZ
Prep solution (cat. no. 950-102; Ventana Medical Systems) at 75°C
for 8 min, antigen retrieval with Cell Conditioning 1 solution
(cat. no. 950-124; Ventana Medical Systems) at 95°C for 64 min,
incubation with primary anti-body (at 37°C for 32 min) followed by
HQ Universal Linker (cat. no. 253-4580; Ventana Medical Systems) at
37°C for 8 min and HRP Multimer (cat. no. 253-4581; Ventana Medical
Systems) at 37°C for 8 min and visualization by DAB. The Optiview
DAB IHC detection kit (cat. no. 760-700; Roche Diagnostics) was
used as a detection system. All sections were counterstained with
Hematoxylin II at 25°C for 8 min (cat. no. 790-2208; Ventana
Medical Systems) and Bluing Reagent at 25°C for 4 min (cat. no.
760-2037; Ventana Medical Systems). Anti-CSE1L (1:50; cat. no.
22219-1-AP; Proteintech Group, Inc.) or anti-p53 (1:50; cat. no.
BS-8687R, Thermo Fisher Scientific, Inc.) were hybridized to detect
target protein. A pathologist (CYL) observed and categorized the
immunohistochemically stained sections using a Nikon Eclipse 80i
fluorescence microscope at ×200 magnification by light microscopy
(Nikon Corporation).
Cell migration assay
The shLuc-HCT116 p53−/− and
shCSE1L-HCT116 p53−/− cells were grown to confluence on
six-well plates before a wound was made by scraping across the cell
monolayer with a 30 gauge needle (outer diameter, 300 µm).
The motility of the cells at the edge of a scratch wound in the
presence or absence of NaB (5 mM) was then analyzed. Cells at the
wound edge were imaged using a bright-field/phase-contrast
microscope at ×200 magnification. Repeat images were taken after
wounding in medium with low serum (DMEM, 1% FBS and 1%
antibiotic/antimyotic solution) at 37°C for 16 h and followed with
complete media for indicated time (0, 4 and 8 h). Serum-free media
was first tested for this assay but HCT p53−/− cells
could not survive in this condition, which necesitated the use of
1% for maintenance followed by complete medium (10% FBS) for the
assay. It is predicted that the extent of interference due to cell
proliferation would be minimal, as the doubling time of HCT116
cells is ~18 h and the maximum duration of the wound healing assay
in the present study was 8 h. ImageJ (version 1.45s; National
Institutes of Health) was used to measure the migration distance at
each time point (42). Next, the
cell migration efficiency after 8 h of cultivation was assessed
using the recovery ratio according to the reduction of wound area
(the percentage of cell area difference, relative to the inititial
time point of 0 h) (43). In
total, three or four independent sets of experiments were performed
for each assay.
Statistical analysis
The difference in gene expression, cell phase and
cell migration between the groups was assessed. A unpaired
student's t test was used to compare two groups whereas one-way
analysis of variance was performed to compare among ≥ three
different groups. All ANOVA analyses were followed with Bonferroni
post hoc testing. The statistical analyses were performed using
SPSS (v. 22.0; IBM Corp). Data are presented as the mean ± standard
error of the mean. P<0.05 was considered to indicate a
statistically significant difference.
Results
CSE1L mRNA expression levels in the
different colonic and CRC cell lines
The expression levels of CSE1L in different colonic
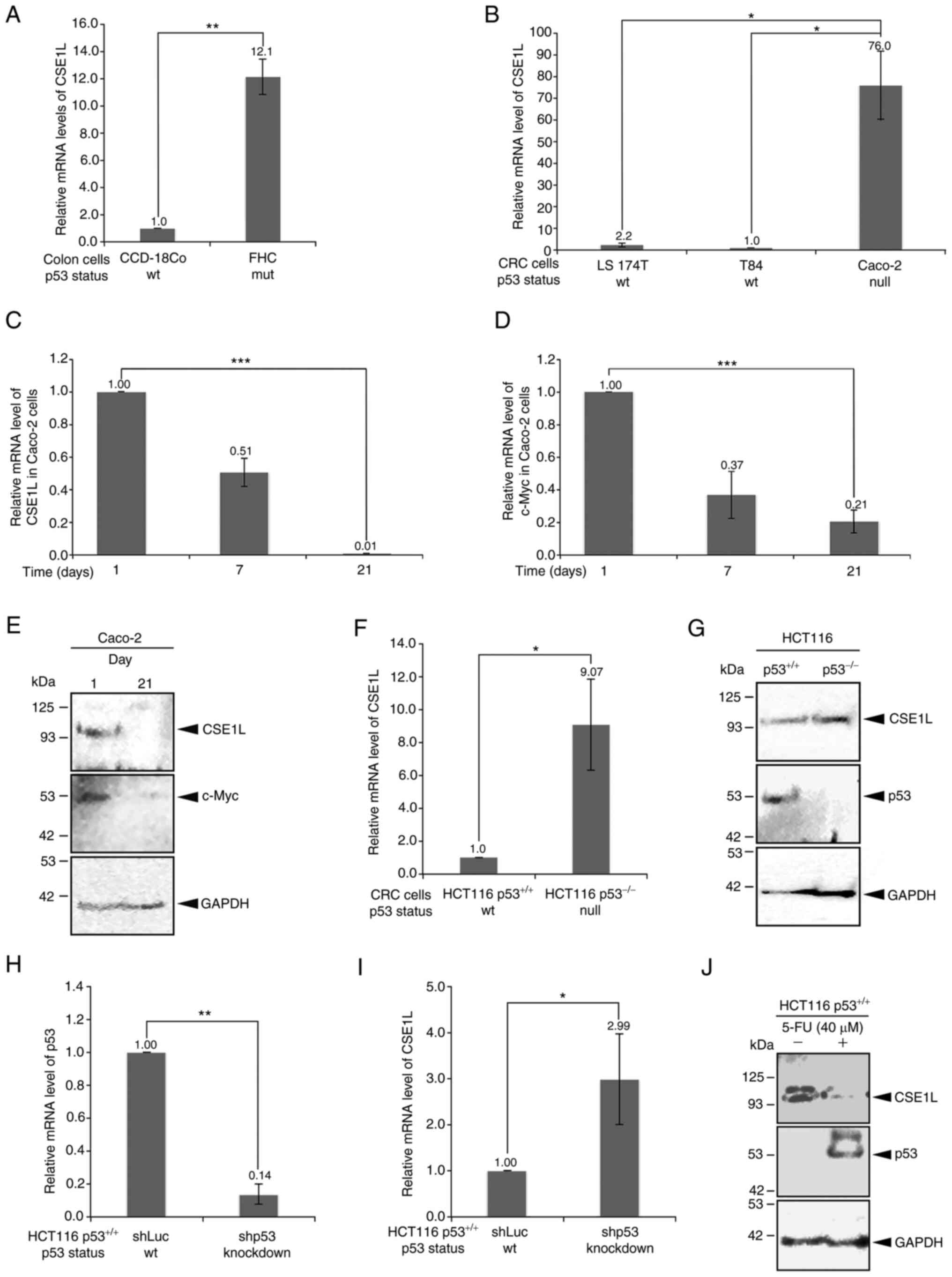
and CRC cell lines were quantified (Fig. 1). CSE1L expression levels in the
two non-transformed cell lines, CCD-18Co and FHC, varied
significantly according to their different p53 mutation statuses
(Fig. 1A). Briefly, the CCD-18Co
cells with the wild-type p53 expressed the lower levels of CSE1L
compared with those in FHC cells with the R273H p53 mutant. Among
the CRC cell lines, Caco-2 cells haboring p53 mutations exhibited
significantly higher CSE1L expression levels compared with those in
LS 174T and T84 cells, both of which express wild-type p53
(Fig. 1B). However, CSE1L and
c-Myc mRNA expression levels both simulataneously and progressively
reduced in Caco-2 cells as their confluency increased (Fig. 1C and D). In addition, the protein
expression levels of CSE1L and c-Myc were markedly decreased in
Caco-2 cells following proliferation to confluence on day 21
compared with those in cells on day 1 (Fig. 1E).
Higher CSE1L protein expression levels were also
observed in HCT116 cells not expressing p53 (Fig. 1F and G) or in HCT116 cells
following p53 knockdown (Fig. 1H and
I). Compared with those in HCT116 p53+/+ cells,
either mRNA (Fig. 1F) or protein
(Fig. 1G) expression levels of
CSE1L were markedly higher in HCT116 p53−/− cells. This
differential expression was also observed in HCT116
p53+/+ cells with p53 expression knocked down. After p53
was significantly knocked down in HCT116 p53+/+ cells
compared with that in shLuc-transfected cells (Fig. 1H), the expression level of CSE1L
mRNA was also significantly increased (Fig. 1I). A similar finding could also be
made on p53 protein expression in HCT116 p53+/+ cells
after 5-FU (40 µM) treatment for 24 h, which was increased
(Fig. 1J). In addition, the
expression of CSE1L was markedly reduced in the 5-FU-treated HCT116
p53+/+ cells compared with that in their untreated
counterparts (Fig. 1J).
Cell cycle regulation of p53-null CRC
cells by CSE1L expression
To understand the molecular significance of CSE1L
expression in CRC cells, CSE1L expression was knocked down in
HCT116 p53−/− cells, which was achieved to significant
levels compared with that in the shLuc-HCT116 p53−/−
cells (Fig. 2A). In addition,
shCSE1L-HCT116 p53−/− cells expressed markedly lower
expression levels of CSE1L protein (Fig. 2B). Compared with those in
shLuc-HCT116 p53−/− cells, the cell populations in
various phases of cell the cycle were altered in the shCSE1L-HCT116
p53−/− cells (Fig.
2C). Specifically, the percentage of shCSE1L-HCT116
p53−/− cells in S phase was significantly decreased,
whereas that in the G1 and G2/M phases was
significantly increased, compared with those of shLuc-HCT116
p53−/− cells (Fig. 2C and
D). Supporting this, shCSE1L-HCT116 p53−/− cells
also expressed markedly lower protein levels of cell cycle
regulators CCNA2, CCNB2 and CCND1 compared with those in
shLuc-HCT116 p53−/− cells (Fig. 2E).
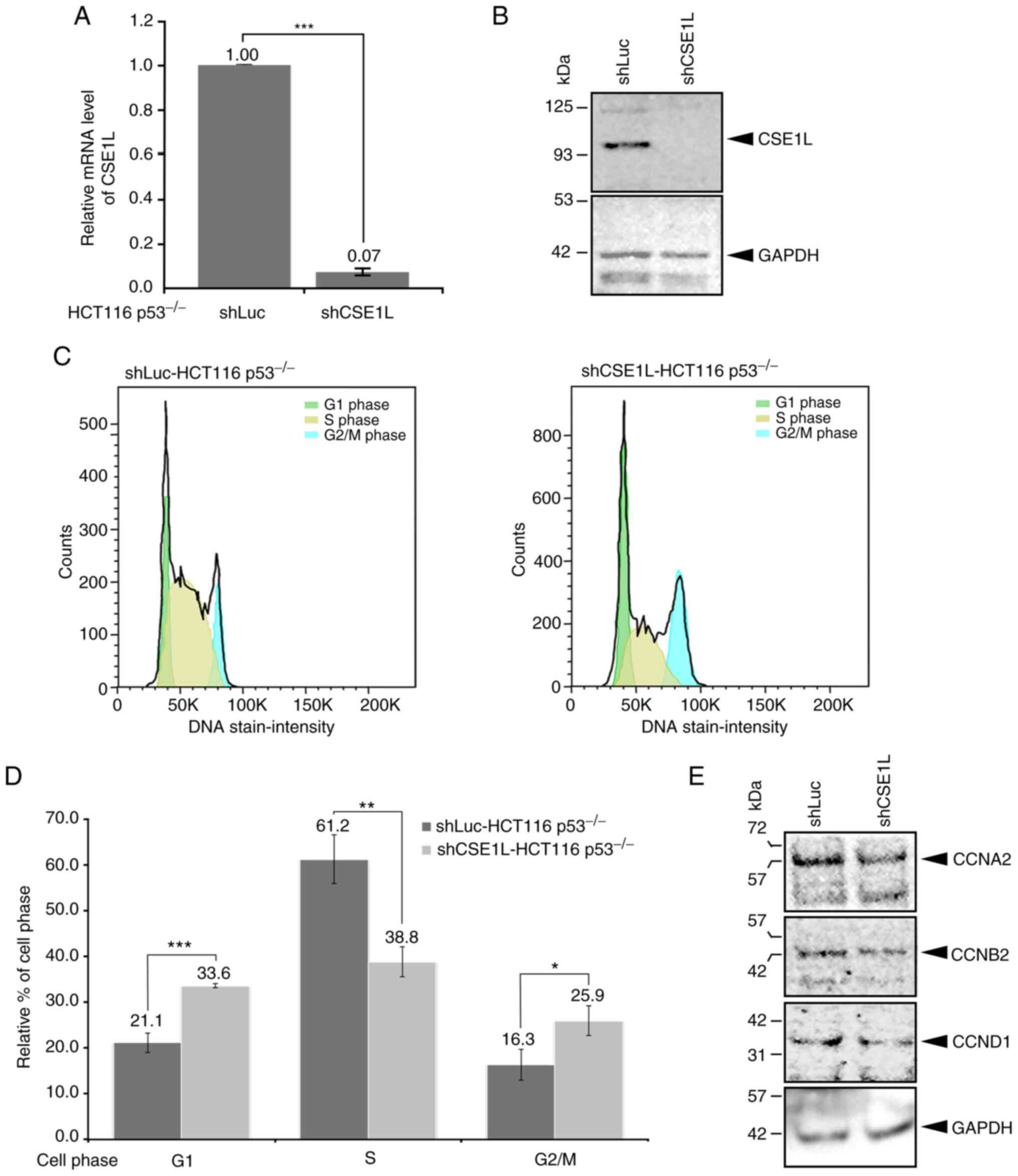 | Figure 2Cellular changes in HCT116
p53−/− cells after knocking down CSE1L expression. (A)
Knockdown efficacy of CSE1L in HCT116 p53−/− cells. (B)
Protein expression levels of CSE1L in HCT116 p53−/−
cells without or with CSE1L knockdown. (C) Population of HCT116
p53−/− cells in the various phases of cell cycle without
or with CSE1L knockdown. (D) Percentages of shCSE1L-HCT116
p53−/− cells in the various phases of cell cycle without
or with knockdown of CSE1L expression were quantified. (E) Protein
expression levels of CCNA2, CCNB2 and CCND1 in HCT116
p53−/− cells without or with CSE1L knockdown.
*P<0.05, **P<0.01 and ***P<0.001.
HCT116 p53−/−, p53-null HCT116 cells. shLuc, lentiviral
construct targeting luciferase; shCSE1L, lentiviral construct
targeting CSE1L; sh, short hairpin; CSE1L, chromosome segregation
1-like protein; CCNA2, cyclin A2; CCNB2, cyclin B2; CCND1, cyclin
D1. |
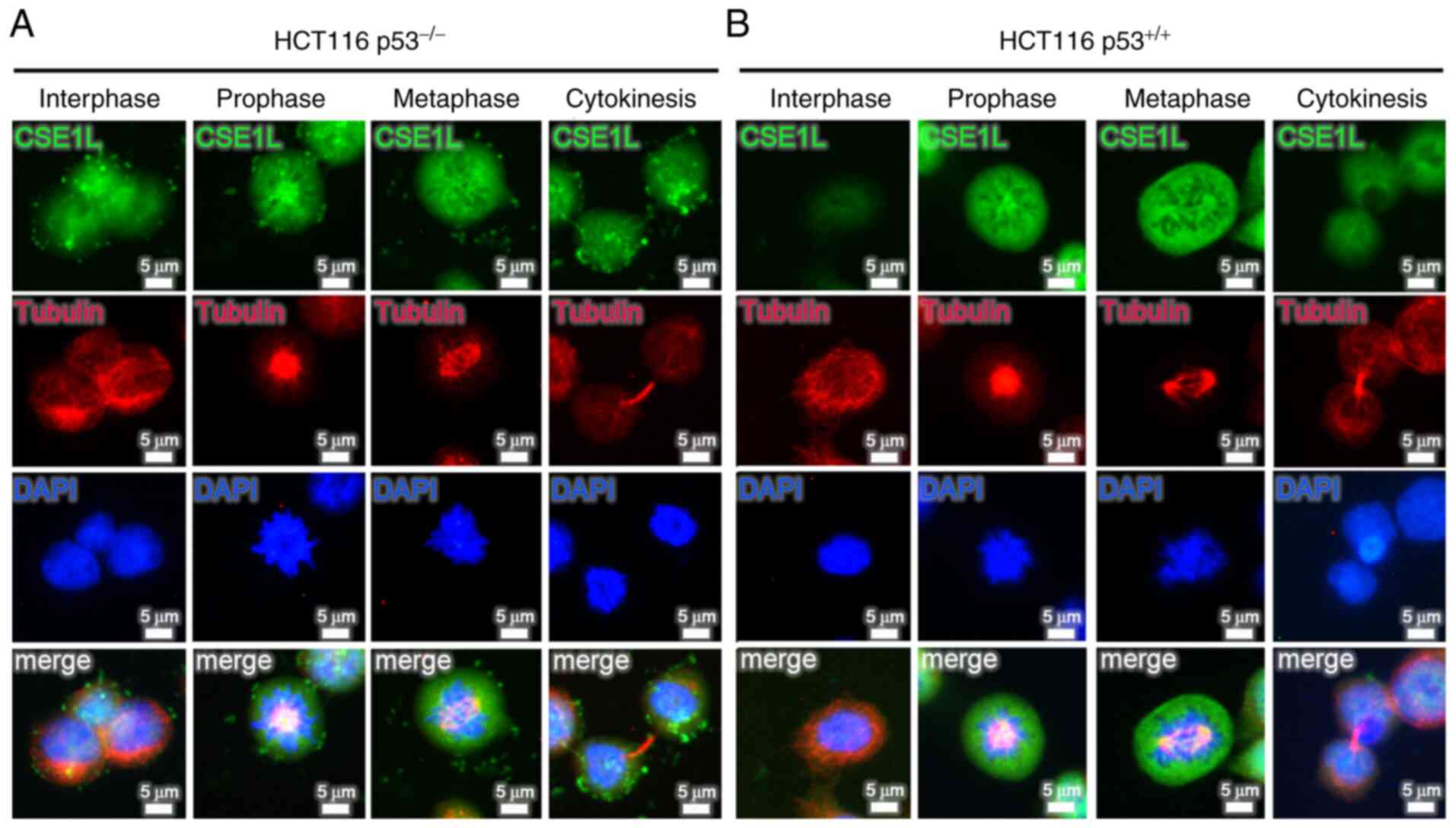
Dynamic expression of CSE1L in HCT116 CRC
cells during mitosis
Analysis of CSE1L expression during different phases
of mitosis in HCT116 cells revealed that expression profile of
CSE1L changed dynamically throughout mitosis (Fig. 3). Both HCT116 p53−/−
cells and HCT116 p53+/+ cells expressed high levels of
CSE1L at prophase and metaphase. However, the signals for CSE1L in
HCT116 p53−/− cells were stronger compared with those in
HCT116 p53+/+ cells during interphase and cytokinesis.
As shown in Fig. 3A for HCT116
p53−/− cells, CSE1L emerged at interphase, increased at
prophase, peaked during metaphase before declining at the
cytokinesis stage. The dynamic expression profiles of CSE1L in
HCT116 p53−/− cells during mitosis was subsequently
analyzed, with the highest levels of CSE1L expression found at
prophase and metaphase (Fig.
S1). By contrast, as shown in Fig. 3B, low levels of CSE1L expression
were detected during interphase and cytokinesis in HCT116
p53+/+ cells, which increased markedly at prophase
before peaking at metaphase.
Reduced CSE1L expression in
butyrate-treated HCT116 p53−/− cells and colon tumors in
mice treated with B. pullicaecorum administration
HCT116 p53−/− cells treated with 5 mM NaB
for 24 h exhibited lower expression levels of of both CSE1L mRNA
(Fig. 4A) and protein in the
total cell lysate (Fig. 4B)
compared with those in cells not treated with NaB. In addition, NaB
treatment reduced the mRNA expression of CSE1L in both the SW480
and SW620 cell lines (with the p53 mutant) after 24 and 48 h
(Fig. S2). In the cytosolic and
nuclear compartments of HCT116 p53−/− cells, the
expression levels of CSE1L also decreased as a result of 5 mM NaB
treatment (Fig. 4B). Furthermore,
the recovery ratio in the migration of shCSE1L-HCT116
p53−/− or NaB-treated-shLuc-HCT116 p53−/−
cells was significantly decreased compared with that in the control
shLuc-HCT116 p53−/− cells (Fig. 4C and D).
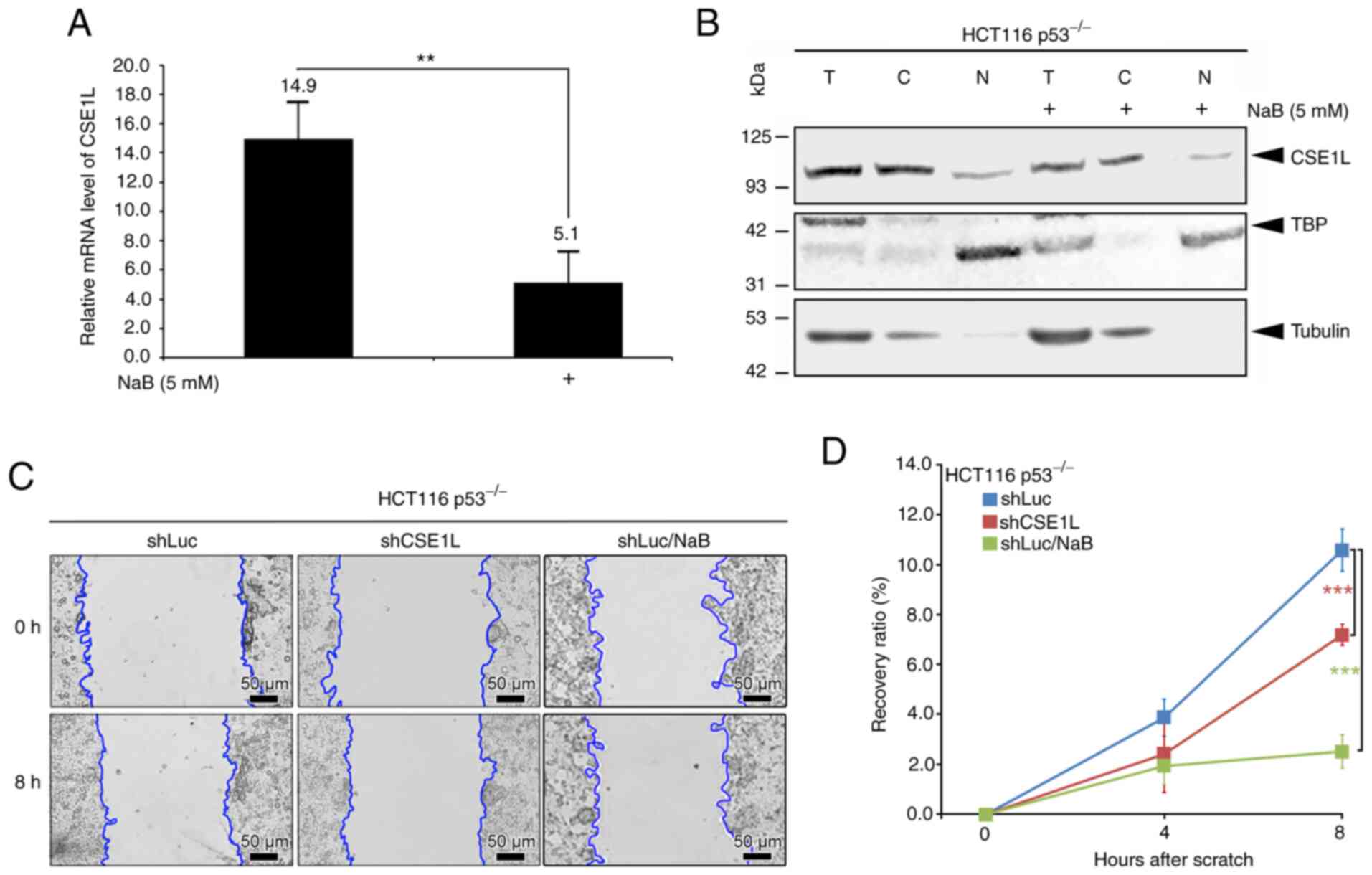 | Figure 4Effects of butyrate on CSE1L
expression in HCT116 p53−/− cells. (A) Relative mRNA
expression levels of CSE1L in cells without or with butyrate
treatment. (B) Protein expression levels of CSE1L in the nucleus or
cytosol of cells following butyrate treatment. (C) Migration
changes and (D) Recovery ratios of cells without or with CSE1L
knockdown or butyrate treatment. Blue line presents the edge of
cell migration. Scale bars, 50 µm. **P<0.01
and ***P<0.001. CSE1L, chromosome segregation 1-like
protein; TBP, TATA-binding protein; NaB, sodium butyrate; T, total
cell lysate; C, cytosolic part; N, nuclear part; shLuc, lentiviral
construct targeting luciferase; shCSE1L, lentiviral construct
targeting CSE1L; sh, short hairpin. |
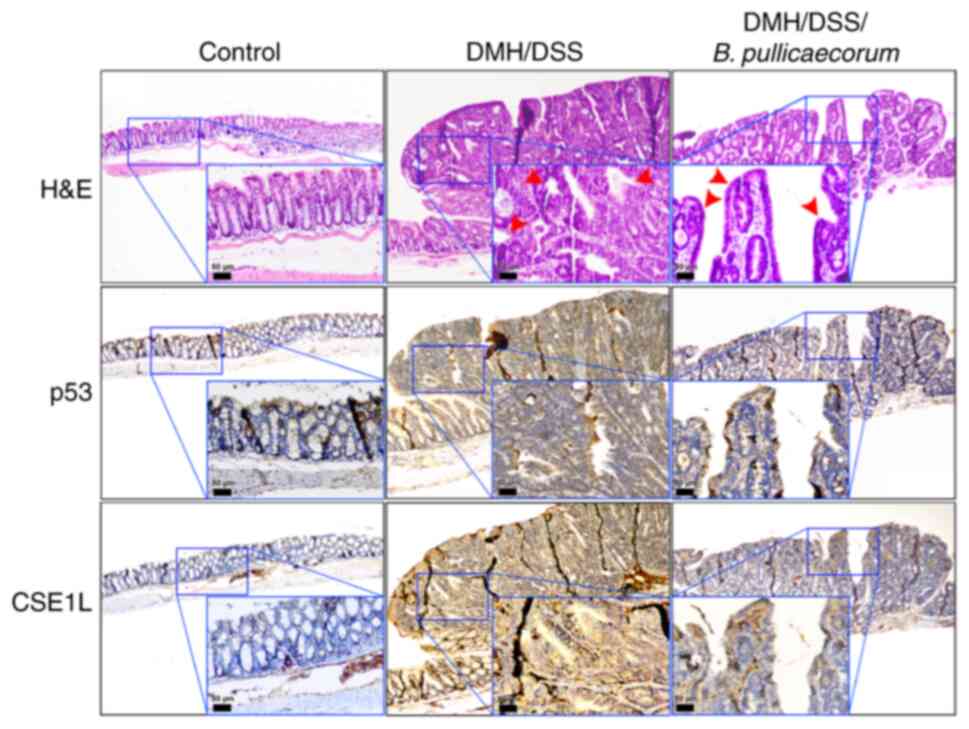
CSE1L expression in colon tumors of mice
with B. pullicaecorum administration
H&E staining and immunohistochemical analysis of
p53 and CSE1L expression was performed in the mouse intestinal
tissues. Reactivity was almost absent in control healthy intestinal
tissues (Fig. 5). Compared with
those in the control mice, colon tumors were induced in mice by
DMH/DSS treatment (Fig. 5). In
the colons of mice following DMH/DSS treatment without B.
pullicaecorum administration, histological sections showed
exophytic tumors exhibiting irregular and complex dysplastic
glands, indicating intramucosal adenocarcinoma (red arrows in the
middle panel of Fig. 5). Weak
nuclear staining of p53 and increased expression of CSE1L were
observed in large tumors with high-grade dysplasia (Fig. 5). By contrast, in mouse tissues
treated with B. pullicaecorum, the histological sections
revealed polypoid lesions consisting mostly of low-grade adenomas,
representing the early stage of neoplasia (red arrows in the right
panel of Fig. 5).
Immunohistochemical analysis showed positive nuclear staining for
p53 and a low intensity CSE1L signal were detected in precancerous
tumors with low-grade dysplasia from DMH/DSS-treated mice that were
administered with butyrate-producing B. pullicaecorum
through oral gavage.
Discussion
CSE1L overexpression was previously found to
associate with the progression of a number of gastrointestinal
cancers, including esophageal cancer, gastric cancer,
hepatocellular carcinoma and CRC (1,2,44,45). Furthermore, CSE1L can promotes the
nuclear distribution of the transcriptional coactivator with
PDZ-binding motif to enhance the malignancy of human cancer tissues
from osteosarcoma, glioma and lung cancer (46). Therefore, understanding the
molecular mechanism underlying the effects of CSE1L may facilitate
the optimization of cancer therapy (47).
CSE1L and p53 serve antagonistic effects on cell
cycle regulation (6,48). In CRC, whilst ~50% all samples
harbor p53 mutations that have been shown to be associated with
poor prognosis and chemoresistance (49,50), others have reported that the
majority of CRC tissues are positive for CSE1L expression (4,8,9).
In the present study, in the colon cell lines tested, which were
either cells from the normal colon or from cancer tissues, they
were found to express varing levels of CSE1L. The colon cell lines
haboring mutant p53 proteins (FHC and Caco-2) exhibited relatively
high CSE1L expression levels. In addition, p53-null HCT116 cells or
HCT116 cells with p53 expression knocked down were found to express
higher levels of CSE1L. Conversely, an overexpression of wild-type
p53 in CRC cells by 5-FU treatment reduced CSE1L expression. These
results provide evidence that changing the functionality of p53 in
CRC cells can alter the expression of CSE1L.
Overexpression of CSE1L in CRC has been associated
with tumor development and malignancy (8,51,52), such that CSE1L knockdown can
inhibit the growth and metastasis of CRC tumors (2,53).
Pimiento et al (8)
previously reported that CSE1L knockdown may represent a potential
target for CRC treatment. This finding is consistent with that in
the present study. Differentiation of Caco-2 cells into a polarized
enterocyte-like monolayer was shown reduce the extent of malignancy
(39). Decreasing CSE1L
expression levels were accompanied by reduced c-Myc expression as
the confluency of Caco-2 cells increased. In addition, CSE1L
knockdown in HCT116 p53−/− cells, specifically
shCSE1L-HCT116 p53−/− cells in the present study, was
found to arrest cell cycle progression at the G1 phase
whilst inhibiting DNA replication at S phase. These results would
agree with immunofluorescent images of cells under different
mitotic phases, which indicated that the CSE1L-expressing HCT116
p53−/− cells would potentiate the expression of CSE1L at
prophase and metaphase. A lack of CSE1L upregulation in the HCT116
p53−/− cells, such as shCSE1L-HCT116 p53−/−
cells or butyrate-treated HCT116 p53−/− cells, thereby
impeded CRC cell cycle progression or migration. Depletion of
cyclins caused by CSE1L knockdown also suggested that the cell
cycle was arrested at the G1 phase. As previously
reported by Ye et al (49)
in breast cancer cells, this form of cell cycle arrest may not be
only caused by reduced cyclin expression but also by the
upregulated expression of the cytochrome P450 family of proteins
(54). It will be necessary to
examine the expression of the cytochrome P450 superfamily of
proteins in the different CRC cell lines following the manipulation
of CSE1L expression to clarify the significance of this
relationship. Taken together, results from the present study imply
that CSE1L knockdown can impede CRC progression. Since CSE1L has
been reported to be a potential target for CRC treatment (8,55),
methods that can decrease the expression of CSE1L in CRC may serve
clinical potential.
Butyrates can regulate intestinal barrier function
and has potential clinical application for human gastrointestinal
diseases (10,56,57). In addition, it has been applied in
combination with other chemotherapeutic agents, such as irinotecan,
for CRC treatment (23). In the
present study, the expression of CSE1L was reduced after the
treatment of CRC cells with butyrate in vitro) or after the
administration of the butyrate-producing B. pullicaecorum to
CRC-bearing mice in vivo). Butyrate also reduced the CSE1L
expression levels in CRC cells carrying p53 mutations, such as
SW480 cells and SW620 cells. Therefore, butyrate may also display
anticancer properties by downregulating the expression of CSE1L, in
addition to butyrate also exhibiting synergistic anticancer effects
with p53 (58,59). In combination with the present
results, CSE1L knockdown may mitigate CRC malignancy and that
butyrate may reduce the expression of CSE1L further.
In the present study, the results demonstrated that
butyrate could reduce the expression of CSE1L in CRC cells in not
only the in vitro cell modesl, but also an in vivo
animal model. Administration of B. pullicaecorum was
previously shown to improve the clinical outcome of CRC and colitis
(17,26). Tumors with more intense nuclear
staining of p53 and weaker CSE1L staining were especially found in
mice bearing DMH/DSS-induced CRC that were administered with B.
pullicaecorum. Pathologically, these tumors were diagnosed to
be precancerous with low-grade dysplasia. However, the present
study may not have completely elucidated the precise mechanism by
which B. pullicaecorum regulates CSE1L expression or how the
differential CSE1L expression can arrest cell cycle progression in
CRC. In the future, a further in vivo study is required to
evaluate the prognosis of mice with CSE1L overexpression after
B. pullicaecorum administration.
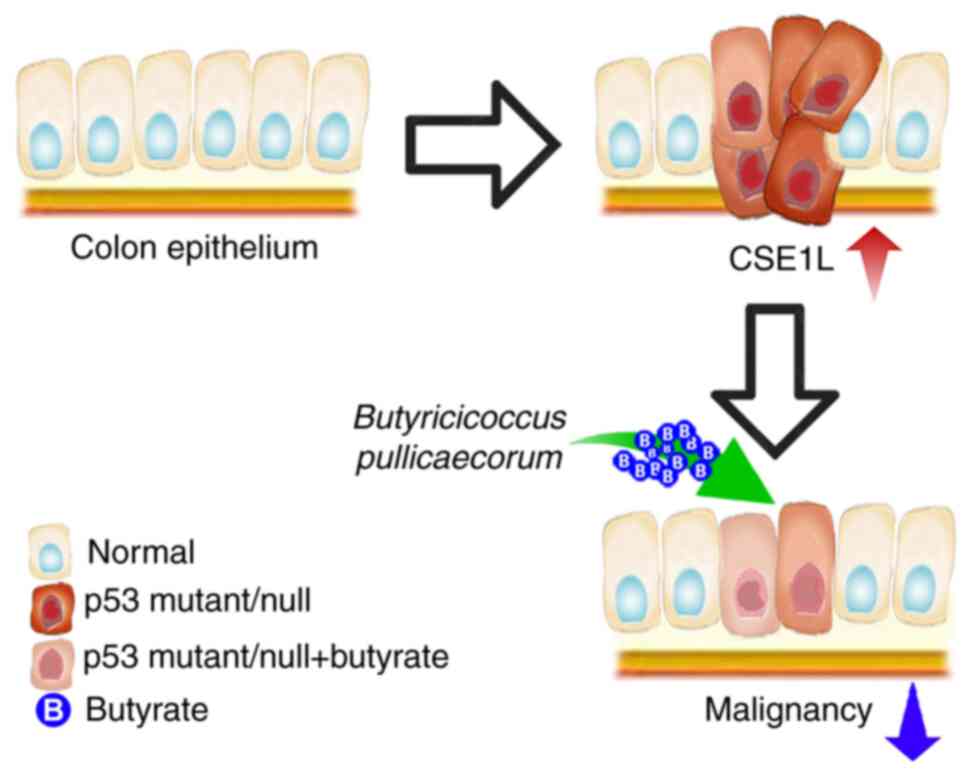
In conclusion, high CSE1L expression levels is
associated with the malignancy of CRC, where reduced CSE1L
expression in CRC cells may hinder proliferation or improve cancer
outcomes. As depicated in Fig. 6,
CSE1L represents a potential target for CRC treatment, such that
the reduction of CSE1L expression or activity can be achieved by
butyrate treatment or B. pullicaecorum administration. This
is because butyrate can repress CSE1L-induced tumorigenic
potential, whereby butyrate-producing microbes, such as B.
pullicaecorum, may reverse the genetic distortion caused by p53
mutations in CRC by regulating CSE1L expression. Therefore,
CSE1L-induced CRC growth may be impaired by butyrate
supplementation or B. pullicaecorum administration.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
CCC, RNY and CJH were involved in the study
conception and design. HHS, YAK, PYL, CYL, KWC and CJH performed
experiments and analysis of data. CCC, CJH, WYK, CYL and WCK were
involved in the study conception and design, performing the
experiments and analysis of data. RNY and CJH can confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The animal experiment in this study was performed
in accordance with the principles of replacement, refinement and
reduction and were approved (approval no. 107-008) by the
Institutional Animal Care and Use Committees of Cathay General
Hospital (Taipei, Taiwan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the Cathay
General Hospital and Taipei Medical University (grant nos.
106CGH-TMU-03 and 107CGH-TMU-02). The funders had no role in the
study design, data collection and analysis, decision to publish, or
preparation of the manuscript.
References
|
1
|
Jiang K, Neill K, Cowden D, Klapman J,
Eschrich S, Pimiento J, Malafa MP and Coppola D: Expression of
CAS/CSE1L, the cellular apoptosis susceptibility protein,
correlates with neoplastic progression in barrett's esophagus. Appl
Immunohistochem Mol Morphol. 26:552–556. 2018. View Article : Google Scholar
|
|
2
|
Li Y, Yuan S, Liu J, Wang Y, Zhang Y, Chen
X and Si W: CSE1L silence inhibits the growth and metastasis in
gastric cancer by repressing GPNMB via positively regulating
transcription factor MITF. J Cell Physiol. 235:2071–2079. 2020.
View Article : Google Scholar
|
|
3
|
Liao CF, Lin SH, Chen HC, Tai CJ, Chang
CC, Li LT, Yeh CM, Yeh KT, Chen YC, Hsu TH, et al: CSE1L, a novel
microvesicle membrane protein, mediates Rastriggered microvesicle
generation and metastasis of tumor cells. Mol Med. 18:1269–1280.
2012. View Article : Google Scholar
|
|
4
|
Tai CJ, Su TC, Jiang MC, Chen HC, Shen SC,
Lee WR, Liao CF, Chen YC, Lin SH, Li LT, et al: Correlations
between cytoplasmic CSE1L in neoplastic colorectal glands and depth
of tumor penetration and cancer stage. J Transl Med. 11:292013.
View Article : Google Scholar
|
|
5
|
Tunccan T, Duzer S, Dilek G, Yuksel UM,
Cetiner H, Kılıc C, Ant A and Duran AB: The role of CSE1L
expression in cervical lymph node metastasis of larynx tumors. Braz
J Otorhinolaryngol. 87:42–46. 2021. View Article : Google Scholar
|
|
6
|
Tanaka T, Ohkubo S, Tatsuno I and Prives
C: hCAS/CSE1L associates with chromatin and regulates expression of
select p53 target genes. Cell. 130:638–650. 2007. View Article : Google Scholar
|
|
7
|
Liao CF, Luo SF, Shen TY, Lin CH, Chien
JT, Du SY and Jiang MC: CSE1L/CAS, a microtubule-associated
protein, inhibits taxol (paclitaxel)-induced apoptosis but enhances
cancer cell apoptosis induced by various chemotherapeutic drugs.
BMB Rep. 41:210–216. 2008. View Article : Google Scholar
|
|
8
|
Pimiento JM, Neill KG, Henderson-Jackson
E, Eschrich SA, Chen DT, Husain K, Shibata D, Coppola D and Malafa
M: Knockdown of CSE1L gene in colorectal cancer reduces
tumorigenesis in vitro. Am J Pathol. 186:2761–2768. 2016.
View Article : Google Scholar
|
|
9
|
Tai CJ, Hsu CH, Shen SC, Lee WR and Jiang
MC: Cellular apoptosis susceptibility (CSE1L/CAS) protein in cancer
metastasis and chemotherapeutic drug-induced apoptosis. J Exp Clin
Cancer Res. 29:1102010. View Article : Google Scholar
|
|
10
|
Huang CC, Shen MH, Chen SK, Yang SH, Liu
CY, Guo JW, Chang KW and Huang CH: Gut butyrate-producing organisms
correlate to placenta specific 8 protein: Importance to colorectal
cancer progression. J Adv Res. 22:7–20. 2020. View Article : Google Scholar
|
|
11
|
Wang Y, Huang D, Chen KY, Cui M, Wang W,
Huang X, Awadellah A, Li Q, Friedman A, Xin WW, et al: Fucosylation
deficiency in mice leads to colitis and adenocarcinoma.
Gastroenterology. 152:193–205. 2017. View Article : Google Scholar
|
|
12
|
Venegas DP, De la Fuente MK, Landskron G,
González MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN and Hermoso
MA: Short chain fatty acids (scfas)-mediated gut epithelial and
immune regulation and its relevance for inflammatory bowel
diseases. Front Immunol. 10:2772019. View Article : Google Scholar
|
|
13
|
Puertollano E, Kolida S and Yaqoob P:
Biological significance of short-chain fatty acid metabolism by the
intestinal microbiome. Curr Opin Clin Nutr Metab Care. 17:139–144.
2014. View Article : Google Scholar
|
|
14
|
Gill PA, van Zelm MC, Muir JG and Gibson
PR: Review article: Short chain fatty acids as potential
therapeutic agents in human gastrointestinal and inflammatory
disorders. Aliment Pharmacol Ther. 48:15–34. 2018. View Article : Google Scholar
|
|
15
|
Kannen V, Parry L and Martin FL: Phages
enter the fight against colorectal cancer. Trends Cancer.
5:577–579. 2019. View Article : Google Scholar
|
|
16
|
Boesmans L, Valles-Colomer M, Wang J,
Eeckhaut V, Falony G, Ducatelle R, Van Immerseel F, Raes J and
Verbeke K: Butyrate producers as potential next-generation
probiotics: Safety assessment of the administration of
butyricicoccus pullicaecorum to healthy volunteers. mSystems.
3:e00094–e00018. 2018. View Article : Google Scholar
|
|
17
|
Chang SC, Shen MH, Liu CY, Pu CM, Hu JM
and Huang CJ: A gut butyrate-producing bacterium Butyricicoccus
pullicaecorum regulates short-chain fatty acid transporter and
receptor to reduce the progression of
1,2-dimethylhydrazine-associated colorectal cancer. Oncol Lett.
20:3272020. View Article : Google Scholar
|
|
18
|
Eeckhaut V, Wang J, Van Parys A,
Haesebrouck F, Joossens M, Falony G, Raes J, Ducatelle R and Van
Immerseel F: The probiotic butyricicoccus pullicaecorum reduces
feed conversion and protects from potentially harmful intestinal
microorganisms and necrotic enteritis in broilers. Front Microbiol.
7:14162016. View Article : Google Scholar
|
|
19
|
Wu X, Wu Y, He L, Wu L, Wang X and Liu Z:
Effects of the intestinal microbial metabolite butyrate on the
development of colorectal cancer. J Cancer. 9:2510–2517. 2018.
View Article : Google Scholar
|
|
20
|
Wang YC, Ku WC, Liu CY, Cheng YC, Chien
CC, Chang KW and Huang CJ: Supplementation of probiotic
butyricicoccus pullicaecorum mediates anticancer effect on bladder
urothelial cells by regulating butyrate-responsive molecular
signatures. Diagnostics (Basel). 11:22702021. View Article : Google Scholar
|
|
21
|
Kazemi Sefat NA, Mohammadi MM, Hadjati J,
Talebi S, Ajami M and Daneshvar H: Sodium butyrate as a histone
deacetylase inhibitor affects toll-like receptor 4 expression in
colorectal cancer cell lines. Immunol Invest. 48:759–769. 2019.
View Article : Google Scholar
|
|
22
|
Ji X, Zhou F, Zhang Y, Deng R, Xu W, Bai
M, Liu Y, Shao L, Wang X and Zhou L: Butyrate stimulates hepatic
gluconeogenesis in mouse primary hepatocytes. Exp Ther Med.
17:1677–1687. 2019.
|
|
23
|
Encarnacao JC, Pires AS, Amaral RA,
Gonçalves TJ, Laranjo M, Casalta-Lopes JE, Gonçalves AC,
Sarmento-Ribeiro AB, Abrantes AM and Botelho MF: Butyrate, a
dietary fiber derivative that improves irinotecan effect in colon
cancer cells. J Nutr Biochem. 56:183–192. 2018. View Article : Google Scholar
|
|
24
|
Nakano K, Mizuno T, Sowa Y, Orita T,
Yoshino T, Okuyama Y, Fujita T, Fujita NO, Matsukawa Y, Tokino T,
et al: Butyrate activates the WAF1/Cip1 gene promoter through Sp1
sites in a p53-negative human colon cancer cell line. J Biol Chem.
272:22199–22206. 1997. View Article : Google Scholar
|
|
25
|
Russo I, Luciani A, De Cicco P, Troncone E
and Ciacci C: Butyrate attenuates lipopolysaccharide-induced
inflammation in intestinal cells and crohn's mucosa through
modulation of antioxidant defense machinery. PLoS One.
7:e328412012. View Article : Google Scholar
|
|
26
|
Eeckhaut V, Machiels K, Perrier C, Romero
C, Maes S, Flahou B, Steppe M, Haesebrouck F, Sas B, Ducatelle R,
et al: Butyricicoccus pullicaecorum in inflammatory bowel disease.
Gut. 62:1745–1752. 2013. View Article : Google Scholar
|
|
27
|
Marteau P: Butyrate-producing bacteria as
pharmabiotics for inflammatory bowel disease. Gut. 62:16732013.
View Article : Google Scholar
|
|
28
|
Eeckhaut V, Ducatelle R, Sas B, Vermeire S
and Van Immerseel F: Progress towards butyrate-producing
pharmabiotics: Butyricicoccus pullicaecorum capsule and efficacy in
TNBS models in comparison with therapeutics. Gut. 63:3672014.
View Article : Google Scholar
|
|
29
|
Kilkenny C, Browne W, Cuthill IC, Emerson
M and Altman DG; NC3Rs Reporting Guidelines Working Group: Animal
research: Reporting in vivo experiments: The ARRIVE guidelines. J
Gene Med. 12:561–563. 2010. View Article : Google Scholar
|
|
30
|
Soucek K, Gajduskova P, Brazdova M,
Hýzd'alová M, Kocí L, Vydra D, Trojanec R, Pernicová Z, Lentvorská
L, Hajdúch M, et al: Fetal colon cell line FHC exhibits tumorigenic
phenotype, complex karyotype, and TP53 gene mutation. Cancer Genet
Cytogenet. 197:107–116. 2010. View Article : Google Scholar
|
|
31
|
Hayashi Y, Tsujii M, Kodama T, Akasaka T,
Kondo J, Hikita H, Inoue T, Tsujii Y, Maekawa A, Yoshii S, et al:
p53 functional deficiency in human colon cancer cells promotes
fibroblast-mediated angiogenesis and tumor growth. Carcinogenesis.
37:972–984. 2016. View Article : Google Scholar
|
|
32
|
Rochette PJ, Bastien N, Lavoie J, Guerin
SL and Drouin R: SW480, a p53 double-mutant cell line retains
proficiency for some p53 functions. J Mol Biol. 352:44–57. 2005.
View Article : Google Scholar
|
|
33
|
Huang CJ, Yang SH, Huang SM, Lin CM, Chien
CC, Chen YC, Lee CL, Wu HH and Chang CC: A predicted protein,
KIAA0247, is a cell cycle modulator in colorectal cancer cells
under 5-FU treatment. J Transl Med. 9:822011. View Article : Google Scholar
|
|
34
|
Ishimine M, Lee HC, Nakaoka H, Orita H,
Kobayashi T, Mizuguchi K, Endo M, Inoue I, Sato K and Yokomizo T:
The Relationship between TP53 Gene status and carboxylesterase 2
expression in human colorectal cancer. Dis Markers.
2018:52807362018. View Article : Google Scholar
|
|
35
|
Abu El Maaty MA, Strassburger W, Qaiser T,
Dabiri Y and Wölfl S: Differences in p53 status significantly
influence the cellular response and cell survival to
1,25-dihydroxyvitamin D3-metformin cotreatment in colorectal cancer
cells. Mol Carcinog. 56:2486–2498. 2017. View Article : Google Scholar
|
|
36
|
Li DD, Sun T, Wu XQ, Chen SP, Deng R,
Jiang S, Feng GK, Pan JX, Zhang XC, Zeng YX and Zhu XF: The
inhibition of autophagy sensitises colon cancer cells with
wild-type p53 but not mutant p53 to topotecan treatment. PLoS One.
7:e450582012. View Article : Google Scholar
|
|
37
|
Bhat UG and Gartel AL: Differential
sensitivity of human colon cancer cell lines to the nucleoside
analogs ARC and DRB. Int J Cancer. 122:1426–1429. 2008. View Article : Google Scholar
|
|
38
|
Kralj M, Husnjak K, Körbler T and Pavelić
J: Endogenous p21WAF1/CIP1 status predicts the response of human
tumor cells to wild-type p53 and p21WAF1/CIP1 overexpression.
Cancer Gene Ther. 10:457–467. 2003. View Article : Google Scholar
|
|
39
|
Huang CJ, Lee CL, Yang SH, Chien CC, Huang
CC, Yang RN and Chang CC: Upregulation of the growth
arrest-specific-2 in recurrent colorectal cancers, and its
susceptibility to chemo-therapy in a model cell system. Biochim
Biophys Acta. 1862:1345–1353. 2016. View Article : Google Scholar
|
|
40
|
Leoni BD, Natoli M, Nardella M, Bucci B,
Zucco F, D'Agnano L and Felsani A: Differentiation of Caco-2 cells
requires both transcriptional and post-translational
down-regulation of Myc. Differentiation. 83:116–127. 2012.
View Article : Google Scholar
|
|
41
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
42
|
Jonkman JE, Cathcart JA, Xu F, Bartolini
ME, Amon JE, Stevens KM and Colarusso P: An introduction to the
wound healing assay using live-cell microscopy. Cell Adh Migr.
8:440–451. 2014. View Article : Google Scholar
|
|
43
|
Ye S, Zhou HB, Chen Y, Li KQ, Jiang SS and
Hao K: Crizotinib changes the metabolic pattern and inhibits ATP
production in A549 non-small cell lung cancer cells. Oncol Lett.
21:612021. View Article : Google Scholar
|
|
44
|
Winkler J, Roessler S, Sticht C, DiGuilio
AL, Drucker E, Holzer K, Eiteneuer E, Herpel E, Breuhahn K, Gretz
N, et al: Cellular apoptosis susceptibility (CAS) is linked to
integrin beta1 and required for tumor cell migration and invasion
in hepatocellular carcinoma (HCC). Oncotarget. 7:22883–22892. 2016.
View Article : Google Scholar
|
|
45
|
Zhu JH, Hong DF, Song YM, Sun LF, Wang ZF
and Wang JW: Suppression of cellular apoptosis susceptibility
(CSE1L) inhibits proliferation and induces apoptosis in colorectal
cancer cells. Asian Pac J Cancer Prev. 14:1017–1021. 2013.
View Article : Google Scholar
|
|
46
|
Nagashima S, Maruyama J, Honda K, Kondoh
Y, Osada H, Nawa M, Nakahama KI, Yuasa MI, Kagechika H, Sugimura H,
et al: CSE1L promotes nuclear accumulation of transcriptional
coactivator TAZ and enhances invasiveness of human cancer cells. J
Biol Chem. 297:1008032021. View Article : Google Scholar
|
|
47
|
Snijders AM and Mao JH: Multi-omics
approach to infer cancer therapeutic targets on chromosome 20q
across tumor types. Adv Mod Oncol Res. 2:215–223. 2016. View Article : Google Scholar
|
|
48
|
Behrens P, Brinkmann U and Wellmann A:
CSE1L/CAS: Its role in proliferation and apoptosis. Apoptosis.
8:39–44. 2003. View Article : Google Scholar
|
|
49
|
Li H, Zhang J, Tong JHM, Chan AWH, Yu J,
Kang W and To KF: Targeting the oncogenic p53 mutants in colorectal
cancer and other solid tumors. Int J Mol Sci. 20:59992019.
View Article : Google Scholar
|
|
50
|
Iacopetta B: TP53 mutation in colorectal
cancer. Hum Mutat. 21:271–276. 2003. View Article : Google Scholar
|
|
51
|
Wang X, Ren Y, Ma S and Wang S: Circular
RNA 0060745, a novel circRNA, promotes colorectal cancer cell
proliferation and metastasis through miR-4736 sponging. Onco
Targets Ther. 13:1941–1951. 2020. View Article : Google Scholar
|
|
52
|
Ma S, Yang D, Liu Y, Wang Y, Lin T, Li Y,
Yang S, Zhang W and Zhang R: LncRNA BANCR promotes tumorigenesis
and enhances adriamycin resistance in colorectal cancer. Aging
(Albany NY). 10:2062–2078. 2018. View Article : Google Scholar
|
|
53
|
Cheng DD, Lin HC, Li SJ, Yao M, Yang QC
and Fan CY: CSE1L interaction with MSH6 promotes osteosarcoma
progression and predicts poor patient survival. Sci Rep.
7:462382017. View Article : Google Scholar
|
|
54
|
Ye M, Han R, Shi J, Wang X, Zhao AZ, Li F
and Chen H: Cellular apoptosis susceptibility protein (CAS)
suppresses the proliferation of breast cancer cells by upregulated
cyp24a1. Med Oncol. 37:432020. View Article : Google Scholar
|
|
55
|
Li KK, Yang L, Pang JC, Chan AKY, Zhou L,
Mao Y, Wang Y, Lau KM, Poon WS, Shi Z and Ng HK: MIR-137 suppresses
growth and invasion, is downregulated in oligodendroglial tumors
and targets CSE1L. Brain Pathol. 23:426–439. 2013. View Article : Google Scholar
|
|
56
|
Beaumont M, Paes C, Mussard E, Knudsen C,
Cauquil L, Aymard P, Barilly C, Gabinaud B, Zemb O, Fourre S, et
al: Gut microbiota derived metabolites contribute to intestinal
barrier maturation at the suckling-to-weaning transition. Gut
Microbes. 11:1–19. 2020. View Article : Google Scholar
|
|
57
|
Silva JPB, Navegantes-Lima KC, Oliveira
ALB, Rodrigues DVS, Gaspar SLF, Monteiro VVS, Moura DP and Monteiro
MC: Protective mechanisms of butyrate on inflammatory bowel
disease. Curr Pharm Des. 24:4154–4166. 2018. View Article : Google Scholar
|
|
58
|
Zhao Y, Shi L, Hu C and Sang S: Wheat bran
for colon cancer prevention: The synergy between phytochemical
alkylresorcinol C21 and intestinal microbial metabolite butyrate. J
Agric Food Chem. 67:12761–12769. 2019. View Article : Google Scholar
|
|
59
|
Pant K, Mishra AK, Pradhan SM, Nayak B,
Das P, Shalimar D, Saraya A and Venugopal SK: Butyrate inhibits HBV
replication and HBV-induced hepatoma cell proliferation via
modulating SIRT-1/Ac-p53 regulatory axis. Mol Carcinog. 58:524–532.
2019. View Article : Google Scholar
|