Introduction
Glioblastoma (GBM; WHO grade IV glioma) is the most
common, malignant and aggressive form of primary brain tumor
affecting adults, accounting for ~50% of diagnosed gliomas each
year (1). Patients with GBM
present with a median survival time of only 15-20 months, with only
5-10% of patients surviving after 5 years (2). Despite the current multimodal
standard of care, which consists of maximal surgical resection
followed by radiotherapy and chemotherapy with the DNA alkylating
agent, temozolomide (TMZ), the overall prognosis of patients
remains poor, underscoring the need for a more in-depth
understanding of tumor biology to inspire the identification of
novel therapeutic targets. Contributing to tumor aggressiveness and
universal recurrence are GBM hallmarks, including but not limited
to: Rapid, diffuse invasion into the surrounding brain parenchyma,
substantial chemo- and radioresistance and rapid adaptation to
microenvironmental stressors, such as hypoxia (3,4).
The amplification of the epidermal growth factor
receptor (EGFR), one of the most common genetic
abnormalities observed in GBM tumors (5), leads to increased proliferative and
anti-apoptotic signaling, as well as invasive behavior (6,7).
Tumors that exhibit EGFR amplification also frequently
present with the constitutively active EGFR variant 3 (EGFRvIII)
mutant (5), which arises from the
deletion of exons 2-7 of the EGFR gene, and results in a
truncated, yet constitutively active, EGFR protein (6). The resultant increased downstream
signaling confers enhanced glioma malignancy through multiple
mechanisms, and, notably, the EGFRvIII mutation is not expressed by
non-malignant tissues (6).
However, although targeting EGFRvIII is a rational strategy to
combat GBM, phase II clinical trials with the EGFR receptor
tyrosine kinase inhibitor, erlotinib, and phase III clinical trials
with the EGFRvIII vaccine, rindopepimut, have failed to robustly
increase patient overall survival (OS), highlighting the immense
plasticity of GBM cell populations, which diminishes treatment
efficacies and hinders tumor management (5,8,9).
The coiled-coil-helix-coiled-coil-helix
domain-containing protein 2 (CHCHD2), initially described as a
regulator of mitochondrial respiration, has recently emerged in the
contexts of non-small cell lung and renal cell carcinoma, as well
as breast cancer (10-12). The CHCHD2 gene is located
proximal to EGFR on chromosome 7p11.2; as such,
CHCHD2 and EGFR are frequently co-amplified in
non-small cell lung carcinoma (NSCLC) (10). CHCHD2 encodes a 16-kDa
protein belonging to a family of nine evolutionarily conserved
small mitochondrial proteins, all containing at least one CHCH
domain (13,14). The CHCH domain, characterized by
two CX9C motifs (two cysteines separated by nine amino
acids), is necessary for simultaneous oxidative folding and protein
import into the mitochondrial intermembrane space via the
CHCHD4-mediated disulfide relay system (15). CHCHD2 canonically functions as a
mitochondrial protein mediating cellular respiration (15-17).
In addition, CHCHD2 regulates other cellular functions across a
variety of biological contexts, including cell migration and the
regulation of apoptosis (10,18,19).
Furthermore, CHCHD2 has been implicated in mitonuclear
communication through its ability to function as a nuclear
transcription factor in response to hypoxia, inducing the
expression of complex IV subunit 4 isoform 2 and itself maximally
at 4% O2 (20).
However, the subcellular localization, distribution and dynamics of
CHCHD2 in GBM cells in response to hypoxia has yet not been
described, at least to the best of our knowledge. Additionally, a
mechanism governing its mitochondrial export and subcellular
redistribution remains elusive. Thus, the functional capabilities
of CHCHD2 in the context of GBM remain unexplored.
The objective of the present study was to
characterize the functional capacity of CHCHD2 in GBM cells
expressing EGFRvIII, as well as investigate the intracellular
dynamics of CHCHD2 in response to metabolic stressors in U87 and
U87vIII GBM cells. The results obtained herein indicate that
subcellular distribution of CHCHD2 between mitochondria and nuclei
is sensitive to the expression of EGFRvIII and hypoxia, and that
CHCHD2 contributes to a number of GBM cell functions representing
disease hallmarks, which may inspire therapeutic strategies
targeting mitochondrial biology to potentially improve GBM tumor
management.
Materials and methods
CHCHD2 gene amplification and mRNA
expression analysis
The analysis of CHCHD2 gene amplification
patterns across GBM tumors was performed on The Cancer Genome Atlas
(TCGA) Provisional GBM database using cBioPortal (http://www.cbioportal.org) (21,22).
The analysis was limited to tumor samples with available copy
number alteration (CNA) data (n=577). The analysis of CHCHD2 mRNA
expression levels (HG-U133A Array) was compared among GBM tumors
using publicly available data via GlioVis (gliovis.bioinfo.cnio.es)
(23). Tumors were stratified by
variables, including tumor grade, tumor vs. non-tumor tissue, GBM
subtype, glioma CpG island methylator phenotype (G-CIMP) status,
isocitrate dehydrogenase 1 (IDH1) mutational status and
O6-methylguanine-DNA methyl-transferase (MGMT)
mutational status.
Cells and cell culture
The human U87 parental GBM cell line, as well as U87
GBM cells transduced to stably express the constitutively active
EGFRvIII mutant (U87vIII) were generously provided by Dr. Nathan
Price (Institute for Systems Biology, Seattle, WA, USA). The
U87vIII cell line used in these studies was authenticated using STR
profiling, as having a >80% identity with U-87MG ATCC
(RRID:CVCL_0022), a glioblastoma of unknown origin. The U87 and
U87vIII cells transfected to express the pDsRed2-Mito fluorescent
mitochondrial marker (RRID:Addgene_52659) were used for a subset of
immunofluorescence experiments. The cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) containing 1 mM sodium
pyruvate, 15 mM HEPES, non-essential amino acids, 10% fetal bovine
serum (FBS) and 1% penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc.). Media devoid of FBS were used for serum
deprivation experiments. Media without phenol red were used for
immunofluorescence experiments. All cell cultures were tested for
mycoplasma contamination prior to orthotopic injection for
experiments using mice.
Patient tissues and clinical
information
GBM patient-derived cells (PDCs) were obtained from
GBM xenografts established with tumor tissue from patients
undergoing surgical treatment at the Mayo Clinic (Rochester, MN,
USA) (24-26). These studies were approved by the
Mayo Clinic Institutional Review Boards and only samples from
patients who had provided prior consent for use of their tissues in
research were included. A full description of the characteristics
of the patients from whom the four GBM xenografts were derived and
from which the PDCs were obtained and used herein, can be found in
the previous study by Sarkaria et al (24).
The panel of PDCs analyzed in the present study
exhibited a disparate EGFR/phosphatase and tensin homolog (PTEN)
status, MGMT methylation state [methylated/unmethylated (M/U)],
molecular subtype [mesenchymal/classical (M/C)], invasive
characteristics in mouse orthotopic xenografts (0, low; 7, high)
and sensitivity to erlotnib (0, not sensitive; 100, sensitive)
(24-26). The PDCs were cultured in 3D
methacrylated gelatin (GelMA) hydrogels (7% wt gelatin) without or
with hyaluronic acid (HA) (6% wt gelatin, 1% wt HA) (the hydrogels
were constructed in the authors' laboratory in collaboration with
JWEC and BACH).
CHCHD2 protein levels in PDCs cultured in
GelMA hydrogels
The total protein levels of CHCHD2 were analyzed in
a panel of GBM PDCs with a disparate EGFR/PTEN status, MGMT
methylation state, molecular subtype, invasive characteristics in
mouse orthotopic xenografts (0, low; 7, high) and sensitivity to
erlotinib (0, not sensitive; 100, sensitive) (24). The PDCs were cultured in GelMA
hydrogels without HA (7% wt GelMA) or with HA (6% wt GelMA, 1% wt
HA).
Hypoxic cell culture
For the hypoxic experiments, medium was
pre-equilibrated overnight in a BioSpherix™ hypoxic incubator
(BioSpherix, Ltd.) at designated oxygen concentrations (7, 4 and
1%) to account for the time required for oxygen-saturated media to
equilibrate with the gas atmosphere (27). Standard culture conditions are
designated as normoxia (20% O2). All cells were
maintained at 37°C and 5% CO2.
Generation of CHCHD2 knockout (KO)
cells
U87vIII CHCHD2 KO cells were derived using
CRISPR-Cas9 genome engineering following published protocols
(28,29). Suitable target sites within exons
of the coding sequence for CHCHD2 were identified using the
online WU-CRISPR design tool (30). Potential guide RNA (gRNA)
oligonucleotides were obtained from Integrated DNA Technologies
(IDT). Each gRNA sequence was 20 nucleotides in length and directly
upstream of the protospacer adjacent motif (PAM) 5′-NGG-3′. A total
of three separate gRNA expression constructs were generated by
cloning phosphorylated and annealed gRNA oligos into the
BbsI (New England Biolabs) site of the pSpCas9(BB)-2A-Puro
expression vector (cat. no. 62988, Addgene) for the co-expression
of each sgRNA with the Cas9 endonuclease. The integrity of the
constructs was confirmed by plasmid sequencing (University of
Illinois Urbana-Champaign Roy J. Carver Biotechnology Center).
The U87vIII cells were transfected with
Cas9-CHCHD2gRNA expression constructs using Lipofectamine
2000® reagent according to the manufacturer's protocol
(Thermo Fisher Scientific, Inc.). Stably transfected cells were
selected with puromycin (10 μg/ml). Successful CHCHD2
protein knockout was confirmed using western blot analysis as
described below. The assessment of genomic mutational status was
conducted using nested PCR of the region containing the induced
double-strand break using two primer sets. The PCR product was
cloned into the pCR2.1-TOPO vector (K450002, Thermo Fisher
Scientific, Inc.) and confirmed by sequencing (UIUC Roy J. Carver
Biotechnology Center).
Measurement of oxygen consumption rate
(OCR)
The mitochondrial respiration of the U87vIII CHCHD2
wild-type (WT) and KO cells was compared using the Seahorse XFp
Extracellular Flux Analyzer (Agilent Technologies, Inc.). The cells
were seeded at 1×104 cells/well 5 h prior to conducting
the mitochondrial stress test, according to the manufacturer's
protocol (Agilent Technologies). Serial applications of oligomycin
(ATP synthase inhibitor, 1 μM), FCCP (protonophore, 0.5
μM), and rotenone and antimycin A (respiratory complex I and
III inhibitor, respectively, 0.5 μM) over time enabled the
calculation of various parameters of mitochondrial respiration in
both cell lines.
Measurement of compartmentalized
glutathione (GSH) redox poise
The U87vIII CHCHD2 WT and KO cells were transfected
with genetically encoded, fluorescent redox biosensors targeted to
the cytosol (cyto-Grx1-roGFP2) or mitochondrial matrix
(mito-Grx1-roGFP2), previously described by the authors' laboratory
(30). Cells expressing cytosolic
or mitochondrial Grx1-roGFP2 were seeded at equal densities in
standard culture medium without phenol red in μ-Slide
eight-well ibiTreat microscopy chambers (ibidi GmbH). Time-lapse
images were collected using a fluorescence-enabled inverted
microscope (Axiovert 200 M, Carl Zeiss AG). The dual-excitation
imaging of live cells used 395 and 494 nm excitation cubes, and an
emission filter at 527 nm was used for both cubes. Exposure times
were set to 100-200 msec, and images were obtained every 15 sec. To
assess the effects of GSH synthesis inhibition on the GSH:GSH
disulfide (oxidized GSH; GSSG) status, the cells were pre-treated
with buthionine sulfoximine (BSO, 100 μM) (6954/100, R&D
Systems, Inc.) for 24, 48, and 72 h prior to time-lapse image
acquisition. Acquired images were processed using Zeiss Axiovision
SE64 Rel6.8 software (AxioVision Imaging System, RRID:SCR_002677;
Carl Zeiss AG), via the manual selection of three to five
individual cells to obtain multiple regions of interest in each
time lapse. The means of emission intensities at 527 nm were
exported to Excel files and corrected by background
subtraction.
Cell proliferation
The proliferation of the U87vIII CHCHD2 WT and KO
cells was compared over a period of 72 h under normoxic (20%
O2) and hypoxic (1% O2) culture conditions
using the sulforhodamine B (SRB) assay (Abcam) according to
previously published protocols (31). Briefly, the cells were seeded at
equal densities in a 96-well plate, followed by fixation with 10%
trichloroacetic acid (MilliporeSigma) for 1 h at 4°C at 0 and 72 h
timepoints. After washing four times with water and air-drying at
room temperature, 0.057% SRB solution (wt/vol in 1% acetic acid)
was applied to each well and incubated for 30 min at room
temperature, followed by washing four times in 1% acetic acid.
After drying, bound SRB was solubilized in 10 mM Tris base solution
(pH 10.5), and the plates were shaken for 30 min at room
temperature. The optical density (OD) of each well was measured at
510 using a BioTek Synergy™ HT microplate reader (BioTek
Instruments). OD values at 72 h were corrected by 0 h OD
subtraction to account for possible variations in initial seeding
densities.
Cell cytotoxicity assays
The sensitivity of the U87vIII CHCHD2 WT and KO
cells to a panel of cytotoxic agents was determined using the SRB
assay according to previously published protocols (31). The cytotoxicity of sulfasalazine
(SSZ, xCT cystine-glutamate antiporter inhibitor; S0883,
MilliporeSigma), erlotinib (Erl, EGFR receptor tyrosine kinase
inhibitor; SML3621, MilliporeSigma), TMZ (DNA alkylating agent;
5.00609, MilliporeSigma) and Pac-1 (procaspase-3 activator;
courtesy of Paul Hergenrother, UIUC) was assessed using doses
derived from the literature (30,32).
Hydrogel preparation and measurement of
cell invasion
The invasive behavior of the U87vIII CHCHD2 WT and
KO cells was examined under normoxic (20% O2) and
hypoxic (1% O2) culture conditions. Invasion was
quantified within GelMA hydrogels using a bead invasion assay as
previously described (33-35). The hydrogels used in this study
were prepared using 5% wt GelMA, ~53% degree of methacrylamide
functionalization determined via H1-NMR (data not
shown), and photopolymerized under UV light (AccuCure LED 365 nm,
7.1 mW cm-2 for 30 sec) in the presence of a lithium
acylphophinate photoinitiator. The compressive modulus of 5% wt
GelMA hydrogels was measured using an Instron 5943 mechanical
tester with Young's modulus obtained from the linear region of the
stress-strain curve (0-10% strain) (35).
To examine invasive behavior, the U87vIII CHCHD2 WT
and KO cells were seeded onto collagen-coated dextran beads (~200
μm diameter, Cytiva) at a density of 2×106 cells
per 5×103 beads in 5 ml DMEM. Cell-bead suspensions were
lightly shaken for 1 min, every 30 min, for 5 h to facilitate cell
adhesion to the beads. The cell-coated beads were then encapsulated
in pre-polymerized GelMA hydrogel solution, and bead-containing
hydrogels were cultured in standard DMEM in either normoxic (20%
O2) or hypoxic (1% O2) culture conditions for
7 days. The cell invasion distance was measured from the bead
surface using ImageJ software (ImageJ, RRID:SCR_003070; National
Institutes of Health) from images acquired using a fluorescence
microscope (Leica Microsystems, Inc.) after fixing and staining
cells with DAPI (Invitrogen; Thermo Fisher Scientific, Inc.) (10
μg/ml in 1X PBS). Cell invasion was reported as the mean
invasion of all cells from the surface of the bead (33).
Western blot analysis
The total protein levels of CHCHD2 (1:500, cat. no.
NBP1-94106, Novus Biologicals, LLC), the glutamate-cystine
antiporter xCT (1:500, cat. no. ab37185, Abcam), GPx-1/2 (1:100,
sc-133160, Santa Cruz Biotechnology), GPx-4 (1:100, cat. no.
NBP2-75511, Novus Biologicals, LLC) and matrix metalloproteinase 2
(MMP-2; 1:500, cat. no. 10373-2-AP, Proteintech Group) were
analyzed using western blot analysis. β-actin (1:1,000, cat. no.
4967, Cell Signaling Technology) was used as the loading control.
Cells were lysed using standard RIPA buffer and total protein
concentrations from whole cell lysates were determined using the
Pierce BCA assay (Thermo Fisher Scientific, Inc.). Protein lysates
were mixed 1:1 with 2X Laemmli Sample Buffer (Bio-Rad Laboratories,
Inc.) (5% β-mercaptoethanol) and heated at 95°C for 5-10 min. The
denatured lysates (20 μg) were loaded into 4-20%
Mini-Protean® TGX™ electrophoresis gels (Bio-Rad
Laboratories), and SDS-PAGE was run at 150 V for 1-1.5 h. The
proteins were transferred onto a nitrocellulose membrane (Amersham;
Cytiva) at 300 mA for 2 h at 4°C. The membranes were then blocked
with either 5% BSA or 5% non-fat dry milk for 1 h at room
temperature, and then incubated with the primary antibodies at the
designated concentrations overnight at 4°C. The membranes were
washed in TBS-T for 5 min three times, then incubated in HRP-linked
goat anti-rabbit secondary antibody (1:2,500, cat. no. 7074, Cell
Signaling Technology) at room temperature for 1.5 h. Following
TBS-T washes (3×5 min each), the membranes were imaged using the
SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo
Fisher Scientific) in an ImageQuant LAS 4010 (Cytiva). Analysis of
the bands was conducted using ImageJ software v1.51 (National
Institutes of Health).
Animals and GBM tumor establishment by
orthotopic injection
NOD.Cg-Prkdcscid
Il2rgtm1Wjl/SzJ mice [NOD scid gamma
(NSG™)] mice (Jackson Laboratory) aged 8-11 weeks (20-30 g) were
used in the present study with food and water at ad libitum
(n=18). Consisting of an equal distribution of sexes in both
groups, 9 mice were inoculated with U87vIII CHCHD2 KO and 9 with
U87vIII WT. Prior to GBM cell induction, the mice were anesthetized
via the intraperitoneal administration of 100 mg/kg ketamine and 10
mg/kg xylazine. The mice were distributed into groups evenly as
regards sex and cell lines used, followed by injection with U87vIII
WT or U87vIII CHCHD2 KO cells. The injection concentration was
1×105/μl, with the desired volume being 0.5
μl per mouse. A 0.5 μl Hamilton syringe was inserted
according to the following coordinates in relation to the bregma:
Rostral 0.5 mm, lateral to right 2.25 mm, and 3.3 mm lowered into
brain tissue. GBM cells were infused for 30-60 sec to ensure
limited injection backflow, followed by a 1-min waiting period
until the removal of the needle. The incision was then closed with
a small amount of 3M VetBond (Amazon.com).
Post-induction, the mice were weighed daily to track percent weight
change and were additionally scored for neurological symptoms of
tumor formation. A weight loss of 20%, severe doming of the skull,
rotational spinning and paralysis were the primary indicators for
the mouse to be sacrificed. Although 16 mice were euthanized, 2
mice in the U87vIII WT group died unexpectedly overnight, possibly
due to severe tumor burden (days 10 and 17 post-injection). The
experiment was concluded when all mice reached the euthanasia
criteria due to tumor burden symptoms (28 days post-injection).
Euthanasia was administered by CO2 asphyxiation with a
fill rate of 30-70% of the chamber volume per minute, verified by
observable breath cessation and followed by cervical dislocation.
Post-euthanasia, the brains were fixed in 4% paraformaldehyde
(MilliporeSigma) for 24 h and sectioned coronally for tumor
histological analysis. All animal care protocols were in accordance
with the National Institutes of Health Guidelines for Care and Use
of Laboratory Animals and were approved by the University of
Illinois Laboratory Animal Care and Use Committee (reference no.
18058).
Statistical analysis
Differences among means were examined using the
unpaired Student's t-test or one-way ANOVA, followed by post hoc
Tukey's analysis where appropriate. A value of P<0.05 was
considered to indicate a statistically significant difference.
Variance is reported as the standard error of the mean. Odds ratios
for CHCHD2 co-amplification with EGFR and log-rank
tests for OS and progression-free survival (PFS) were conducted in
cBioPortal (cBioPortal,RR ID:SCR_014555) (21,22).
Tukey's honestly significant difference or pairwise t-tests were
conducted to compare mRNA expression levels in GlioVis (23).
Results
CHCHD2 is amplified in a subset of GBM
tumors and is associated with a decreased patient survival
The analysis of CHCHD2 amplification patterns
across GBM tumors was performed on The Cancer Genome Atlas (TCGA)
Provisional GBM database using cBioPortal (http://www.cbioportal.org) (21,22).
The analysis was performed on tumor samples with available CNA data
(n=577). The amplification of CHCHD2 was observed in 9% of
GBM tumors (Fig. 1A). Of the
tumors with EGFR amplification, CHCHD2 was
co-amplified in 20% of cases, with a significant tendency to
co-occur (log2 odds ratio >3, P<0.001). Of the
nine proteins in the CHCH domain-containing protein family, only
CHCHD2 was amplified at an appreciable frequency in GBM
tumors, with the next most frequently amplified CHCH protein being
CHCHD3 (1.2%) (Fig. 1A).
Additionally, patients with CHCHD2-amplified tumors
exhibited a decreased OS (12.48 vs. 14.45 months; log-rank
P=0.0223) (Fig. 1B) and PFS (4.86
vs. 7.82 months; log-rank P=0.0420) (Fig. 1C). While this effect could
potentially be explained by the propensity for EGFR to
co-amplify with CHCHD2, the amplification of EGFR was
not solely associated with a decreased patient survival (Fig. S1). These data indicate that the
amplification of the CHCHD2 gene occurs in a subset of GBM
tumors, is associated with a decreased OS and PFS, and is almost
always accompanied by EGFR amplification. The GlioVis data
indicated that CHCHD2 mRNA expression was increased in grade IV
gliomas (GBM) relative to grade II and III gliomas (Fig. 1D), as well as in IHD1-wt
tumors (primary GBM) compared to IDH1-mutant (secondary GBM)
(Fig. 1E). CHCHD2 expression was
also increased in GBM tumors compared to non-tumor tissue (Fig. 1F). Additionally, CHCHD2 expression
was increased in classical subtype tumors (Fig. 1G), likely due to the location of
CHCHD2 on chromosome 7, the amplification of which defines
classical GBM tumors and is accompanied by a focused predilection
for EGFR amplification (36). Tumors exhibiting genome-wide
promoter hypermethylation, i.e., glioma-CpG island methylator
phenotype (G-CIMP), exhibited a decreased CHCHD2 mRNA expression
(Fig. 1H), consistent with the
epigenetic methylation-induced silencing of CHCHD2. The
CHCHD2 expression levels were similar in MGMT methylated vs.
unmethylated tumors (Fig. 1I).
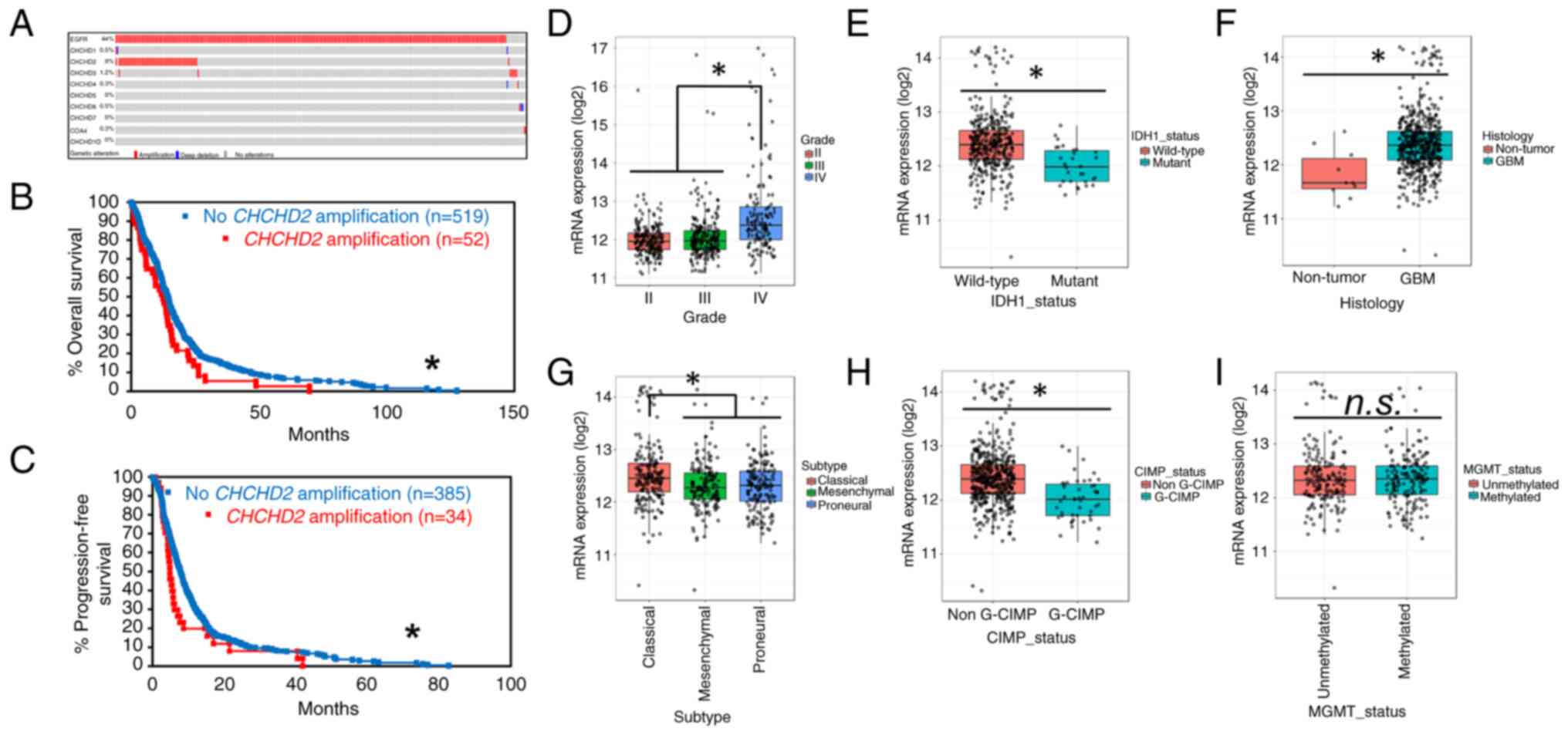 | Figure 1Amplification patterns of CHCHD2
across GBM tumors. (A) Oncoprint from cBioPortal (www.cbioportal.org) representing tumors with
amplification (red), deep deletion (blue), or no alteration (gray)
of query genes. Percentages represent the percentage of samples
analyzed (tumor samples with copy number alteration data, n=577)
with alterations in the given gene. COA4 encodes CHCHD8. (B)
Overall survival of patients with (red) and without (blue) CHCHD2
amplification. (C) Progression-free survival of patients with (red)
and without (blue) CHCHD2 amplification. *P<0.05.
Using GlioVis, CHCHD2 mRNA expression was determined using Human
Genome U133A Array was compared across (D) glioma grade, (E) IDH1
mutational status, (F) non-tumor vs. tumor, (G) GBM subtype
(classical, mesenchymal, proneural), (H) G-CIMP status, which
stratifies tumors based on genome-wide DNA methylation status, and
(I) MGMT methylation status. Data were analyzed using Tukey's
honestly significant difference or the pairwise t-test in GlioVis.
*P<0.05. CHCHD2, coil ed-coil-helix-coiled-coil-helix
domain-containing protein 2; GBM, glioblastoma. |
CHCHD2 protein levels vary across GBM
PDCs samples
The total protein levels of CHCHD2 were analyzed in
a panel of GBM PDCs cultured in GelMA hydrogels (7% wt gelatin).
PDCs were characterized by a disparate EGFR/PTEN status, MGMT
methylation state, molecular subtype, invasive characteristics in
mouse orthotopic xenografts (0: low; 7, high) and sensitivity to
erlotinib 0, not sensitive; 100, sensitive) (Fig. 2A). Included in this panel were:
GBM10 (EGFR-/PTEN-), GBM44
(EGFR-/PTEN-), GBM12
(EGFR+/PTENwt), GBM39
(EGFRvIII/PTENwt) and GBM6 (EGFRvIII/PTENwt).
Notably, of the PDCs analyzed in the present study, the CHCHD2
levels were greatest in GBM12 and GBM6. Notably, GBM6 expresses
EGFRvIII, is the most invasive, is relatively resistant to
erlotinib and is of the classical subtype (Fig. 2B). These results are in accordance
with those obtained for the mRNA levels of CHCHD2, being highest in
classical subtype tumors (Fig.
1G).
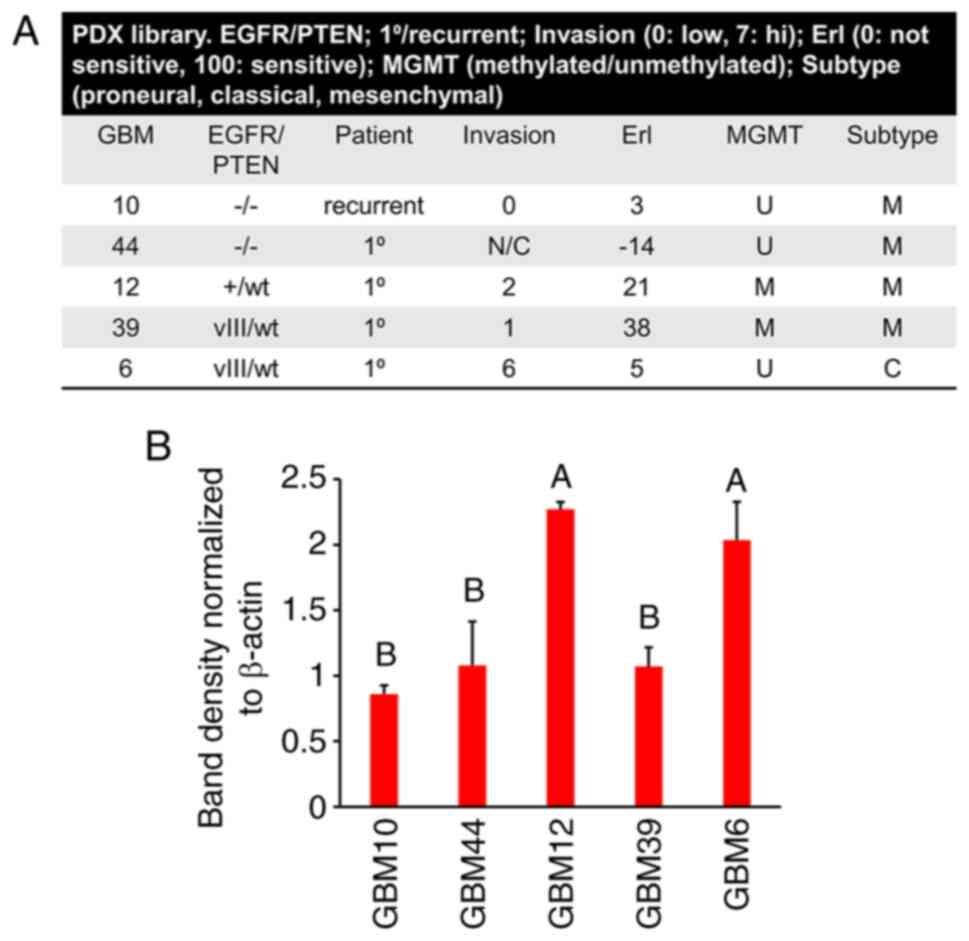 | Figure 2CHCHD2 protein levels in GBM
patient-derived cells. (A) The PDX subset analyzed in the present
study, with descriptors including EGFR/PTEN status, patient status,
invasion in mouse orthotopic xenografts, erlotinib sensitivity,
MGMT methylation status and subtype. (B) CHCHD2 total protein
levels in patient-derived cell samples from each PDX cultured in
GelMA hydrogels containing matrix-immobilized HA (6% wt GelMA, 1%
wt HA). Post-hoc multiple comparisons were conducted comparing the
mean of each group to every other group. Groups with the same
letter in the bar graph (e.g., A) did not differ significantly from
each other, but differed significantly (P<0.05) from the groups
labeled 'B'. CHCHD2, coiled-coil-he lix-coiled-coil-helix
domain-containing protein 2; GBM, glioblastoma PDX, patient-derived
xenograft; EGFR, epidermal growth factor receptor; PTEN,
phosphatase and tensin homolog. |
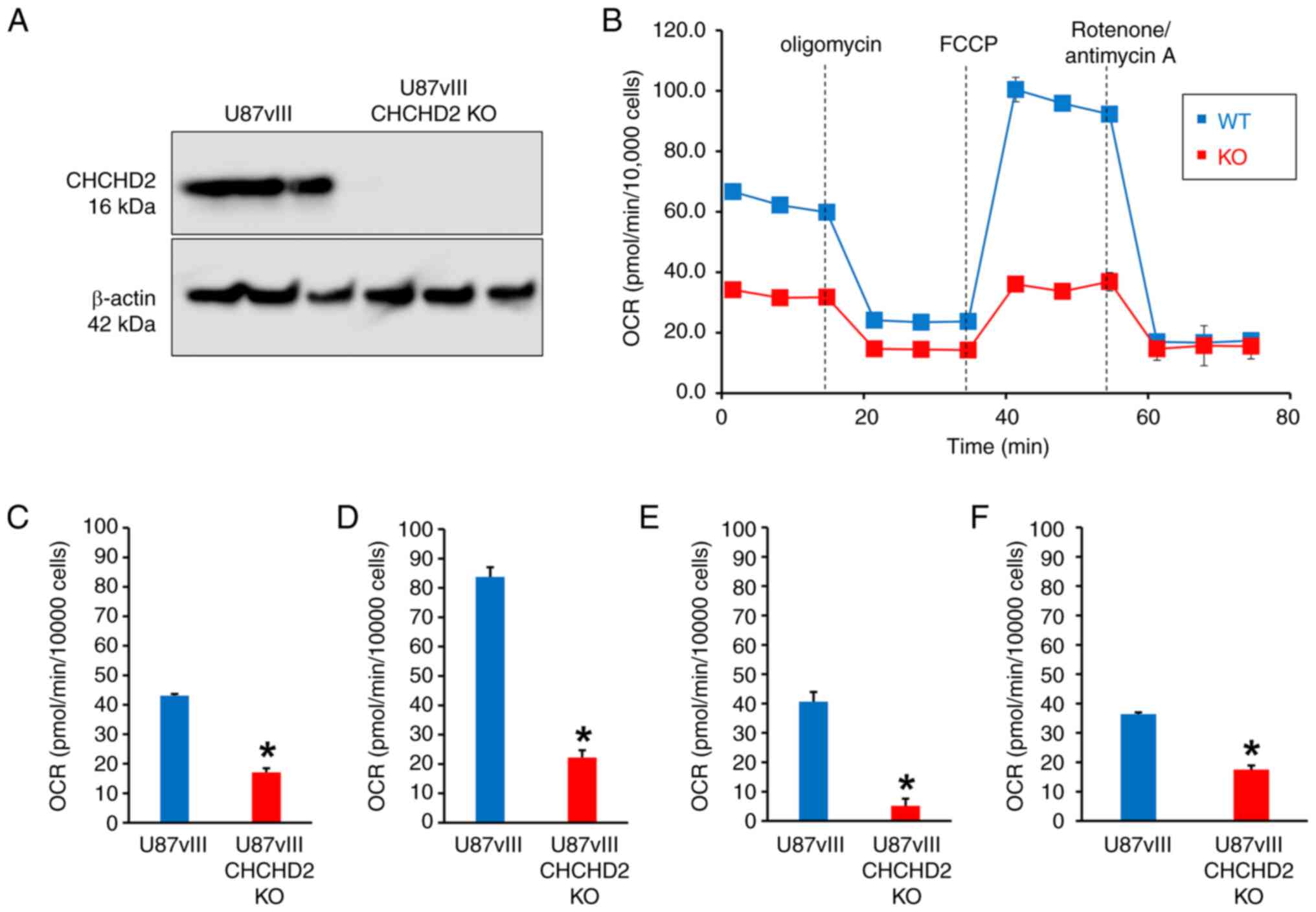
Knockout of CHCHD2 alters mitochondrial
respiration in U87vIII cells
The gene amplification, mRNA and protein expression
patterns of CHCHD2 observed across clinically relevant GBM patient
samples suggested a biologically relevant role for CHCHD2 in
mediating GBM cell phenotypes. To examine the essential
functionality of CHCHD2 in the context of GBM, the U87vIII CHCHD2
KO cells were derived using CRISPR-Cas9 following previously
published protocols (28,29) (Fig.
S2), and protein knockout was validated at the protein level
using western blot analysis (Fig.
3A) (28,29). CHCHD2 was first described during a
computational screen as a mediator of mitochondrial respiration
(16). Herein, to determine
whether CHCHD2 KO affected mitochondrial respiration in GBM cells,
a mitochondrial stress test was conducted on U87vIII CHCHD2 WT and
KO cells using a Seahorse XFp Extracellular Flux Analyzer (Fig. 3B) (37). The CHCHD2 KO cells displayed a
decreased basal and maximal OCR (Fig.
3B-D). The spare respiratory capacity was also significantly
lower in the CHCHD2 KO cells (Fig.
3E), indicating a decreased ability of the U87vIII CHCHD2 KO
cells to respond to the increased energy demand. Additionally, the
amount of oxygen consumed coupled to ATP production by ATP synthase
was significantly lower in CHCHD2 KO cells (Fig. 3F). Collectively, these data
indicate that CHCHD2 is indeed required for efficient mitochondrial
respiration in U87 GBM cells expressing EGFRvIII.
Knockout of CHCHD2 leads to a more
reduced GSH redox pool in the mitochondrial matrix
Upon observing defects in mitochondrial respiration,
it was further hypothesized that deficient electron transport chain
function would result in a more oxidized intracellular redox
environment manifested by increased amounts of GSSG. GSH, an
enzymatically produced tripeptide of cysteine, glycine and
glutamate, is the main intracellular redox buffer, which, along
with thioredoxins and glutaredoxins, maintains thiol redox status
(38). The GSH redox couple
(reduced and oxidized glutathione, GSH and GSSG, respectively),
along with glutathione peroxidase (GPx) and glutathione reductase
(GR), comprises the glutathione system, which maintains thiol redox
homeostasis and functions in antioxidant defense (39). The balance of reduced to oxidized
glutathione (GSH:GSSG) thus represents the intracellular redox
status, and can be interrogated within live cells using genetically
encoded, fluorescent redox biosensors (Grx1-roGFP2) (Fig. 4A). Additionally, such probes can be
targeted to various subcellular compartments, including cytosol
(cyto-Grx1-roGFP2) or mitochondrial matrix (mito-Grx1-roGFP2) to
measure the compartmentalized GSH redox status in live cells in
real time via ratiometric fluorescence intensity measurements
(Fig. 4B) (30). The percent of oxidized
mito-Grx1-roGFP2 in U87vIII CHCHD2 KO cells was decreased by 15.8%
compared to that measured in CHCHD2 WT cells, indicating that the
pool of glutathione in the mitochondrial matrix of KO cells was
more reduced than the WT counterparts (Fig. 4C). This effect was confined to the
mitochondrial matrix, as the cytosolic glutathione pool was not
affected by CHCHD2 KO (Fig. 4D).
Treatment with BSO, an inhibitor of glutamate-cysteine ligase (the
rate-limiting enzyme in GSH synthesis), led to similar oxidation of
the glutathione pool over time in the mitochondrial matrix of both
U87vIII CHCHD2 WT and KO cells (Fig.
4E). Additionally, the levels of components of the glutathione
system, including the glutamate-cystine antiporter xCT, GPx-1/2 and
GPx-4, were all unaltered in the CHCHD2 KO cells (Fig. 4F). These data demonstrate a role
for CHCHD2 in mediating the GSH redox balance specifically in the
mitochondrial matrix, albeit through a mechanism not involving GSH
synthesis, cystine import through xCT, or reduced GSH flux through
glutathione peroxidases 1, 2 or 4.
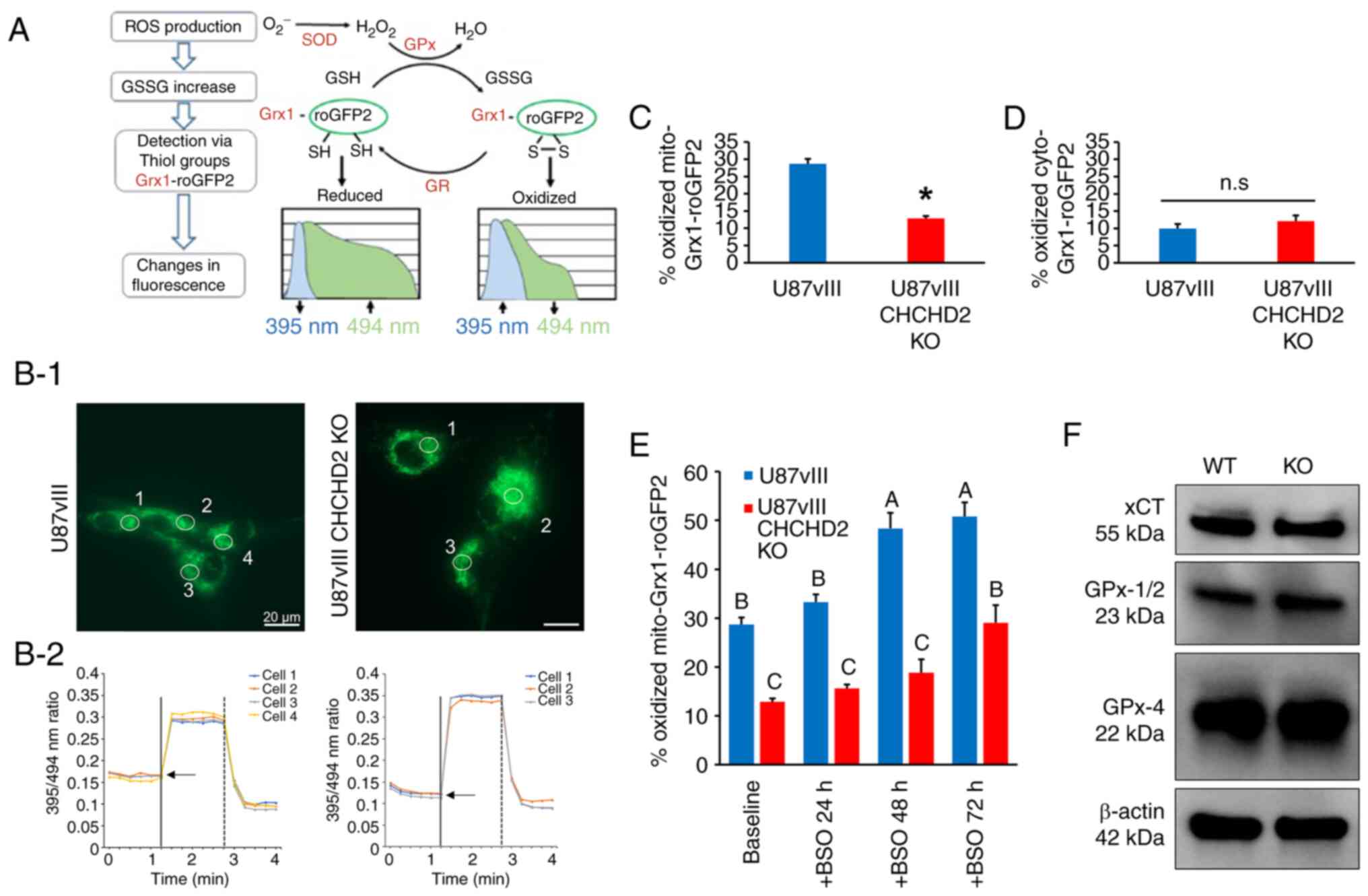 | Figure 4Glutathione redox poise in U87vIII
CHCHD2 WT and KO cells. (A) Schematic diagram of the molecular
mechanism of the Grx1-roGFP2 sensor and redox response of the
compartmentalized probe to exogenous oxidant and reductant.
Superoxide (•O2-) is rapidly converted by SOD into
H2O2, which is then reduced by GPx to water.
Grx fused to roGFP2 efficiently and rapidly equilibrates the probe
with alterations in the local GSH:GSSG ratio. Additionally,
thiol-disulfide equilibration is reversible, as GSH reductase
catalyzes the reduction of GSSG to GSH. (B-1) Representative
fluorescence images demonstrate the sensor targeted to mitochondria
of U87vIII CHCHD2 WT (left) and KO cells (right). (B-2)
Corresponding time-lapse responses of the 395/494 nm ratio to
treatment with 1 mM diamide (vertical solid line) to the fully
oxidized state and 10 mM DTT (vertical dashed line) to the fully
reduced state. Each trace designates a separate cell. Arrows
represent basal oxidation level of probe. (C) Percentage oxidized
mito-Grx1-roGFP2 and (D) cyto-Grx1-roGFP2 in U87vIII CHCHD2 WT and
KO cells. (E) Percentage oxidized mito-Grx1-roGFP2 in U87vIII
CHCHD2 WT and KO cells at baseline, and after treatment with GSH
synthesis inhibitor BSO for 24, 48 and 72 h. (F) Western blot
analysis of xCT, GPx-1/2 and GPx-4 in U87vIII CHCHD2 WT and KO
cells. Data are presented as the mean ± SE. *P<0.05,
vs. U87vIII cells. Post-hoc multiple comparisons were conducted
comparing the mean of each group to every other group. Groups with
the same letter in the bar graph (e.g., A) did not differ
significantly from each other, but differed significantly
(P<0.05) from the groups labeled with other letters, e.g., 'B'.
SOD, superoxide dismutase; GPx, glutathione peroxidase; Grx,
glutaredoxin; GR, glutathione reductase; GSSG, oxidized
glutathione; CHCHD2, coiled-coil-helix-coiled-coil-helix
domain-containing protein 2; KO, knockout; BSO, buthionine
sulfoximine. |
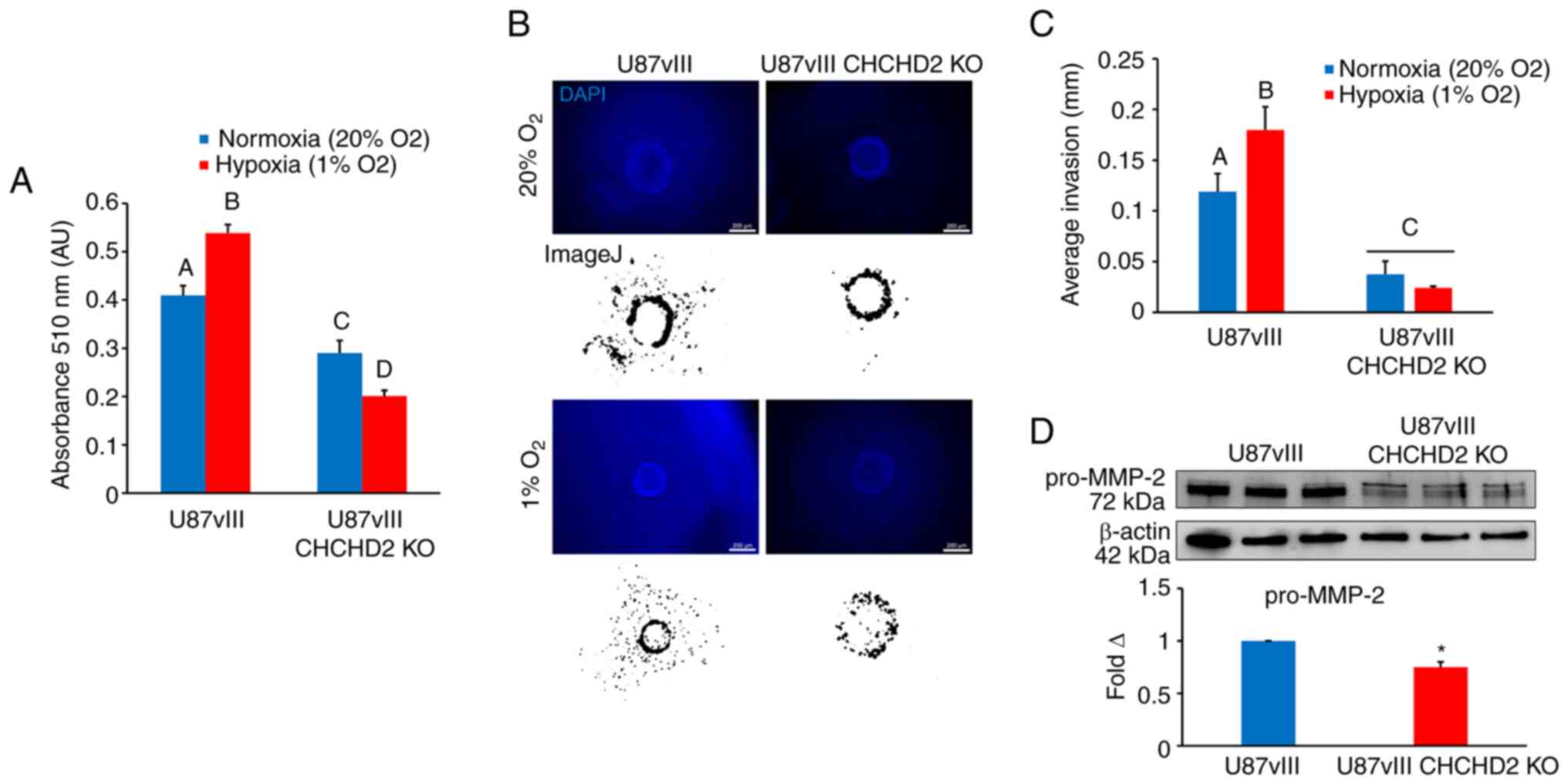
U87vIII cell growth and invasion are
negatively affected by CHCHD2 KO under both normoxic and hypoxic
conditions
The observed deficiencies in mitochondrial
respiration led to the hypothesis that U87vIII GBM cell growth
would be negatively affected by CHCHD2 KO. To examine this
hypothesis, the U87vIII CHCHD2 WT and KO cells were incubated in
either standard oxygen culture conditions (normoxia, 20%
O2) or pathophysiologically relevant hypoxia (1%
O2). Utilizing the SRB colorimetric assay (31) to assess cell growth over a period
of 72 h, CHCHD2 WT cell growth was observed to be significantly
increased under hypoxic conditions (Fig. 5A). CHCHD2 KO cell growth was
decreased compared to the normoxic control (Fig. 5A). Additionally, the
growth-inducing effect of hypoxia on U87vIII cells was abrogated
upon CHCHD2 KO (Fig. 5A). These
results demonstrate that CHCHD2 is involved not only in mediating
U87vIII cell growth under normoxic conditions, but also plays a
role in the initial increased cell proliferation observed in
hypoxic cells.
The rapid, diffuse invasion of GBM cells into tumor
margins and into the surrounding brain parenchyma represents a
major obstacle impeding effective tumor treatment. Herein, to
determine whether CHCHD2 knockout affects GBM cell invasion, a bead
invasion assay in GelMA hydrogels (Young's modulus 2.9±0.45 kPa)
was used as a three-dimensional culture platform to model
biophysical aspects of the brain parenchyma and GBM cell invasion
(33,34). This technique enables the
monitoring of cell invasion from a defined starting point in a
spatiotemporal manner in normal as well as hypoxic
three-dimensional culture conditions (Fig. 5B). Hypoxia (1% O2)
stimulated U87vIII cell invasion over long-term culture (7 days;
Fig. 5C). In contrast, the U87vIII
CHCHD2 KO cells displayed minimal cell invasion under both normoxic
and hypoxic conditions, with the hypoxia-induced invasion observed
in CHCHD2 WT cells being abrogated (Fig. 5C). Furthermore, the CHCHD2 KO cells
displayed decreased basal levels of pro-MMP-2 (Fig. 5D), a key protein involved in the
breakdown of extracellular matrix. These data demonstrate a role
for CHCHD2 in mediating U87vIII cell invasion potentially involving
MMP-2, particularly in response to hypoxia.
CHCHD2 knockout enhances U87vIII
sensitivity to a variety of cytotoxic drugs
To determine the effects of CHCHD2 KO on cellular
resistance to various chemotherapeutic agents, the cells were
treated with increasing concentrations of a panel of drugs, and
cytotoxicity was assessed using the SRB assay (31). Included in this panel were: TMZ, a
DNA-alkylating agent and the standard-of-care chemotherapy
administered to patients with GBM (38); Erl, a receptor tyrosine kinase
inhibitor that inhibits EGFR and EGFRvIII tyrosine kinase activity
(8); SSZ, an inhibitor of the cell
membrane xCT antiporter, which couples the export of the amino acid
glutamate with the import of cystine, thus depriving cells of the
rate-limiting substrate to synthesize reduced GSH (40-42);
and Pac-1, a novel activator of apoptosis which acts on
procaspase-3 (32). The results
(Fig. 6) demonstrated that the
CHCHD2 KO cells were more susceptible to treatment with TMZ, Erl,
and most significantly, SSZ. However, CHCHD2 KO had no effect on
cellular sensitivity to treatment with Pac-1 (Fig. 6D), consistent with the findings of
a previous study, demonstrating that CHCHD2 regulates apoptosis
upstream of procaspase-3 activity in the apoptotic cascade
(19). These results demonstrate a
role for CHCHD2 in mediating cell sensitivity to various drugs
relevant to GBM treatment, highlighting CHCHD2 as a promising
avenue for future investigations.
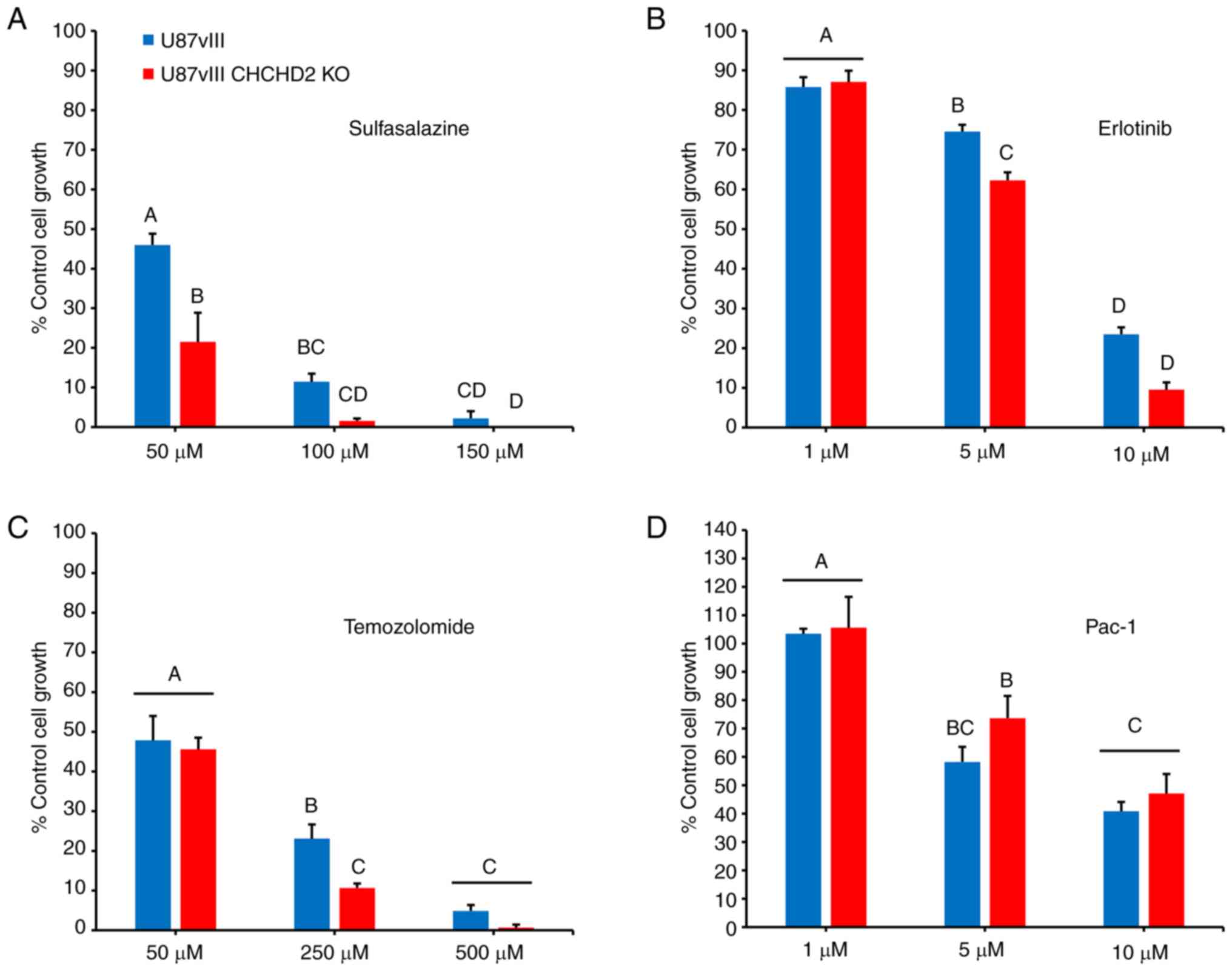 | Figure 6Therapeutic sensitivity of U87vIII
and U87vIII CHCHD2KO cells. Sensitivity of U87vIII and CHCHD2KO
cells to (A) sulfasalazine, (B) erlotinib, (C) temozolomide, and
(D) Pac-1 determined using the sulforhodamine B assay with the
indicated doses for a duration of 72 h. Data are presented as a
percentage of the untreated control, as the mean ± SE. Post-hoc
multiple comparisons were conducted comparing the mean of each
group to every other group. Groups with the same letter in the bar
graph (e.g., A) did not differ significantly from each other, but
differed significantly (P<0.05) from the groups labeled with
other letters, e.g., 'B'. CHCHD2,
coiled-coil-helix-coiled-coil-helix domain-containing protein 2;
KO, knockout. |
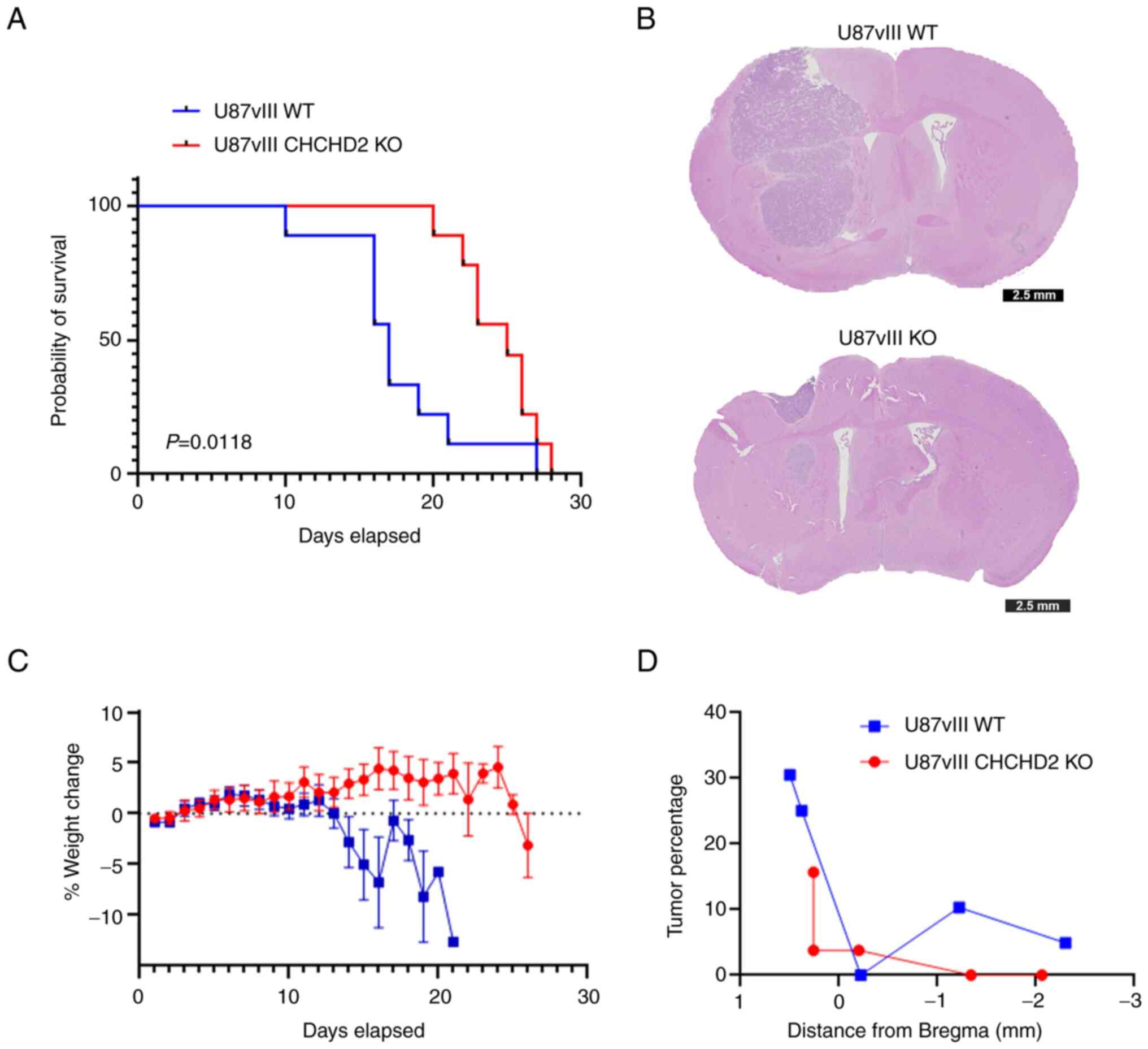
CHCHD2 knockout in GBM tumors affects the
overall survival of mice
To determine the effects of CHCHD2 on tumor growth
in vivo, NSG mice were orthotopically injected with U87vIII
WT and CHCHD2 KO cells. Mice bearing U87vIII WT tumors presented
with a median survival of 17 days vs. 25 days for U87vIII CHCHD2 KO
tumor-bearing mice (log-rank P=0.0118) (Fig. 7A). Representative coronal sections
of mice sacrificed on day 16 post-injection demonstrated
substantial tumor growth and infiltration into the surrounding
brain (Fig. 7B). The mice injected
with U87vIII WT cells exhibited a rapidly diminishing health by day
13 of the study, as evidenced by a decrease in weight (Fig. 7C). Furthermore, the U87vIII WT
cells consistently produced tumors occupying a larger percentage of
parenchyma compared to the CHCHD2 KO-derived tumors (Fig. 7D). Overall, these data indicate a
role for CHCHD2 in promoting the progression of GBM tumors in
mice.
Discussion
Mitochondria, in addition to regulating cellular
energy conservation, serve as signaling organelles. The vast
majority of mitochondrial proteins are encoded by nuclear genes,
necessitating the ability for mitochondria to communicate their
status to the nucleus in response to metabolic perturbations.
Disturbances in ATP and ROS production, damage to mitochondrial
DNA, and aberrations in mitochondrial protein folding induce a
mitonuclear retrograde signaling pathway by which mitochondria
communicate with the nucleus to induce changes in nuclear gene
expression in order to maintain metabolic homeostasis (43). Several proteins have been
demonstrated to participate in inter-organelle signaling between
mitochondria and the nucleus, including p53, fumarase, the pyruvate
dehydrogenase complex and CHCHD2 (15,44-46).
CHCHD2 presented as a promising target to investigate, for the
following reasons: i) Its proximity to and frequency of
co-amplification with EGFR with NSCLC and GBM (10), both of which have been
characterized as relatively oxidative vs. fermentative tumors
(47); ii) its mRNA expression
patterns across glioma grade and tumor vs. non-tumor tissue; iii)
its reported oxygen-sensitive transcription factor activity
(15,20); and iv) its pleiotropic roles
mediating cellular functions reminiscent of cancer hallmarks,
including proliferation, migration and invasion, and the inhibition
of apoptosis (15,18,19).
The present study demonstrated that CHCHD2 was involved in
mediating therapeutic sensitivity, as well as cell growth and
invasion in in vitro and in vivo models of GBM.
The knockout of CHCHD2 in U87vIII GBM cells
resulted in decreased baseline respiration as well as spare
respiratory capacity, an effect corroborated by multiple studies
which have characterized CHCHD2 as a canonical regulator of
mitochondrial respiration (10,15-17).
While metabolic reprogramming towards increased glycolytic flux to
favor increased cell proliferation is a recognized hallmark of
cancer, functional mitochondria remain essential in maintaining
malignant cell bioenergetics in particular tumors, including those
of lung and brain (47-49). Notably, mitochondrial spare
respiratory capacity has been positively associated with glioma
stem cell resistance to radiotherapy (50). Thus, the decreased spare
respiratory capacity measured in U87vIII CHCHD2 KO cells may
partially contribute to their increased sensitivity to treatment
with TMZ, Erl, and SSZ.
Additionally, CHCHD2 has ben demonstrated to
mediate mitochondrial outer membrane permeabilization (MOMP)
(19), the 'point of no return'
during the intrinsic pathway of apoptosis. Indeed, in the present
study, the U87vIII CHCHD2 KO cells exhibited an enhanced
sensitivity to TMZ, Erl and SSZ. The observation that no increase
in cytotoxicity was found in the CHCHD2 KO cells treated with
Pac-1, an activator of procaspase-3, which acts downstream of MOMP
in the apoptotic cascade (32),
further corroborates the demonstrated role of CHCHD2 early in the
regulation of apoptosis.
Notably, as evidenced by the use of
mito-Grx1-roGFP2, the mitochondrial GSH pool was more reduced in
response to CHCHD2 KO compared to WT cells, an effect independent
of GSH biosynthesis. It should be noted that roGFP2 itself is not
directly oxidized by reactive oxygen species (ROS), but rather
equilibrates with the local GSH redox potential, which is
influenced by the GSH:GSSG ratio, and in turn is influenced by
activity of various ROS-scavenging enzymes that use GSH as a
cofactor. As previously demonstrated, in 293/293T cells, CHCHD2
knockdown was accompanied by the decreased expression of superoxide
dismutase 2 and the loss of glutathione peroxidase expression
(15). However, herein, no changes
were observed in the protein levels of GPx-1/2, Gpx-4, nor the
glutamate-cystine antiporter xCT upon CHCHD2 knockdown in U87vIII
cells.
Incubation under hypoxic conditions increased
U87vIII CHCHD2 WT cell proliferation over a period of 3 days, and
increased invasion over a period of 7 days. Using the GelMA
hydrogel platforms described herein, a previous study demonstrated
that hypoxic U87vIII cells exhibited an increased cell
proliferation until day 5, at which point cell proliferation
stalled, while invasion continued to increase up to day 7 (35). Notably, CHCHD2 KO abrogated the
increased cell growth and invasion in hypoxia exhibited by WT
cells. The observed decrease in U87vIII CHCHD2 KO cell growth is
not likely due to an increase in apoptosis, as evidence has
indicated that shRNA-mediated CHCHD2 knockdown does not alter the
cellular levels of poly (ADP-ribose) polymerase (PARP) (15). Deficiencies in mitochondrial
respiration and ATP production are likely responsible for hampering
cell proliferative capacity, and may also be implicated in the
suppression of cell invasion, as migration and invasion throughout
parenchyma is an energy-expensive process, particularly when moving
through dense extracellular matrices (51). The observed decrease in pro-MMP-2
expression in CHCHD2 KO cells provides further explanation for
their decrease in invasive capacity. Another study highlighted
CHCHD2 as an activator of NIH3T3 fibroblast migration via the
AKT-RhoA/ROCK-JNK cascade (18).
Critically, the effects of CHCHD2 observed in vitro were
closely mirrored in vivo, as mice orthotopically injected
with U87vIII CHCHD2 KO cells presented with a decreased tumor
burden and an improved survival time.
To the best of our knowledge, the present study is
the first to describe the functional relevance of CHCHD2 in GBM.
The data obtained indicate that CHCHD2 is essential for
mitochondrial respiration and maintenance of the mitochondrial GSH
status in U87vIII cells. Additionally, these findings demonstrate a
critical role for CHCHD2 in mediating cell growth and invasion
under normoxic and hypoxic conditions, and resistance to various
cytotoxic agents, underscoring CHCHD2 as a mediator of the GBM
malignant phenotype. The functional outcomes investigated herein
relate primarily to the mitochondrial functions of CHCHD2. As a
protein implicated in mitonuclear communication in response to
hypoxia, the nuclear functions of CHCHD2 in GBM cells and the panel
of nuclear genes it regulates are of equal importance and comprise
topics for future research. The nuclear function of CHCHD2 has been
previously demonstrated to function in concert with other
transcription factors, namely recombining binding protein
suppressor of hairless (RBPJ) (20), the main downstream effector protein
of the Notch signaling pathway, which itself has been implicated in
the maintenance of glioma stem cell maintenance and viability
(52). Future studies are required
to identify the complement of genes regulated by CHCHD2 in both
normal oxygen conditions, as well as under hypoxic conditions.
Limitations of the present study include the use of
a single cell line to determine the effects of knocking out the
expression of CHCHD2 via CRISPR-Cas9 genome engineering. Consistent
with GBM tumors, the PDCs include multiple cell types, thereby
precluding the knockout of CHCHD2 in the heterogeneous
patient-derived samples. Regardless, the combined cell line and
patient-derived data clearly indicate that a more in-depth
understanding of both the nuclear and mitochondrial functions of
CHCHD2 may identify the mechanisms through which this intriguing
protein involved in mitonuclear retrograde signaling may be
manipulated to improve the outcomes of patients with GBM.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
JCL, BACH, AJS and HRG were involved in the
conception and design of the study. JCL, VLK, JWEC and JNS were
involved in the development of the study methodology. JCL, PLH,
VLK, JEC, JS, AR, MS, AJS and HRG were involved in the acquisition
of data (provided animals, provided facilities and performed
experiments). JCL, PLH, VLK, MS, BACH, AJS and HRG were involved in
the analysis and interpretation of data (e.g., statistical
analysis, biostatistics, computational analysis). JCL, PLH, BACH,
AJS and HRG were involved in the writing, reviewing and/or revision
of the manuscript. HRG supervised the study. JCL and HRG confirm
the authenticity of the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The use of human tumor tissues was were approved by
the Mayo Clinic Institutional Review Boards and only samples from
patients who had provided prior consent for use of their tissues in
research were included. All animal care protocols were in
accordance with National Institutes of Health Guidelines for Care
and Use of Laboratory Animals and were approved by the University
of Illinois Laboratory Animal Care and Use Committee (reference no.
18058).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Romana
Nowak (University of Illinois Urbana-Champaign) for the use of the
BioSpherixTM hypoxic incubator and Professor Zeynep Madak-Erdogan
(University of Illinois Urbana-Champaign) for the use of the
Seahorse XFp Extracellular Flux Analyzer.
Funding
The present study was partially supported by the National Cancer
Institute of the National Institutes of Health (grant nos. R01
CA256481 and R01 CA197488). The authors also acknowledge additional
funding provided by the Department of Chemical and Biomolecular
Engineering, as well as the Cancer Center at Illinois at the
University of Illinois Urbana-Champaign.
References
|
1
|
Barnholtz-Sloan JS, Ostrom QT and Cote D:
Epidemiology of brain tumors. Neurol Clin. 36:395–419. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Taillibert S, Kanner A, Read W,
Steinberg D, Lhermitte B, Toms S, Idbaih A, Ahluwalia MS, Fink K,
et al: Effect of tumor-treating fields plus maintenance
temozolomide vs maintenance temozolomide alone on survival in
patients with glioblastoma: A randomized clinical trial. JAMA.
318:2306–2316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ceccarelli M, Barthel FP, Malta TM,
Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh
A, Pagnotta SM, et al: Molecular profiling reveals biologically
discrete subsets and pathways of progression in diffuse glioma.
Cell. 164:550–563. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weller M, Butowski N, Tran DD, Recht LD,
Lim M, Hirte H, Ashby L, Mechtler L, Goldlust SA, Iwamoto F, et al:
Rindopepimut with temozolomide for patients with newly diagnosed,
EGFRvIII-expressing glioblastoma (ACT IV): A randomised,
double-blind, international phase 3 trial. Lancet Oncol.
18:1373–1385. 2017. View Article : Google Scholar
|
|
6
|
Gan HK, Kaye AH and Luwor RB: The EGFRvIII
variant in glioblastoma multiforme. J Clin Neurosci. 16:748–754.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Parker JJ, Canoll P, Niswander L,
Kleinschmidt-DeMasters BK, Foshay K and Waziri A: Intratumoral
heterogeneity of endogenous tumor cell invasive behavior in human
glioblastoma. Sci Rep. 8:180022018. View Article : Google Scholar :
|
|
8
|
Raizer JJ, Abrey LE, Lassman AB, Chang SM,
Lamborn KR, Kuhn JG, Yung WK, Gilbert MR, Aldape KA, Wen PY, et al:
A phase II trial of erlotinib in patients with recurrent malignant
gliomas and nonprogressive glioblastoma multiforme postradiation
therapy. Neuro Oncol. 12:95–103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Raizer JJ, Giglio P, Hu J, Groves M,
Merrell R, Conrad C, Phuphanich S, Puduvalli VK, Loghin M,
Paleologos N, et al: A phase II study of bevacizumab and erlotinib
after radiation and temozolomide in MGMT unmethylated GBM patients.
J Neurooncol. 126:185–192. 2016. View Article : Google Scholar :
|
|
10
|
Wei Y, Vellanki R, Coyaud É, Ignatchenko
V, Li L, Krieger J, Taylor P, Tong J, Pham NA, Liu G, et al: CHCHD2
is coamplified with EGFR in NSCLC and regulates mitochondrial
function and cell migration. Mol Cancer Res. 13:1119–1129. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng Q, Qu D, Lu Z and Zhang L: Knockdown
of CHCHD2 inhibits migration and angiogenesis of human renal cell
carcinoma: A potential molecular marker for treatment of RCC. Oncol
Lett. 17:765–772. 2019.PubMed/NCBI
|
|
12
|
Aras S, Maroun MC, Song Y, Bandyopadhyay
S, Stark A, Yang Z, Long MP, Grossman LI and Fernández-Madrid F:
Mitochondrial autoimmunity and MNRR1 in breast carcinogenesis. BMC
Cancer. 19:4112019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Modjtahedi N, Tokatlidis K, Dessen P and
Kroemer G: Mitochondrial proteins containing
Coiled-Coil-Helix-Coiled-Co il-Helix (CHCH) domains in health and
disease. Trends Biochem Sci. 41:245–260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou ZD, Saw WT and Tan EK: Mitochondrial
CHCHD-containing proteins: Physiologic functions and link with
neurodegenerative diseases. Mol Neurobiol. 5:5547–5549. 2017.
View Article : Google Scholar
|
|
15
|
Aras S, Bai M, Lee I, Springett R,
Hüttemann M and Grossman LI: MNRR1 (formerly CHCHD2) is a
bi-organellar regulator of mitochondrial metabolism. Mitochondrion.
20:43–51. 2015. View Article : Google Scholar
|
|
16
|
Baughman JM, Nilsson R, Gohil VM, Arlow
DH, Gauhar Z and Mootha VK: A computational screen for regulators
of oxidative phosphorylation implicates SLIRP in mitochondrial RNA
homeostasis. PLoS Genet. 5:e10005902009. View Article : Google Scholar :
|
|
17
|
Meng H, Yamashita C, Shuba-Fukushima K,
Inoshita T, Funayama M, Sato S, Hatta T, Natsume T, Umitsu M,
Takagi J, et al: Loss of Parkinson's disease-associated protein
CHCHD2 affects mitochondrial crista structure and destabilizes
cytochrome c. Nat Commun. 8:155002017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seo M, Lee WH and Suk K: Identification of
novel cell migration-promoting genes by a functional genetic
screen. FASEB J. 24:464–478. 2010. View Article : Google Scholar
|
|
19
|
Liu Y, Clegg HV, Leslie PL, Di J, Tollini
LA, He Y, Kim TH, Jin A, Graves LM, Zheng J and Zhang Y: CHCHD2
inhibits apoptosis by interacting with Bcl-x L to regulate Bax
activation. Cell Death Differ. 22:1035–1046. 2015. View Article : Google Scholar :
|
|
20
|
Aras S, Pak O, Sommer N, Finley R Jr,
Hüttemann M, Weissmann N and Grossman LI: Oxygen-dependent
expression of cytochrome c oxidase subunit 4-2 gene expression is
mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic
Acids Res. 41:2255–2266. 2013. View Article : Google Scholar
|
|
21
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar
|
|
22
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:p112013. View Article : Google Scholar
|
|
23
|
Bowman RL, Wang Q, Carro A, Verhaak RGW
and Squatrito M: GlioVis data portal for visualization and analysis
of brain tumor expression datasets. Neuro Oncol. 19:139–141. 2017.
View Article : Google Scholar
|
|
24
|
Sarkaria JN, Carlson BL, Schroeder MA,
Grogan P, Brown PD, Giannini C, Ballman KV, Kitange GJ, Guha A,
Pandita A and James CD: Use of an orthotopic xenograft model for
assessing the effect of epidermal growth factor receptor
amplification on glioblastoma radiation response. Clin Cancer Res.
12:2264–2271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sarkaria JN, Yang L, Grogan PT, Kitange
GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB
and James CD: Identification of molecular characteristics
correlated with glioblastoma sensitivity to EGFR kinase inhibition
through use of an intracranial xenograft test panel. Mol Cancer
Ther. 6:1167–1674. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vaubel RA, Tian S, Remonde D, Schroeder
MA, Mladek AC, Kitange GJ, Caron A, Kollmeyer TM, Grove R, Peng S,
et al: Genomic and phenotypic characterization of a broad panel of
patient-derived xenografts reflects the diversity of glioblastoma.
Clin Cancer Res. 26:1094–1104. 2020. View Article : Google Scholar
|
|
27
|
Hüttemann M, Lee I, Liu J and Grossman LI
and Grossman LI: Transcription of mammalian cytochrome c oxidase
subunit IV-2 is controlled by a novel conserved oxygen responsive
element. FEBS J. 274:5737–5748. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ran FA, Hsu PD, Wright J, Agarwala V,
Scott DA and Zhang F: Genome engineering using the CRISPR-Cas9
system. Nat Protoc. 8:2281–2308. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wong N, Liu W and Wang X: WU-CRISPR:
Characteristics of functional guide RNAs for the CRISPR/Cas9
system. Genome Biol. 16:2182015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kolossov VL, Beaudoin JN, Ponnuraj N,
DiLiberto S, Hanafin WP, Kenis PJA and Gaskins HR: Thiol-based
antioxidants elicit mitochondrial oxidation via respiratory complex
III. Am J Physiol Cell Physiol. 309:C81–C91. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vichai V and Kirtikara K: Sulforhodamine B
colorimetric assay for cytotoxicity screening. Nat Protoc.
1:1112–1116. 2006. View Article : Google Scholar
|
|
32
|
Joshi AD, Botham RC, Schlein LJ, Roth HS,
Mangraviti A, Borodovsky A, Tyler B, Joslyn S, Looper JS, Podell M,
et al: Synergistic and targeted therapy with a procaspase-3
activator and temozolomide extends survival in glioma rodent models
and is feasible for the treatment of canine malignant glioma
patients. Oncotarget. 8:80124–80138. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen JE, Lumibao J, Blazek A, Gaskins HR
and Harley B: Hypoxia activates enhanced invasive potential and
endogenous hyaluronic acid production by glioblastoma cells.
Biomater Sci. 6:854–862. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen JE, Pedrons S and Harley BAC: The
combined influence of hydrogel stiffness and matrix-bound
hyaluronic acid content on glioblastoma invasion. Macromol Biosci.
17: View Article : Google Scholar : 2017.
|
|
35
|
Chen JE, Pedron S, Shyu P, Hu Y, Sarkaria
JN and Harley BAC: Influence of hyaluronic acid transitions in
tumor microenvironment on glioblastoma malignancy and invasive
hehavior. Front Mater. 5:392018. View Article : Google Scholar
|
|
36
|
Brennan CW, Verhaak RG, McKenna A, Campos
B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ,
Berman SH, et al: The somatic genomic landscape of glioblastoma.
Cell. 155:462–477. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Divakaruni AS, Paradyse A, Ferrick DA,
Murphy AN and Jastroch M: Analysis and interpretation of
microplate-based oxygen consumption and pH data. Methods Enzymol.
547:309–354. 2014. View Article : Google Scholar
|
|
38
|
Hanschmann EM, Godoy JR, Berndt C,
Hudemann C and Lillig CH: Thioredoxins, glutaredoxins, and
peroxiredoxins-molecular mechanisms and health significance: From
cofactors to antioxidants to redox signaling. Antioxid Redox
Signal. 19:1539–1605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C,
Bogdahn U, et al: Radiotherapy plus concomitant and adjuvant
temozolomide for glioblastoma. N Engl J Med. 352:987–996. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen L, Li X, Liu L, Yu B, Xue Y and Liu
Y: Erastin sensitizes glioblastoma cells to temozolomide by
restraining xCT and cystathionine-γ-lyase function. Oncol Rep.
33:1465–1474. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huberfeld G and Vecht CJ: Seizures and
gliomas-towards a single therapeutic approach. Nat Rev Neurol.
12:204–216. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Eisenberg-Bord M and Schuldiner M: Ground
control to major TOM: Mitochondria-nucleus communication. FEBS J.
284:196–210. 2017. View Article : Google Scholar
|
|
44
|
Yogev O, Naamati A and Pines O: Fumarase:
A paradigm of dual targeting and dual localized functions. FEBS J.
278:4230–4242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhuang J, Wang PY, Huang X, Chen X, Kang
JG and Hwang PM: Mitochondrial disulfide relay mediates
translocation of p53 and partitions its subcellular activity. Proc
Natl Acad Sci USA. 110:17356–17361. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sutendra G, Kinnaird A, Dromparis P,
Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E and
Michelakis ED: A nuclear pyruvate dehydrogenase complex is
important for the generation of acetyl-CoA and histone acetylation.
Cell. 158:84–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Courtney KD, Bezwada D, Mashimo T,
Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P,
Lotan Y, et al: Isotope tracing of human clear cell renal cell
carcinomas demonstrates suppressed glucose oxidation in vivo. Cell
Metab. 28:793–800.e2. 2018. View Article : Google Scholar :
|
|
48
|
Marin-Valencia I, Yang C, Mashimo T, Cho
S, Baek H, Yang XI, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z,
et al: Analysis of tumor metabolism reveals mitochondrial glucose
oxidation in genetically diverse human glioblastomas in the mouse
brain in vivo. Cell Metab. 15:827–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vlashi E, Lagadec C, Vergnes L, Matsutani
T, Masui K, Poulou M, Popescu R, Della Donna L, Evers P, Dekmezian
C, et al: Metabolic state of glioma stem cells and nontumorigenic
cells. Proc Natl Acad Sci USA. 108:16062–16067. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zanotelli MR, Goldblatt ZE, Miller JP,
Bordeleau F, Li J, VanderBurgh JA, Lampi MC, King MR and
Reinhart-King CA: Regulation of ATP utilization during metastatic
cell migration by collagen architecture. Mol Biol Cell. 29:1–9.
2018. View Article : Google Scholar :
|
|
52
|
Xie Q, Wu Q, Kim L, Miller TE, Liau BB,
Mack SC, Yang K, Factor DC, Fang X, Huang Z, et al: RBPJ maintains
brain tumor-initiating cells through CDK9-mediated transcriptional
elongation. J Clin Invest. 126:2757–2772. 2016. View Article : Google Scholar :
|