Introduction
Lung cancer is the leading cause of cancer
incidence, morbidity and mortality worldwide (1). Non-small cell lung cancer (NSCLC) is
the most common subtype of lung cancer that accounts for ~80% of
lung cancer cases and is associated with low 5-year overall
survival (OS) (2). Nearly 60% of
patients with NSCLC are diagnosed with advanced or metastatic
cancer (stages III and IV) and are not amenable for surgical
treatment (3). Conventional
radiotherapy and chemotherapy are the primary treatment strategies
for NSCLC patients with advanced cancer. However, the 5-year OS
rates of patients with advanced or metastatic NSCLC are low because
of chemotherapy resistance, which is associated with driver gene
mutations, epigenetic alterations and tumor heterogeneity (4). Therefore, there is an urgent need to
develop novel chemotherapeutic drugs with fewer adverse effects to
improve the survival outcomes in NSCLC.
Kinesin family member 11 (KIF11) is a mitotic
kinesin belonging to the kinesin-like family of proteins. It is
involved in the formation of the bipolar spindle and maintenance of
prophase and prometaphase during mitosis (5). KIF11 plays a crucial role in cell
division and is required for cell proliferation. KIF11 is
overexpressed in several cancer types and is considered as a
potential diagnostic and prognostic biomarker due to its
significant association with neoplasia, tumor progression and poor
survival outcomes (6-8). High KIF11 expression correlates with
poor OS in NSCLC patients with lung adenocarcinoma (LUAD) (9,10).
KIF11 is a promising diagnostic biomarker and therapeutic target in
NSCLC (11,12). Therefore, targeted inhibition of
KIF11 is a potential therapeutic strategy for NSCLC patients.
Natural plant compounds (NPCs) including
resveratrol, curcumin and baicalin show antitumor properties
including suppression of cancer cell proliferation and upregulation
of cellular apoptosis, autophagy and cell cycle arrest (13-15).
These anticancer NPCs inhibit epithelial-to-mesenchymal transition
(EMT) by altering different cancer-related signal transduction
pathways (16).
Cyclovirobuxine D (CVB-D, molecular formula:
C26H46N2O) is the main
pharmaceutically active steroidal alkaloid that is isolated from
the Chinese medicinal herb, Buxus microphylla (B.
microphylla). CVB-D is widely used for the treatment of
cardiovascular diseases as it demonstrates significant antioxidant,
anti-inflammatory and autophagy-regulating properties (17). CVB-D also shows promising
anticancer effects in hepatocellular carcinoma (HCC), colorectal
cancer and glioblastoma multiforme cells (18-20).
Zeng et al (21) showed
that CVB-D-induced mitophagy and apoptosis in the lung cancer cells
by activating the p65/BNIP3/LC3 signaling axis. However, the
mechanisms by which CVB-D inhibits the growth and progression of
lung cancer cells, particularly NSCLC cells are not well
characterized. Therefore, in the present study, the therapeutic
effects of CVB-D on the NSCLC cells and the underlying molecular
mechanisms were investigated using the in vivo xenograft
NSCLC model in nude mice and NSCLC cell lines.
Materials and methods
NSCLC cell lines, reagents and culture
conditions
The NSCLC cell lines, A549 (cat no. SCSP-503) and
H1299 (cat no. SCSP-589), and BEAS-2B (cat no. KCB200922YJ) were
purchased from the Cell bank of the Chinese Academy of Sciences.
The A549 and H1299 cells were cultured in RPMI-1640 medium (cat no.
01-100-1ACS; Biological Industries) containing 10% fetal bovine
serum (FBS) (cat no. 04-001-1A; Biological Industries), 100 U/ml
penicillin, and 100 mg/ml streptomycin. BEAS-2B cells were cultured
in the serum-free medium for BEAS-2B cells purchased from the Cell
bank of the Chinese Academy of Sciences (cat no. KCB M006). All the
cell lines were cultured in a humidified chamber maintained at 37°C
and 5% CO2. The stock solution of CVB-D (100 mmol/l;
cat. no. C117989; Shanghai Aladdin Biochemical Technology Co.,
Ltd.) was prepared in methanol.
MTT assay
The cytotoxic effects of CVB-D on the NSCLC and
BEAS-2B cells were analyzed using the MTT assay. The A549, H1299
and BEAS-2B cells (5×103 cells per well) were cultured
for 24 h in 96-well plates, and treated with different
concentrations of CVB-D (0-120 µM) for 24, 48 and 72 h.
Then, 20 µl of 5 mg/ml MTT solution was added to each well
and the plates were further incubated at 37°C for 4 h. The formazan
crystals were solubilized by adding 100 µl DMSO per well.
The absorbance was recorded in a microplate reader at 490 nm. The
assay was repeated thrice.
Colony formation assay
NSCLC cells (1×103/cells/well) were
cultured in six-well plates for 24 h. Then, the cell culture medium
was replaced with RPMI-1640 medium containing different doses of
CVB-D (0-80 µM). The cells were further incubated at 37°C
for 24 h. Then, the culture supernatants were removed, and fresh
culture medium (2 ml) was added to each well. The culture plates
were incubated for 2 weeks to allow formation of visible colonies.
The number of cells for a colony formation was >50 cells. The
cells were then fixed with 4% paraformaldehyde (PFA) for 20 min and
stained with 0.5% crystal violet at room temperature for 20 min.
The colonies were counted manually and images were captured using a
smartphone. The assay was repeated thrice.
Cell cycle assay
The A549 and H1299 cells (2.5×105 cells
per well) were cultured in six-well plates for 24 h. Then, they
were incubated with medium containing 40 µM CVB-D at 37°C
for a further 24 h. The cells were washed with phosphate-buffered
saline (PBS) and fixed with 75% ethanol at 4°C for 20 h. The fixed
cells were washed twice with pre-chilled PBS and stained with
Propidium iodide/RNaseA solution (cat no. MA0220; Dalian Meilun
Biology Technology Co., Ltd.) at 4°C for 30 min. Flow cytometry
(FCM) was performed in a Novocyte flow cytometer (Agilent
Technologies, Inc.) and the data was analyzed using the FlowJo VX
software (FlowJo LLC) to determine the proportion of cells in
multiple phases of the cell cycle.
Apoptosis assay
Cellular apoptosis was analyzed using the Annexin
V-FITC/PI Apoptosis Detection Kit (cat no. MA0220; Dalian Meilun
Biology Technology Co., Ltd.) according to the manufacturer's
protocol. Briefly, the NSCLC cells were cultured overnight in
six-well plates (2.5×105 cells per well) and treated
with different doses of CVB-D (0-80 µM) at 37°C for 24 h.
Then, the cells (1×106 cells per sample) were washed
with PBS, resuspended in Annexin V staining buffer, and stained
with the Annexin V-FITC/PI cocktail in the dark at room temperature
for 15 min. FCM was performed using the NovoCyte flow cytometer
(Agilent Technologies, Inc.) and the data was analyzed using the
FlowJo VX software to determine the proportion of apoptotic cells.
The experiments were performed thrice.
Wound healing assay
NSCLC cells (2.5×105 cells/well) were
cultured in six-well culture plates at 37°C to generate a
monolayer. The monolayers were wounded by scratching with a
200-µl micropipette tip. The cellular debris was removed by
gently washing with PBS. The cells were then cultured in serum-free
media containing different doses of CVB-D (0-20 µM) for 24
and 48 h. Then, the images of the monolayer were acquired and the
migration of cells into the wounded areas was analyzed using the
ImageJ software (version 1.8.0; National Institutes of Health). The
assay was repeated thrice times.
Transwell migration and Matrigel invasion
assay
Transwell and Matrigel assays were used to analyze
the migration and invasion, respectively, of NSCLC cells that were
pre-treated with different concentrations of CVB-D (0-20 µM)
for 48 h. Briefly, 200 µl serum-free medium containing
untreated NSCLC cells (5×104 cells per well) or NSCLC
cells pre-treated with 10 or 20 µM CVB-D (7.5×104
cells per well to compensate for cell death) was added in the upper
chambers of the Transwell (cat no. 353097; BD) and 500 µl of
RPMI-1640 medium supplemented with 10% FBS was added in the lower
chambers of the 24-well plate. The polycarbonate membrane of the
upper chamber was coated at 37°C for 30 min with 100 µl of
the Matrigel (cat no. BD356234; BD Biosciences) for the Matrigel
invasion assay. The plates were incubated at 37°C for 48 h in a
humidified incubator. Then, the non-migratory or non-invasive cells
were removed from the upper layer. The migratory or invasive cells
were fixed with 4% PFA and stained with 0.5% crystal violet for 20
min. The stained cells were counted in three random visual fields
under a light microscope (IX51; Olympus Corporation) and images
were captured. The assay was performed thrice.
RNA-sequencing (RNA-seq) protocol
NSCLC cells (2.5×105 cells) were cultured
overnight in six-cm culture dishes until they reached exponential
growth. Then, they were cultured in RPMI-1640 medium with or
without 50 µM CVB-D at 37°C for 24 h. The cells were then
digested with 1.0 ml RNAiso Plus reagent (Takara Biotechnology Co.,
Ltd.) and the cellular extract was stored at -80°C. Total mRNA
extraction and sequencing libraries were generated by the Shanghai
Personal Biotechnology Co., Ltd. The concentration, quality and
integrity of the mRNA samples was estimated using the NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). The
concentrations of mRNA samples were determined at 260 nm
wavelength. The absorbance ratio of 260/280 nm (1.8-2.0) was used
as an indicator of nucleic acid purity. Oligo dT-coupled magnetic
beads were used to purify mRNAs with poly(A) tails from the total
RNA samples. RNA fragmentation was performed using the NEBNext
Ultra Directional RNA Library Prep Kit for Illumina (New England
BioLabs, Inc.) with divalent cations in an Illumina proprietary
fragmentation buffer at elevated temperature. The first strand cDNA
was synthesized using random oligonucleotides and SuperScript II
reverse transcriptase. The second strand was synthesized using DNA
polymerase I and RNase H. The overhangs were converted into blunt
ends using the exonuclease/polymerase activities. The
aforementioned reagents including oligo dT-coupled magnetic beads,
SuperScript II reverse transcriptase, DNA polymerase I, RNase H and
exonuclease/polymerase were contained in the NEBNext Ultra II RNA
Library Prep Kit for Illumina (cat no. E7530L; New England Biolabs,
Inc.). Then, the 3'ends of the DNA fragments were adenylated and
the Illumina PE adapter oligonucleotides were ligated. The cDNA
fragments were purified using the AMPure XP system (Beckman
Coulter, Inc.) and selectively enriched by PCR amplification. The
PCR products were purified (AMPure XP system) and quantified using
the Agilent high-sensitivity DNA assay in the Bioanalyzer 2100
system (Agilent 2100). The library was then sequenced using the
NovaSeq 6000 platform (Illumina, Inc.) and a series of strict
quality control steps were performed. The data from the final
library was trimmed in comparison with the reference genome
(Genome: Homo_sapiens.GRCh38.dna.primary_assembly.fa.; Database
version: GRCh38.100; Genebuild by http://asia.ensembl.org/Homo_sapiens/Info/Index).
Bioinformatics analyses
The online DAVID 6.8 software (https://david.ncifcrf.gov/conversion.jsp) was used for
gene identity (ID) conversion. Gene Expression Profiling
Interactive Analysis (GEPIA)2 (http://gepia2.cancer-pku.cn) database was used to
identify differentially expressed genes (DEGs) between the tumor
and normal tissues, survival analysis and comparative analysis of
the expression levels of multiple genes between datasets. The
potential NSCLC therapeutic target genes were identified using the
GeneCards database (https://www.genecards.org/). The Human Protein Atlas
(HPA) database was used to estimate the KIF11 protein levels in the
NSCLC LUAD tissues (http://www.proteinatlas.org). The potential CVB-D
target genes were identified using the Swisstarget prediction
database (http://www.swisstargetprediction.ch/index.php). All
the DEGs from different datasets were imported into the Venny 2.1
online tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html)
to identify the overlapping genes. The String online database
(https://cn.string-db.org/cgi/input?sessionId=buvVxyvCwXVT&input_page_show_search=on)
was used to analyze the protein-protein association networks. The
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway enrichment analyses of the downregulated DEGs was
performed using the http://www.genome.jp/kegg and http://www.geneontology.org websites. The CVB-D
targets were screened based on the following criteria: i)
Oncogenes; ii) overexpressed in LUAD; iii) overexpressed oncogenes
associated with lower OS; and iv) predicted CVB-D target genes.
Molecular docking
The canonical 3D structure of CVB-D (CAS No.
860-79-7) was obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/). The crystal
structures of the KIF11, CDC25C and CDK1/cyclinB1 complex (PDB ID:
1X88, 3OP3 and 6GU2) were downloaded from the RCSB PDB database
(http://www.rcsb.org/). AutoDock Tools (version
1.5.6) software was used to convert the PDB file format of CVB-D,
KIF11, CDC25C, CDK1 and cyclinB1 to the PDBQT format and AutoDock
Vina to perform the virtual molecular docking. Then, we selected
the best docking pose was selected according to the score of
binding energy. The docking image was acquired using Discovery
Studio (version 2021).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from NSCLC cells using the
RNAiso Plus reagent (cat no. 9109) and reverse transcribed into
cDNA using the Reverse transcription kit (cat no. RR047A; both from
Takara Biotechnology Co., Ltd.) according to the manufacturer's
instructions. Subsequently, cDNAs were amplified using the TB
Green™ Premix Ex Taq™ II (cat no. RR820A; Takara, Biotechnology
Co., Ltd.) with the ABI Step One Plus Real-Time PCR system (Thermo
Fisher Scientific, Inc.). The thermocycling conditions for qPCR
amplification were as follows: Initial denaturation at 95°C for 30
sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec.
The primer sequences used in the present study are listed in
Table SI. The relative gene
expression levels were estimated using the 2−∆∆Cq method
(22). GAPDH was used as the
internal control. The experiment was repeated thrice.
Western blotting
NSCLC cells (2.5×105 cells/well) were
incubated with CVB-D (0-80 µM) in six-well plates for 24 h.
Then, after removing the medium, the cells were rinsed with
pre-chilled PBS and lysed with RIPA lysis buffer (cat no. P0013B;
Beyotime Institute of Biotechnology) containing protease and
phosphatase inhibitors (cat nos. P6730 and P1260; Beijing Solarbio
Science & Technology Co., Ltd.). The concentration of protein
lysates was estimated using the BCA assay kit (cat. no. C503051;
Sangon Biotech Co., Ltd.). Equal amounts (50 µg) of protein
lysates were separated on 10% SDS-PAGE gels and then transferred
onto PVDF membranes. The membranes were blocked with 5% BSA (cat
no. A8020; Beijing Solarbio Science & Technology Co., Ltd.) at
room temperature for 2 h containing buffer. Then, the membranes
were incubated overnight at 4°C with the following primary
antibodies (diluted with 5% BSA): anti-proliferating cell nuclear
antigen (PCNA; 1:2,000; cat no. 2586S), anti-Bax (1:1,000; cat no.
2772S), anti-Bcl-2 (1:1,000; cat no. 15071T), anti-Vimentin
(1:1,000; cat no. 5741SS), anti-N-Cadherin (1:1,000; cat no.
4061S), anti-E-Cadherin (1:1,000; cat no. 3195S), anti-Slug
(1:1,000; cat no. 9585S), anti-Snail (1:1,000; cat no. 3879S),
anti-phosphorylated (p)-p65 (1:1,000; cat no. 3033S), anti-p65
(1:1,000; cat no. 8242S), anti-p-JNK (1:1,000; cat no. 9251S) and
anti-JNK (1:1,000; cat no. 9252S); all from Cell Signaling
Technology, Inc.; anti-CDK1 (1:500; cat no. AF6108), anti-CDC25c
(1:500; cat no. AF6258), anti-CyclinB1 (1:500; cat no. AF6168) and
anti-KIF11 (1:500; cat no. AF4745); all from Affinity Biosciences,
Ltd.; and anti-GAPDH (1:1,000; cat no. abs830030; Absin Bioscience,
Inc.). Then, the blots were incubated with one of the following
secondary antibodies (diluted with 2% BSA): Goat anti-rabbit
IgG-HRP (1:10,000; cat no. abs20040ss) and goat anti-mouse IgG-HRP
(1:10,000; cat no. abs20039ss; both from from Absin Bioscience,
Inc.). at room temperature for 1 h. The protein bands were
developed using the electrochemiluminescence (ECL) reagent (cat no.
32106; Thermo Fisher Scientific, Inc. and detected using the Mini
Chemiluminescent Imaging and Analysis System (MiniChemiTM 610;
Beijing SageCreation Science & Technology Co., Ltd.; http://www.sinsitech.com/). The relative grayscale
values of the protein bands were estimated using the ImageJ
software. Western blotting experiments were repeated thrice.
Tumor xenograft experiments
A total of six female BALB/c nude mice (mean weight,
20±0.5 g; age, 5-6 weeks-old) were purchased from the Department of
Experimental Animal Center, HFK Bioscience Co. Ltd (Beijing,
China). The animal experiments were approved (approval no.
2021-114) by the Animal Ethics Committee of the First Hospital of
Shanxi Medical University (Taiyuan, China). The mice were housed in
a pathogen-free environment under controlled temperature (20-25°C)
and humidity (53-58%) with a 12/12-h light/dark cycle and provided
with food and water ad libitum. Equal volumes (100
µl) of 1×107 A549 cells resuspended in culture
medium were injected subcutaneously into the flanks of the nude
mice to establish tumor xenografts. After 7 days, the mice were
randomly divided into the control and CVB-D intervention groups
(n=3 per group). Physiological saline or CVB-D (20 mg/kg) were
injected intraperitoneally into the control and CVB-D intervention
group, respectively, once a day for 4 weeks. The xenograft tumor
weights and volumes and the body weights of the xenograft tumor
model mice in the control and CVB-D treatment groups were recorded
during the process. In the process of the establishment of animal
model in the present study, none of the mice succumbed. The mice
were then sacrificed by CO2 aspiration with 30% volume
displacement rate per min, until the behavioral signs of pain or
distress to inhaled CO2 including jumps and paws at the
nose and face, ataxia, labored breathing were disappeared (23). Histopathological analysis of the
tumor tissues was performed to determine the effects of CVB-D on
the growth and progression of the xenografted lung cancer
cells.
Ultrasonic imaging (USI)
USI (Philips Healthcare) was used to determine the
in vivo therapeutic efficacy of CVB-D. The tumor growth was
evaluated using the Bmode ultrasound. The degree of tumor hardness
was estimated using ultrasonic elastosonography (USE). Tumor
angiogenesis was evaluated using Color Doppler Flow Imaging (CDFI)
and Color Power Angiography (CPA). Ultrasonic MicroPure Imaging
(MI) was used to detect microcalcifications.
Histopathological examination
Hematoxylin and eosin (H&E) staining was
performed to determine the histopathological changes in the
xenograft tumors. The xenograft tumor model mice were euthanized on
day 29 after CVB-D treatment. Tumor tissues were harvested, fixed
at room temperature for >24 h with 4% PFA, embedded in paraffin,
sectioned at 5-µm thick preparations with a microtome and
stained with H&E at room temperature (hematoxylin for 5 min and
eosin for 3-5 min). Images of the stained sections were captured
using a light microscope.
Immunohistochemistry (IHC)
IHC was performed using the UltraSensitive™ SP IHC
kit (cat no. 9720; Fuzhou Maixin Biotech Co., Ltd.) according to
the manufacturer's protocols. The paraffin-embedded xenograft tumor
tissues were sectioned at 5-µm thick preparations, dewaxed
with xylene (Tianjin Zhiyuan Chemical Reagents Co., Ltd.), and
incubated for 20 min with sodium citrate buffer (pH 6.0) at 95°C
for antigen retrieval. Then, the slices were blocked for 30 min at
37°C with goat serum (included in the UltraSensitive™ SP IHC kit)
and incubated with primary antibodies (diluted with 5% BSA)
overnight at 4°C against the target proteins: anti-KIF11 (1:100;
cat no. 23333-1-AP; from Proteintech Group, Inc.), anti-CDC25C
(1:100; cat no. AF6258), anti-CDK1 (1:100; cat no. AF6108) and
anti-cyclinB1 (1:100; cat no. AF6168; all from Affinity
Biosciences, Ltd.); anti-Ki67 (1:100; cat no. A20018), anti-Bcl2
(1:100; cat no. A19693) and anti-N-Cadherin (1:100; cat no. A0433;
all from ABclonal Biotech Co., Ltd.). The slices were then
incubated with the streptavidin-peroxidase complex at 37°C for 30
min and color development was performed with the DAB detection kit
(cat no. 0031; Fuzhou Maixin Biotech Co., Ltd.). The distribution,
localization, and expression level of the target proteins were
visualized and images were captured using a light microscope
equipped with a digital camera. The integrated optical density was
analyzed using the Image pro plus 6.0 software (Media Cybernetics,
Inc.).
TUNEL assay
TUNEL Apoptosis Detection Kit (cat no. 11684817910;
Roche Diagnostics) was used to determine the proportion of
apoptotic cells in the xenograft tumor tissues. The dewaxed
sections of the tumor tissues were processed according to the
manufacturer's protocol. Images of six randomly selected visual
fields were captured per slide using a light microscope and the
proportions of apoptotic cells were estimated.
Transient transfection of small
interfering (si)RNA
A549 and H1299 cells were transfected for 48 h with
the negative control siRNA (si-NC), or siRNAs against KIF-11
(si-KIF11; cat no. SIGS0001287-4) or cyclinB1 (si-CyclinB1; cat no.
SIGS0003277-4) using the RiboFECT CP Transfection Kit (C10511-05;
all from Guangzhou RiboBio Co., Ltd.) according to the
manufacturer's instructions. The concentration of siRNAs used in
the present study was 50 nM. Subsequent experiments were performed
after transfection for 48 h at 37°C. The control, KIF11-silenced
cells and cyclinB1-silenced cells were used for subsequent
experiments. The transfection controls used for each siRNA
experiment were contained in the corresponding commercial kits. The
sequences for si-KIF11 and si-cyclinB1 are listed in Table SII. The sequence for si-NC was not
disclosed because the RiboBio Co., Ltd. applied for patent
protections for their commercial siRNA kits.
Statistical analysis
Statistical analysis was performed using the SPSS
13.0 statistical software (SPSS, Inc.). All data are presented as
the mean ± standard deviation and the results were presented using
the GraphPad Prism version 7.0 software (GraphPad software Inc.).
The differences in data between two groups were analyzed using the
paired Student's t-test after testing for normality and homogeneity
of variance. The differences in data between multiple groups were
analyzed using the one-way analysis of variance (ANOVA) and the
Bonferroni method as post hoc. Correlation among the DEGs was
evaluated using Pearson's correlation analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
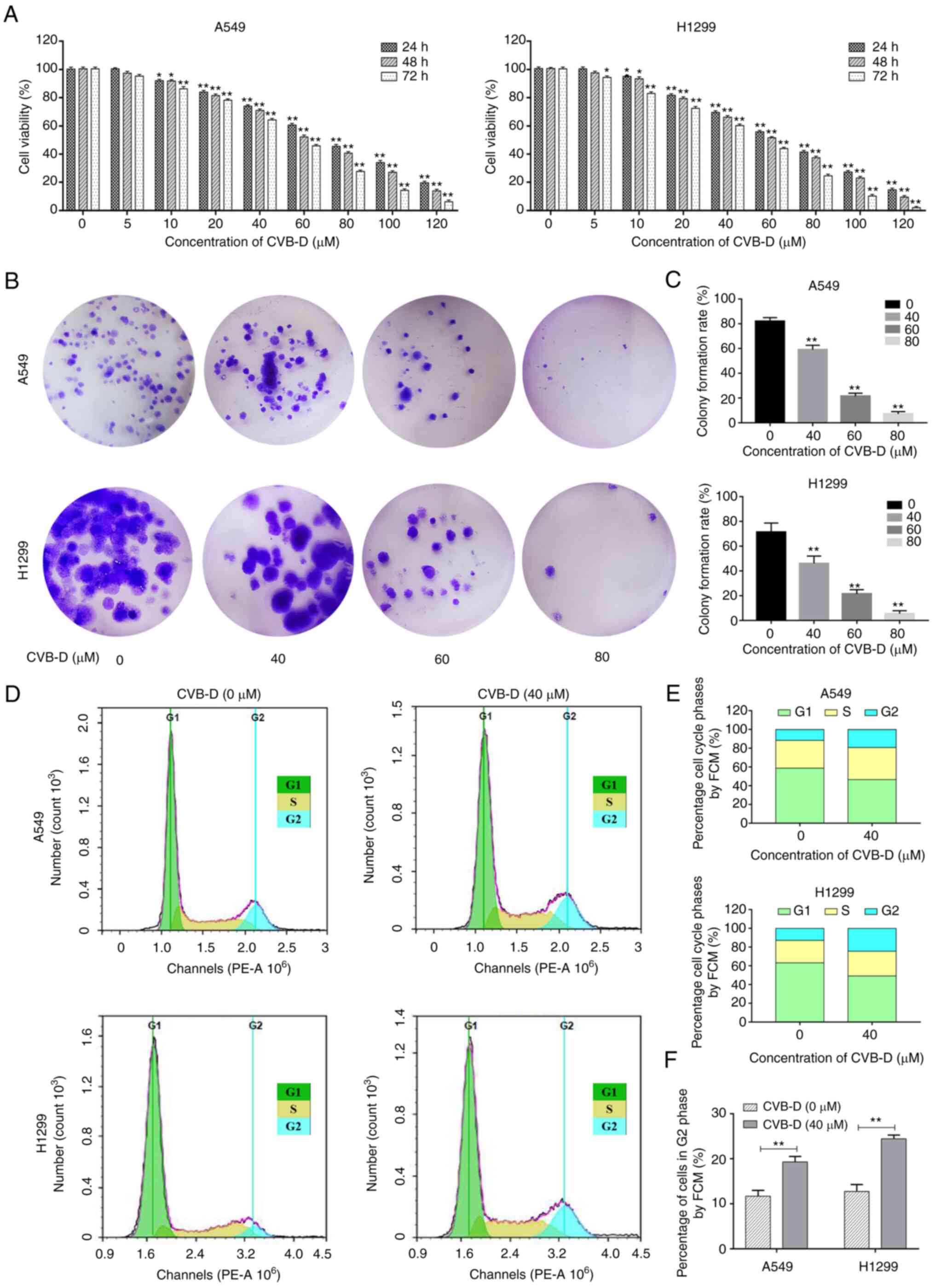
CVB-D inhibits in vitro proliferation of
NSCLC cells by inducing cell cycle arrest in the G2/M
phase
MTT assay results showed that CVB-D treatment
significantly reduced the viability of A549 and H1299 cells in a
dose- and time-dependent manner (Fig.
1A). The 50% inhibitory concentration (IC50) values
of CVB-D for the A549 and H1299 cells based on the Probit
regression analysis were 68.73 and 61.16 µM at 24 h, 59.46
and 54.99 µM at 48 h, and 47.78 and 41.7 µM at 72 h,
respectively. Furthermore, at the concentrations used in this
assay, CVB-D treatment did not induce cytotoxicity in the BEAS-2B
cells (Fig. S1). This data
demonstrated the therapeutic potential of CVB-D to treat resistant
lung cancer. CVB-D treatment significantly reduced the colony
formation ability of the NSCLC cells in a dose-dependent manner
(Fig. 1B and C). Furthermore,
CVB-D treatment induced G2/M cell cycle arrest of the
NSCLC cells (Fig. 1D-F). The
percentages of CVB-D-treated A549 and H1299 cells in the
G2/M phase were significantly higher than those in the
control groups (Fig. 1E and F).
CyclinB1 is a checkpoint protein that regulates G2/M
phase progression of the cell cycle (24). Western blot results demonstrated
that CVB-D treatment significantly decreased expression levels of
the cyclinB1 protein in the NSCLC cells (Fig. S2A and B). These results suggested
that CVB-D inhibited proliferation of NSCLC cells by inducing cell
cycle arrest in the G2/M phase.
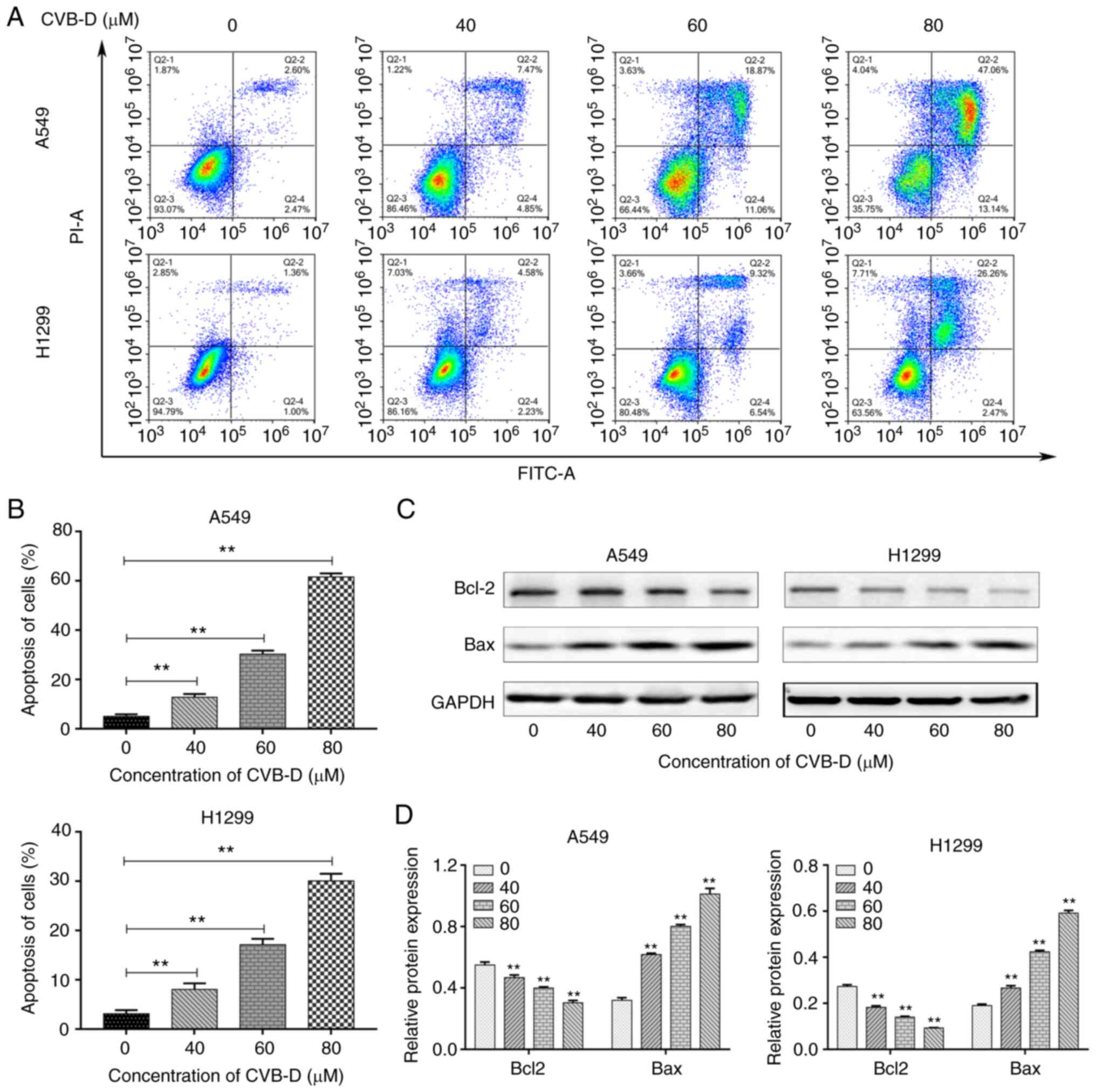
CVB-D treatment induces apoptosis of
NSCLC cells
It was then analyzed whether CVB-D promoted
apoptosis of the NSCLC cells. FACS analysis revealed that CVB-D
treatment induced apoptosis of A549 and H1299 cells in a
concentration-dependent manner (Fig.
2A and B). Furthermore, western blot analysis showed that CVB-D
treatment significantly decreased Bcl-2 protein levels and
increased Bax protein levels in the NSCLC cells (Fig. 2C and D). This demonstrated that
CVB-D treatment induced in vitro apoptosis of the NSCLC
cells.
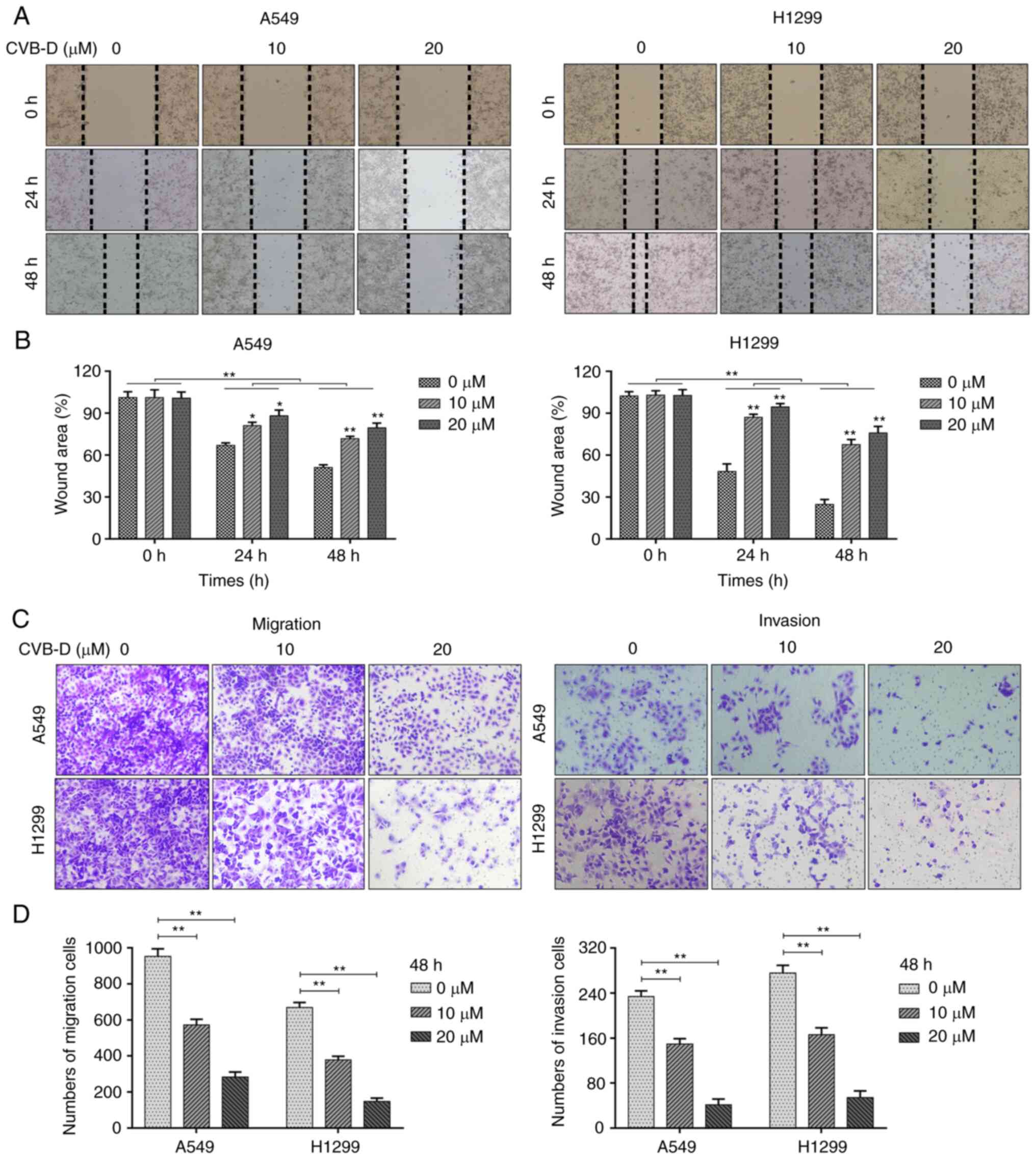
CVB-D inhibits migration and invasion of
NSCLC cells in a dose-dependent manner
Next, the effects of CVB-D on the migration and
invasiveness of the NSCLC cells were analyzed. The A549 and H1299
cells were treated with sub-lethal concentrations of CVB-D (0-20
µM) in the wound healing assays to eliminate cell viability
effects. Wound healing assay results showed that CVB-D treatment
significantly decreased the wound healing rate or the migration
rate of the NSCLC cells in a concentration-dependent manner
(Fig. 3A and B). Furthermore,
Transwell assays demonstrated that CVB-D treatment significantly
decreased the number of migratory and invasive NSCLC cells in a
concentration-dependent manner (Fig.
3C and D). Overall, these results demonstrated that treatment
with sub-lethal concentrations of CVB-D inhibited the migration and
invasion of NSCLC cells in a dose-dependent manner.
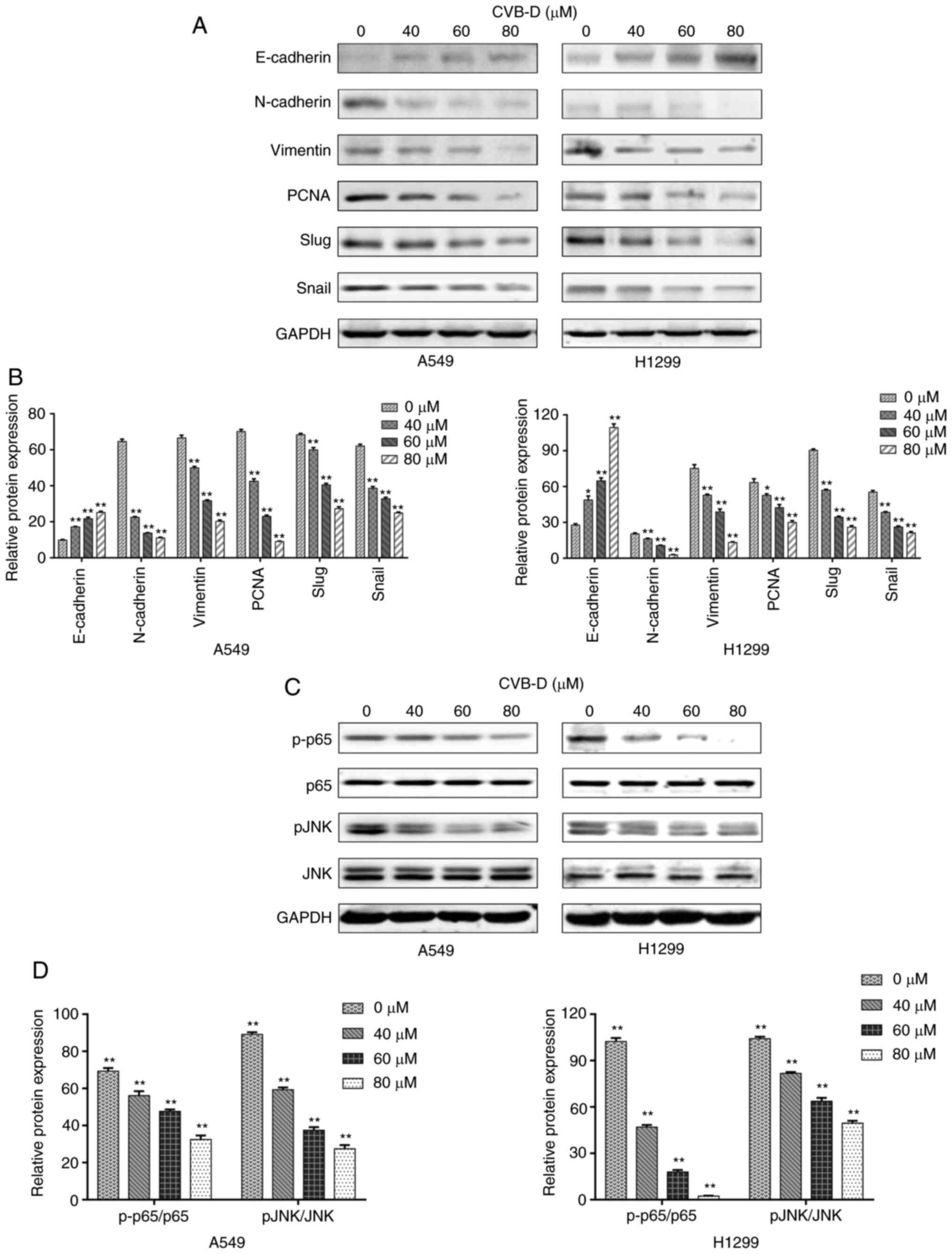
CVB-D treatment inhibits EMT, invasion
and migration of NSCLC cells
Western blot assays were then performed to determine
the levels of EMT-related biomarkers including E-cadherin, vimentin
and N-cadherin in the CVB-D treated NSCLC cells. CVB-D treatment
significantly increased the expression levels of E-cadherin and
decreased the expression levels of N-cadherin and vimentin
(Fig. 4A and B). This demonstrated
that CVB-D inhibited EMT in the NSCLC cells. PCNA is a known
proliferation biomarker due to its critical role in DNA metabolism
and synthesis, replication and repair (25). Furthermore, Snail and Slug are
essential transcriptional factors that promote cell migration
during tumorigenesis and metastasis in multiple cancers (26). Western blot analysis demonstrated
that CVB-D treatment significantly decreased the expression levels
of PCNA, Snail and Slug proteins in the NSCLC cells in a
dose-dependent manner (Fig. 4A and
B). These results demonstrated that CVB-D inhibited
proliferation, G2/M phase cell cycling, survival,
migration and invasion of the NSCLC cells.
CVB-D treatment inhibits NF-κB/JNK
signaling pathway in the NSCLC cells
Next, RNA-seq analysis was performed to identify the
molecular mechanisms underlying the effects of CVB-D treatment on
the NSCLC cells (Fig. 5A). Before
performing the differential expression analysis, Pearson's
correlation analysis was performed between the control and
CVB-D-treated samples. Pearson's correlation coefficients were
close to 1, thereby indicating high correlation in the expression
patterns between the samples (Fig.
5B). Principal component analysis demonstrated satisfactory
repeatability in the treatment and the selection of samples
(Fig. 5C). The heatmap revealed
clustering of DEGs between the control and CVB-D treatment groups
(Fig. 5D). Bidirectional cluster
analysis was performed to identify DEGs between the CVB-D-treated
NSCLC cells and the control untreated NSCLC cells. Volcano plots
showed 4244 DEGs including 2115 upregulated DEGs and 2129
downregulated DEGs in the CVB-D-treated NSCLC cells (Fig. 5E and F). Functional enrichment
analysis of the downregulated DEGs demonstrated enrichment of GO
terms related to molecular functions (MF) including 'DNA binding'
and 'ion binding', GO terms related to cellular components (CC)
including 'intracellular' and 'intracellular part' and GO terms
related to biological processes (BP) including 'RNA biosynthetic
process' and 'heterocycle biosynthetic process' (Fig. 5G). Furthermore, KEGG signaling
pathways such as classical pathway in cancer and MAPK pathways
including NF-κB and JNK were also enriched (Fig. 5H). Western blotting assays
confirmed that CVB-D treatment significantly decreased the
expression levels of p-p65 (NF-kB) and p-JNK in a
concentration-dependent manner (0, 40, 60, and 80 µM for 24
h) without altering total p65 and JNK expression levels (Fig. 4C and D). This suggested that CVB-D
treatment inhibited the activation of NF-κB/JNK signaling pathway
in the NSCLC cells.
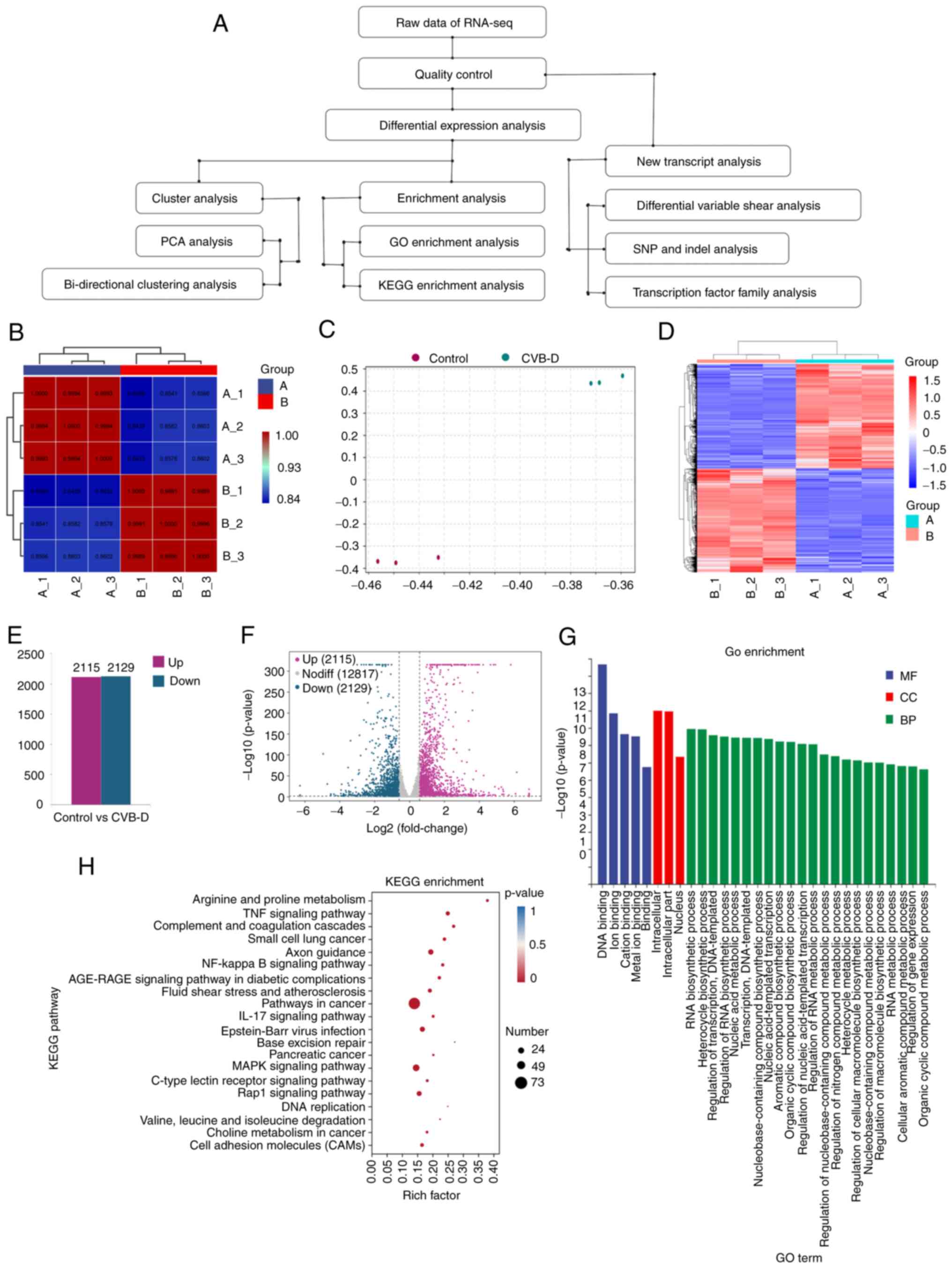 | Figure 5Transcriptome analysis of the
RNA-sequencing data from the control and CVB-D-treated A549 cells.
(A) Flowchart of the transcriptome analysis. (B) Pearson's
correlation analysis of the gene expression patterns between the
control and CVB-D-treated NSCLC samples. (C) PCA showed the
grouping of control and CVB-D-treated NSCLC samples based on the
transcriptome data. (D) Heatmap revealed the bidirectional cluster
analysis of the DEGs between control and CVB-D-treated A549 cells.
The upregulated and downregulated genes are highlighted in red and
blue, respectively. (E) Distribution of the 2115 upregulated DEGs
and 2129 downregulated DEGs in the CVB-D-treated A549 cells. (F)
Volcano plots demonstrated the expression profiles of DEGs in the
CVB-D treatment group compared with the control group, including
the upregulated (purple), downregulated (green) and normally (gray)
expressed genes. (G) GO enrichment analysis showed the enriched
molecular function MF, CC and BP terms associated with the
downregulated DEGs. (H) The bubble chart revealed enriched KEGG
pathways related to the downregulated DEGs. CVB-D, cyclovirobuxine
D; NSCLC, non-small cell lung cancer; PCA, principal component
analysis; DEGs, differentially expressed genes; GO, Gene Ontology;
MF, molecular function; CC, cellular component; BP, biological
process; KEGG, Kyoto Encyclopedia of Genes and Genomes. |
CVB-D suppresses the
KIF11-CDK1-CDC25C-CyclinB1 G2/M phase transition
regulatory network in the NSCLC cells
GEPIA 2 database analysis was performed based on two
threshold parameters, namely, |log2FoldChange|>1 and P<0.05.
The results showed that 1103 genes out of the 4245 overexpressed
genes correlated with LUAD and 500 prognosis-associated DEGs
correlated with OS and/or disease-free survival (DFS) rates of
patients with LUAD. Furthermore, 5593 potential therapeutic target
genes related to NSCLC were identified from the GeneCards database.
The Ensembl-gene-IDs (beginning with 'ENSG') for the 2129
downregulated genes from the RNA-seq study were converted into
standard-gene-symbols using the online gene ID conversion tool in
the DAVID 6.8 database and the ENSEMBL database. Standard gene
symbols were identified for 2111 out of 2129 downregulated genes
but the gene symbols for the remaining 18 downregulated genes could
not be identified. Comparative analysis of the 2111 downregulated
genes, 1103 overexpressed genes in LUAD, and 5593 potential
therapeutic target genes related to NSCLC identified 66 overlapping
genes. In addition, 10 genes that correlated with the OS and/or DFS
rates of LUAD patients were identified by comparative analysis of
the 66 overlapping genes and the 500 prognosis-associated DEGs
using the Venny online tool (Fig.
6A). The overexpression of these 10 genes in the GEPIA 2 LUAD
patients' dataset and their associations with OS and/or DFS rates
in the LUAD patients are shown in Figs. S3-S5.
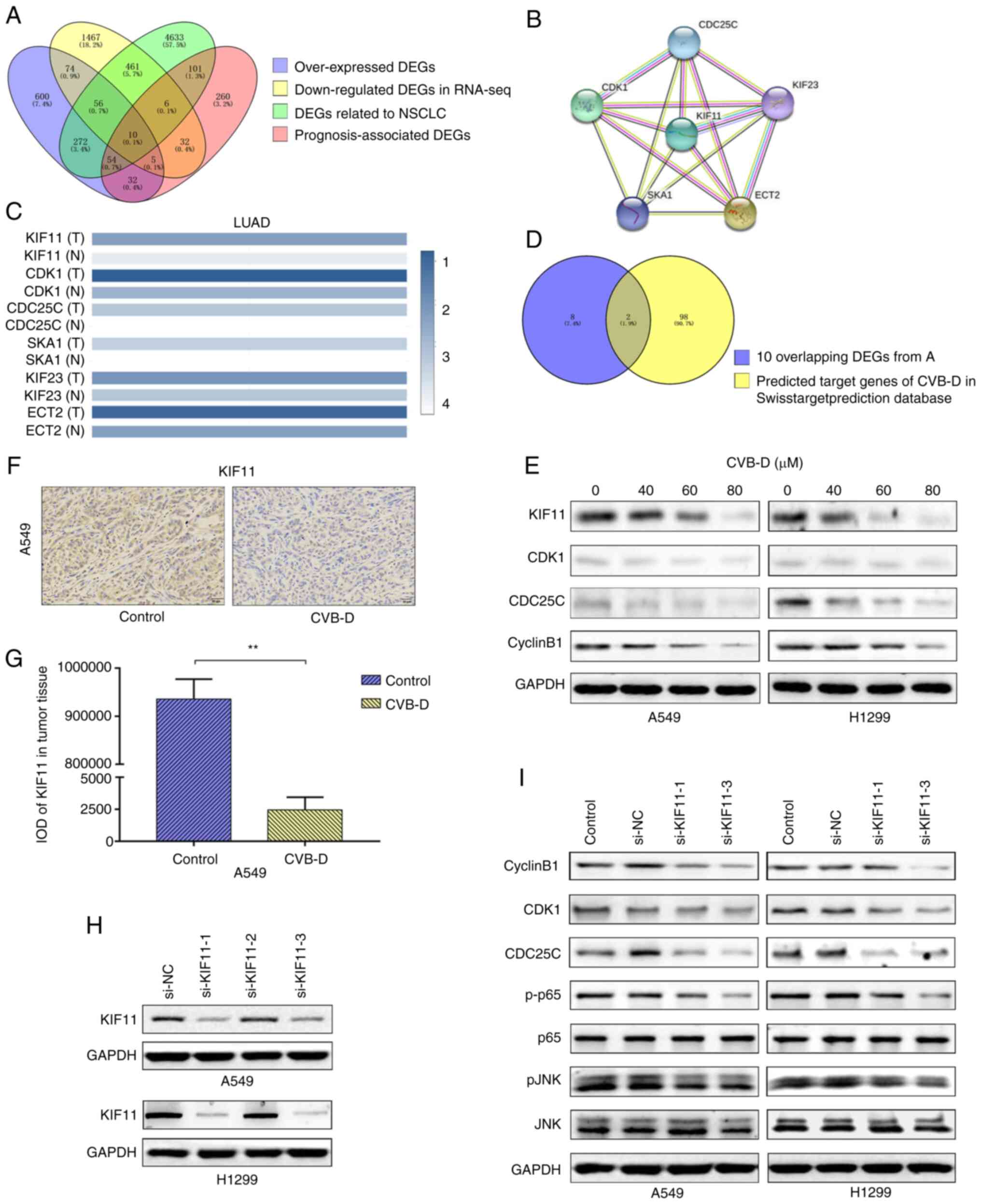 | Figure 6KIF11, KIF11-CDK1-CDC25C-cyclinB1
oncogenic network and NF-kB/JNK signaling pathway are the potential
CVB-D therapeutic targets in the NSCLC cells. (A) Venn diagram
showed the overlap number of potential CVB-D target genes in NSCLC
cells by intersecting four LUAD datasets. (B) Protein-protein
interaction network analysis revealed interaction between the 10
overlapping DEGs. The disconnected nodes are hidden. (C) The
interactive heatmap from GEPIA 2 database showed expression of the
6 DEGs in LUAD and normal lung tissues. KIF11 is the most
differentially expressed gene between LUAD and normal lung tissues.
(D) Venn diagram revealed 2 common genes (KIF11 and CDK1) based on
intersection between the 10 overlapping DEGs and the 100 predicted
CVB-D target genes extracted from the Swisstarget prediction
database. (E) Western blot analysis identified the expression of
KIF11, CDK1, CDC25C and cyclinB1 proteins in the control and
CVB-D-treated NSCLC cells. (F and G) Immunohistochemical assay
results showed the expression levels and localization of the KIF11
protein in the control and CVB-D-treated A549 xenograft tumor
tissues. (H) Western blot assay results revealed the KIF11 protein
levels in the si-NC and si-KIF11-transfected NSCLC cells. (I)
Western blot analysis demonstrated the expression levels of the
oncogenic signaling network proteins (KIF11, CDK1, CDC25C and
CyclinB1) and the NF-κB/JNK signaling pathway proteins (p65, p-p65,
JNK and p-JNK) in the si-NC- and si-KIF11-transfected NSCLC cells.
**P<0.01 vs. control group. KIF11, kinesin family
member 11; CVB-D, cyclovirobuxine D; NSCLC, non-small cell lung
cancer; LUAD, lung adenocarcinoma; DEGs, differentially expressed
genes; si, small interfering; NC, negative control; p-,
phosphorylated. |
A protein-protein interaction (PPI) network was
constructed among these 10 DEG-encoded proteins using the String
online database (Fig. 6B). It
revealed close interactions between six proteins but the remaining
four proteins did not show significant interactions. Therefore, the
non-interacting nodes were excluded from the PPI network in
Fig. 6B. The six interacting
proteins were KIF11, CDK1, CDC25C, SKA1, KIF23 and ECT2. GeneCards
database analysis demonstrated that the GeneCards Inferred
Functionality Scores (GIFtS) for KIF11, CDK1 and CDC25C (46, 43 and
46, respectively) were higher than those GIFtS for SKA1, KIF23 and
ECT2 (35, 42 and 39, respectively). Higher GIFtS predicted stronger
degree of functionality. The 3 genes with higher GIFtS were
associated with lower OS and/or DFS in patients with LUAD compared
with those with lung squamous carcinoma. GEPIA 2 database analysis
showed significantly higher correlation between KIF11 and CDK1
(r=0.8; P<0.001), KIF11 and CDC25C (r=0.74; P<0.001) and CDK1
and CDC25C (r=0.67; P<0.001) (Fig.
S6). The correlation between KIF11 and CDK1 (r=0.8; P<0.001)
was higher than the correlations between CDK1 and ECT2 (r=0.66;
P<0.001), CDK1 and SKA1 (r=0.68; P<0.001), and CDK1 and KIF23
(r=0.77; P<0.001) (Fig. S7A).
The correlation between KIF11 and CDC25C (r=0.74; P<0.001) was
higher than the correlations between ECT2 and CDC25C (r=0.63;
P<0.001) and KIF23 and CDC25C (r=0.72; P<0.001), and lower
than the correlation between SKA1 and CDC25C (r=0.75; P<0.001)
(Fig. S7B). The activation of
CDK1 and its regulator CDC25C showed close correlation with
G2/M phase cell cycle arrest. The G2
phase-related cyclinB1 is associated with the activation of CDK1
and CDC25C, and G2/M cell cycle arrest. Therefore, in
addition to CDC25C and CDK1, KIF11, KIF23, SKA1 and ECT2 were
identified as potential CVB-D targets based on their correlation
with cyclin B1.
The correlations between KIF23 and cyclinB1 (r=0.81;
P<0.001), SKA1 and cyclin B1 (r=0.8; P<0.001), and ECT2 and
cyclinB1 (r=0.67, P<0.001) were lower than the correlation
between KIF11 and cyclinB1 (r=0.82; P<0.001) (Fig. S7C). Furthermore, the correlations
between other transcription factors and the four potential CVB-D
target genes, KIF11, KIF23, SKA1 and ECT2, were analyzed. The
correlation of KIF11 with Bax, Bcl-2, PCNA, N-cadherin, E-cadherin,
Slug, and Snail was low and there was no correlation between these
factors and the other three genes, namely, KIF23, SKAI and ECT2
(Fig. S8). The expression levels
of these six DEGs were then compared using the interactive heatmap
from the GEPIA 2 database. KIF11 expression levels showed
significant differences among the LUAD tumors and the normal lung
tissues, but the differences in the expression levels of KIF23,
SKAI and ECT2 between the LUAD tumors and the normal lung tissues
were not significantly different (Fig.
6C). Therefore, KIF23, SKA1 and ECT2 were excluded.
Furthermore, KIF11 and CDK1 were identified as the potential CVB-D
target genes by comparative analysis of the 10 overlapping genes
and the 100 predicted CVB-D-target genes from the Swisstarget
prediction database (Fig. 6D).
Bioinformatics data analysis of the lung cancer
patient data from several online databases demonstrated that the
KIF11 protein and mRNA levels were significantly higher in the LUAD
tissues compared with the adjacent normal lung tissues (Figs. S3 and S9A and B). HPA database
analysis showed that KIF11 was an unfavorable prognostic marker in
lung cancer; KIF11 protein expression was closely related to cell
cycle regulation and KIF11 protein was mostly located in the
cytosol and the mitotic spindle (Fig.
S9C). KIF11 mRNA levels in the LUAD tissues ranked 17th among
31 types of tumors (Fig. S9D).
KIF11 overexpression was associated with worse OS in LUAD (Fig. S9E). HPA and GEPIA 2 database
analyses also demonstrated that CDK1, CDC25C, and cyclinB1 mRNA
levels were significantly higher in the lung cancer tissues and
were considered as unfavorable prognostic markers in patients with
lung cancer (Figs. S3 and S9F).
In patients with lung cancer, high expression levels of CDK1,
CDC25C and cyclinB1 were associated with worse OS and DFS (Fig. S9E). The correlations between
KIF11, CDK1, cyclinB1 genes and their 15 closest neighbors based on
the tissue RNA-seq data are shown in Table SIII. Spearman's correlation
coefficients between KIF11, CDK1, and cyclinB1 were close to 1.
This demonstrated strong correlation between KIF11, CDK1 and
cyclinB1 in cell cycle regulation.
Western blot analysis demonstrated that CVB-D
treatment decreased the expression levels of KIF11, CDK1, CDC25C
and cyclinB1 proteins in the A549 and H1299 cells in a
concentration-dependent manner (Figs.
6E and S10A). IHC results
demonstrated that the expression levels of KIF11 in the CVB-D group
were significantly reduced compared with the control group
(Fig. 6F and G). Western blot and
RT-qPCR analyses revealed that transfection of si-KIF11-1 and
si-KIF11-3 significantly reduced the levels of KIF11 protein and
mRNA in the NSCLC cells (Figs. 6H;
S10B and C). KIF11 silencing
significantly reduced the expression levels of CDC25C, CDK1,
p-p65/p65, p-JNK/JNK and cyclinB1 in the NSCLC cells (Figs. 6I and S10D). This suggested that KIF11 was
upstream of the NF-κB/JNK signaling pathway. KIF11 knockdown
partially rescued the growth inhibition of NSCLC cells by CVB-D
(Fig. S10E). Then, the expression
levels of KIF11, CDC25C and CDK1 were further analyzed in the
cyclinB1-silenced NSCLC cells. Western blot results showed that
transfection of si-cyclinB1-1 and si-cyclinB1-3 significantly
reduced the levels of cyclinB1 protein in the NSCLC cells (Fig. S11A and B). CyclinB1 silencing
significantly reduced the expression levels of KIF11, CDC25C and
CDK1 protein levels in the NSCLC cells (Fig. S11C and D). This suggested that
cyclinB1 regulated the KIF11-CDC25C-CDK1 G2/M phase
transition regulatory axis. Furthermore, these results suggested
that CVB-D targeted the KIF11-CDC25C-CDK1-cyclinB1 G2/M
phase transition regulatory network in the NSCLC cells.
CVB-D inhibits in vivo progression of
NSCLC tumors
Next, the in vivo therapeutic effects of
CVB-D were evaluated in the A549 xenograft nude mice model
(Fig. 7A). CVB-D treatment
significantly inhibited xenograft tumor growth in vivo. The
xenograft tumor weights and volumes were significantly lower in the
CVB-D treatment group compared with the control group after 4 weeks
(Fig. 7B-E). H&E staining
demonstrated that CVB-D treatment significantly inhibited tumor
growth and angiogenesis (Fig. 7F).
TUNEL assay demonstrated that CVB-D treatment significantly
increased apoptosis in the A549 xenograft tumors (Fig. 7F and G). The control and
CVB-D-treated xenograft tumor-bearing nude mice did not exhibit any
significant differences in the body weights (Fig. 7H). IHC staining analysis showed
that CVB-D treatment significantly decreased the proportion of
Ki67-positive cells in the xenograft tumors compared with the
controls (Fig. 7I and J). This
suggested that CVB-D treatment inhibited in vivo NSCLC cell
proliferation. Furthermore, the expression levels of the
anti-apoptotic Bcl-2 protein and the EMT-related N-cadherin protein
were significantly decreased in the xenograft tumor tissues of the
CVB-D treatment group compared with the controls (Fig. 7I and J). IHC analysis revealed
reduced expression of KIF11, CDK1, CDC25C and cyclinB1 in the
xenograft tumor tissues of the CVB-D treatment group compared with
the controls (Figs. 6F, 7I and J). This suggested that CVB-D
induced in vivo G2/M phase cell cycle arrest of
the NSCLC cells. The in vivo CVB-D treatment exerted
significant anti-NSCLC effects with low cytotoxicity in the normal
tissues; moreover, malignant lesions were not observed in the major
organs including brain, heart, lung, liver, kidney spleen, gut and
stomach during the 4-week CVB-D treatment in mice (Fig. S12).
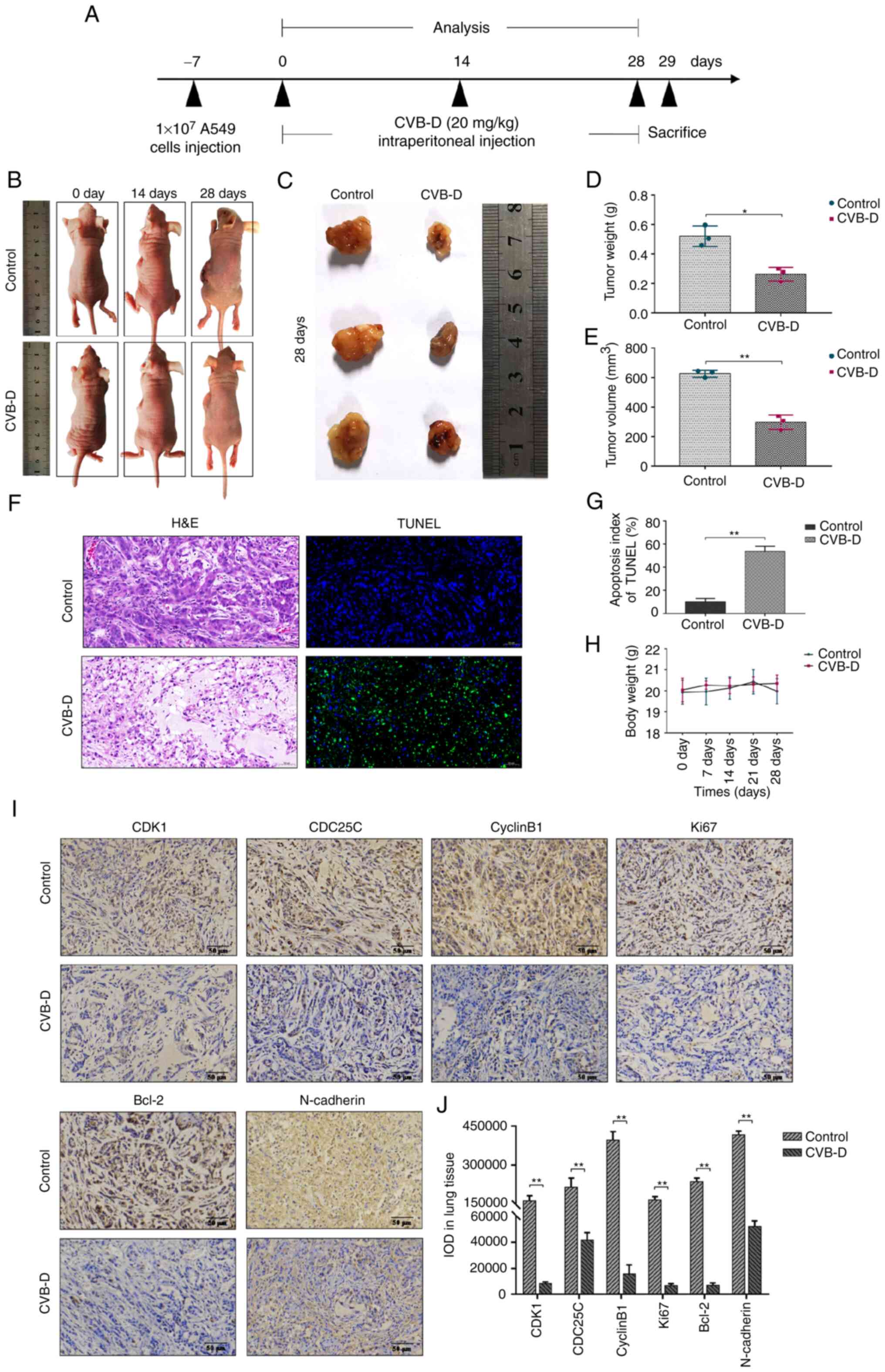 | Figure 7CVB-D inhibits in vivo
progression of A549 xenografts tumors. (A) Experimental design for
tumor xenograft experiments of NSCLC in nude mice to evaluate the
in vivo therapeutic effects of CVB-D. Specific time points
for CVB-D treatment and experimental analysis are indicated. (B)
Representative images of the NSCLC xenograft tumor models with or
without CVB-D treatment. (C) Representative images of the NSCLC
xenograft tumor tissues from control and CVB-D-treated mice. (D and
E) NSCLC xenograft tumor weights and volumes in the control and
CVB-D treatment groups of nude mice. (F) Representative H&E and
TUNEL staining images of the A549 xenograft tumor sections in the
control and CVB-D treatment groups of nude mice (magnification,
×200; scale bar, 50 µm). (G) The proportion of apoptotic
cells in the A549 xenograft tumors derived from the control and
CVB-D-treatment groups of nude mice based on TUNEL staining. (H)
The body weights of the xenograft tumor model mice in the control
and CVB-D treatment groups. (I and J) Immunohistochemical assay
data revealed the expression levels and distribution of CDK1,
CDC25C, cyclinB1, Ki67, Bcl-2 and N-cadherin proteins in the A549
xenograft tumors derived from the control and CVB-D treatment
groups (magnification, ×200; scale bar, 50 µm).
*P<0.05 and **P<0.01 vs. control.
CVB-D, cyclovirobuxine D; NSCLC, non-small cell lung cancer;
H&E, hematoxylin and eosin. |
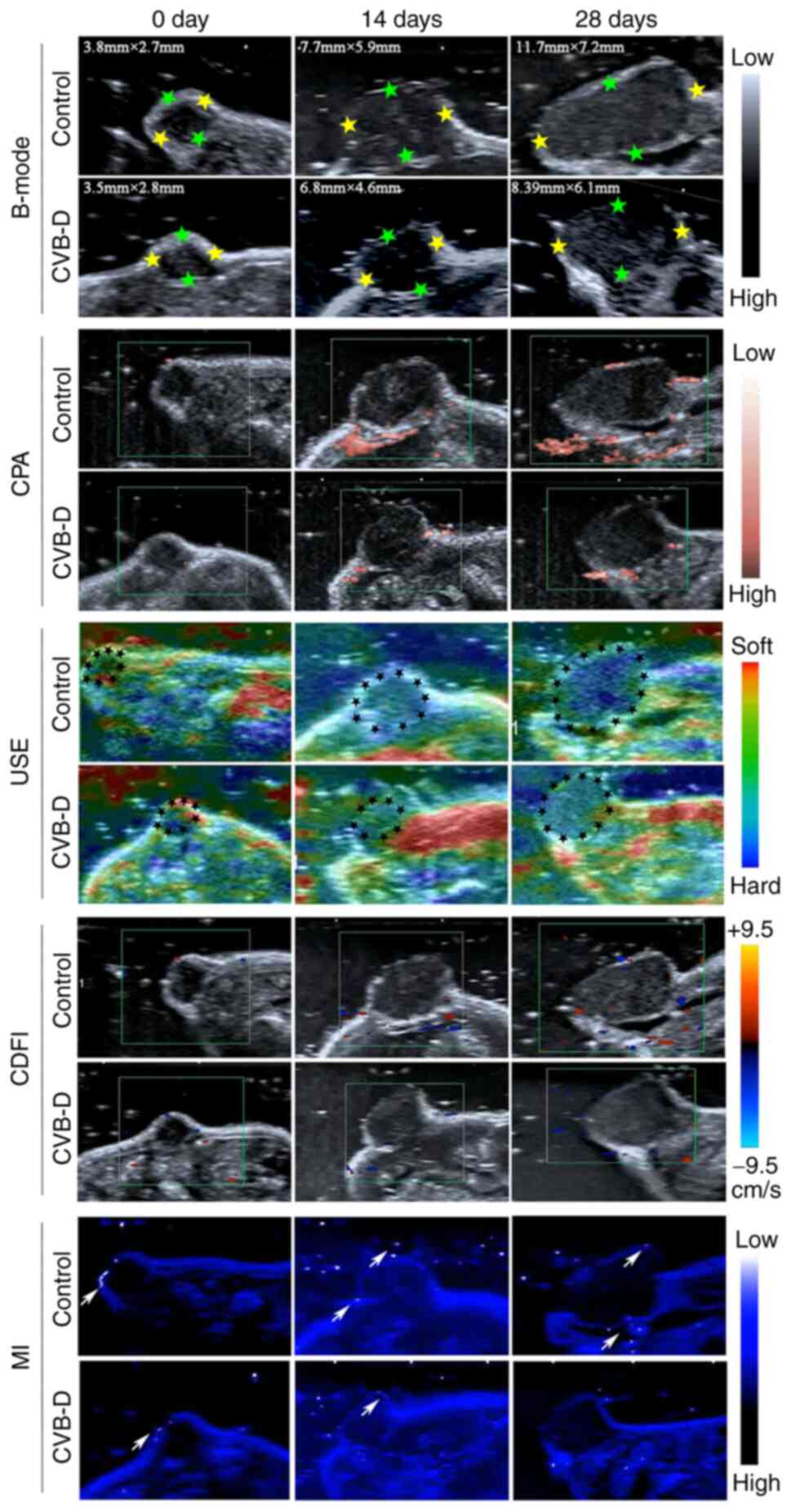
USI is a non-invasive clinical imaging technology
without any side effects and can systematically provide reliable
information regarding the in vivo progression and
therapeutic efficacy of tumor (27). Therefore, USI was used to evaluate
the anti-NSCLC effects of CVB-D in the A549 xenograft nude mice.
B-mode ultrasonography, USE, CDFI, CPA in the USI mode and MI were
used to determine the tumor size, the hardness degree, the presence
of angiogenesis, and microcalcifications, respectively. USI results
(Fig. 8) showed that the average
tumor size of the xenograft tumors was significantly decreased
after CVB-D treatment. CDFI and CPA scans demonstrated that
angiogenesis was reduced in the CVB-D treatment group compared with
the control group. USE data demonstrated that the degree of tumor
hardness, which is closely correlated with tumor malignancy,
decreased after CVB-D treatment. Ultrasonic MI technique was used
to characterize microcalcifications in the A549 xenograft nude
mice. Ultrasonic MI data showed firefly signs and the presence of
multiple hypoechoic areas indicating malignancy in the control
group. However, firefly signs and hyperechoic areas were
significantly reduced in the CVB-D group. These results
demonstrated that CVB-D significantly inhibited growth and
progression of NSCLC cell derived xenograft tumors in the nude
mice.
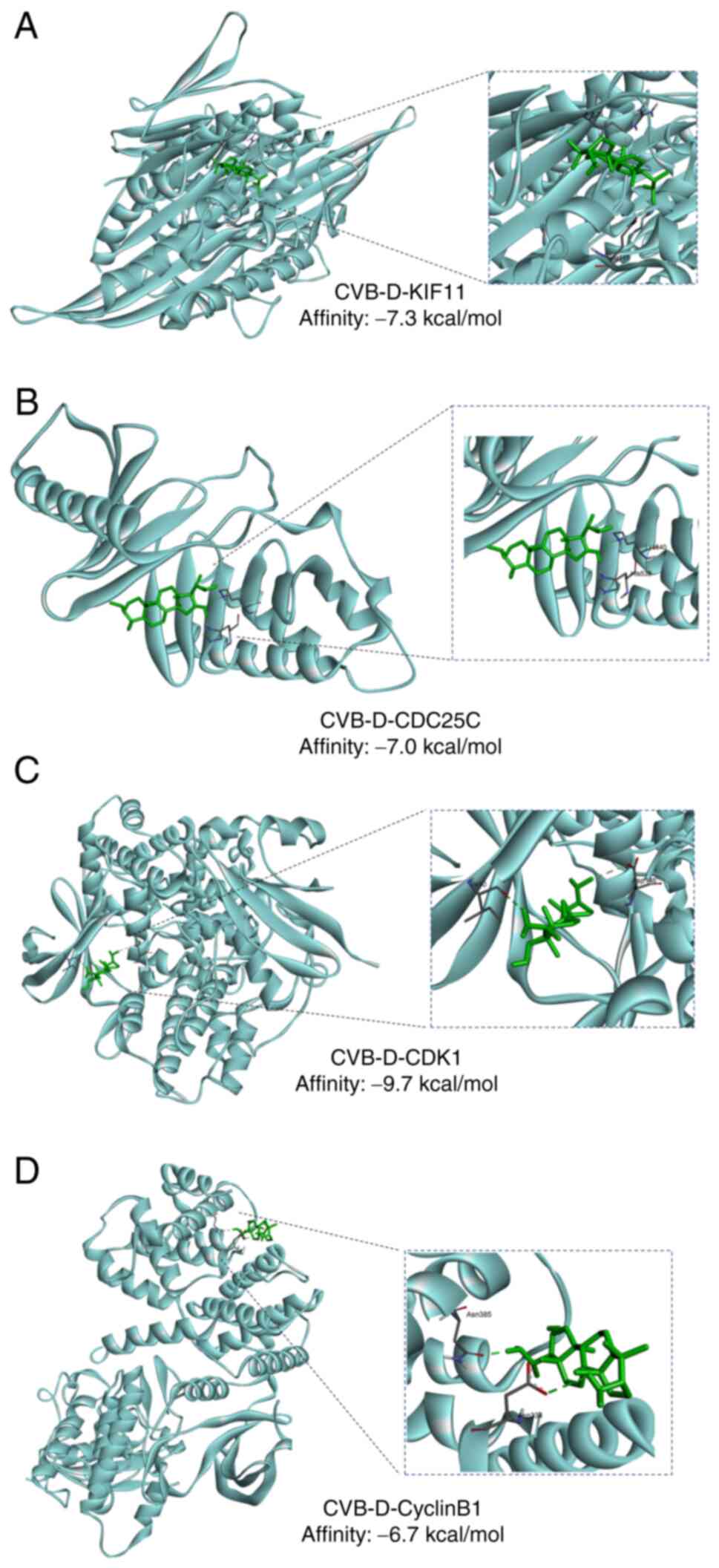
Molecular docking indicates the binding
between CVB-D and KIF11-CDC25C-CDK1-cyclinB1 network
Molecular docking, an established in silico
structure-based method, has been widely used in drug discovery and
prediction of the binding interfaces between a target protein
(enzyme) and drug molecules (ligands) through molecular techniques
(28). AutoDock Vina was used to
perform molecular docking of CVB-D into the crystal structures of
KIF11, CDC25C, CDK1 and cyclinB1. The affinity of the docking pose
was -7.3, -7.0, -9.7 and -6.7 kcal/mol, respectively, which
indicated that there were strong bindings between CVB-D and KIF11,
CDC25C, CDK1 and cyclinB1 (Fig.
9).
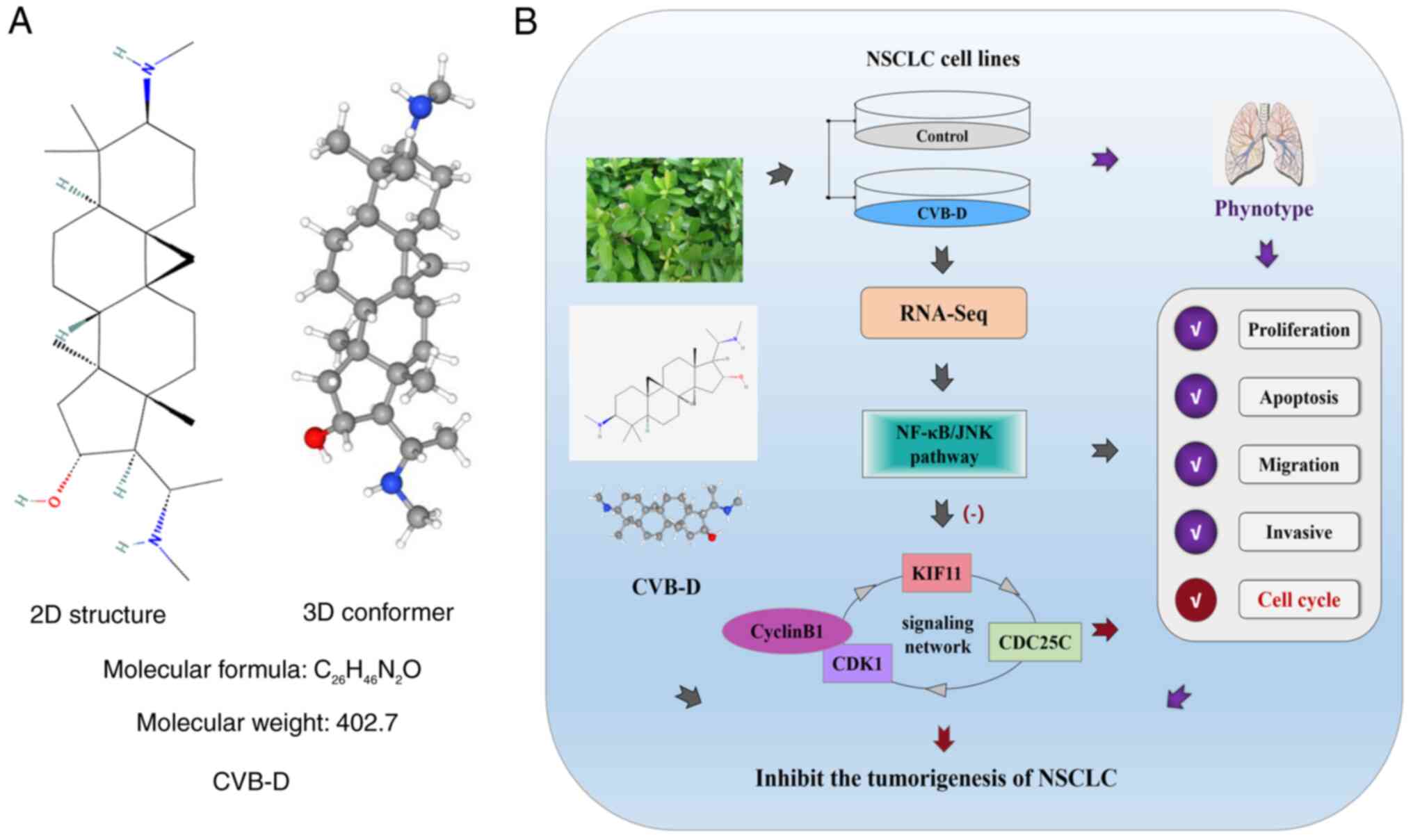
Chemical structure and mechanism of
action of CVB-D in NSCLC cells
The chemical structure and molecular formula of
CVB-D is shown in Fig. 10. The
experimental data in the present study demonstrated that CVB-D
significantly reduced the survival, proliferation, migration and
invasion of the NSCLC cells by negatively regulating the NF-κB/JNK
signaling pathway. Furthermore, CVB-D induced G2/M phase
cell cycle arrest of the NSCLC cells through targeted inhibition of
the oncogenic KIF11-CDC25C-CDK1-cyclinB1 G2/M phase
transition regulatory network. The schematic presentation of the
anti-NSCLC effects of CVB-D and the underlying molecular mechanisms
are demonstrated in Fig. 10.
Discussion
Lung cancer is the leading cause of cancer-related
morbidity and mortality worldwide. Therefore, lung cancer research
is a major focus area of research. NSCLC accounts for >80% of
the lung cancer cases (29). Most
patients with NSCLC are diagnosed with locally advanced or
metastatic disease. Currently, there are no effective screening
measures for diagnosing lung cancer in the preliminary stages.
Furthermore, the prognosis of patients with advanced NSCLC is poor
(30). In the last two decades,
advances in medical technology have significantly improved our
understanding of the pathogenic mechanisms underlying NSCLC growth
and progression and have resulted in the development of novel
therapies including small molecule-targeted antitumor drugs
including tyrosine kinase inhibitors and immunotherapy. Despite
these advances, the survival rates of patients with NSCLC remain
significantly low, particularly for those with metastatic disease
(31). Therefore, there is an
urgent need for continued research and development of novel
chemotherapeutic drugs and alternative agents with fewer side
effects to improve the survival outcomes of patients with NSCLC. In
the present study, the antitumor efficacy and underlying mechanisms
of CVB-D, an NPC derived from B. microphylla, were
investigated using the NSCLC nude mice model. Recent studies have
demonstrated anticancer activities of CVB-D in multiple cancers
(18-20). Furthermore, CVB-D induced mitophagy
in the lung cancer cells by modulating the p65/BNIP3/LC3 signaling
axis (21). However, to the best
of our knowledge, the targets of CVB-D in the lung cancer cells
have not been reported. In the present study, it was demonstrated
that CVB-D inhibited NSCLC growth and progression by downregulating
the MAPK/NF-κB signaling pathway. CVB-D also suppressed growth and
progression of NSCLC cells by suppressing the
KIF11-CDC25C-CDK1-cyclinB1 G2/M phase transition
regulatory network in the NSCLC cells.
EMT is closely associated with tumor invasion and
metastasis, and resistance to chemotherapy and radiotherapy
(32) in various cancers (16). The present study showed that CVB-D
treatment inhibited EMT in the NSCLC cells based on the increased
expression of E-cadherin and decreased expression of mesenchymal
markers including N-cadherin and vimentin. Therefore, CVB-D is a
potential therapeutic drug for inhibiting EMT and progression of
NSCLC. Tumor metastasis is a characteristic feature of malignant
tumors and is associated with poor prognosis since the cancer cells
acquire migratory and invasive characteristics, leave the primary
tumor site and establish metastasis at distant sites (33). In the current study, it was
revealed that CVB-D suppressed migration and invasion of NSCLC
cells in a concentration-dependent manner. the effects of CVB-D on
the lung cancer cells, were evaluated both in vitro and
in vivo, using different experimental approaches. CVB-D
significantly reduced in vitro NSCLC cell proliferation,
colony formation, survival and cell cycle progression in a
dose-dependent manner. USI data showed that CVB-D treatment
decreased in vivo angiogenesis. FCM analysis demonstrated
that CVB-D induced G2/M phase cell cycle arrest of the
NSCLC cells. The expression levels of several pro-apoptotic and
cell cycle-related transcription factors including Bcl-2, Bax and
cyclinB1 were reduced by CVB-D treatment. Bioinformatics analysis
identified that the expression levels of cyclinB1 were
significantly higher in the LUAD tissues compared with the adjacent
para-cancerous tissues. Moreover, high cyclinB1 expression was
associated with tumor progression and shorter OS time. CyclinB1
plays a key role in mitosis by regulating cell cycle progression
through the G2/M phase (34). These results demonstrated that
CVB-D was a potential therapy for NSCLC because it effectively
inhibited EMT, migration and invasion, and G2/M cell
cycling of NSCLC cells.
RNA-seq data is extensively used in clinical
research and therapy to identify pathogenic mechanisms in various
cancers (35). RNA-seq data
analysis and mining is used to identify drug targets and their
mechanisms of action in the human cancer cells including
identification of the oncogenic signaling pathways that are
targeted by the antitumor agents (36). In the present study, RNA-seq data
analysis showed that CVB-D treatment downregulated KIF11, CDC25C
and CDK1 expression levels in the NSCLC cells.
RNA-seq data analysis identified KIF11 was the
potential therapeutic target of CVB-D in the NSCLC cells. KIF11
plays a significant role in spindle formation. Previous studies
have reported that KIF11 is an oncogene in glioblastoma, HCC,
breast, gallbladder and colorectal cancers (6-8,37,38).
Furthermore, high expression of KIF11 is associated with poor
survival outcomes in NSCLC patients (11,39).
Recently, Yang et al (40)
performed robust rank aggregation analysis of several LUAD datasets
and reported that KIF11 was a potential prognostic biomarker
associated with LUAD development. Furthermore, RNA-seq analysis of
paired LUAD and para-cancerous lung tissue samples resulted in the
identification of eight enriched genes including KIF11 belonging to
pathways related to cell division that were potential diagnostic
and prognostic biomarkers for LUAD (41). Schneider et al (9) reported that five mitosis-related
genes including KIF11 correlated with poor prognosis in NSCLC
patients. Liu et al (39)
evaluated The Cancer Genome Atlas-NSCLC datasets and demonstrated
that high KIF11 expression was associated with lymph node
metastases, advanced pathological stages and worse prognosis in
patients with LUAD and NSCLC; moreover, multivariate analysis
demonstrated that KIF11 was an independent prognostic factor for OS
in patients with NSCLC. Wang et al (42) demonstrated that the expression
levels of five DEGs including KIF11 correlated with the prognosis
and progression of bladder cancer. Meng et al (43) and Gong et al (44) compared gene expression profiles of
LUAD and NSCLC datasets in the Gene Expression Omnibus, Oncomine
and GEPIA databases using functional enrichment, PPI network,
DAVID, String, HPA and TfactS analyses and demonstrated that key
cell cycle-related genes including KIF11 were potential biomarkers
in lung cancer. Furthermore, several cell cycle-related genes
including CDKs and KIF11 were downregulated by THZ1 treatment in
the nasopharyngeal carcinoma cell lines; KIF11 expression
correlated with THZ1-induced inhibition of nasopharyngeal carcinoma
cell proliferation (45).
The inhibition of KIF11 by DHPMs significantly
reduced growth, survival and progression of several breast cancer
cell lines (46). Triple-negative
breast cancer (TNBC) is an aggressive and highly heterogenous
subtype that frequently develops resistance to chemotherapy and
lacks effective treatment options. Jiang et al (47) reported that high expression of
KIF11 in the CD44+/CD24− subpopulation of
docetaxel-resistant TNBC cells correlated with shorter DFS; KIF11
silencing suppressed TNBC cell proliferation, migration and
invasion by inducing G2/M cell cycle arrest and
apoptosis in the docetaxel-resistant TNBC xenograft models;
moreover, KIF11 inhibitor SB743921 significantly reduced
docetaxel-resistant TNBC xenograft tumor growth. In another study,
knockdown of KIF11 and Aurora Kinase A induced G2/M cell
cycle arrest in the TNBC cells; moreover, targeted inhibition of
the Id1-KIF11 pathway combined with conventional chemotherapy
achieved improved therapeutic effects for patients with TNBC
(48). KIF11 knockdown
significantly reduced proliferation of malignant pleural
mesothelioma cells (49). The high
expression of KIF11 in colorectal cancer correlated with advanced
clinical tumor stages and vessel invasion; moreover, KIF11
knockdown inhibited growth and increased oxaliplatin sensitivity of
colorectal tumor cells by activating p53 signaling and inhibiting
GSK3β signaling (50).
Furthermore, KIF11 is an independent prognostic factor in LUAD and
is associated with poor OS and worse progression-free survival;
KIF11 silencing significantly reduced proliferation, migration and
invasion of LUAD cells and induced G2/M cell cycle and
apoptosis (11). Therefore, these
data suggested that KIF11 is a potential oncogene and a promising
therapeutic target in NSCLC. However, it remains to be evaluated if
KIF11 inhibitors can be used as an alternative therapy to overcome
resistance to conventional chemotherapeutics.
RNA-seq data also showed that CVB-D reduced CDC25C
levels in the NSCLC cells. CDC25C is an important cyclin that
participates in the regulation of G2/M progression by
mediating DNA damage repair (51).
Activation of the CDK1/cyclinB1 complex triggers mitosis and
progression of the G2/M cell cycle phase is regulated by
CDC25C (52). DNA damage elicits
signals that block mitosis by inhibiting the activation and nuclear
import of CDK1/cyclinB1 (51,52).
CDC25C plays a significant role in regulating checkpoint proteins
and subsequent signal transduction to ensure accurate transmission
of DNA information to the daughter cells (51,52).
Downregulation of CDC25C in response to DNA damage induces cell
cycle arrest in the G2/M phase (51). Several studies have reported that
aberrantly high expression of CDC25C in the lung, gastric, colon,
breast and prostate cancers, is closely related with tumor growth
and metastases, and poor prognosis; therefore, CDC25C is a useful
diagnostic and prognostic biomarker in several malignant tumors
(51).
RNA-seq data also revealed that CVB-D downregulated
CDK1 in the NSCLC cells. CDK1 is an essential cell cycle regulatory
protein that plays a key role in tumorigenesis and is considered as
a promising target for cancer therapy. CyclinB1 is a regulatory
subunit of CDK1, which promotes transition of cells from the
G2 phase into the mitosis (M) phase. The association of
cyclinB1 with CDK1 is required for G2 phase cells to
enter into mitosis (24). The
CDK1/cyclinB1 complex was considered as key regulatory proteins
driving G2 to M phase (24,53).
CDC25C activates the CDK1/cyclinB1 complex and regulates the
progression of the G2/M cell cycle. Furthermore, in the
G2/M phase, positive feedback activation between CDC25C
and CDK1/cyclinB1 promotes cell cycle progression through the
G2 checkpoint (51).
The association between KIF11, CDC25C, CDK1 and
cyclinB1 was analyzed. The Venny online tool was used to analyze
gene expression data of patients with NSCLC from the GEPIA 2 and
GeneCards database and 10 overlapping DEGs were selected, which
were all downregulated by CVB-D treatment in the RNA-seq data,
overexpressed in LUAD tissues and associated with poor OS and/or
DFS rates in patients with LUAD, and were recognized as therapeutic
targets for NSCLC according to the GeneCards database. PPI network
analysis of these 10 overlapping DEGs identified 6 closely related
proteins including 3 oncogenes, namely, KIF11, CDK1 and CDC25C that
exhibited high correlation with LUAD growth and progression. The
present study demonstrated significant correlation between the
expression levels of KIF11 and cyclinB1 in the NSCLC tissues and
cells. Furthermore, Swisstarget prediction database analysis
demonstrated that KIF11 and CDK1 were potential target genes of
CVB-D. And then, these were verified by molecular docking. Finally,
the KIF11-CDC25C-CDK1-cyclinB1 G2/M transition
regulatory network was identified as a potential target of CVB-D in
the NSCLC cells.
RNA-seq data analysis demonstrated that CVB-D
significantly inhibited JNK and NF-κB signaling pathways in the
NSCLC cells. JNKs are MAPKs that transduce extracellular and
intracellular stress and stimulatory signals that regulate cellular
proliferation, differentiation, apoptosis, invasion, cell cycle
arrest and malignant transformation (54-56).
JNK activation promotes survival, proliferation and motility of
HCC, ovarian cancer, and NSCLC cells (57-59).
The inhibitors of JNK signaling pathway suppress cancer cell
proliferation, survival, motility and malignancy. Therefore, JNK
signaling pathway is an attractive target for cancer therapy.
Previous studies have shown that inhibition of the JNK pathway
suppressed the tumor cell viability, growth and progression of
tumor cells by inducing cell cycle arrest and apoptosis (60,61).
JNK pathway inhibitors such as SP600125 promote G2/M
cell cycle arrest and/or apoptosis of human breast carcinoma, HCC,
NSCLC and melanoma cells (56,59,62-64).
JNK activation is associated with cell cycle progression (60). NF-κB is a pleiotropic transcription
factor and a key regulator of innate immune responses and multiple
biological processes involved in cancer initiation, growth and
progression (65,66). NF-κB signaling is constitutively
activated in breast, gastric carcinoma, prostate, cervical, lung
and pancreatic cancer tissues (67). Activation of the NF-κB signaling
pathway correlates with increased proliferation, metastasis,
angiogenesis and survival of cancer cells (67,68).
Furthermore, increased NF-κB activity promotes cancer cell survival
and drug resistance by upregulating anti-apoptotic genes (69).
The combination therapy with NF-κB pathway
inhibitors and classical chemotherapeutics is more effective in
treating malignancies compared with treatment with traditional
chemotherapy (70). Therefore,
NF-κB activation is an important target for effective treatment of
several cancers. NF-κB inhibition improves the efficacy of several
chemotherapeutics (67). NF-κB
signaling pathway is the therapeutic target of several NPCs
including poly phenols, flavonoids, terpenes, phenolics and
alkaloids, which demonstrate anti-proliferative, pro-apoptotic,
anti-angiogenic and anti-metastatic effects (67,68,71).
Plant-derived anticancer agents including curcumin, baicalein and
triptolide strongly inhibit NF-κB activation in the lung cancer
cells, thereby confirming NF-κB is a key therapeutic target in lung
cancer (67,71-73).
Inhibition of NF-κB activity was shown by reduced levels of nuclear
p-p65, the active form of NF-κB.
The results of the bioinformatics analysis were
also confirmed by showing the inhibitory effects of CVB-D on the
expression levels of KIF11, CDC25C, CDK1 and cyclinB1 proteins in
the NSCLC cell lines using western blot analysis and in the
subcutaneous tumor xenografts from the nude mouse model using IHC.
The present study demonstrated that CVB-D inhibited NSCLC
proliferation, metastasis, survival and cell cycle by suppressing
NF-κB/JNK signaling, upregulating pro-apoptotic proteins such as
Bax, inhibition of EMT-related proteins, downregulation of
proliferation-related proteins including PCNA and cyclinB1, and
suppression of anti-apoptotic factors such as Bcl-2. The present
results also suggested that NF-κB and JNK signaling pathways acted
upstream of the CDC25C-CDK1-cyclinB1 axis in the NSCLC cells and
were consistent with the induction of G2/M cell cycle
arrest by CVB-D.
In the current study, it was also identified that
si-RNA-mediated knockdown of KIF11 expression in the NSCLC cell
lines decreased the levels CDC25C, CDK1 and cyclinB1, as well as
p-JNK and p-p65. This suggested that KIF11 was upstream of the
NF-κB/JNK pathway. Furthermore, to ascertain whether KIF11 mediated
the therapeutic functions of CVB-D, rescue experiments were
performed and it was revealed that the therapeutic effects of CVB-D
were more effective in the KIF11-silenced NSCLC cells. Moreover,
si-RNA-mediated silencing of KIF11 partially reversed CVB-D-induced
growth inhibition in the NSCLC cells. These results suggested that
the antitumor effects of CVB-D in the NSCLC cells were mediated by
suppression of the KIF11-CDC25C-CDK1-cyclinB1 G2/M phase
transition regulatory axis. Furthermore, the antitumor mechanism of
CVB-D was a KIF11-dependent mechanism with CDC25C/CDK1/cyclinB1
acting as could be downstream targets of KIF11.
Lastly, the antitumor effects of CVB-D were further
corroborated through the KIF11-CDC25C-CDK1-cyclinB1 G2/M
phase transition regulatory axis by evaluating the expression
levels of KIF11, CDC25C and CDK1 in cyclinB1-silenced NSCLC cells.
CyclinB1 silencing significantly reduced the expression levels of
KIF11 CDC25C, and CDK1. This was consistent with the results of a
previous study by Saijo et al (74) that showed significant correlation
between the expression levels of KIF11 and cyclinB1. It was
postulated by the authors that the knockdown of cyclinB1 disrupted
the formation of the CDK1/cyclinB1 complex that was required for
transitioning from G2 phase into the mitotic phase,
thereby resulting in G2/M phase arrest. Moreover,
cyclinB1 silencing downregulated the expression of a key
G2/M regulatory protein CDC25C. Hence, KIF11, CDC25C and
CDK1 contributed to the G2/M phase cell cycle arrest of
cyclinB1-silenced NSCLC cells. Additionally, this suggested that
the KIF11-CDC25C-CDK1 signaling axis was regulated by cyclinB1.
These findings also showed the positive regulatory loop of the
KIF11-CDC25C-CDK1-cyclinB1 signaling axis, which is a potential
therapeutic target of CVB-D in the NSCLC cells.
Currently, platinum compounds including cisplatin
and carboplatin are the first-line chemo therapeutics for patients
with NSCLC either alone or in combination with other therapies.
However, resistance to platinum therapeutics is a major cause of
relapse in a large proportion of patients with NSCLC.
Chemotherapeutic resistance is caused by alterations in pathways
that regulate apoptosis, hypoxia, EMT and metabolism (75). Therefore, therapeutic strategies
for relapsed NSCLC patients include therapies that induce tumor
cell apoptosis through upregulation of Bax and inhibition of Bcl-2,
inhibition of hypoxia, glycolytic metabolism and EMT, as well as
suppression of oncogenic signaling pathways including MAPK/JNK and
NF-κB (75-77), and increase the sensitivity to
cisplatin. Paclitaxel (PTX) is a first-line drug for the treatment
of advanced patients with NSCLC. However, advanced stage NSCLC
patients eventually develop resistance to prolonged treatment with
PTX. Previous studies reported that PTX resistance of NSCLC cells
can be overcome by downregulating the anti-apoptotic proteins,
upregulating the pro-apoptotic proteins, and inhibiting the
expression of drug-resistance genes and EMT-related genes, which
are regulated by the NF-κB/MAPKs signaling pathways (78-80).
Furthermore, interference with spindle formation improves the PTX
susceptibility of NSCLC cells (78). KIF11 plays a significant role in
spindle formation. KIF11 inhibitors overcome adverse effects
associated with classical microtubule-targeting agents such as PTX,
and show potential to overcome resistance to PTX (5). KIF11 inhibitors also sensitize NSCLC
cells that are resistant to trametinib (MEK1/2 inhibitor) and
induce anti-proliferative and anti-angiogenesis effects by
inhibiting spindle formation during cell division (5). These data suggested that KIF11 is a
promising therapeutic target. Therefore, future pre-clinical and
clinical studies are required to test the efficacy of KIF11
inhibitors as later-line therapy of relapsed NSCLC patients or in
combination with first-line chemotherapeutics for the treatment of
newly diagnosed NSCLC patients.
This is an era of precision medicine and
significant advances have been reported regarding the use of
nanoparticles (NPs) to ensure efficient and safe delivery of
chemotherapeutic antitumor drugs to specific tumor sites (81). Therefore, in the future, NPs loaded
with CVB-D may be effective for treating a broad category of
patients with NSCLC.
There are certain limitations to the present study.
First, the sample size of the lung cancer xenograft nude mice model
was small due to animal ethics limitations. However, despite this
issue, the experimental results were consistent. Second, the
differences in the efficiency of the siRNAs against KIF11 and
cyclinB1 may be due to off-target effects.
In summary, it was demonstrated that CVB-D, a
natural steroidal alkaloid, was highly effective in suppressing the
in vitro and in vivo growth, survival and progression
of NSCLC cells by inducing G2/M phase cell cycle arrest
and apoptosis. RNA-seq data analysis and in vitro and in
vivo experiments demonstrated that CVB-D inhibited the
KIF11-CDC25C-CDK1-cyclinB1 G2/M phase transition
regulatory network and the NF-κB/JNK signaling pathway. Therefore,
the present study demonstrated that CVB-D was a promising targeted
therapy for lung cancer.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed in the present
study are available from the corresponding author on reasonable
request. The original RNA-seq data in the present study has been
deposited in the NCBI Sequence Read Archive (SRA) with the
associated accession number PRJNA861972.
Authors' contributions
TX and YC conceived and designed the study. TX
performed most of the experiments. YC and TX performed the USI
experiments. JX guided the experiments related to the phenotype of
cells. WD and TX performed the pathology experiments. PK guided the
experiments related to IHC. TX analyzed the data and wrote the
manuscript. XZ and PK reviewed and revised the manuscript. TX, YC
and XZ supervised the project and provided funding. TX and YC
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All the animal experiments and protocols were
approved (approval no. 2021-114) by the Animal Ethics Committee of
the First Hospital of Shanxi Medical University (Taiyuan, China)
and were conducted according to the national regulations in
China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the Basic Research Projects
of Natural Sciences in Shanxi (grant. no. 20210302124039), the
Scientific and Technological Innovation Programs of Higher
Education Institutions in Shanxi (grant nos. 2020L0201 and
2020L0223), the National Natural Scientific Foundation of China
(grant. nos. 32200168 and 82001850) and the Start-up Foundation for
Doctoral Scientific Research of Shanxi Medical University (grant.
no. XD1905).
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Inamura K: Lung Cancer: Understanding its
molecular pathology and the 2015 WHO Classification. Front Oncol.
7:1932017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Travis WD, Brambilla E, Burke AP, Marx A
and Nicholson AG: Introduction to the 2015 World Health
Organization Classification of tumors of the lung, pleura, thymus,
and heart. J Thorac Oncol. 10:1240–1242. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu WJ, Du Y, Wen R, Yang M and Xu J: Drug
resistance to targeted therapeutic strategies in Non-small cell
lung cancer. Pharmacol Ther. 206:1074382020. View Article : Google Scholar
|
|
5
|
Garcia-Saez I and Skoufias DA: Eg5
targeting agents: From new anti-mitotic based inhibitor discovery
to cancer therapy and resistance. Biochem Pharmacol.
184:1143642021. View Article : Google Scholar
|
|
6
|
Wei D, Rui B, Qingquan F, Chen C, Ping HY,
Xiaoling S, Hao W and Jun G: KIF11 promotes cell proliferation via
ERBB2/PI3K/AKT signaling pathway in gallbladder cancer. Int J Biol
Sci. 17:514–526. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Neska-Dlugosz I, Buchholz K, Durslewicz J,
Gagat M, Grzanka D, Tojek K and Klimaszewska-Wisniewska A:
Prognostic impact and functional annotations of KIF11 and KIF14
expression in patients with colorectal cancer. Int J Mol Sci.
22:97322021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li TF, Zeng HJ, Shan Z, Ye RY, Cheang TY,
Zhang YJ, Lu SH, Zhang Q, Shao N and Lin Y: Overexpression of
kinesin super-family members as prognostic biomarkers of breast
cancer. Cancer Cell Int. 20:1232020. View Article : Google Scholar
|
|
9
|
Schneider MA, Christopoulos P, Muley T,
Warth A, Klingmueller U, Thomas M, Herth FJ, Dienemann H, Mueller
NS, Theis F and Meister M: AURKA, DLGAP5, TPX2, KIF11 and CKAP5:
Five specific mitosis-associated genes correlate with poor
prognosis for non-small cell lung cancer patients. Int J Oncol.
50:365–372. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu F, Zhang Y, Gao Z, Zhao Y, Wen Z, Han
H, Li Y and Chen H: Development and validation of a five-gene model
to predict post-operative brain metastasis in operable lung
adenocarcinoma. Int J Cancer. 147:584–592. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Z, Yu B, Qi F and Li F: KIF11 Serves as
an independent prognostic factor and therapeutic target for
patients with lung adenocarcinoma. Front Oncol. 11:6702182021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Liu X, Li J and Ren F:
Identification and integrated analysis of key biomarkers for
diagnosis and prognosis of non-small cell lung cancer. Med Sci
Monit. 25:9280–9289. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Ma L, Su M, Zhou Y, Mao K, Li C,
Peng G, Zhou C, Shen B and Dou J: Baicalin induces cellular
senescence in human colon cancer cells via upregulation of DEPP and
the activation of Ras/Raf/MEK/ERK signaling. Cell Death Dis.
9:2172018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pricci M, Girardi B, Giorgio F, Losurdo G,
Ierardi E and Di Leo A: Curcumin and colorectal cancer: From basic
to clinical evidences. Int J Mol Sci. 21:23642020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rauf A, Imran M, Butt MS, Nadeem M, Peters
DG and Mubarak MS: Resveratrol as an anti-cancer agent: A review.
Crit Rev Food Sci Nutr. 58:1428–1447. 2018. View Article : Google Scholar
|
|
16
|
Erin N, Grahovac J, Brozovic A and Efferth
T: Tumor microenvironment and epithelial mesenchymal transition as
targets to overcome tumor multidrug resistance. Drug Resist Updat.
53:1007152020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang Z, Fu L, Xu Y, Hu X, Yang H, Zhang
Y, Luo H, Gan S, Tao L, Liang G and Shen X: Cyclovirobuxine D
protects against diabetic cardiomyopathy by activating
Nrf2-mediated antioxidant responses. Sci Rep. 10:64272020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou L, Tang H, Wang F, Ou S, Wu T, Fang
Y, Xu J and Guo K: Cyclovirobuxine D inhibits cell proliferation
and migration and induces apoptosis in human glioblastoma
multiforme and lowgrade glioma. Oncol Rep. 43:807–816.
2020.PubMed/NCBI
|
|
19
|
Zhang J, Chen Y, Lin J, Jia R, An T, Dong
T, Zhang Y and Yang X: Cyclovirobuxine D exerts anticancer effects
by suppressing the EGFR-FAK-AKT/ERK1/2-slug signaling pathway in
human hepatocellular carcinoma. DNA Cell Biol. 39:355–367. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang F, Chen Y, Ren S, Li Z, Sun K, Xing
Y, Zhu Y and Piao D: Cyclovirobuxine D inhibits colorectal cancer
tumorigenesis via the CTHRC1AKT/ERKSnail signaling pathway. Int J
Oncol. 57:183–196. 2020.PubMed/NCBI
|
|
21
|
Zeng C, Zou T, Qu J, Chen X, Zhang S and
Lin Z: Cyclovirobuxine D Induced-mitophagy through the
p65/BNIP3/LC3 axis potentiates its apoptosis-inducing effects in
lung cancer cells. Int J Mol Sci. 22:58202021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
23
|
Creamer-Hente MA, Lao FK, Dragos ZP and
Waterman LL: Sex- and strain-related differences in the stress
response of mice to CO2 euthanasia. J Am Assoc Lab Anim
Sci. 57:513–519. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie B, Wang S, Jiang N and Li JJ: Cyclin
B1/CDK1-regulated mitochondrial bioenergetics in cell cycle
progression and tumor resistance. Cancer Lett. 443:56–66. 2019.
View Article : Google Scholar :
|
|
25
|
Cardano M, Tribioli C and Prosperi E:
Targeting proliferating cell nuclear antigen (PCNA) as an effective
strategy to inhibit tumor cell proliferation. Curr Cancer Drug
Targets. 20:240–252. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brabletz S, Schuhwerk H, Brabletz T and
Stemmler MP: Dynamic EMT: A multi-tool for tumor progression. EMBO
J. 40:e1086472021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen Y, Liu P, Sun P, Jiang J, Zhu Y, Dong
T, Cui Y, Tian Y, An T, Zhang J, et al: Oncogenic MSH6-CXCR4-TGFB1
feedback loop: A novel therapeutic target of photothermal therapy
in glioblastoma multiforme. Theranostics. 9:1453–1473. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pinzi L and Rastelli G: Molecular docking:
Shifting paradigms in drug discovery. Int J Mol Sci. 20:43312019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Socinski MA, Obasaju C, Gandara D, Hirsch
FR, Bonomi P, Bunn P, Kim ES, Langer CJ, Natale RB, Novello S, et
al: Clinicopathologic features of advanced squamous NSCLC. J Thorac
Oncol. 11:1411–1422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Duma N, Santana-Davila R and Molina JR:
Non-small cell lung cancer: Epidemiology, screening, diagnosis, and
treatment. Mayo Clin Proc. 94:1623–1640. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Herbst RS, Morgensztern D and Boshoff C:
The biology and management of non-small cell lung cancer. Nature.
553:446–454. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pastushenko I and Blanpain C: EMT
Transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar
|
|
33
|
Majidpoor J and Mortezaee K: Steps in
metastasis: An updated review. Med Oncol. 38:32021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gavet O and Pines J: Progressive
activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell.
18:533–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hong M, Tao S, Zhang L, Diao LT, Huang X,
Huang S, Xie SJ, Xiao ZD and Zhang H: RNA sequencing: New
technologies and applications in cancer research. J Hematol Oncol.
13:1662020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Paczkowska M, Barenboim J, Sintupisut N,
Fox NS, Zhu H, Abd-Rabbo D, Mee MW, Boutros PC, Drivers P; PCAWG
Drivers and Functional Interpretation Working Group; et al:
Integrative pathway enrichment analysis of multivariate omics data.
Nat Commun. 11:7352020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Venere M, Horbinski C, Crish JF, Jin X,
Vasanji A, Major J, Burrows AC, Chang C, Prokop J, Wu Q, et al: The
mitotic kinesin KIF11 is a driver of invasion, proliferation, and
self-renewal in glioblastoma. Sci Transl Med. 7:304ra1432015.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hu ZD, Jiang Y, Sun HM, Wang JW, Zhai LL,
Yin ZQ and Yan J: KIF11 promotes proliferation of hepatocellular
carcinoma among patients with liver cancers. Biomed Res Int.
2021:26767452021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu J, Tian Y, Yi L, Gao Z, Lou M and Yuan
K: High KIF11 expression is associated with poor outcome of NSCLC.
Tumori. 108:40–46. 2022. View Article : Google Scholar
|
|
40
|
Yang Y, Zhang S and Guo L:
Characterization of cell cycle-related competing endogenous RNAs
using robust rank aggregation as prognostic biomarker in lung
adenocarcinoma. Front Oncol. 12:8073672022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li S, Xuan Y, Gao B, Sun X, Miao S, Lu T,
Wang Y and Jiao W: Identification of an eight-gene prognostic
signature for lung adenocarcinoma. Cancer Manag Res. 10:3383–3392.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang L, Chen S, Luo Y, Yuan L, Peng T,
Qian K, Liu X, Xiao Y and Wang X: Identification of several cell
cycle relevant genes highly correlated with the progression and
prognosis of human bladder urothelial tumor. J Cell Physiol.
234:13439–13451. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Meng F, Zhang L, Ren Y and Ma Q:
Transcriptome analysis reveals key signature genes involved in the
oncogenesis of lung cancer. Cancer Biomark. 29:475–482. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gong K, Zhou H, Liu H, Xie T, Luo Y, Guo
H, Chen J, Tan Z, Yang Y and Xie L: Identification and integrate
analysis of key biomarkers for diagnosis and prognosis of non-small
cell lung cancer based on bioinformatics analysis. Technol Cancer
Res Treat. 20:153303382110602022021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gao L, Xia S, Zhang K, Lin C, He X and
Zhang Y: Gene expression profile of THZ1-treated nasopharyngeal
carcinoma cell lines indicates its involvement in the inhibition of
the cell cycle. Transl Cancer Res. 10:445–460. 2021. View Article : Google Scholar
|
|
46
|
Guido BC, Ramos LM, Nolasco DO, Nobrega
CC, Andrade BY, Pic-Taylor A, Neto BA and Correa JR: Impact of
kinesin Eg5 inhibition by 3,4-dihydropyrimidin-2(1H)-one
derivatives on various breast cancer cell features. BMC Cancer.
15:2832015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jiang M, Zhuang H, Xia R, Gan L, Wu Y, Ma
J, Sun Y and Zhuang Z: KIF11 is required for proliferation and
self-renewal of docetaxel resistant triple negative breast cancer
cells. Oncotarget. 8:92106–92118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Thankamony AP, Murali R, Karthikeyan N,
Varghese BA, Teo WS, McFarland A, Roden DL, Holliday H, Konrad CV,
Cazet A, et al: Targeting the Id1-Kif11 Axis in triple-negative
breast cancer using combination therapy. Biomolecules. 10:12952020.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kato T, Lee D, Wu L, Patel P, Young AJ,
Wada H, Hu HP, Ujiie H, Kaji M, Kano S, et al: Kinesin family
members KIF11 and KIF23 as potential therapeutic targets in
malignant pleural mesothelioma. Int J Oncol. 49:448–456. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhou Y, Yang L, Xiong L, Wang K, Hou X, Li
Q, Kong F, Liu X and He J: KIF11 is upregulated in colorectal
cancer and silencing of it impairs tumor growth and sensitizes
colorectal cancer cells to oxaliplatin via p53/GSK3beta signaling.
J Cancer. 12:3741–3753. 2021. View Article : Google Scholar :
|
|
51
|
Liu K, Zheng M, Lu R, Du J, Zhao Q, Li Z,
Li Y and Zhang S: The role of CDC25C in cell cycle regulation and
clinical cancer therapy: A systematic review. Cancer Cell Int.
20:2132020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Takizawa CG and Morgan DO: Control of
mitosis by changes in the subcellular location of cyclin-B1-Cdk1
and Cdc25C. Curr Opin Cell Biol. 12:658–665. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang JN, Zhang ZR, Che Y, Yuan ZY, Lu ZL,
Li Y, Li N, Wan J, Sun HD, Sun N, et al: Acetyl-macrocalin B, an
ent-kaurane diterpenoid, initiates apoptosis through the
ROS-p38-caspase 9-dependent pathway and induces G2/M phase arrest
via the Chk1/2-Cdc25C-Cdc2/cyclin B axis in non-small cell lung
cancer. Cancer Biol Ther. 19:609–621. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu Q, Wu W, Jacevic V, Franca TCC, Wang X
and Kuca K: Selective inhibitors for JNK signalling: A potential
targeted therapy in cancer. J Enzyme Inhib Med Chem. 35:574–583.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Bubici C and Papa S: JNK signalling in
cancer: In need of new, smarter therapeutic targets. Br J
Pharmacol. 171:24–37. 2014. View Article : Google Scholar :
|
|
56
|
Abdelrahman KS, Hassan HA, Abdel-Aziz SA,
Marzouk AA, Narumi A, Konno H and Abdel-Aziz M: JNK signaling as a
target for anticancer therapy. Pharmacol Rep. 73:405–434. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Song W, Ma Y, Wang J, Brantley-Sieders D
and Chen J: JNK signaling mediates EPHA2-dependent tumor cell
proliferation, motility, and cancer stem cell-like properties in
non-small cell lung cancer. Cancer Res. 74:2444–2454. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Dou Y, Jiang X, Xie H, He J and Xiao S:
The Jun N-terminal kinases signaling pathway plays a 'seesaw' role
in ovarian carcinoma: A molecular aspect. J Ovarian Res. 12:992019.
View Article : Google Scholar
|
|
59
|
Wang J and Tai G: Role of C-Jun N-terminal
kinase in hepatocellular carcinoma development. Target Oncol.
11:723–738. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gutierrez GJ, Tsuji T, Cross JV, Davis RJ,
Templeton DJ, Jiang W and Ronai ZA: JNK-mediated phosphorylation of
Cdc25C regulates cell cycle entry and G(2)/M DNA damage checkpoint.
J Biol Chem. 285:14217–14228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Udden SN, Kwak YT, Godfrey V, Khan MAW,
Khan S, Loof N, Peng L, Zhu H and Zaki H: NLRP12 suppresses
hepatocellular carcinoma via downregulation of cJun N-terminal
kinase activation in the hepatocyte. Elife. 8:e403962019.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Han YH, Mun JG, Jeon HD, Park J, Kee JY
and Hong SH: Gomisin A ameliorates metastatic melanoma by
inhibiting AMPK and ERK/JNK-mediated cell survival and metastatic
phenotypes. Phytomedicine. 68:1531472020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Okada M, Shibuya K, Sato A, Seino S,
Watanabe E, Suzuki S, Seino M and Kitanaka C: Specific role of JNK
in the maintenance of the tumor-initiating capacity of A549 human
non-small cell lung cancer cells. Oncol Rep. 30:1957–1964. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cheng J, Li M, Tzeng CM, Gou X and Chen S:
Suppression of tumorigenicity 5 ameliorates tumor characteristics
of invasive breast cancer cells via ERK/JNK Pathway. Front Oncol.
11:6215002021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Baud V and Karin M: Is NF-kappaB a good
target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov.
8:33–40. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Hoesel B and Schmid JA: The complexity of
NF-κB signaling in inflammation and cancer. Mol Cancer. 12:862013.
View Article : Google Scholar
|
|
67
|
Rasmi RR, Sakthivel KM and Guruvayoorappan
C: NF-κB inhibitors in treatment and prevention of lung cancer.
Biomed Pharmacother. 130:1105692020. View Article : Google Scholar
|
|
68
|
Sarkar FH and Li Y: NF-kappaB: A potential
target for cancer chemoprevention and therapy. Front Biosci.
13:2950–2959. 2008. View
Article : Google Scholar
|
|
69
|
Fouani L, Kovacevic Z and Richardson DR:
Targeting oncogenic nuclear factor Kappa B Signaling with
redox-active agents for cancer treatment. Antioxid Redox Signal.
30:1096–1123. 2019. View Article : Google Scholar
|
|
70
|
Suhail M, Tarique M, Muhammad N, Naz H,
Hafeez A, Zughaibi TA, Kamal MA and Rehan M: A critical
transcription factor NF-κB as a cancer therapeutic target and its
inhibitors as cancer treatment options. Curr Med Chem.
28:4117–4132. 2021. View Article : Google Scholar
|
|
71
|
Khan H, Ullah H, Castilho P, Gomila AS,
D'Onofrio G, Filosa R, Wang F, Nabavi SM, Daglia M, Silva AS, et
al: Targeting NF-κB signaling pathway in cancer by dietary
polyphenols. Crit Rev Food Sci Nutr. 60:2790–2800. 2020. View Article : Google Scholar
|
|
72
|
Rajagopal C, Lankadasari MB, Aranjani JM
and Harikumar KB: Targeting oncogenic transcription factors by
polyphenols: A novel approach for cancer therapy. Pharmacol Res.
130:273–291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ashrafizadeh M, Najafi M, Makvandi P,
Zarrabi A, Farkhondeh T and Samarghandian S: Versatile role of
curcumin and its derivatives in lung cancer therapy. J Cell
Physiol. 235:9241–9268. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Saijo T, Ishii G, Ochiai A, Yoh K, Goto K,
Nagai K, Kato H, Nishiwaki Y and Saijo N: Eg5 expression is closely
correlated with the response of advanced non-small cell lung cancer
to antimitotic agents combined with platinum chemotherapy. Lung
Cancer. 54:217–225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Lv P, Man S, Xie L, Ma L and Gao W:
Pathogenesis and therapeutic strategy in platinum resistance lung
cancer. Biochim Biophys Acta Rev Cancer. 1876:1885772021.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang LH, Li Y, Yang SN, Wang FY, Hou Y,
Cui W, Chen K, Cao Q, Wang S, Zhang TY, et al: Gambogic acid
synergistically potentiates cisplatin-induced apoptosis in
non-small-cell lung cancer through suppressing NF-κB and MAPK/HO-1
signalling. Br J Cancer. 110:341–352. 2014. View Article : Google Scholar
|
|
77
|
Wang J, Tian L, Khan MN, Zhang L, Chen Q,
Zhao Y, Yan Q, Fu L and Liu J: Ginsenoside Rg3 sensitizes hypoxic
lung cancer cells to cisplatin via blocking of NF-κB mediated
epithelial-mesenchymal transition and stemness. Cancer Lett.
415:73–85. 2018. View Article : Google Scholar
|
|
78
|
Cui H, Arnst K, Miller DD and Li W: Recent
advances in elucidating paclitaxel resistance mechanisms in
non-small cell lung cancer and strategies to overcome drug
resistance. Curr Med Chem. 27:6573–6595. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jiang N, Dong XP, Zhang SL, You QY, Jiang
XT and Zhao XG: Triptolide reverses the Taxol resistance of lung
adenocarcinoma by inhibiting the NF-κB signaling pathway and the
expression of NF-κB-regulated drug-resistant genes. Mol Med Rep.
13:153–159. 2016. View Article : Google Scholar
|
|
80
|
Li DD, Qin XC, Yang Y, Chu HX, Li RL, Ma
LX, Ding HW and Zhao QC: Daurinoline suppressed the migration and
invasion of chemo-resistant human non-small cell lung cancer cells
by reversing EMT and Notch-1 and sensitized the cells to Taxol.
Environ Toxicol Pharmacol. 66:109–115. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li Y and Zhang H: Nanoparticle-based drug
delivery systems for enhanced tumor-targeting treatment. J Biomed
Nanotechnol. 15:1–27. 2019. View Article : Google Scholar
|