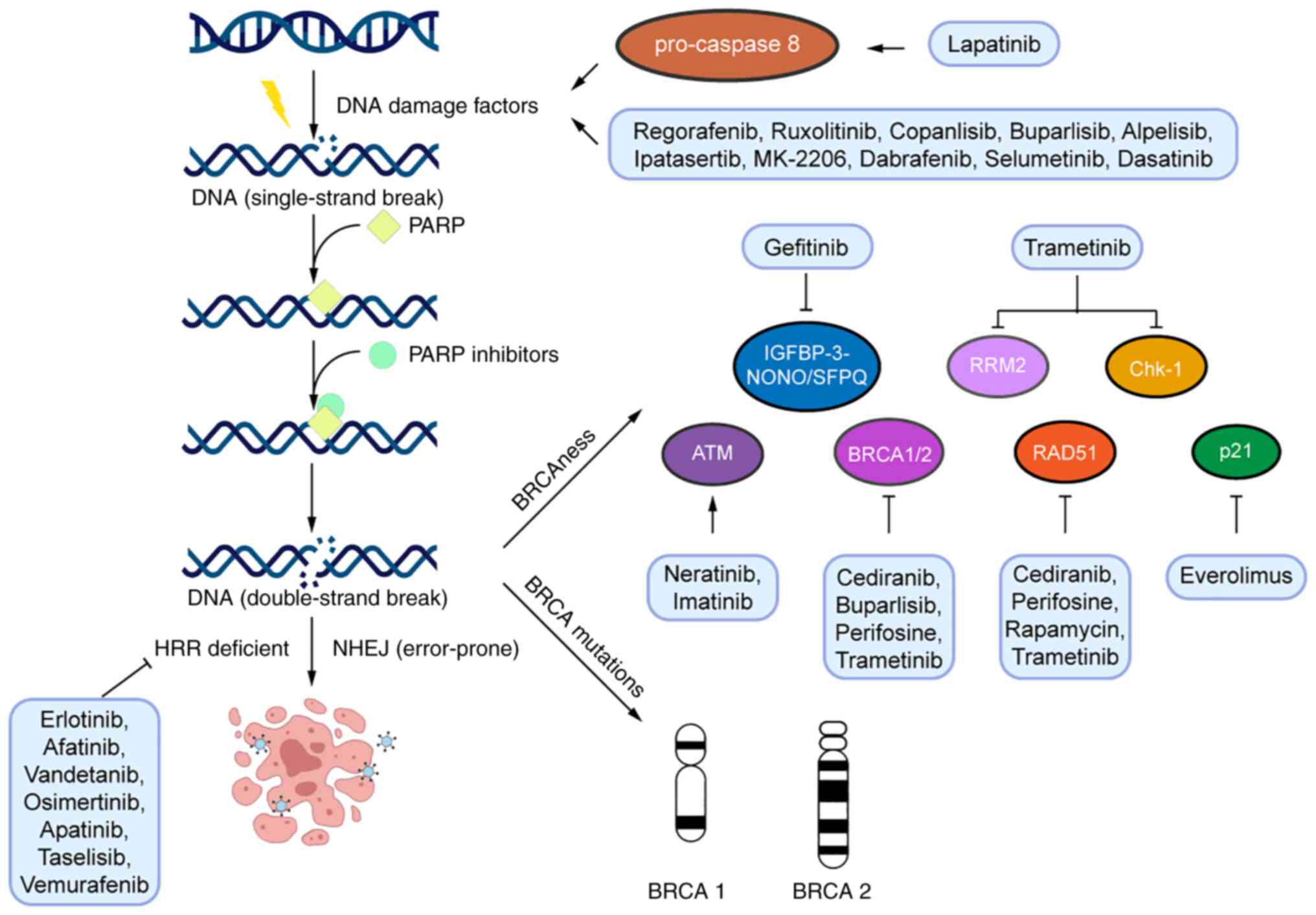
PARP inhibitors (PARPi) achieved a major
breakthrough as targeted antitumor agents in the past decades; they
are critical clinical drugs designed to cause cancer cell death by
targeting PARP (1). An
understanding of the roles of the PARP1 and PARP2 enzymes in the
DNA damage response (DDR) led to long-term efforts to develop
PARP1i/PARP2i (2-7). Cancer cells that suffer DNA damage
caused by reactive oxygen species (ROS), chemotherapy, etc., are
able to repair damage through single-strand break (SSB) repair. Due
to the tumor-specific genetic defects, PARPi may induce the killing
of cancer cells while sparing normal cells. The PARP family
comprises a subset of nuclear proteins that detect SSBs and
subsequently recruit DNA repair effectors, remodel chromatin and
eventually repair DNA by PARylation of PARP substrate proteins.
Based on the pivotal role of PARP in the DDR, PARPi were indicated
to trap PARP at the site of damage by binding to the ADP
ribosyltransferase catalytic domain, causing conversion to
double-strand breaks (DSBs) and impairing the progression of
replication forks. Two primary repair models, homologous
recombination (HR) and nonhomologous end-joining (NHEJ), are
involved in DSB responses in healthy and unmutated cells. However,
in abnormal cells with BRCA1/2 deficiency or HR deficiency (HRD),
the HR repair pathway may be inhibited and turn into error-prone
NHEJ repair, eventually leading to cytotoxic DSBs. In addition,
PARPi may suppress the classic NHEJ pathway (8,9). The
above machanisms eventually lead to cell death (10-18).
Based on this mechanism, four PARPi, olaparib, niraparib, rucaparib
and talazoparib, have been approved by the FDA to be applied in
human tumors with deleterious BRCA mutations or HRD in ovarian,
breast, pancreatic and prostate cancer.
Although great successes have been achieved in the
discovery and development of PARPi with satisfactory clinical
benefits, new issues regarding PARPi are emerging during clinical
practice. Drug resistance is the first problem that affects the
clinical response of patients receiving PARPi. The resistance to
PARPi generated in BRCA1/2-deficient tumor cells mainly arises from
five aspects: Somatic reversion or restoration of BRCA1/2 open
reading frame (19), epigenetic
reversion of BRCA1 promoter hypermethylation, hypomorphic BRAC1/2
allele (20), deficiency of PARP1
expression (21) and loss of end
resection regulation (22,23). In addition, only patients
harbouring BRCA mutations or HRD may benefit from PARPi therapy;
however, these patients account for only a small proportion of the
total cancer patient population. For instance, in ovarian cancer
(OC), which benefits from PARPi the most, less than half of the
patients with high-grade serous epithelial OC have alterations in
HR repair genes (17). PAPRi
resistance and the BRCA or HR status limit the clinical application
of PAPRi. In these circumstances, combined therapy is a feasible
strategy to improve the clinical benefit and expand the application
of PARPi. Small molecular inhibitors are, in certain aspects, ideal
candidates for combination treatment.
Small molecular inhibitors are agents with a
molecular weight of 500-900 Da that target biomolecules. To date,
various small molecular inhibitors have been approved by the food
and drug administration (FDA) as targeted therapies and applied in
multiple haematological cancers and solid tumors, such as epidermal
growth factor receptor inhibitors (EGFRi), Janus kinase
(JAK)-signal transducer and activator of transcription (STAT)
pathway inhibitors and phosphoinositide-3 kinase
(PI3K)-AKT-mammalian target of rapamycin complex (mTORC) pathway
inhibitors. As reported, the anti-tumor mechanisms of these
inhibitors include increasing DNA damage, suppressing cellular DNA
repair and affecting the expression of HRD factors (the major
mechanisms are presented in Table
I and Fig. 1). These
mechanisms offer the possibility that small molecular inhibitors
may have synergistic effects with PARPi and minimize PAPRi
resistance. As mentioned above, biomolecules regulate processes
including metabolism, transcription and transfer signalling within
cells, and serve as components of cells (24). In addition, small molecular
inhibitors have better oral bioavailability, higher tissue and
tumor microenvironment penetration and higher selective toxicity
profiles compared to therapeutic antibodies (25,26);
tremendous research efforts have been made in this field.
In the present review, the roles of small molecular
inhibitors in DNA damage were summarized, particularly those
targeting biomolecules functions in HR. Based on these molecular
mechanisms, the effects of the combination of PARPi and small
molecular inhibitors were further demonstrated in cancer treatment
studies and clinical trials. The present review aimed to promote
the application of PARPi for treating cancer. It remains to be
interpreted whether combined therapies may improve the prognosis of
patients with BRCA1/2 deficiency and whether patients with HR
repair defection may gain a clinical benefit compared to PARPi
monotherapy.
RTKs, which act as membrane receptor proteins and
subsequently activate intracellular signalling, have been reported
to participate in a variety of biological processes, such as
growth, motility, differentiation and metabolism (27,28).
The expression and activation of RTKs are of vital importance in
tumor diagnosis and treatment; hence, the determination of the RTK
concentration and phosphorylation degree is a focal spot in therapy
development, particularly for metastatic breast cancer,
gastrointestinal stromal tumor and non-small cell lung cancer
(NSCLC) (29,30). Emerging evidence implies that
disturbance of RTK signalling impacts DDR systems and subsequently
exhibits a synergism with DNA-damaging agents in cancer treatment
(31).
EGFR is a transmembrane protein with cytoplasmic
kinase activity that transduces growth signalling from the
extra-cellular environment into the cell. The EGFR family has been
verified to comprise four members: EGFR, human EGFR-related 2
(HER2; also known as neu/ERBB2), kinase-impaired HER3 and HER4
(32). These signalling pathways
are involved in tumor growth and angiogenesis, as well as the
activation of transcription factors related to cell growth and
mitosis (33-35). Evidence has indicated that small
molecular inhibitors, such as EGFRi, may act on healthy cells,
which would cause specific toxicity. However, several small
molecular inhibitors have been approved by the FDA as targeted
therapies and applied in multiple haematological cancers and solid
tumors, each of which has specific indications and individual
toxicity profiles. The astonishing antitumor effect and adverse
events underscore the importance of having a comprehensive
reference to help guide clinical decisions when treating patients.
With the desire for higher potency and overcoming drug resistance,
EGFRi are constantly being developed and have been proven to
suppress tumor growth in vitro and in vivo. However,
single-agent EGFRi therapy is prone to adverse events, modest
efficacy and drug resistance (36-38).
EGFR amplification was indicated to be associated with DNA repair
pathways and rendered glioma sphere-forming cells susceptible to
PARPi (39,40). Further studies revealed that the
intrinsic mechanism was Rad51 and Mre-11 upregulation mediated by
EGFR, supporting the synergistic effect of EGFRi and PARPi
(41,42). Certain EGFRi, such as gefitinib and
cediranib, have undergone clinical trials in combination with
PARPi.
Gefitinib, a classic agent of the first-generation
EGFRi, is a synthetic low-molecular-weight anilinquinazoline
compound that selectively targets HER1 or ErbB1 (43). In 2015, gefitinib was approved by
the FDA for the treatment of patients with metastatic EGFR
mutation-positive NSCLC.
In NSCLC, gefitinib treatment markedly reduced
phosphorylated (p)-EGFR, p-AKT and p-MAPK levels, and increased
cleaved PARP (44). In MDA-MB-468
and HCC1806 cells, pretreatment with gefitinib prevented the
synthesis of IGFBP-3-NONO/SFPQ complexes, which comprise a
multi-protein DNA repair complex and modulate DSB repair by NHEJ
(45). In addition, in patients
with EGFR-mutant NSCLC treated with gefitinib, the low mRNA levels
of BRCA1 resulted in a relatively longer progression-free survival
(PFS) and PARPi attenuated BRCA1 expression (46). In 2021, a case study reported that
a 62-year-old patient with lung carcinoma with a BRCA2 germline
mutation benefited from combination therapy (47). These studies on gefitinib and PARPi
provide a basis for the combination of drugs in clinical
trials.
A multicentre, randomized phase IB/IIB study, GOAL,
recruited pathologically confirmed patients with stage IV NSCLC in
Spain and Mexico. Eligible patients were randomly allocated (1:1)
to receive gefitinib 250 mg daily or gefitinib 250 mg daily plus
olaparib 200 mg three times daily in a 28-day cycle. However,
comparing the PFS, overall survival, response rate, safety and
tolerability, no significant differences were observed. The use of
next-generation sequencing is supposed to identify PARP and BRCA1
expression in the subgroup of EGFR-mutant patients who may benefit
from adjunctive therapy (46).
Lapatinib interacts with the ATP-binding site of
HER1 (EGFR1/ErbB1) and HER2/c-neu (ErbB2) and further inhibits
downstream signalling cascades (48). Lapatinib reduced EGFR protein
expression, increased the population of apoptotic cells and
increased TP53 gene signals (49).
To further investigate the association between lapatinib and
DNA-damaging agents, a study in MCF-7 and MDA-MB-468 cells revealed
that pretreatment with lapatinib or erlotinib promoted
pro-caspase-8 dimerization, which subsequently enhanced the
efficacy of DNA-damaging agents (50). Based on these results, a new
treatment was proposed, namely the combination of lapatinib and
PARPi, which was tested in vivo and in vitro. Under
both circumstances, an anti-tumor effect was verified in
triple-negative breast cancer (TNBC) and an increase in cytosolic
BRCA1 and EGFR was found to be distant from their nuclear DNA
repair substrates (51).
Erlotinib is also a selective inhibitor of tyrosine
kinase and functions in tumor cell division, cell cycle arrest and
apoptosis (52). Although it has
adverse effects, erlotinib has been approved by the FDA for the
treatment of NSCLC and pancreatic cancer. It has been observed that
in HER2 short hairpin RNA-transfected bladder cancer cells, cell
growth was inhibited and more DNA was damaged after treatment with
erlotinib (53). Similarly, in
CRL-5876, human lung adenocarcinoma cells underwent DNA DSBs
(54), and human breast cancer
cells exhibited suppression of HR repair of chromosomal breaks
(55). In an ovarian tumor
xenograft model, treatment combining erlotinib and AZD2281
(olaparib, a potent inhibitor of PARP) exhibited a better ability
to reduce tumor size compared to any monotherapy through
downregulation of p-ERK1/2 and p-AKT (56). Furthermore, the gain-of-function
mutation of EGFR was reported to induce PARPi resistance, thus
supporting the combined therapy of PARPi (veliparib) and EGFRi
(erlotinib) for lung cancer (57).
Afatinib binds irreversibly to cysteine 797 of EGFR
and cysteines 805 and 803 in HER2 and HER4. It also inhibits the
transphosphorylation of HER3 protein. Phosphorylation within the
ErbB dimer is blocked and downstream signalling pathways are
disrupted when cells are treated with afatinib, leading to cell
apoptosis both in vitro and in vivo. Afatinib was
revealed to significantly increase PFS in patients with advanced
NSCLC resistant to gefitinib or erlotinib (58,59).
In a previous study, when cotreatment with radiotherapy was
applied, afatinib possessed a better ability to kill cells, cause
apoptosis and damage DNA than erlotinib (53). In addition, in a
gefitinib-resistant cell subline of NSCLC (PC-9-GR), afatinib was
indicated to increase apoptosis and inhibit the DDR (60).
Vandetanib is approved by the FDA as a once-daily
oral multikinase inhibitor targeting the rear-ranged during
transfection (RET) tyrosine kinase, vascular endothelial growth
factor receptor (VEGFR) and EGFR for the treatment of progressive
medullary thyroid cancer, as well as NSCLC and breast cancer
(61). In the CAL-27 oral squamous
cell carcinoma cell line, vandetanib interfered with cellular DNA
repair to enhance the efficacy of photodynamic therapy (PDT)
(62). Apart from synergistic
effects with PDT, vandetanib enhanced the activity of DNA-damaging
agents as a result of G1 phase accumulation (63).
Neratinib is a pan-TKI targeting HER1, HER2 and HER4
that is primarily applied in HER2-positive breast cancer treatment
and was approved in the USA in 2017. As monotherapy or a component
of combination therapy, it is undergoing clinical trials in
metastatic breast cancer, advanced breast cancer, NSCLC, colorectal
cancer and glioblastoma (64). In
a 115-cancer cell line panel, BRCA2 mutations were correlated with
the response to neratinib and high expression of ATM, BRCA2 and
BRCA1 was associated with neratinib resistance (65). Neratinib has also been indicated to
cause DNA damage by γH2AX phosphorylation and ATM activation
(66). This result offers support
for further fundamental and clinical studies exploring the
combination therapy effect of neratinib and PARPi.
Osimertinib is a third-generation EGFRi that
inhibits cell proliferation by binding to cysteine-797 in the EGFR
ATP-binding sites. Compared with other EGFRis, osimertinib is able
to penetrate the blood-brain barrier to reach the central nervous
system and attack brain metastasis (67). A study revealed that osimertinib
functioned in a concentration-dependent and time-dependent manner
by means of proliferation inhibition and DDR delay in EGFR T790M
mutant NSCLC (68).
Apatinib is a highly selective inhibitor of VEGFR-2,
which inhibits c-kit, c-src and RET tyrosine kinase (69). Apatinib is the second
anti-angiogenetic drug approved in China for advanced metastatic
gastric cancer (GC). However, it has limited efficacy for
chemotherapy-experienced patients with other advanced cancers
(70). Colony formation assays
revealed that apatinib suppressed the repair of radiation-induced
DNA DSBs in hepatocellular carcinoma (HCC) in a PI3K/AKT-dependent
manner (71).
Cediranib is a potent and selective inhibitor of
VEGFR-1, -2 and -3 and is metabolized via flavin-containing
monooxygenase (FMO)1, FMO3 and uridine
5′-diphospho-glucuronosyltransferase 1A4 (72). Previous studies have suggested that
cediranib was able to induce hypoxia and thus suppress the
expression of HRD factors BRCA1/2 and RAD51 recombinase (73). Based on these theories, a mouse
model injected with epithelial ovarian cancer (EOC) cell lines was
established and cediranib was verified to abrogate prosurvival
signaling in the antiapoptotic AKT pathway and subsequently
enhanced the efficacy of olaparib (74).
In a phase I formulation bridging trial
(NCT01116648), the combination therapy of cediranib and PARPi was
used to generate preliminary evidence of anticancer activity in
high-grade serous ovarian cancer (HGSOC) (75). Furthermore, females with recurrent
platinum-sensitive ovarian cancer were recruited in a phase II
trial (NCT01116648) to compare the effects of cediranib and
olaparib in combination with those of olaparib alone. Among the 90
subjects enrolled, 46 received olaparib monotherapy at 400 mg twice
daily (bid) and 44 received combination therapy with cediranib 30
mg once daily (qd) and olaparib 200 mg BID. The median PFS
increased from 9.0 to 17.7 months in the group cotreated with
cediranib. In addition, in subjects with deleterious germline
BRCA1/2 mutation (gBRCAm) status, the median PFS increased from
16.5 to 19.4 months (P=0.06), while in those with non-gBRCAm or
unknown status, an increase from 5.7 to 16.5 months (P=0.008) was
observed (76,77). Another phase I clinical trial
(NCT02484404) recruited patients with advanced breast cancer or
gynaecological malignancies with gBRCAm. The trial tested the
3-drug combination in a 3 + 3 dose escalation. Cediranib was taken
discontinuously (5 days on/2 days off) at 15 or 20 mg with
durvalumab 1,500 mg intravenously (iv) every 4 weeks, and olaparib
tablets 300 mg bid. The primary end-point was the recommended phase
2 dose (RP2D), while secondary end-points were response rate,
pharmacokinetics and correlative analyses. The recommended RP2D was
cediranib 20 mg daily (5 days on/2 days off) with durvalumab 1,500
mg iv every 4 weeks and olaparib tablets 300 mg bid (78-80).
Furthermore, two more vital phase III trials (NCT02446600 and
NCT02502266) have been performed in ovarian cancer (81); both are three-armed studies and
recruited patients with recurrent platinum-sensitive HGSOC and
platinum-resistant HGSOC, respectively. PFS was the primary
end-point in both trials. The ICON9 trial (NCT03278717), sponsored
by Cancer Research UK, plans to investigate platinum-sensitive
recurrent HGSOC, endometrial histology or clear-cell ovarian
cancer. The EVOLVE trial (NCT02681237) recruited 34 heavily
pretreated patients assigned to three cohorts: Platinum-sensitive
after PARPi; platinum-resistant after PARPi; or progression on
standard chemotherapy after progression on PARPi. Patients received
olaparib 300 mg twice daily with cediranib 20 mg once daily until
disease progression or unacceptable toxicity. The primary
end-points were the objective response rate (RECIST v1.1) and PFS
at 16 weeks (82). However, these
clinical trials are currently in progress and no data have been
published, yet.
Imatinib is a 2-phenylaminopyrimidine derivative
primarily used in the treatment of myeloid leukaemia (83). Imatinib was indicated to be related
to DNA damage by reducing RAD51 protein levels in various previous
studies (84). To further
determine the mechanism of the anti-leukaemic action of imatinib,
the inhibition of DNA damage checkpoint arrest was demonstrated to
have a pivotal role in an ATM/ATR-dependent manner (85), and the downregulation of key DNA
repair genes was also observed after imatinib treatment (86). In addition, ATM kinase-dependent
phosphorylation of Nbs1, a member of the Mre11-RAD50-Nbs1 complex,
was enhanced to improve the efficacy of imatinib (87). A stepwise study used a primary
culture of ovarian cancer cells in 96-well plate assays and
revealed that imatinib had a synergistic effect with olaparib and
protected against olaparib cytotoxicity (88).
Regorafenib is a small molecular inhibitor of
kinases and targets various pathologic processes, including
oncogenesis, tumor angiogenesis and tumor microenvironment
formation. It was approved by the FDA for metastatic colorectal
cancer in 2012 and advanced HCC in 2017 (89). A preclinical study to determine the
potential of regorafenib in solid paediatric malignancy treatment
has been performed both in vitro and in vivo. The
results suggested that DNA damaging agents, such as a topoisomerase
I inhibitor and irradiation, had efficacy in platelet-derived
growth factor receptor-amplified tumors (90). To further validate this phenomenon,
another study was conducted in TNBC MDA-MB-231, SUM159PT and MCF10a
cell lines. Angiogenesis was inhibited and γH2AX was assessed to
confirm the existence of the DDR after regorafenib treatment
(91).
The TKIs introduced above are reciprocal receptors
on the cell membrane for the interconnection of cytokines or growth
factors. The JAK family has four primary members: JAK1, JAK2, JAK3
and Tyk2 (92). Of the four
members, JAK1, JAK2 and Tyk2 are expressed ubiquitously, while JAK3
is expressed mainly in haematopoietic cells (93,94).
The STAT family comprises seven members: STAT1, STAT2, STAT3,
STAT4, STAT5A, STAT5B and STAT6, and is a downstream target of JAK
that functions in signal activation as well as transduction
(95,96). Among these members, research
focuses on STAT3 and STAT5, which may have roles in disease
treatment resistance and are associated with multiple cancer types,
such as leukaemia and lymphoma (97). The JAK/STAT pathway, which is also
known as the IL-6 signalling pathway, was discovered >20 years
ago and has been further investigated recently (98). This signalling pathway is involved
in multiple important cellular activities, such as cell
proliferation, differentiation, apoptosis, immune regulation and
haematopoiesis (99). In a
case-control cohort study conducted in New Zealand, carrying a risk
allele associated with the STAT-JAK pathway was indicated to
predispose patients to DNA damage (100). In A549 cells, the mechanism of
X-ray-induced DNA damage was found to be related to the activation
of the JAK/STAT pathway (101).
JAK inhibitors (jakinibs) have been recognized as
safe and efficient therapies for diseases generated by
inflammation, which have been mentioned above (102). Type I and type II cytokine
receptors are a family of receptors comprised of >50 cytokines,
interleukins, interferons, colony-stimulating factors and hormones.
These receptors activate or suppress downstream signalling pathways
in a JAK-dependent manner. Thus, interfering with JAKs may result
in an immunomodulatory therapy with several adverse effects, such
as cytopenia and infection (103). JAKs were reported to activate
ATM/checkpoint kinase 2 (Chk2)/H2AX and ATR/Chk1 DDR, implying that
JAKi may enhance the efficacy of PARPi by disrupting the DDR
(104-106).
Ruxolitinib competes with the ATP binding domain in
the catalytic site of JAK1/JAK2 tyrosine kinase and was approved by
the FDA for the treatment of myelofibrosis in 2011 and by the
European Medicines Agency (EMA) in 2012 (107). Previous studies revealed an
increased tendency of DNA damage and genomic instability in
JAK2V617F expression models. As the underlying mechanism remains
unclear, numerous studies are attempting to establish in
vitro and in vivo models to answer this question. First,
JAK2V617F-overexpressing cells were indicated to promote DNA damage
and genomic instability via ROS accumulation (108,109). Furthermore, preclinical data
suggest that the molecules involved in the DDR were deficient in
JAK2V617F-expressing cells. Similar results have been observed in
patients (110). In addition,
JAK2 mutation has been linked to deficiencies in various DDR
pathways (111). Conversely,
another study established JAK2V617F-positive myeloproliferative
neoplasms (MPNs) and indicated that the patients remained
clinically and cytogenetically stable for numerous years (104). Based on these findings, a study
reported a synergistic inhibition of MPNs with the combination of
ruxolitinib and PARPi both in vitro and in vivo
(112).
In a study investigating the role of innate immune
regulators in human papillomavirus (HPV) pathogenesis, STAT5 was
indicated to be activated in HPV-positive cells and regulate HPV
genome amplification through activation of ATM in part via
peroxisome proliferator-activated receptor-γ. In addition, STATs
function as activators of DNA damage and ROS production in
TNFα-mediated senescence (113),
providing evidence of the feasibility of cotreatment with STATi and
PARPi.
The PI3K family of enzymes is recruited once growth
factor receptors are activated and generates 3′ phosphoinositide
lipids as second messengers to induce various cellular targeting
proteins (114). Among the
various second messengers, the serine/threonine kinase AKT is of
vital significance (115). When
AKT is stimulated, the rapamycin-sensitive mTORC1 signalling
pathway is triggered. In addition, rapamycin-sensitive mTORC2
contributes to AKT phosphorylation at critical sites (116). As crucial kinases during the
cellular lifespan, the PI3K-AKT-mTOR pathway contributes to cell
proliferation, transcription, translation, survival and growth
(117). This pathway is also
related to autophagy and apoptosis (118). Therefore, once disturbed, various
human malignancies occur (119).
Thus, this pathway may serve as a pivotal antitumor therapeutic
target for further research. In an HCC cell line model, PKI-587
promoted oxaliplatin sensitivity by suppressing the DDR pathway in
a PI3K-AKT-mTOR-dependent manner (120). Huanget al (121) summarized the studies related to
the PI3K pathway and DDR in ovarian cancer, which represents a
novel targeted treatment in cancer as well as a combined treatment
with PARPi.
Emerging data suggest that the PI3K pathway has a
role in DNA replication and genome stability, making DDR system
inhibitors, such as PARPi, potential combination therapies for PI3K
pathway pharmacologic inhibitors (121,122). For instance, copanlisib,
buparlisib and alpelisib have been investigated in clinical trials
combined with PARPi.
Copanlisib is a panclass I PI3K inhibitor developed
by Bayer that specifically targets the α and δ isoforms. In May
2017, copanlisib was approved by the FDA for the treatment of
relapsed follicular lymphoma in adult patients with at least two
prior therapies, which was further supported by the phase II study,
CHRONOS-1. Copanlisib has been reported to inhibit the
proliferation of various human cancer cell lines, inhibit cell
cycle progression and induce apoptosis in multiple myeloma cells
(123), inhibit BCR-independent
activation of NF-κB in diffuse large B-cell lymphoma cell lines and
inhibit growth in several lymphoma cell lines (124).
A phase Ib clinical trial (NCT03586661) sponsored by
the M.D. Anderson Cancer Center is currently recruiting patients
with recurrent endometrial and recurrent ovarian, primary
peritoneal or fallopian tube cancer to study the effects of the
synergy of niraparib with copanlisib. The included patients
received oral niraparib qd on days 1-28 and copanlisib iv on days
1, 8 and 15 to determine the maximum tolerated dose (125).
Buparlisib is an inhibitor targeting all isoforms of
panclass I PI3K in a comparative ATP-binding manner. In a
BRCA1-linked TNBC mouse model, carbon flux studies indicated an
impaired nonoxidative pentose phosphate pathway and subsequent
contribution to nucleotide synthesis suppression and DNA damage
(126). In another study
performed in Ishikawa, AN3CA and Nou-1 cells, DNA damage was
observed with γ-H2AX accumulation after buparlisib treatment, while
in Hec-108 cells, the HR repair system was interfered with
(127). These results enhance the
understanding of the effect of buparlisib on DNA damage (128). Due to the interaction between
buparlisib and DNA damage, the synergy of buparlisib with PARPi was
superior to either agent alone in in vitro and in
vivo models (126-129). In addition to breast and prostate
cancers, similar results have been observed in ovarian cancer
cells, particularly with PIK3CA mutation. BRCA was downregulated
after the cotreatment and may serve as a biomarker to recognize the
response to PARPi (130).
Based on preclinical studies, a phase Ib trial was
carried out among 24 patients with high-grade ovarian carcinoma and
46 patients with TNBC. The purpose was to investigate the maximum
tolerated dose, toxicities, pharmacokinetics and biomarkers of the
responses of combination treatment of PI3K inhibitors and PARPi.
The recommended dose is 50 mg BKM120 qd with 300 mg olaparib bid. A
synergistic effect was proven in phase I clinical trials regardless
of the status of gBRCA; therefore, further clinical studies should
be performed (131).
Alpelisib targets PI3Kα and its application in
breast cancer is currently under investigation. Mutation or
amplification of the PIK3CA gene that encodes the p110α subunit of
PI3K (134) occurs frequently in
solid tumors and therefore provides a new therapeutic target for
tumors (135). Cotreatment of
alpelisib with fulvestrant has been approved for application in
postmenopausal females with hormone receptor-positive,
HER-2-negative, PIK3CA-mutated, advanced or metastatic breast
cancer (136). Kimet al
(137) performed an in
vitro study using eight GC cell lines, three of which were
PIK3CA mutants. However, regardless of the PIK3CA mutation status,
all eight cell lines exhibited decreased AKT and S6K1
phosphorylation levels and induced G0/G1-phase arrest when treated
with alpelisib. Furthermore, the combination of alpelisib and
paclitaxel produced a synergistic anti-tumor effect via increased
DNA damage and apoptosis (137).
Based on these preclinical works, it is presumed that the
combination of PARPi with alpelisib may contribute to tumor
therapies. To assess the safety and recommended dose of olaparib
combined with alpelisib, a multicentre, open-label, phase Ib trial
enrolling 34 patients was established following a 3+3
dose-escalation design (NCT01623349). Of the 28 patients in the
dose-escalation cohort, 10 (36%) achieved a partial response and 14
(50%) had stable disease, which suggested that synergy of olaparib
and alpelisib offers a feasible strategy for tumor treatment
without insufferable adverse effects (138).
AKT was reported to inhibit TOPBP1, a DNA
repair/replication fork origin firing regulator, implying impacts
on DNA damage and synergistic effects with PARPi (139). Based on this molecular mechanism,
capivasertib has been studied in clinical trials to investigate its
efficacy and safety when combined with PARPi.
Ipatasertib is a potent inhibitor targeting the
ATP-binding domain of all three isoforms of AKT kinase (140). Strong antitumor activity of
ipatasertib has been indicated in various cancer types, including
breast, prostate, lung and colon cancer (141). Studies have revealed that AKT
kinase contributes to DDR, DSB repair and apoptosis; however, the
mechanisms have remained to be fully elucidated. Two activated
mutations in AKT1-TDSD and AKT1-E17K have been demonstrated to
accelerate DSB repair through a genetic approach (142). In an in vitro study,
Yuet al (143) observed
increased intracellular ROS levels and subsequently increased DNA
damage after treatment with ipatasertib.
Capivasertib is a potent selective pan-Akt kinase
inhibitor. The efficacy of capivasertib monotherapy has been
verified in various preclinical studies (144,145). This may be associated with
signalling crosstalk as well as feedback loop disruption. A phase I
trial enrolled 64 patients with advanced solid tumors to assess the
efficacy of capivasertib with olaparib. In the first trial to
combine PARPi and AKT inhibitor, 24 (44.6%) of 56 evaluable
patients achieved a clinical benefit, including patients with
gBRCA1/2m or BRCA1/2 wild-type (146). This observation suggests the
requirement for further cotreatment therapy. Currently, an active
but not recruiting nonrandomized open-label phase Ib study
(NCT02208375) aims to investigate the oral PARPi olaparib with the
oral AKT inhibitor capivasertib among patients with recurrent
endometrial cancer, TNBC or ovarian, primary peritoneal or
fallopian tube cancer. However, the maximum tolerated dose,
toxicity profiles, response rate and PFS remain to be fully
determined.
Perifosine is an oral alkylphospholipid that
inhibits AKT kinase activity by interfering with the pleckstrin
homology domain and impairing its membrane localization and
phosphorylation (147). Flow
cytometry suggested arrested cell cycle progression at the G2 phase
and western blot analysis indicated PARP activation upon perifosine
treatment (148). In TNBC cells,
perifosine was observed to induce RAD51 ubiquitination, block the
RAD51-BRCA2 interaction and decrease HR-mediated DNA DSB repair.
Based on this mechanism, research has explored the efficacy of the
combination of perifosine and olaparib and revealed a synergistic
antitumor activity in vivo (149).
MK-2206 is an orally active allosteric inhibitor
targeting AKT1 and AKT2 enzymes and is under investigation for the
treatment of solid tumors (150).
In EOC cells, higher AKT activity was reported to impact DNA
damage, which implied a synergism between MK-2206 and cisplatin or
olaparib (151).
Rapamycin has acute activity on mTORC1 but chronic
activity on mTORC2, the balance of which may be of vital importance
in ageing research (154).
Studies have revealed its capacities, such as cancer cell
proliferation inhibition and lifespan promotion (155).
In a study including 35 patients with kidney
transplant, DNA damage was analysed in peripheral blood
lymphocytes. The decrease in DNA damage in lymphocytes after
rapamycin treatment offered a new strategy for antitumor therapies
(156). In addition to the DNA
damage in lymphocytes, sperm DNA damage has also been measured
among infertile patients with and without varicocele, which
indicated a positive correlation with mTOR gene expression
(157). Furthermore, in in
vivo and in vitro TNBC models, rapamycin inhibited Rad51
focus formation induced by olaparib, suggesting the synergistic
effect of cotreatment via DNA DSB or SSB repair (157).
Everolimus is a selective oral inhibitor of the
mTORC1 complex, which is frequently activated in human
malignancies. As a result, everolimus is supposed to slow tumor
growth instead of inducing cell death (158).
Treatment with everolimus inhibited the increase in
p21 and the expression of DNA repair genes and mitotic checkpoint
regulators, which has been observed in multiple myeloma cells
(159), hepatocytes with chronic
liver injury (160) and isogenic
tumor cell lines (161). Studies
have further suggested that the combination of everolimus and
olaparib inhibited the growth of tumors, and were performed in
clone A, U87-MG xenografts and BRCA2-mutated patient-derived
xenografts of breast cancer (162,163).
A phase I open-label clinical study (NCT03154281) is
recruiting 24 patients to investigate the safety and tolerability
of niraparib in combination with everolimus in advanced
gynaecological malignancies and breast cancer. The outcome of this
study may offer fundamental support for the next phase of clinical
studies.
RAS was first identified as a downstream signalling
molecule of EGF in 1984. EGF activates EGFR on the membrane and
subsequently initiates guanine exchange factor to load RAS with GTP
(164). The RAS-GTP dimer
recruits RAF or RAF/MEK heterodimers to the plasma membrane and
contributes to RAF activation via back-to-back dimerization, while
a face-to-face homodimer facilitates the activation of MEK
(165). An early study explored
the downstream activity of MEK and revealed that MEK1/2 was able to
regulate ERK1/2 by phosphorylating the conserved Thr/Tyr in the
activation loop (166). This
pathway has been confirmed to be related to DNA damage in
vitro and in vivo and its inhibition promotes DNA damage
(167).
The active site of RAF is located at the interface
of the N-terminal lobe and C-terminal lobe. RAF inhibitors form
imperfect dimers in various positions of the αC-helix within each
promoter. Unlike most small molecular inhibitors targeting all
cells, RAF inhibitors selectively suppress RAF activity and
downstream signalling pathways in BRAF-mutant cells (168).
Dabrafenib is an oral drug approved by the FDA and
EMA alone or in combination with trametinib for the treatment of
BRAF-mutant unresectable or metastatic melanoma and advanced NSCLC
(169). Based on The Cancer
Genome Atlas and GTEx databases, Jianget al (170) determined that the expression of
the MUC gene was altered by dabrafenib treatment, which facilitates
DNA damage. Another study investigated the ROS levels in melanoma
models and indicated elevated ROS levels, as well as increased DNA
damage both in vivo and in vitro (171).
Vemurafenib is a selective BRAF V600E kinase
inhibitor that binds to its ATP-binding sites and therefore
inhibits cell proliferation in cells with BRAF V600E mutations
(172). It is known that
nonmelanoma skin cancer exhibits an ultraviolet radiation-induced
DDR. Using south-western blotting, DNA damage and repair capacity
were analysed and the results revealed that vemurafenib hampered
the DDR (173). This finding is
consistent with the previous study exploring the relationship
between vemurafenib treatment and the DDR (174).
MEK, encoded by 7 genes, is a downstream protein of
RAF. Instead of targeting the ATP binding sites directly, MEK
inhibitors bind to the pocket adjacent to them. This subset of
therapies has been under investigation in phase I-III clinical
trials in patients with various cancer types, such as advanced
NSCLC, melanoma, colon cancer, ovarian cancer and papillary thyroid
cancer (175-178).
As a potent ATP-noncompetitive MEK1/2 inhibitor,
selumetinib suppresses ERK phosphorylation and has been approved as
an adjuvant treatment for thyroid cancer and as monotherapy in
neurofibromatosis type 1 (179).
To identify cofactors that may enhance the antitumor capacity of
selumetinib, human tumor xenograft models were utilized. Research
has indicated an improved antitumor effect compared to monotherapy
and an increased level of γH2AX compared to temozolomide, a
DNA-alkylating agent, when applied as a cotreatment of selumetinib
and temozolomide. The data suggested a potential mechanism of the
combination of selumetinib with PARPi, which may suppress tumor
growth and proliferation in a DDR-inhibitory manner (180). Encouraged by these findings, a
nonrandomized clinical trial is recruiting patients with
endometrial, ovarian and other solid tumors with RAS pathway
alterations and ovarian tumors with PARPi resistance.
Trametinib is a second-generation small molecular
inhibitor of MEK kinase, which is an ATP noncompetitive inhibitor
against both MEK1 and MEK2 with a longer half-life and small
peak-to-trough ratios. An in vitro study indicated that
trametinib contributed to cell proliferation deceleration, cell
cycle arrest in G1 phase and apoptosis (181).
As a nonreceptor protein tyrosine kinase related to
malignancy formation, Src has been under investigation for three
decades. Src is a nonprimary protein that contributes to tumor
generation but rather participates in numerous signalling pathways
associated with cell division and survival. Therefore, Src
inhibitor monotherapy is not sufficient for tumor suppression
(183). In BRCA2-null prostate
cancer cell lines, upregulation of Src phosphorylation and a
synergistic effect of Src inhibitors with PARPis were observed
(184).
Dasatinib is a potent multikinase inhibitor that
targets Src family kinase and therefore blocks cell duplication,
migration and invasion. In addition, it promotes apoptosis of tumor
cells, suppresses metastatic spread of tumor cells and sensitizes
or resensitizes tumor cells to multiple therapies (185). Among studies performed in 6 HNSCC
cell lines as well as NSCLC cell lines with kinase-inactivating
BRAF mutation (KIBRAF), dasatinib suppressed the radiation-induced
DDR in HN-5 cells and induced DNA damage to senescence dependent on
Chk1 and p21 in KIBRAF (186,187). Based on the crosstalk associated
with DNA damage, the effect of the combination therapy of dasatinib
with olaparib has been evaluated in 18 cell lines representative of
the most frequent solid tumors, which exhibited synergism in
treatment (188).
The DNA damage repair system provides a genome-wide
surveillance mechanism to preserve chromosome integrity by
recognizing and repairing both exogenous and endogenous DNA
defects. Impairment of these systems results in mutations and
subsequently leads to tumorigenesis. However, no significant
clinical responses were observed among patients with BRCA mutations
or HRD. Limited monotherapy efficacy and drug resistance have
become major concerns in certain targeted therapies, such as PARPi.
There may be a risk of secondary tumors and therefore, the clinical
management and efficacy of PARPi require to be further investigated
in the future. Based on these aspects, combination therapies are
currently under urgent investigation. Small molecular inhibitors
have been approved by the FDA as targeted therapies. Their roles in
the DDR were described above and are summarized in Table I, Fig.
1; cotreatment with PARPi and small molecular inhibitors may be
performed to improve the limited efficacy of monotherapy. In
Table II, the phase I/II clinical
trials were summarized. The safety and efficacy have been confirmed
and the tolerable doses have been determined through these trials.
In the future, it is pivotal to identify patients who will benefit
the most from the synergistic treatments. Furthermore, indicators
to determine the likelihood of a clinical benefit may be obtained
from matched tissue and blood samples. In addition to targeted
therapies, immune therapies have become hot research topics and a
prominent synergism has been confirmed in the cotreatment of
anti-programmed death ligand 1 and PARPi. It remains to be
determined whether small molecular inhibitors are able to sensitize
cancers to immune therapy.
Data sharing is not applicable.
NJ performed the literature search and drafted the
manuscript. YX and QLG conceived the review and revised the
manuscript. All authors have read and approved the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
No funding was received.
|
1
|
Pilié PG, Tang C, Mills GB and Yap TA:
State-of-the-art strategies for targeting the DNA damage response
in cancer. Nat Rev Clin Oncol. 16:81–104. 2019. View Article : Google Scholar
|
|
2
|
Ali SO, Khan FA, Galindo-Campos MA and
Yelamos J: Understanding specific functions of PARP-2: New lessons
for cancer therapy. Am J Cancer Res. 6:1842–1863. 2016.PubMed/NCBI
|
|
3
|
Ma W, Halweg CJ, Menendez D and Resnick
MA: Differential effects of poly(ADP-ribose) polymerase inhibition
on DNA break repair in human cells are revealed with Epstein-Barr
virus. Proc Natl Acad Sci USA. 109:6590–6595. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Satoh MS and Lindahl T: Role of
poly(ADP-ribose) formation in DNA repair. Nature. 356:356–358.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ramakrishnan Geethakumari P, Schiewer MJ,
Knudsen KE and Kelly WK: PARP inhibitors in prostate cancer. Curr
Treat Options Oncol. 18:372017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Park SH, Jang KY, Kim MJ, Yoon S, Jo Y,
Kwon SM, Kim KM, Kwon KS, Kim CY and Woo HG: Tumor suppressive
effect of PARP1 and FOXO3A in gastric cancers and its clinical
implications. Oncotarget. 6:44819–44831. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mihailidou C, Karamouzis MV, Schizas D and
Papavassiliou AG: Co-targeting c-Met and DNA double-strand breaks
(DSBs): Therapeutic strategies in BRCA-mutated gastric carcinomas.
Biochimie. 142:135–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patel AG, Sarkaria JN and Kaufmann SH:
Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP)
inhibitor lethality in homologous recombination-deficient cells.
Proc Natl Acad Sci USA. 108:3406–3411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
De Lorenzo SB, Patel AG, Hurley RM and
Kaufmann SH: The elephant and the blind men: Making sense of PARP
inhibitors in homologous recombination deficient tumor cells. Front
Oncol. 3:2282013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lord CJ and Ashworth A: PARP inhibitors:
Synthetic lethality in the clinic. Science. 355:1152–1158. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Francica P and Rottenberg S: Mechanisms of
PARP inhibitor resistance in cancer and insights into the DNA
damage response. Genome Med. 10:1012018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rouleau M, Patel A, Hendzel MJ, Kaufmann
SH and Poirier GG: PARP inhibition: PARP1 and beyond. Nat Rev
Cancer. 10:293–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dellomo AJ, Baer MR and Rassool FV:
Partnering with PARP inhibitors in acute myeloid leukemia with
FLT3-ITD. Cancer Lett. 454:171–178. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
D'Andrea AD: Mechanisms of PARP inhibitor
sensitivity and resistance. DNA Repair (Amst). 71:172–176. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang F and Mazin AV: Targeting the
homologous recombination pathway by small molecule modulators.
Bioorg Med Chem Lett. 24:3006–3013. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Konstantinopoulos PA, Ceccaldi R, Shapiro
GI and D'Andrea AD: Homologous recombination deficiency: Exploiting
the fundamental vulnerability of ovarian cancer. Cancer Discov.
5:1137–1154. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Byrum AK, Vindigni A and Mosammaparast N:
Defining and modulating 'BRCAness'. Trends Cell Biol. 29:740–751.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Quigley D, Alumkal JJ, Wyatt AW, Kothari
V, Foye A, Lloyd P, Aggarwal R, Kim W, Lu E, Schwartzman J, et al:
Analysis of circulating cell-free DNA identifies multiclonal
heterogeneity of BRCA2 reversion mutations associated with
resistance to PARP inhibitors. Cancer Discov. 7:999–1005. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Drost R, Bouwman P, Rottenberg S, Boon U,
Schut E, Klarenbeek S, Klijn C, van der Heijden I, van der Gulden
H, Wientjens E, et al: BRCA1 RING function is essential for tumor
suppression but dispensable for therapy resistance. Cancer Cell.
20:797–809. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pettitt SJ, Krastev DB, Brandsma I, Dréan
A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell
J, et al: Genome-wide and high-density CRISPR-Cas9 screens identify
point mutations in PARP1 causing PARP inhibitor resistance. Nat
Commun. 9:18492018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boersma V, Moatti N, Segura-Bayona S,
Peuscher MH, van der Torre J, Wevers BA, Orthwein A, Durocher D and
Jacobs JJL: MAD2L2 controls DNA repair at telomeres and DNA breaks
by inhibiting 5′ end resection. Nature. 521:537–540. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu GT, Chapman JR, Brandsma I, Yuan J,
Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M,
et al: REV7 counteracts DNA double-strand break resection and
affects PARP inhibition. Nature. 521:541–544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McNerney MP and Styczynski MP: Small
molecule signaling, regulation, and potential applications in
cellular therapeutics. Wiley Interdiscip Rev Syst Biol Med. 10.
View Article : Google Scholar : 2018
|
|
25
|
Zhu HF and Li Y: Small-molecule targets in
tumor immunotherapy. Nat Prod Bioprospect. 8:297–301. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng B, Yuan WE, Su J, Liu Y and Chen J:
Recent advances in small molecule based cancer immunotherapy. Eur J
Med Chem. 157:582–598. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du Z and Lovly CM: Mechanisms of receptor
tyrosine kinase activation in cancer. Mol Cancer. 17:582018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Robinson DR, Wu YM and Lin SF: The protein
tyrosine kinase family of the human genome. Oncogene. 19:5548–5557.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gschwind A, Fischer OM and Ullrich A: The
discovery of receptor tyrosine kinases: Targets for cancer therapy.
Nat Rev Cancer. 4:361–370. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kinase Inhibitors: Methods and Protocols.
Kinase Inhibitors: Methods and Protocols. 795:Kuster B: (Methods in
Molecular Biology). 2012. View Article : Google Scholar
|
|
31
|
Bensimon A, Koch JP, Francica P, Roth SM,
Riedo R, Glück AA, Orlando E, Blaukat A, Aebersold DM, Zimmer Y, et
al: Deciphering MET-dependent modulation of global cellular
responses to DNA damage by quantitative phosphoproteomics. Mol
Oncol. 14:1185–1206. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bronte G, Rolfo C, Giovannetti E, Cicero
G, Pauwels P, Passiglia F, Castiglia M, Rizzo S, Vullo FL,
Fiorentino E, et al: Are erlotinib and gefitinib interchangeable,
opposite or complementary for non-small cell lung cancer treatment?
Biological, pharmacological and clinical aspects. Crit Rev Oncol
Hematol. 89:300–313. 2014. View Article : Google Scholar
|
|
33
|
Hirsch FR, Varella-Garcia M, Bunn PA Jr,
Di Maria MV, Veve R, Bremmes RM, Barón AE, Zeng C and Franklin WA:
Epidermal growth factor receptor in non-small-cell lung carcinomas:
Correlation between gene copy number and protein expression and
impact on prognosis. J Clin Oncol. 21:3798–3807. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bartlett JM, Langdon SP, Simpson BJ,
Stewart M, Katsaros D, Sismondi P, Love S, Scott WN, Williams AR,
Lessells AM, et al: The prognostic value of epidermal growth factor
receptor mRNA expression in primary ovarian cancer. Br J Cancer.
73:301–306. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Blume-Jensen P and Hunter T: Oncogenic
kinase signalling. Nature. 411:355–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gordon AN, Finkler N, Edwards RP, Garcia
AA, Crozier M, Irwin DH and Barrett E: Efficacy and safety of
erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR)
tyrosine kinase inhibitor, in patients with advanced ovarian
carcinoma: Results from a phase II multicenter study. Int J Gynecol
Cancer. 15:785–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu Y, Gao Z, Hu R, Wang Y, Wang Y, Su Z,
Zhang X, Yang J, Mei M, Ren Y, et al: PD-L2 glycosylation promotes
immune evasion and predicts anti-EGFR efficacy. J Immunother
Cancer. 9:e0026992021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tanaka K, Yu HA, Yang S, Han S, Selcuklu
SD, Kim K, Ramani S, Ganesan YT, Moyer A, Sinha S, et al: Targeting
Aurora B kinase prevents and overcomes resistance to EGFR
inhibitors in lung cancer by enhancing BIM- and PUMA-mediated
apoptosis. Cancer Cell. 39:1245–1261.e6. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu S, Gao F, Zheng S, Zhang C,
Martinez-Ledesma E, Ezhilarasan R, Ding J, Li X, Feng N, Multani A,
et al: EGFR amplification induces increased DNA damage response and
renders selective sensitivity to talazoparib (PARP inhibitor) in
glioblastoma. Clin Cancer Res. 26:1395–1407. 2020. View Article : Google Scholar
|
|
40
|
Rodemann HP, Dittmann K and Toulany M:
Radiation-induced EGFR-signaling and control of DNA-damage repair.
Int J Radiat Biol. 83:781–791. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Volman Y, Hefetz R, Galun E and
Rachmilewitz J: DNA damage alters EGFR signaling and reprograms
cellular response via Mre-11. Sci Rep. 12:57602022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rajput M, Singh R, Singh N and Singh RP:
EGFR-mediated Rad51 expression potentiates intrinsic resistance in
prostate cancer via EMT and DNA repair pathways. Life Sci.
286:1200312021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rawluk J and Waller CF: Gefitinib. Recent
Results Cancer Res. 211:235–246. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bokobza SM, Jiang Y, Weber AM, Devery AM
and Ryan AJ: Short-course treatment with gefitinib enhances
curative potential of radiation therapy in a mouse model of human
non-small cell lung cancer. Int J Radiat Oncol Biol Phys.
88:947–954. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
de Silva HC, Lin MZ, Phillips L, Martin JL
and Baxter RC: IGFBP-3 interacts with NONO and SFPQ in
PARP-dependent DNA damage repair in triple-negative breast cancer.
Cell Mol Life Sci. 76:2015–2030. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Garcia-Campelo R, Arrieta O, Massuti B,
Rodriguez-Abreu D, Granados ALO, Majem M, Vicente D, Lianes P,
Bosch-Barrera J, Insa A, et al: Combination of gefitinib and
olaparib versus gefitinib alone in EGFR mutant non-small-cell lung
cancer (NSCLC): A multicenter, randomized phase II study (GOAL).
Lung Cancer. 150:62–69. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang L, Wang J, Cui LZ, Wang K, Yuan MM,
Chen RR and Zhang LK: Successful treatment of refractory lung
adenocarcinoma harboring a germline BRCA2 mutation with olaparib: A
case report. World J Clin Cases. 9:7498–7503. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Voigtlaender M, Schneider-Merck T and
Trepel M: Lapatinib. Recent Results Cancer Res. 211:19–44. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Abo-Zeid MAM, Abo-Elfadl MT and
Gamal-Eldeen AM: Evaluation of lapatinib cytotoxicity and
genotoxicity on MDA-MB-231 breast cancer cell line. Environ Toxicol
Pharmacol. 71:1032072019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li YT, Qian XJ, Yu Y, Li ZH, Wu RY, Ji J,
Jiao L, Li X, Kong PF, Chen WD, et al: EGFR tyrosine kinase
inhibitors promote pro-caspase-8 dimerization that sensitizes
cancer cells to DNA-damaging therapy. Oncotarget. 6:17491–17500.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nowsheen S, Cooper T, Stanley JA and Yang
ES: Synthetic lethal interactions between EGFR and PARP inhibition
in human triple negative breast cancer cells. PLoS One.
7:e466142012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Abdelgalil AA, Al-Kahtani HM and
Al-Jenoobi FI: Erlotinib. Profiles Drug Subst Excip Relat Methodol.
45:93–117. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tsai YC, Ho PY, Tzen KY, Tuan TF, Liu WL,
Cheng AL, Pu YS and Cheng JC: Synergistic blockade of EGFR and HER2
by new-generation EGFR tyrosine kinase inhibitor enhances radiation
effect in bladder cancer cells. Mol Cancer Ther. 14:810–820. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Keta O, Bulat T, Golić I, Incerti S, Korać
A, Petrović I and Ristić-Fira A: The impact of autophagy on cell
death modalities in CRL-5876 lung adenocarcinoma cells after their
exposure to γ-rays and/or erlotinib. Cell Biol Toxicol. 32:83–101.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li L, Wang H, Yang ES, Arteaga CL and Xia
F: Erlotinib attenuates homologous recombinational repair of
chromosomal breaks in human breast cancer cells. Cancer Res.
68:9141–9146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Sui H, Shi C, Yan Z and Li H: Combination
of erlotinib and a PARP inhibitor inhibits growth of A2780 tumor
xenografts due to increased autophagy. Drug Des Devel Ther.
9:3183–3190. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dong Q, Liu M, Chen B, Zhao Z, Chen T,
Wang C, Zhuang S, Li Y, Wang Y, Ai L, et al: Revealing biomarkers
associated with PARP inhibitors based on genetic interactions in
cancer genome. Comput Struct Biotechnol J. 19:4435–4446. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wecker H and Waller CF: Afatinib. Small
Molecules in Oncology. Springer; New York, NY, USA: pp. 199–215.
2018, View Article : Google Scholar
|
|
59
|
Miller VA, Hirsh V, Cadranel J, Chen YM,
Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al: Afatinib
versus placebo for patients with advanced, metastatic
non-small-cell lung cancer after failure of erlotinib, gefitinib,
or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase
2b/3 randomised trial. Lancet Oncol. 13:528–538. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang S, Zheng X, Huang H, Wu K, Wang B,
Chen X and Ma S: Afatinib increases sensitivity to radiation in
non-small cell lung cancer cells with acquired EGFR T790M mutation.
Oncotarget. 6:5832–5845. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Doan HQ, Hu MI, Goldstein J, Piha-Paul SA,
Subbiah V and Patel AB: Vandetanib photoinduced cutaneous
toxicities. Cutis. 103:E24–E29. 2019.PubMed/NCBI
|
|
62
|
Chu PL, Shihabuddeen WA, Low KP, Poon DJJ,
Ramaswamy B, Liang ZG, Nei WL, Chua KLM, Thong PSP, Soo KC, et al:
Vandetanib sensitizes head and neck squamous cell carcinoma to
photodynamic therapy through modulation of EGFR-dependent DNA
repair and the tumour microenvironment. Photodiagnosis Photodyn
Ther. 27:367–374. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Macy ME, DeRyckere D and Gore L:
Vandetanib mediates anti-leukemia activity by multiple mechanisms
and interacts synergistically with DNA damaging agents. Invest New
Drugs. 30:468–479. 2012. View Article : Google Scholar
|
|
64
|
Deeks ED: Neratinib: First global
approval. Drugs. 77:1695–1704. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Conlon NT, Kooijman JJ, van Gerwen SJC,
Mulder WR, Zaman GJR, Diala I, Eli LD, Lalani AS, Crown J and
Collins DM: Comparative analysis of drug response and gene
profiling of HER2-targeted tyrosine kinase inhibitors. Br J Cancer.
124:1249–1259. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Booth L, Roberts JL, Samuel P,
Avogadri-Connors F, Cutler RE, Lalani AS, Poklepovic A and Dent P:
The irreversible ERBB1/2/4 inhibitor neratinib interacts with the
PARP1 inhibitor niraparib to kill ovarian cancer cells. Cancer Biol
Ther. 19:525–533. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lazzari C, Gregorc V, Karachaliou N,
Rosell R and Santarpia M: Mechanisms of resistance to osimertinib.
J Thorac Dis. 12:2851–2858. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang N, Wang L, Meng X, Wang J, Zhu L, Liu
C, Li S, Zheng L, Yang Z, Xing L and Yu J: Osimertinib (AZD9291)
increases radio-sensitivity in EGFR T790M non-small cell lung
cancer. Oncol Rep. 41:77–86. 2019.
|
|
69
|
Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y,
Li J and Lou L: YN968D1 is a novel and selective inhibitor of
vascular endothelial growth factor receptor-2 tyrosine kinase with
potent activity in vitro and in vivo. Cancer Sci. 102:1374–1380.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Aoyama T and Yoshikawa T: Apatinib-new
third-line option for refractory gastric or GEJ cancer. Nat Rev
Clin Oncol. 13:268–270. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liao J, Jin H, Li S, Xu L, Peng Z, Wei G,
Long J, Guo Y, Kuang M, Zhou Q and Peng S: Apatinib potentiates
irradiation effect via suppressing PI3K/AKT signaling pathway in
hepatocellular carcinoma. J Exp Clin Cancer Res. 38:4542019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Tang W, McCormick A, Li J and Masson E:
Clinical pharmacokinetics and pharmacodynamics of cediranib. Clin
Pharmacokinet. 56:689–702. 2017. View Article : Google Scholar
|
|
73
|
Kaplan AR, Gueble SE, Liu YF, Oeck S, Kim
H, Yun Z and Glazer PM: Cediranib suppresses homology-directed DNA
repair through down-regulation of BRCA1/2 and RAD51. Sci Transl
Med. 11:eaav45082019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lin ZP, Zhu YL, Lo YC, Moscarelli J, Xiong
A, Korayem Y, Huang PH, Giri S, LoRusso P and Ratner ES:
Combination of triapine, olaparib, and cediranib suppresses
progression of BRCA-wild type and PARP inhibitor-resistant
epithelial ovarian cancer. PLoS One. 13:e02073992018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Liu JF, Tolaney SM, Birrer M, Fleming GF,
Buss MK, Dahlberg SE, Lee H, Whalen C, Tyburski K, Winer E, et al:
A Phase 1 trial of the poly(ADP-ribose) polymerase inhibitor
olaparib (AZD2281) in combination with the anti-angiogenic
cediranib (AZD2171) in recurrent epithelial ovarian or
triple-negative breast cancer. Eur J Cancer. 49:2972–2978. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Liu JF, Barry WT, Birrer M, Lee JM,
Buckanovich RJ, Fleming GF, Rimel B, Buss MK, Nattam S, Hurteau J,
et al: Combination cediranib and olaparib versus olaparib alone for
women with recurrent platinum-sensitive ovarian cancer: A
randomised phase 2 study. Lancet Oncol. 15:1207–1214. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liu JF, Barry WT, Birrer M, Lee JM,
Buckanovich RJ, Fleming GF, Rimel BJ, Buss MK, Nattam SR, Hurteau
J, et al: Overall survival and updated progression-free survival
outcomes in a randomized phase II study of combination cediranib
and olaparib versus olaparib in relapsed platinum-sensitive ovarian
cancer. Ann Oncol. 30:551–557. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zimmer AS, Nichols E, Cimino-Mathews A,
Peer C, Cao L, Lee MJ, Kohn EC, Annunziata CM, Lipkowitz S, Trepel
JB, et al: A phase I study of the PD-L1 inhibitor, durvalumab, in
combination with a PARP inhibitor, olaparib, and a VEGFR1-3
inhibitor, cediranib, in recurrent women's cancers with biomarker
analyses. J Immunother Cancer. 7:1972019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Thomas A, Vilimas R, Trindade C,
Erwin-Cohen R, Roper N, Xi L, Krishnasamy V, Levy E, Mammen A,
Nichols S, et al: Durvalumab in combination with olaparib in
patients with relapsed SCLC: Results from a phase II Study. J
Thorac Oncol. 14:1447–1457. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Karzai F, VanderWeele D, Madan RA, Owens
H, Cordes LM, Hankin A, Couvillon A, Nichols E, Bilusic M, Beshiri
ML, et al: Activity of durvalumab plus olaparib in metastatic
castration-resistant prostate cancer in men with and without DNA
damage repair mutations. J Immunother Cancer. 6:1412018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Tattersall A, Ryan N, Wiggans AJ,
Rogozińska E and Morrison J: Poly(ADP-ribose) polymerase (PARP)
inhibitors for the treatment of ovarian cancer. Cochrane Database
Syst Rev. 2:CD0079292022.PubMed/NCBI
|
|
82
|
Lheureux S, Oaknin A, Garg S, Bruce JP,
Madariaga A, Dhani NC, Bowering V, White J, Accardi S, Tan Q, et
al: EVOLVE: A multicenter open-label single-arm clinical and
translational phase II trial of cediranib plus olaparib for ovarian
cancer after PARP inhibition progression. Clin Cancer Res.
26:4206–4215. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Waller CF: Imatinib mesylate. Recent
Results Cancer Res. 212:1–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Choudhury A, Zhao H, Jalali F, Al Rashid
S, Ran J, Supiot S, Kiltie AE and Bristow RG: Targeting homologous
recombination using imatinib results in enhanced tumor cell
chemosensitivity and radiosensitivity. Mol Cancer Ther. 8:203–213.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Morii M, Fukumoto Y, Kubota S and
Yamaguchi N, Nakayama Y and Yamaguchi N: Imatinib inhibits
inactivation of the ATM/ATR signaling pathway and recovery from
adriamycin/doxorubicin-induced DNA damage checkpoint arrest. Cell
Biol Int. 39:923–932. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Benito R, Lumbreras E, Abáigar M,
Gutiérrez NC, Delgado M, Robledo C, García JL, Rodríguez-Vicente
AE, Cañizo MC and Rivas JM: Imatinib therapy of chronic myeloid
leukemia restores the expression levels of key genes for DNA damage
and cell-cycle progression. Pharmacogenet Genomics. 22:381–388.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Rink L, Slupianek A, Stoklosa T,
Nieborowska-Skorska M, Urbanska K, Seferynska I, Reiss K and
Skorski T: Enhanced phosphorylation of Nbs1, a member of DNA
repair/checkpoint complex Mre11-RAD50-Nbs1, can be targeted to
increase the efficacy of imatinib mesylate against BCR/ABL-positive
leukemia cells. Blood. 110:651–660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Mukhopadhyay A, Drew Y, Matheson E,
Salehan M, Gentles L, Pachter JA and Curtin NJ: Evaluating the
potential of kinase inhibitors to suppress DNA repair and sensitise
ovarian cancer cells to PARP inhibitors. Biochem Pharmacol.
167:125–132. 2019. View Article : Google Scholar
|
|
89
|
Ettrich TJ and Seufferlein T: Regorafenib.
Recent Results Cancer Res. 211:45–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Daudigeos-Dubus E, Le Dret L,
Lanvers-Kaminsky C, Bawa O, Opolon P, Vievard A, Villa I, Pagès M,
Bosq J, Vassal G, et al: Regorafenib: Antitumor activity upon mono
and combination therapy in preclinical pediatric malignancy models.
PLoS One. 10:e01426122015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mehta M, Griffith J, Panneerselvam J, Babu
A, Mani J, Herman T, Ramesh R and Munshi A: Regorafenib sensitizes
human breast cancer cells to radiation by inhibiting multiple
kinases and inducing DNA damage. Int J Radiat Biol. 97:1109–1120.
2021. View Article : Google Scholar :
|
|
92
|
Cai B, Cai JP, Luo YL, Chen C and Zhang S:
The specific roles of JAK/STAT signaling pathway in sepsis.
Inflammation. 38:1599–1608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
O'Shea JJ, Pesu M, Borie DC and Changelian
PS: A new modality for immunosuppression: Targeting the JAK/STAT
pathway. Nat Rev Drug Discov. 3:555–564. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Jaime-Figueroa S, De Vicente J, Hermann J,
Jahangir A, Jin S, Kuglstatter A, Lynch SM, Menke J, Niu L, Patel
V, et al: Discovery of a series of novel
5H-pyrrolo[2,3-b]pyrazine-2-phenyl ethers, as potent JAK3 kinase
inhibitors. Bioorg Med Chem Lett. 23:2522–2526. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yu H, Pardoll D and Jove R: STATs in
cancer inflammation and immunity: A leading role for STAT3. Nat Rev
Cancer. 9:798–809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Boengler K, Hilfiker-Kleiner D, Drexler H,
Heusch G and Schulz R: The myocardial JAK/STAT pathway: From
protection to failure. Pharmacol Ther. 120:172–185. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Abroun S, Saki N, Ahmadvand M, Asghari F,
Salari F and Rahim F: STATs: An old story, Yet mesmerizing. Cell J.
17:395–411. 2015.PubMed/NCBI
|
|
98
|
Stark GR and Darnell JE Jr: The JAK-STAT
pathway at twenty. Immunity. 36:503–514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Bolli R, Dawn B and Xuan YT: Role of the
JAK-STAT pathway in protection against myocardial
ischemia/reperfusion injury. Trends Cardiovasc Med. 13:72–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Ferguson LR, Han DY, Fraser AG, Huebner C,
Lam WJ, Morgan AR, Duan H and Karunasinghe N: Genetic factors in
chronic inflammation: Single nucleotide polymorphisms in the
STAT-JAK pathway, susceptibility to DNA damage and Crohn's disease
in a New Zealand population. Mutat Res. 690:108–115. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Peng LQ, Li CH and Mao B: Activation of
the JAK/STAT signal pathway may be involved in DNA damage of A549
cells induced by X-ray. Sheng Li Xue Bao. 71:698–704. 2019.In
Chinese. PubMed/NCBI
|
|
102
|
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou
X, Ma H, Wei D and Sun S: The role of JAK/STAT signaling pathway
and its inhibitors in diseases. Int Immunopharmacol. 80:1062102020.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Schwartz DM, Kanno Y, Villarino A, Ward M,
Gadina M and O'Shea JJ: JAK inhibition as a therapeutic strategy
for immune and inflammatory diseases. Nat Rev Drug Discov.
16:843–862. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Karantanos T and Moliterno AR: The roles
of JAK2 in DNA damage and repair in the myeloproliferative
neoplasms: Opportunities for targeted therapy. Blood Rev.
32:426–432. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Khashab F, Al-Saleh F, Al-Kandari N, Fadel
F and Al-Maghrebi M: JAK inhibition prevents DNA damage and
apoptosis in testicular ischemia-reperfusion injury via modulation
of the ATM/ATR/Chk pathway. Int J Mol Sci. 22:133902021. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Reddig A, Voss L, Guttek K, Roggenbuck D,
Feist E and Reinhold D: Impact of different JAK inhibitors and
methotrexate on lymphocyte proliferation and DNA damage. J Clin
Med. 10:14312021. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Becker H, Engelhardt M, von Bubnoff N and
Wäsch R: Ruxolitinib. Martens UM: Small Molecules in Oncology.
Recent Results in Cancer Research. 201. Springer; Berlin,
Heidelberg: pp. 249–257. 2014, View Article : Google Scholar
|
|
108
|
Ahn JS, Li J, Chen E, Kent DG, Park HJ and
Green AR: JAK2V617F mediates resistance to DNA damage-induced
apoptosis by modulating FOXO3A localization and Bcl-xL deamidation.
Oncogene. 35:2235–2246. 2016. View Article : Google Scholar
|
|
109
|
Kagoya Y, Yoshimi A, Tsuruta-Kishino T,
Arai S, Satoh T, Akira S and Kurokawa M: JAK2V617F+
myeloproliferative neoplasm clones evoke paracrine DNA damage to
adjacent normal cells through secretion of lipocalin-2. Blood.
124:2996–3006. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Chen E, Ahn JS, Massie CE, Clynes D,
Godfrey AL, Li J, Park HJ, Nangalia J, Silber Y, Mullally A, et al:
JAK2V617F promotes replication fork stalling with
disease-restricted impairment of the intra-S checkpoint response.
Proc Natl Acad Sci USA. 111:15190–15195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Nakatake M, Monte-Mor B, Debili N,
Casadevall N, Ribrag V, Solary E, Vainchenker W and Plo I:
JAK2(V617F) negatively regulates p53 stabilization by enhancing
MDM2 via La expression in myeloproliferative neoplasms. Oncogene.
31:1323–1333. 2012. View Article : Google Scholar
|
|
112
|
Nieborowska-Skorska M, Maifrede S,
Dasgupta Y, Sullivan K, Flis S, Le BV, Solecka M, Belyaeva EA,
Kubovcakova L, Nawrocki M, et al: Ruxolitinib-induced defects in
DNA repair cause sensitivity to PARP inhibitors in
myeloproliferative neoplasms. Blood. 130:2848–2859. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Kandhaya-Pillai R, Miro-Mur F,
Alijotas-Reig J, Tchkonia T, Kirkland JL and Schwartz S:
TNFα-senescence initiates a STAT-dependent positive feedback loop,
leading to a sustained interferon signature, DNA damage, and
cytokine secretion. Aging (Albany NY). 9:2411–2435. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Katso R, Okkenhaug K, Ahmadi K, White S,
Timms J and Waterfield MD: Cellular function of phosphoinositide
3-kinases: Implications for development, homeostasis, and cancer.
Annu Rev Cell Dev Biol. 17:615–675. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Liu PX, Cheng HL, Roberts TM and Zhao JJ:
Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev
Drug Discov. 8:627–644. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Guertin DA and Sabatini DM: The
pharmacology of mTOR inhibition. Sci Signal. 2:pe242009. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Datta SR, Brunet A and Greenberg ME:
Cellular survival: A play in three Akts. Genes Dev. 13:2905–2927.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G,
Liu J and Zhang J: Targeting PI3K/AKT/mTOR-mediated autophagy for
tumor therapy. Appl Microbiol Biotechnol. 104:575–587. 2020.
View Article : Google Scholar
|
|
119
|
Porta C and Figlin RA:
Phosphatidylinositol-3-kinase/Akt signaling pathway and kidney
cancer, and the therapeutic potential of
phosphatidylinositol-3-kinase/Akt inhibitors. J Urol.
182:2569–2577. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Zhang Y, Xie C, Li A, Liu X, Xing Y, Shen
J, Huo Z, Zhou S, Liu X, Xie Y, et al: PKI-587 enhances
chemosensitivity of oxaliplatin in hepatocellular carcinoma through
suppressing DNA damage repair pathway (NHEJ and HR) and
PI3K/AKT/mTOR pathway. Am J Transl Res. 11:5134–5149.
2019.PubMed/NCBI
|
|
121
|
Huang TT, Lampert EJ, Coots C and Lee JM:
Targeting the PI3K pathway and DNA damage response as a therapeutic
strategy in ovarian cancer. Cancer Treat Rev. 86:1020212020.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Pal S, Kozono D, Yang X, Fendler W, Fitts
W, Ni J, Alberta JA, Zhao J, Liu KX, Bian J, et al: Dual HDAC and
PI3K inhibition abrogates NFκB- and FOXM1-mediated DNA damage
response to radiosensitize pediatric high-grade gliomas. Cancer
Res. 78:4007–4021. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Glauer J, Pletz N, Schön M, Schneider P,
Liu N, Ziegelbauer K, Emmert S, Wulf GG and Schön MP: A novel
selective small-molecule PI3K inhibitor is effective against human
multiple myeloma in vitro and in vivo. Blood Cancer J. 3:e1412013.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Markham A: Copanlisib: First global
approval. Drugs. 77:2057–2062. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Sun C, Fang Y, Labrie M, Li X and Mills
GB: Systems approach to rational combination therapy: PARP
inhibitors. Biochem Soc Trans. 48:1101–1108. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Juvekar A, Hu H, Yadegarynia S, Lyssiotis
CA, Ullas S, Lien EC, Bellinger G, Son J, Hok RC, Seth P, et al:
Phosphoinositide 3-kinase inhibitors induce DNA damage through
nucleoside depletion. Proc Natl Acad Sci USA. 113:E4338–E4347.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bian X, Gao J, Luo F, Rui C, Zheng T, Wang
D, Wang Y, Roberts TM, Liu P, Zhao JJ and Cheng H: PTEN deficiency
sensitizes endometrioid endometrial cancer to compound PARP-PI3K
inhibition but not PARP inhibition as monotherapy. Oncogene.
37:341–351. 2018. View Article : Google Scholar :
|
|
128
|
DuRoss AN, Neufeld MJ, Landry MR, Rosch
JG, Eaton CT, Sahay G, Thomas CR Jr and Sun C: Micellar formulation
of talazoparib and buparlisib for enhanced DNA damage in breast
cancer chemoradiotherapy. ACS Appl Mater Interfaces.
11:12342–12356. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Juvekar A, Burga LN, Hu H, Lunsford EP,
Ibrahim YH, Balmañà J, Rajendran A, Papa A, Spencer K, Lyssiotis
CA, et al: Combining a PI3K inhibitor with a PARP inhibitor
provides an effective therapy for BRCA1-related breast cancer.
Cancer Discov. 2:1048–1063. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Wang D, Wang M, Jiang N, Zhang Y, Bian X,
Wang X, Roberts TM, Zhao JJ, Liu P and Cheng H: Effective use of
PI3K inhibitor BKM120 and PARP inhibitor olaparib to treat PIK3CA
mutant ovarian cancer. Oncotarget. 7:13153–13166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Matulonis UA, Wulf GM, Barry WT, Birrer M,
Westin SN, Farooq S, Bell-McGuinn KM, Obermayer E, Whalen C,
Spagnoletti T, et al: Phase I dose escalation study of the
PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose)
polymerase (PARP) inhibitor olaparib for the treatment of
high-grade serous ovarian and breast cancer. Ann Oncol. 28:512–518.
2017. View Article : Google Scholar
|
|
132
|
Zumsteg ZS, Morse N, Krigsfeld G, Gupta G,
Higginson DS, Lee NY, Morris L, Ganly I, Shiao SL, Powell SN, et
al: Taselisib (GDC-0032), a potent β-sparing small molecule
inhibitor of PI3K, radiosensitizes head and neck squamous
carcinomas containing activating PIK3CA alterations. Clin Cancer
Res. 22:2009–2019. 2016. View Article : Google Scholar
|
|
133
|
Ndubaku CO, Heffron TP, Staben ST,
Baumgardner M, Blaquiere N, Bradley E, Bull R, Do S, Dotson J,
Dudley D, et al: Discovery of
2-{3-[2-(1-isopropyl-3-methyl-1H-1,2-4-triazol-
5-yl)-5,6-dihydrobenzo[f]imidazo[1,2-d][1,4]oxazepin-9-yl]-
1H-pyrazol-1-yl}-2-methylpropanamide (GDC-0032): A β-sparing
phosphoinositide 3-kinase inhibitor with high unbound exposure and
robust in vivo antitumor activity. J Med Chem. 56:4597–4610. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Juric D, Rodon J, Tabernero J, Janku F,
Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M,
Gil-Martin M, et al: Phosphatidylinositol 3-kinase α-selective
inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors:
Results from the first-in-human study. J Clin Oncol. 36:1291–1299.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Pereira B, Chin SF, Rueda OM, Vollan HK,
Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ,
et al: The somatic mutation profiles of 2,433 breast cancers
refines their genomic and transcriptomic landscapes. Nat Commun.
7:114792016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Markham A: Alpelisib: First global
approval. Drugs. 79:1249–1253. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Kim KJ, Kim JW, Sung JH, Suh KJ, Lee JY,
Kim SH, Lee JO, Kim JW, Kim YJ, Kim JH, et al: PI3K-targeting
strategy using alpelisib to enhance the antitumor effect of
paclitaxel in human gastric cancer. Sci Rep. 10:123082020.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Konstantinopoulos PA, Barry WT, Birrer M,
Westin SN, Cadoo KA, Shapiro GI, Mayer EL, O'Cearbhaill RE, Coleman
RL, Kochupurakkal B, et al: Olaparib and α-specific PI3K inhibitor
alpelisib for patients with epithelial ovarian cancer: A
dose-escalation and dose-expansion phase 1b trial. Lancet Oncol.
20:570–580. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Conduit SE, Davies EM, Ooms LM, Gurung R,
McGrath MJ, Hakim S, Cottle DL, Smyth IM, Dyson JM and Mitchell CA:
AKT signaling promotes DNA damage accumulation and proliferation in
polycystic kidney disease. Hum Mol Genet. 29:31–48. 2020.
|
|
140
|
Han C, Savage S, Al-Sayah M, Yajima H,
Remarchuk T, Reents R, Wirz B, Iding H, Bachmann S, Fantasia SM, et
al: Asymmetric synthesis of Akt kinase inhibitor ipatasertib. Org
Lett. 19:4806–4809. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Saura C, Roda D, Roselló S, Oliveira M,
Macarulla T, Pérez-Fidalgo JA, Morales-Barrera R, Sanchis-García
JM, Musib L, Budha N, et al: A first-in-human phase I study of the
ATP-competitive AKT inhibitor ipatasertib demonstrates robust and
safe targeting of AKT in patients with solid tumors. Cancer Discov.
7:102–113. 2017. View Article : Google Scholar :
|
|
142
|
Oeck S, Al-Refae K, Riffkin H, Wiel G,
Handrick R, Klein D, Iliakis G and Jendrossek V: Activating Akt1
mutations alter DNA double strand break repair and
radiosensitivity. Sci Rep. 7:427002017. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Yu L, Liu Z, Qiu L, Hao L and Guo J:
Ipatasertib sensitizes colon cancer cells to TRAIL-induced
apoptosis through ROS-mediated caspase activation. Biochem Biophys
Res Commun. 519:812–818. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Capivasertib active against AKT1-mutated
cancers. Cancer Discov. 9:OF72019. View Article : Google Scholar
|
|
145
|
Nasrazadani A and Brufsky AM: Capivasertib
inhibits a key pathway in metastatic breast cancer. Lancet Oncol.
21:318–319. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yap TA, Kristeleit R, Michalarea V,
Pettitt SJ, Lim JSJ, Carreira S, Roda D, Miller R, Riisnaes R,
Miranda S, et al: Phase I trial of the PARP inhibitor olaparib and
AKT inhibitor capivasertib in patients with BRCA1/2- and
Non-BRCA1/2-mutant cancers. Cancer Discov. 10:1528–1543. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Gills JJ and Dennis PA: Perifosine: Update
on a novel Akt inhibitor. Curr Oncol Rep. 11:102–110. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Fei HR, Chen G, Wang JM and Wang FZ:
Perifosine induces cell cycle arrest and apoptosis in human
hepatocellular carcinoma cell lines by blockade of Akt
phosphorylation. Cytotechnology. 62:449–460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Song Z, Tu X, Zhou Q, Huang J, Chen Y, Liu
J, Lee S, Kim W, Nowsheen S, Luo K, et al: A novel UCHL3
inhibitor, perifosine, enhances PARP inhibitor cytotoxicity through
inhibition of homologous recombination-mediated DNA double strand
break repair. Cell Death Dis. 10:3982019. View Article : Google Scholar
|
|
150
|
Hirai H, Sootome H, Nakatsuru Y, Miyama K,
Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS and
Kotani H: MK-2206, an allosteric Akt inhibitor, enhances antitumor
efficacy by standard chemotherapeutic agents or molecular targeted
drugs in vitro and in vivo. Mol Cancer Ther. 9:1956–1967. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Whicker ME, Lin ZP, Hanna R, Sartorelli AC
and Ratner ES: MK-2206 sensitizes BRCA-deficient epithelial ovarian
adenocarcinoma to cisplatin and olaparib. BMC Cancer. 16:5502016.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Ma Y, Vassetzky Y and Dokudovskaya S:
mTORC1 pathway in DNA damage response. Biochim Biophys Acta Mol
Cell Res. 1865:1293–1311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Danesh Pazhooh R, Rahnamay Farnood P,
Asemi Z, Mirsafaei L, Yousefi B and Mirzaei H: mTOR pathway and DNA
damage response: A therapeutic strategy in cancer therapy. DNA
Repair (Amst). 104:1031422021. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Lamming DW, Ye L, Katajisto P, Goncalves
MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima
RS, et al: Rapamycin-induced insulin resistance is mediated by
mTORC2 loss and uncoupled from longevity. Science. 335:1638–1643.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Arriola Apelo SI and Lamming DW:
Rapamycin: An InhibiTOR of aging emerges from the soil of Easter
Island. J Gerontol A Biol Sci Med Sci. 71:841–849. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Chebel A, Catallo R, Mabon C, Bachy E,
Wenner T, Salles G, Pouteil-Noble C and Ffrench M: Rapamycin
safeguards lymphocytes from DNA damage accumulation in vivo. Eur J
Cell Biol. 95:331–341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Mahran AM, Mosad E, Abdel-Raheem MA, Ahmed
EH, Abdel Motaleb AA and Hofny ER: The correlation between
mammalian target of rapamycin (mTOR) gene expression and sperm DNA
damage among infertile patients with and without varicocele.
Andrologia. 51:e133412019. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Hasskarl J: Everolimus. RecRecent Results
Cancer Res. 211:101–123. 2018. View Article : Google Scholar
|
|
159
|
Beider K, Bitner H, Voevoda-Dimenshtein V,
Rosenberg E, Sirovsky Y, Magen H, Canaani J, Ostrovsky O, Shilo N,
Shimoni A, et al: The mTOR inhibitor everolimus overcomes
CXCR4-mediated resistance to histone deacetylase inhibitor
panobinostat through inhibition of p21 and mitotic regulators.
Biochem Pharmacol. 168:412–428. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Buitrago-Molina LE, Pothiraju D, Lamlé J,
Marhenke S, Kossatz U, Breuhahn K, Manns MP, Malek N and Vogel A:
Rapamycin delays tumor development in murine livers by inhibiting
proliferation of hepatocytes with DNA damage. Hepatology.
50:500–509. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Beuvink I, Boulay A, Fumagalli S,
Zilbermann F, Ruetz S, O'Reilly T, Natt F, Hall J, Lane HA and
Thomas G: The mTOR inhibitor RAD001 sensitizes tumor cells to
DNA-damaged induced apoptosis through inhibition of p21
translation. Cell. 120:747–759. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
El Botty R, Coussy F, Hatem R, Assayag F,
Chateau-Joubert S, Servely JL, Leboucher S, Fouillade C, Vacher S,
Ouine B, et al: Inhibition of mTOR downregulates expression of DNA
repair proteins and is highly efficient against BRCA2-mutated
breast cancer in combination to PARP inhibition. Oncotarget.
9:29587–29600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Mattoo AR, Joun A and Jessup JM:
Repurposing of mTOR complex inhibitors attenuates MCL-1 and
sensitizes to PARP inhibition. Mol Cancer Res. 17:42–53. 2019.
View Article : Google Scholar
|
|
164
|
Kamata T and Feramisco JR: Epidermal
growth factor stimulates guanine nucleotide binding activity and
phosphorylation of ras oncogene proteins. Nature. 310:147–150.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Degirmenci U, Wang M and Hu J: Targeting
aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells.
9:1982020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Crews CM and Erikson RL: Purification of a
murine protein-tyro-sine/threonine kinase that phosphorylates and
activates the Erk-1 gene product: Relationship to the fission yeast
byr1 gene product. Proc Natl Acad Sci USA. 89:8205–8209. 1992.
View Article : Google Scholar
|
|
167
|
Dai Y, Chen S, Pei XY, Almenara JA, Kramer
LB, Venditti CA, Dent P and Grant S: Interruption of the
Ras/MEK/ERK signaling cascade enhances Chk1 inhibitor-induced DNA
damage in vitro and in vivo in human multiple myeloma cells. Blood.
112:2439–2449. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Karoulia Z, Gavathiotis E and Poulikakos
PI: New perspectives for targeting RAF kinase in human cancer. Nat
Rev Cancer. 17:676–691. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Puszkiel A, Noé G, Bellesoeur A, Kramkimel
N, Paludetto MN, Thomas-Schoemann A, Vidal M, Goldwasser F,
Chatelut E and Blanchet B: Clinical pharmacokinetics and
pharmacodynamics of dabrafenib. Clin Pharmacokinet. 58:451–467.
2019. View Article : Google Scholar
|
|
170
|
Jiang Z, Wang H, Li L, Hou Z, Liu W, Zhou
T, Li Y and Chen S: Analysis of TGCA data reveals genetic and
epigenetic changes and biological function of MUC family genes in
colorectal cancer. Future Oncol. 15:4031–4043. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Yuan L, Mishra R, Patel H, Alanazi S, Wei
X, Ma Z and Garrett JT: BRAF mutant melanoma adjusts to BRAF/MEK
inhibitors via dependence on increased antioxidant SOD2 and
increased reactive oxygen species levels. Cancers (Basel).
12:16612020. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Garbe C and Eigentler TK: Vemurafenib.
Recent Results Cancer Res. 211:77–89. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Kimeswenger S, Mann U, Hoeller C,
Foedinger D and Jantschitsch C: Vemurafenib impairs the repair of
ultraviolet radiation-induced DNA damage. Melanoma Res. 29:134–144.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Peacock M, Brem R, Macpherson P and Karran
P: DNA repair inhibition by UVA photoactivated fluoroquinolones and
vemurafenib. Nucleic Acids Res. 42:13714–13722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Kim C and Giaccone G: MEK inhibitors under
development for treatment of non-small-cell lung cancer. Expert
Opin Investig Drugs. 27:17–30. 2018. View Article : Google Scholar
|
|
176
|
Sarkisian S and Davar D: MEK inhibitors
for the treatment of NRAS mutant melanoma. Drug Des Devel Ther.
12:2553–2565. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Jin J, Guo Q, Xie J, Jin D and Zhu Y:
Combination of MEK inhibitor and the JAK2-STAT3 pathway inhibition
for the therapy of colon cancer. Pathol Oncol Res. 25:769–775.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Arend RC, Davis AM, Chimiczewski P,
O'Malley DM, Provencher D, Vergote I, Ghamande S and Birrer MJ: EMR
20006-012: A phase II randomized double-blind placebo controlled
trial comparing the combination of pimasertib (MEK inhibitor) with
SAR245409 (PI3K inhibitor) to pimasertib alone in patients with
previously treated unresectable borderline or low grade ovarian
cancer. Gynecol Oncol. 156:301–307. 2020. View Article : Google Scholar
|
|
179
|
Markham A and Keam SJ: Selumetinib: First
approval. Drugs. 80:931–937. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Holt SV, Logié A, Odedra R, Heier A,
Heaton SP, Alferez D, Davies BR, Wilkinson RW and Smith PD: The
MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances
anti-tumour efficacy when combined with conventional
chemotherapeutic agents in human tumour xenograft models. Br J
Cancer. 106:858–866. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Zeiser R, Andrlová H and Meiss F:
Trametinib (GSK1120212). Recent Results Cancer Res. 211:91–100.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Estrada-Bernal A, Chatterjee M, Haque SJ,
Yang L, Morgan MA, Kotian S, Morrell D, Chakravarti A and Williams
TM: MEK inhibitor GSK1120212-mediated radiosensitization of
pancreatic cancer cells involves inhibition of DNA double-strand
break repair pathways. Cell Cycle. 14:3713–3724. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Roskoski R Jr: Src protein-tyrosine kinase
structure, mechanism, and small molecule inhibitors. Pharmacol Res.
94:9–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Chakraborty G, Patail NK, Hirani R,
Nandakumar S, Mazzu YZ, Yoshikawa Y, Atiq M, Jehane LE, Stopsack
KH, Lee GM, et al: Attenuation of SRC kinase activity augments PARP
inhibitor-mediated synthetic lethality in BRCA2-altered prostate
tumors. Clin Cancer Res. 27:1792–1806. 2021. View Article : Google Scholar
|
|
185
|
Lindauer M and Hochhaus A: Dasatinib.
Recent Results Cancer Res. 212:29–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Peng S, Sen B, Mazumdar T, Byers LA, Diao
L, Wang J, Tong P, Giri U, Heymach JV, Kadara HN and Johnson FM:
Dasatinib induces DNA damage and activates DNA repair pathways
leading to senescence in non-small cell lung cancer cell lines with
kinase-inactivating BRAF mutations. Oncotarget. 7:565–579. 2016.
View Article : Google Scholar :
|
|
187
|
Raju U, Riesterer O, Wang ZQ, Molkentine
DP, Molkentine JM, Johnson FM, Glisson B, Milas L and Ang KK:
Dasatinib, a multi-kinase inhibitor increased radiation sensitivity
by interfering with nuclear localization of epidermal growth factor
receptor and by blocking DNA repair pathways. Radiother Oncol.
105:241–249. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Corrales-Sánchez V, Noblejas-López MDM,
Nieto-Jiménez C, Pérez-Peña J, Montero JC, Burgos M, Galán-Moya EM,
Pandiella A and Ocaña A: Pharmacological screening and
transcriptomic functional analyses identify a synergistic
interaction between dasatinib and olaparib in triple-negative
breast cancer. J Cell Mol Med. 24:3117–3127. 2020. View Article : Google Scholar : PubMed/NCBI
|