Introduction
To date, pancreatic cancer is the third-highest
cause of cancer-related deaths (~48,000) in the U.S (1). Its incidence is gradually increasing,
and it is likely to become the second-highest cause of
cancer-related deaths by 2030 (2).
The treatment of pancreatic cancer includes radical methods such as
surgical resection. However, the prognosis for such treatment
remains poor because pancreatic cancer is usually advanced at the
time of diagnosis. Furthermore, within two years of curative
resection, 80% of patients develop a recurrence of pancreatic
cancer (3).
Since 80% of patients have regional or metastatic
cancer at diagnosis, chemotherapy or radiation therapy are the
primary treatment options (1).
Previous studies have investigated the use of combination
chemotherapy regimens, including gemcitabine and nab-paclitaxel
(GnP) or 5-fluorouracil, oxaliplatin, and irinotecan (FOLFIRINOX).
These regimens have an important role in the treatment of advanced
pancreatic cancer (4,5). Despite these advanced treatments, the
prognosis of advanced pancreatic cancer remains poor. This may be
related to drug resistance (6).
The mechanism of drug resistance is considered to
include various pathological factors specific to pancreatic cancer,
including a peritumor microenvironment containing dense fibrous
stroma and an abnormal vascular network around the tumor (7,8). In
pancreatic cancer, the tumor microenvironment consists of a rigid
extracellular matrix (ECM) formed from collagen I, elastin and
fibronectin (8,9). Pancreatic cancer causes specific
changes in the ECM close to the tumor, resulting in a
microenvironment that promotes tumor growth and multi-organ
metastasis (10,11). Desmoplastic reaction, a large
deposition of ECM, and pancreatic cancer cells have biochemical
effects that increase the interstitial fluid pressure, which
reduces tumor perfusion and inhibits the delivery of antitumor
agents (12-15). This is exacerbated by the lowered
density of the tumor vasculature, reducing the effect of cytotoxic
therapy against pancreatic cancer cells (13).
Ribonucleotide reductase large subunit M1 (RRM1), a
rate-limiting enzyme in the DNA synthesis pathway, requires the
conversion of ribonucleotides to dNTPs (16). RRM1 has an effect on the poor
disease prognosis of several cancers, including pancreatic cancer
(17-19). Furthermore, RRM1 has been
associated with resistance to gemcitabine, an important drug in the
treatment of pancreatic cancer (19-21).
However, the way in which RRM1 is involved in the biology of
pancreatic cancer, particularly related to cell motility, is poorly
understood.
In the present study, the relationship between
epithelial-mesenchymal transition (EMT), motility invasion
capacity, histone acetylation and drug resistance in
gemcitabine-resistant (GEM-R) cell lines was investigated. In
vitro overexpression or siRNA experiments were also performed
to examine the functions of RRM1, concentrating on the relationship
between invasiveness and changes in the expression of factors such
as N-cadherin. It was found that RRM1 contributed to the malignant
phenotype of cancer cells, including increased motility and
invasiveness, particularly during gemcitabine resistance.
Therefore, RRM1 may be a therapeutic target for the treatment of
pancreatic cancer.
Materials and methods
Materials
Anti-RRM1 (1:1,000; D12F12; cat. no. 8637),
anti-acetyl histone H3 (Lys9) (1:1,000; C5B11; cat. no. 9649),
anti-acetyl histone H3 (Lys27) (1:1,000; D5E4; cat. no. 8173),
anti-histone H3 (1:2,000; D1H2; cat. no. 4499) and GAPDH (1:2,000;
D16H11; cat. no. 5174) antibodies were obtained from Cell Signaling
Technology, Inc. Anti-N-cadherin (1:1,000, cat. no. 610920)
antibody was purchased from BD Biosciences. Tenascin-C (TNC)
(1:200; E-9; cat. no. sc-25328) and fibronectin (1:200; P5F3; cat.
no. sc-18827) antibodies were obtained from Santa Cruz
Biotechnology, Inc. Anti-COL11A (1:200; cat. no. ab64883) antibody
was purchased from Abcam. HAT inhibitor C646 (cat. no. SML0002) was
obtained from MilliporeSigma.
Cell cultures
The human pancreatic cancer cell lines MIAPaCa2 and
Panc1 were obtained from the American Type Culture Collection in
August 2016. The cancer cell lines were authenticated by short
tandem repeat analysis for DNA profiling, and all experiments were
performed with mycoplasma-free cells. Cancer cells were maintained
in high-glucose DMEM medium containing 10% FBS (FUJIFILM Wako Pure
Chemical Corporation) and 1% penicillin/streptomycin in a
humidified 5% CO2 chamber at 37°C.
Cell viability assay
Cell numbers were evaluated by WST-8 assay (Cell
Counting Kit-8; Dojindo Molecular Technologies, Inc.), as
previously described (19).
Briefly, 5.0-7.5×103 cells per well were seeded onto
96-well plates and incubated overnight at 37°C. After 72 h of
gemcitabine treatment at each concentration from 1 nM to 10
µM, cell viability was determined according to the
manufacturer's instructions. The absorbance of each well was
measured at 450 nm, using an iMark™ microplate reader (Bio-Rad
Laboratories, Inc.), and was determined to be within the linear
range of the assay.
Establishment of a GEM-R cancer cell
line
GEM-R pancreatic cancer cell subclones were
generated using Panc1 cells in our laboratory, as previously
described (22). Briefly, the
Panc1 cells were serially cultured with exposure to incrementally
increasing gemcitabine concentrations (10-100 nM) for two
months.
Overexpression of RRM1 by stable
transfection
A human RRM1 expression plasmid (pCMV6-RRM1;
NM_001033) was purchased from Origene Technologies, Inc. A total of
1 µg RRM1 of expression vectors or corresponding empty
vectors (pCMV6-Entry) were transfected into Panc1 cells using
Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions onto 12-well plates.
After 72 h of transfection, the cells were incubated in culture
medium containing 800 µg/ml G418. After culturing in the
selection medium containing G418 for two weeks, stably transfected
cells were selected and the expression of RRM1 protein was
confirmed by western blotting.
Western blotting
Western blotting was performed as previously
described (23). Protein bands
were visualized and their intensities quantified using ImageQuant
LAS 4000 mini (GE Healthcare). Western blot analyses were conducted
at least three times with similar results, and representative blots
are presented. A densitometric analysis was performed using
ImageQuant TL (GE Healthcare) to calculate the intensity of each
protein band. Figures for western blotting are cropped and
displayed according to the appropriate molecular weight.
Gene silencing by small interfering
(si)RNA
Loss-of-function analysis was performed using siRNAs
specific for RRM1 (siRRM1#1: cat. no. HSS109388, Invitrogen; Thermo
Fisher Scientific, Inc.; sense, 5′-CCC AGU UAC UGA AUA AGC AGA UCU
U-3′ and antisense, 5′-AAG AUC UGC UUA UUC AGU AAC UGG G-3′) or
scrambled negative control (siNC: #12935300; Stealth RNAi™ Negative
Control Med GC Duplex #2; Invitrogen; Thermo Fisher Scientific,
Inc.). An alternative sequence of siRNA targeting RRM1 (siRRM1#2;
cat. no. HSS184469; Invitrogen; Thermo Fisher Scientific, Inc.;
sense, 5′-CAG AAG CUU UGU UAU GGA CUC AAU A-3′ and antisense,
5′-UAU UGA GUC CAU AAC AAA GCU UCU G-3′) was also used. Each siRNA
(20 nM) was transfected into pancreatic cancer cells using
Lipofectamine RNA iMAX (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Cancer cells
transfected with each siRNA were cultured at 37°C for 72 h. The
knockdown of RRM1 was confirmed by western blotting.
Reverse transcription-quantitative PCR
(RT-qPCR)
Extracted RNA using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) was reverse transcribed into
first-strand cDNA using SuperScript VILO cDNA Synthesis kits
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The expression of mRNA was determined
using the following TaqMan Gene Expression Assays (Applied
Biosystems; Thermo Fisher Scientific, Inc.): CDH2, Hs00983056_m1;
COL11A, Hs01097664_m1; and TNC, Hs01115665_m1. Gene expression
values are presented as ratios between genes of interest and an
internal reference gene (Hs99999901_s1 for eukaryotic 18S). These
were then normalized against the value for the control (relative
expression level) and analyzed by the comparative 2−ΔΔCq
method (24). Each assay was
performed in duplicate for each sample.
Clonogenic assay
Long-term cell survival was evaluated by clonogenic
assays, as previously described (25). Cells were seeded at
1.0×103 per well into six-well plates, in triplicate.
After overnight incubation, adherent cells were treated with
gemcitabine for 24 h, at which point the culture medium was changed
to fresh medium without gemcitabine. Cells were incubated for at
least one week, and colonies were stained with a 0.3% crystal
violet solution for 10 min at room temperature. A cluster of 50 or
more stained cells was counted as one colony.
Transwell migration and invasion
assays
Transwell migration and invasion assays were
performed in 24-well modified Boyden chambers precoated already
with (invasion) or without (migration) Matrigel (Transwell chamber;
BD Biosciences) with 8-µm pore size membrane of Transwell
chambers. Pancreatic cancer cells (Panc1, 5×104 cells;
and MIAPaCa2, 7.5×104 cells per well) in serum-free
medium were seeded onto the trans-membrane in the upper chamber,
with 10% FBS in the lower chamber as a chemoattractant. After a
24-h incubation, cells that had migrated through the membrane were
fixed and stained with a Diff-Quik Stain Set (Siemens AG) for 10
min at room temperature and counted under magnification (×100) in
five randomly selected high-power fields. Each assay was performed
in triplicate.
Scratch wound-healing assay
Cancer cells were cultured in six-well plates until
confluent. Confluent cell layers were carefully scratched using a
sterile 200-µl tip, washed twice with fresh medium, and
cultured for 24 h. Images of the scratched cell layers were
acquired with a phase contrast microscope linked to a charge
coupled device camera, and the wound area was evaluated.
Cell adhesion assay
Cell adhesion assays were performed using the
CytoSelect 48-Well Cell Adhesion Assay (ECM array; CBA-070,
CellBiolabs) according to the manufacturer's protocol. Cancer cells
(1.0×105 cells) were plated in triplicate on a precoated
plate (Fibronectin, Collagen I, Collagen IV and Laminin I). After
60 min of incubation at 37°C, adherent cells were fixed and stained
by incubating for 10 min at room temperature. The stained solution
was then removed and quantified colorimetrically at 560 nm, using a
plate reader.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assaying was
performed using a ChIP-IT Express Kit (Active Motif, Inc.)
according to the manufacturer's protocol, as previously described
(26). Cells were fixed with 1%
formaldehyde/PBS for 10 min. DNA was sonicated to 500-1,000 bp for
all experiments. Immunoprecipitated DNA enrichment was normalized
to the input. The antibodies used were anti-H3K9ac (1:50; cat. no.
9649), anti-H3K27ac (1:50; cat. no. 8173; both from Cell Signaling
Technology, Inc.), and anti-histone H3 (1 µg; cat. no.
07-690; MilliporeSigma). Normal rabbit IgG (1:50; cat. no. 2729;
Cell Signaling Technology, Inc.) was used as a negative control for
each assay. The primer set for qPCR was as follows: RPM1 sense,
5′-GCC TCT GCT CTG AAG AAA GTG-3′ and antisense, GAC AGA GTG CGA
AGG GTT AGG-3′.
Immunofluorescence staining
Cells were cultured on four-chamber CultureSlides
(Becton, Dickinson and Company). The cells were then fixed in 4%
formaldehyde in PBS for 15 min, blocked with blocking buffer (PBS
with 5% BSA and 0.2% Triton X-100) for 1 h, and incubated overnight
with the primary antibody at 4°C (anti-N-cadherin; 1:500). Alexa
Fluor 488-conjugated goat anti-mouse IgG antibodies (1:1,000; cat.
no. 4408; Cell Signaling Technology, Inc.) were used as secondary
antibodies. The cells were viewed under a fluorescence microscope
(BZ-700; Keyence Corporation).
RNA sequencing
Total RNA was quantified and qualified using a Qubit
RNA Assay (Thermo Fisher Scientific, Inc.) and TapeStation RNA
ScreenTape (Agilent Technologies, Inc.). cDNA synthesis followed by
transcriptome libraries were performed using a NEBNext Ultra II
Directional RNA Library Prep Kit for Illumina (New England BioLabs,
Inc.), where dUTP was incorporated during second strand cDNA
synthesis, instead of dTTP, which blocks PCR amplification against
the second strand templates, enabling strand-specific library
preparation. The resulting transcriptome sequencing libraries were
quantified by Qubit DNA Assay (Thermo Fisher Scientific, Inc.) and
their fragment size distributions were confirmed by TapeStation
D1000 ScreenTape (Agilent). The transcriptome libraries established
were loaded onto a next-generation sequencing platform, HiSeq 4000
or equivalent (Illumina, Inc.). Sequencing was performed according
to the manufacturer's instructions. Image analysis and base calling
were performed using software on the HiSeq instrument. GOSeq
(27) and TopGO software
(https://bioconductor.org/packages/release/bioc/html/topGO.html)
were utilized in the Gene Ontology (GO) enrichment analysis for
differentially expressed genes. All processes were conducted at
Azenta Life Sciences (formerly GENEWIZ).
Statistical analysis
The drawing of figures, fitting of curves,
IC50 calculations and statistical analyses were
performed with GraphPad Prism 7 software (GraphPad Software, Inc.).
Unless otherwise specified, independent experiments were conducted
in triplicate and the values presented are their means, compared
using the Student's t-test for single comparison or a one-way ANOVA
with a Tukey's post hoc test and a two-way ANOVA with Sidak's post
hoc test for multiple comparisons, as appropriate. P<0.05 was
considered to indicate a statistically significant difference.
Results
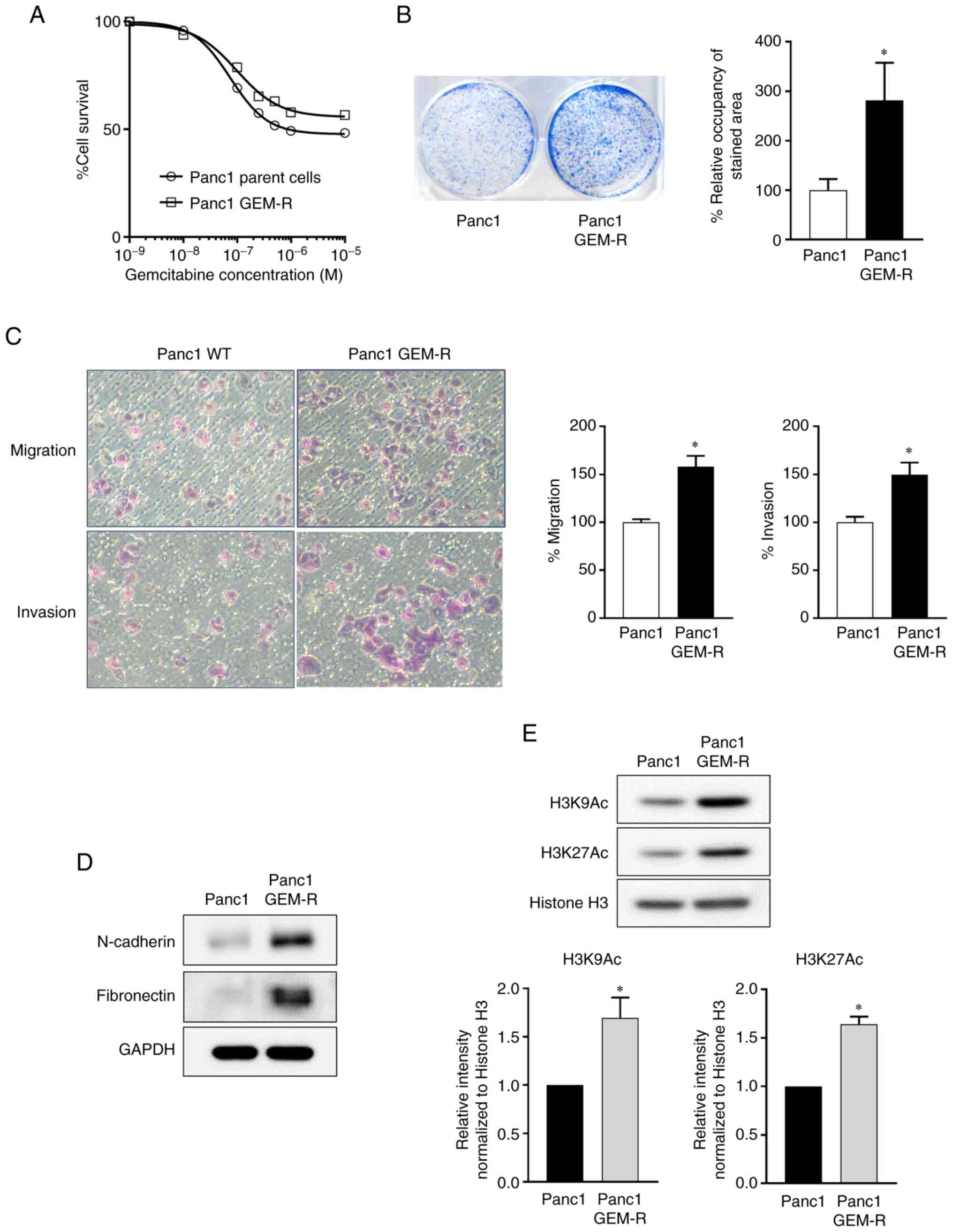
Cell motility and histone H3 acetylation
are increased in GEM-R Panc1 cells
During the acquisition of drug resistance, cancer
cells refractory to drugs are considered to be involved in a
variety of malignant processes, including the acquiring of stem
cell properties, increased motility, and the induction of EMT.
GEM-R Panc1 cells were generated and their effects on cancer
motility, including migration and invasion, and their relevance to
EMT and drug resistance were evaluated.
The IC50 of the parent cells was 75 nM at
72 h, whereas it was 100 nM for GEM-R Panc1 cells, 1.3 times higher
than the parent cells (Fig. 1A). A
colony formation assay for gemcitabine treatment revealed a
2.8-fold increase in the length of cell survival (Fig. 1B), demonstrating that Panc1 cells
had acquired drug resistance to gemcitabine.
Panc1 GEM-R cells exhibited significantly enhanced
motility in migration and invasion assays, compared with control
cells (migration: 1.6-fold; invasion: 1.5-fold; P<0.05 in both
cases) (Fig. 1C). The expression
levels of N-cadherin and fibronectin, which are mesenchymal markers
in EMT, were upregulated (Fig.
1D). Notably, histone H3 acetylation was enhanced in Panc1
GEM-R cells (H3K9Ac:1.7-fold; H3K27Ac: 1.6-fold; P<0.05 in both
cases) (Fig. 1E). Thus, Panc1
cells that acquired gemcitabine resistance showed enhanced drug
resistance, increased migration and invasion, EMT and histone H3
acetylation.
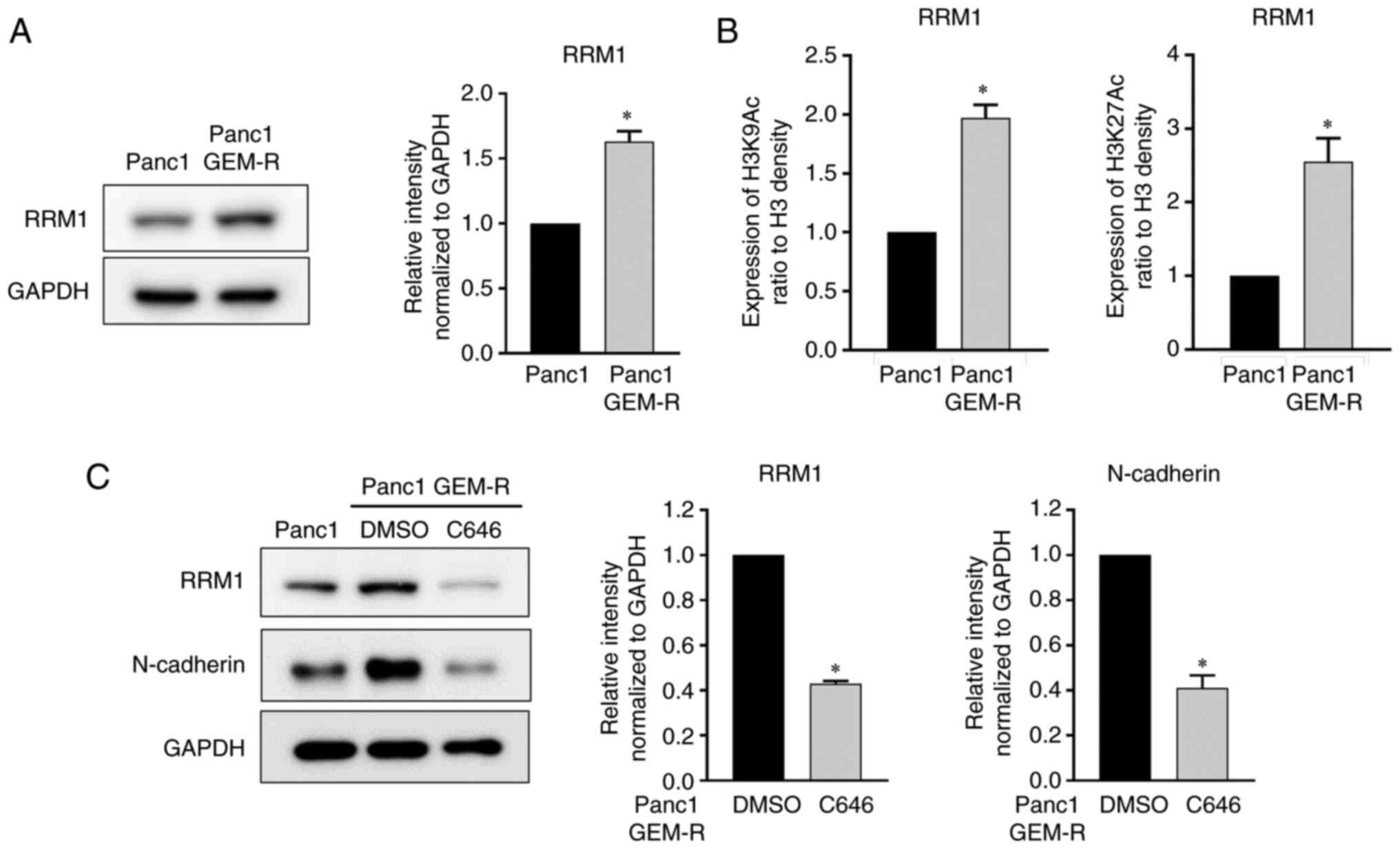
Increased RRM1 expression in GEM-R Panc1
cells is regulated by histone acetylation
The RRM1 gene is upregulated when gemcitabine
resistance is acquired, and may be associated with a worse
prognosis in pancreatic cancer, as previously reported by the
authors (19,20). Indeed, the protein level of RRM1
was increased 1.6-fold in the Panc1 GEM-R cell line (Fig. 2A). Next, the relationship between
upregulated histone H3 acetylation in cells with gemcitabine
resistance and the increased expression of RRM1 was examined.
Chip-PCR experiments showed that the RRM1 gene transcription level
was significantly enhanced by H3K9 and H3K27 acetylation,
indicating that RRM1 transcriptional activity was partly regulated
by histone H3 acetylation (Fig.
2B).
The increased expression levels of RRM1 and
N-cadherin as well as histone H3 acetylation were observed in the
GEM-R cells. Notably, C646, a histone acetylation inhibitor,
inhibited histone H3 acetylation at concentrations sufficient to
inhibit the acetylation of histone H3 (Fig. S1) and concurrently decreased the
expression of RRM1 to 43%, compared with controls. Furthermore,
histone acetylation inhibition reduced N-cadherin protein levels to
41%, compared with controls (Fig.
2C).
The aforementioned findings indicated that histone
H3 acetylation was upregulated after the acquisition of gemcitabine
resistance, leading to the increased expression of RRM1. N-cadherin
was also involved in histone acetylation.
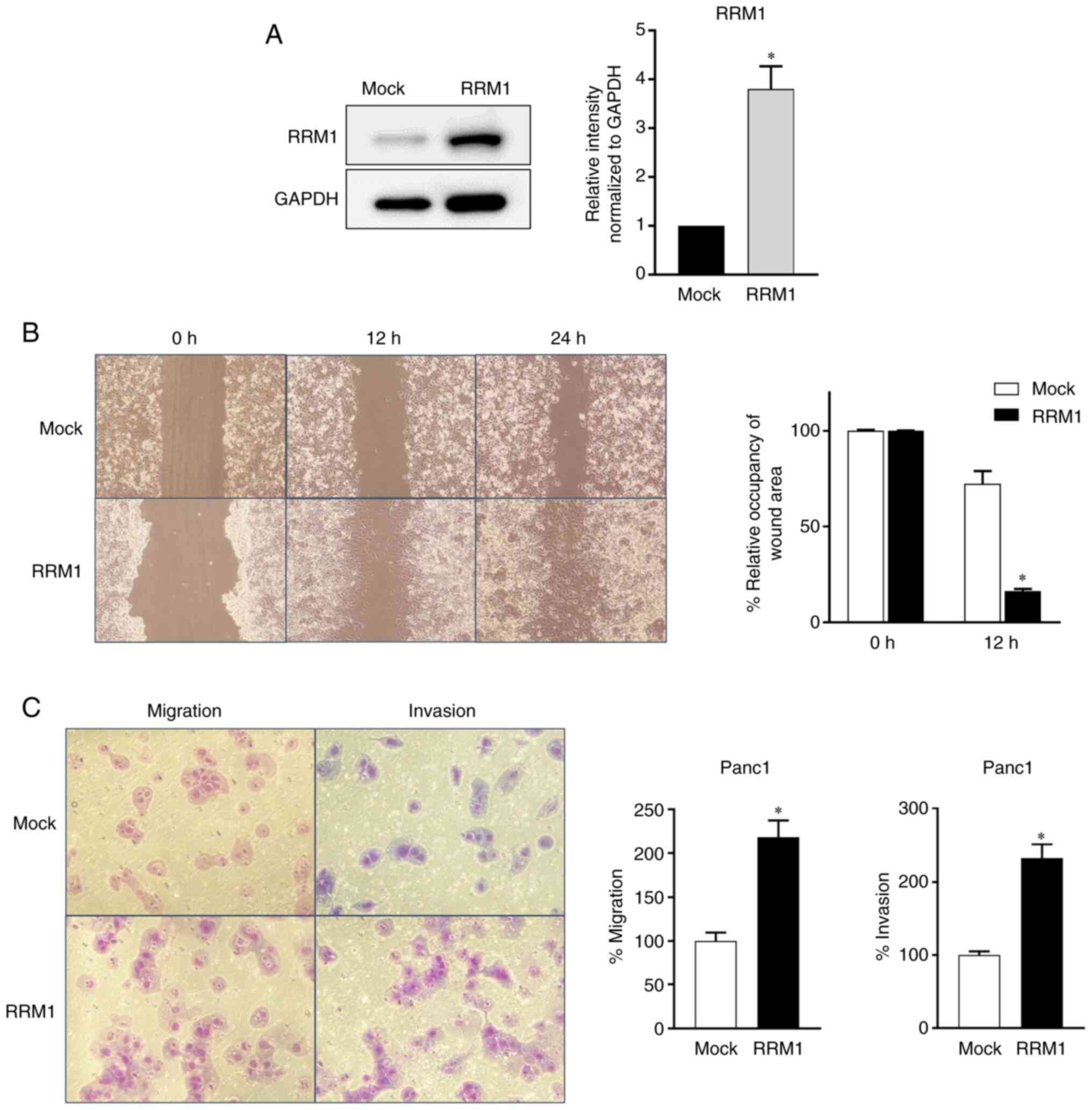
RRM1 expression is associated with the
migration and invasion of pancreatic cancer cells
Next, the effect of increased RRM1 expression on
motility, including migration and invasion, was investigated. A
Panc1 cell line overexpressing RRM1 was generated, which was 3.8
times more active than the parent cell line, as revealed by western
blotting (Fig. 3A). The increased
RRM1 expression significantly enhanced the migration ability of
Panc1 cells in a wound-healing assay, compared with normal
counterparts (Fig. 3B), and their
migration and invasion abilities were significantly increased, 2.2
and 2.3-fold respectively, in a chamber assay (Fig. 3C).
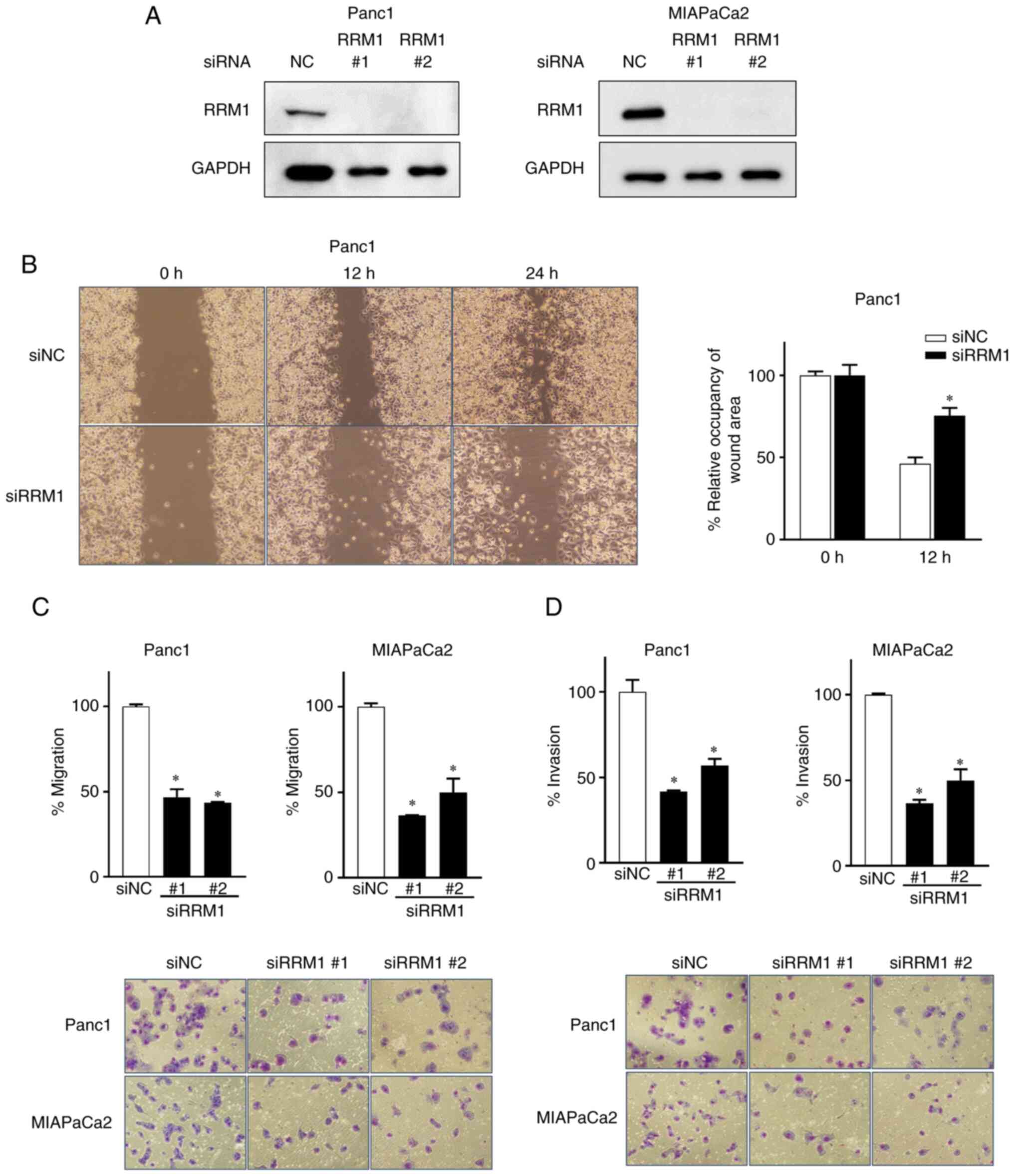
By contrast, the functional suppression of
endogenous RRM1 levels in MIAPaCa2 and Panc1 cells by siRNA
suppressed the motility of cancer cells. A total of two different
RRM1 siRNAs were used and it was found that the downregulation of
RRM1 expression significantly reduced the migration and invasion of
MIAPaCa2 and Panc1 cells, as assessed by wound-healing assays
(Fig. 4A and B). Therefore, RRM1
may promote the malignant transformation of cancer cells by
enhancing their motility. Furthermore, the suppression of RRM1
reduced the levels of N-cadherin (Fig. S2) and the migratory and invasive
capacity of cancer cell lines (Fig. 4C
and D). Therefore, the RRM1/N-cadherin axis may be associated
with the acquisition of malignant traits by pancreatic cancer
cells.
RRM1 overexpression increases the
expression of ECM-related genes
Next, the effects of RRM1 overexpression were
compared with empty vector transfected in Panc1 cells, using RNA
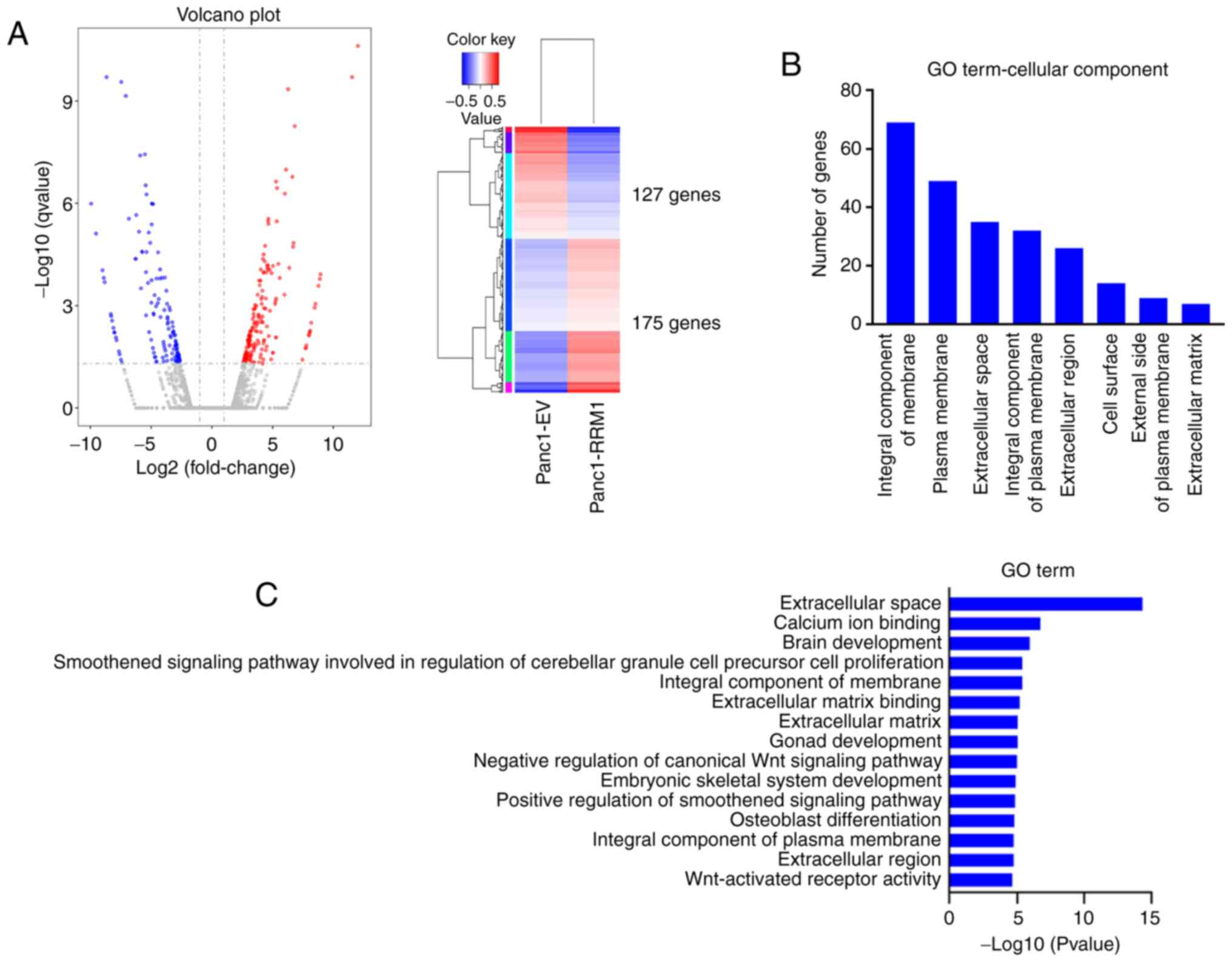
sequencing. A total of 302 statistically significant altered genes
were identified. A volcano plot and heat map showing the 302 genes
significantly associated with RRM1 overexpression also revealed
that more upregulated than downregulated genes (log2FC; 2.5) were
observed (Fig. 5A). RRM1
overexpression resulted in 175 upregulated genes and 127
downregulated genes.
Using GO analysis, with a focus on cellular
components, the top differentially-expressed hallmark gene sets
enriched in RRM1-overexpressing Panc1 cells were analyzed. RRM1
induced genes that encoded proteins that are an integral component
of membranes, plasma membranes and the extracellular space
(Fig. 5B). A group of 14 genes
associated with the extracellular space had the most significant
expression changes, compared with RRM1 expression. Other notable
expression changes were related to the cytoskeleton, including the
ECM and ECM binding (Fig. 5C). The
expression of N-cadherin was also upregulated in this gene set.
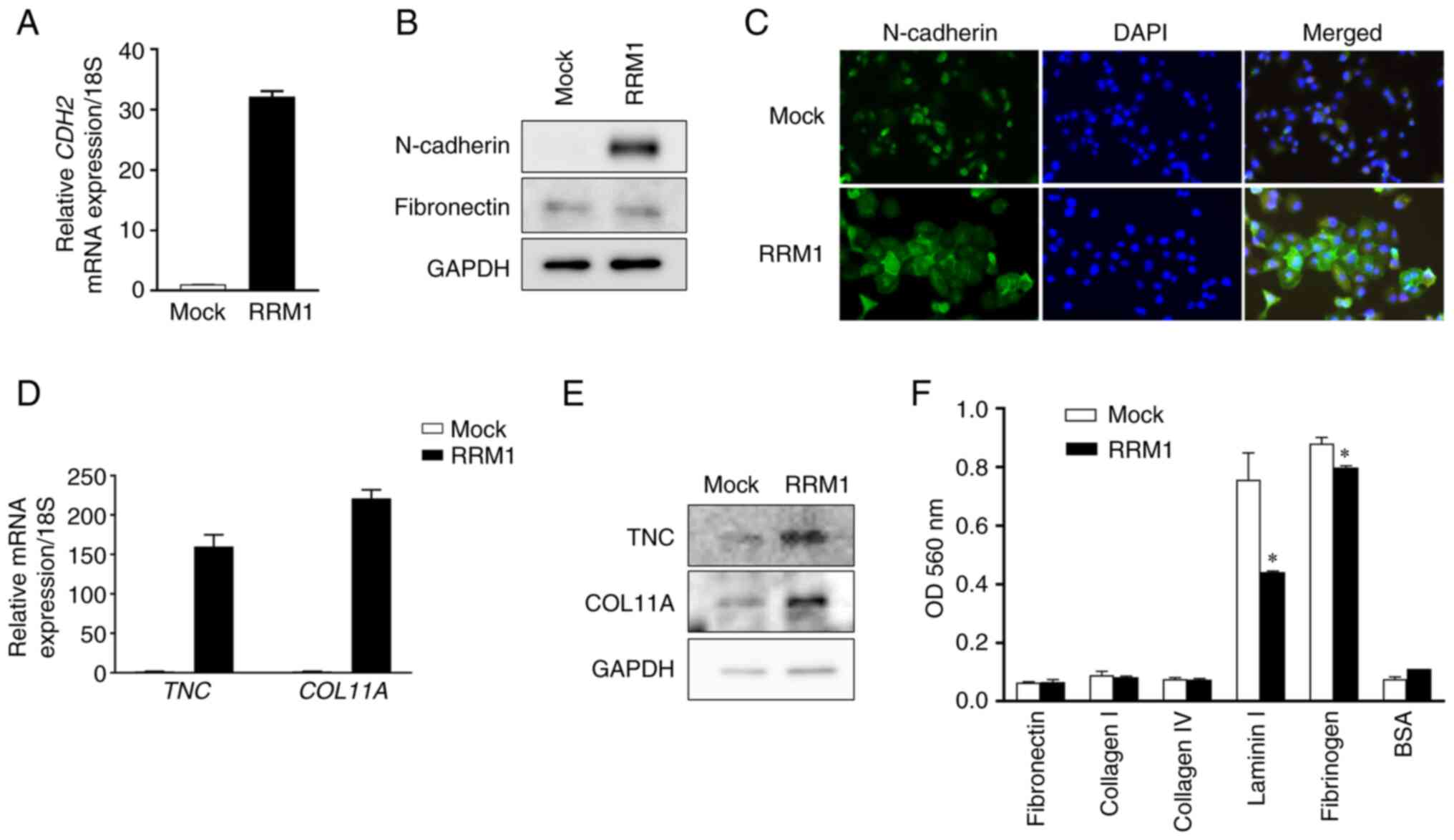
The increased N-cadherin mRNA expression after RRM1
overexpression was confirmed by RT-qPCR. RRM1 overexpression also
increased N-cadherin protein levels. N-cadherin expression changes
were confirmed by western blotting and fluorescence staining.
However, there was no change in the expression of fibronectin
(Fig. 6A-C).
In addition to N-cadherin, the increased expression
of RRM1 altered the expression levels of genes involved in the ECM,
including TNC and COL11A (Fig. 6D and
E). Finally, to evaluate the relevance of EMT, relative cell
attachment activity was assessed by an adhesion assay (28). The increased expression of RRM1
decreased the adhesion of cells to substrates, including laminin
and fibrinogen, and induced EMT, which may contribute to the
increased motility and malignant transformation of cancer cells
(Fig. 6F). It was previously
reported by the authors that cytoplasmic RRM1 activation was
associated with gemcitabine resistance (19). In the present study, a gene
expression analysis approach was used to confirm that RRM1
activation was characterized by changes in the cytoplasmic skeleton
associated with remodeling of the ECM.
Discussion
Gemcitabine is a key drug in the treatment of
pancreatic cancer. However, the molecular mechanisms involved in
the susceptibility of cells to gemcitabine are poorly understood
although its pharmacological effects are clear (16). Detailed analyses of the global
molecular mechanisms of gemcitabine resistance have shown that the
acquisition of drug resistance by cancer cells is associated with a
variety of phenotypic changes (29).
If cancer cells survive their exposure to anticancer
drugs, various molecular programs are initiated during the
acquisition of drug resistance, leading to the promotion of
malignant properties by EMT (30,31).
The current study confirmed findings of previous
studies that cells resistant to gemcitabine, a key drug in the
treatment of pancreatic cancer, induced EMT and promoted the
migration and invasion of cancer cells (31,32).
When the GEM-R strain was created, it was judged that a 2-month
treatment period was appropriate to generate GEM-R cells, based on
previous studies (22,33). Our GEM-R Panc1 cells were less
altered than those reported in previous studies. However, as
revealed in the present study, the newly generated GEM-R cell line
does demonstrate GEM-R traits, including increased expression of
EMT markers such as N-cadherin and fibronectin and increased motor
invasiveness, indicating that gemcitabine exposure does indeed
induce malignant traits. One additional finding was that histone H3
acetylation was necessary for the acquisition of drug resistance to
gemcitabine. It was previously reported by the authors that p300, a
histone acetylation cofactor, bound to chromatin during the acute
phase of gemcitabine exposure, and that its suppression enhanced
the sensitivity of pancreatic cancer cells to gemcitabine (25). Meidhof et al (34) reported that HDAC inhibitors induced
MET and enhanced gemcitabine sensitivity, which is consistent with
our finding that increased histone acetylation is associated with
resistance to gemcitabine.
RRM1 is an enzyme necessary for the conversion of
ribonucleotides to deoxyribonucleosides, and may be involved in DNA
synthesis and repair. A previous study reported that RRM1 gene
expression was correlated with worse prognosis in various cancers
and was associated with gemcitabine resistance (17-19).
The survival rate of pancreatic cancer patients with high RRM1
expression treated with gemcitabine was significantly lower than
that of patients with low RRM1 expression (18,19).
A meta-analysis of pancreatic cancer prognosis in patients treated
with gemcitabine indicated the prognostic relevance of RRM1 for
pancreatic cancer (20). However,
the functional importance of RRM1 is poorly understood. The current
study investigated the association of RRM1, which is upregulated in
GEM-R cells, with cancer cell migration and invasion. It was found
that the stable expression of RRM1 increased motility-related
traits such as invasion.
Wang et al (17) reported that RRM1 was present in the
cytoplasm and nucleus of gastric cancer cells. During serum
deprivation it was predominant in the cytoplasm, and upon serum
repletion it translocated to the nucleus, confirming the functional
changes of RRM1 under certain stress signals, such as hypoxia. It
was previously reported that RRM1 cytoplasmic expression was
upregulated further after exposure to gemcitabine (19). In the current study, it was
identified that the functional significance of the increased
cytoplasmic expression of RRM1 is associated with global changes in
cellular matrix remodeling, including the induction of EMTs
including N-cadherin, TNC and COL11A.
The association of N-cadherin with motility has been
reported in numerous carcinomas. The suppression of RRM2 in lung
cancer cells reduced their migration and invasion, similar to the
reduction of N-cadherin expression (35). RRM1 expression in cervical cancer
was associated with EMT by upregulating N-cadherin, which altered
the cytoplasmic expression of p53 (36). p53R2, a ribonuclease
subunit-related protein, binds to RRM1. Its inhibition in cervical
cancer cells induced EMT and increased the expression of N-cadherin
through the Akt signaling pathway (37). The association of N-cadherin to
prognosis was also reported in pancreatic cancer (38).
Global changes in the ECM, including N-cadherin and
the cytoskeleton, may enhance the motility of cancer cells. COL11A
is upregulated in pancreatic cancer and was correlated with
numerous clinical parameters as well as increased motility and
invasiveness via EMT (39).
Similarly, TNC is upregulated in pancreatic cancer and contributes
to enhanced motility and invasion (40,41).
Furthermore, COL11A and TNC were associated with resistance to
gemcitabine by modulating BAX/BCL-2 functions and activating
ERK/NF-κB signaling, respectively (42,43).
These ECM-related genes were previously reported to be associated
with the prognosis of pancreatic cancer patients (44,45),
indicating that RRM1 may be a master gene located upstream of these
genes and involved in regulating their expression. Thus, the
mechanisms involved and how they regulate the expression levels of
ECM genes require further study.
When the HAT inhibitor C646 was used to inhibit
increased histone acetylation in Panc1 GEM-resistant cells and
migration and invasion assays were performed, the migratory ability
of these cells was not affected. This result is considered a
limitation to the present study. The main reason for this result is
that C646 comprehensively suppresses histone acetylation, which has
certain effects on the regulation of gene expression, and thus
affects the expression of various genes other than the RRM1 gene,
meaning that its effects on motor activity such as migration and
invasion cannot be accurately assessed. In the current study, only
the motor migration activity associated with the RRM1 gene was
investigated.
As aforementioned, RRM1 expression has been shown to
be associated with worse prognosis in pancreatic cancer (19). However, the biological significance
of the RRM1 gene has not yet been clarified. In the present study,
the biological significance of RRM1 expression associated with cell
motility was reported. It was demonstrated that RRM1 transition is
activated by histone H3 acetylation, and induces activation of
biologically significant related genes such as N-cadherin, TNC, and
COL11A, which involves ECM remodeling. Therefore, RRM1 is critical
for the acquisition of a malignant phenotype, including increased
invasiveness, in pancreatic cancer cells. It could not be
demonstrated that the expression levels of ECM genes are regulated
by histone acetylation. It is possible that ECM expression may be
regulated by downstream factors of RRM1 signaling, such as through
some transcription factors.
Supplementary Data
Availability of data and materials
The data generated in the present study are
available from the corresponding author on reasonable request and
may be found in the DDBJ Sequenced Read Archive repository
(https://www.ddbj.nig.ac.jp/dra/index-e.html) with
accession number DRA015584.
Authors' contributions
HO conceived the study, performed the experiments
and wrote and edited the manuscript. YM, TK and YA performed the
experiments. SW, HY and KO were involved in data curation. DA, YI,
HU and KA contributed to the conception of the study and analysis
and interpretation of data. AK, ST, and MT supervised and directed
the research and edited the manuscript. YM, TK and MT confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by JSPS KAKENHI Grant-in-Aid for
Scientific Research (C) (grant. no. 21K08793).
Abbreviations:
|
RRM1
|
ribonucleotide reductase large subunit
M1
|
|
TME
|
tumor microenvironment
|
|
ECM
|
extracellular matrix
|
|
EMT
|
epithelial-mesenchymal transition
|
|
TNC
|
Tenascin-C
|
References
|
1
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kleeff J, Reiser C, Hinz U, Bachmann J,
Debus J, Jaeger D, Friess H and Büchler MW: Surgery for recurrent
pancreatic ductal adenocarcinoma. Ann Surg. 245:566–572. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang C, Liu B, Xu X, Zhuang B, Li H, Yin
J, Cong M, Xu W and Lu A: Toward targeted therapy in
chemotherapy-resistant pancreatic cancer with a smart triptolide
nanomedicine. Oncotarget. 7:8360–8372. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murakami T, Hiroshima Y, Matsuyama R,
Homma Y, Hoffman RM and Endo I: Role of the tumor microenvironment
in pancreatic cancer. Ann Gastroenterol Surg. 3:130–137. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ferrara B, Pignatelli C, Cossutta M, Citro
A, Courty J and Piemonti L: The extracellular matrix in pancreatic
cancer: Description of a complex network and promising therapeutic
options. Cancers (Basel). 13:44422021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cukierman E and Bassi DE:
Physico-mechanical aspects of extracellular matrix influences on
tumorigenic behaviors. Semin Cancer Biol. 20:139–145. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pickup MW, Mouw JK and Weaver VM: The
extracellular matrix modulates the hallmarks of cancer. EMBO Rep.
15:1243–1253. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Laklai H, Miroshnikova YA, Pickup MW,
Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer
DD, Mouw JK, et al: Genotype tunes pancreatic ductal adenocarcinoma
tissue tension to induce matricellular fibrosis and tumor
progression. Nat Med. 22:497–505. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of Hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jacobetz MA, Chan DS, Neesse A, Bapiro TE,
Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, et
al: Hyaluronan impairs vascular function and drug delivery in a
mouse model of pancreatic cancer. Gut. 62:112–120. 2013. View Article : Google Scholar
|
|
15
|
Weniger M, Honselmann KC and Liss AS: The
extracellular matrix and pancreatic cancer: A complex relationship.
Cancers (Basel). 10:3162018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ueno H, Kiyosawa K and Kaniwa N:
Pharmacogenomics of gemcitabine: Can genetic studies lead to
tailor-made therapy? Br J Cancer. 97:145–151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Q, Liu X, Zhou J, Huang Y, Zhang S,
Shen J, Loera S, Yuan X, Chen W, Jin M, et al: Ribonucleotide
reductase large subunit M1 predicts poor survival due to modulation
of proliferative and invasive ability of gastric cancer. PLoS One.
8:e701912013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie H, Jiang W, Jiang J, Wang Y, Kim R and
Liu X and Liu X: Predictive and prognostic roles of ribonucleotide
reductase M1 in resectable pancreatic adenocarcinoma. Cancer.
119:173–181. 2013. View Article : Google Scholar
|
|
19
|
Kato T, Ono H, Fujii M, Akahoshi K, Ogura
T, Ogawa K, Ban D, Kudo A, Tanaka S and Tanabe M: Cytoplasmic RRM1
activation as an acute response to gemcitabine treatment is
involved in drug resistance of pancreatic cancer cells. PLoS One.
16:e02529172021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han QL, Zhou YH, Lyu Y, Yan H and Dai GH:
Effect of ribonucleotide reductase M1 expression on overall
survival in patients with pancreatic cancer receiving gemcitabine
chemotherapy: A literature-based meta-analysis. J Clin Pharm Ther.
43:163–169. 2018. View Article : Google Scholar
|
|
21
|
Nakano Y, Tanno S, Koizumi K, Nishikawa T,
Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T and Kohgo
Y: Gemcitabine chemoresistance and molecular markers associated
with gemcitabine transport and metabolism in human pancreatic
cancer cells. Br J Cancer. 96:457–463. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ono H, Basson MD and Ito H: PTK6
potentiates gemcitabine-induced apoptosis by prolonging S-phase and
enhancing DNA damage in pancreatic cancer. Mol Cancer Res.
13:1174–1184. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ono H, Basson MD and Ito H: PTK6 promotes
cancer migration and invasion in pancreatic cancer cells dependent
on ERK signaling. PLoS One. 9:e960602014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Ono H, Basson MD and Ito H: P300
inhibition enhances gemcitabine-induced apoptosis of pancreatic
cancer. Oncotarget. 7:51301–51310. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watanabe S, Shimada S, Akiyama Y, Ishikawa
Y, Ogura T, Ogawa K, Ono H, Mitsunori Y, Ban D, Kudo A, et al: Loss
of KDM6A characterizes a poor prognostic subtype of human
pancreatic cancer and potentiates HDAC inhibitor lethality. Int J
Cancer. 145:192–205. 2019. View Article : Google Scholar
|
|
27
|
Young MD, Wakefield MJ, Smyth GK and
Oshlack A: Gene ontology analysis for RNA-seq: Accounting for
selection bias. Genome Biol. 11:R142010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou B, Guo W, Sun C, Zhang B and Zheng F:
Linc00462 promotes pancreatic cancer invasiveness through the
miR-665/TGFBR1-TGFBR2/SMAD2/3 pathway. Cell Death Dis. 9:7062018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang G, Guan W, Cao Z, Guo W, Xiong G,
Zhao F, Feng M, Qiu J, Liu Y, Zhang MQ, et al: Integrative genomic
analysis of gemcitabine resistance in pancreatic cancer by
patient-derived xenograft models. Clin Cancer Res. 27:3383–3396.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Arumugam T, Ramachandran V, Fournier KF,
Wang H, Marquis L, Abbruzzese JL, Gallick GE, Logsdon CD, McConkey
DJ and Choi W: Epithelial to mesenchymal transition contributes to
drug resistance in pancreatic cancer. Cancer Res. 69:5820–5828.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Li Y, Kong D, Banerjee S, Ahmad A,
Azmi AS, Ali S, Abbruzzese JL, Gallick GE and Sarkar FH:
Acquisition of epithelial-mesenchymal transition phenotype of
gemcitabine-resistant pancreatic cancer cells is linked with
activation of the notch signaling pathway. Cancer Res.
69:2400–2407. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang R, Cheng L, Xia J and Wang Z, Wu Q
and Wang Z: Gemcitabine resistance is associated with
epithelial-mesenchymal transition and induction of HIF-1α in
pancreatic cancer cells. Curr Cancer Drug Targets. 14:407–417.
2014. View Article : Google Scholar
|
|
33
|
Duxbury MS, Ito H, Zinner MJ, Ashley SW
and Whang EE: Inhibition of SRC tyrosine kinase impairs inherent
and acquired gemcitabine resistance in human pancreatic
adenocarcinoma cells. Clin Cancer Res. 10:2307–2318. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meidhof S, Brabletz S, Lehmann W, Preca
BT, Mock K, Ruh M, Schüler J, Berthold M, Weber A, Burk U, et al:
ZEB1-associated drug resistance in cancer cells is reversed by the
class I HDAC inhibitor mocetinostat. EMBO Mol Med. 7:831–847. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang X, Li Y, Zhang N, Gao Y, Han L, Li
S, Li J, Liu X, Gong Y and Xie C: RRM2 silencing suppresses
malignant phenotype and enhances radiosensitivity via activating
cGAS/STING signaling pathway in lung adenocarcinoma. Cell Biosci.
11:742021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wen D, Huang Z, Li Z, Tang X, Wen X, Liu J
and Li M: LINC02535 co-functions with PCBP2 to regulate DNA damage
repair in cervical cancer by stabilizing RRM1 mRNA. J Cell Physiol.
235:7592–7603. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang C, Xu R, Li XX, Wang YY, Liang WQ,
Zeng JD, Zhang SS, Xu XY, Yang Y, Zhang MY, et al: p53R2
overexpression in cervical cancer promotes AKT signaling and EMT,
and is correlated with tumor progression, metastasis and poor
prognosis. Cell Cycle. 16:1673–1682. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakajima S, Doi R, Toyoda E, Tsuji S, Wada
M, Koizumi M, Tulachan SS, Ito D, Kami K, Mori T, et al: N-cadherin
expression and epithelial-mesenchymal transition in pancreatic
carcinoma. Clin Cancer Res. 10:4125–4133. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang H, Zhou H, Ni H and Shen X:
COL11A1-driven epithelial-mesenchymal transition and stemness of
pancreatic cancer cells induce cell migration and invasion by
modulating the AKT/GSK-3β/snail pathway. Biomolecules. 12:3912022.
View Article : Google Scholar
|
|
40
|
Paron I, Berchtold S, Vörös J, Shamarla M,
Erkan M, Höfler H and Esposito I: Tenascin-C enhances pancreatic
cancer cell growth and motility and affects cell adhesion through
activation of the integrin pathway. PLoS One. 6:e216842011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Juuti A, Nordling S, Louhimo J, Lundin J
and Haglund C: Tenascin C expression is upregulated in pancreatic
cancer and correlates with differentiation. J Clin Pathol.
57:1151–1155. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang H, Ren R, Yang Z, Cai J, Du S and
Shen X: The COL11A1/Akt/CREB signaling axis enables
mitochondrial-mediated apoptotic evasion to promote chemoresistance
in pancreatic cancer cells through modulating BAX/BCL-2 function. J
Cancer. 12:1406–1420. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shi M, He X, Wei W, Wang J, Zhang T and
Shen X: Tenascin-C induces resistance to apoptosis in pancreatic
cancer cell through activation of ERK/NF-κB pathway. Apoptosis.
20:843–857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Park H, Lee Y, Lee H, Kim JW, Hwang JH,
Kim J, Yoon YS, Han HS and Kim H: The prognostic significance of
cancer-associated fibroblasts in pancreatic ductal adenocarcinoma.
Tumour Biol. 39:10104283177184032017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Leppänen J, Lindholm V, Isohookana J,
Haapasaari KM, Karihtala P, Lehenkari PP, Saarnio J, Kauppila JH,
Karttunen TJ, Helminen O and Huhta H: Tenascin C, fibronectin, and
tumor-stroma ratio in pancreatic ductal adenocarcinoma. Pancreas.
48:43–48. 2019. View Article : Google Scholar
|