Introduction
According to Global Cancer Incidence, Mortality and
Prevalence (GLOBOCAN) 2020, hepatocellular carcinoma (HCC) is the
third highest cause of cancer-related mortality globally, and a
devastating disease with a high prevalence and unsatisfactory
prognosis. There are ~906,000 new cases and 830,000 deaths of
primary liver cancer in 2020, of which HCC accounted for 75-85% of
cases (1,2). The risk factors of HCC are chronic
hepatitis, hepatitis B virus (HBV)/HCV infection-triggered
cirrhosis, alcoholic cirrhosis, dietary aflatoxin exposure,
non-alcoholic steatohepatitis, α-1-antitrypsin deficiency and
hemochromatosis (3). This disease
may be cured through liver transplantation or resection, but these
surgical schemes are not suitable for the majority of patients late
in the disease course (4). The
5-year survival rates for HCC in China are <12.5%, thus
effective treatment schemes for HCC require further investigation
(5).
Glypican-3 (GPC3), a member of the heparan sulfate
proteoglycan family, is a cell-surface
glycophosphatidylinositol-anchored protein (6,7).
GPC3 is highly expressed in at least 70% of HCC patients but not in
normal adult tissues (8-10). GPC3 has been suggested to be an
important diagnostic biomarker and immunotherapeutic target for HCC
(11-13). Preclinical studies performed by Shi
et al (14) confirmed the
potential of GPC3-chimeric antigen receptor (CAR)-T cell therapy
for HCC.
CAR comprises an extracellular antigen recognition
domain (ARD), an intra-cellular signaling domain (i.e. CD3ζ) with
or without 1 or 2 costimulatory molecules, and a transmembrane
domain (15). CAR-T cells can
distinguish the exact tumour-associated antigens (TAAs) in a manner
independent of the major histocompatibility complex (16-18).
Nowadays, although the curative effect of CAR-T cell therapy has
been proven to treat hematological malignancies (19), it remains unsatisfactory in
treating solid tumours (20,21).
In contrast to blood tumours, solid tumours have an
immunosuppressive tumor microenvironment (TME), which induces the
expression of PD-1 on CAR-T cells. PD-1/PD-L1 axis is a well-known
immune checkpoint inhibitor pathway. The combination of PD-L1 and
PD-1 generates an inhibitory signal that prevents T cell
activation, enabling tumour cells to escape from the monitoring of
the immune system (22,23). Hence, CAR-T cells should be
somewhat modified to avoid this inhibitory signal of the PD-1/PD-L1
pathway in HCC.
A double-target CAR with an extracellular ARD
containing anti-PD-1 single-chain fragment variable (scFv) and
anti-GPC3 scFv was established to help CAR-T cells persistently
resistant to PD-1 inhibitory signals. It was hypothesized that the
double-target CAR is capable of targeting tumour cells via
anti-GPC3 scFv and blocking PD-1 which was expressed on para-tumour
CAR-T cells via anti-PD-1 scFv. It appears that the
newly-established double-target CAR-T cells were more effective in
reducing tumor burden and prolonging the survival of tumor
xenograft models than traditional single-target CAR-T cells,
representing an effective strategy for applying CAR-T cell therapy
to solid tumours.
Materials and methods
Cells and cell culture
HCCLM3 and HuH7 cells were obtained from China
Center for Type Culture Collection, and SNU423, SNU182 and 293T
cells were obtained from the American Type Culture Collection.
Primary T cells from humans were cultured in a 37°C cell incubator
(5% CO2) containing RPMI-1640 medium (MilliporeSigma)
containing recombinant human IL-2 (30 IU/ml; cat. no. Z00368-1;
GenScript), 10% fetal bovine serum inactivated by heat and 1%
penicillin-streptomycin (Cytiva).
Preparation and transduction of vectors
and lentiviruses
In the present study, the established CAR
encompassed an extracellular ARD, a CD8 hinge, a CD28 transmembrane
domain, a CD28 combined with or without 4-1BB domain, and a
CD3ζ-derived signal transduction domain. A pKC lentiviral vector
(BioVector NTCC, Inc.) was sub-cloned with discrepant CAR sequences
in the frame.
Then this vector, together with packaging plasmid
r-8.91, enveloping protein plasmid vesicular stomatitis virus G (1
µg: 900 ng: 100 ng), was used for 293T cell transfection
using a PEIpro® transfection reagent (Getong Technology
Co., Ltd.). The virus-containing supernatants were collected 48 and
72 h later, and an Amicon Ultra-15 centrifugal filter from
MilliporeSigma was used to enrich the virus. In addition, a human T
cell enrichment cocktail (cat. no. 15061; RosetteSep™; Stemcell
Technologies, Inc.) was used for primary T cell isolation from the
peripheral blood of tumor-free volunteers. The volunteers were
health examiners in our hospital (samples were collected between
March-April 2021), including 2 males and 2 females, aged 32-50
years, with an average of (40±7.8) years. The use of human
peripheral blood was approved (approval no. 2021-081) by the Ethics
Committee of Peking University Shenzhen Hospital (Shenzhen, China)
and all donors provided informed written consent. Next, monoclonal
antibodies (mAbs; 5 µg/ml) against pre-enveloped CD3 (cat.
no. 05121-25-500) and dissolvable CD28 (cat. no. 10311-25-500; both
from PeproTech, Inc.) were used to activate the obtained cells for
48 h prior to lentiviral infection. Next, T cell treatment with
polybrene (8 µg/ml) was conducted for 4 h, followed by
transduction with lentivirus enriched on the plate enveloped by
NovoNectin (cat. no. CH38; Novoprotein Scientific, Inc.) plates for
8 h at 37°C (multiplicity of infection: 3). After 48 h, CAR
expression was examined. Empty lentivirus was used as control.
Reverse transcription-quantitative (RT-q)
PCR
Total RNA was isolated using Qiagen RNeasy Mini kit
(Qiagen, Inc.), according to the manufacturer's instructions and
cDNA synthesis was completed using the High Capacity cDNA Reverse
Transcription kit (Applied Biosystems; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. qPCR was performed
under the following thermocycling conditions: 10 min at 95°C, 40
cycles of 15 sec at 95°C and 1 min at 60°C. qPCR was performed
using 2X SYBR-Green PCR Master mix (Beijing Solarbio Science &
Technology Co., Ltd.) and 200 nM forward and reverse primers for
Bcl-2 and caspase-3. GAPDH was used as a reference gene. The
sequences of the primers used were as follows: Bcl-2 forward,
5′-AAA AAT ACA ACA TCA CAG AGG AAG T-3′ and reverse, 5′-GTT TCC CCC
TTG GCA TGA GA-3′; caspase-3 forward, 5′-TGC TAT TGT GAG GCG GTT
GT-3′ and reverse, 5′-TTA ACG AAA ACC AGA GCG CC-3′; and GAPDH,
forward, 5′-CTG GGC TAC ACT GAG CAC C-3′ and reverse, 5′-AAG TGG
TCG TTG AGG GCA ATG-3′. Each assay was run on an Applied Biosystems
7300 Real-Time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.) in triplicate, and the fold-changes of gene
expression were derived using the comparative 2−ΔΔCq
method, as previously described (24).
Flow cytometry (FC)
mAbs against FITC-PD1 (1:20; cat. no. 329903),
PE-PD-L1 (1:20, cat. no. 329705), FITC-CD69 (1:20; cat. no.
310903), FITC-CD3 (1:20; cat. no. 317305), FITC-Annexin V (1:20;
cat. no. 640905), FITC-CD62L (1:20; cat. no. 304803), FITC-CD45RA
(1:20; cat. no. 304105), FITC-CD45 (1:20; cat. no. 304006),
APC-Granzyme B (1:20; GrB; cat. no. 372203), FITC-perforin (1:20;
cat. no. 353309; all from BioLegend, Inc.), and FITC-GPC3 (1:20;
cat. no. 100393-R024; Sino Biological) were used. Following rinsing
twice with PBS, the cells were dyed and underwent 20-min mAb
incubation (5 µl per million cells in 100 µl staining
volume) at 4°C protected from light, and they were assessed after
they were immobilized in PBS.
To detect CAR expression on CAR-T cells, the cells
were stained with biotinylated protein L (cat. no. M00097;
GenScript) and then stained with streptavidin-PE at 37°C for 10 min
(cat. no. 405203; BioLegend, Inc.). Flow cytometry data were
acquired on a Gallios flow cytometer (Beckman Coulter, Inc.) and
analyzed using the FlowJo software (Tree Star, Inc.). The level of
expression of CAR on each type of CAR-T cell was adjusted to the
same level by un-transduced T cells before use.
PD-1+ CAR-T cells were obtained by
stimulating CAR-T cells with pre-coated anti-CD3 and soluble
anti-CD28 anti-bodies for one week to induce PD-1 expression.
Finally, the obtained cells were dyed at 4°C for 15 min. using
FITC-PD-1 mAb and distinguished from their PD-1−
counterparts using FITC fluorescence.
Cytokine investigation
ELISA (cat. nos. KGEHC102g, KGEHC003 and KGEHC154;
Nanjing KeyGen Biotech Co., Ltd.; and cat. no. K4279-100; AmyJet
Scientific, Inc.) was performed after gathering the
supernatants/sera from mice, in order to clarify whether granzyme B
(GrB), interferons (IFN)-γ, perforin and IL-2 exist.
In vitro CAR-T cell proliferation
assays
HuH7 cells were stimulated with IFN-γ (40
µg/ml) for 8 h to induce the expression of PD-L1 and then
inactivated with mitomycin C (100 µg/ml; cat. no. MB1164;
Dalian Meilun Biology Technology Co., Ltd.) at 37°C for 2 h. Every
4 days, the cells were collected following inactivation to provoke
each group of CAR-T cells (105/well), and the CAR-T
cells were counted. Next, uninfected T cells (control) were
cultured using 30 IU/ml recombinant IL-2. FC was ultimately used to
distinguish the CAR-T cell phenotype.
Cell toxicity and death rate
A lactate dehydrogenase (LDH) assay was used to
measure CAR-T cell toxicity using the corresponding kit (cat. no.
C0016; Beyotime Institute of Biotechnology). CAR-T cells
(1×105) were co-cultured with the target cells at
various effector to target (E:T) ratios (4:1, 8:1, 16:1 and 32:1).
The working concentration of the anti-PD-1 mAb (cat. no. 201905014;
TopAlliance Biosciences) combined with GPC3-CAR-PD-1+ T
cell was 10 µg/ml. The overall volume of the cultured system
was 100 µl and was incubated for 12 h at 37°C in 96-well
plates. Cell toxicity was calculated as follows: Cell toxicity
(%)=(mixture cell experiment - effector cell spontaneous-target
cell spontaneous-medium control)/(target cell maximum-target cell
spontaneous-medium control) ×100%.
Subsequently, the corresponding cells were cultured
in media comprising GrB (cat. no. ENZ-855; ProSpec-Tany TechnoGene,
Ltd.) with/without perforin (cat. no. APB317Mu01; Cloud-Clone
Corp.) for 12 h. Next, an LDH assay kit was used to calculate the
cell death rate as follows: Cell death rate (%)=(cell
experiment-cell spontaneous-medium control)/(cell maximum-cell
spontaneous-medium control) ×100%.
TX assays
All experimental procedures in the present study
were approved (approval no. 2021-081) by the Ethics Committee of
Peking University Shenzhen Hospital (Shenzhen, China). A total of
24 female NOD/SCID mice (4-6 weeks old, 6 in each group) were
housed at the Laboratory Animal Center of Peking University
Shenzhen Hospital. Mice were housed in a sterile room under a 12-h
light/dark cycle at ~23°C and 50% humidity, with ad libitum
access to food and water. A total of 5×106 HuH7 cells in
100 µl PBS were subcutaneously injected into the right flank
of mice to establish the TX model. Once the average tumor size
reached 100-200 mm3, the mice were randomly assigned to
different groups. A total of 1×106 CAR-T cells in 100
µl PBS were injected intratumorally into each mouse and the
tumors were measured weekly post-injection. In the CAR-T cells
combined with anti-PD-1 group, each mouse was intraperitoneally
injected with 150 mg anti-PD-1 mAb once a week (a total of 5
times). The tumor volume was calculated according to the formula
V=(length x width2)/2. The health and behaviour
condition of mice was monitored daily. On day 42 or when a humane
endpoint had been reached (e.g., >25% body weight loss, signs of
illness or distress including ruffled fur, difficulty with diet, or
abnormal posture), mice were euthanized and dissected for tumor
tissue analysis. Euthanasia was performed using an intravascular
administration of an overdose of sodium pentobarbital (200 mg/kg)
followed by cervical dislocation. Euthanasia was confirmed by the
loss of vital signs, such as respiration and heartbeat
cessation.
Immunohistochemistry (IHC)
Tumor tissue was fixed with 4% formaldehyde at 37°C
for 24 h, embedded in paraffin and sectioned into a thickness of
2-µm. Following dewaxing with xylene, hydration in alcohol
with different concentrations, tissue incubation in hydrogen
peroxide (3%) was performed to quench endogenous peroxidase and
sodium citrate buffer (0.01 M, pH 6.0) was used to retrieve
antigens at 95°C. Using 1% bovine serum albumin (Azer Scientific,
Inc.), the slide blocking lasted for 30 min at room temperature.
The sections were sequentially incubated with primary antibodies at
4°C overnight, including anti-Ki-67 (1:5,000; cat. no.
27309-1-AP;), anti-VEGF-A (cat. no. 19003-1-AP; 1:500; both from
ProteinTech Group, Inc.), anti-MMP-9 (cat. no. 13667; 1:500; Cell
Signaling Technology, Inc.). On the next day, slides were incubated
with HRP-conjugated secondary antibodies (1:1,000; cat. no. 7074S;
Cell Signaling Technology, Inc.) for 40 min at room temperature.
After scanning IHC sections, images were captured using CaseViewer
2.2 (3DHISTECH Kft).
Western blot analysis
The cell lysate was obtained using 2% SDS and then
centrifuged (4°C, 12,000 × g, 15 min) to obtain the supernatants.
The protein concentration was detected using a BCA protein assay
kit (cat. no. 23225; Thermo Fisher Scientific, Inc.). A total of 20
µg protein was separated using a 3% SDS-PAGE and transferred
to a PVDF membrane. The transferred PVDF membrane was blocked using
5% skimmed milk for 1 h at 25°C and washed with Tris-buffered
saline with Tween 20 (TBS-T) (1% Tween 20) (cat. no. 170-6435;
Bio-Rad Laboratories, Inc.), then incubated with primary antibodies
for 10 h at 4°C and incubated with the secondary antibodies at room
temperature for 1 h. Finally, the membrane was visualized using the
ImageQuant™ LAS 4000 system (Cytiva). Anti-GPC3 (1:1,000; cat. no.
ab124829), anti-PD-L1 (1:1,000; cat. no. ab243877), Goat
Anti-Rabbit IgG H&L (HRP) (1:5,000; cat. no. ab6721) and rabbit
anti-human GAPDH (1:2,500; cat. no. ab9485; all from Abcam) were
used. The protein bands were analyzed using ImageJ software
(version 1.48; National Institutes of Health).
Data assessment
All data analyses were performed using GraphPad
Prism version 7.0 (GraphPad Software, Inc.). One-way analysis of
variance (ANOVA) followed by Bonferroni test or unpaired t-tests
were used to compare different groups. The Kaplan-Meier method and
log-rank test were used to assess the survival curves of the mice.
P<0.05 was considered to indicate a statistically significant
difference.
Results
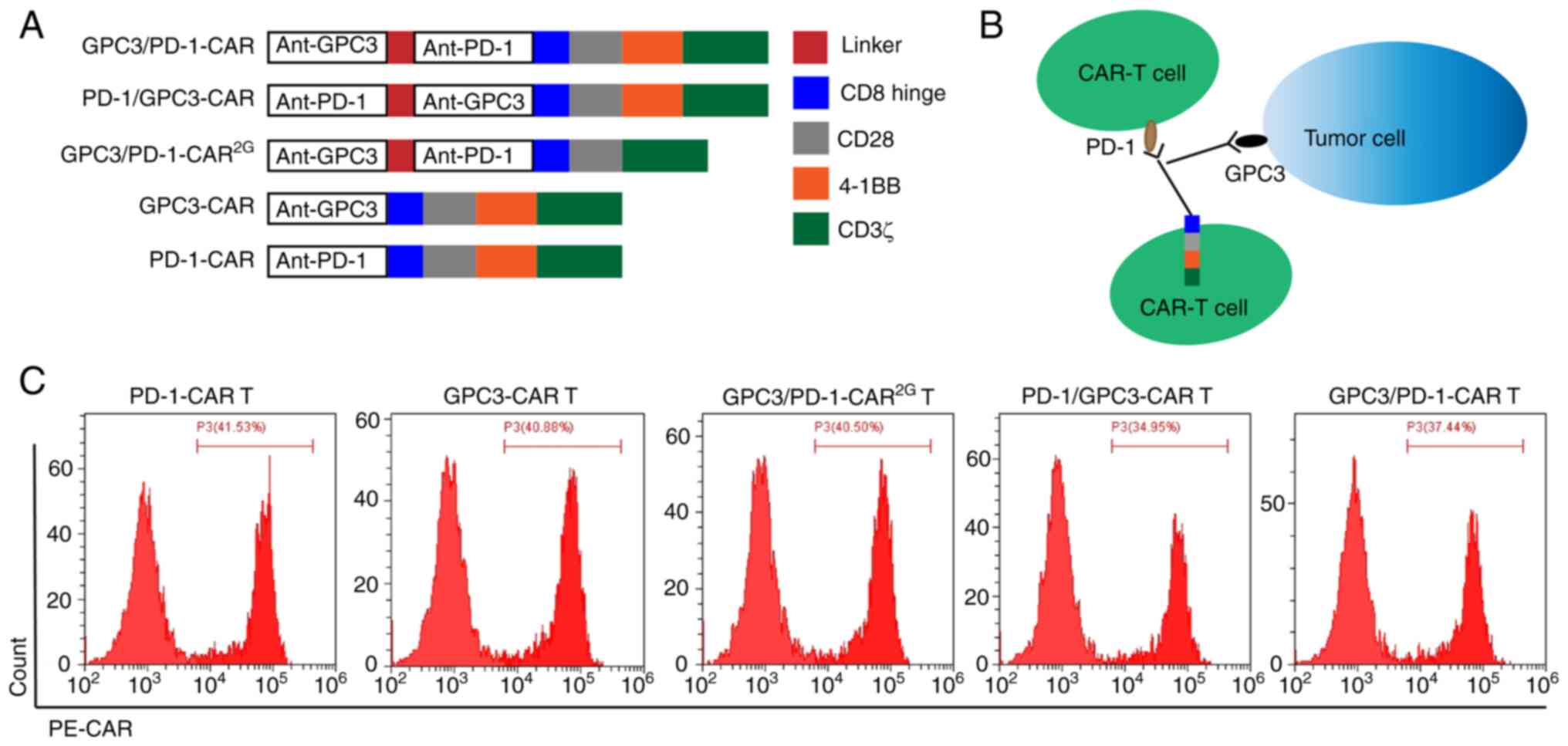
Establishment and expression of
double-target CAR molecules
Double-target CAR, known as GPC3/PD-1-CAR, which
could recognize PD-1 in T cells and GPC3 in tumour cells, was
established. Next, anti-GPC3 scFv was connected to anti-PD-1 scFv
using a GGGGS linker, which is used to bind the antigen outside the
cell. Through a CD8 hinge and a CD28 transmembrane domain, the
extracellular antigen binding domain (ABD) is connected to the
intra-cellular domain. The intra-cellular domain consisted of the
CD3ζ-chain, CD28 and 4-1BB (2 costimulatory domains). To
investigate the effects of the sequential order of anti-PD-1 scFv
and anti-GPC3 scFv on the function of dual-function CAR, the
sequential order of the two scFvs in GPC3/PD-1-CAR was changed,
constructed as PD-1/GPC3-CAR. Anti-GPC3 scFv and anti-PD-1 scFv
were extracellular ABDs of GPC3-CAR and PD-1/CAR (two single-target
CARs), and their other structures were the same as those of
double-target CARs. The aforementioned CAR structures are all
three-generation CARs containing two co-stimulation domains of CD28
and 4-1BB. GPC3/PD-1-CAR2G, a 2G double-target CAR, was
also established, which comprised only CD28, for the purpose of
comparing the functions of 2G and 3G CARs. Except where noted, the
CARs in the present study were of the third generation
(CAR3G). Fig. 1A is a
diagram of the role of established CARs. Fig. 1B is a diagram of the role of
double-target CAR-T cells. FC was used to measure differences in
the expression of CARs in T cells. As revealed in Fig. 1C, the FC results exhibited a
positive CAR expression on T cells (positive rate
34.95-41.53%).
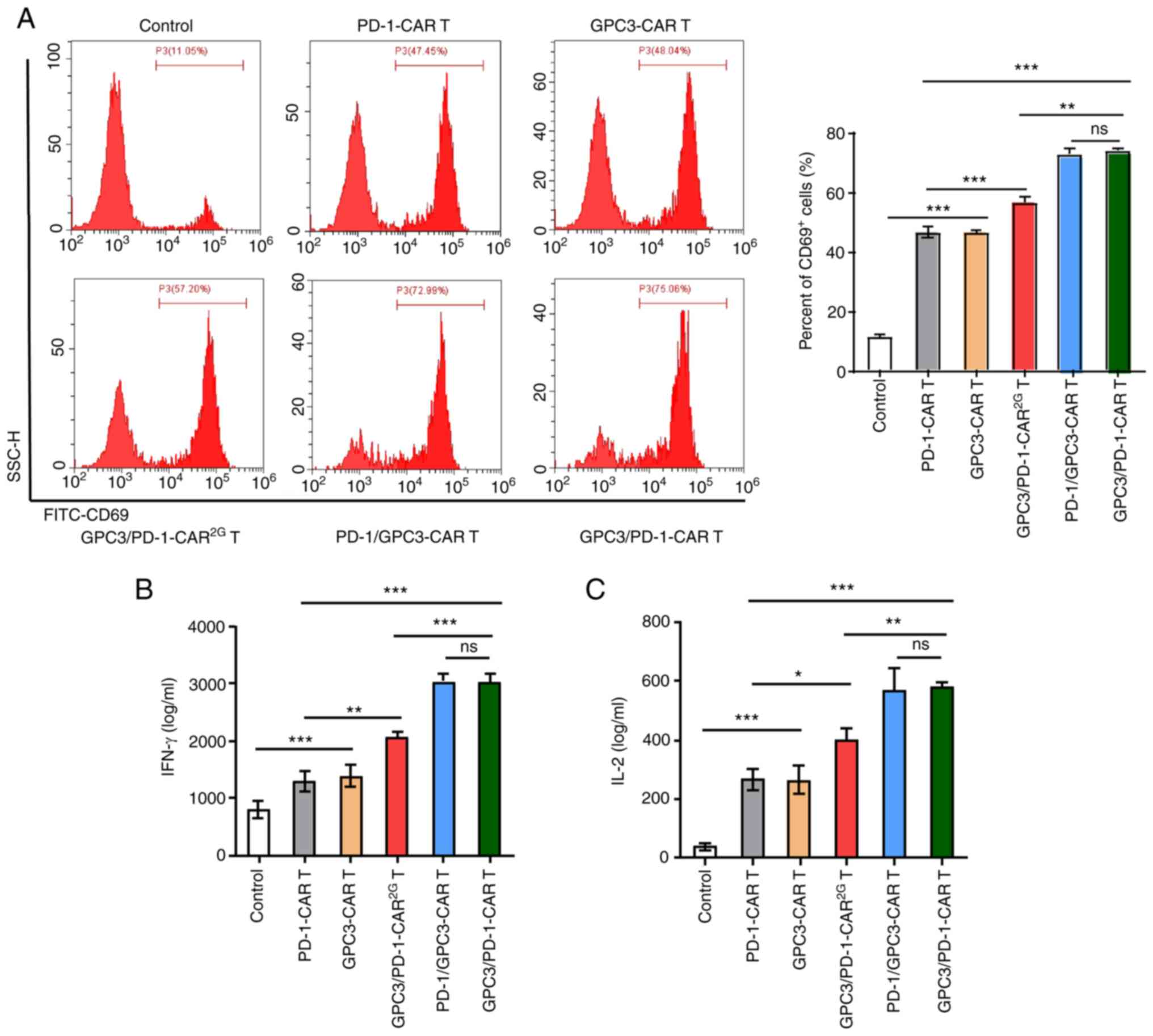
The established CARs have specific
reactions to target antigens
Recombinant PD-1 and GPC3 proteins were used to
provoke CAR-T cells, in order to clarify the function of the
established CARs on certain reactions to target antigens. Provoked
by target antigens, the expression level of CD69 (an earliest
marker elevated after T cell activation) in CAR-T cells was
examined. Following stimulation by GPC3 and PD-1, a pronounced
elevation of CD69 was discovered in GPC3/PD-1-CAR (positive rate
75.06%) and PD-1/GPC3-CAR (positive rate 72.99%) T cells, and a
modest elevation was observed in PD-1-CAR (positive rate 47.45%)
and GPC3-CAR (positive rate 48.04%) T cells (Fig. 2A). This provoking also resulted in
a higher IFN-γ and IL-2 secretion in CAR-T cells compared with
control cells (Fig. 2B and C).
Beyond that, GPC3-CAR-T and PD-1-CAR-T cells displayed a lower CD69
expression level and lower cytokine secretion compared with
double-target CAR-T cells (Fig.
2A-C).
The aforementioned results showed the targeted
control of the established CARs on active signals, and the
provoking effect of the 2 targets on cells, i.e., under the
provoking of the 2 targets, dual-function CAR-T cells mediated a
stronger active signal.
Double-target CAR-T cells display
targeted toxicity to HCC cells
Western blotting was performed to measure the
expression of GPC3 and PD-L1 in SNU182, HCCLM3, HuH7 and SNU423
cells. It appeared that the two targets (GPC3 and PD-L1) were
expressed in all four HCC cell lines (Fig. S1A).
As specified in the 'Cell toxicity and death rate'
section, the LDH assay revealed that with un-transduced T cells as
controls, GPC3/PD-1-CAR-T cells successfully eliminated SNU182,
HCCLM3, HuH7 and SNU423 cells at discrepant E:T ratios (Fig. 3A). Next, it was explored whether
the expression level of GPC3 impacted the function of
GPC3/PD-1-CAR-T cells. It was revealed that GPC3 expression was
knocked down in HuH7 cells via GPC3 shRNA transfection (Fig. S1B); these cells were called
HuH7(sh-GPC3). Next, HuH7(sh-GPC3) and HuH7 cells were cultured
with GPC3/PD-1-CAR-T cells at different E:T ratios, and cell
toxicity was determined using LDH assay. GPC3/PD-1-CAR-T cells were
more efficient in eliminating HuH7 than HuH7(sh-GPC3) cells
(Fig. 3B).
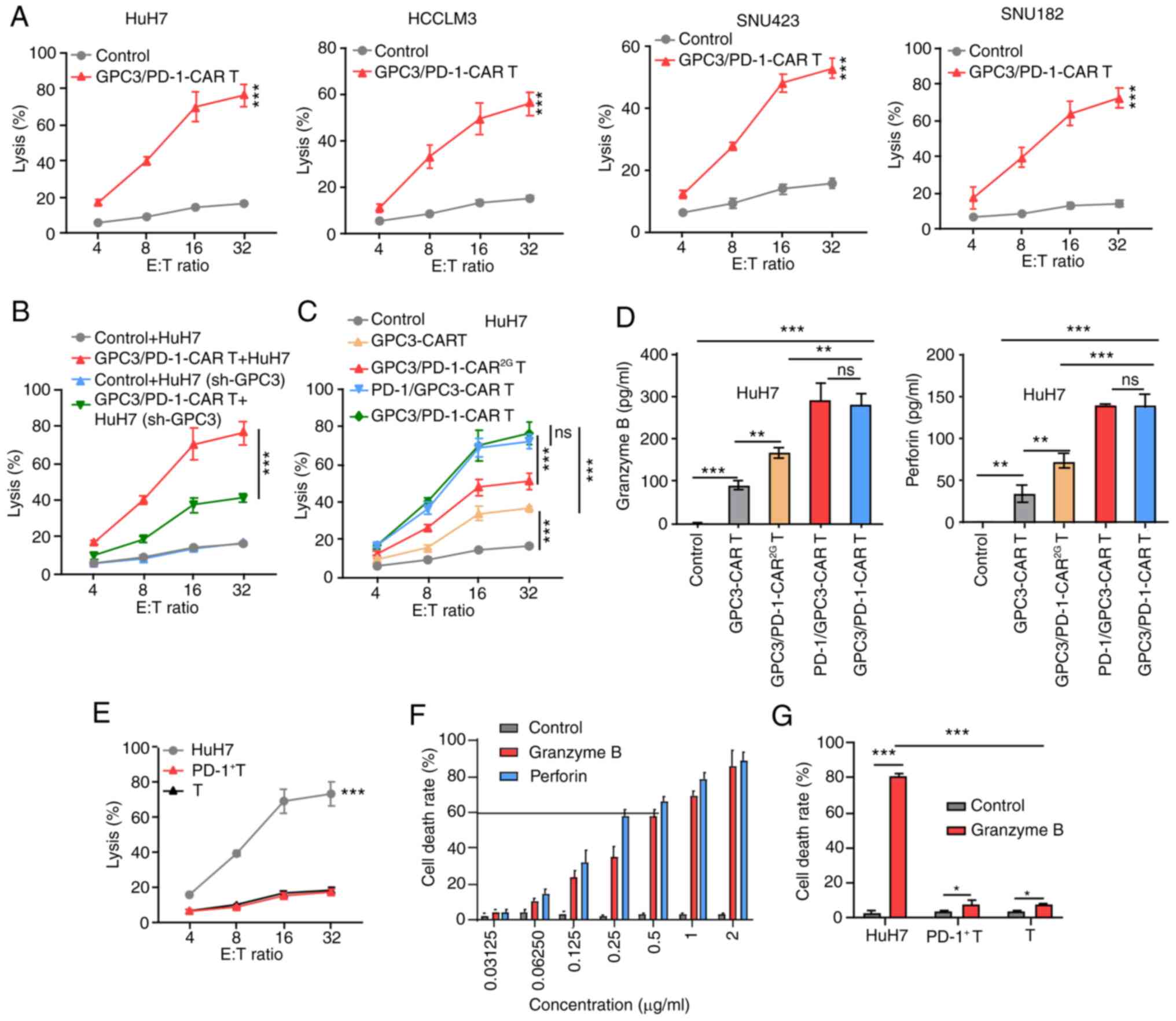 | Figure 3Double-target CAR-T cells show
cytolytic potency to target HCC cells. (A) Cytotoxicity of
GPC3/PD-1-CAR-T cells to the four indicated HCC cell lines at
various E:T ratios evaluated by an LDH cytotoxicity assay. (B)
Comparison of the toxicity of GPC3/PD-1-CAR-T cells to HuH7 cells
with different GPC3 levels at E:T ratios of 4:1, 8:1, 16:1 and
32:1. Un-transduced T cells served as control. (C) Comparison of
the toxicity of various CAR-T cells against HuH7 at different E:T
ratios. Un-transduced T cells served as control. (D) Levels of GrB
and perforin measured using ELISA after co-culturing different
CAR-T cells with HuH7 at an E:T ratio of 1:1 for 24 h. (E)
Comparison between the cytotoxicity of GPC3/PD-1-CAR-T cells and
that of HuH7, PD-1+ T and T cells at various E:T ratios, as
assessed by an LDH assay. (F) The death rate of HuH7 cells under a
series of concentrations of GrB and perforin for 12 h. (G) The
death rates of HuH7, PD-1+ T and T cells after co-culturing in the
indicated culture medium for 12 h. Data are presented as the mean ±
SD. *P<0.05, **P<0.01 and
***P<0.001. ns, not significant; CAR, chimeric
antigen receptor; HCC, hepatocellular carcinoma; GPC3, glypican-3;
PD-1, programmed death 1; E:T, effector-to-target; LDH, lactate
dehydrogenase; GrB, granzyme B. |
Furthermore, the differences among the activity of
the four types of CAR-T cells co-cultured with HuH7 cells showed
that toxicity and cytokine secretion were analogous for the two 3G
double-target CAR-T cells, but they were superior to those of
GPC3-CAR-T and GPC3/PD-1-CAR2G-T cells (Fig. 3C and D).
These results indicated that T cells of
dual-functional CAR and GPC3-CAR can mediate robust cytotoxicity to
tumor cells in a target-dependent manner and the killing efficiency
of these cells is positively correlated with GPC3 expression.
GPC3/PD-1-CAR were selected for later research in light of the
analogous activity of the two classes of double-target CARs.
To determine the presence of targeted toxicity of
double-target CAR-T cells to T cells expressing PD-1, T cells
underwent 1 week of incubation with pre-coated anti-CD3 and soluble
anti-CD28 antibodies to obtain PD-1+ T cells. Next,
PD-1+ T cells, unprovoked T cells (T cells), and HuH7
cells were co-cultured with double-target CAR-T cells,
respectively, at discrepant E:T ratios. As shown by the results of
the LDH assay, double-target CAR-T cells exhibited no strong
killing activity against T cells with different PD-1 expression
levels (Fig. 3E).
The cause of insignificant toxicity of double-target
CAR-T cells to T cells expressing PD-1 was examined by culturing
HuH7 cells in a cascade of media containing different
concentrations of GrB and perforin. When the cell death rate
reached ~60%, GrB and perforin concentrations reached 0.5 and 0.25
µg/ml, respectively (Fig.
3F). Next, medium containing 0.5 µg/ml GrB and 0.25
µg/ml perforin was utilized to compare the death rates of
HuH7, PD-1+ T and T cells, with a normal medium used as
the control. In the GrB- and perforin-containing culture medium,
PD-1+ T cell and T cell death rates exhibited slight
upward trends, which were significantly lower than that of HuH7
cells (Fig. 3G). A higher
tolerance of T cells to GrB and perforin were observed, and both
indices are markers for the killing of target cells by CAR-T cells.
This deciphered the cause of insignificant toxicity of
double-target CAR-T cells to PD-1+ T cells.
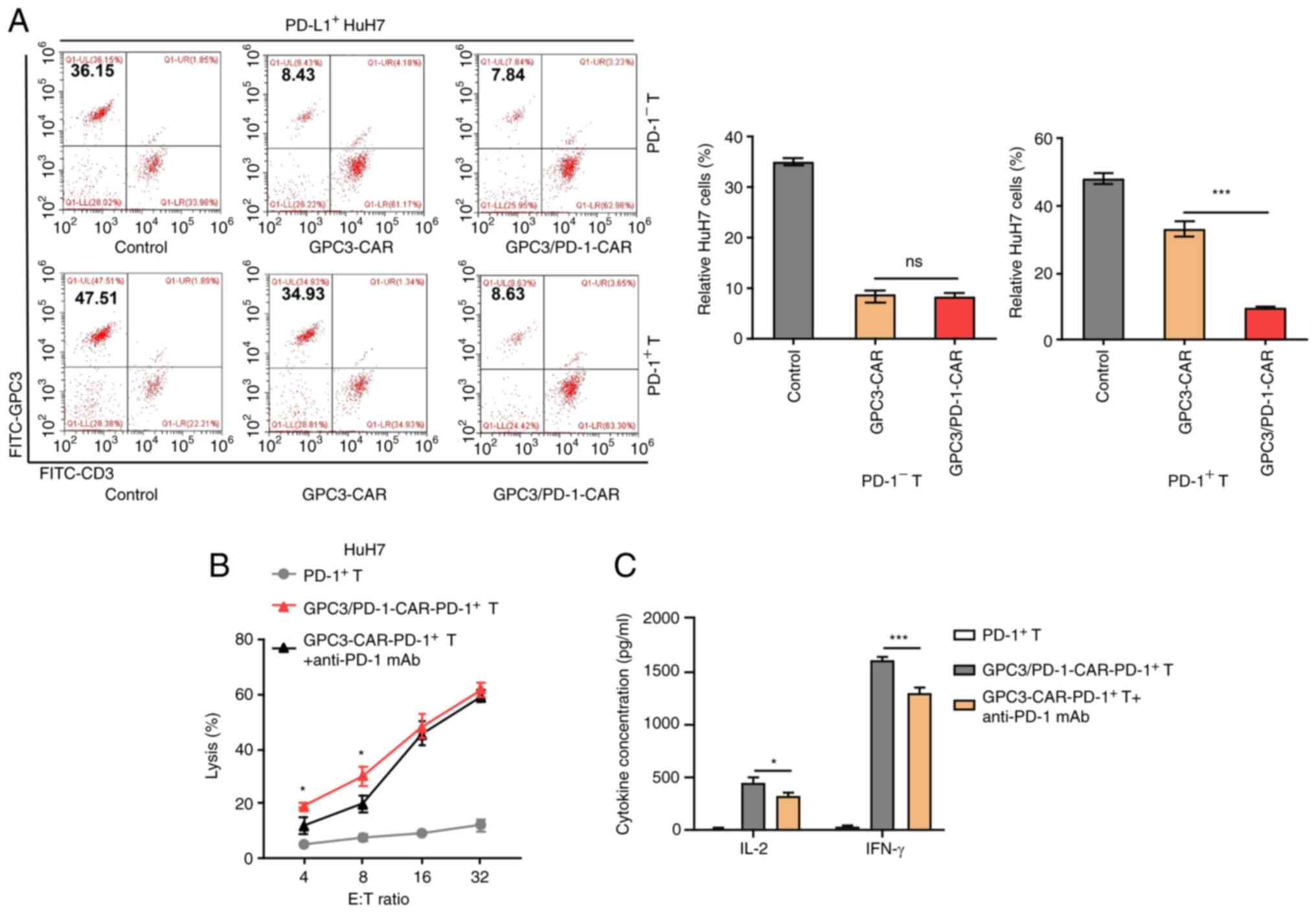
Double-target CAR-PD-1+ T cells have
enhanced toxicity to tumour cells highly expressing PD-L1
CAR-T cells were stimulated with precoated anti-CD3
and soluble anti-CD28 antibodies for 1 week to construct
CAR-PD-1+-T cells, and then FACS was adopted to get
CAR-PD-1+-T and CAR-PD-1−-T cells. HuH7 cells
were also provoked using IFN-γ to induce PD-L1 expression, and
PD-L1+-HuH7 cells were subsequently used to determine
cell toxicity.
The dual-function and single-target
CAR-PD-1+ -T cells were co-cultured with
PD-L1+ -HuH7 cells at 1:1 ratio for 3 days, and the
residual targeted cells were examined using FC. The marker was GPC3
for HuH7 cells and CD3 for CAR-T cells. The PD-L1+ -HuH7
cells continued to grow in co-culture with
GPC3-CAR-PD-1+-T cells, suggesting that the activation
of the CAR-T cells may be dampened by the PD-1/PD-L1 pathway
(Fig. 4A). Conversely,
double-target CAR-PD-1+-T cells were capable of
successfully limiting the target tumours despite PD-1
expression.
Through the combination of an mAb against human PD-1
with GPC3-CAR-PD-1+-T cells, it was sought to interrupt
the PD-1/PD-L1 pathway in eliminating PD-L1+ -HuH7 cells
and this combination strategy was compared with double-target
CAR-PD-1+-T cells. It appeared that double-target
CAR-PD-1+-T cells had stronger cytolytic effects at E:T
ratios of 4:1 and 8:1 to PD-L1+-HuH7 cells, as compared
with the combination strategy (Fig.
4B). In addition, double-target CAR-PD-1+-T cells
secreted more cytokines at an E:T ratio of 4:1 compared with the
secretion observed following the combination regimen (Fig. 4C).
Double-target CAR-T cells exhibit a
decreased inhibitory receptor (IR) expression, increased
proliferation and subdue terminal differentiation in long-time
antigen stimulation
Inactivated tumour cells were used to provoke
GPC3-CAR-T and GPC3/PD-1-CAR-T cells every 4 days for 24 days under
no other stimuli, and the expression, differentiation and
proliferation of IRs was measured at 8, 16 and 24 days to determine
the effect of PD-1 blocking on double-target CAR-T cells, using
un-infected T cells as controls.
Although there was no difference in PD-1 expression
between the two classes of CAR-T cells, less lymphocyte activation
gene 3 and T cell immunoglobulin and mucin-domain containing-3 were
expressed in double-target CAR-T cells than in GPC3-CAR-T cells
(Fig. 5A), confirming that PD-1
blockade prevents CAR-T cells from entering an exhausted state.
CAR-T cell apoptosis during stimulation was then measured. When
PD-1 was blocked, less double-target CAR-T cells were subjected to
apoptosis (25.41% on day 24) (Fig.
5B), confirming the pro-survival effect of blocking PD-1 on
CAR-T cells.
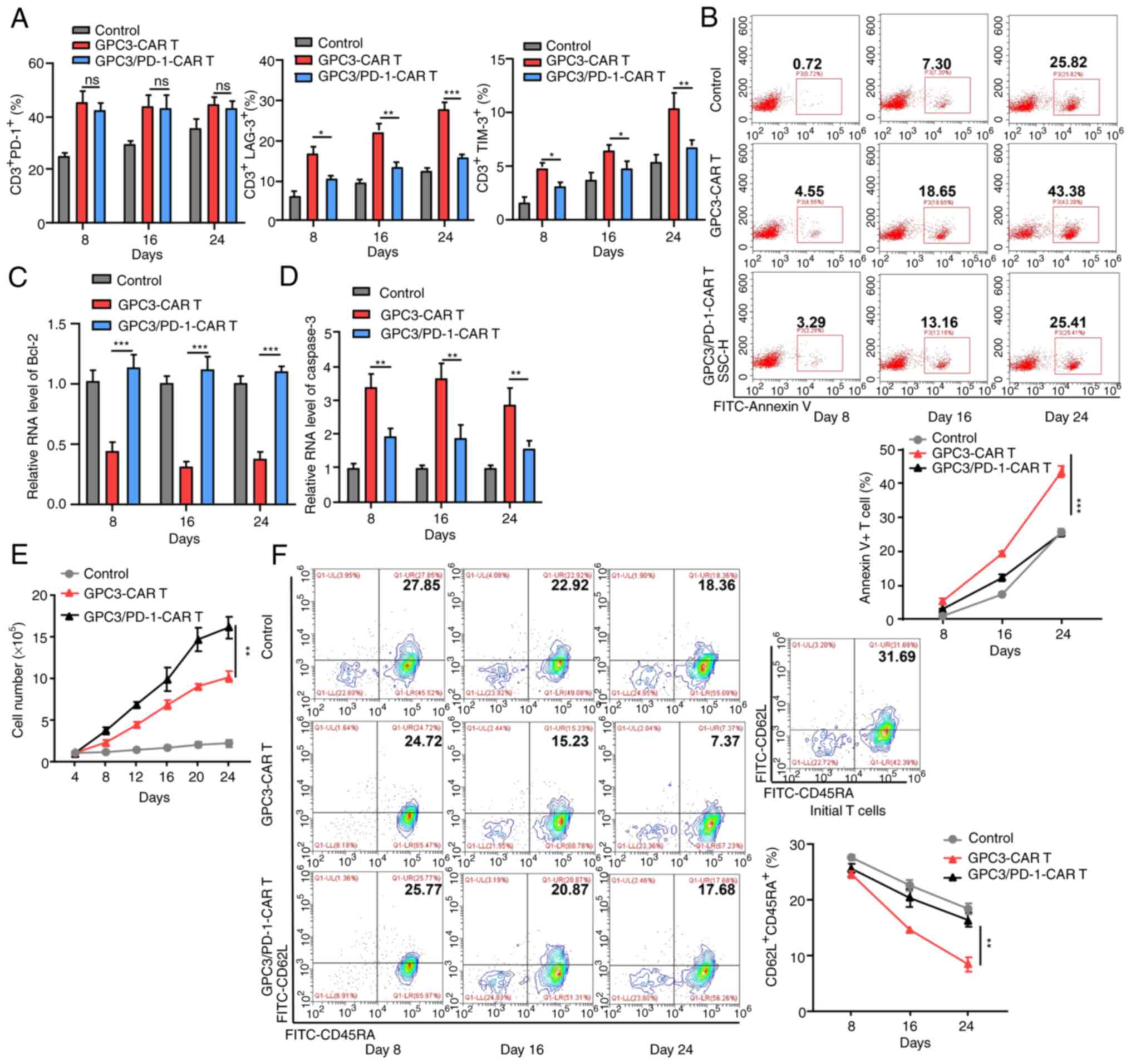 | Figure 5Double-target CAR-T cells display
downgraded IR expression, strengthened proliferation capability,
and subdued terminal differentiation in long time antigen
provoking. (A) Expression levels of PD-1, LAG-3 and TIM-3 in each
group at 8, 16 and 24 days in the long-run provoking process. (B)
Annexin V+ cell percentage in each group at 8, 16 and 24 days in
the long-run provoking process. (C and D) Levels of Bcl-2 and
caspase-3 in CAR-T cells on days 8, 16 and 24 in the long-run
provoking process examined using reverse transcription-quantitative
PCR. (E) Alterations in the total cell number in each group during
long-run provoking every 4 days (two-way ANOVA). (F) The expression
of CD62L and CD45RA on the CAR-T cells and initial T cells in each
group in the long-run provoking process. Data are presented as the
mean ± SD. *P<0.05, **P<0.01 and
***P<0.001. ns, not significant. CAR, chimeric
antigen receptor; IR, inhibitory receptor; PD-1, programmed death
1; LAG-3, lymphocyte-activation gene 3; TIM-3, T cell
immunoglobulin and mucin-domain containing-3. |
As revealed by RT-qPCR, double-target CAR-T cells
expressed more Bcl-2 (anti-apoptotic; Fig. 5C) and less caspase-3
(pro-apoptotic; Fig. 5D) compared
with GPC3-CAR-T cells, which is indicative of the resistance of
double-target CAR-T cells to apoptosis. Double-target CAR-T cells
also exhibited enhanced long-term proliferation capacity throughout
the extended culture, as compared with GPC3-CAR-T cells (Fig. 5E). Beyond that, as the provoking
increased, a higher proportion of double-target CAR-T cells
presented stem-like-memory (CD62L+CD45RA+)
phenotype (17.68% on Day 24) than that of GPC3-CAR-T cells (7.37%
on day 24; Fig. 5F).
In conclusion, the blocking of PD-1 provides CAR-T
cell exhaustion resistance, antiapoptotic, and low terminal
differentiation properties in long-term antigen stimulation.
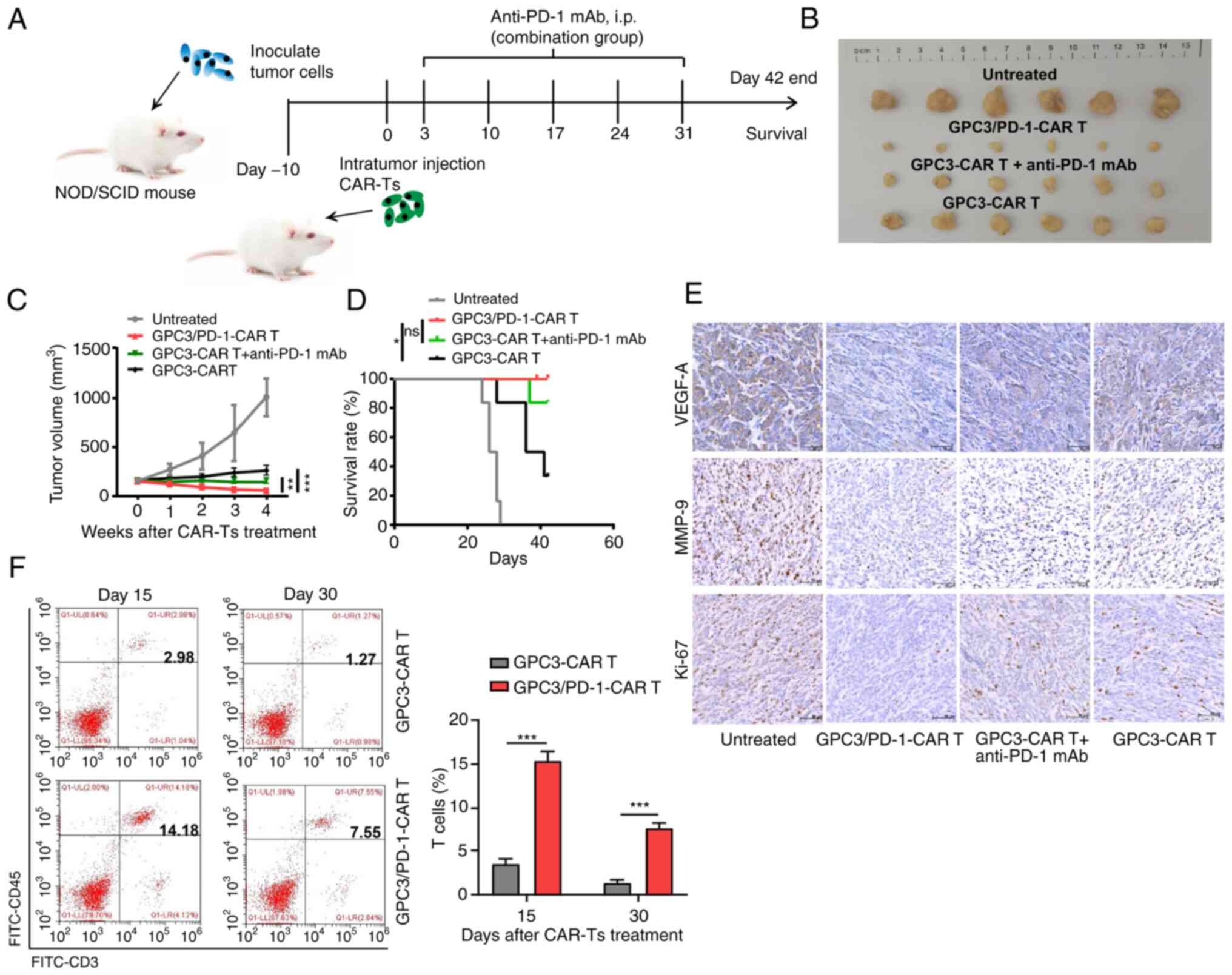
Double-target CAR-T cells have superior
anti-tumor effects in the TX model mice
The tumour-resistant property of double-target CAR-T
cells was assessed in vivo by modeling HuH7 tumour-bearing
NOD/SCID mice. As detailed in Fig.
6A, each mouse received an intra-tumoral injection of
106 CAR-T cells. In the group composed of CAR-T cells
combined with anti-PD-1 antibody, 150 mg mAbs against PD-1 were
intraperitoneally injected into each mouse for 5 weeks (once a
week). It was revealed that tumors in mice undergoing double-target
CAR-T cell treatment grew slower than those in mice receiving
combined regimen and GPC3-CAR-T cells (Fig. 6B and C). In addition, a more
notable survival benefit was observed in double-target CAR-T cells
compared with GPC3-CAR-T cells (Fig.
6D).
IHC was then performed to measure Ki-67, VEGF-A and
MMP-9 expression in the tumour tissues. Tumor tissue treated with
the dual-function CAR-T cells expressed lower levels of Ki-67,
VEGF-A and MMP-9, indicating that the dual-function CAR-T cells
have greater activation in suppressing the proliferation,
angiogenesis and metastasis of the tumor cells (Fig. 6E). Furthermore, the group treated
with dual-function CAR-T cells exhibited a higher frequency of
total T cells within tumor tissue (Fig. 6F).
Discussion
Anti PD-1/PD-L1 monoclonal antibodies have been used
in clinical research and the results are very promising. In
particular, atezolizumab (anti-PD-L1 mAb), nivolumab (anti-PD1
mAb), and pembrolizumab (anti-PD1 mAb) have already been approved
with durable clinical response and prolonged overall survival,
reaching clinics for the treatment of melanoma, non-small cell lung
cancer, and renal cell carcinoma (24,25).
However, this treatment is greatly limited by its low response
rates in certain types of cancer, lack of known biomarkers,
immune-related toxicity, innate and acquired drug resistance
(26). Precisely speaking, the
response rate of most cancers is not greater than 30%, which
results in a limited therapeutic efficacy (27). As a new type of immunotherapy,
CAR-T has attracted much attention due to its specific killing of
tumor cells. Jiang et al (28) established a bispecific CAR
targeting tyrosine-protein kinase Met and PD-L1 and proved that
these bispecific CAR-T cells have enhanced therapeutic effects on
HCC. Yuan et al (29)
established a bispecific CAR targeting c-Met and PD-1 and proved
that these bispecific CAR-T cells exhibited potent anti-tumor
efficacy in solid tumors. In the present study, a new-class
double-target CAR that recognizes GPC3 and blocks PD-1 was
established. Compared with c-Met, GPC3 is more specifically
upregulated in HCC (30,31). Blocked PD-1 markedly increased the
toxicity of CAR-T cells in vitro. On the other hand, in
vivo assays revealed the hindering effect of double-target
CAR-T cells on tumour growth and their promoting effect in
extending the survival of tumour-bearing mice, as compared with
their single-target counterparts. Beyond that, double-target CAR-T
cells exhibited enhanced persistence, limited inhibitory receptor
expression, and less differentiated phenotypes in tumour tissues,
giving rise to more potent tumour-resisting effects than their
single-target counterparts.
CAR-T therapy has achieved effective responses in
relapsed B-cell leukemia and lymphoma (32-34).
However, there are still many difficulties in the application of
CAR-T in the therapy of solid tumors (35-37).
Following in-depth research, the PD-1/PD-L1 pathway has been
accepted as a pivotal hallmark in checkpoint blockade treatment
(38,39). It has been shown by pre-clinical
studies that PD-1/PD-L1 mAbs combined with CAR-T cells can jointly
suppress tumours (40-42). Impacted by PD-1 blocking, the newly
established double-target CAR-T cells displayed resistance to the
suppression of the PD-1/PD-L1 pathway, and their toxicity remained
unchanged in PD-L1+ tumors. On the other hand, secreting
perforin and GrB is one of primary ways of CAR-T cell toxicity
(43,44). It was evidenced herein that, in
contrast to tumour cells, T cells were more tolerant to GrB and
perforin, providing one explanation for the targeting effect of
double-target CAR-T cells on PD-1+ tumour cells.
The limitations of the TX model are evident. The
findings of in-vivo experiments were principally based on
the interplay between CAR-T and tumour cells. However, certain
immune cells in the TME, such as endogenous tumour-infiltrating T
cells, myeloid-derived suppressor cells and dendritic cells, also
express PD-L1, exerting markedly affecting tumour outcome. In
addition, nude mice deficient in normal immune function were
selected as research objects; other breeds will be used for future
modelling. In addition, the lack of comparison between CAR-T cell
therapy and PD-1/PD-L1 antibodies is a limitation to the present
study. Furtermore, the lack of CAR-T cell proliferation detection
is another limitation.
In conclusion, impacted by PD-1 blocking, the newly
constructed double-target CAR-T cells exhibit stronger
tumour-suppressing effects on HCC than common single-target CAR-T
cells. The present study provided new ideas for the successful
treatment of solid tumors by CAR-T cell therapy.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JC and WVZ conceived the study. TZ, YL and XC
curated the data. TZ, YL and ZC reanalyzed the data. DL and JQ were
responsible for the study methodology. DL was responsible for the
resources. JC and WVZ supervised the study. All authors wrote,
reviewed and edited the original draft. All authors read and
approved the final manuscript. JC and WVZ confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
All experimental procedures in the present study
were approved (approval no. 2021-081) by the Ethics Committee of
Peking University Shenzhen Hospital (Shenzhen, China). Written
informed consent was obtained from all human peripheral blood
donors.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Sanming Project of
Medicine in Shenzhen (grant no. SZSM201612071), the Shenzhen Key
Medical Discipline Construction Fund (grant no. SZXK078) and the
The Cell Technology Center and Transformation Base, Innovation
Center of Guangdong-Hong Kong-Macao Greater Bay Area, Ministry of
Science and Technology of China [grant no. YCZYPT (2018)03-1].
References
|
1
|
Yin H, Sun L, Pu Y, Yu J, Feng W, Dong C,
Zhou B, Du D, Zhang Y, Chen Y and Xu H: Ultrasound-Controlled
CRISPR/Cas9 system augments sonodynamic therapy of hepatocellular
carcinoma. ACS Cent Sci. 7:2049–2062. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lai JP, Sandhu DS, Yu C, Han T, Moser CD,
Jackson KK, Guerrero RB, Aderca I, Isomoto H, Garrity-Park MM, et
al: Sulfatase 2 up-regulates glypican 3, promotes fibroblast growth
factor signaling, and decreases survival in hepatocellular
carcinoma. Hepatology. 47:1211–1222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cabibbo G, Enea M, Attanasio M, Bruix J,
Craxi A and Camma C: A meta-analysis of survival rates of untreated
patients in randomized clinical trials of hepatocellular carcinoma.
Hepatology. 51:1274–1283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wei Y, Tang X, Ren Y, Yang Y, Song F, Fu
J, Liu S, Yu M, Chen J, Wang S, et al: An RNA-RNA crosstalk network
involving HMGB1 and RICTOR facilitates hepatocellular carcinoma
tumorigenesis by promoting glutamine metabolism and impedes
immunotherapy by PD-L1+ exosomes activity. Signal Transduct Target
Ther. 6:4212021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao W, Kim H, Feng M, Phung Y, Xavier CP,
Rubin JS and Ho M: Inactivation of Wnt signaling by a human
antibody that recognizes the heparan sulfate chains of glypican-3
for liver cancer therapy. Hepatology. 60:576–587. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Knelson EH, Gaviglio AL, Nee JC, Starr MD,
Nixon AB, Marcus SG and Blobe GC: Stromal heparan sulfate
differentiates neuroblasts to suppress neuroblastoma growth. J Clin
Invest. 124:3016–3031. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cui X, Li Z, Gao PJ, Gao J and Zhu JY:
Prognostic value of glypican-3 in patients with HBV-associated
hepatocellular carcinoma after liver transplantation. Hepatobiliary
Pancreat Dis Int. 14:157–163. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fu SJ, Qi CY, Xiao WK, Li SQ, Peng BG and
Liang LJ: Glypican-3 is a potential prognostic biomarker for
hepatocellular carcinoma after curative resection. Surgery.
154:536–544. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Q, Han Z, Tao J, Zhao M, Zhang W, Li
P, Tang L and Gu Y: An innovative peptide with high affinity to
GPC3 for hepatocellular carcinoma diagnosis. Biomater Sci.
7:159–167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia L, Teng Q, Chen Q and Zhang F:
Preparation and characterization of anti-GPC3 nanobody against
hepatocellular carcinoma. Int J Nanomedicine. 15:2197–2205. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Du K, Li Y, Liu J, Chen W, Wei Z, Luo Y,
Liu H, Qi Y, Wang F and Sui J: A bispecific antibody targeting GPC3
and CD47 induced enhanced antitumor efficacy against dual
antigen-expressing HCC. Mol Ther. 29:1572–1584. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu M, Luo H, Fan M, Wu X, Shi B, Di S, Liu
Y, Pan Z, Jiang H and Li Z: Development of GPC3-specific chimeric
antigen receptor-engineered natural killer cells for the treatment
of hepatocellular carcinoma. Mol Ther. 26:366–378. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi
J, Lu Q, Gao H, Jiang H, Wang H, et al: Chimeric antigen
receptor-glypican-3 T-cell therapy for advanced hepatocellular
carcinoma: Results of phase I trials. Clin Cancer Res.
26:3979–3989. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kang CH, Kim Y, Lee DY, Choi SU and Lee
HK: Park CH. c-Met-Specific chimeric antigen receptor T cells
demonstrate anti-tumor effect in c-met positive gastric cancer.
Cancers (Basel). 13:57382021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mansilla-Soto J, Eyquem J, Haubner S,
Hamieh M, Feucht J, Paillon N, Zucchetti AE, Li Z, Sjostrand M,
Lindenbergh PL, et al: HLA-independent T cell receptors for
targeting tumors with low antigen density. Nat Med. 28:345–352.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang SS, Luong K, Gracey FM, Jabar S,
McColl B, Cross RS and Jenkins MR: A novel peptide-MHC targeted
chimeric antigen receptor T cell forms a T cell-like immune
synapse. Biomedicines. 9:18752021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsimberidou AM, Van Morris K, Vo HH, Eck
S, Lin YF, Rivas JM and Andersson BS: T-cell receptor-based
therapy: An innovative therapeutic approach for solid tumors. J
Hematol Oncol. 14:1022021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Consonni M, Garavaglia C, Grilli A, de
Lalla C, Mancino A, Mori L, De Libero G, Montagna D, Casucci M,
Serafini M, et al: Human T cells engineered with a leukemia
lipid-specific TCR enables donor-unrestricted recognition of
CD1c-expressing leukemia. Nat Commun. 12:48442021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu J, Yang Q, Zhang W, Du H, Chen Y, Zhao
Q, Dao L, Xia X, Natalie Wall F, Zhang Z, et al: Cell
membrane-anchored and tumor-targeted IL-12 (attIL12)-T cell therapy
for eliminating large and heterogeneous solid tumors. J Immunother
Cancer. 10:e0036332022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meyran D, Terry RL, Zhu JJ, Haber M,
Ziegler DS, Ekert PG, Trapani JA, Darcy PK and Neeson PJ:
Early-phenotype CAR-T cells for the treatment of pediatric cancers.
Ann Oncol. 32:1366–1380. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lemoine J, Ruella M and Houot R: Born to
survive: How cancer cells resist CAR T cell therapy. J Hematol
Oncol. 14:1992021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Greenbaum U, Dumbrava EI, Biter AB,
Haymaker CL and Hong DS: Engineered T-cell receptor T cells for
cancer immunotherapy. Cancer Immunol Res. 9:1252–1261. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Sharma P and Allison JP: Immune checkpoint
targeting in cancer therapy: Toward combination strategies with
curative potential. Cell. 161:205–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y
and Xia Y: Improvement of the anticancer efficacy of PD-1/PD-L1
blockade via combination therapy and PD-L1 regulation. J Hematol
Oncol. 15:242022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen N, Yang C, Zhang X, Tang Z and Chen
X: Cisplatin nanoparticles possess stronger anti-tumor synergy with
PD1/PD-L1 inhibitors than the parental drug. Acta Biomater.
135:543–555. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang W, Li T, Guo J, Wang J, Jia L, Shi
X, Yang T, Jiao R, Wei X, Feng Z, et al: Bispecific c-Met/PD-L1
CAR-T cells have enhanced therapeutic effects on hepatocellular
carcinoma. Front Oncol. 11:5465862021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yuan X, Sun Z, Yuan Q, Hou W, Liang Q,
Wang Y, Mo W, Wang H and Yu M: Dual-function chimeric antigen
receptor T cells targeting c-Met and PD-1 exhibit potent anti-tumor
efficacy in solid tumors. Invest New Drugs. 39:34–51. 2021.
View Article : Google Scholar
|
|
30
|
Zheng X, Liu X, Lei Y, Wang G and Liu M:
Glypican-3: A novel and promising target for the treatment of
hepatocellular carcinoma. Front Oncol. 12:8242082022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun L, Gao F, Gao Z, Ao L, Li N, Ma S, Jia
M, Li N, Lu P, Sun B, et al: Shed antigen-induced blocking effect
on CAR-T cells targeting Glypican-3 in hepatocellular carcinoma. J
Immunother Cancer. 9:e0018752021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jacoby E, Shahani SA and Shah NN: Updates
on CAR T-cell therapy in B-cell malignancies. Immunol Rev.
290:39–59. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gauthier J, Bezerra ED, Hirayama AV,
Fiorenza S, Sheih A, Chou CK, Kimble EL, Pender BS, Hawkins RM,
Vakil A, et al: Factors associated with outcomes after a second
CD19-targeted CAR T-cell infusion for refractory B-cell
malignancies. Blood. 137:323–335. 2021. View Article : Google Scholar :
|
|
34
|
Frigault MJ, Dietrich J, Martinez-Lage M,
Leick M, Choi BD, DeFilipp Z, Chen YB, Abramson J, Crombie J,
Armand P, et al: Tisagenlecleucel CAR T-cell therapy in secondary
CNS lymphoma. Blood. 134:860–866. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song
SX and Ma CQ: Improving the ability of CAR-T cells to hit solid
tumors: Challenges and strategies. Pharmacol Res. 175:1060362022.
View Article : Google Scholar
|
|
36
|
Shen L, Xiao Y, Tian J and Lu Z:
Remodeling metabolic fitness: Strategies for improving the efficacy
of chimeric antigen receptor T cell therapy. Cancer Lett.
529:139–152. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dana H, Chalbatani GM, Jalali SA, Mirzaei
HR, Grupp SA, Suarez ER, Raposo C and Webster TJ: CAR-T cells:
Early successes in blood cancer and challenges in solid tumors.
Acta Pharm Sin B. 11:1129–1147. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Akbari P, Huijbers EJM, Themeli M,
Griffioen AW and van Beijnum JR: The tumor vasculature an
attractive CAR T cell target in solid tumors. Angiogenesis.
22:473–475. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dammeijer F, van Gulijk M, Mulder EE,
Lukkes M, Klaase L, van den Bosch T, van Nimwegen M, Lau SP,
Latupeirissa K, Schetters S, et al: The PD-1/PD-L1-checkpoint
restrains T cell immunity in tumor-draining lymph nodes. Cancer
Cell. 38:685–700.e8. 2020. View Article : Google Scholar
|
|
40
|
Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu
Y, Yang Q, Nie J, Tang N, Zhang X, et al: Phase I study of CAR-T
cells with PD-1 and TCR disruption in mesothelin-positive solid
tumors. Cell Mol Immunol. 18:2188–2198. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kato D, Yaguchi T, Iwata T, Katoh Y, Morii
K, Tsubota K, Takise Y, Tamiya M, Kamada H, Akiba H, et al: GPC1
specific CAR-T cells eradicate established solid tumor without
adverse effects and synergize with anti-PD-1 Ab. Elife.
9:e493922020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shi X, Zhang D, Li F, Zhang Z, Wang S,
Xuan Y, Ping Y and Zhang Y: Targeting glycosylation of PD-1 to
enhance CAR-T cell cytotoxicity. J Hematol Oncol. 12:1272019.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Restifo NP, Dudley ME and Rosenberg SA:
Adoptive immunotherapy for cancer: harnessing the T cell response.
Nat Rev Immunol. 12:269–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
O'Connell J, O'Sullivan GC, Collins JK and
Shanahan F: The Fas counterattack: Fas-mediated T cell killing by
colon cancer cells expressing Fas ligand. J Exp Med. 184:1075–1082.
1996. View Article : Google Scholar : PubMed/NCBI
|