Introduction
Hepatocellular carcinoma (HCC) constitutes ~80% of
liver malignancies, making it the third most lethal cancer and the
sixth most prevalent malignancy worldwide (1,2).
HCC pathogenesis and progression are influenced by numerous factors
(3,4). Despite recent advances in surgical
resection, targeted pharmacotherapy, precision radiotherapy and
immunotherapy, a significant proportion of HCC patients face
unsatisfactory survival outcomes, attributable to the inherently
aggressive nature of HCC (5-7).
Hence, there is a pressing need to investigate key molecules
involved in HCC development and identify associated diagnostic
markers and treatment targets for enhancing patient prognosis.
The ribosomal protein lateral stalk subunit P0
(RPLP0), an integral component of the RPLP family and a key
ribosomal protein in the 60S subunit, markedly influences cell fate
determination and is intricately involved in tumorigenesis. RPLP0
expression is markedly upregulated in gastric, breast, ovarian,
endometrial and cervical carcinomas and its overexpression is
associated with suboptimal survival outcomes in patients (8-11).
Wang et al (12)
discovered that RPLP0 interacts with NONO independently of the
ribosome and can anchor to damaged DNA, thereby promoting the
autophosphorylation of DNA-dependent protein kinase at the Thr2609
site, enhancing the repair of double-strand breaks and conferring
resistance to radiotherapy. The knockdown of RPLP0 has been
observed to enhance autophagy, a cellular degradation mechanism
targeting cytoplasmic components (13,14) and to trigger G2/M cell
cycle arrest in breast cancer (9). Although RPLP0 abnormal expression
markedly affects tumor progression, its pathophysiological role in
HCC remains to be elucidated.
While prior research has established the involvement
of RPLP0 in HCC development (15), the specific regulatory pathways
and mechanisms governing its regulation remain underexplored.
Therefore, the present study performed an in-depth analysis to
unravel the intricate mechanisms underlying the formation of
regulatory loops through which RPLP0 contributes to HCC
progression. By integrating multiple transcriptomic datasets and
clinical data, coupled with rigorous in vitro and in
vivo experimental procedures, a clinically relevant RPLP0
profile was established in HCC. These results collectively position
RPLP0 as a prognostic marker and therapeutic target, representing a
significant advancement in our understanding of HCC biology and
offering a promising strategy for improving clinical outcomes.
Materials and methods
Cell culture
The cell lines THLE-2, SNU-182, Li-7, Huh-7,
PLC/PRF/5, MHCC97-H and HCCLM3 were sourced from the Chinese
Academy of Sciences and maintained at 37°C with 5% CO2
in a humidified incubator. THLE-2 cells were cultured in BEGM
(Gibco; Thermo Fisher Scientific, Inc.), SNU-182 cells were
propagated in RPMI 1640 (Gibco; Thermo Fisher Scientific, Inc.),
Li-7 cells were maintained in MEM (Gibco; Thermo Fisher Scientific,
Inc.), and Huh-7, PLC/PRF/5, MHCC97-H, and HCCLM3 cells were grown
in DMEM (Gibco; Thermo Fisher Scientific, Inc.). Each of these
media was supplemented with 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.), and the incubator was supplied by Gibco (Thermo
Fisher Scientific, Inc.). For subsequent experiments, Huh-7 and
MHCC97-H cell lines were selected based on their demonstrated
phenotypic stability and experimental reproducibility, making them
suitable for detailed investigation, as well as their
well-characterized properties in HCC research (16). The properties of the cell lines
are given in Table SI.
Tissue samples
Between October 2024 and February 2025, a cohort of
44 paired specimens, comprising tumor and adjacent non-tumor
tissues (designated at sites >5 cm from the tumor margin), was
procured from patients diagnosed with HCC (clinicopathological
features are provided in Table
SII). The median age was 62 years (range, 36-78). These tissue
specimens were exclusively sourced from individuals with untreated
primary HCC who had provided their written informed consent. The
study was performed in strict compliance with the Declaration of
Helsinki and received approval from the Institutional Review Board
under protocol number (2024) CDYFYYLK (07-004).
Bioinformatics analysis
The expression profile and clinical significance of
RPLP0 were systematically evaluated using an integrative
bioinformatics approach that used data from multiple
high-throughput repositories. Specifically, this analysis
incorporated comprehensive datasets from The Cancer Genome Atlas
(TCGA; https://portal.gdc.cancer.gov/),
which includes 371 HCC samples and 50 non-tumor controls; the
Genotype-Tissue Expression (GTEx) project (https://gtexportal.org/home/index.html), encompassing
110 healthy liver tissues; the Gene Expression Profiling
Interactive Analysis (GEPIA)2 repository (http://gepia2.cancer-pku.cn/#index); and the Gene
Expression Omnibus (GEO) platform (https://www.ncbi.nlm.nih.gov/), using datasets
GSE57957, GSE36411, GSE39791 and GSE76427. R software (version
4.3.2 https://cran.r-project.org/) was used to
assess correlations between RPLP0 expression, histological grading
and tumor staging based on TCGA data. Additionally, pathway
enrichment analysis was performed by stratifying samples into high
and low RPLP0 expression groups. Transcription factors and specific
promoter sequences for RPLP0 were identified using predictive tools
such as the Cistrome Data Browser (http://cistrome.org/db/#/) and JASPAR databases
(https://jaspar.elixir.no/).
Reverse transcription-quantitative (RT-q)
PCR
Following the respective manufacturers'
instructions, RNA was extracted with TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), then reverse
transcription and qPCR were performed using the cDNA Synthesis
SuperMix and qPCR SYBR Green Master Mix, respectively (both from
Shanghai Yeasen Biotechnology Co., Ltd.). The thermocycling
protocol consisted of an initial denaturation at 95°C for 2 min,
followed by 40 cycles of 95°C for 10 sec and 60°C for 30 sec.
Melting curve analysis was carried out as follows: 95°C for 15 sec,
60°C for 1 min, and 95°C for 15 sec. Relative mRNA expression
levels were quantified using the comparative 2−ΔΔCq
method (17), with GAPDH serving
as the endogenous control. All experiments were independently
repeated three times. Primer sequences are listed in Table SIII.
Western blotting
To ensure high-fidelity protein extraction, samples
were lysed using RIPA buffer (Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with protease and phosphatase inhibitors to
preserve post-translational modifications and prevent protein
degradation. The extracted proteins were quantified using a BCA kit
(Thermo Fisher, Inc.). Subsequently, depending on the molecular
weight of the proteins of interest, SDS-PAGE electrophoresis was
executed on gels with either 10 or 12.5% polyacrylamide
concentration. Each lane was loaded with a standardized amount of
total protein (20 μg). Following electrophoresis, proteins
were transferred to 0.45 μm PVDF membranes (MilliporeSigma)
using a wet transfer system. The membrane was then blocked with 5%
non-fat milk in TBST (0.1% Tween-20) for 1 h at room temperature to
prevent non-specific binding. The membrane was incubated overnight
at 4°C with primary antibodies specific to the proteins of
interest, diluted according to the manufacturer's recommendations.
After washing with TBST (0.1% Tween-20), the membrane was incubated
with an HRP-conjugated secondary antibody for 1 h at room
temperature. The target proteins were ultimately clearly and
precisely detected using an ECL detection system (Bio-Rad
Laboratories, Inc.). The resulting band intensities were quantified
by densitometry using ImageJ software (version 1.53m; National
Institutes of Health). Antibodies details are in Table SIV.
Immunohistochemistry
Tumor specimens were fixed in 10% neutral buffered
formalin for 48 h at room temperature, followed by routine
dehydration through a graded ethanol series and embedding in
paraffin. Consecutive sections were cut at a thickness of 4
μm. Following deparaffinization and rehydration, antigen
retrieval was performed using citrate buffer (pH 6.0). Endogenous
peroxidase activity was quenched by incubation with 3% hydrogen
peroxide for 10 min. Sections were then permeabilized with 0.2%
Triton X-100 and blocked with 5% bovine serum albumin (BSA; both
from Beyotime Institute of Biotechnology) for 1 h at room
temperature. After being placed in contact with the primary
antibody at room temperature for 2 h to promote optimal
antigen-antibody binding, the sections underwent a 30-min
incubation at 37°C with the secondary antibody. Antibodies details
are in Table SIV. Chromogenic
visualization was achieved using a 0.02% diaminobenzidine (DAB)
solution for 5 min, providing clear staining of target proteins.
Subsequently, the sections were counterstained with hematoxylin for
30 sec to provide nuclear contrast at room temperature, dehydrated
through graded alcohols, cleared in xylene, and air-dried. The
slides were then mounted and examined under light microscopy for
detailed immunohistochemical analysis.
Small interfering (si)RNA, plasmid
transfection and short hairpin (sh)RNA
In the present study, the target cell lines, Huh7
and MHCC97-H cells, were seeded in 6-well plates. Cells were
subjected to transfection using siRPLP0 (150 nM; Shanghai
GenePharma Co., Ltd.) and pECMV-RPLP0 (2.5 μg; MiaoLing
Biology) to knock down and overexpress RPLP0, respectively, using
Lipofectamine® 3000 transfection reagent (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Parallel transfections with si-NC (150 nM) or oe-NC (2.5 μg)
served as negative controls. A c-Myc-specific siRNA (150 nM;
Shanghai GenePharma Co., Ltd.) and the plasmid pCMV-c-Myc (2.5
μg; MiaoLing Biology) were employed to knock down and
overexpress c-Myc, respectively. The transfection mixture was
incubated with the cells for 6 h at 37°C, after which it was
replaced with fresh complete medium. Subsequent functional
experiments were typically performed 48 h post-transfection. To
achieve stable RPLP0 knockdown, MHCC97-H cells were transduced with
shRNA specifically targeting RPLP0 (shRPLP0; Hanbio Biotechnology
Co., Ltd.) at a multiplicity of infection (MOI) of 50. Following a
48-h incubation at 37°C, stable transductants were selected and
maintained in complete medium supplemented with 2.0 μg/ml
puromycin (Biosharp Life Sciences) for two weeks. Detailed
sequences are listed in Table
SV.
Cell proliferation assay
After successful transfection, the cells were
trypsinized and subsequently resuspended in single-cell
suspensions. For CCK-8 assays, 1,500 cells was dispensed into each
well of a 96-well plate and absorbance was measured at specific
intervals. In the colony-forming efficiency test, 1,000 isolated
cells were uniformly distributed in each well of 6-well plates.
After 12 days, colonies became macroscopically visible and they
were sequentially fixed, stained, air-dried, and photographed.
Transwell assay
Cells were transfected for 48 h, after which 200
μl of a suspension containing 4×104 cells was
introduced into the upper chamber of a Matrigel-coated (incubated
at 37°C for 2 h for gel formation) Transwell system featuring 8
μm pores. Meanwhile, the lower chamber was replenished with
600 μl of DMEM supplemented with 20% FBS. At 48 h, samples
were fixed with 4% paraformaldehyde for 30 min at room temperature,
stained with 0.1% crystal violet for 20 min at room temperature and
imaged under a light microscope. For migration assays, Transwell
chambers that had not been pretreated with Matrigel were used.
Apoptosis and cell cycle assay
Apoptosis and cell cycle distribution were analyzed
using the Annexin V-PE/7-AAD apoptosis kit and the Cell Cycle
Staining Kit (MultiSciences Biotech Co., Ltd.), respectively,
according to the manufacturer's guidelines. After staining at room
temperature for 30 min in the dark, the samples were analyzed using
a flow cytometer (Beckman Coulter, Inc.). Data were processed with
FlowJo software (version 10.1; BD FlowJo) (18). The apoptotic rate was defined as
the combined percentage of early and late apoptotic cells in the
total population.
Reactive oxygen species (ROS) assay
Intracellular ROS levels were determined employing
the Reactive Oxygen Species Assay Kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. Cells
underwent staining with 1X Hoechst for 20 min at ambient
temperature and subsequently were incubated in the dark at 37°C
with 10 μM DCFH-DA for 30 min. After incubation, the excess
DCFH-DA was removed by washing the cells three times with
serum-free culture medium to minimize non-specific extracellular
fluorescence. The assessment of ROS levels was performed using
either fluorescence microscopy (Leica Microsystems GmbH) or a flow
cytometer (CytoFLEX; Beckman Coulter, Inc.). During flow cytometry,
the excitation and emission wavelengths were set at 488 nm and 525
nm, respectively, and a total of 10,000 events were acquired per
sample. Data were analyzed using FlowJo software (version 10.1; BD
FlowJo) (18).
Monodansylcadaverine (MDC) staining
To assess the levels of intracellular
autophagosomes, the present study used an MDC staining kit
(Beyotime Institute of Biotechnology). Cells underwent a 20-min
incubation period in dim conditions at ambient temperature with 1X
Hoechst solution. This was followed by another 30 min of incubation
at 37°C in 500 μl of 1X MDC solution, with darkness
maintained throughout. After washing three times with assay buffer,
the cells were examined under a fluorescence microscope (Leica
Microsystems GmbH).
Transmission electron microscopy
(TEM)
The collected cells were fixed with a specialized
electron microscopy fixative (Wuhan Servicebio Technology Co.,
Ltd.) at 4°C overnight, followed by dehydration through an ethanol
gradient. Subsequently, the samples underwent infiltration with
propylene oxide and were embedded in resin using a progressive
temperature protocol (37°C for 12 h, 45°C for 12 h, and 60°C for 24
h). Ultrathin sections (70 nm) were prepared using an
ultramicrotome, then double-stained with uranyl acetate for 15 min
and lead citrate for 5 min at room temperature. After overnight
drying, the sections were examined utilizing a transmission
electron microscope (Hitachi High-Technologies Corporation).
Chromatin immunoprecipitation (ChIP)
assay
The ChIP procedure was performed in strict
accordance with the guidelines furnished by the ChIP assay kit
procured from Cell Signaling Technology, Inc. Cells grown in 10 cm
dishes at 80-90% confluence were initially fixed with 1%
formaldehyde for 10 min at room temperature. After quenching with
125 mM glycine, cells were lysed and the chromatin was fragmented
using a non-contact ultrasonicator (Little Scientific Instruments
Co., Ltd.) set at 100% power for 25 min with cycles of 10 sec ON
and 5 sec OFF at 4°C, yielding DNA fragments of 200-600 bp. A 10
μl aliquot of the lysate was reserved and stored at −80°C
for subsequent use as an input. Next, 500 μl of lysate
underwent immunoprecipitation through overnight incubation at 4°C
using protein A/G beads with 5 μl of either anti-c-Myc (4
μg) or anti-IgG antibodies (4 μg). The beads were
washed sequentially with low salt, high salt and LiCl wash buffers
provided in the ChIP assay kit. The purified DNA fragments
subsequently underwent qPCR. Purified DNA fragments were amplified
via PCR using reagents acquired from Vazyme Biotech Co., Ltd. and
the following cycling conditions: initial denaturation at 95°C for
3 min; 35 cycles of denaturation at 95°C for 15 sec, annealing at
60°C for 15 sec, and extension at 72°C for 30 sec; and final
extension at 72°C for 5 min. PCR products were separated on 2%
agarose gels and visualized with GelStain (Bio-Rad Laboratories,
Inc.) under UV illumination. The specific primer sequences used for
qPCR in the ChIP assay are presented in Table SVI.
Dual luciferase assay
The promoter sequences of both the native
(wild-type; WT) and altered (mutant; MUT) RPLP0 were cloned into
pGL4 plasmids (Promega Corporation). Afterward, cells seeded in
24-well plates at 70-80% confluence were co-transfected using
Lipofectamine® 3000 reagent (Thermo Fisher Scientific,
Inc.) with either pGL4-RPLP0-WT or pGL4-RPLP0-MUT, together with
the pGL4.74 internal control plasmid and either pCMV-c-Myc or the
corresponding empty vector. After a 48-h incubation period,
luciferase activity was quantified employing the Dual Luciferase
Assay Kit (Promega Corporation).
Xenograft tumor model
A subcutaneous xenograft model was generated using
16 BALB/c nude female mice (4-5 weeks old, 15-16 g; Sibeifu
Laboratory Animal Co. Ltd.) and maintained under strict SPF
conditions (22±1°C; relative humidity 50±10%; a 12/12-h light/dark
cycle, and ad libitum access to sterilized food and water).
Lentivirus-infected MHCC97-H cells, targeting shNC and shRPLP0,
were successfully screened to establish stable cell lines.
Subsequently, an inoculation of 6×106 cells, comprising
either shNC or shRPLP0, was administered at the specified
subcutaneous site in each mouse. The dimensions of the subcutaneous
tumors were precisely assessed and recorded every four days. Tumor
volume was calculated using the formula: length × width2
×0.5. The largest tumor observed in the present study measured
10.82 mm in length and 7.28 mm in width, with a maximum volume of
286.72 mm3. In compliance with animal ethics guidelines,
tumor size was limited to a mean diameter of 20 mm and a volume of
2,000 mm3. Sacrifice was via carbon dioxide
(CO2) inhalation at a chamber displacement rate of
30-50% per min prior to tumor excision, with mortality confirmed by
absent heartbeat and respiration. Animal studies were performed
following ethical guidelines and were approved by the Animal Ethics
Committee (approval no. CDYFY-IACUC-202407QR259).
Statistical analysis
Data analysis was conducted using GraphPad Prism 9
(Dotmatics). For comparisons of RPLP0 mRNA expression between tumor
and adjacent non-tumor tissues from HCC patients, a paired
Student's t-test was applied. Other comparisons between two groups
were conducted using an unpaired Student's t-test. For multiple
group comparisons, one-way ANOVA followed by Bonferroni's post hoc
test was applied. The Spearman correlation coefficient was
determined to assess relationships between variables. Kaplan-Meier
curves and log-rank tests were used to analyze survival prognosis.
Data were presented as mean ± SEM. P<0.05 was considered to
indicate a statistically significant difference.
Results
RPLP0 is markedly upregulated in HCC and
its high expression is associated with poorer prognosis
Analyses based on the TCGA and GTEx databases
demonstrated that RPLP0 transcriptome levels were markedly elevated
in HCC samples (Fig. 1A).
Consistent findings were observed in the GSE57957, GSE36411,
GSE39791 and GSE76427 datasets, providing further validation of the
heightened expression of RPLP0 in HCC (Fig. 1B). The GEPIA 2 database revealed
that individuals with elevated RPLP0 levels experienced decreased
overall survival (OS) and disease-free survival (DFS) rates
(Fig. 1C and D). TCGA database
analysis showed that high RPLP0 expression levels were associated
with unfavorable histological grades and tumor stages (Fig. 1E and F). Furthermore, comparative
studies of mRNA and protein levels in 44 matched tissues clearly
indicated a marked elevation in RPLP0 in HCC (Fig. 1G and H). After stratifying samples
by median RPLP0 expression, a Kaplan-Meier analysis demonstrated
that higher RPLP0 expression was markedly associated with inferior
outcomes in HCC patients (Fig.
1I). Immunohistochemical analysis of the collected samples
further validated the upregulation of RPLP0 expression in HCC
tissues (Fig. 1J). Compared with
immortalized hepatocytes, RPLP0 expression was notably higher in
HCC cell lines at both mRNA and protein levels (Fig. 1K and L). Overall, these results
suggest that RPLP0 may be utilized as a diagnostic and prognostic
biomarker in HCC.
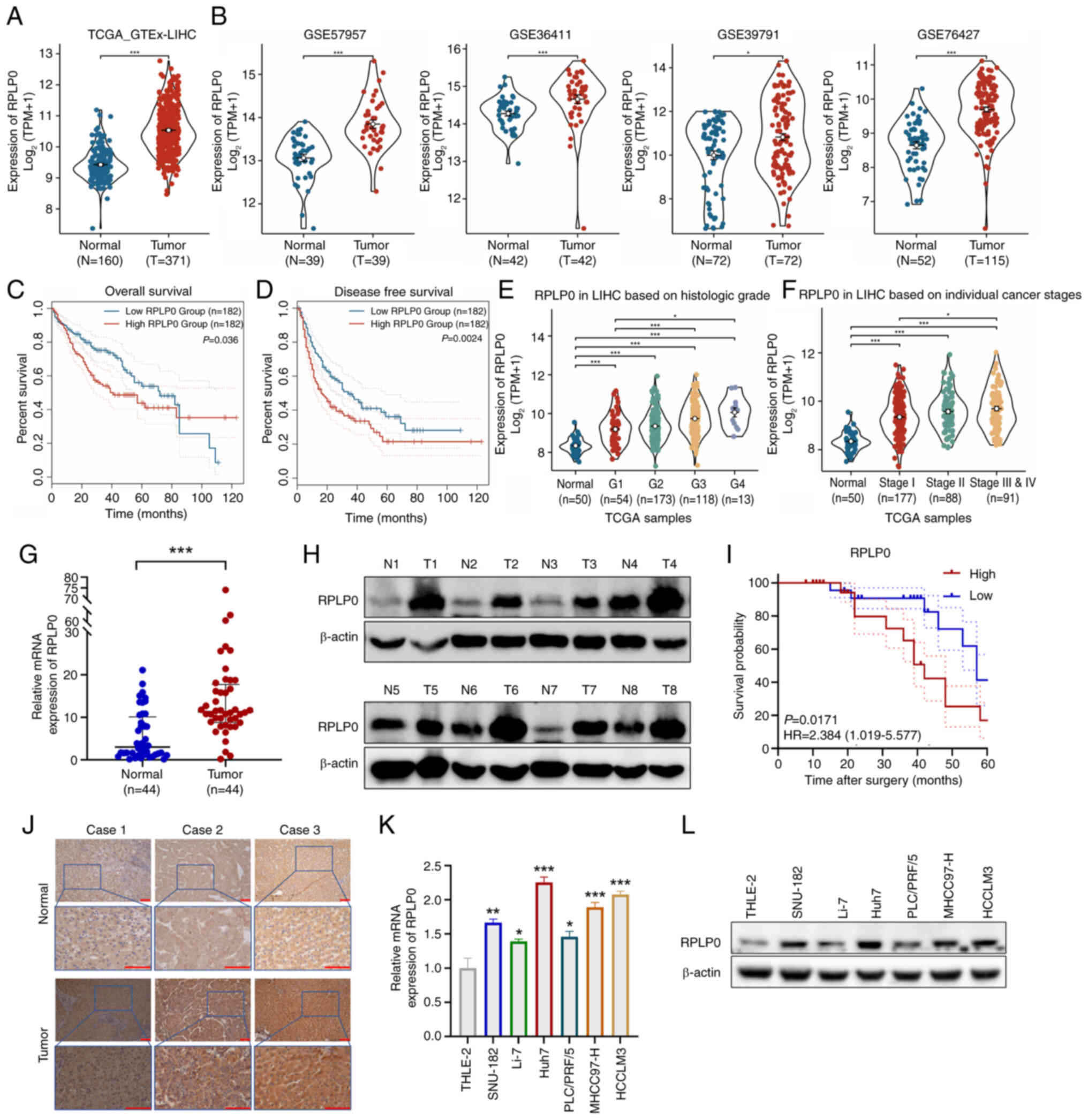 | Figure 1Elevated RPLP0 expression is
associated with unfavorable survival outcomes in HCC. (A) RPLP0
expression levels derived from the TCGA_GTEx database. (B)
Confirmation of RPLP0 expression in the GSE57957, GSE36411,
GSE39791 and GSE76427 datasets. Association between RPLP0
expression status and (C) OS and (D) DFS among HCC patients in the
GEPIA2 database. The relationship between RPLP0 expression and (E)
histological grade and (F) cancer stage in the TCGA database. The
expression levels of (G) mRNA and (H) protein for RPLP0 were
analyzed in 44 paired HCC samples. (I) The influence of RPLP0
expression on the survival outcomes of HCC patients. (J)
Immunohistochemical analysis of RPLP0 in HCC and adjacent
non-cancerous tissues (scale bar, 100 μm). RPLP0 (K) mRNA
and (L) protein levels were examined in immortalized hepatocytes
and HCC cell lines. *P<0.05, **P<0.01,
***P<0.001. RPLP0, ribosomal protein lateral stalk
subunit P0; HCC, hepatocellular carcinoma; TCGA, the cancer genome
atlas; GTEx, the genotype-tissue expression; N, normal; T, tumor;
GEO, gene expression omnibus; GEPIA2, gene expression profiling
interactive analysis; OS, overall survival; DFS, disease-free
survival. |
RPLP0 downregulation suppresses the
malignant phenotype of HCC cells, inducing apoptosis and
G2/M cell cycle arrest
To further explore the biological function of RPLP0
in HCC, its expression was modulated in Huh7 and MHCC97-H cell
lines using siRPLP0 and oeRPLP0, respectively and their controls.
This caused changes in RPLP0 levels at both the mRNA and protein
(Fig. 2A-D) levels. The findings
obtained through the CCK-8, colony formation efficiency test and
Transwell assay showed that RPLP0 downregulation suppressed the
proliferative, invasive and migratory potentials of HCC cells. By
contrast, the upregulation of RPLP0 enhanced these cellular
properties in HCC cells (Fig.
2E-G). Western blot analysis indicated that the siRPLP0 group
exhibited markedly increased levels of E-cadherin in comparison to
the si-NC group, accompanied by notably decreased levels of
Vimentin and Snail. Conversely, the oeRPLP0 group demonstrated
markedly reduced levels of E-cadherin in comparison to the oe-NC
group, with concurrently elevated levels of Vimentin and Snail
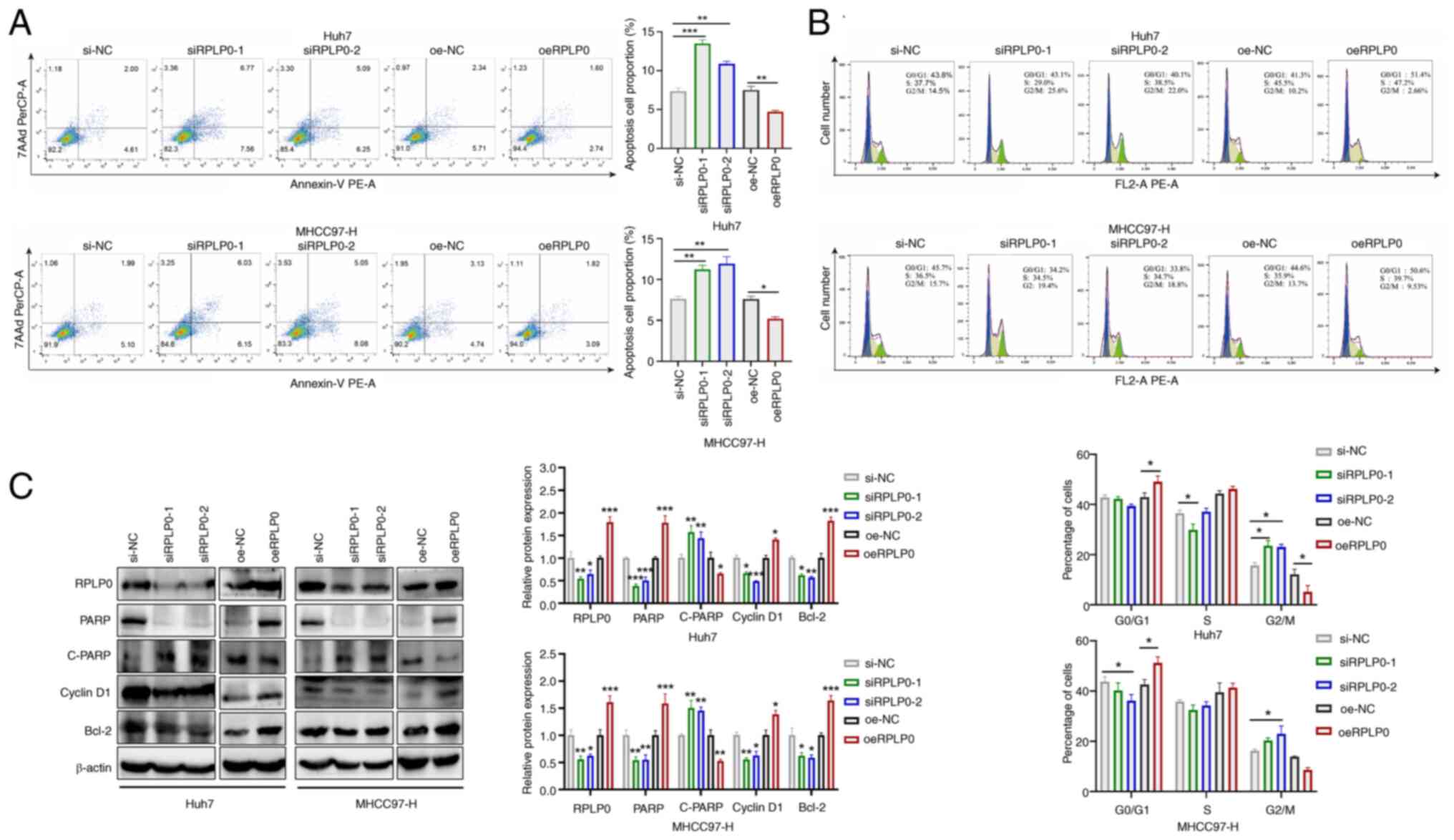
(Fig. 2H). Flow cytometry
analysis indicated that downregulation of RPLP0 promoted apoptosis
and mediated G2/M cell cycle arrest, whereas
upregulation of RPLP0 inhibited apoptosis and promoted
G2/M cell cycle transition (Fig. 3A-B). Subsequently, the influence
of RPLP0 modulation on the expression patterns of proteins
implicated in apoptosis and cell cycle regulation was examined.
Western blot analysis showed that Poly (ADP-ribose) polymerase
(PARP), Bcl-2 and Cyclin D1 levels were markedly decreased in the
siRPLP0 group whereas cleaved (C-)PARP expression levels were
elevated. Conversely, the oeRPLP0 group displayed a marked
up-regulation in PARP, Bcl-2 and Cyclin D1 levels, accompanied by
the downregulation of C-PARP expression (Fig. 3C). These findings conjointly
indicate that the knockdown of RPLP0 delays malignant progression
in HCC cells.
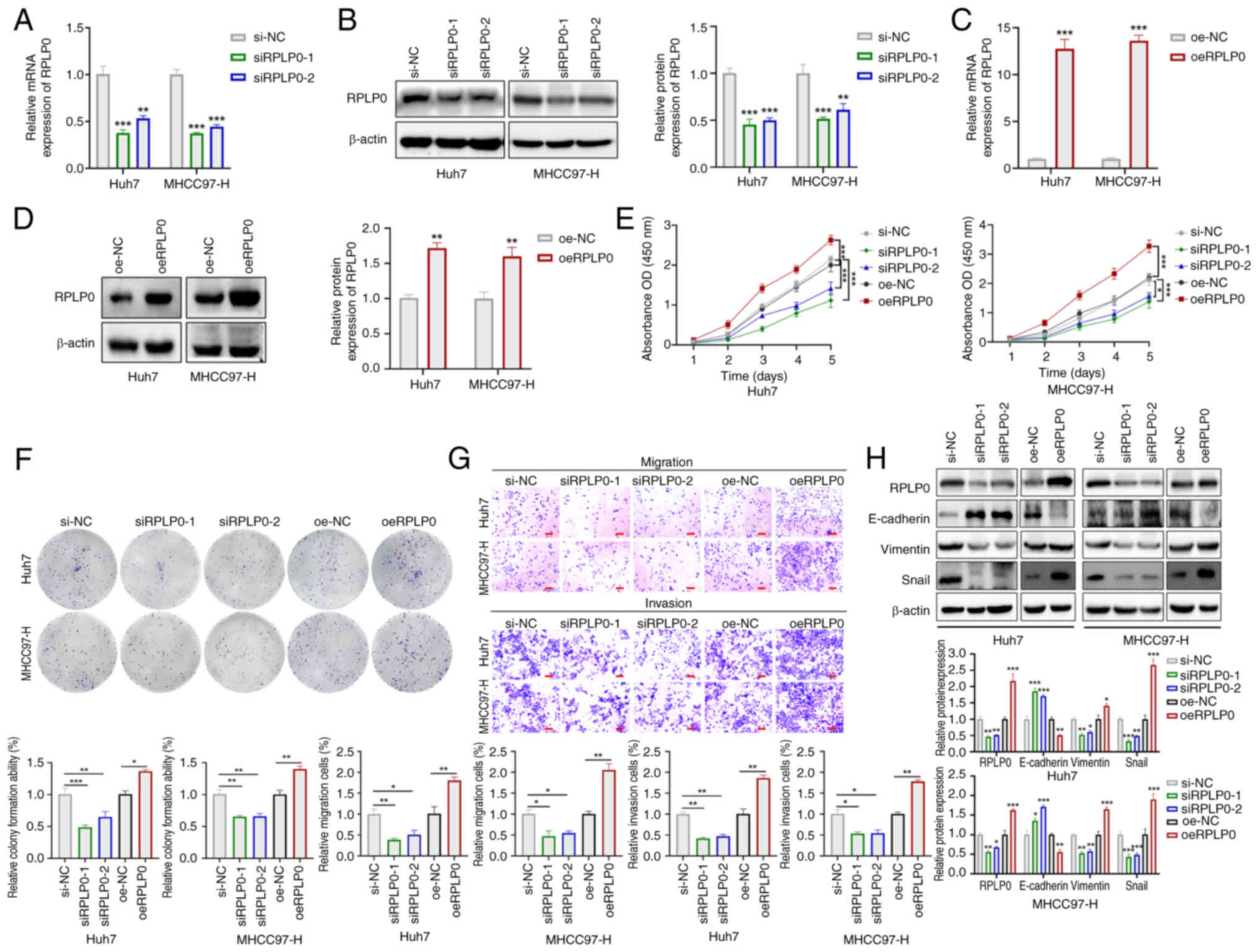 | Figure 2RPLP0 overexpression promotes
malignant phenotypes in HCC cells. Alterations in mRNA and protein
levels following (A and B) RPLP0 knockdown and (C and D)
overexpression in HCC cells. (E) CCK-8, (F) colony formation and
(G) Transwell assays were conducted to assess HCC cell
proliferation, invasion and migration (scale bar, 150 μm).
(H) EMT-related protein expression changes (E-cadherin, Vimentin
and Snail) were observed in treated HCC cells.
*P<0.05, **P<0.01,
***P<0.001. RPLP0, ribosomal protein lateral stalk
subunit P0; HCC, hepatocellular carcinoma; CCK-8, cell counting
kit-8; EMT, epithelial-mesenchymal transition. |
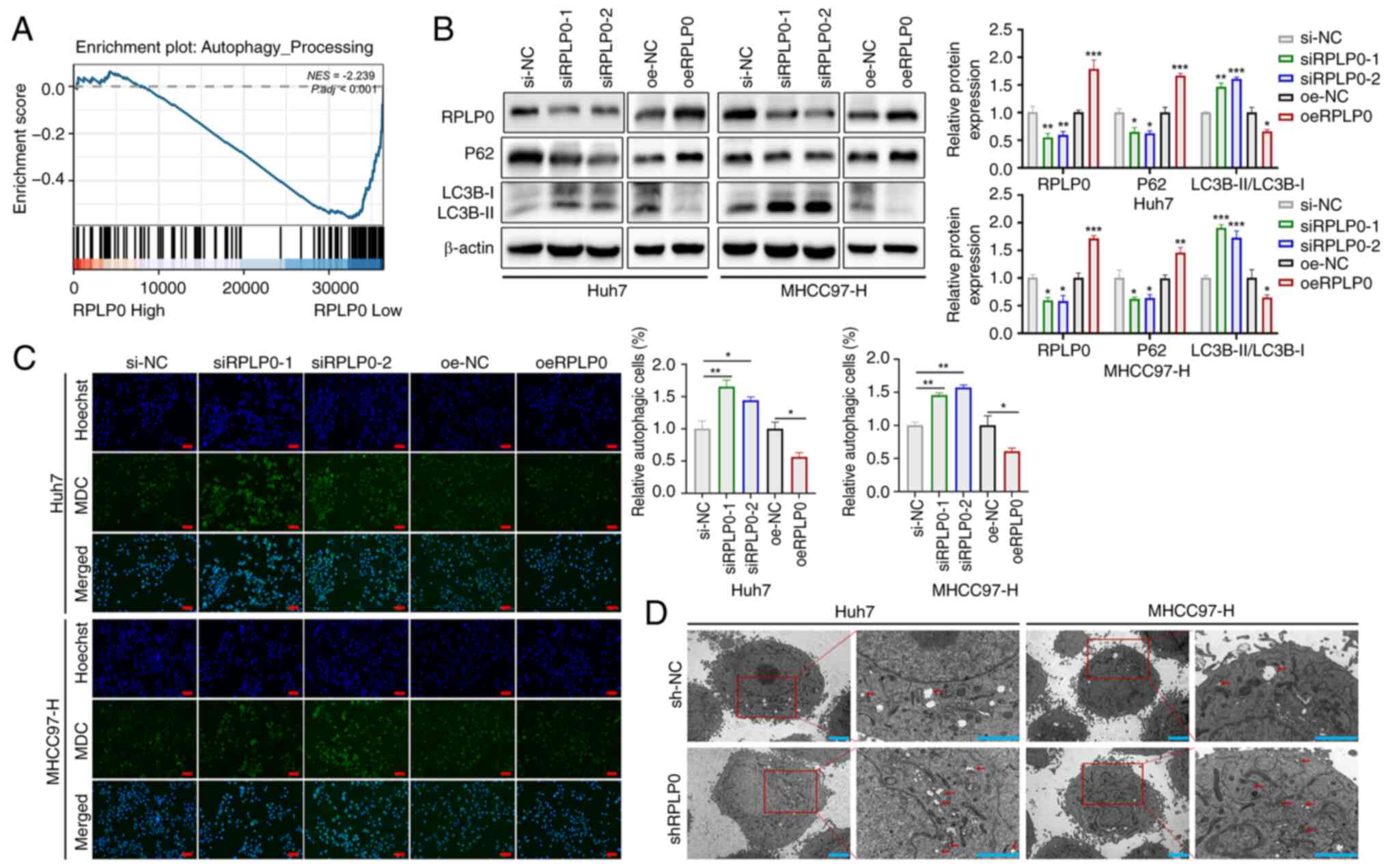
RPLP0 knockdown induces autophagy in
HCC
Autophagy is an intracellular regulatory mechanism
that preserves cellular homeostasis by eliminating superfluous or
dysfunctional cellular components and recycling metabolic
substrates (19,20). To assess the effect of RPLP0
expression on autophagy levels in HCC, GSEA was conducted,
revealing a statistically significant correlation between decreased
RPLP0 expression and the augmentation of autophagic processes
(Fig. 4A). Importantly, further
validation through western blot analysis demonstrated that the
suppression of RPLP0 resulted in a substantial elevation in the
LC3B-II/LC3B-I ratio and a concurrent decrease in P62 levels
(Fig. 4B). Subsequently, the
presence of autophagosome vacuoles was evaluated employing MDC
staining and transmission electron microscopy. The analysis
unveiled that the downregulation of RPLP0 led to an increased
accumulation of autophagosome vacuoles in HCC cells (Fig. 4C and D). Autophagy was inhibited
by treating Huh7 and MHCC97-H cells that had been knocked down with
3-methyladenine (3-MA) (21). The
outcomes signaled that 3-MA reversed the elevation in the
LC3B-II/LC3B-I ratio induced by RPLP0 downregulation, indicating
that RPLP0 knockdown indeed triggered autophagy (Fig. 5A). Given that the accumulation of
autophagosomes may signify either enhanced autophagic flux or
impaired autophagic processes (22), the effect of RPLP0 knockdown on
autophagic flux was investigated by the administration of
chloroquine (CQ), a known inhibitor of autophagy-lysosome fusion
(23). Treatment with CQ resulted
in an increase in the LC3B-II/LC3B-I ratio at both 12 and 24 h,
suggesting that RPLP0 knockdown promoted autophagic flux (Fig. 5B). These findings unequivocally
demonstrate that the downregulation of RPLP0 induces autophagy in
HCC cells.
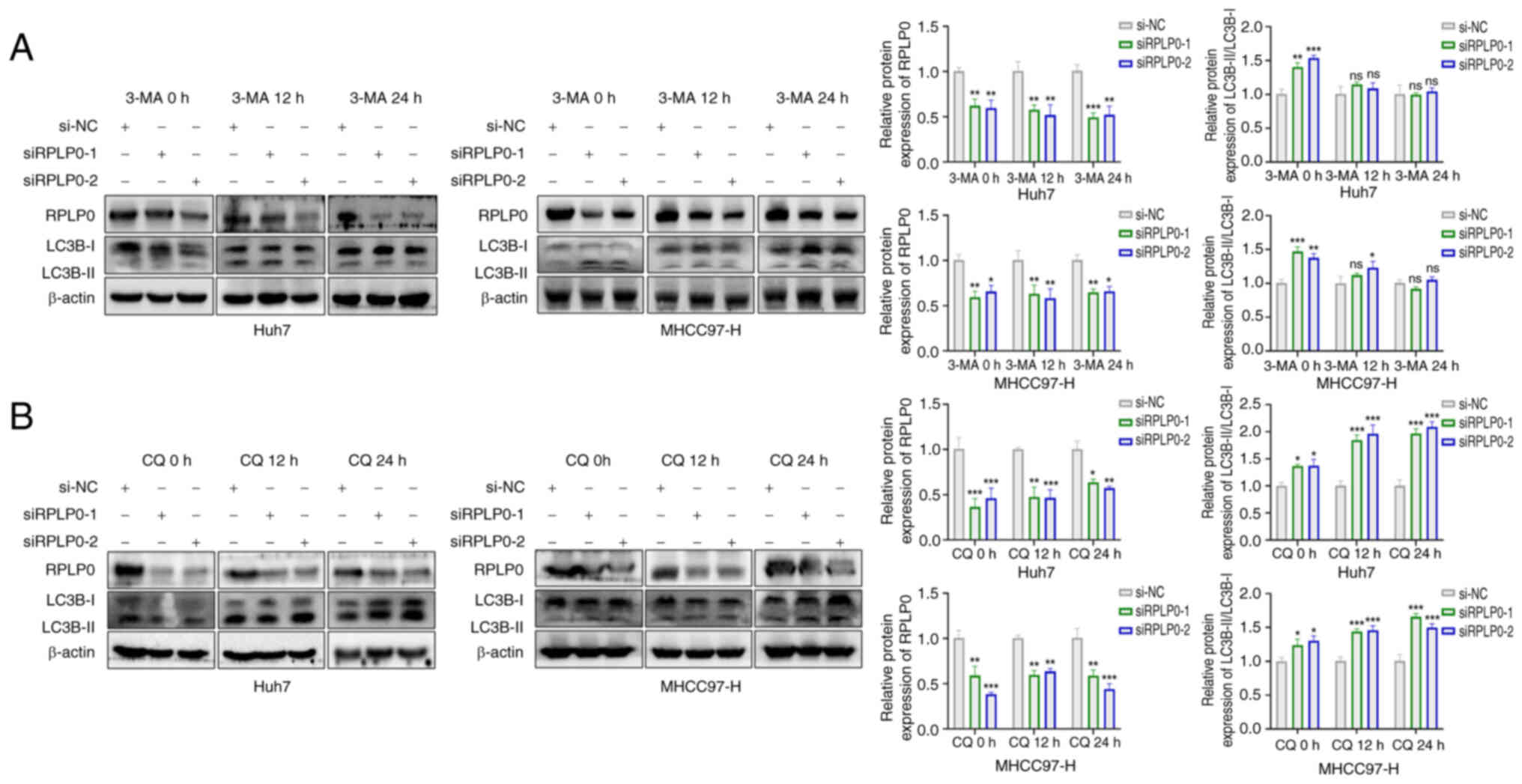 | Figure 5RPLP0 knockdown stimulates autophagic
flux in HCC cells, as shown by 3-MA and CQ. (A) Western blotting
analyzed LC3B expression in HCC cells pretreated with transfection
and subsequently exposed to 5 mM 3-MA for 0, 12 and 24 h. (B) HCC
cells were treated with 20 μM CQ for 0, 12 and 24 h,
respectively, followed by RPLP0 knockdown, and western blotting
evaluated LC3B levels. *P<0.05,
**P<0.01, ***P<0.001. RPLP0, ribosomal
protein lateral stalk subunit P0; HCC, hepatocellular carcinoma;
3-MA, 3-methyladenine; CQ, chloroquine. |
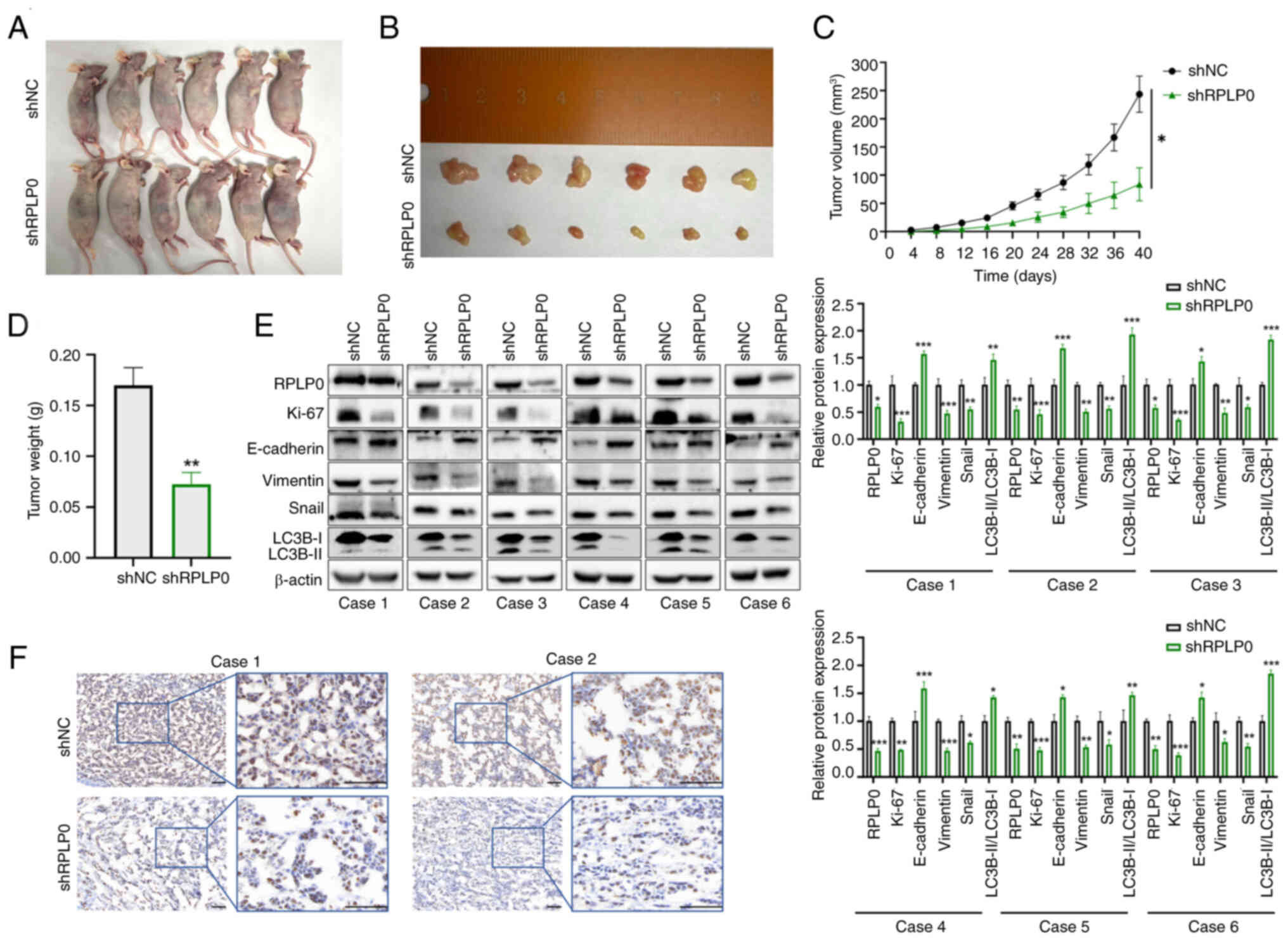
Suppression of RPLP0 expression impedes
the progression of xenograft tumors
To explore the influence of RPLP0 knockdown on the
progression of subcutaneous xenograft tumors, MHCC97-H cells were
genetically modified using lentiviral vectors encoding either a
non-targeting control short hairpin RNA (shNC) or an RPLP0-targeted
short hairpin RNA (shRPLP0). Throughout the experimental period,
the tumor volumes were meticulously monitored and documented at
regular 4-day intervals. After 40 days, mouse xenograft models were
successfully established, xenograft tumors were successfully
isolated and tumor weights were statistically analyzed. The shRPLP0
group exhibited notably reduced tumor volumes and weights in
comparison to the shNC group (Fig.
6A-D). Western blotting of excised tumors indicated that the
shRPLP0 group demonstrated increased expression in E-cadherin and
the LC3B-II/LC3B-I ratio relative to the shNC group. Meanwhile, the
expression levels of Ki-67, Vimentin and Snail were markedly
decreased in the shRPLP0 group (Fig.
6E). Furthermore, immunohistochemical analysis of the xenograft
tumors demonstrated lower Ki-67 expression levels in the shRPLP0
group (Fig. 6F).
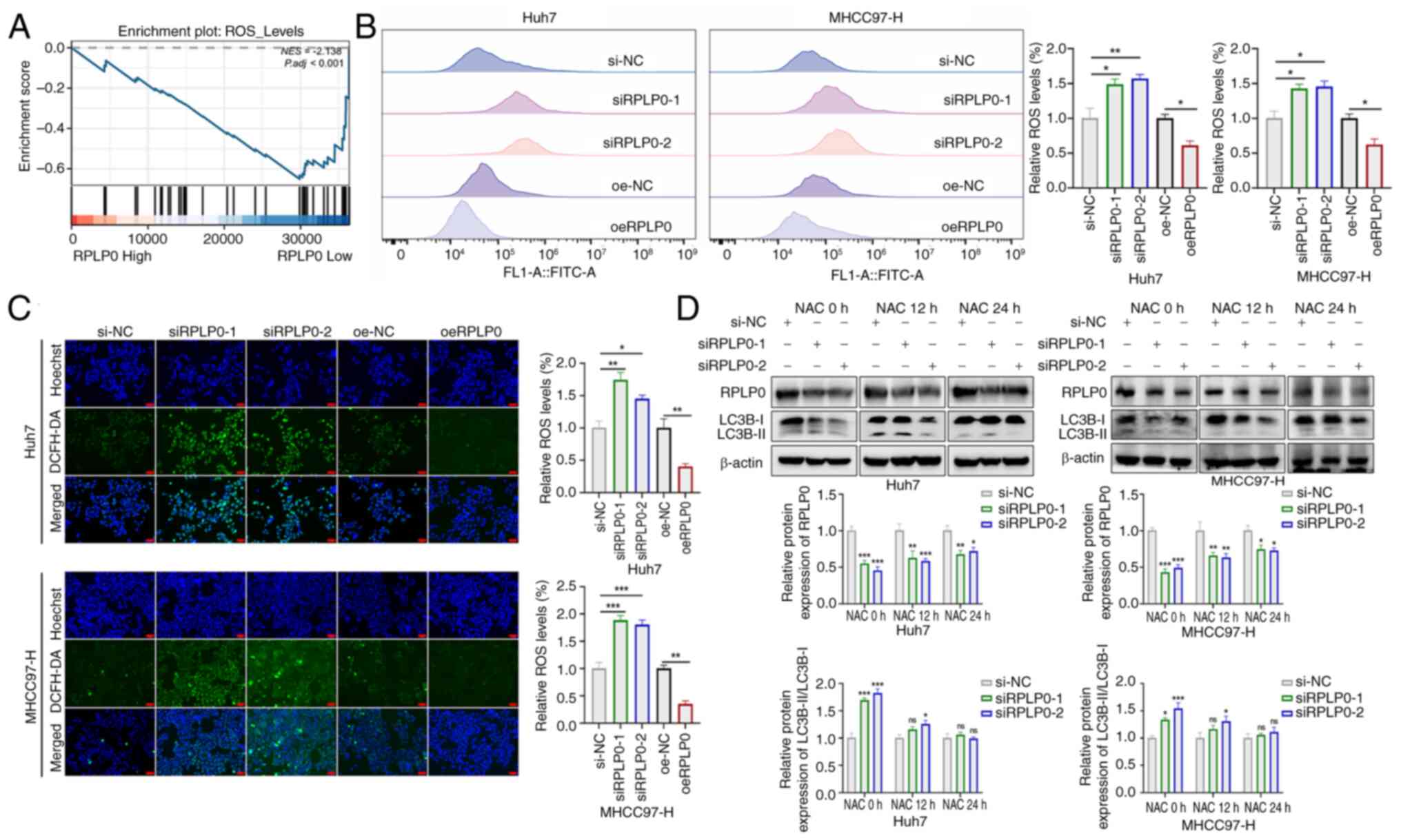
Suppression of RPLP0 inactivates the
JAK2/STAT3 signaling pathway via ROS accumulation
Deficiency of the human ribosomal P complex in
breast cancer has been shown to result in significant dysregulation
of the TXN protein that neutralizes the effects of oxidative damage
(9,24). As previously documented, RPLP0
knockdown promoted autophagy (Fig.
4A-D), which is closely related to intracellular oxidative
stress (25,26). To explore the effect of RPLP0
knockdown on ROS production, GSEA was performed. The findings
revealed significant enrichment of ROS accumulation in the
low-RPLP0 expression group (Fig.
7A). DCFH-DA staining can directly reflect ROS levels (27). Accordingly, DCFH-DA staining was
performed on transfected Huh7 and MHCC97-H cells and subsequent
flow cytometry and fluorescence microscopy showed that RPLP0
knockdown markedly stimulated ROS production (Fig. 7B and C). To investigate the
relationship between ROS and autophagy, ROS levels were decreased
using N-acetyl-l-cysteine (NAC) (28). As depicted in Fig. 7D, a marked reduction in the
LC3B-II/LC3B-I ratio was observed following 12 and 24 h of NAC
treatment, suggesting that the progression of autophagy was
contingent upon ROS production. Moreover, the results showed that
RPLP0 knockdown inhibited JAK2/STAT3 pathway activation (Fig. 8A). IL-6 is well-documented as an
activator of the JAK/STAT3 pathway (29-31). Following a 48-h transfection
pretreatment in Huh7 and MHCC97-H cell lines, the administration of
40 ng/ml IL-6 for 24 h reverted alterations in RPLP0,
phosphorylated (p-) JAK2/JAK2 and p-STAT3/STAT3 expression,
implying that the activation of the JAK2/STAT3 pathway is at least
partially dependent on RPLP0 (Fig.
8B). To further investigate the effects of RPLP0 knockdown on
ROS-mediated alterations in the JAK2/STAT3 pathway, NAC was
administered. The findings revealed that the levels of RPLP0,
p-JAK2/JAK2 and p-STAT3/STAT3 exhibited time-dependent increases at
12 and 24 h post-treatment, suggesting that RPLP0 knockdown induces
ROS accumulation, which mediates the inactivation of the JAK2/STAT3
pathway (Fig. 8C). Notably, the
mRNA levels of RPLP0 time-dependently increased following NAC
treatment (Fig. S1). In summary,
RPLP0 knockdown suppressed the JAK2/STAT3 signaling pathway by
promoting ROS accumulation, consequently delaying the progression
of HCC.
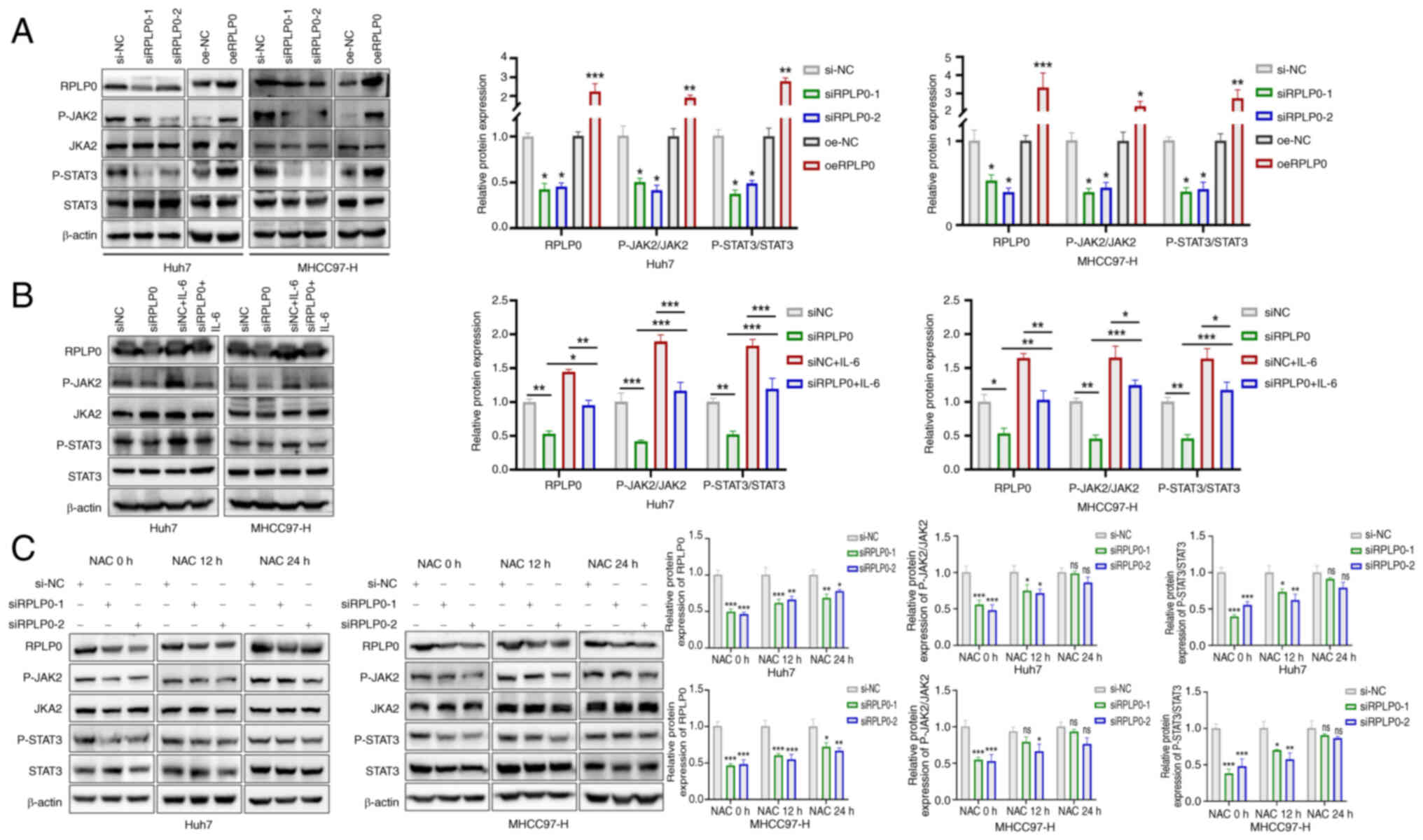 | Figure 8Suppressing RPLP0 deactivates the
JAK2/STAT3 pathway by accumulating ROS. (A) Alterations in RPLP0,
p-JAK2/JAK2 and p-STAT3/STAT3 expression were assessed in HCC cells
following transfection. (B) After 48 h of transfection, Huh7 and
MHCC97-H cells were exposed to 40 ng/ml IL-6 for an additional 24
h, followed by western blot analysis of RPLP0, p-JAK2/JAK2 and
p-STAT3/STAT3 expression. (C) HCC cells underwent a 48-h
transfection pretreatment and were then treated with 5 mM NAC for
0, 12 and 24 h, followed by western blot analysis of RPLP0,
p-JAK2/JAK2 and p-STAT3/STAT3 expression. *P<0.05,
**P<0.01, ***P<0.001. RPLP0, ribosomal
protein lateral stalk subunit P0; ROS, reactive oxygen species; p-,
phosphorylated; HCC, hepatocellular carcinoma; NAC,
N-acetyl-L-cysteine. |
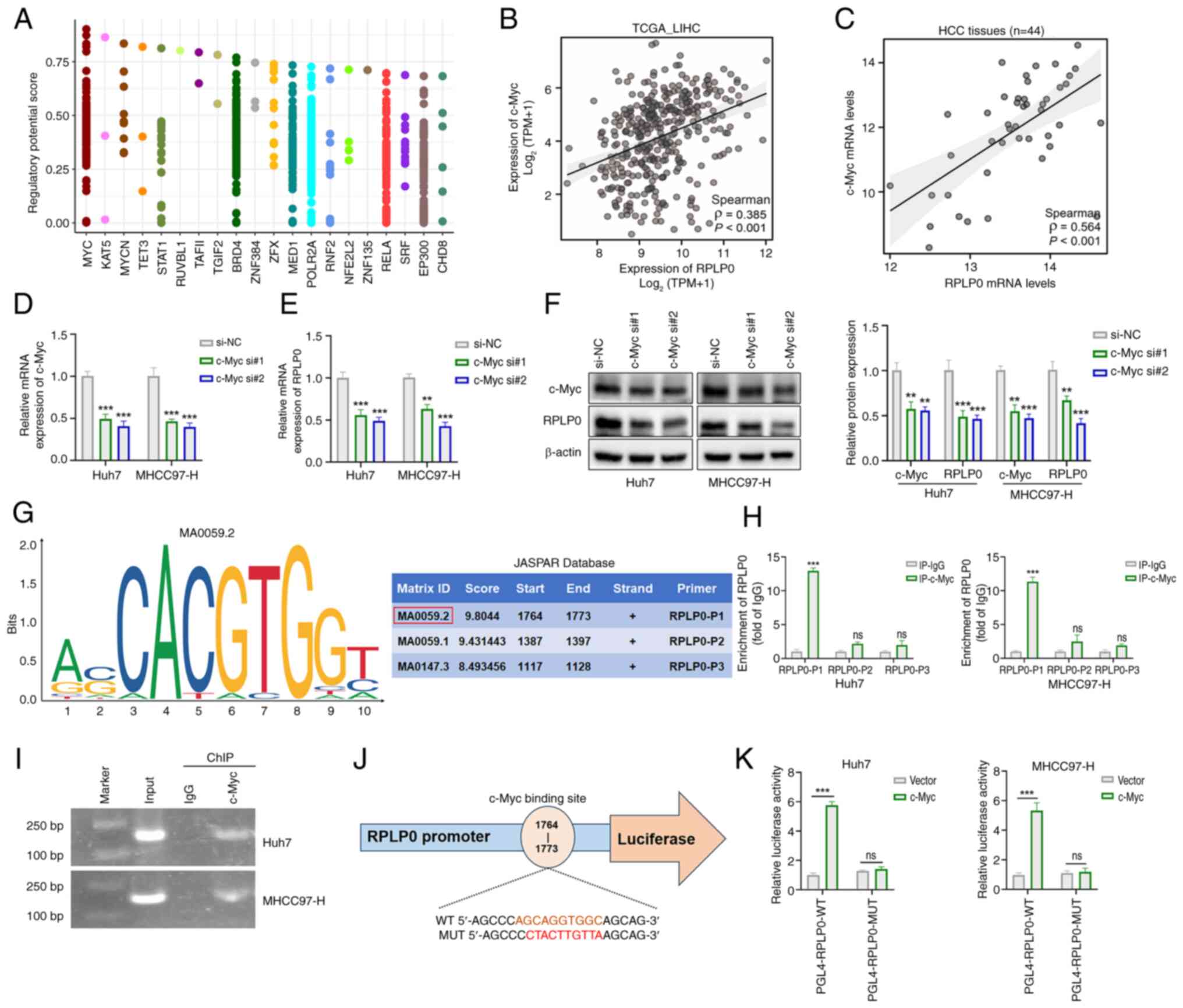
c-Myc activates RPLP0 transcription
To identify upstream regulators that activate RPLP0
transcription, predictions were made using the Cistrome Data
Browser. As depicted in Fig. 9A,
c-Myc had the highest relative score as a potential transcription
factor for RPLP0. Correlation analyses of TCGA data and 44 HCC
tissue samples demonstrated a statistically significant positive
association between c-Myc and RPLP0 expression levels (Fig. 9B and C). Furthermore, c-Myc
knockdown in HCC cells yielded a marked reduction in the mRNA and
protein levels of both c-Myc and RPLP0 (Fig. 9D-F). The DNA binding motifs for
c-Myc were retrieved from the JASPAR database and the corresponding
primers (RPLP0-P1/P2/P3) were designed by selecting different
binding sites with high relative scores (Fig. 9G). The direct interaction of c-Myc
with the RPLP0 promoter region was validated through ChIP-qPCR and
agarose gel electrophoresis of ChIP products (Fig. 9H and I). Moreover, luciferase
reporter vectors for RPLP0-WT and RPLP0-MUT were constructed,
incorporating specific c-Myc binding sites within the RPLP0
promoter sequence (Fig. 9J). The
dual luciferase reporter assay results indicated that c-Myc
enhanced the transcriptional activity of RPLP0-WT, whereas the
luciferase activity of RPLP0-MUT remained unaffected by c-Myc
(Fig. 9K). In summary, RPLP0
functioned as a direct target of c-Myc-mediated transcription.
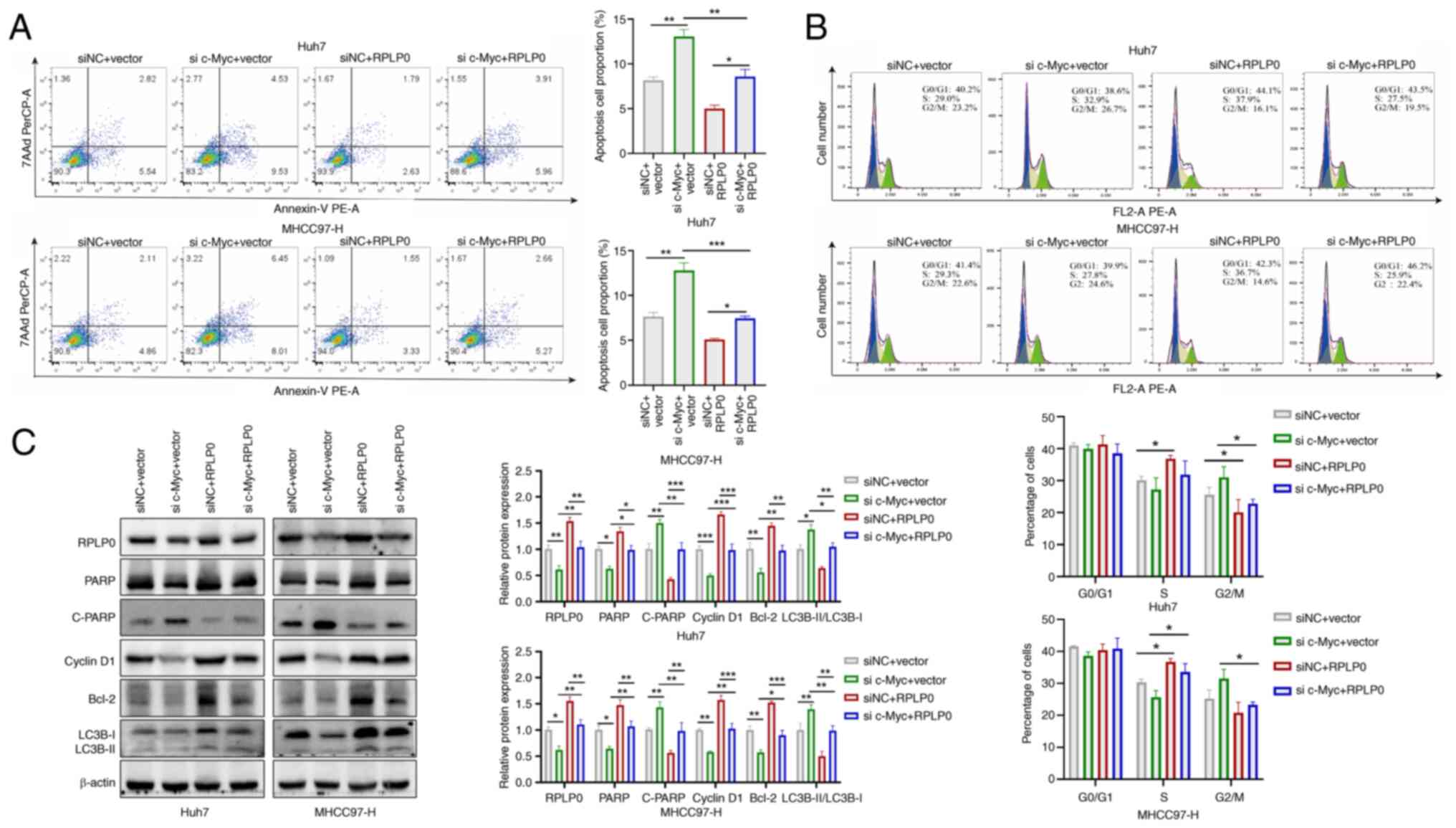
RPLP0 facilitates the malignant
progression of HCC through reliance on c-Myc regulation
To investigate whether the involvement of RPLP0 in
advancing HCC development is contingent upon c-Myc regulation, Huh7
and MHCC97-H cell lines were used in co-transfection experiments.
Downregulation of c-Myc reversed the increases in HCC cell
proliferation, invasion and migration induced by RPLP0
overexpression (Fig. 10A-C).
Furthermore, RPLP0 overexpression restored the levels of Vimentin
and Snail while concomitantly downregulating E-cadherin expression
(Fig. 10D). Flow cytometry
analysis revealed that c-Myc knockdown effectively mitigated the
suppression of apoptosis and the G2/M cell cycle arrest
induced by RPLP0 overexpression (Fig. 11A and B). In addition, western
blot analysis revealed that downregulation of c-Myc partially
counteracted the influence of RPLP0 overexpression on the protein
levels of PARP, C-PARP, Cyclin D1 and Bcl-2, as well as the
LC3B-II/LC3B-I ratio (Fig. 11C).
The present observations provide supporting evidence for the
involvement of RPLP0 in facilitating HCC progression, with
modulation of c-Myc serving as a key mechanism in this process.
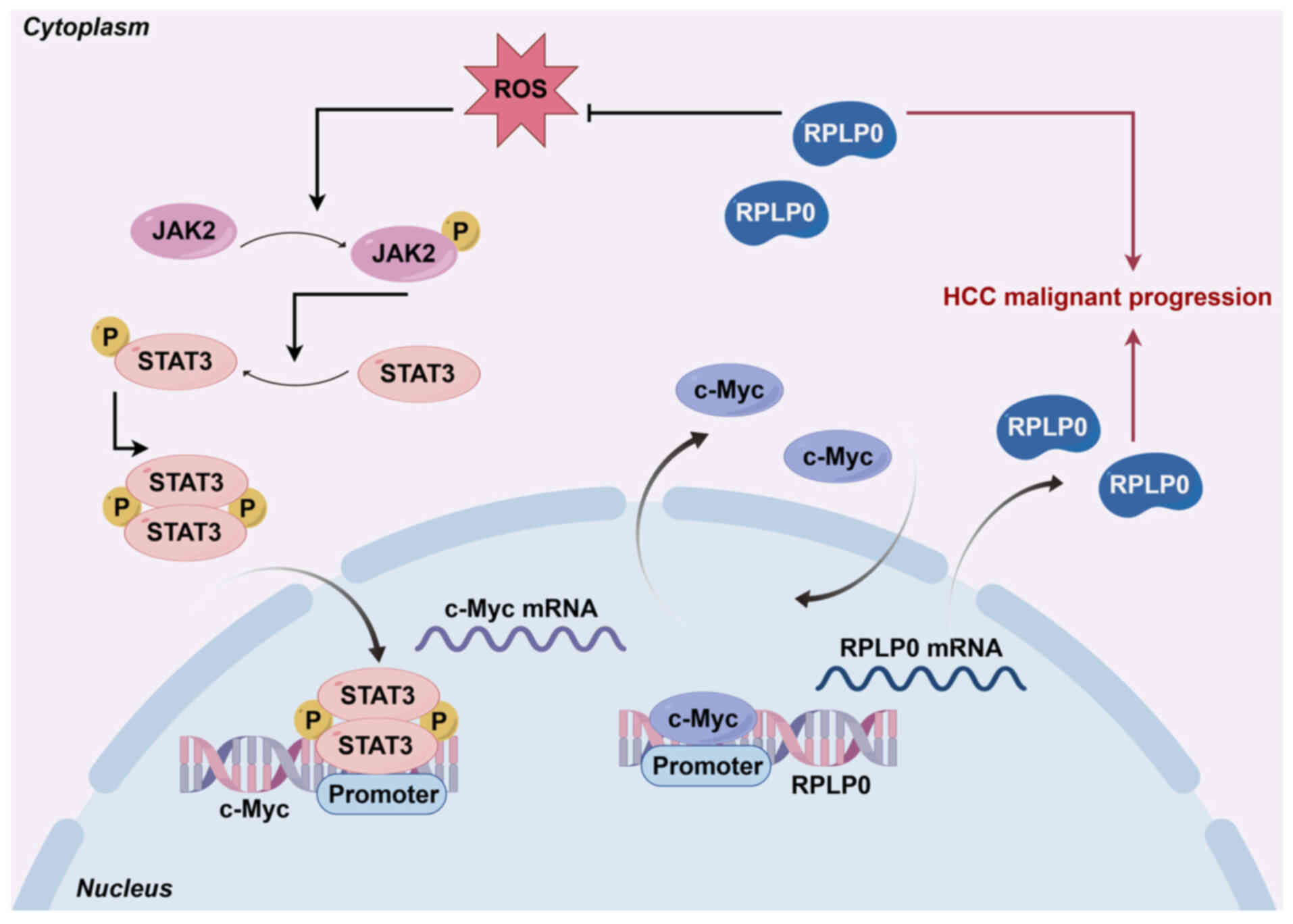
Collectively, these results indicate that RPLP0, regulated by
c-Myc, activates the JAK2/STAT3 signaling pathway by reducing ROS
levels, thereby facilitating the malignant progression of HCC
(Fig. 12).
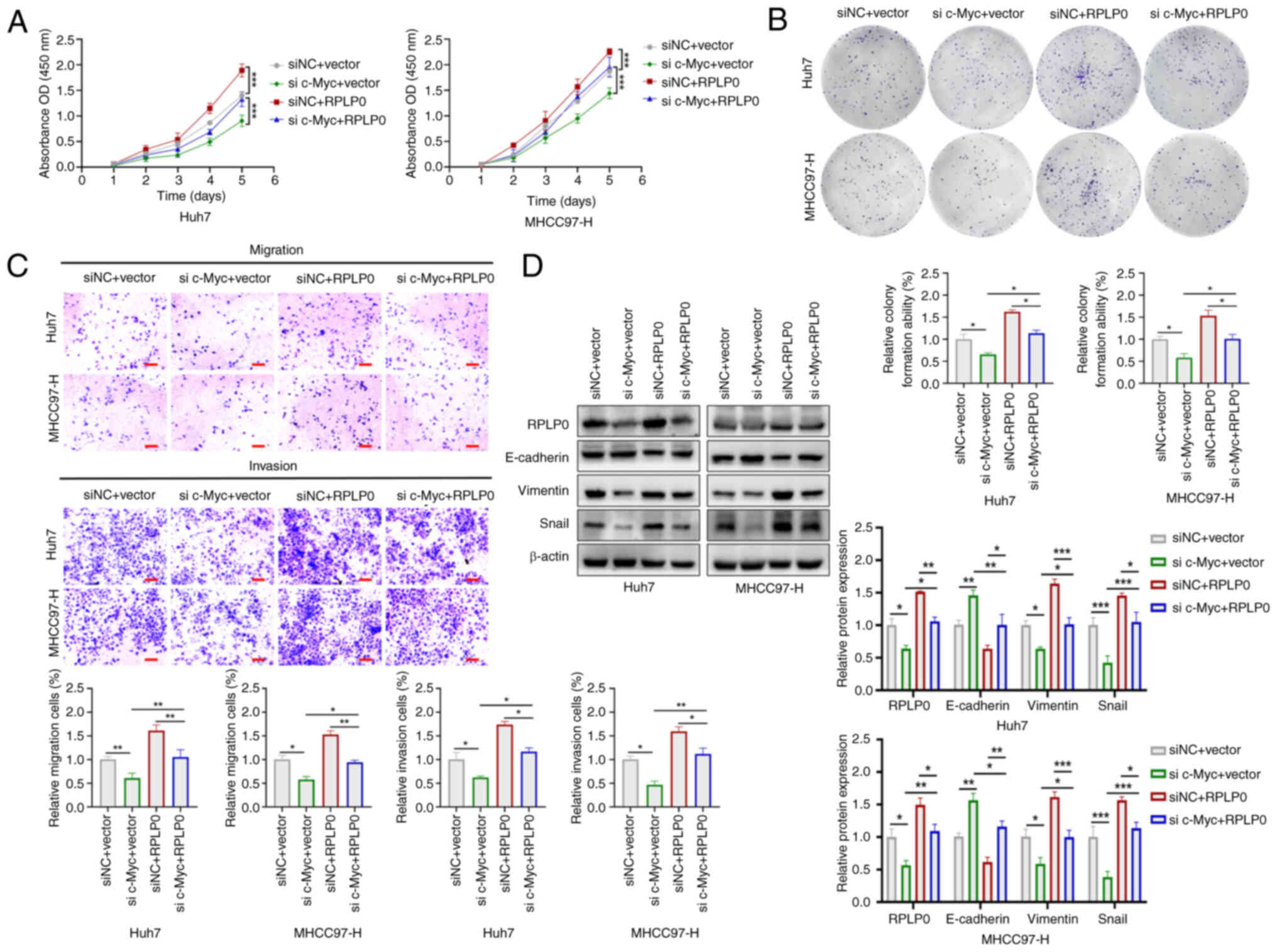 | Figure 10RPLP0 facilitates the proliferation
and metastasis of HCC cells through the regulation of c-Myc. (A)
CCK-8, (B) colony formation and (C) Transwell assays were used to
assess cell proliferation, migration and invasion (scale bar, 150
μm). (D) EMT-related protein levels, such as E-cadherin,
Vimentin and Snail, were examined in co-transfected HCC cells.
*P<0.05, **P<0.01,
***P<0.001. RPLP0, ribosomal protein lateral stalk
subunit P0; HCC, hepatocellular carcinoma; EMT,
epithelial-mesenchymal transition. |
Discussion
Oncogene activation and tumor suppressor gene
inactivation are critical determinants in the malignant progression
of HCC (32,33). The aberrant activation of
pro-carcinogenic genes can drive the abnormal proliferation and
malignant transformation of hepatocytes (34,35). Consequently, a pressing
requirement exists to discover potential biological markers for
diagnostic and prognostic evaluation. The present study
comprehensively analyzed bioinformatics databases, diverse HCC cell
lines and tissue samples and concluded that elevated RPLP0 levels
in HCC are closely linked to unfavorable patient prognosis. These
findings indicated that RPLP0 may function as a tumorigenic factor
in HCC progression. A series of functional experiments on HCC cells
showed that downregulating RPLP0 expression substantially hindered
cellular proliferation, invasion and migration capabilities and
induced G2/M cell cycle arrest, along with the
triggering of apoptotic and autophagic processes. The results of
analogous xenograft tumor experiments indicated that the knockdown
of RPLP0 effectively suppressed tumor growth.
Cellular autophagy can be induced by a range of
factors, such as organelle damage, nutritional deficiencies and
oxidative stress (36). ROS
functions as highly reactive oxygenates and their excessive
production leads to cell cycle arrest and mitochondria-dependent
apoptosis (37). The present
investigation revealed that the reduction of RPLP0 expression
facilitated the accumulation of ROS, supported by the results of
both GSEA analysis and DCFH-DA staining. In addition, the results
indicated that RPLP0 knockdown inhibited JAK2/STAT3 pathway
activation. Subsequently, in-depth investigations revealed that the
supplementation of NAC, following the knockdown of RPLP0,
time-dependently increased RPLP0, p-JAK2/JAK2 and p-STAT3/STAT3
levels, suggesting that RPLP0 silencing mediates JAK2/STAT3
signaling pathway inactivation by increasing ROS levels. Prior
investigations have identified a significant association between
elevated ROS concentrations and the suppression of the JAK2/STAT3
pathway, accompanied by increased autophagic activity (38-42). In the present study, RPLP0
knockdown resulted in increased ROS levels, which in turn repressed
JAK2/STAT3 pathway activation and stimulated autophagic processes,
consequently impeding the malignant progression of HCC.
Notably, both mRNA and protein levels of RPLP0
exhibited a time-dependent upregulation following the
administration of NAC at 12 and 24 h, suggesting that RPLP0 may be
regulated by transcription factors. Furthermore, the present study
demonstrated that NAC effectively counteracted the downregulation
in p-JAK2/JAK2 and p-STAT3/STAT3 expression induced by RPLP0
knockdown. Previous investigations have established that p-STAT3
possesses the ability to translocate into the nucleus, modulating
the transcription of various target genes, such as c-Myc, BCL-2,
Vimentin and MMP2/9, among others (43-45). It is worthwhile emphasizing that
the Cistrome Data Browser database was used to predict potential
transcription factors for RPLP0 and the results demonstrated that
c-Myc exhibited the highest relative score. An in-depth analysis of
data sourced from TCGA database, coupled with an examination of HCC
tissue samples, revealed a significant positive correlation between
the expression levels of RPLP0 and c-Myc. As a prevalent oncogene,
c-Myc facilitates the onset and progression of various tumors by
enhancing cellular growth and proliferation, modulating the
transcription of downstream target genes, inducing genomic
instability and reprogramming cellular metabolism (46-48). Subsequently, CHIP and dual
luciferase reporter assays revealed that c-Myc interacts with the
promoter sequence of RPLP0 to promote its transcription.
The present study demonstrated that RPLP0 knockdown
elevated intracellular ROS levels, thereby inducing autophagy.
However, the precise molecular mechanisms by which RPLP0 regulates
ROS generation remain unclear. Elucidating the upstream signals and
downstream effectors of RPLP0-mediated oxidative stress is a key
objective of our ongoing research.
In summary, the present study offered a new
perspective on the function of RPLP0 as a pro-oncogenic factor in
the progression of HCC. The results indicated that RPLP0 forms a
feedback loop with c-Myc via the ROS-mediated JAK2/STAT3 pathway.
Concurrently, c-Myc reciprocally activates RPLP0, thereby
perpetuating the circuit and contributing to the progression of
HCC. The findings of the present study provided innovative
perspectives on potential therapeutic approaches for the management
of HCC.
Supplementary Data
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
YQM and LBY conceived and designed the study. GRM,
HXH, and XBH performed most of the experiments, analyzed the data,
and wrote the manuscript. XPX and XDP supervised the entire
project. YQM and XDP confirm the authenticity of all raw data. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Nanchang University
approval no. (2024)CDYFYYLK (07-004) and all patients provided
informed consent. The animal experiments were also approved by the
animal ethics committee (approval no. CDYFY-IACUC-202407QR259).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors wish to express their sincere gratitude
to Professor Fanglin Zheng, Institute of Respiratory Diseases, The
First Affiliated Hospital, Jiangxi Medical College, Nanchang
University, for his invaluable advice.
Funding
The present study was supported by Jiangxi Kang-shen
Biotechnology Company (grant no. 1210702001), Wu Jieping Medical
Foundation (grant no. 320.6750.2024-13-32) and the Innovative and
Entrepreneurial Youth Talents Project of Jiangxi (grant no.
JXSQ2018106040).
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Wang X, Zhang L and Dong B: Molecular
mechanisms in MASLD/MASH-related HCC. Hepatology. 8:1303–1324.
2025. View Article : Google Scholar
|
|
3
|
Foerster F, Gairing SJ, Müller L and Galle
PR: NAFLD-driven HCC: Safety and efficacy of current and emerging
treatment options. J Hepatol. 76:446–457. 2022. View Article : Google Scholar
|
|
4
|
Toh MR, Wong EYT, Wong SH, Ng AWT, Loo LH,
Chow PK and Ngeow J: Global epidemiology and genetics of
hepatocellular carcinoma. Gastroenterology. 164:766–782. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Childs A, Aidoo-Micah G, Maini MK and
Meyer T: Immunotherapy for hepatocellular carcinoma. JHEP Rep.
6:1011302024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Medavaram S and Zhang Y: Emerging
therapies in advanced hepatocellular carcinoma. Exp Hematol Oncol.
7:172018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rimassa L, Pressiani T and Merle P:
Systemic treatment options in hepatocellular carcinoma. Liver
Cancer. 8:427–446. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Artero-Castro A, Castellvi J, García A,
Hernández J, Ramón Y, Cajal S and Lleonart ME: Expression of the
ribosomal proteins Rplp0, Rplp1, and Rplp2 in gynecologic tumors.
Hum Pathol. 42:194–203. 2011. View Article : Google Scholar
|
|
9
|
Artero-Castro A, Perez-Alea M, Feliciano
A, Leal JA, Genestar M, Castellvi J, Peg V, Ramón Y, Cajal S and
Lleonart ME: Disruption of the ribosomal P complex leads to
stress-induced autophagy. Autophagy. 11:1499–1519. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Teller A, Jechorek D, Hartig R, Adolf D,
Reißig K, Roessner A and Franke S: Dysregulation of apoptotic
signaling pathways by interaction of RPLP0 and cathepsin X/Z in
gastric cancer. Pathol Res Pract. 211:62–70. 2015. View Article : Google Scholar
|
|
11
|
Wang CH, Wang LK, Wu CC, Chen ML, Lee MC,
Lin YY and Tsai FM: The ribosomal protein RPLP0 mediates
PLAAT4-induced cell cycle arrest and cell apoptosis. Cell Biochem
Biophys. 77:253–260. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang YL, Zhao WW, Bai SM, Ma Y, Yin XK,
Feng LL, Zeng GD, Wang F, Feng WX, Zheng J, et al: DNA
damage-induced paraspeckle formation enhances DNA repair and tumor
radioresistance by recruiting ribosomal protein P0. Cell Death Dis.
13:7092022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller DR and Thorburn A: Autophagy and
organelle homeostasis in cancer. Dev Cell. 56:906–918. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Meng Y, Huang X, Zhang G, Fu S, Li Y, Song
J, Zhu Y, Xu X and Peng X: MicroRNA-450b-5p modulated RPLP0
promotes hepatocellular carcinoma progression via activating
JAK/STAT3 pathway. Transl Oncol. 50:1021502024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng P, Xu D, Cai Y, Zhu L, Xiao Q, Peng
W and Chen B: A multi-omic analysis reveals that Gamabufotalin
exerts anti-hepatocellular carcinoma effects by regulating amino
acid metabolism through targeting STAMBPL1. Phytomedicine.
135:1560942024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Qian J, Wang Q, Xiao L, Xiong W, Xian M,
Su P, Yang M, Zhang C, Li Y, Zhong L, et al: Development of
therapeutic monoclonal antibodies against DKK1 peptide-HLA-A2
complex to treat human cancers. J Immunother Cancer.
12:e0081452024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizushima N and Levine B: Autophagy in
human diseases. N Engl J Med. 383:1564–1576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan LS, Zhang SF, Luo G, Cheng BC, Zhang
C, Wang YW, Qiu XY, Zhou XH, Wang QG, Song XL, et al: Schisandrin B
mitigates hepatic steatosis and promotes fatty acid oxidation by
inducing autophagy through AMPK/mTOR signaling pathway. Metabolism.
131:1552002022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao YG, Codogno P and Zhang H: Machinery,
regulation and pathophysiological implications of autophagosome
maturation. Nat Rev Mol Cell Biol. 22:733–750. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miao CC, Hwang W, Chu LY, Yang LH, Ha CT,
Chen PY, Kuo MH, Lin SC, Yang YY, Chuang SE, et al: LC3A-mediated
autophagy regulates lung cancer cell plasticity. Autophagy.
18:921–934. 2022. View Article : Google Scholar :
|
|
24
|
Bradford HF, McDonnell TCR, Stewart A,
Skelton A, Ng J, Baig Z, Fraternali F, Dunn-Walters D, Isenberg DA,
Khan AR, et al: Thioredoxin is a metabolic rheostat controlling
regulatory B cells. Nat Immunol. 25:873–885. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ornatowski W, Lu Q, Yegambaram M, Garcia
AE, Zemskov EA, Maltepe E, Fineman JR, Wang T and Black SM: Complex
interplay between autophagy and oxidative stress in the development
of pulmonary disease. Redox Biol. 36:1016792020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Russell RC and Guan KL: The multifaceted
role of autophagy in cancer. EMBO J. 41:e1100312022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang R, Gao W, Wang Z, Jian H, Peng L, Yu
X, Xue P, Peng W, Li K and Zeng P: Polyphyllin I induced
ferroptosis to suppress the progression of hepatocellular carcinoma
through activation of the mitochondrial dysfunction via
Nrf2/HO-1/GPX4 axis. Phytomedicine. 122:1551352024. View Article : Google Scholar
|
|
28
|
Tsai HY, Bronner MP, March JK, Valentine
JF, Shroyer NF, Lai LA, Brentnall TA, Pan S and Chen R: Metabolic
targeting of NRF2 potentiates the efficacy of the TRAP1 inhibitor
G-TPP through reduction of ROS detoxification in colorectal cancer.
Cancer Lett. 549:2159152022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lai SY and Johnson FM: Defining the role
of the JAK-STAT pathway in head and neck and thoracic malignancies:
Implications for future therapeutic approaches. Drug Resist Updat.
13:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: new and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen L, Zhang C, Xue R, Liu M, Bai J, Bao
J, Wang Y, Jiang N, Li Z, Wang W, et al: Deep whole-genome analysis
of 494 hepatocellular carcinomas. Nature. 627:586–593. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ng CKY, Dazert E, Boldanova T,
Coto-Llerena M, Nuciforo S, Ercan C, Suslov A, Meier MA, Bock T,
Schmidt A, et al: Integrative proteogenomic characterization of
hepatocellular carcinoma across etiologies and stages. Nat Commu.
13:24362022. View Article : Google Scholar
|
|
34
|
Calderaro J, Ziol M, Paradis V and
Zucman-Rossi J: Molecular and histological correlations in liver
cancer. J Hepatol. 71:616–630. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Villanueva A: Hepatocellular carcinoma. N
Engl J Med. 380:1450–1462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li X, He S and Ma B: Autophagy and
autophagy-related proteins in cancer. Mol Cancer. 19:122020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan J, Ren D, Wang J, Liu X, Zhang H, Wu M
and Yang G: Bruceine D induces lung cancer cell apoptosis and
autophagy via the ROS/MAPK signaling pathway in vitro and in vivo.
Cell Death Dis. 11:1262020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cao Y, Wang J, Tian H and Fu GH:
Mitochondrial ROS accumulation inhibiting JAK2/STAT3 pathway is a
critical modulator of CYT997-induced autophagy and apoptosis in
gastric cancer. J Exp Clin Cancer Res. 39:1192020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim J, Park A, Hwang J, Zhao X, Kwak J,
Kim HW, Ku M, Yang J, Kim TI, Jeong KS, et al: KS10076, a chelator
for redox-active metal ions, induces ROS-mediated STAT3 degradation
in autophagic cell death and eliminates ALDH1+ stem cells. Cell
Rep. 40:1110772022. View Article : Google Scholar
|
|
40
|
Liang JR and Yang H: Ginkgolic acid (GA)
suppresses gastric cancer growth by inducing apoptosis and
suppressing STAT3/JAK2 signaling regulated by ROS. Biomed
Pharmacother. 125:1095852020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhao Z, Wang Y, Gong Y, Wang X, Zhang L,
Zhao H, Li J, Zhu J, Huang X, Zhao C, et al: Celastrol elicits
antitumor effects by inhibiting the STAT3 pathway through ROS
accumulation in non-small cell lung cancer. J Transl Med.
20:5252022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou H, Li J, He Y, Xia X, Liu J and Xiong
H: SLC25A17 inhibits autophagy to promote triple-negative breast
cancer tumorigenesis by ROS-mediated JAK2/STAT3 signaling pathway.
Cancer Cell Int. 24:852024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garg M, Shanmugam MK, Bhardwaj V, Goel A,
Gupta R, Sharma A, Baligar P, Kumar AP, Goh BC, Wang L and Sethi G:
The pleiotropic role of transcription factor STAT3 in oncogenesis
and its targeting through natural products for cancer prevention
and therapy. Med Res Rev. December 1–2020.Epub ahead of print.
PubMed/NCBI
|
|
44
|
Hu Y, Dong Z and Liu K: Unraveling the
complexity of STAT3 in cancer: Molecular understanding and drug
discovery. J Exp Clin Cancer Res. 43:232024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zou S, Tong Q, Liu B, Huang W, Tian Y and
Fu X: Targeting STAT3 in cancer immunotherapy. Mol Cancer.
19:1452020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dhanasekaran R, Deutzmann A,
Mahauad-Fernandez WD, Hansen AS, Gouw AM and Felsher DW: The MYC
oncogene-the grand orchestrator of cancer growth and immune
evasion. Nat Rev Clin Oncol. 19:23–36. 2022. View Article : Google Scholar
|
|
47
|
Duffy MJ, O'Grady S, Tang M and Crown J:
MYC as a target for cancer treatment. Cancer Treat Rev.
94:1021542021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lourenco C, Resetca D, Redel C, Lin P,
MacDonald AS, Ciaccio R, Kenney TMG, Wei Y, Andrews DW, Sunnerhagen
M, et al: MYC protein interactors in gene transcription and cancer.
Nat Rev Cancer. 21:579–591. 2021. View Article : Google Scholar : PubMed/NCBI
|