Triple-negative breast cancer (TNBC) represents a
unique and challenging BC subset, characterized by the absence of
estrogen receptor (ER), progesterone receptor (PR) and human
epidermal growth factor receptor 2 (HER2) (1), as the lack of these established
hormonal and HER2 targets renders TNBC unresponsive to some of the
most effective therapies available for other BC subtypes, such as
endocrine therapy and HER2-targeting treatments (2). Consequently, TNBC is often
associated with poor prognosis characterized by high rates of early
recurrence, metastasis and shorter overall survival (OS) than other
BC subtypes (3,4).
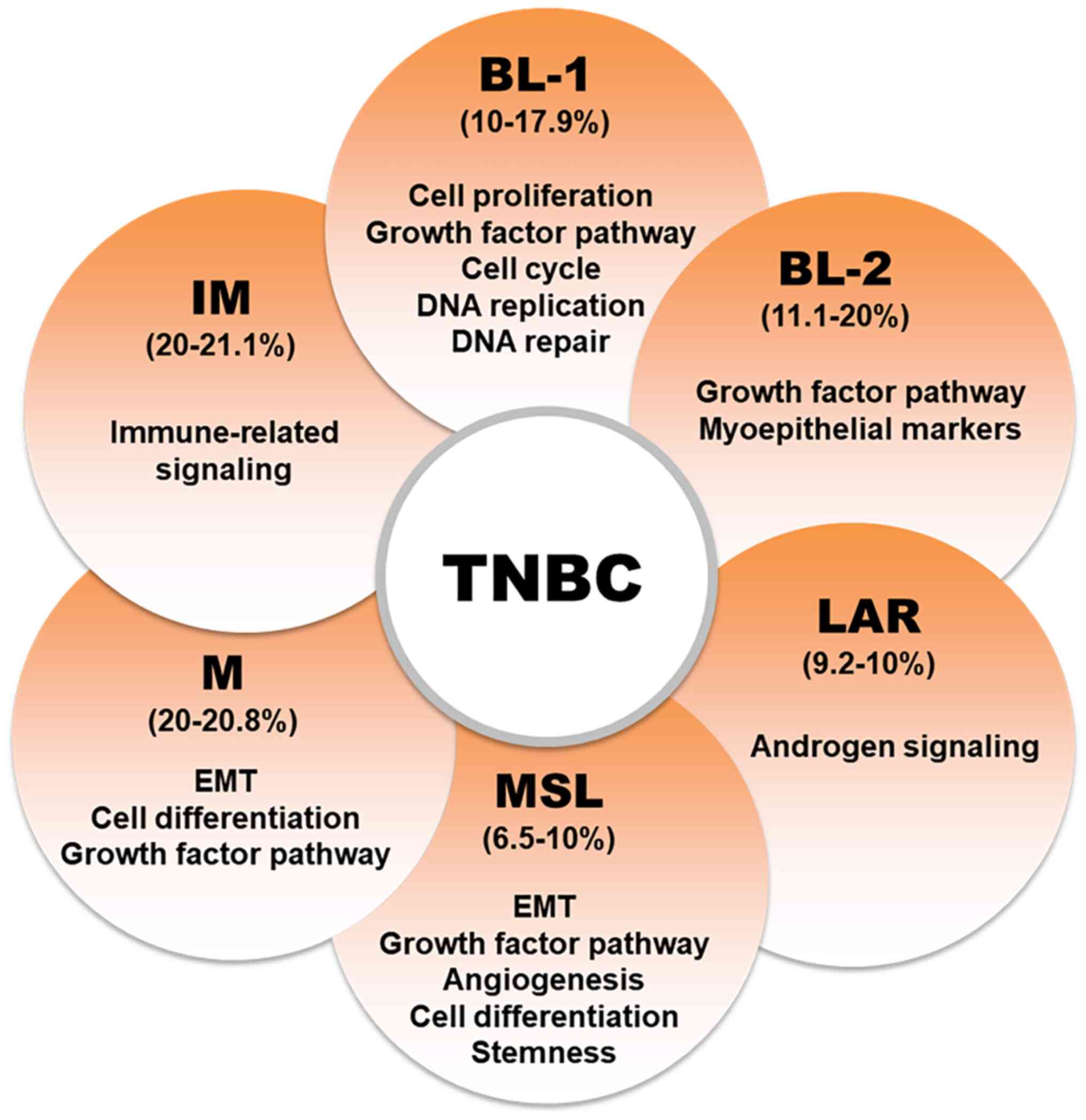
TNBC was initially classified into six subtypes
based on gene expressions profiles (Fig. 1): Luminal androgen receptor (LAR),
mesenchymal (M), basal-like 1 (BL1), BL2, immunomodulatory and
mesenchymal stem-like (MSL). However, subsequent studies showed
that the IM and MSL subtypes are primarily characterized by high
levels of tumor-infiltrating lymphocytes (TILs) and stromal cells
and do not constitute distinct cancer cell subtypes. Consequently,
the TNBC classification was refined to include four main subtypes
(BL1, BL2, LAR and MSC) (1),
which differ significantly in treatment response, prognosis and
survival rates. Notably, BL1 and BL2 represent ~75% of TNBC cases
(5).
The incidence of TNBC depends on demographic factors
including age, ethnicity, genetic predisposition and sex. In
particular, its prevalence is higher in younger women, African
American women and those harboring breast cancer susceptibility
gene 1 (BRCA1) gene mutations (6). The aggressive nature of TNBC, in
combination with its molecular heterogeneity, complicates treatment
and underscores the need to understand underlying biology in depth
(7). Over the last two decades,
considerable efforts have been directed toward mapping the
molecular landscape of TNBC, and as a result, several TNBC
molecular subtypes have been identified with unique biological
behaviors and therapeutic vulnerabilities. The heterogeneity of
TNBC is evidenced by diverse genetic alterations, epigenetic
modifications and dysregulated signaling pathways (8). Key molecular aberrations in TNBC
include defects in DNA repair mechanisms, alterations in cell cycle
regulation and activations of oncogenic signaling pathways,
including the phosphoinositide 3-kinase (PI3K)/protein kinase B
(AKT)/mammalian target of rapamycin (mTOR) and JAK/STAT pathways
(9). These molecular
characteristics not only contribute to the aggressive phenotype of
TNBC but also highlight potential targets for therapeutic
intervention.
One of the most promising areas of TNBC research
involves exploring DNA damage response pathways, particularly in
tumors with BRCA1 or BRCA2 mutations (10). Nonetheless, not all TNBCs feature
BRCA mutations and resistance to therapies targeting these
mutations remains challenging. Thus, the prognosis of patients with
TNBC remains poor despite the progress made, particularly in
advanced disease stages. Current treatment options are limited and
the emergence of resistance to conventional chemotherapies further
complicates TNBC management (11). Consequently, there is an urgent
requirement to identify novel molecular targets and develop more
efficacious therapeutic strategies.
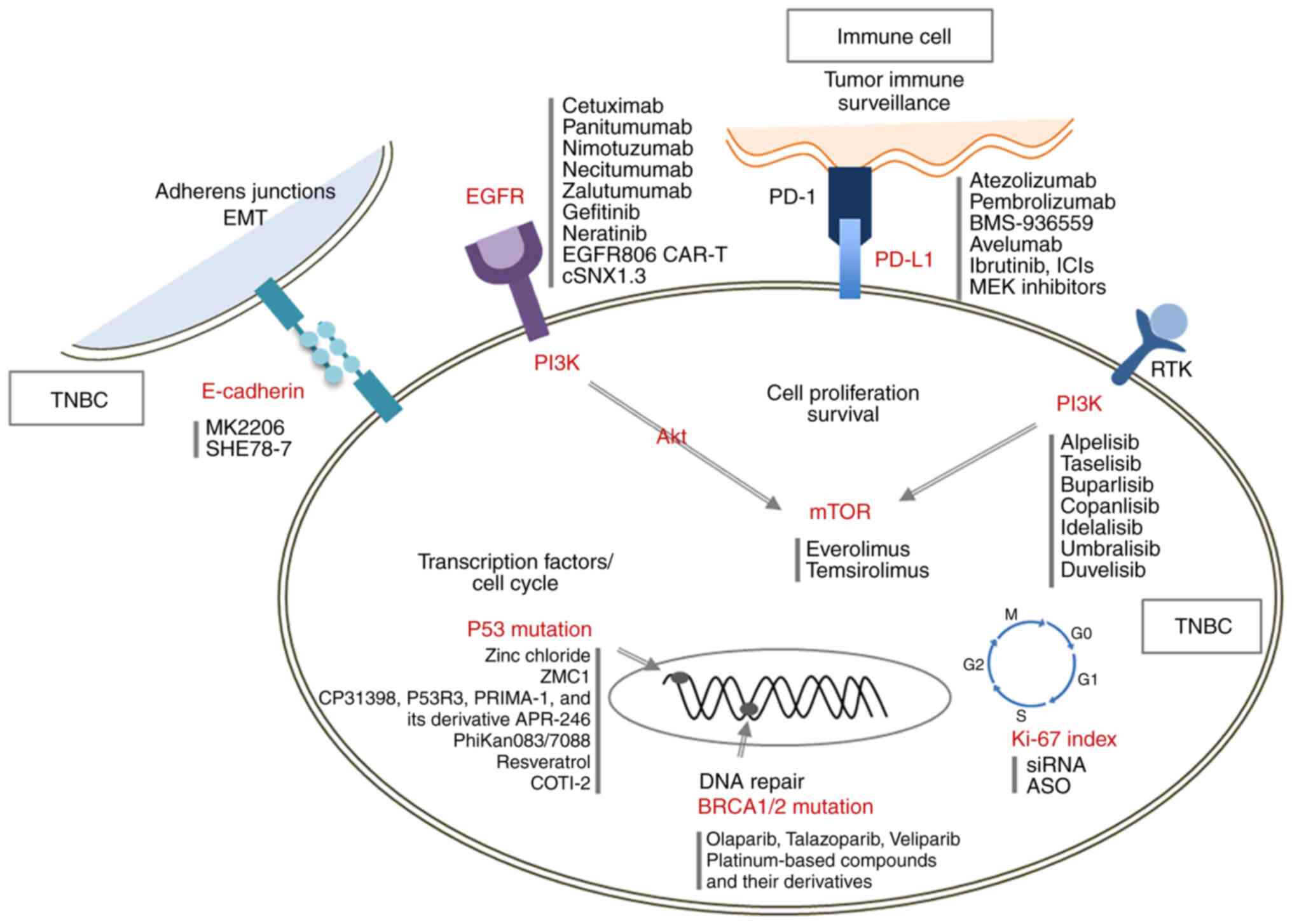
This review aims to provide a comprehensive overview
of seven key molecular targets (Table
I) related to the poor prognosis of TNBC and to describe
current and emerging therapeutic strategies designed to improve
patient outcomes (Fig. 2,
Table II). By exploring the
complex molecular landscape of TNBC and the therapeutic challenges
it presents, the current review suggests potential routes for
developing more effective treatments for this aggressive form of
BC.
Ki-67 is considered one of the most promising
targets and prognostic markers in TNBC (12). This non-histone nuclear protein
was discovered by Gerdes et al (13) in 1980 and its encoding gene is
situated on the long arm of human chromosome 10q26 (14). Ki-67 is a recognized crucial cell
proliferation marker and has been extensively investigated. The
expression of this protein provides a reliable and rapid means of
assessing malignant cell growth and is strongly correlated with
tumor progression, metastasis and local recurrence in various
malignancies (15). Thus,
evaluations of Ki-67 expression are crucial for assessing tumor
cell proliferation, understanding tumor biology and estimating
potential risks.
Ki-67 is expressed in all active phases of the cell
cycle but is absent in quiescent cells, which makes it a dependable
marker of tumor proliferation (16). Furthermore, its expression is
tightly regulated by the cell cycle, particularly by E2F
transcription factors during the G1 phase. Ki-67 mRNA levels
increase during G1, while Ki-67 protein is degraded by the
ubiquitin-proteasome system (17,18). Thus, Ki-67 levels vary in cells
transitioning from quiescence back into the cell cycle.
Immunohistochemical (IHC) analysis-determined Ki-67
labeling indices have been used as potential predictive and
prognostic biomarkers of pathological complete response (pCR)
following neoadjuvant chemotherapy in patients with TNBC (19,20). pCR is a critical indicator of a
positive response to BC treatment, as it is strongly linked to
improved cancer-free and OS outcomes (21). MIB1 (anti-Ki67 antibody) is widely
regarded as the 'gold standard' clone for Ki-67 assessment
(22,23), though other clones, such as SP6,
30-9, K2 and MM1, are also used (24,25). Notably, rabbit anti-human Ki-67
monoclonal antibody SP6 recognizes the same Ki-67 epitope as MIB1
and has been reported to enhance the sensitivity of quantitative
image analysis (26,27).
Elevated Ki-67 levels are linked to increased tumor
aggressiveness and have been associated with poorer outcomes in
patients with TNBC (28). Studies
indicate that patients with TNBC expressing high levels of Ki-67
have poorer prognoses, as evidenced by lower disease-free survival
(DFS) and OS rates. According to a study by Goldhirsch et al
(29), Ki-67 expression of ≥40%
above normal significantly increases the risk of recurrence and
mortality in TNBC. They proposed a Ki-67 expression cutoff value of
14% for classifying BCs as having a favorable or unfavorable
prognosis, such as luminal A BC, and those with a poor prognosis,
specifically luminal B BC. However, the applicability of this Ki-67
cutoff to TNBC remains uncertain (30). These findings underscore the
significance of Ki-67 as a predictive and prognostic marker in
TNBC.
PD-L1 serves as a primary ligand for PD-1, a
co-inhibitory receptor that can be constitutively expressed or
induced in different cell types, including myeloid, lymphoid,
normal epithelial cells and cancer cells (31). Under physiological conditions, the
interaction between programmed death 1 (PD-1) and PD-L1 plays a
critical role in establishing immune tolerance and facilitating the
regulation of immune cell activity to prevent tissue damage and
autoimmune responses (32).
PD-L1 is a pivotal biomarker in TNBC and importantly
influences cancer cell immune evasion (33). In patients with TNBC, the
expression of PD-L1 on tumor and immune cells within the tumor
microenvironment has been linked to prognosis and treatment
outcomes (34). Oner et al
(35) recently observed PD-L1
expression in a subset of TNBC cases and found it to be associated
with the ability of tumors to suppress immune responses, and thus,
to contribute to the aggressive nature of TNBC. PD-L1 expression is
particularly crucial to enable the activities of immune checkpoint
inhibitors (ICIs), such as pembrolizumab and atezolizumab, which
target the PD-1/PD-L1 pathway. Interestingly, these therapies
produced promising results, particularly in PD-L1-positive patients
with TNBC, by enhancing the ability of the immune system to
identify and eradicate cancer cells (34,36).
PD-L1 regulation is of considerable research
interest and is involved in interferon gamma-induced tumor cell
surface expression, which inhibits immune responses and oncogenic
signaling via the phosphatase and tensin homolog (PTEN)/PI3K
pathway (37). PTEN loss or
silencing is common in glioblastoma and BC, promotes PD-L1
expression and is associated with PI3K pathway activation (38). In BC, PIK3CA mutations and PTEN
loss, which are associated with ER/PR-negative tumors, are present
in 30-40% of primary tumors (39). Furthermore, The Cancer Genome
Atlas data indicate that basal-like tumors, primarily TNBCs,
display PTEN mutation or loss in 35% of cases and that these are
correlated with PI3K activation (40).
Clinical studies have shown that patients with TNBC
with high PD-L1 expression, particularly those treated with ICIs
plus chemotherapy, tend to have better DFS and OS (33,35,36). However, response to ICIs can vary
significantly and not all PD-L1-positive patients achieve long-term
benefits (36). This variability
highlights the need for ongoing research to identify additional
biomarkers that predict which patients are likely to benefit from
these therapies.
Furthermore, PD-L1 expression has been found to
correlate with a higher rate of pCR following neoadjuvant
chemotherapy in patients with TNBC and to serve as a surrogate
marker of long-term survival (45). While high PD-L1 expression in TNBC
is linked to a more favorable response to standard treatments, its
role as a standalone prognostic marker has yet to be
determined.
BRCA1 and BRCA2 were identified in the 1990s as
components of the normal human genome, but their mutations are
positively associated with the risk of developing breast and
ovarian cancer (46,47). BRCA1 is expressed in endocrine
tissues and various cell types, including neuroepithelial cells,
during the early developmental stage. Likewise, BRCA2 expression is
observed in numerous tissue types, and it is higher in breast and
thymus tissues and lower in lung, ovary and spleen tissues
(48,49). Of note, BRCA1 and BRCA2 mutations
are strongly associated with TNBC. Women harboring BRCA1 mutations
are at higher risk of TNBC development because of the role BRCA1
plays in DNA repair. In fact, 15-20% of patients with TNBC harbor
mutations in the BRCA1 or BRCA2 genes (50).
BRCA1 and BRCA2 dysfunctions, such as mutations,
promoter methylation and diminished protein expression, are
alternative causes that impair their activities and contribute to
the 'BRCAness' phenotype (51,52). These dysfunctions result in
deficiencies in homologous recombination-mediated DNA double-strand
break repair. Furthermore, in the absence of functional BRCA1 or 2,
cells may depend on more error-prone repair pathways (53). BRCA1 mutations are linked with
reduced short- and long-term survival, whereas BRCA2 mutations do
not appear to significantly affect survival (52). In addition, BRCA1 methylation has
been associated with poor survival in patients with BC (54). However, studies have provided
mixed results for different TNBC subtypes, i.e., BRCA1 or 2
mutation carriers had similar or poorer survival than non-mutation
carriers (55,56).
BRCA1 mutations are associated with earlier TNBC
onset and a more aggressive disease course (57). In addition, patients with BRCA1
mutations exhibit a higher frequency of basal-like TNBC, a more
aggressive subtype of TNBC characterized by the absence of
estrogen, progesterone and HER2 receptors, making it particularly
challenging to treat using conventional therapies. Nevertheless,
individuals harboring BRCA mutations may respond better to
DNA-damaging agents, such as platinum-based chemotherapies or
poly(ADP-ribose) polymerase inhibitors (PARPi), due to defects in
homologous recombination repair pathways caused by the mutations
(58). Although less frequent
than BRCA1 mutations in patients with TNBC, BRCA2 mutations are
still linked to TNBC. Researchers are now focusing on more
personalized management strategies (59).
Epithelial cadherin (E-cadherin) is a vital
component of adherens junctions, which are crucial for cell
adhesion and maintaining the epithelial phenotype. The homophilic
binding of adjacent cells by E-cadherin is essential for
controlling proliferation contact inhibition, which occurs upon
reaching confluence (60).
Disruptions or abnormal expressions of E-cadherin/β-catenin complex
can lead to epithelial-mesenchymal transition (EMT) and contribute
to various malignancies (61).
E-cadherin is a pivotal player in TNBC due to its role in cell
adhesion and influence on tumor progression. In TNBC, loss of
E-cadherin promotes cancer cell detachment, migration and invasion
by activating EMT-associated transcription factors such as Snail,
Twist and Zeb1. In addition, E-cadherin loss is correlated with
elevations in the expression of mesenchymal markers like N-cadherin
and vimentin, and thus, further promotes metastasis (62,63).
On the other hand, reduced E-cadherin expression in
TNBC is associated with higher tumor grades, larger tumor sizes,
increased risk of lymph node metastasis and poorer prognosis
(64). A study by Shen et
al (61) showed that 67.5% of
TNBC cases exhibited loss of E-cadherin expression and found it to
be correlated with more aggressive tumor features and poorer
prognosis. Furthermore, studies suggest that the role of E-cadherin
as a prognostic marker in TNBC may be influenced by its
interactions with molecular pathways, such as the Wnt/β-catenin
signaling pathway (65,66). These findings underscore the
importance of E-cadherin as a diagnostic marker and a potential
therapeutic target in TNBC.
Several mechanisms have been identified that
contribute to the inactivation of E-cadherin in BC, particularly
through mutations in the E-cadherin 1 gene (67,68). These mutations are frequently
detected in invasive lobular BC (ILC) and were originally found in
55-56% of ILC cases (69).
However, subsequent studies reported lower frequencies with
mutation rates of 15-27%. These mutations often coincide with loss
of heterozygosity (LOH) at the E-cadherin locus, which supports the
suggestion it acts as a tumor suppressor gene. Approximately 50% of
ILC cases exhibit LOH, leading to protein dysfunction and loss of
E-cadherin expression (69).
E-cadherin is a promising biomarker of the responses
of specific BC subtypes to mitogen-activated protein kinase kinase
(MEK) inhibition (71). Routine
breast biopsies aimed at differentiating invasive ductal carcinoma
(IDC) from lobular carcinoma (72,73) revealed that E-cadherin is
expressed in 90% of IDC cases, which account for 80% of all BC
cases (74). The pivotal role
played by E-cadherin in tumor progression, metastasis and rapid
growth underscores its potential as an oncogene and a critical
target for BC therapies (75,76). However, E-cadherin expression is
linked with poorer survival in patients with BC, which challenges
its classification as a tumor suppressor gene. Studies have also
indicated that E-cadherin interacts with epidermal growth factor
receptor (EGFR), activates the MEK/ERK pathway and fosters
aggressive, proliferative tumor phenotypes in IDC (76). Although MEK inhibitors, such as
PD0325901, effectively counter this hyper-proliferative effect
in vitro and in vivo, significant hurdles must be
overcome before their clinical deployment.
EGFR is present on the surfaces of normal epithelial
cells and overexpressed in certain tumors, including brain, lung
and ovarian cancer. Furthermore, increased EGFR expression has been
associated with tumor cell migration, invasion and prognosis
(77). The binding of ligands to
EGFR promotes receptor homodimerization or heterodimerization,
leading to autophosphorylation of its tyrosine kinase domains and,
subsequently, to the stimulation of downstream signaling pathways
such as those of PI3K/AKT and Ras/Raf/MEK/ERK (78,79).
EGFR mutations are closely associated with
ethnicity, gender, adenocarcinoma and smoking status (80-83). The rarity of activating EGFR
mutations in TNBC has been documented in numerous studies that
reported different mutation frequencies in East Asians and
Caucasians (84,85). These studies conducted in Europe
and Australia reported no or low mutation rates, whereas certain
Asian studies reported frequencies of 7.7-11.4% in TNBC or
basal-like cancers (84,85). However, studies conducted on
Japanese and Chinese cohorts detected no activating EGFR mutations.
The presence of regional differences in EGFR mutations in TNBC,
akin to those detected in lung cancer, remains uncertain. Thus,
further investigations are required to determine whether certain
patients with TNBC may benefit from tyrosine kinase inhibitor (TKI)
therapy.
EGFR mutations occur predominantly in exons 18-21.
The most common are exon 19 deletion mutations, the exon 21 L858R
point mutation and the exon 20 T790M mutation. These mutations
constitute 50-90% of all EGFR mutations and are collectively termed
classical mutations (83,86). Other mutations considered rare but
clinically meaningful include G719X in exon 18 and L861Q and G719C
in exon 21 (87,88). Advances in sequencing and
polymerase chain reaction technologies have identified EGFR
mutations as predictors of the success of EGFR-TKI targeted
therapies (89). For patients
with non-small cell lung cancer (NSCLC) with EGFR mutations, the
use of EGFR-TKI therapy resulted in an objective response rate
(ORR) of 70-80%, a median progression-free survival (PFS) of 9-12
months and an OS of 20-32 months, which established EGFR-TKI as the
preferred first-line treatment (90). Clinical trials of EGFR-targeted
TKIs in TNBC as monotherapy or in combination with chemotherapy
have generally been unfruitful. This contrasts their efficacies in
lung cancer, in which activating EGFR mutations drive therapeutic
response.
TNBC frequently exhibits EGFR overexpression, which
is associated with poor clinical outcomes (91). The prevalence of EGFR
overexpression in TNBC varies significantly across studies from 13
to 78% (92). Furthermore, the
rate of EGFR protein overexpression in TNBC, as evaluated by IHC,
ranges from 13 to 76%, depending on the evaluation methods and
antibodies used (93,94). For instance, rates of EGFR
expression were reported at 13 and 37% using Novocastra antibodies,
52% with Zymed antibodies and 76% with the EGFR PharmDx Kit (Dako)
(95,96). The study that used the PharmDx Kit
reported an EGFR protein overexpression rate of 72%, but EGFR mRNA
levels varied considerably (97),
suggesting post-transcriptional regulation contributes to EGFR
protein overexpression in TNBC.
EGFR overexpression has also been associated with
poorer DFS and OS in patients with TNBC (98). The use of specific EGFR
inhibitors, such as erlotinib, demonstrated promise in preclinical
models by inhibiting tumor growth and metastasis, particularly in
terms of reducing the EMT frequently observed in aggressive cancer
phenotypes (99). However, the
clinical effectiveness of EGFR as a therapeutic target in TNBC
remains uncertain due to variable patient responses. This
variability may be due to differences in EGFR expression levels and
the presence of co-expressed markers that interfere with the
efficacy of EGFR-targeted therapies (98). Therefore, optimizing the
effectiveness of EGFR-targeted therapy necessitates a patient
selection strategy that considers EGFR expression levels and the
molecular characteristics and signaling pathways associated with
EGFR. Furthermore, additional research is required to assess
whether combination therapies involving EGFR inhibitors and other
treatments such as immunotherapy or chemotherapy could lead to more
favorable outcomes than monotherapy.
The PIK3CA gene is located on chromosome 3q26.3 and
encodes the catalytic subunit of PI3K, a key enzyme in the
PI3K/AKT/mTOR signaling pathway (100). PI3Ks are intracellular signaling
enzymes and are divided into three classes in mammals (101). Class I PI3K is the most
significant in terms of promoting tumor development and has four
catalytic isoforms, which each contain a regulatory subunit (p85α,
β or γ) and a catalytic subunit (p110α, β, δ or γ) (100,102). Upon activation by binding of
growth factors, such as EGFR and VEGFR, to their respective
receptors, a series of downstream signaling molecules are
activated. Located in the cytoplasm, PI3K catalyzes the conversion
of phosphatidylinositol 4,5-bisphosphate into phosphatidylinositol
(3,4,5)-trisphosphate (PIP3) (102,103), which then recruits AKT to the
cell membrane, where it becomes activated by phosphorylation. The
activated AKT then phosphorylates downstream targets, including
mTOR, to initiate a cascade that augments protein synthesis and
cell growth (104).
The PI3K/AKT/mTOR signaling pathway plays a vital
role in the progression and aggressiveness of TNBC. This pathway
contributes to tumor growth, survival, motility and therapy
resistance and is frequently dysregulated in TNBC (108,109), and this dysregulation is
correlated with poor clinical outcomes in patients with TNBC.
Common mutations and alterations in TNBC include the loss of the
tumor suppressor PTEN and mutations in PIK3CA, which result in
hyperactivation of the PI3K/AKT/mTOR pathway. This hyperactivation
promotes oncogenic processes, such as cell proliferation and
survival, and facilitates metastasis and chemoresistance (9,110). Furthermore, the pathway has been
suggested to contribute to chemoresistance and tumor relapse
through an association with cancer stem cells in TNBC (111). Doxorubicin resistance is also
promoted by this pathway due to its impact on EMT and the cancer
stem cell population (112).
Targeting the PI3K/AKT/mTOR signaling pathway has
become a focus for the development of therapeutic strategies for
TNBC. However, although PI3K/AKT/mTOR pathway inhibitors produced
promising results in preclinical studies, their clinical
effectiveness is hindered by obstacles such as drug resistance and
toxicity (9,108). Consequently, current research
emphasizes refining drug combinations and pinpointing biomarkers to
identify patients most likely to benefit from these treatments.
p53 is a nuclear transcription factor that activates
a range of target genes crucial for initiating cell cycle arrest or
apoptosis (113,114). Under ordinary conditions, p53 is
present at minimal levels due to proteasomal degradation, chiefly
controlled by the RING-finger E3 ubiquitin ligase murine double
minute 2 (MDM2) (115). When DNA
is damaged, p53 accumulates in the nucleus through
post-translational modifications like phosphorylation and
acetylation, potentially disrupting its interaction with MDM2 and
activating p53 (114,116). Once activated, p53 either
triggers cell cycle arrest or induces apoptosis, depending on the
severity and nature of the damage (114,116). Cell cycle arrest, mediated by
p53, provides time for DNA repair, thereby enabling cells to resume
normal functioning if the damage is repaired. Alternatively, when
the damage is severe, p53 initiates apoptosis to prevent the spread
of defective genetic material (117).
P53 is primarily affected by missense mutations or
non-synonymous single-nucleotide variants, which frequently occur
in the core domain and result in full-length mutant p53 proteins
(118). Other mutations, such as
synonymous, frameshift, silent and post-translational modifications
in TP53, are also observed in various tumors (119). Missense mutations in p53,
involving amino acid replacement, are associated with poor
prognosis and the pathogenesis of >50% of malignant tumors.
Mutated p53 often forms aggregates and is associated with loss of
function (LOF), dominant-negative and gain-of-function (GOF)
mutations at hotspot regions (120,121). In particular, GOF mutations
enhance cancer traits, such as migration, invasion and metastasis,
while LOF mutations enhance chemoresistance. There are two
categories of missense mutations: Contact mutations (e.g., R248Q,
R273H) that enable the protein to retain its conformation but lose
its DNA binding ability, and conformational mutations (e.g., R175H,
Y220C) that disrupt protein structure and stability (118,122). Furthermore, zinc ion loss
resulting from mutations, such as C176F or Y220C, destabilizes the
p53 DNA-binding domain, inhibiting tetramer formation and promoting
misfolding and aggregation (119,122). For instance, the Y220C mutation
exposes hydrophobic regions, thus facilitating amyloid-like
aggregation. These changes adequately emphasize the structural and
functional instability of mutant p53 during cancer progression
(123).
The absence or mutation of p53 in human cancers is
linked to increased tumor growth and treatment resistance (122). Mutant misfolded p53 proteins,
resulting from dominant-negative TP53 mutations under abnormal
conditions, can aggregate with themselves and wild-type p53 and
cause endoplasmic reticulum stress and the formation of prion-like
amyloid fibrils. These aggregates bear a GOF phenotype, lack
tumor-suppressor activity and promote cancer progression (119,120,124). Mutations in the p53 gene are
observed in ~80% of TNBC cases and play a crucial role in driving
the aggressive nature and progression of this cancer subtype
(125). In addition, they
abrogate the tumor-suppressing function of p53, which results in
unchecked cell growth, resistance to standard therapies, increased
risk of metastasis and poorer prognoses (125,126).
Mutant p53 interacts with other proteins, such as
p63 and p73, which also function as tumor suppressors, inhibits
their activities and promotes metastasis (127,128). These interactions modulate
critical signaling pathways, including the transforming growth
factor-β pathway, and facilitate tumor cell migration and invasion
through receptors like EGFR and hepatocyte growth factor receptor
(129,130). In addition, when affected by p53
mutations, the PI3K/Akt/mTOR pathway enhances tumor growth and
invasion (131).
Research into the downstream effects of these
mutations is evolving with a focus on ICIs and molecular inhibitors
(132). The prognostic
significance of p53 expression in TNBC remains under investigation
(133). However, studies suggest
that BC exhibiting IHC-determined p53 positivity is associated with
a more aggressive and metastatic phenotype and unfavorable patient
outcomes (134,135). Ongoing research aims to target
the p53 pathway in TNBC specifically. Efforts are directed toward
developing novel therapeutic strategies that restore wild-type p53
function or target the downstream effects of p53 mutations.
Ki-67 has emerged as a significant therapeutic
target and characteristically exhibits high expression in malignant
cells and a minimal presence in normal tissues. Retrospective
studies explored the possibility of using Ki-67 as a potential
marker for assessing cell proliferation and predicting the outcomes
of various cancers (136,137).
Clinical research increasingly supports the diagnostic utility of
Ki-67 in cancer (138). IHC
remains the standard method for evaluating Ki-67 expression, and a
10-14% positivity threshold is commonly used to classify patients
at higher risk (139). Ki-67
also serves as a biomarker of treatment response and long-term
clinical outcome in patients with BC undergoing neoadjuvant
endocrine therapy and has been the subject of several prospective
trials (140-142). Further, Ki-67 aids the
classification of patients with partial responses and identifies
those who may benefit from extended systemic therapy or are ready
for primary surgery (143).
However, variations in Ki-67 measurement limit its consistent
application in standard BC IHC workups.
Studies that utilized antisense RNA and RNA
interference (RNAi) indicated that Ki-67 knockdown reduces cell
proliferation. Furthermore, RNAi has emerged as an effective tool
for cancer therapy and small interfering (si)RNAs have been used to
target multiple genes and enhance antitumor efficacy (144,145). Recent research demonstrated that
siRNA targeting Ki-67 effectively and specifically inhibited the
proliferation of human renal cell carcinoma (RCC) cells more than
antisense strategies (146). In
addition, a pSilencerKi-67 construct using short hairpin RNAs
against Ki-67 was developed to overcome the transient effects of
siRNAs, and this construct better inhibited cell proliferation and
apoptosis induction in 786-O RCC cells than synthetic siRNAs
(147).
The clinical application of siRNAs is hindered by
off-target effects and immune activation via toll-like receptors
(148). To mitigate these
issues, approaches such as RNA modification, optimized delivery
systems and tumor-specific targeting have been explored (149,150). Among these approaches, oncolytic
adenoviruses, which selectively replicate in tumor cells, emerged
as promising vehicles for gene therapies, such as siRNA delivery.
Conditionally replicative adenoviruses (CRAds) have been engineered
to target tumor cells using two strategies, viz. deleting viral
elements essential for replication in normal cells or utilizing
tumor-specific promoters (151,152). CRAds based on Ad5 have been
demonstrated to be effective and safe cancer treatments (153). In this context, researchers
developed ZD55-Ki-67, an oncolytic adenovirus that delivers
Ki-67-shRNA (154). This
construct effectively induced apoptosis in renal cancer cells and
curtailed tumor growth in preclinical models (155). To enhance safety, G250-Ki-67
virus with a renal cancer-specific G250 promoter was engineered to
ensure replication occurred solely in renal cancer cells. This
virus successfully downregulated Ki-67, reduced cell proliferation
and induced apoptosis, which highlighted its potential for treating
renal clear cell carcinoma (156).
Antisense oligonucleotides (ASOs) delivered
systemically without formulation have demonstrated efficacy in
treating several non-oncology diseases and are currently being
investigated for cancer therapy (157-159). Research has shown that
Ki-67-specific ASOs can suppress cancer cell proliferation and
reduce tumor growth (160,161). Schlüter et al (14) discovered that Ki-67 antisense
oligodeoxynucleotides (ASODNs) inhibited the proliferation of human
myeloma cells, while Kausch et al (162,163) observed similar effects on cancer
cells in vitro and in vivo. Ki-67 ASODNs were also
subjected to a phase I clinical trial for bladder cancer (164). Furthermore, recent studies also
indicate that methylated oligonucleotides targeting Ki-67 can
impede renal carcinoma cell proliferation and induce apoptosis
(20).
The clinical application of ASOs is constrained by
issues such as low affinity, susceptibility to nuclease degradation
and non-specific binding. Peptide nucleic acids (PNAs), i.e.,
synthetic DNA analogs with a modified backbone, bind to
complementary targets with enhanced stability and specificity
(165). PNAs have been developed
as antisense and antigene agents to control gene expression and
have proved to be more effective than ASOs in suppressing human
telomerase activity (166).
Additional research is needed to improve the clinical utility of
these therapies, which may constitute a new paradigm for cancer
treatment.
PD-L1 is expressed in invasive lobular and ductal
BCs, in which it localizes in CD8+ T lymphocytes (167). Overexpression of PD-L1 mRNA is
associated with poor prognostic factors, including hormone receptor
negativity, HER2 positivity, higher tumor grade, advanced stage and
elevated proliferation rates (168). About 20% of TNBC cases exhibit
PD-L1 expression due to PTEN loss, which is characterized by
significant cytotoxic T-cell infiltration and improved response to
neoadjuvant chemotherapy (34).
These findings underscore the potential of anti-PD-1 and PD-L1
inhibitors as treatments for TNBC (169).
PD-1 and PD-L1 inhibitors have demonstrated
significant clinical efficacy in lung, kidney, bladder, skin and
breast malignancies (170-173). TNBC may exhibit a more favorable
response to immunotherapy than other BC subtypes due to higher TIL
levels, a greater mutational burden and higher PD-L1 expression
(174). Furthermore, elevated
TIL levels correlate with enhanced outcomes in patients with TNBC
(175). By enhancing immune
clearance, ICIs targeting the PD-1/PD-L1 pathway offer a promising
therapeutic strategy for TNBC (176). As a result, immunotherapies
targeting the PD-1/PD-L1 pathway, which counteract
immunosuppression in the TNBC tumor environment, have been
developed. In 2019, the US Food and Drug Administration (FDA)
approved atezolizumab (an anti-PD-L1 antibody) in combination with
nanoparticle albumin-bound paclitaxel as a first-line treatment for
TNBC based on the results of the IMpassion130 trial (NCT02425891).
This combination therapy has since become the standard of care for
patients with PD-L1-positive, unresectable, locally advanced or
metastatic TNBC (36).
Additionally, in 2017, pembrolizumab (an anti-PD-1 antibody) was
approved as a histology-agnostic therapy for tumors with high
microsatellite instability or mismatch repair deficiency, marking
the first FDA approval of a cancer treatment based solely on a
tumor biomarker independently of the primary site (177,178).
Drugs designed to inhibit PD-1 signaling have shown
prolonged clinical efficacy against various advanced solid tumors
(179). In a phase I clinical
trial, the monoclonal antibody BMS-936559 (MDX 1105), which targets
PD-L1, demonstrated objective response rates ranging from 6 to 17%
in 160 patients with an advanced solid tumor (180). The JAVELIN study evaluated the
effectiveness of avelumab, an anti-PD-L1 antibody, across different
BC subtypes, regardless of PD-L1 expression levels. In the TNBC
group of 58 ER+/HER2-patients, the response rate was 8.6%, and
among 72 ER+/HER2-patients and 26 HER2+ patients, the response
rates were 2.8 and 3.8%, respectively. Early findings suggest that
tumors positive for PD-L1 expression are more likely to respond to
treatment (181).
Combinations of MEK inhibitors and PD-L1/PD-1
inhibitors have been reported to enhance antitumor immune response
in a BC mouse model (182). A
study by Sagiv-Barfi et al (183) reported that when ibrutinib plus
anti-PD-L1 antibody were administered to mice with TNBC that lacked
inherent sensitivity to ibrutinib; tumor progression was
significantly decreased and survival increased compared to the
effects of either drug used independently.
Patients with TNBC show a significantly higher
prevalence of BRCA1/2 mutations (15-20%) than other BC subtypes
(50). The PARPi olaparib, which
induces synthetic lethality in tumors deficient in homologous
recombination, specifically targets and eliminates tumor cells
harboring BRCA1/2 mutations. Results from the phase III olympiAD
trial (184) demonstrated that
olaparib monotherapy significantly improved PFS in patients with
HER2-negative metastatic BC with BRCA1/2 mutations. The ORR in the
olaparib-treated group was 59.9%.
Talazoparib, a PARP inhibitor, was approved by the
FDA in 2018, as a monotherapy for adult patients with germline
BRCA-mutated, HER2-negative, locally advanced or metastatic BC,
including TNBC (185). This is
the first single-agent therapy to achieve pCR in germline
BRCA-positive, HER2-negative early BC (186). Common adverse events included
anemia and nausea, which are typical of PARPi (186). These favorable results led to
the phase II confirmatory NEOTALA study (NCT03499353), which
contains a larger patient cohort.
PARPi has produced promising outcomes when combined
with other cytotoxic agents, due to its synergistic effects and
potential to enhance chemotherapy regimens (187). For instance, Veliparib has been
reported to increase the cytotoxicity of temozolomide and achieve
complete response in 50% of women with germline BRCA-associated BC
and a response rate of 22% (188). Additionally, combinations of
olaparib with cisplatin, carboplatin and topotecan have yielded
response rates of up to 73%, where response included stable
disease, partial and complete responses (189,190). However, significant hematologic
toxicity, including grade 3 neutropenia, was reported when olaparib
was combined with paclitaxel (191).
Platinum-based compounds and their derivatives
target tumor cells by inducing DNA strand breaks. These compounds
also eradicate cancer cells by promoting oxidative stress, altering
microRNA regulation and activating protein kinase C (192-194). Cells with BRCA1/2 mutations,
which are deficient in DNA repair functionality, are particularly
susceptible to DNA-damaging agents (195). Consequently, the relationship
between BRCA1/2 mutations and platinum sensitivity in patients with
TNBC has emerged as a research topic of particular interest. A
meta-analysis of 22 trials indicated that platinum-based therapies
are highly effective for patients with BRCA1/2-mutated TNBC. The
inclusion of platinum significantly enhanced treatment outcomes,
particularly in the neoadjuvant setting and for advanced-stage
disease (196).
SHE78-7 is a monoclonal antibody that targets
E-cadherin-mediated cell adhesion (200). In HT29 colorectal adenocarcinoma
spheroids, SHE78-7 disrupted E-cadherin interactions and thereby
enhanced the sensitivity of these spheroids to chemotherapeutics,
such as 5-fluorouracil, paclitaxel and etoposide, but not to
cisplatin. Furthermore, this antibody diminishes the activity of
specific protein kinase C isoforms associated with chemoresistance
and facilitates intracellular drug accumulation (201). This study supports the notion
that targeting cell adhesion mechanisms may provide a means of
overcoming solid tumor resistance. However, further studies are
needed to assess its clinical applicability, given the vital role
played by E-cadherin in healthy tissue adhesion.
The therapeutic efficacies of anti-EGFR monoclonal
antibodies, including cetuximab and panitumumab, and small-molecule
TKIs like gefitinib and neratinib, have been explored in clinical
trials for TNBC. However, these therapies often yield limited
patient responses or are associated with resistance development
(93,202), which highlights the need for
novel EGFR-targeted therapies.
Cetuximab, the first FDA-approved EGFR-targeted
therapeutic antibody (in 2004), inhibits EGFR activation by
obstructing its ligand-binding pocket (203). When combined with chemotherapy
or radiotherapy, cetuximab has reportedly prolonged median OS by ~3
months in colorectal cancer (204), ~5 months in head and neck cancer
(205) and ~8 months in NSCLC
(206). Other EGFR-targeting
antibodies, such as panitumumab, nimotuzumab, necitumumab and
zalutumumab, have demonstrated efficacy in colorectal, head and
neck, and biliary cancer, as well as NSCLC (207,208). While most antibodies block EGFR
ligand binding, nimotuzumab also elicits immune responses, which
enhance its therapeutic impact despite reduced binding efficiency
(209). However, these
antibodies have only had limited success in TNBC despite achieving
high EGFR expression.
EGFR-targeting therapies, including monoclonal
antibodies and small molecule TKIs, have been employed to treat
TNBC, but numerous patients display poor responses or develop
resistance (93). Chimeric
antigen receptor-T (CAR-T) technology has recently shown great
promise as an immunotherapy for solid cancers by enabling T cells
to target tumor-specific antigens through scFv binding domains. In
addition, EGFR-specific CAR-T cells exhibited enhanced recognition
and cytotoxicity in TNBC cells expressing high levels of EGFR
compared to controls, as evidenced by increased cytokine secretion
and enhanced cell lysis in vitro (210). Furthermore, these cytotoxic
effects were significantly increased by promoting EGFR
dimerization. These findings indicate that the efficacy of
EGFR-specific CAR-T cells is closely associated with EGFR
expression levels and underscore their potential as a targeted
therapy for TNBC (211).
On the other hand, EGFR CAR-T cells have shown
promise in preclinical models of TNBC, but their reliance on
cetuximab-based scFv raises concerns about on-target off-tumor
toxicity, as they affect EGFR expression in both tumor cells and
normal keratinocytes (212,213). To overcome this limitation, a
modified CAR-T construct that employs the tumor-specific EGFR
mAb806 antibody was developed (214). This antibody selectively targets
EGFR, which is linked to oncogene amplification, and in clinical
trials, has demonstrated efficacy in eliminating TNBC cells in
vitro while showing minimal toxicity (215). EGFR806 CAR-T cells are also
being investigated in clinical studies for the treatment of
pediatric brain tumors (NCT03638167), and no dose-limiting toxicity
has been reported to date (216). Also, low EGFR expression in
normal adult brain tissue reduces the risk of undesired off-tumor
effects, which further highlights its therapeutic potential
(217).
The Schroeder lab recently developed a peptide
named cSNX1.3 to mimic the interaction domain between endosomal
trafficking protein SNX1 and EGFR (218). This end-capped peptide binds to
EGFR with an efficiency comparable to SNX1 and reduces nEGFR levels
in TNBC cells overexpressing EGFR. Treatment with cSNX1.3
selectively impacts EGFR-dependent oncogenic behaviors such as
proliferation, survival, migration and mammosphere formation but
has no effect on normal immortalized breast epithelial cells
(218).
In precision medicine, therapeutic strategies that
target PIK3CA in TNBC aim to inhibit abnormal signaling pathways
that drive cancer progression. Significant progress has been made,
notably in patients with BC demonstrating heightened sensitivity to
PI3K inhibition. A small molecule inhibitor of PI3K p110α,
alpelisib, has demonstrated efficacy in clinical trials by
effectively suppressing tumors with PIK3CA mutations (219,220). Additionally, dual inhibitors
like taselisib, which target PI3K and its downstream effector AKT,
have exhibited potential in preclinical studies (221). However, these dual-blockade
agents have adverse effects that may limit their clinical benefits.
For instance, although AZD2014 (an mTOR catalytic inhibitor)
exhibited notable activity in preclinical studies, it was less
effective than everolimus in patients with homologous
recombination-positive BC in clinical settings (222). Recent clinical studies, such as
the BELLE-2 and BELLE-3 studies, have underscored the efficiency of
PI3K inhibitors like buparlisib (a pan-class I inhibitor) for
treating PIK3CA-mutant homologous recombination-positive BC
(223,224). The exploration of combination
therapies involving PI3K inhibitors and chemotherapy or
immunotherapy is now underway with the aim of improving efficacy.
Despite issues like resistance and tumor heterogeneity, continued
research into therapies targeting PIK3CA provides a promising route
to improving outcomes (219).
The complexity of PIK3CA-driven TNBC underscores the need for
innovative strategies, though current studies offer a potential
means of addressing this aggressive cancer.
Combination therapies are widely recognized as
potential means of addressing TNBC, the molecular heterogeneity of
which poses significant treatment challenges. Targeting multiple
proteins, such as mTOR, AKT and PI3K, often results in feedback
mechanisms that restrict the effectiveness of these treatments
(225). For instance, mTOR
inhibition can lead to the upregulation of downstream receptor
tyrosine kinases (RTKs), a class of cell surface receptors that
mediate signal transduction through phosphorylation of tyrosine
residues, and cause rebound activation of AKT. Similarly, AKT
inhibition can trigger forkhead box O-mediated transcription and
subsequent RTK activation (223). Furthermore, PI3K inhibition not
only prevents AKT activation but also enhances MAPK signaling.
These intricate interactions highlight the complexities of
precisely targeting cancer pathways and the potential for
developing resistance (226). To
address these challenges, research is underway to combine PI3K
inhibitors with conventional chemotherapy or ICIs to achieve
synergistic therapeutic benefits. Currently, five FDA-approved PI3K
inhibitors are available (copanlisib, idelalisib, umbralisib,
duvelisib and alpelisib), which paves the way for further
investigation into new, more effective inhibitors with improved
safety profiles (227).
mTOR inhibitors, including everolimus and
temsirolimus, have been demonstrated to be effective BC treatments.
Studies like BOLERO-2, PrE0102 and GINECO have demonstrated that
combining everolimus with endocrine therapy significantly improves
PFS in postmenopausal patients with homologous
recombination-positive, HER2-negative advanced BC (228,229). Furthermore, the BOLERO-4 and
BOLERO-5 studies confirmed that everolimus plus letrozole or
exemestane prolonged PFS, and the MIRACLE study reported their
effectiveness in premenopausal patients with metastatic BC
(230). Additionally, a phase I
trial of temsirolimus and everolimus in combination with liposomal
doxorubicin and bevacizumab in patients with metaplastic TNBC
demonstrated an ORR of 21% [complete response (CR)=4 (8%); partial
response (PR)=7 (13%)], with enhanced efficacy observed in patients
with PI3K pathway activation. However, subsequent trials were
discontinued (231).
Developing p53-targeted drugs poses significant
challenges due to the necessity to selectively target mutant p53 in
cancer cells while sparing wild-type p53 in healthy cells and is
further complicated by the structural diversity of mutant p53
proteins due to various mutations (232). Therapeutic strategies targeting
p53 are generally classified as strategies that restore wild-type
p53 function or eliminate mutant p53 (233).
Several studies have focused on the use of zinc
ions to reactivate mutant p53 function. When mutant p53 binds zinc,
it regains its wild-type conformation and DNA-binding abilities,
which trigger target gene activation and inhibit tumor growth
(234). Zinc supplements, such
as zinc chloride, have been shown to suppress p53 oncogenicity,
restore binding to target promoters and activate apoptotic pathways
in mutants, including H175 and H273 (235). Zinc ion treatments also enhance
the functions of p53 and p73 by restoring their interactions with
gene promoters (122). In
addition, zinc metallochaperones (ZMCs), such as ZMC1, have been
developed to reactivate zinc-deficient mutant p53, and thus,
promote the apoptosis in R175H p53 mutations and restore a
wild-type-like structure (236).
Furthermore, targeting p53 and BRCA1 using ZMCs has been suggested
as a promising therapeutic strategy for BCs in which these tumor
suppressors are inactivated (237). Albumin nano vector formulations
have also been suggested for the effective delivery of ZMC-based
drugs (237). Studies show
combining ZMC1 with zinc ions can suppress tumor growth and enhance
survival, particularly in zinc-deficient and BRCA1-deficient BC
models (238). In addition, ZMC1
acts synergistically with PARPi olaparib, even in
olaparib-resistant tumor cells. Despite these encouraging results,
further investigations are required to evaluate the safety and
efficacy of these strategies in clinical settings (238).
The structural reversibility of mutant p53,
particularly in temperature-sensitive variants, enables the
restoration of wild-type activity under specific conditions
(122). DNAJA1 (DnaJ heat shock
protein family member A1), a chaperone protein, prevents the
proteasomal degradation of misfolded mutant p53 and contributes to
tumor suppression (239). It has
also been suggested inhibiting DNAJA1 would promote the degradation
of mutant p53 and reduce the malignant potential of cancer cells
(240). In addition, compounds
such as PLINH, derived from plumbagin, have been shown to suppress
the growth and migration of cancer cells by depleting mutant p53
through DNAJA1 inhibition. However, DNAJA1 inhibitors are not yet
clinically available and further research is required to confirm
their safety and efficacy (240).
Compounds such as CP31398, P53R3 and PRIMA-1, and a
derivative APR-246 (Eprenetapopt), can stabilize mutations in the
DNA-binding domain (DBD) of p53, prevent p53 aggregation and
restore its tumor suppressor function (241). APR-246 is the most advanced and
extensively studied of these compounds and exhibits pronounced
pro-apoptotic effects, particularly in mutated p53-containing
cancers like TNBC (242).
APR-246 interacts synergistically with chemotherapeutics such as
eribulin but with cell-dependent variable efficacy. Reactivation of
p53 by PRIMA-1 and its active metabolite,
2-methylene-3-quinuclidinone, can restore misfolded p53 mutants to
their native conformation, thereby inducing apoptosis and
activating multiple p53 target genes. Furthermore, anti-amyloid
oligomer antibody assays demonstrated that PRIMA-1 reduces the
accumulation of mutant p53 aggregates in breast and ovarian cancer
cell lines (242,243). These compounds represent
promising therapeutic options for targeting p53 mutations in
TNBC.
Resveratrol, a natural compound sourced from plants
such as berries, peanuts and grapes, has demonstrated the ability
to target cancer signaling pathways and inhibit amyloid protein
aggregation, including mutant p53 in BC models (247). A study from 2018 demonstrated
that resveratrol prevents aggregation in the DBD region of
wild-type and mutant p53 (R248Q) dose-dependently and that mutant
p53 BC cell lines (HCC-70 and MDA-MB-231) were more responsive to
treatment than wild-type p53 cells (247). Furthermore, resveratrol reduced
p53 aggregation and amyloid colocalization in mutant cell lines and
tumor models (248).
Additionally, small-molecule inhibitors targeting the
inositol-requiring enzyme 1α and PRKR-like ER kinase enzyme active
sites, particularly those based on salicylaldehyde, have been shown
to effectively enhance response to chemotherapy and reduce tumor
cell secretions in TNBC xenograft models (248).
Synthetic siRNA delivery offers a promising
strategy for p53-targeted genetic therapy by mitigating the GOF
effects of mutant p53 while preserving wild-type p53 functionality
(249). Interestingly, specific
siRNAs designed to target hotspot p53 mutations have effectively
reduced tumor viability in patient-derived xenografts without
impacting wild-type p53 or inducing organ toxicity (250). In TNBC cells, siRNA-mediated
suppression of mutant p53 expression led to the activation of
pro-apoptotic genes, such as caspases, BCL-2 family members and
death receptors, and the downregulation of anti-apoptotic genes,
thus promoting cell death (251). Although the development of
siRNA-based therapies is in the preliminary stage, advancements in
RNA delivery technology hold substantial promise for the treatment
of mutant-specific cancer (249).
TNBC remains one of the most aggressive and
challenging subtypes of BC due to its molecular heterogeneity, lack
of specific therapeutic targets and poor clinical outcomes. Despite
the significant progress made in understanding the biology of TNBC,
including the identification of key prognostic markers, such as
Ki-67, PD-L1, BRCA1/2 mutations, E-cadherin loss and EGFR
alterations, therapeutic advancements have been constrained by
issues such as drug resistance and tumor complexity. The
PI3K/AKT/mTOR pathway and mutant p53 remain critical molecular
targets for intervention, and ongoing research continues to explore
their potential to enhance patient outcomes. Innovative approaches
such as ICIs, CAR-T cell therapies, siRNA-based treatments and zinc
metallochaperones offer promise for the management of TNBC. These
strategies leverage advancements in molecular biology to tackle the
unique challenges posed by the heterogeneity and resistance
mechanisms of TNBC. Nonetheless, their clinical efficacies and
safety profiles require further validation through comprehensive
trials. Combination therapies that target multiple pathways,
including DNA damage repair and immune modulation, are gaining
traction to counteract treatment resistance and improve therapeutic
efficacy. Furthermore, the integration of targeted therapies with
conventional treatments, such as chemotherapy and radiotherapy,
also holds promise due to observed synergistic effects.
Although TNBC remains a challenging subtype,
ongoing advancements in immunotherapy, targeted therapy and
antibody-drug conjugate treatments are promising. The development
of novel biomarkers, the application of precision medicine and the
optimization of combination therapies are anticipated to enhance
TNBC treatment outcomes. Overcoming treatment resistance and
establishing personalized therapeutic strategies are pivotal in
advancing the treatment of this formidable disease.
Not applicable.
Writing of the original draft was conducted by ESK.
The author has read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The author declares that they have no competing
interests.
Not applicable.
This Research was supported by a Research Grant from Duksung
Women's University (grant no. 2024-3000009365).
|
1
|
Lehmann BD, Jovanović B, Chen X, Estrada
MV, Johnson KN, Shyr Y, Moses HL, Sanders ME and Pietenpol JA:
Refinement of triple-negative breast cancer molecular subtypes:
Implications for neoadjuvant chemotherapy selection. PLoS One.
11:e01573682016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ismail-Khan R and Bui MM: A review of
triple-negative breast cancer. Cancer Control. 17:173–176. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perou CM: Molecular stratification of
triple-negative breast cancers. Oncologist. 16(Suppl 1): S61–S70.
2011. View Article : Google Scholar
|
|
4
|
Bernardi R and Gianni L: Hallmarks of
triple negative breast cancer emerging at last? Cell Res.
24:904–905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mehanna J, Haddad FG, Eid R, Lambertini M
and Kourie HR: Triple-negative breast cancer: Current perspective
on the evolving therapeutic landscape. Int J Womens Health.
11:431–437. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prakash O, Hossain F, Danos D, Lassak A,
Scribner R and Miele L: Racial disparities in triple negative
breast cancer: A review of the role of biologic and non-biologic
factors. Front Public Health. 8:5769642020. View Article : Google Scholar
|
|
7
|
Asleh K, Riaz N and Nielsen TO:
Heterogeneity of triple negative breast cancer: Currentadvances in
subtyping and treatment implications. J Exp Clin Cancer Res.
41:2652022. View Article : Google Scholar
|
|
8
|
Newton EE, Mueller LE, Treadwell SM,
Morris CA and Machado HL: Molecular targets of triple-negative
breast cancer: Where do we stand? Cancers (Basel). 14:4822022.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang HP, Jiang RY, Zhu JY, Sun KN, Huang
Y, Zhou HH, Zheng YB and Wang XJ: PI3K/AKT/mTOR signaling pathway:
An important driver and therapeutic target in triple-negative
breast cancer. Breast Cancer. 31:539–551. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Atchley DP, Albarracin CT, Lopez A, Valero
V, Amos CI, Gonzalez-Angulo AM, Hortobagyi GN and Arun BK: Clinical
and pathologic characteristics of patients with BRCA-positive and
BRCA-negative breast cancer. J Clin Oncol. 26:4282–4288. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Porta FM, Sajjadi E, Venetis K,
Frascarelli C, Cursano G, Guerini-Rocco E, Fusco N and Ivanova M:
Immune biomarkers in triple-negative breast cancer: Improving the
predictivity of current testing methods. J Pers Med. 13:11762023.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ricciardi GR, Adamo B, Ieni A, Licata L,
Cardia R, Ferraro G, Franchina T, Tuccari G and Adamo V: Androgen
receptor (AR), E-cadherin, and Ki-67 as emerging targets and novel
prognostic markers in triple-negative breast cancer (TNBC)
patients. PLoS One. 10:e01283682015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gerdes J, Li L, Schlueter C, Duchrow M,
Wohlenberg C, Gerlach C, Stahmer I, Kloth S, Brandt E and Flad HD:
Immunobiochemical and molecular biologic characterization of the
cell proliferation-associated nuclear antigen that is defined by
monoclonal antibody Ki-67. Am J Pathol. 138:867–873.
1991.PubMed/NCBI
|
|
14
|
Schlüter C, Duchrow M, Wohlenberg C,
Becker MH, Key G, Flad HD and Gerdes J: The cell
proliferation-associated antigen of antibody Ki-67: A very large,
ubiquitous nuclear protein with numerous repeated elements,
representing a new kind of cell cycle-maintaining proteins. J Cell
Biol. 123:513–522. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Selz J, Stevens D, Jouanneau L, Labib A
and Le Scodan R: Prognostic value of molecular subtypes, ki67
expression and impact of postmastectomy radiation therapy in breast
cancer patients with negative lymph nodes after mastectomy. Int J
Radiat Oncol Bio Phys. 84:1123–1132. 2012. View Article : Google Scholar
|
|
16
|
Sobecki M, Mrouj K, Camasses A, Parisis N,
Nicolas E, Llères D, Gerbe F, Prieto S, Krasinska L, David A, et
al: The cell proliferation antigen Ki-67 organises heterochromatin.
Elife. 5:e137222016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ishida S, Huang E, Zuzan H, Spang R, Leone
G, West M and Nevins JR: Role for E2F in control of both DNA
replication and mitotic functions as revealed from DNA microarray
analysis. Mol Cell Biol. 21:4684–4699. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sobecki M, Mrouj K, Colinge J, Gerbe F,
Jay P, Krasinska L, Dulic V and Fisher D: Cell-cycle regulation
accounts for variability in Ki-67 expression levels. Cancer Res.
77:2722–2734. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Keam B, Im SA, Lee KH, Han SW, Oh DY, Kim
JH, Lee SH, Han W, Kim DW, Kim TY, et al: Ki-67 can be used for
further classification of triple negative breast cancer into two
subtypes with different response and prognosis. Breast Cancer Res.
13:R222011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li XQ, Pei DS, Qian GW, Yin XX, Cheng Q,
Li LT and Zheng JN: The effect of methylated oligonucleotide
targeting Ki-67 gene in human 786-0 renal carcinoma cells. Tumour
Biol. 32:863–873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scholl SM, Pierga JY, Asselain B, Beuzeboc
P, Dorval T, Garcia-Giralt E, Jouve M, Palangié T, Remvikos Y,
Durand JC, et al: Breast tumour response to primary chemotherapy
predicts local and distant control as well as survival. Eur J
Cancer. 31A:1969–1975. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Benini E, Rao S, Daidone MG, Pilotti S and
Silvestrini R: Immunoreactivity to MIB-1 in breast cancer:
Methodological assessment and comparison with other proliferation
indices. Cell Prolif. 30:107–115. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dowsett M, Nielsen TO, A'Hern R, Bartlett
J, Coombes RC, Cuzick J, Ellis M, Henry NL, Hugh JC, Lively T, et
al: Assessment of Ki67 in breast cancer: Recommendations from the
International Ki67 in Breast Cancer working group. J Natl Cancer
Inst. 103:1656–1664. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Urruticoechea A, Smith IE and Dowsett M:
Proliferation marker Ki-67 in early breast cancer. J Clin Oncol.
23:7212–7220. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cattoretti G, Becker MH, Key G, Duchrow M,
Schlüter C, Galle J and Gerdes J: Monoclonal antibodies against
recombinant parts of the Ki-67 antigen (MIB 1 and MIB 3) detect
proliferating cells in microwave-processed formalin-fixed paraffin
sections. J Pathol. 168:357–363. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Muftah AA, Aleskandarany MA, Al-Kaabi MM,
Sonbul SN, Diez-Rodriguez M, Nolan CC, Caldas C, Ellis IO, Rakha EA
and Green AR: Ki67 expression in invasive breast cancer: The use of
tissue microarrays compared with whole tissue sections. Breast
Cancer Res Treat. 164:341–348. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Viale G, Hanlon Newell AE, Walker E,
Harlow G, Bai I, Russo L, Dell'Orto P and Maisonneuve P: Ki-67
(30-9) scoring and differentiation of luminal A- and luminal B-like
breast cancer subtypes. Breast Cancer Res Treat. 178:451–458. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Q, Ma G, Deng Y, Luo W, Zhao Y, Li W
and Zhou Q: Prognostic value of Ki-67 in patients with resected
triple-negative breast cancer: A meta-analysis. Front Oncol.
9:10682019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goldhirsch A, Winer EP, Coates AS, Gelber
RD, Piccart-Gebhart M, Thürlimann B and Senn HJ; Panel members:
Personalizing the treatment of women with early breast cancer:
Highlights of the St Gallen international expert consensus on the
primary therapy of early breast cancer 2013. Ann Oncol.
24:2206–2223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Penault-Llorca F and Radosevic-Robin N:
Ki67 assessment in breast cancer: An update. Pathology. 49:166–171.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Boussiotis VA: Molecular and biochemical
aspects of the PD-1 checkpoint pathway. N Engl J Med.
375:1767–1778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kythreotou A, Siddique A, Mauri FA, Bower
M and Pinato DJ: PD-L1. J Clin Pathol. 71:189–194. 2018. View Article : Google Scholar
|
|
33
|
Thomas R, Al-Khadairi G and Decock J:
Immune checkpoint inhibitors in triple negative breast cancer
treatment: Promising future prospects. Front Oncol. 10:6005732021.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mittendorf EA, Philips AV, Meric-Bernstam
F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM,
Akcakanat A, et al: PD-L1 expression in triple-negative breast
cancer. Cancer Immunol Res. 2:361–370. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oner G, Önder S, Karatay H, Ak N, Tükenmez
M, Müslümanoğlu M, İğci A, Dincçağ A, Özmen V, Aydiner A, et al:
Correction: Clinical impact of PD-L1 expression in triplenegative
breast cancer patients with residual tumor burden after neoadjuvant
chemotherapy. World J Surg Oncol. 21:542023. View Article : Google Scholar
|
|
36
|
Schmid P, Adams S, Rugo HS, Schneeweiss A,
Barrios CH, Iwata H, Diéras V, Hegg R, Im SA, Shaw Wright G, et al:
Atezolizumab and nab-paclitaxel in advanced triple-negative breast
cancer. N Engl J Med. 379:2108–2121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dong H, Strome SE, Salomao DR, Tamura H,
Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al:
Tumor-associated B7-H1 promotes T-cell apoptosis: A potential
mechanism of immune evasion. Nat Med. 8:793–800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parsa AT, Waldron JS, Panner A, Crane CA,
Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et
al: Loss of tumor suppressor PTEN function increases B7-H1
expression and immunoresistance in glioma. Nat Med. 13:84–88. 2007.
View Article : Google Scholar
|
|
39
|
Gonzalez-Angulo AM, Ferrer-Lozano J,
Stemke-Hale K, Sahin A, Liu S, Barrera JA, Burgues O, Lluch AM,
Chen H, Hortobagyi GN, et al: PI3K pathway mutations and PTEN
levels in primary and metastatic breast cancer. Mol Cancer Ther.
10:1093–1101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Loi SM: Tumor-infiltrating lymphocytes,
breast cancer subtypes and therapeutic efficacy. OncoImmunology.
2:e247202013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dieci MV, Tsvetkova V, Orvieto E,
Piacentini F, Ficarra G, Griguolo G, Miglietta F, Giarratano T,
Omarini C, Bonaguro S, et al: Immune characterization of breast
cancer metastases: Prognostic implications. Breast Cancer Res.
20:622018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yeong J, Lim JCT, Lee B, Li H, Ong CCH,
Thike AA, Yeap WH, Yang Y, Lim AYH, Tay TKY, et al: Prognostic
value of CD8 + PD-1+ immune infiltrates and PDCD1 gene expression
in triple negative breast cancer. J Immunother Cancer. 7:342019.
View Article : Google Scholar
|
|
44
|
Lotfinejad P, Asghari Jafarabadi M, Abdoli
Shadbad M, Kazemi T, Pashazadeh F, Sandoghchian Shotorbani S,
Jadidi Niaragh F, Baghbanzadeh A, Vahed N, Silvestris N and
Baradaran B: Prognostic role and clinical significance of
tumor-infiltrating lymphocyte (TIL) and programmed death ligand 1
(PD-L1) expression in triple-negative breast cancer (TNBC): A
systematic review and meta-analysis study. Diagnostics (Basel).
10:7042020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mittendorf EA, Zhang H, Barrios CH, Saji
S, Jung KH, Hegg R, Koehler A, Sohn J, Iwata H, Telli ML, et al:
Neoadjuvant atezolizumab in combination with sequential
nab-paclitaxel and anthracycline-based chemotherapy versus placebo
and chemotherapy in patients with early-stage triple-negative
breast cancer (IMpassion031): A randomised, double-blind, phase 3
trial. Lancet. 396:1090–1100. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Baretta Z, Mocellin S, Goldin E, Olopade
OI and Huo D: Effect of BRCA germline mutations on breast cancer
prognosis: A systematic review and meta-analysis. Medicine
(Baltimore). 95:e49752016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hughes DJ, Ginolhac SM, Coupier I Corbex
M, Bressac-de-Paillerets B, Chompret A, Bignon YJ, Uhrhammer N,
Lasset C, Giraud S, et al: Common BRCA2 variants and modification
of breast and ovarian cancer risk in BRCA1 mutation carriers.
Cancer Epidemiol Biomarkers Prev. 14:265–267. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu K, Yang S and Zhao Y: Prognostic
significance of BRCA mutations in ovarian cancer: An updated
systematic review with meta-analysis. Oncotarget. 8:285–302. 2017.
View Article : Google Scholar :
|
|
49
|
Nanda R, Schumm LP, Cummings S, Fackenthal
JD, Sveen L, Ademuyiwa F, Cobleigh M, Esserman L, Lindor NM,
Neuhausen SL and Olopade OI: Genetic testing in an ethnically
diverse cohort of high-risk women: A comparative analysis of BRCA1
and BRCA2 mutations in American families of European and African
ancestry. JAMA. 294:1925–1933. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wong-Brown MW, Meldrum CJ, Carpenter JE,
Clarke CL, Narod SA, Jakubowska A, Rudnicka H, Lubinski J and Scott
RJ: Prevalence of BRCA1 and BRCA2 germline mutations in patients
with triple-negative breast cancer. Breast Cancer Res Treat.
150:71–80. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bianchini G, Balko JM, Mayer IA, Sanders
ME and Gianni L: Triple-negative breast cancer: Challenges and
opportunities of a heterogeneous disease. Nat Rev Clin Oncol.
13:674–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lee EH, Park SK, Park B, Kim SW, Lee MH,
Ahn SH, Son BH, Yoo KY and Kang D; KOHBRA Research Group; Korean
Breast Cancer Society: Effect of BRCA1/2 mutation on short-term and
long-term breast cancer survival: A systematic review and
meta-analysis. Breast Cancer Res Treat. 122:11–25. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stoppa-Lyonnet D: The biological effects
and clinical implications of BRCA mutations: Where do we go from
here? Eur J Hum Genet. 24(Suppl 1): S3–S9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wu L, Wang F, Xu R, Zhang S, Peng X, Feng
Y, Wang J and Lu C: Promoter methylation of BRCA1 in the prognosis
of breast cancer: A meta-analysis. Breast Cancer Res Treat.
142:619–627. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang C, Zhang J, Wang Y, Ouyang T, Li J,
Wang T, Fan Z, Fan T, Lin B and Xie Y: Prevalence of BRCA1
mutations and responses to neoadjuvant chemotherapy among BRCA1
carriers and non-carriers with triple-negative breast cancer. Ann
Oncol. 26:523–528. 2015. View Article : Google Scholar
|
|
56
|
Paluch-Shimon S, Friedman E, Berger R,
Papa M, Dadiani M, Friedman N, Shabtai M, Zippel D, Gutman M, Golan
T, et al: Neo-adjuvant doxorubicin and cyclophosphamide followed by
paclitaxel in triple-negative breast cancer among BRCA1 mutation
carriers and non-carriers. Breast Cancer Res Treat. 157:157–165.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fu X, Tan W, Song Q, Pei H and Li J: BRCA1
and breast cancer: Molecular mechanisms and therapeutic strategies.
Front Cell Dev Biol. 10:8134572022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Turk AA and Wisinski KB: PARP inhibitors
in breast cancer: Bringing synthetic lethality to the bedside.
Cancer. 124:2498–2506. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Meyer P, Landgraf K, Högel B, Eiermann W
and Ataseven B: BRCA2 mutations and triple-negative breast cancer.
PLoS One. 7:e383612012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mendonsa AM, Na TY and Gumbiner BM:
E-cadherin in contact inhibition and cancer. Oncogene.
37:4769–4780. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shen T, Zhang K, Siegal GP and Wei S:
Prognostic value of E-cadherin and β-catenin in triple-negative
breast cancer. Am J Clin Pathol. 146:603–610. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu JB, Feng CY, Deng M, Ge DF, Liu DC, Mi
JQ and Feng XS: E-cadherin expression phenotypes associated with
molecular subtypes in invasive non-lobular breast cancer: Evidence
from a retrospective study and meta-analysis. World J Surg Oncol.
15:1392017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fang Y, Wang Y, Ma H, Guo Y, Xu R, Chen X,
Chen X, Lv Y, Li P and Gao Y: TFAP2A downregulation mediates
tumor-suppressive effect of miR-8072 in triple-negative breast
cancer via inhibiting SNAI1 transcription. Breast Cancer Res.
26:1032024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tang D, Xu S, Zhang Q and Zhao W: The
expression and clinical significance of the androgen receptor and
E-cadherin in triple-negative breast cancer. Med Oncol. 29:526–533.
2012. View Article : Google Scholar
|
|
65
|
Merikhian P, Eisavand MR and Farahmand L:
Triple-negative breast cancer: Understanding Wnt signaling in drug
resistance. Cancer Cell Int. 21:4192021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Loh CY, Chai JY, Tang TF, Wong WF, Sethi
G, Shanmugam MK, Chong PP and Looi CY: The E-cadherin and
N-cadherin switch in epithelial-to-mesenchymal transition:
Signaling, therapeutic implications, and challenges. Cells.
8:11182019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
De Leeuw WJ, Berx G, Vos CB, Peterse JL,
Van de Vijver MJ, Litvinov S, Van Roy F, Cornelisse CJ and
Cleton-Jansen AM: Simultaneous loss of E-cadherin and catenins in
invasive lobular breast cancer and lobular carcinoma in situ. J
Pathol. 183:404–411. 1997. View Article : Google Scholar
|
|
68
|
Corso G, Figueiredo J, De Angelis SP,
Corso F, Girardi A, Pereira J, Seruca R, Bonanni B, Carneiro P,
Pravettoni G, et al: E-cadherin deregulation in breast cancer. J
Cell Mol Med. 24:5930–5936. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Droufakou S, Deshmane V, Roylance R, Hanby
A, Tomlinson I and Hart IR: Multiple ways of silencing E-cadherin
gene expression in lobular carcinoma of the breast. Int J Cancer.
92:404–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Brouxhon SM, Kyrkanides S, Teng X,
O'Banion MK, Clarke R, Byers S and Ma L: Soluble-E-cadherin
activates HER and IAP family members in HER2+ and TNBC human breast
cancers. Mol Carcinog. 53:893–906. 2014. View Article : Google Scholar
|
|
71
|
Kuhn PM, Russo GC, Crawford AJ,
Venkatraman A, Yang N, Starich BA, Schneiderman Z, Wu PH, Vo T,
Wirtz D and Kokkoli E: Local, sustained, and targeted co-delivery
of MEK inhibitor and doxorubicin inhibits tumor progression in
E-cadherin-positive breast cancer. Pharmaceutics. 16:9812024.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
De Schepper M, Vincent-Salomon A,
Christgen M, Van Baelen K, Richard F, Tsuda H, Kurozumi S, Brito
MJ, Cserni G, Schnitt S, et al: Results of a worldwide survey on
the currently used histopathological diagnostic criteria for
invasive lobular breast cancer. Mod Pathol. 35:1812–1820. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pai K, Baliga P and Shrestha BL:
E-cadherin expression: A diagnostic utility for differentiating
breast carcinomas with ductal and lobular morphologies. J Clin
Diagn Res. 7:840–844. 2013.PubMed/NCBI
|
|
74
|
Curtis C, Shah SP, Chin SF, Turashvili G,
Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et
al: The genomic and transcriptomic architecture of 2,000 breast
tumours reveals novel subgroups. Nature. 486:346–352. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Padmanaban V, Krol I, Suhail Y, Szczerba
BM, Aceto N, Bader JS and Ewald AJ: E-cadherin is required for
metastasis in multiple models of breast cancer. Nature.
573:439–444. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Russo GC, Crawford AJ, Clark D, Cui J,
Carney R, Karl MN, Su B, Starich B, Lih TS, Kamat P, et al:
E-cadherin interacts with EGFR resulting in hyper-activation of ERK
in multiple models of breast cancer. Oncogene. 43:1445–1462. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Song H, Wu T, Xie D, Li D, Hua K, Hu J and
Fang L: WBP2 downregulation inhibits proliferation by blocking YAP
transcription and the EGFR/PI3K/Akt signaling pathway in triple
negative breast cancer. Cell Physiol Biochem. 48:1968–1982. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kim S, You D, Jeong Y, Yu J, Kim SW, Nam
SJ and Lee JE: Berberine down-regulates IL-8 expression through
inhibition of the EGFR/MEK/ERK pathway in triple-negative breast
cancer cells. Phytomedicine. 50:43–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al: Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): An open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010. View Article : Google Scholar
|
|
82
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Arrieta O, Cardona AF, Federico Bramuglia
G, Gallo A, Campos-Parra AD, Serrano S, Castro M, Avilés A, Amorin
E, Kirchuk R, et al: Genotyping non-small cell lung cancer (NSCLC)
in Latin America. J Thorac Oncol. 6:1955–1959. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tilch E, Seidens T, Cocciardi S, Reid LE,
Byrne D, Simpson PT, Vargas AC, Cummings C, Fox SB, Lakhani SR and
Chenevix Trench G: Mutations in EGFR, BRAF and RAS are rare in
triple-negative and basal-like breast cancers from Caucasian women.
Breast Cancer Res Treat. 143:385–392. 2014. View Article : Google Scholar
|
|
85
|
Jacot W, Lopez-Crapez E, Thezenas S, Senal
R, Fina F, Bibeau F, Romieu G and Lamy PJ: Lack of EGFR-activating
mutations in European patients with triple-negative breast cancer
could emphasise geographic and ethnic variations in breast cancer
mutation profiles. Breast Cancer Res. 13:R1332011. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rosell R, Moran T, Queralt C, Porta R,
Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M,
et al: Screening for epidermal growth factor receptor mutations in
lung cancer. N Engl J Med. 361:958–967. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Shigematsu H, Lin L, Takahashi T, Nomura
M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et
al: Clinical and biological features associated with epidermal
growth factor receptor gene mutations in lung cancers. J Natl
Cancer Inst. 97:339–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Pallis AG, Voutsina A, Kalikaki A,
Souglakos J, Briasoulis E, Murray S, Koutsopoulos A, Tripaki M,
Stathopoulos E, Mavroudis D and Georgoulias V: 'Classical' but not
'other' mutations of EGFR kinase domain are associated with
clinical outcome in gefitinib-treated patients with non-small cell
lung cancer. Br J Cancer. 97:1560–1566. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yamane H, Ochi N, Yasugi M, Tabayashi T,
Yamagishi T, Monobe Y, Hisamoto A, Kiura K and Takigawa N:
Docetaxel for non-small-cell lung cancer harboring the activated
EGFR mutation with T790M at initial presentation. Onco Targets
Ther. 6:155–160. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lee HJ, Kim YT, Kang CH, Zhao B, Tan Y,
Schwartz LH, Persigehl T, Jeon YK and Chung DH: Epidermal growth
factor receptor mutation in lung adenocarcinomas: Relationship with
CT characteristics and histologic subtypes. Radiology. 268:254–264.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gupta GK, Collier AL, Lee D, Hoefer RA,
Zheleva V, Siewertsz van Reesema LL, Tang-Tan AM, Guye ML, Chang
DZ, Winston JS, et al: Perspectives on triple-negativebreast
cancer: Current treatment strategies, unmet needs, and potential
targets for future therapies. Cancers (Basel). 12:23922020.
View Article : Google Scholar
|
|
92
|
Gumuskaya B, Alper M, Hucumenoglu S,
Altundag K, Uner A and Guler G: EGFR expression and gene copy
number in triple-negative breast carcinoma. Cancer Genet Cytogenet.
203:222–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Nakai K, Hung MC and Yamaguchi H: A
perspective on anti-EGFR therapies targeting triple-negative breast
cancer. Am J Cancer Res. 6:1609–1623. 2016.PubMed/NCBI
|
|
94
|
Choi J, Jung WH and Koo JS:
Clinicopathologic features of molecular subtypes of triple negative
breast cancer based on immunohistochemical markers. Histol
Histopathol. 27:1481–1493. 2012.PubMed/NCBI
|
|
95
|
Tan DSP, Marchió C, Jones RL, Savage K,
Smith IE, Dowsett M and Reis-Filho JS: Triple negative breast
cancer: Molecular profiling and prognostic impact in adjuvant
anthracycline-treated patients. Breast Cancer Res Treat. 111:27–44.
2008. View Article : Google Scholar
|
|
96
|
Martin V, Botta F, Zanellato E, Molinari
F, Crippa S, Mazzucchelli L and Frattini M: Molecular
characterization of EGFR and EGFR-downstream pathways in triple
negative breast carcinomas with basal like features. Histol
Histopathol. 27:785–792. 2012.PubMed/NCBI
|
|
97
|
Meseure D, Vacher S, Drak Alsibai K,
Trassard M, Susini A, Le Ray C, Lerebours F, Le Scodan R, Spyratos
F, Marc Guinebretiere J, et al: Profiling of EGFR mRNA and protein
expression in 471 breast cancers compared with 10 normal tissues: A
candidate biomarker to predict EGFR inhibitor effectiveness. Int J
Cancer. 131:1009–1010. 2012. View Article : Google Scholar
|
|
98
|
Medić-Milijić N, Jovanić I, Nedeljković M,
Marković I, Spurnić I, Milovanović Z, Ademović N, Tomić T and Tanić
N and Tanić N: Prognostic and clinical significance of PD-L1, EGFR
and androgen receptor (AR) expression in triple-negative breast
cancer (TNBC) patients. Life (Basel). 14:6822024.
|
|
99
|
Ueno NT and Zhang D: Targeting EGFR in
Triple negative breast cancer. J Cancer. 2:324–328. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Miricescu D, Totan A, Stanescu-Spinu II,
Badoiu SC, Stefani C and Greabu M: PI3K/AKT/mTOR signaling pathway
in breast cancer: From molecular landscape to clinical aspects. Int
J Mol Sci. 22:1732020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Pascual J and Turner NC: Targeting the
PI3-kinase pathway in triple-negative breast cancer. Ann Oncol.
30:1051–1060. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Fruman DA, Chiu H, Hopkins BD, Bagrodia S,
Cantley LC and Abraham RT: The PI3K pathway in human disease. Cell.
170:605–635. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yoshida T and Delafontaine P: Mechanisms
of IGF-1-mediated regulation of skeletal muscle hypertrophy and
atrophy. Cells. 9:19702020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Shi X, Wang J, Lei Y, Cong C, Tan D and
Zhou X: Research progress on the PI3K/AKT signaling pathway in
gynecological cancer (Review). Mol. Med. Rep. 19:4529–4535.
2019.PubMed/NCBI
|
|
105
|
Revathidevi S and Munirajan AK: Akt in
cancer: Mediator and more. Semin Cancer Biol. 59:80–91. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Carpten JD, Faber AL, Horn C, Donoho GP,
Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage
S, et al: A transforming mutation in the pleckstrin homology domain
of AKT1 in cancer. Nature. 448:439–444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Chin YR, Yoshida T, Marusyk A, Beck AH,
Polyak K and Toker A: Targeting Akt3 signaling in triple-negative
breast cancer. Can Res. 74:964–973. 2014. View Article : Google Scholar
|
|
108
|
Li H, Prever L, Hirsch E and Gulluni F:
Targeting PI3K/AKT/mTOR signaling pathway in breast cancer. Cancers
(Basel). 13:35172021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Costa RLB, Han HS and Gradishar WJ:
Targeting the PI3K/AKT/mTOR pathway in triple-negative breast
cancer: A review. Breast Cancer Res Treat. 169:397–406. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Carey LA: Finding the positive in
triple-negative breast cancer. Nat Cancer. 2:476–478. 2021.
View Article : Google Scholar
|
|
111
|
Xia P and Xu XY: PI3K/Akt/mTOR signaling
pathway in cancer stem cells: From basic research to clinical
application. Am J Cancer Res. 5:1602–1609. 2015.PubMed/NCBI
|
|
112
|
Karami Fath M, Ebrahimi M, Nourbakhsh E,
Zia Hazara A, Mirzaei A, Shafieyari S, Salehi A, Hoseinzadeh M,
Payandeh Z and Barati G: PI3K/Akt/mTOR signaling pathway in cancer
stem cells. Pathol Res Pract. 237:1540102022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Reinhardt HC and Schumacher B: The p53
network: Cellular and systemic DNA damage responses in aging and
cancer. Trends Genet. 28:128–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Sionov RV and Haupt Y: The cellular
response to p53: The decision between life and death. Oncogene.
18:6145–6157. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Kubbutat MH, Jones SN and Vousden KH:
Regulation of p53 stability by MDM2. Nature. 387:299–303. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Lacroix M, Toillon RA and Leclercq G: p53
and breast cancer, an update. Endocr Relat Cancer. 13:293–325.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Ozaki T and Nakagawara A: Role of p53 in
cell death and human cancers. Cancers (Basel). 3:994–1013. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Babikir HA, Afjei R, Paulmurugan R and
Massoud TF: Restoring guardianship of the genome: Anticancer drug
strategies to reverse oncogenic mutant p53 misfolding. Cancer Treat
Rev. 71:19–31. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Costa DCF, de Oliveira GAP, Cino EA,
Soares IN, Rangel LP and Silva JL: Aggregation and prion-like
properties of misfolded tumor suppressors: Is cancer a prion
disease? Cold Spring Harbor Perspect Biol. 8:a0236142016.
View Article : Google Scholar
|
|
120
|
Silva JL, De Moura Gallo CV, Costa DCF and
Rangel LP: Prion-like aggregation of mutant p53 in cancer. Trends
Biochem Sci. 39:260–267. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Eriksson SE, Ceder S, Bykov VJN and Wiman
KG: p53 as a hub in cellular redox regulation and therapeutic
target in cancer. J Mol Cell Biol. 11:330–341. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
D'Orazi G and Givol D: p53 reactivation:
The link to zinc. Cell Cycle. 11:2581–2582. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wang G and Fersht AR: First-order
rate-determining aggregation mechanism of p53 and its implications.
Proc Natl Acad Sci USA. 109:13590–13595. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ghosh S, Salot S, Sengupta S, Navalkar A,
Ghosh D, Jacob R, Das S, Kumar R, Jha NN, Sahay S, et al: p53
amyloid formation leading to its loss of function: Implications in
cancer pathogenesis. Cell Death Differ. 24:1784–1798. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Li JP, Zhang XM, Zhang Z, Zheng LH, Jindal
S and Liu YJ: Association of p53 expression with poor prognosis in
patients with triple-negative breast invasive ductal carcinoma.
Medicine (Baltimore). 98:e154492019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View Article : Google Scholar
|
|
127
|
Neilsen PM, Noll JE, Suetani RJ, Schulz
RB, Al-Ejeh F, Evdokiou A, Lane DP and Callen DF: Mutant p53 uses
p63 as a molecular chaperone to alter gene expression and induce a
pro-invasive secretome. Oncotarget. 2:1203–1217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Lim LY, Vidnovic N, Ellisen LW and Leong
CO: Mutant p53 mediates survival of breast cancer cells. Br J
Cancer. 101:1606–1612. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Adorno M, Cordenonsi M, Montagner M,
Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo
V, et al: A mutant-p53/Smad complex opposes p63 to empower
TGFbeta-induced metastasis. Cell. 137:87–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Muller PAJ, Trinidad AG, Timpson P, Morton
JP, Zanivan S, van den Berghe PVE, Nixon C, Karim SA, Caswell PT,
Noll JE, et al: Mutant p53 enhances MET trafficking and signalling
to drive cell scattering and invasion. Oncogene. 32:1252–1265.
2013. View Article : Google Scholar :
|
|
131
|
Huang G, Zhong X, Yao L, Ma Q, Liao H, Xu
L, Zou J, Sun R, Wang D and Guo X: MicroRNA-449a inhibits cell
proliferation and migration by regulating mutant p53 in MDA-MB-468
cells. Exp Ther Med. 22:10202021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Marvalim C, Datta A and Lee SC: Role of
p53 in breast cancer progression: An insight into p53 targeted
therapy. Theranostics. 13:1421–1442. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Shah SP, Roth A, Goya R, Oloumi A, Ha G,
Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al: The clonal
and mutational evolution spectrum of primary triple-negative breast
cancers. Nature. 486:395–399. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Bae SY, Nam SJ, Jung Y, Lee SB, Park BW,
Lim W, Jung SH, Yang HW and Jung SP: Differences in prognosis and
efficacy of chemotherapy by p53 expression in triple-negative
breast cancer. Breast Cancer Res Treat. 172:437–444. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Jasar D, Smichkoska S, Kubelka K,
Filipovski V and Petrushevska G: Expression of p53 protein product
in triple negative breast cancers and relation with clinical and
histopathological parameters. Pril (Makedon Akad Nauk Umet Odd Med
Nauki). 36:69–79. 2015.PubMed/NCBI
|
|
136
|
Karamitopoulou E, Perentes E, Tolnay M and
Probst A: Prognostic significance of MIB-1, p53, and bcl-2
immunoreactivity in meningiomas. Hum Pathol. 29:140–145. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Geyer FC, Rodrigues DN, Weigelt B and
Reis-Filho JS: Molecular classification of estrogen
receptor-positive/luminal breast cancers. Adv Anat Pathol.
19:39–53. 2012. View Article : Google Scholar
|
|
138
|
Zizi-Sermpetzoglou A, Moustou E,
Petrakopoulou N, Arkoumani E, Tepelenis N and Savvaidou V: Atypical
polypoid adenomyoma of the uterus. A case report and a review of
the literature. Eur J Gynaecol Oncol. 33:118–121. 2012.PubMed/NCBI
|
|
139
|
Ibrahim T, Farolfi A, Scarpi E, Mercatali
L, Medri L, Ricci M, Nanni O, Serra L and Amadori D: Hormonal
receptor, human epidermal growth factor receptor-2, and Ki67
discordance between primary breast cancer and paired metastases:
Clinical impact. Oncology. 84:150–157. 2013. View Article : Google Scholar
|
|
140
|
Niikura N, Masuda S, Kumaki N, Xiaoyan T,
Terada M, Terao M, Iwamoto T, Oshitanai R, Morioka T, Tuda B, et
al: Prognostic significance of the Ki67 scoring categories in
breast cancer subgroups. Clin Breast Cancer. 14:323–329.e3. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Polley MYC, Leung SCY, McShane LM, Gao D,
Hugh JC, Mastropasqua MG, Viale G, Zabaglo LA, Penault-Llorca F,
Bartlett JMS, et al: An international Ki67 reproducibility study. J
Natl Cancer Inst. 105:1897–1906. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Polley MYC, Leung SCY, Gao D, Mastropasqua
MG, Zabaglo LA, Bartlett JMS, McShane LM, Enos RA, Badve SS, Bane
AL, et al: An international study to increase concordance in Ki67
scoring. Mod Pathol. 28:778–786. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Baum M, Buzdar A, Cuzick J, Forbes J,
Houghton J, Howell A and Sahmoud T; ATAC (Arimidex Tamoxifen Alone
or in Combination) Trialists' Group: Anastrozole alone or in
combination with tamoxifen versus tamoxifen alone for adjuvant
treatment of postmenopausal women with early-stage breast cancer:
Results of the ATAC (Arimidex, Tamoxifen Alone or in Combination)
trial efficacy and safety update analyses. Cancer. 98:1802–1810.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wyatt CA, Geoghegan JC and Brinckerhoff
CE: Short hairpin RNA-mediated inhibition of matrix
metalloproteinase-1 in MDA-231 cells: Effects on matrix destruction
and tumor growth. Cancer Res. 65:11101–11108. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Zuckerman JE and Davis ME: Clinical
experiences with systemically administered siRNA-based therapeutics
in cancer. Nat Rev Drug Discov. 14:843–856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zheng JN, Ma TX, Cao JY, Sun XQ, Chen JC,
Li W, Wen RM, Sun YF and Pei DS: Knockdown of Ki-67 by small
interfering RNA leads to inhibition of proliferation and induction
of apoptosis in human renal carcinoma cells. Life Sci. 78:724–729.
2006. View Article : Google Scholar
|
|
147
|
Zheng JN, Sun YF, Pei DS, Liu JJ, Ma TX,
Han RF, Li W, Zheng DB, Chen JC and Sun XQ: Treatment with
vector-expressed small hairpin RNAs against Ki67 RNA-induced cell
growth inhibition and apoptosis in human renal carcinoma cells.
Acta Biochim Biophys Sin (Shanghai). 38:254–261. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Burnett JC and Rossi JJ: RNA-based
therapeutics: Current progress and future prospects. Chem Biol.
19:60–71. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
de Carvalho Vicentini FTM,
Borgheti-Cardoso LN, Depieri LV, de Macedo Mano D, Abelha TF and
Petrilli R: Delivery systems and local administration routes for
therapeutic siRNA. Pharm Res. 30:915–931. 2013. View Article : Google Scholar
|
|
150
|
Conde J, Edelman ER and Artzi N:
Target-responsive DNA/RNA nanomaterials for microRNA sensing and
inhibition: The jack-of-all-trades in cancer nanotheranostics? Adv
Drug Deliv Rev. 81:169–183. 2015. View Article : Google Scholar
|
|
151
|
Bischoff JR, Kirn DH, Williams A, Heise C,
Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A and
McCormick F: An adenovirus mutant that replicates selectively in
p53-eficient human tumor cells. Science. 274:373–376. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Yu DC, Chen Y, Dilley J, Li Y, Embry M,
Zhang H, Nguyen N, Amin P, Oh J and Henderson DR: Antitumor synergy
of CV787, a prostate cancer-specific adenovirus, and paclitaxel and
docetaxel. Cancer Res. 61:517–525. 2001.PubMed/NCBI
|
|
153
|
Rajecki M, Kanerva A, Stenman UH, Tenhunen
M, Kangasniemi L, Särkioja M, Ala-Opas MY, Alfthan H, Sankila A,
Rintala E, et al: Treatment of prostate cancer with Ad5/3Delta24hCG
allows non-invasive detection of the magnitude and persistence of
virus replication in vivo. Mol Cancer Ther. 6:742–751. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Chen RF, Li YY, Li LT, Cheng Q, Jiang G
and Zheng JN: Novel oncolytic adenovirus sensitizes renal cell
carcinoma cells to radiotherapy via mitochondrial apoptotic cell
death. Mol Med Rep. 11:2141–2146. 2015. View Article : Google Scholar
|
|
155
|
Toth K and Wold WSM: Increasing the
efficacy of oncolytic adenovirus vectors. Viruses. 2:1844–1866.
2010. View Article : Google Scholar
|
|
156
|
Liu J, Fang L, Cheng Q, Li L, Su C, Zhang
B, Pei D, Yang J, Li W and Zheng J: Effects of G250 promoter
controlled conditionally replicative adenovirus expressing
Ki67-siRNA on renal cancer cell. Cancer Sci. 103:1880–1888. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Gaudet D, Alexander VJ, Baker BF, Brisson
D, Tremblay K, Singleton W, Geary RS, Hughes SG, Viney NJ, Graham
MJ, et al: Antisense inhibition of apolipoprotein C-III in patients
with hypertriglyceridemia. N Engl J Med. 373:438–447. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Kennedy BWC: Mongersen, an oral SMAD7
antisense oligonucleotide, and Crohn's disease. N Engl J Med.
372:24612015. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Wheeler TM, Leger AJ, Pandey SK, MacLeod
AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF and Thornton CA:
Targeting nuclear RNA for in vivo correction of myotonic dystrophy.
Nature. 488:111–115. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Natale R, Blackhall F, Kowalski D, Ramlau
R, Bepler G, Grossi F, Lerchenmüller C, Pinder-Schenck M, Mezger J,
Danson S, et al: Evaluation of antitumor activity using change in
tumor size of the survivin antisense oligonucleotide LY2181308 in
combination with docetaxel for second-line treatment of patients
with non-small-cell lung cancer: a randomized open-label phase II
study. J Thorac Oncol. 9:1704–1708. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Sen M, Thomas SM, Kim S, Yeh JI, Ferris
RL, Johnson JT, Duvvuri U, Lee J, Sahu N, Joyce S, et al:
First-in-human trial of a STAT3 decoy oligonucleotide in head and
neck tumors: Implications for cancer therapy. Cancer Discov.
2:694–705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Kausch I, Lingnau A, Endl E, Sellmann K,
Deinert I, Ratliff TL, Jocham D, Sczakiel G, Gerdes J and Böhle A:
Antisense treatment against Ki-67 mRNA inhibits proliferation and
tumor growth in vitro and in vivo. Int J Cancer. 105:710–716. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Kausch I, Jiang H, Ewerdwalbesloh N, Doehn
C, Krüger S, Sczakiel G and Jocham D: Inhibition of Ki-67 in a
renal cell carcinoma severe combined immunodeficiency disease mouse
model is associated with induction of apoptosis and tumour growth
inhibition. BJU Int. 95:416–420. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Lingnau A, Steiner U, Kurzidim H, Jocham D
and Kausch I: Phase I dose-escalation study of intravesical
instillation of antisense oligonucleotide FFC15-01 against Ki-67 in
patients with non-muscle invasive bladder cancer. Debates Bladder
Cancer. 2:12010.
|
|
165
|
Ratilainen T, Holmén A, Tuite E, Nielsen
PE and Nordén B: Thermodynamics of sequence-specific binding of PNA
to DNA. Biochemistry. 39:7781–7791. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Thomas SM, Sahu B, Rapireddy S, Bahal R,
Wheeler SE, Procopio EM, Kim J, Joyce SC, Contrucci S, Wang Y, et
al: Antitumor effects of EGFR antisense guanidine-based peptide
nucleic acids in cancer models. ACS Chem Biol. 8:345–352. 2013.
View Article : Google Scholar :
|
|
167
|
Thompson ED, Taube JM, Asch-Kendrick RJ,
Ogurtsova A, Xu H, Sharma R, Meeker A, Argani P, Emens LA and
Cimino-Mathews A: PD-L1 expression and the immune microenvironment
in primary invasive lobular carcinomas of the breast. Mod Pathol.
30:1551–1560. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Sabatier R, Finetti P, Mamessier E,
Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D and
Bertucci F: Prognostic and predictive value of PDL1 expression in
breast cancer. Oncotarget. 6:5449–5464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Baracco EE, Pietrocola F, Buqué A, Bloy N,
Senovilla L, Zitvogel L, Vacchelli E and Kroemer G: Inhibition of
formyl peptide receptor 1 reduces the efficacy of anticancer
chemotherapy against carcinogen-induced breast cancer.
Oncoimmunology. 5:e11392752016. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Rey-Cárdenas M, Guerrero-Ramos F, Gómez de
Liaño Lista A, Carretero-González A, Bote H, Herrera-Juárez M,
Carril-Ajuria L, Martín-Soberón M, Sepulveda JM, Billalabeitia EG,
et al: Recent advances in neoadjuvant immunotherapy for urothelial
bladder cancer: What to expect in the near future. Cancer Treat
Rev. 93:1021422021. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Reck M, Remon J and Hellmann MD:
First-line immunotherapy for non-small-cell lung cancer. J Clin
Oncol. 40:586–597. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Huang AC and Zappasodi R: A decade of
checkpoint blockade immunotherapy in melanoma: Understanding the
molecular basis for immune sensitivity and resistance. Nat Immunol.
23:660–670. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Sendur MAN: Adjuvant immunotherapy for
renal cell carcinoma. Lancet Oncol. 23:1110–1111. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Romero D: Benefit in patients with
PD-L1-positive TNBC. Nat Rev Clin Oncol. 16:62019.
|
|
175
|
Loi S, Drubay D, Adams S, Pruneri G,
Francis PA, Lacroix-Triki M, Joensuu H, Dieci MV, Badve S, Demaria
S, et al: Tumor-infiltrating lymphocytes and prognosis: A pooled
individual patient analysis of early-stage triple-negative breast
cancers. J Clin Oncol. 37:559–569. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Yarchoan M, Johnson BR, Lutz ER, Laheru DA
and Jaffee EM: Targeting neoantigens to augment antitumour
immunity. Nat Rev Cancer. 17:209–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Dudley JC, Lin MT, Le DT and Eshleman JR:
Microsatellite instability as a biomarker for PD-1 blockade. Clin
Cancer Res. 22:813–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Bonneville R, Krook MA, Kautto EA, Miya J,
Wing MR, Chen HZ, Reeser JW, Yu L and Roychowdhury S: Landscape of
microsatellite instability across 39 cancer types. JCO Precis
Oncol. 2017:PO.17.000732017.PubMed/NCBI
|
|
179
|
Lipson EJ, Forde PM, Hammers HJ, Emens LA,
Taube JM and Topalian SL: Antagonists of PD-1 and PD-L1 in cancer
treatment. Semin Oncol. 42:587–600. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ,
Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Pusztai L, Karn T, Safonov A, Abu-Khalaf
MM and Bianchini G: New strategies in breast cancer: Immunotherapy.
Clin Cancer Res. 22:2105–2110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Loi S, Dushyanthen S, Beavis PA, Salgado
R, Denkert C, Savas P, Combs S, Rimm DL, Giltnane JM, Estrada MV,
et al: RAS/MAPK activation is associated with reduced
tumor-infiltrating lymphocytes in triple-negative breast cancer:
Therapeutic cooperation between MEK and PD-1/PD-L1 immune
checkpoint inhibitors. Clin Cancer Res. 22:1499–1509. 2016.
View Article : Google Scholar :
|
|
183
|
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng
PP, Chang BY and Levy R: Therapeutic antitumor immunity by
checkpoint blockade is enhanced by ibrutinib, an inhibitor of both
BTK and ITK. Proc Natl Acad Sci USA. 112:E966–E972. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Hodgson D, Lai Z, Dearden S, Barrett JC,
Harrington EA, Timms K, Lanchbury J, Wu W, Allen A, Senkus E, et
al: Analysis of mutation status and homologous recombination
deficiency in tumors of patients with germline BRCA1 or BRCA2
mutations and metastatic breast cancer: OlympiAD. Ann Oncol.
32:1582–1589. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
U.S. Food and Drug Administration (FDA):
TALZENNA® (talazoparib) prescribing information. FDA; Silver
Spring, MD: 2025, https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/217439s000lbl.pdf
(May 2025). Accessed on May 9 2025.
|
|
186
|
Litton JK, Scoggins ME, Hess KR, Adrada
BE, Murthy RK, Damodaran S, DeSnyder SM, Brewster AM, Barcenas CH,
Valero V, et al: Neoadjuvant talazoparib for patients with operable
breast cancer with a germline BRCA pathogenic variant. J Clin
Oncol. 38:388–394. 2020. View Article : Google Scholar :
|
|
187
|
Pop L, Suciu ID, Ionescu P and Ionescu OD:
The dual blockade in the neoadjuvant setting of HER-2 positive
early-stage breast cancer. J Med Life. 12:329–331. 2019. View Article : Google Scholar
|
|
188
|
Isakoff SJ, Overmoyer B, Tung NM, Gelman
RS, Giranda VL, Bernhard KM, Habin KR, Ellisen LW, Winer EP and
Goss PE: A phase II trial of the PARP inhibitor veliparib (ABT888)
and temozolomide for metastatic breast cancer. J Clin Oncol. 28(15
Suppl): S10192010. View Article : Google Scholar
|
|
189
|
Rodler ET, Gralow J, Kurland BF, Griffin
M, Yeh R, Thompson JA, Porter P, Swisher EM, Gadi VK, Korde LA, et
al: Phase I: Veliparib with cisplatin (CP) and vinorelbine (VNR) in
advanced triple-negative breast cancer (TNBC) and/or BRCA
mutation-associated breast cancer. J Clin Oncol. 32(15 Suppl):
S25692014. View Article : Google Scholar
|
|
190
|
Samol J, Ranson M, Scott E, Macpherson E,
Carmichael J, Thomas A and Cassidy J: Safety and tolerability of
the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib
(AZD2281) in combination with topotecan for the treatment of
patients with advanced solid tumors: A phase I study. Invest New
Drugs. 30:1493–1500. 2012. View Article : Google Scholar
|
|
191
|
Dent RA, Lindeman GJ, Clemons M, Wildiers
H, Chan A, McCarthy NJ, Singer CF, Lowe ES, Watkins CL and
Carmichael J: Phase I trial of the oral PARP inhibitor olaparib in
combination with paclitaxel for first- or second-line treatment of
patients with metastatic triple-negative breast cancer. Breast
Cancer Res. 15:R882013. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Marullo R, Werner E, Degtyareva N, Moore
B, Altavilla G, Ramalingam SS and Doetsch PW: Cisplatin induces a
mitochondrial-ROS response that contributes to cytotoxicity
depending on mitochondrial redox status and bioenergetic functions.
PLoS One. 8:e811622013. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Song X, Kong F, Zong ZF, Ren M, Meng Q, Li
Y and Sun Z: miR-124 and miR-142 enhance cisplatin sensitivity of
non-small cell lung cancer cells through repressing autophagy via
directly targeting SIRT1. RSC Adv. 9:5234–5243. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Pabla N and Dong Z: Curtailing side
effects in chemotherapy: A tale of PKCδ in cisplatin treatment.
Oncotarget. 3:107–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
McCabe N, Turner NC, Lord CJ, Kluzek K,
Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka
MZ, et al: Deficiency in the repair of DNA damage by homologous
recombination and sensitivity to poly(ADP-ribose) polymerase
inhibition. Cancer Res. 66:8109–8115. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Jia X, Wang K, Xu L, Li N, Zhao Z and Li
M: A systematic review and meta-analysis of BRCA1/2 mutation for
predicting the effect of platinum-based chemotherapy in
triple-negative breast cancer. Breast. 66:31–39. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Teo K, Gómez-Cuadrado L, Tenhagen M, Byron
A, Rätze M, van Amersfoort M, Renes J, Strengman E, Mandoli A,
Singh AA, et al: E-cadherin loss induces targetable autocrine
activation of growth factor signalling in lobular breast cancer.
Sci Rep. 8:154542018. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Bajrami I, Marlow R, van de Ven M, Brough
R, Pemberton HN, Frankum J, Song F, Rafiq R, Konde A, Krastev DB,
et al: E-cadherin/ROS1 inhibitor synthetic lethality in breast
cancer. Cancer Discov. 8:498–515. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Mateus AR, Simões-Correia J, Figueiredo J,
Heindl S, Alves CC, Suriano G, Luber B and Seruca R: E-cadherin
mutations and cell motility: A genotype-phenotype correlation. Exp
Cell Res. 315:1393–1402. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Watabe M, Nagafuchi A, Tsukita S and
Takeichi M: Induction of polarized cell-cell association and
retardation of growth by activation of the E-cadherin-catenin
adhesion system in a dispersed carcinoma line. J Cell Biol.
127:247–256. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Green SK, Francia G, Isidoro C and Kerbel
RS: Antiadhesive antibodies targeting E-cadherin sensitize
multicellular tumor spheroids to chemotherapy in vitro. Mol Cancer
Ther. 3:149–159. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Masuda H, Zhang D, Bartholomeusz C,
Doihara H, Hortobagyi GN and Ueno NT: Role of epidermal growth
factor receptor in breast cancer. Breast Cancer Res Treat.
136:331–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ,
Kussie P and Ferguson KM: Structural basis for inhibition of the
epidermal growth factor receptor by cetuximab. Cancer Cell.
7:301–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Qin S, Li J, Wang L, Xu J, Cheng Y, Bai Y,
Li W, Xu N, Lin LZ, Wu Q, et al: Efficacy and tolerability of
first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin
(FOLFOX-4) versus FOLFOX-4 in patients with RAS wild-type
metastatic colorectal cancer: The open-label, randomized, phase III
TAILOR trial. J Clin Oncol. 36:3031–3039. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Bonner JA, Harari PM, Giralt J, Azarnia N,
Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Hirsch FR, Redman MW, Moon J, Agustoni F,
Herbst RS, Semrad TJ, Varella-Garcia M, Rivard CJ, Kelly K, Gandara
DR and Mack PC: EGFR high copy number together with high EGFR
protein expression predicts improved outcome for cetuximab-based
therapy in squamous cell lung cancer: Analysis from SWOG S0819, a
phase III trial of chemotherapy with or without cetuximab in
advanced NSCLC. Clin Lung Cancer. 23:60–71. 2022. View Article : Google Scholar :
|
|
207
|
Cai WQ, Zeng LS, Wang LF, Wang YY, Cheng
JT, Zhang Y, Han ZW, Zhou Y, Huang SL, Wang XW, et al: The latest
battles between EGFR monoclonal antibodies and resistant tumor
cells. Front Oncol. 10:12492020. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Xu MJ, Johnson DE and Grandis JR:
EGFR-targeted therapies in the post-genomic era. Cancer Metastasis
Rev. 36:463–473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Mazorra Z, Lavastida A, Concha-Benavente
F, Valdés A, Srivastava RM, García-Bates TM, Hechavarría E,
González Z, González A, Lugiollo M, et al: Nimotuzumab induces NK
cell activation, cytotoxicity, dendritic cell maturation and
expansion of EGFR-specific T cells in head and neck cancer
patients. Front Pharmacol. 8:3822017. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Benmebarek MR, Karches CH, Cadilha BL,
Lesch S, Endres S and Kobold S: Killing mechanisms of chimeric
antigen receptor (CAR) T cells. Int J Mol Sci. 20:12832019.
View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Byrd TT, Fousek K, Pignata A, Szot C,
Samaha H, Seaman S, Dobrolecki L, Salsman VS, Oo HZ, Bielamowicz K,
et al: TEM8/ANTXR1-specific CAR T cells as a targeted therapy for
triple-negative breast cancer. Cancer Res. 78:489–500. 2018.
View Article : Google Scholar :
|
|
212
|
Xia L, Zheng Z, Liu JY, Chen YJ, Ding J,
Hu GS, Hu YH, Liu S, Luo WX, Xia NS and Liu W: Targeting
triple-negative breast cancer with combination therapy of EGFR CAR
T cells and CDK7 inhibition. Cancer Immunol Res. 9:707–722. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Hübner J, Raschke M, Rütschle I, Gräßle S,
Hasenberg T, Schirrmann K, Lorenz A, Schnurre S, Lauster R,
Maschmeyer I, et al: Simultaneous evaluation of anti-EGFR-induced
tumour and adverse skin effects in a microfluidic human 3D
co-culture model. Sci Rep. 8:150102018. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Jungbluth AA, Stockert E, Huang HJ,
Collins VP, Coplan K, Iversen K, Kolb D, Johns TJ, Scott AM,
Gullick WJ, et al: A monoclonal antibody recognizing human cancers
with amplification/overexpression of the human epidermal growth
factor receptor. Proc Natl Acad Sci USA. 100:639–644. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Scott AM, Lee FT, Tebbutt N, Herbertson R,
Gill SS, Liu Z, Skrinos E, Murone C, Saunder TH, Chappell B, et al:
A phase I clinical trial with monoclonal antibody ch806 targeting
transitional state and mutant epidermal growth factor receptors.
Proc Natl Acad Sci USA. 104:4071–4076. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Vitanza N, Gust J, Wilson A, Huang W,
Perez F, Wright J, Leary S, Cole B, Albert C, Pinto N, et al:
IMMU-03. Updates on brainchild-01, -02, and -03: Phase 1
locoregional car T cell trials targeting HER2, EGFR, and B7-H3 for
children with recurrent CNS tumors and DIPG. Neuro Oncol. 22(Suppl
3): iii3602020. View Article : Google Scholar :
|
|
217
|
Huerta JJ, Diaz-Trelles R, Naves FJ,
Llamosas MM, Del Valle ME and Vega JA: Epidermal growth factor
receptor in adult human dorsal root ganglia. Anat Embryol (Berl).
194:253–257. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Atwell B, Chen CY, Christofferson M,
Montfort WR and Schroeder J: Sorting nexin-dependent therapeutic
targeting of oncogenic epidermal growth factor receptor. Cancer
Gene Ther. 30:267–276. 2023. View Article : Google Scholar :
|
|
219
|
André F, Ciruelos E, Rubovszky G, Campone
M, Loibl S, Rugo HS, Iwata H, Conte P, Mayer IA, Kaufman B, et al:
Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced
breast cancer. N Engl J Med. 380:1929–1940. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Cataldo ML, De Placido P, Esposito D,
Formisano L, Arpino G, Giuliano M, Bianco R, De Angelis C and
Veneziani BM: The effect of the alpha-specific PI3K inhibitor
alpelisib combined with anti-HER2 therapy in HER2+/PIK3CA mutant
breast cancer. Front Oncol. 13:11082422023. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Juric D, Krop I, Ramanathan RK, Wilson TR,
Ware JA, Sanabria Bohorquez SM, Savage HM, Sampath D, Salphati L,
Lin RS, et al: Phase I dose-escalation study of taselisib, an oral
PI3K inhibitor, in patients with advanced solid tumors. Cancer
Discov. 7:704–715. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Schmid P, Zaiss M, Harper-Wynne C,
Ferreira M, Dubey S, Chan S, Makris A, Nemsadze G, Brunt AM,
Kuemmel S, et al: Abstract GS2-07: MANTA-a randomized phase II
study of fulvestrant in combination with the dual mTOR inhibitor
AZD2014 or everolimus or fulvestrant alone in estrogen
receptor-positive advanced or metastatic breast cancer. Cancer Res.
78(4 Suppl): GS2–07. 2018. View Article : Google Scholar
|
|
223
|
Dent S, Cortés J, Im YH, Diéras V, Harbeck
N, Krop IE, Wilson TR, Cui N, Schimmoller F, Hsu JY, et al: Phase
III randomized study of taselisib or placebo with Fulvestrant in
estrogen receptor-positive, PIK3CA-mutant, HER2-negative, advanced
breast cancer: The SANDPIPER trial. Ann Oncol. 32:197–207. 2021.
View Article : Google Scholar
|
|
224
|
Baselga J, Im SA, Iwata H, Cortés J, De
Laurentiis M, Jiang Z, Arteaga CL, Jonat W, Clemons M, Ito Y, et
al: Buparlisib plus fulvestrant versus placebo plus fulvestrant in
postmenopausal, hormone receptor-positive, HER2-negative, advanced
breast cancer (BELLE-2): A randomised, double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 18:904–916. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Mallick S, Duttaroy AK and Dutta S: The
PIK3CA gene and its pivotal role in tumor tropism of
triple-negative breast cancer. Transl Oncol. 50:1021402024.
View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Xu S, Li S, Guo Z, Luo J, Ellis MJ and Ma
CX: Combined targeting of mTOR and AKT is an effective strategy for
basal-like breast cancer in patient-derived xenograft models. Mol
Cancer Ther. 12:1665–1675. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Mishra R, Patel H, Alanazi S, Kilroy MK
and Garrett JT: PI3K inhibitors in cancer: Clinical implications
and adverse effects. Int J Mol Sci. 22:34642021. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Bachelot T, Bourgier C, Cropet C,
Ray-Coquard I, Ferrero JM, Freyer G, Abadie-Lacourtoisie S, Eymard
JC, Debled M, Spaëth D, et al: Randomized phase II trial of
everolimus in combination with tamoxifen in patients with hormone
receptor-positive, human epidermal growth factor receptor
2-negative metastatic breast cancer with prior exposure to
aromatase inhibitors: A GINECO study. J Clin Oncol. 30:2718–2724.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Kornblum N, Zhao F, Manola J, Klein P,
Ramaswamy B, Brufsky A, Stella PJ, Burnette B, Telli M, Makower DF,
et al: Randomized phase II trial of fulvestrant plus everolimus or
placebo in postmenopausal women with hormone receptor-positive,
human epidermal growth factor receptor 2-negative metastatic breast
cancer resistant to aromatase inhibitor therapy: Results of
PrE0102. J Clin Oncol. 36:1556–1563. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Carlino F, Diana A, Terminiello M,
Ventriglia A, Piccolo A, Bruno V, Lobianco L, Caterino M,
Ciardiello F, Danielee B, et al: 302P Clinical implication of
tissue re-biopsy in metastatic breast cancer (MBC) patients: A
single centre retrospective analysis. Ann Oncol. 32:S495–S496.
2021. View Article : Google Scholar
|
|
231
|
Basho RK, Gilcrease M, Murthy RK, Helgason
T, Karp DD, Meric-Bernstam F, Hess KR, Herbrich SM, Valero V,
Albarracin C, et al: Targeting the PI3K/AKT/mTOR pathway for the
treatment of mesenchymal triple-negative breast cancer: Evidence
from a phase 1 trial of mTOR inhibition in combination with
liposomal doxorubicin and bevacizumab. JAMA Oncol. 3:509–515. 2017.
View Article : Google Scholar
|
|
232
|
Kastenhuber ER and Lowe SW: Putting p53 in
context. Cell. 170:1062–1078. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Joerger AC and Fersht AR: Structural
biology of the tumor suppressor p53 and cancer-associated mutants.
Adv Cancer Res. 97:1–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Loh SN: Follow the mutations: Toward
class-specific, small-molecule reactivation of p53. Biomolecules.
10:3032020. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Puca R, Nardinocchi L, Porru M, Simon AJ,
Rechavi G, Leonetti C, Givol D and D'Orazi G: Restoring p53 active
conformation by zinc increases the response of Mutant p53 tumor
cells to anticancer drugs. Cell Cycle. 10:1679–1689. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Kwan K, Castro-Sandoval O, Gaiddon C and
Storr T: Inhibition of p53 protein aggregation as a cancer
treatment strategy. Curr Opin Chem Biol. 72:1022302023. View Article : Google Scholar
|
|
237
|
P SS, Naresh P, A J, Wadhwani A, M SK and
Jubie S: Dual modulators of p53 and cyclin D in ER alpha signaling
by albumin nanovectors bearing zinc chaperones for ER-positive
breast cancer therapy. Mini Rev Med Chem. 21:792–802. 2012.
View Article : Google Scholar
|
|
238
|
Yu X, Na B, Zaman S, Withers T, Gilleran
J, Blayney AJ, Bencivenga AF, Blanden AR, Liu Y, Boothman DA, et
al: Abstract 3432: Zinc metallochaperones for mutant p53
reactivation in cancer therapeutics. Cancer Res. 80(16 Suppl):
S34322020. View Article : Google Scholar
|
|
239
|
Parrales A, Ranjan A, Iyer SV, Padhye S,
Weir SJ, Roy A and Iwakuma T: DNAJA1 controls the fate of misfolded
mutant p53 through the mevalonate pathway. Nat Cell Biol.
18:1233–1243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Alalem M, Bhosale M, Ranjan A, Yamamoto S,
Kaida A, Nishikawa S, Parrales A, Farooki S, Anant S, Padhye S and
Iwakuma T: Mutant p53 depletion by novel inhibitors for
HSP40/J-domain proteins derived from the natural compound
plumbagin. Cancers (Basel). 14:41872022. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Kamada R, Toguchi Y, Nomura T, Imagawa T
and Sakaguchi K: Tetramer formation of tumor suppressor protein
p53: Structure, function, and applications. Biopolymers.
106:598–612. 2016. View Article : Google Scholar
|
|
242
|
Synnott NC, Murray A, McGowan PM, Kiely M,
Kiely PA, O'Donovan N, O'Connor DP, Gallagher WM, Crown J and Duffy
MJ: Mutant p53: A novel target for the treatment of patients with
triple-negative breast cancer? Int J Cancer. 140:234–246. 2017.
View Article : Google Scholar
|
|
243
|
Rangel LP, Ferretti GDS, Costa CL, Andrade
SMMV, Carvalho RS, Costa DCF and Silva JL: p53 reactivation with
induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid
aggregation of mutant p53 in cancer cells. J Biol Chem.
294:3670–3682. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Duffy MJ, Synnott NC and Crown J: Mutant
p53 in breast cancer: Potential as a therapeutic target and
biomarker. Breast Cancer Res Treat. 170:213–219. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Reza MN, Ferdous N, Emon MTH, Islam MS,
Mohiuddin AKM and Hossain MU: Pathogenic genetic variants from
highly connected cancer susceptibility genes confer the loss of
structural stability. Sci Rep. 11:192642021. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Tonsing-Carter E, Bailey BJ, Saadatzadeh
MR, Ding J, Wang H, Sinn AL, Peterman KM, Spragins TK, Silver JM,
Sprouse AA, et al: Potentiation of carboplatin-mediated DNA damage
by the Mdm2 modulator nutlin-3a in a humanized orthotopic
breast-to-lung metastatic model. Mol Cancer Therapeut.
14:2850–2863. 2015. View Article : Google Scholar
|
|
247
|
da Costa DCF, Campos NPC, Santos RA,
Guedes-da-Silva FH, Martins-Dinis MMDC, Zanphorlin L, Ramos C,
Rangel LP and Silva JL: Resveratrol prevents p53 aggregation in
vitro and in breast cancer cells. Oncotarget. 9:29112–29122. 2018.
View Article : Google Scholar
|
|
248
|
Chen X and Cubillos-Ruiz JR: Endoplasmic
reticulum stress signals in the tumour and its microenvironment.
Nat Rev Cancer. 21:71–88. 2021. View Article : Google Scholar :
|
|
249
|
Hassin O and Oren M: Drugging p53 in
cancer: One protein, many targets. Nat Rev Drug Discov. 22:127–144.
2023. View Article : Google Scholar
|
|
250
|
Ubby I, Krueger C, Rosato R, Qian W, Chang
J and Sabapathy K: Cancer therapeutic targeting using
mutant-p53-specific siRNAs. Oncogene. 38:3415–3427. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Braicu C, Pileczki V, Irimie A and
Berindan-Neagoe I: p53siRNA therapy reduces cell proliferation,
migration and induces apoptosis in triple negative breast cancer
cells. Mol Cell Biochem. 381:61–68. 2013. View Article : Google Scholar : PubMed/NCBI
|