Introduction
Pancreatic cancer stands among the leading causes of
cancer-related mortality globally, with its incidence having more
than doubled over the past 25 years. Predominant in regions such as
North America, Europe and Australia, pancreatic cancer accounts for
over 90% of cases classified as pancreatic ductal adenocarcinomas,
which exhibit an extremely poor prognosis; boasting an overall
5-year survival rate of merely ~9% (1). This dismal outcome is largely
attributed to late-stage diagnosis and resistance to current
therapies, underscoring the urgent need to unravel the underlying
mechanisms driving disease progression.
A key biological process implicated in pancreatic
cancer pathogenesis is the epithelial-mesenchymal transition (EMT),
whereby epithelial cells lose characteristic features such as
polarity and tight junctions, while acquiring mesenchymal traits
including spindle-shaped morphology and enhanced migratory
capacity. This dynamic shift occurs along a spectrum of
intermediate states rather than as an abrupt switch and plays a
pivotal role in embryonic development, fibrotic disorders and tumor
progression (2). Functionally,
EMT is categorized into three subtypes: Type 1, involved in
embryogenesis and organ development without pathological
consequences; Type 2, driven by inflammatory injury and
contributing to tissue fibrosis; and Type 3, which occurs in
neoplastic cells and directly facilitates cancer invasion and
metastasis (3).
The aggressive biology of pancreatic cancer,
characterized by early invasion, metastasis and chemoresistance,
stems partly from such EMT-mediated processes, which contribute to
its notoriously late diagnosis and dismal prognosis (4). Established risk factors (including
smoking, obesity, diabetes and genetic mutations such as BRCA2)
exert their pathogenic effects through pathways such as chronic
inflammation and metabolic disorders, which in turn drive EMT
activation (4). Notably,
modifiable risk factors account for 65.6% of cases in patients aged
≤60 years, markedly higher than the 17.2% in older cohorts
(5). This disparity highlights
the potential of targeting EMT pathways regulated by these
modifiable factors for early intervention, thereby providing a
strategy to address the 'advanced-stage diagnosis' dilemma that
plagues pancreatic cancer management.
The present review systematically elaborates on the
molecular mechanisms governing EMT in pancreatic cancer, dissect
targeted therapeutic strategies and discuss challenges in clinical
translation. By synthesizing these insights, it aimed to provide a
theoretical framework for the development of novel therapeutic
regimens.
Multidimensional mechanisms of EMT driving
the malignant phenotype of pancreatic cancer
EMT is a dynamic and reversible cellular process
that plays a central role in driving the unique pathophysiology of
pancreatic ductal adenocarcinoma (PDAC). Mutant Kirsten rat sarcoma
virus oncogene homologue (KRAS), an oncogenic driver, serves as
central driving force in pancreatic ductal adenocarcinoma. Its
sustained signaling propels crucial malignant programs, including
EMT, via downstream pathways such as MAPK and PI3K. Specifically,
it upregulates transcription factors such as Snail and ZEB and
synergizes with signals such as transforming growth factor-β
(TGF-β) to suppress epithelial markers and promote a mesenchymal
phenotype, thereby endowing cancer cells with invasive and
metastatic capabilities (6).
Unlike a number of other solid tumors, PDAC is characterized by an
extremely dense desmoplastic reaction; a microenvironment rich in
extracellular matrix (ECM) and stromal cells. This microenvironment
not only acts as a physical barrier to tumor progression but also
serves as a key signaling hub for inducing and maintaining EMT
(7). In PDAC, EMT does not
involve a simple binary switch between epithelial and mesenchymal
phenotypes; instead, it is a continuous process encompassing
multiple stable intermediate states (that is, partial EMT or hybrid
E/M states), in which cells co-express both epithelial and
mesenchymal markers. This phenotypic plasticity is critical to the
malignant behavior of PDAC, endowing cancer cells with advantages
in invasion, metastasis and tumor initiation (8). These effects are triggered by
TME-derived signals [such as TGF-β and hepatocyte growth factor
(HGF)], which activate a regulatory cascade involving EMT
transcription factors (EMT-TFs) such as Snail and ZEB1: On one
hand, enhancing cytoskeletal remodeling and matrix degradation to
promote invasion and metastasis; on the other hand, activating stem
cell signaling pathways (such as Notch and Wnt) and upregulating
drug resistance-related molecules [such as ATP-Binding Cassette
Subfamily G Member 2 (ABCG2)] to confer stemness and treatment
resistance. Collectively, these processes systematically drive the
malignant progression of PDAC (9,10).
At the molecular level, TGF-β in the TME activates transcription
factors of the Snail and ZEB families via Sma and MAD homologs
(SMAD)-dependent and non-SMAD pathways [such as
phosphatidylinositol 3-kinase (PI3K) and mitogen-activated protein
kinase (MAPK)], while HGF induces the expression of factors such as
Slug via extracellular signal-regulated kinase (ERK) signaling
mediated by the c-mesenchymal-epithelial transition (MET) receptor.
These core EMT-TFs [Snail, Slug, Zinc finger E-box-binding homeobox
1 (ZEB1/2), Twist] synergistically suppress epithelial markers
(such as E-cadherin and Claudins) and activate mesenchymal
molecules (such as Vimentin and N-cadherin), driving plastic
phenotypic transitions in cells (2,11).
By remodeling cell identity, these transcription factors enable
PDAC cells to detach from the original ductal structure, invade the
dense stroma, enter the circulatory system and form lethal
metastatic lesions in distant organs (12,13).
Core programs of EMT-regulated invasion
and metastasis
The most lethal feature of PDAC is the early onset
of invasion and metastasis; even at the pre-cancerous pancreatic
intraepithelial neoplasia (PanIN) stage, TME-derived signals such
as TGF-β and HGF can induce EMT. EMT drives cells to detach from
ductal structures, degrade the dense stroma and enter the
circulatory system, initiating the metastatic cascade and serving
as the core engine of this multi-step process (14). In this process, EMT not only
reshapes the cellular phenotype in response to TME signals but also
endows cells with migratory and invasive capabilities, laying the
foundation for the early metastasis of PDAC (14). Ultimately, EMT provides PDAC cells
(originally adherent to ductal structures) with a complete toolkit
to break free from constraints and cross the dense stromal barrier
unique to PDAC.
Disruption of cell adhesion and
acquisition of motility
A classic hallmark of EMT is the downregulation or
loss of function of E-cadherin, a core component of adherens
junctions between epithelial cells that maintains junction
stability through binding to p120. The loss of E-cadherin disrupts
intercellular junctions in pancreatic ductal epithelial cells,
relieves inhibition of Rho family GTPases and enables cells to
acquire individualistic characteristics and detach from the primary
tumor, representing the critical first step in initiating
metastasis (15). In PDAC,
EMT-TFs (particularly SNAIL1 and ZEB1) activated by TME factors
such as TGF-β can directly bind to the promoter of the CDH1 gene
(which encodes E-cadherin). By recruiting epigenetic modification
complexes [such as histone deacetylases (HDACs) and DNA
methyltransferases (DNMTs)], these TFs strongly and stably repress
CDH1 transcription (16,17). This breakage of the adhesion chain
allows cancer cells to escape from the primary tumor mass, creating
a prerequisite for invasion (18). Concurrently, EMT is often
accompanied by cadherin switching: While inhibiting CDH1
expression, EMT-TFs upregulate N-cadherin (CDH2) expression,
thereby achieving functional substitution between the two
cadherins. This switch is not a simple replacement of expression
patterns but rather remodels cell adhesion properties; shifting
from E-cadherin-mediated homophilic adhesion between epithelial
cells to N-cadherin-mediated heterophilic adhesion with stromal
cells [such as cancer-associated fibroblasts (CAFs)] and activating
the α-ctenin/vinculin-FAK/Src signaling pathway to enhance directed
adhesion strength. laying the molecular foundation for subsequent
interactions with stromal cells (19). In the PDAC TME, CAFs highly
express N-cadherin, which forms mechanically active heterotypic
adhesions with residual E-cadherin on the surface of
EMT-experienced cancer cells. Through α-catenin/vinculin-mediated
force transmission, this interaction activates the focal adhesion
kinase (FAK)/Src signaling pathway to enhance directed migration.
Meanwhile, this stable interaction provides mechanical support and
survival signals for cancer cells to traverse the dense stroma,
helping them adapt to TME stress (20).
Alongside changes in cell adhesion patterns, EMT
drives cytoskeletal remodeling to meet migratory demands via a
TGF-β-mediated alternative splicing regulatory network: alternative
splicing variants of genes such as ARHGEF11 and CTTN activate Rho
family GTPase signaling, prompting the reorganization of actin from
a cortical network (which maintains polarity) into stress fibers
that traverse the cell. This structural transition provides the
mechanical basis for directed cell movement (21). These stress fibers anchor to the
ECM via integrin-mediated focal adhesions and myosin contraction
generates an intracellular tension gradient. This gradient, in
coordination with traction forces transmitted by integrin-focal
adhesions, enables cells to overcome the resistance of the dense
stroma. The precise regulation of polarized reorganization of the
actin cytoskeleton and dynamic balance of focal adhesions
ultimately achieves morphological adaptation to the
mesenchymal-like migration mode (22). Among Rho family GTPases (key
molecules regulating cell functions), RhoA and Rac1 show opposite
activity changes during EMT: Rac1 activity increases markedly,
while RhoA activity decreases. These two enzymes regulate the
contractility and stiffness of the actin cortex (a structure
maintaining cell shape) with the cell cycle; in the interphase
(when cells do not divide), they reduce cortical tension to adapt
to shape changes caused by the environment; in mitosis (when cells
divide), they enhance cortical mechanical strength to support cell
shape adjustment. This regulation enables the mechanical transition
of the cytoskeleton to accurately match EMT-related phenotypes
(23,24).
Degradation of extracellular matrix and
creation of invasive pathways
Another pathological challenge of PDAC is its
abnormally dense fibrotic stroma, primarily composed of type I, III
and IV collagen, fibronectin and hyaluronan. This stroma is
synergistically deposited by tissue-resident macrophages of
embryonic origin, activated pancreatic stellate cells (PSCs) and
CAFs, driven by TGF-β and matrix stiffness via FAK signaling,
forming a robust physical barrier (8,25,26). To breach this barrier for invasion
and intravasation, cancer cells must actively degrade ECM
components. Notably, the ECM in PDAC exhibits a paradoxical role:
Although it is produced via synergistic deposition by pancreatic
stellate cells (PSCs), cancer-associated fibroblasts CAFs and other
cells, induced by tumor stimuli in the tumor microenvironment (such
as TGF-β signaling, matrix stiffness), it forms a physical barrier
that hinders the initial spread of tumors. Therefore, cancer cells
need to overcome this barrier through EMT: EMT-related
transcription factors (such as Snail, ZEB1) recruit co-activators
with histone acetyltransferase activity (such as CBP/p300), thereby
activating the expression of proteolytic enzymes such as matrix
metalloproteinases (MMPs). These enzymes can specifically degrade
ECM components such as collagen, creating pathways for cancer cell
invasion (26). The MMP family
plays a central role in this process: via MMP-14-mediated cascade
activation (such as activating MMP-2 and MMP-9), combined with
spatiotemporally specific degradation regulated by integrin/FAK
signaling, MMPs act as key effectors for breaking through the dense
stromal barrier (27). MMP-14 is
particularly critical: it anchors to the cell membrane via its
transmembrane domain and palmitoylation of Cys574 in its
cytoplasmic tail further stabilizes membrane localization.
Trafficking mediated by the LLY573 motif targets MMP-14 precisely
to invadopodia; meanwhile, its homodimerization enhances
collagenase activity, efficiently cleaving type I collagen. In
coordination with the spatiotemporal degradation pattern regulated
by FAK-p130cas signaling, MMP-14 creates invasive channels for
cells (28).
More importantly, in the complex TME of PDAC, the
EMT program establishes a destructive synergistic relationship
between cancer cells and stromal cells: EMT-experienced cancer
cells secrete TGF-β and platelet-derived growth factor (PDGF),
which activate the transformation of PSCs into CAFs via the Smad
pathway and MAPK/ERK signaling, respectively. Activated CAFs, in
turn, become the primary source of matrix-degrading enzymes through
high MMP expression (29,30). This bidirectional crosstalk forms
a malignant positive feedback loop: EMT cells induce a CAF-mediated
fibrotic TME via secretion of TGF-β, while excessive TGF-β secreted
by CAFs in this microenvironment further reinforces the EMT
phenotype via the SMAD pathway and non-SMAD signals (such as
MAPK/ERK). Simultaneously, mechanical tension generated by ECM
remodeling amplifies invasion signals via integrin/FAK signaling,
paving the way for collective invasion of tumor cells (31). Thus, EMT endows cancer cells with
autonomous motility and autophagy-regulated matrix degradation
capabilities. Through synergistic interactions with CAFs (via
TGF-β/SMAD and MAPK/ERK signaling), EMT drives PDAC progression
from in situ carcinoma to invasive cancer. Additionally, via
autophagy-dependent exosome release and immune evasion (to avoid
immune surveillance), EMT ultimately promotes efficient metastasis
to distant organs such as the liver (32).
Following EMT-mediated invasion through the dense
stroma, cancer cells further acquire the ability to intravasate
into the vascular system. This process is facilitated by
EMT-induced upregulation of MMPs (such as MMP-14) that degrade
vascular basement membrane components, as well as enhanced
interactions with endothelial cells via N-cadherin-mediated
adhesion (20,28). EMT thus acts as a key driver
linking tumor detachment, stromal invasion and vascular entry,
laying the foundation for subsequent metastatic dissemination.
EMT is the core engine driving PDAC invasion and
metastasis. TGF-β-induced E-cadherin suppression (via SNAIL1/ZEB1)
and N-cadherin upregulation enable adhesion switching, conferring
migratory capacity. Concurrently, EMT activates MMP-14 to degrade
collagen and pave invasion routes. Crucially, EMT and CAFs form a
TGF-β-driven positive feedback loop: EMT cells activate CAFs, which
secrete TGF-β to reinforce EMT, collectively breaching the dense
stroma and enabling metastasis (Fig.
1).
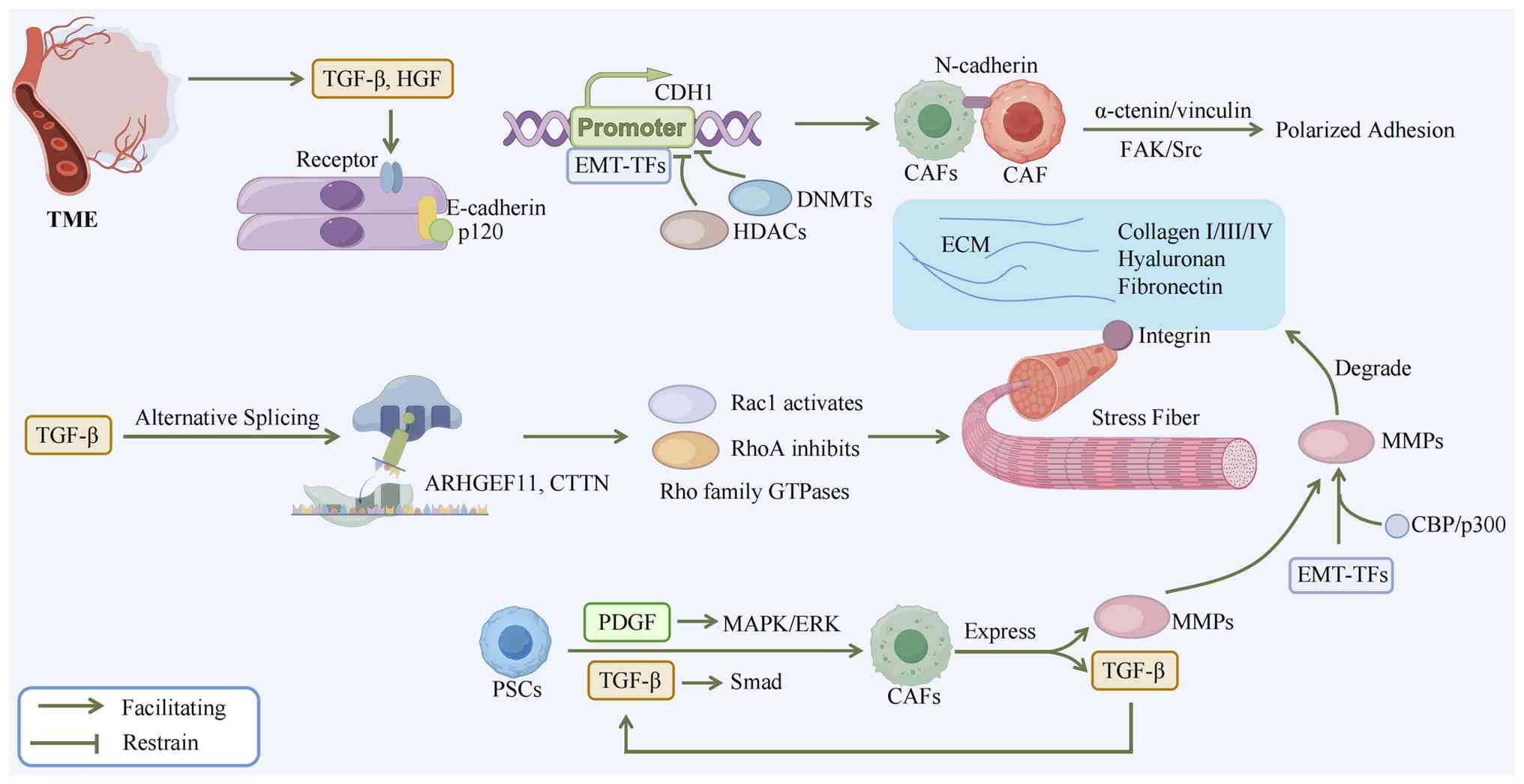 | Figure 1Mechanism of TME-induced EMT and the
CAF-cancer cell feedback loop in PDAC progression. During the PanIN
stage, signals derived from the TME, such as TGF-β and HGF,
activate the EMT program via their cognate receptors. As a core
component of epithelial adherens junctions, E-cadherin maintains
junctional stability by binding to p120 catenin. The core executors
of EMT-TFs bind to the promoter region of the CDH1 gene and recruit
epigenetic modification complexes (such as HDACs, DNMTs) to
suppress the expression of the epithelial marker E-cadherin, while
simultaneously upregulating the mesenchymal marker N-cadherin. This
'cadherin switching' liberates cancer cells from their original
cell-cell connections, allowing them to anchor to the stroma via
heterotypic adhesion with CAFs and enhances directed migration by
activating the α-catenin/vinculin-FAK/Src signaling pathway.
Concurrently, TGF-β signaling modulates the activity of Rho GTPases
(such as activating Rac1, inhibiting RhoA) by mediating the
alternative splicing of genes such as ARHGEF11and CTTN. This drives
cytoskeletal remodeling and stress fiber formation. The
intracellular tension gradient generated by myosin contraction,
coordinated with traction forces transmitted via integrin-mediated
focal adhesions, helps cells overcome stromal resistance, thereby
powering cell migration. To pave the way for invasion,
EMT-transformed cancer cells secrete factors such as TGF-β and
PDGF, which induce the transformation of PSCs into CAFs via the
SMAD pathway and MAPK/ERK signaling, respectively. Activated CAFs
not only highly express matrix metalloproteinases, becoming a major
source of matrix-degrading enzymes, but also secrete excess TGF-β.
This excess TGF-β further reinforces the EMT phenotype in cancer
cells through both SMAD and non-SMAD pathways (such as MAPK/ERK).
Simultaneously, mechanical tension generated by ECM remodeling
amplifies invasion signals via integrin/FAK signaling, ultimately
forming a malignant positive-feedback loop that drives progressive
invasion. TME, tumor microenvironment; EMT, epithelial-mesenchymal
transition; CAF, cancer-associated fibroblast; PDAC, pancreatic
ductal adenocarcinoma; PanIN, precancerous pancreatic
intraepithelial neoplasia; TGF-β, transforming growth factor-β;
HGF, hepatocyte growth factor; EMT-TFs, EMT-EMT transcription
factors; HDACs, histone deacetylases; DNMTs, DNA
methyltransferases; FAK, focal adhesion kinase; PDGF,
platelet-derived growth factor; PSCs, pancreatic stellate
cells. |
Role of EMT/MET plasticity in circulating
tumor cell colonization
After intravasation, EMT continues to regulate the
survival and metastatic potential of circulating tumor cells (CTCs)
during circulation and mediates successful colonization at distant
organs via mesenchymal-epithelial transition (MET) reversal.
After PDAC cells invade the vascular system, they
disseminate in the blood or lymphatic fluid as CTCs, the essential
carriers of distant metastasis. The pre-metastatic precursor cell
properties enriched in CTCs have been confirmed in PDAC studies
(33,34). The harsh environment of the
circulatory system requires CTCs to resist blood shear stress,
activate the YAP1 pathway via platelets to resist anoikis and evade
immune surveillance with the help of neutrophil extracellular traps
(33). EMT enhances the survival
and metastatic capacity of CTCs by downregulating epithelial
markers (such as EpCAM), upregulating mesenchymal phenotypes (such
as Vimentin) and activating the JNK signaling pathway, making it a
key mechanism for CTCs to adapt to the circulatory environment
(33,35).
Studies have found that CTCs in the peripheral blood
of PDAC patients often exhibit varying degrees of EMT
characteristics and their phenotypic heterogeneity is associated
with TGFβ/Smad pathway activation and Snail-mediated downregulation
of epithelial markers (36,37). Although cells that have fully
undergone EMT exhibit enhanced motility via the RhoA/ROCK pathway:
Activation of this pathway regulates actin filament contraction and
focal adhesion dynamic assembly, thereby driving cytoskeletal
reorganization to promote migration, while their single-cell state
makes them more vulnerable to damage from blood shear stress and
attack by natural killer (NK) cells (37,38). In recent years, the 'partial' EMT
or 'hybrid' EMT state has been confirmed to be the phenotype with
the highest metastatic efficiency: such cells acquire mesenchymal
properties via Twist1/MAPK pathway activation to resist apoptosis,
while retaining E-cadherin-mediated cell junctions; their
plasticity is maintained by the Notch-Jagged1 signaling pathway,
which sustains intercellular fate consistency through lateral
induction (38).
Cells in this state achieve dual functions through
unique phenotypic plasticity: On the one hand, they acquire
mesenchymal characteristics (such as enhanced motility and
anti-apoptotic potential) via EMT, enabling them to detach from the
primary tumor and survive in the circulation (39); on the other hand, the retained
E-cadherin mediates intercellular junctions, providing a structural
basis for the formation of CTC clusters, which have markedly higher
metastatic potential and circulatory survival rate than single CTCs
(40,41).
CTC clusters enhance metastatic efficiency through
synergistic mechanisms: Their aggregate structure, together with
protective microthrombi formed by platelets, reduces direct damage
to internal cells from fluid shear stress; meanwhile, by expressing
molecules such as PD-L1 and CD47, CTC clusters inhibit T-cell
function via the PD-1/PD-L1 signaling axis and block phagocytosis
through the interaction between CD47 and macrophage SIRPα, thereby
reducing the probability of elimination by immune surveillance
(42); simultaneously,
intercellular signal communication within CTC clusters (such as
E-cadherin-mediated adhesion and Notch-Jagged1 signaling
maintaining phenotypic plasticity) and interactions with platelets
(such as TGF-β released by platelets activating Smad pathway to
sustain mesenchymal properties and ADP promoting TGF-β release via
platelet P2Y12 receptor) further enhance their survival capacity,
making them more likely to be retained and colonized in distal
capillaries (43,44).
Resisting anoikis is critical for CTC survival and
EMT endows this capacity by activating pathways such as PI3K/Akt
and MEK/ERK. For example, TWIST1 can upregulate Akt phosphorylation
to inhibit the apoptotic program triggered by matrix detachment,
while Snail enhances cell survival by regulating the expression of
the anti-apoptotic protein Bcl-2 (45,46). Additionally, mesenchymal
phenotype-related receptors (such as integrins) can bind to
platelets or soluble matrix proteins (such as PDGF and VEGF) in the
circulation, forming a protective microenvironment that further
reduces the risk of apoptosis and lays the foundation for
metastasis (47,48).
The final step of the metastatic cascade, and the
key to the formation of metastatic lesions, is the successful
colonization of CTCs in distant organs. To proliferate in the new
microenvironment and form macroscopically visible secondary tumors,
disseminated cancer cells must undergo MET, the reverse process of
EMT. This process restores epithelial cell polarity and
intercellular adhesion capacity, enabling cells to integrate into
new tissues and proliferate in an orderly manner (49,50). This phenotypic plasticity depends
not only on the dynamic regulation of transcription factors (such
as Snail and Twist) but also on epigenetic reprogramming (such as
histone modification and DNA methylation) and the regulation of
mRNA stability by RNA-binding proteins (51,52). For example, lncRNA H19 maintains
the balance of cell plasticity during EMT/MET switching by sponging
miR-200b/c and let-7b (49).
The core of phenotypic plasticity lies in the
dynamic adaptation to the pressure of the metastatic
microenvironment: sustained TGF-β signals in the primary tumor
induce an EMT-mediated mesenchymal phenotype (that is, a phenotype
with low E-cadherin expression and high vimentin expression) in
CTCs, enabling them to resist shear stress and immune attack in the
circulation; when CTCs reach the metastatic site, epithelial
differentiation signals (such as BMPs) in the microenvironment can
activate the SMAD pathway, triggering MET to rebuild epithelial
structures and support colonization and proliferation (53,54). This bidirectional switching
mechanism allows cancer cells to resist shear stress and immune
attack in the circulation with a mesenchymal phenotype and rebuild
epithelial structures via MET to support proliferation during
colonization. Histological analysis of PDAC liver metastases
confirms that metastatic tumor cells transmit the CD44v6/C1QBP
complex via exosomes, remodeling the hepatic fibrotic
microenvironment and maintaining an epithelial-like differentiated
state, providing direct evidence for the occurrence of MET
(55). In summary, from driving
invasion of the primary tumor, to endowing treatment resistance and
stem cell phenotypes and finally to ensuring the circulatory
survival of CTCs and their eventual distant colonization, EMT and
its reverse process MET together form a core axis the entire
metastatic process of PDAC. This remarkable cellular plasticity
allows PDAC cells to flexibly adapt to various environmental
pressures, making it one of the fundamental reasons for the
difficulty in radical treatment-and thus an extremely attractive
therapeutic target (Fig. 2).
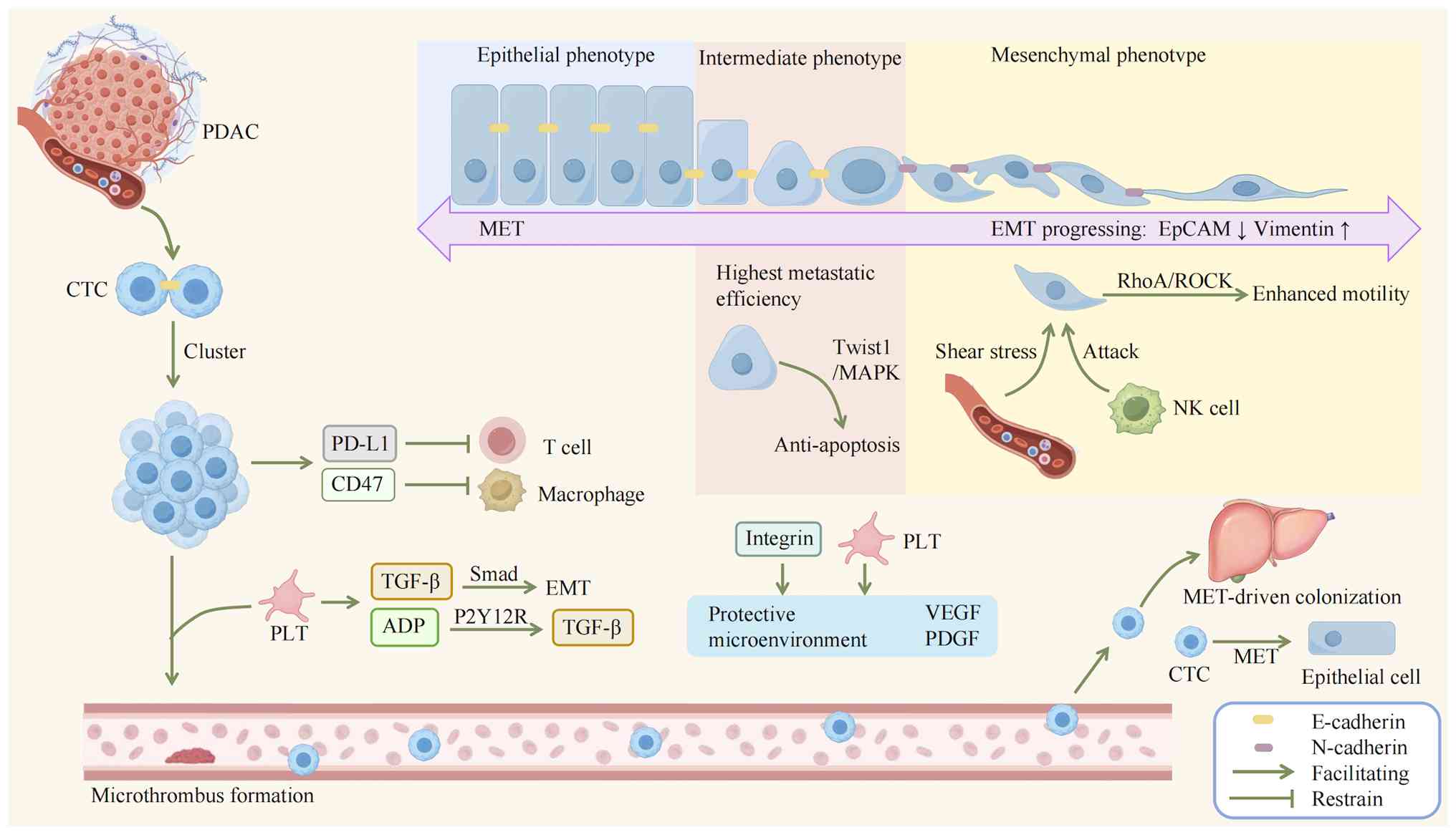 | Figure 2Mechanisms of circulating tumor cell
survival and metastasis in PDAC. After detaching from the primary
tumor and entering the bloodstream as CTCs, PDAC cells exhibit
distinct metastatic behaviors: cells undergoing complete EMT,
despite gaining enhanced motility via the RhoA/ROCK pathway, are
vulnerable as single cells to shear stress and NK cell-mediated
attacks. In contrast, the 'partial'mixed EMT/ phenotype
demonstrates the highest metastatic efficiency, acquiring
anti-apoptotic capabilities through the Twist1/MAPK pathway. CTCs
retaining E-cadherin can form clusters, which markedly enhance
circulatory survival and metastatic potential via multidimensional
cooperative mechanisms. These aggregates, shielded by platelet-rich
microthrombi, reduce direct shear stress damage and facilitate
immune evasion through PD-L1 expression (suppressing T cells) and
CD47 expression (blocking macrophage phagocytosis). Interactions
with platelets, such as TGF-β release activating SMAD signaling and
ADP-P2Y12 receptor engagement promoting further TGF-β secretion,
augment survival, while adhesion via integrins to platelets or
soluble matrix proteins (such as PDGF and VEGF) creates a
protective microenvironment that minimizes apoptosis. The final
critical step in the metastatic cascade, successful colonization at
distant organs, hinges on MET, which reestablishes epithelial
polarity and intercellular adhesion capabilities. PDAC, pancreatic
ductal adenocarcinoma; CTCs, circulating tumor cells; EMT,
epithelial-mesenchymal transition; MAPK, mitogen-activated protein
kinase; PD-L1, programmed cell death ligand 1; PDGF,
platelet-derived growth factor; VEGF, vascular endothelial growth
factor; MET, mesenchymal-epithelial transition. |
Mechanisms of the EMT-stemness-therapy
resistance axis
Beyond driving the entire metastatic cascade
(detachment, invasion, intravasation, circulation and
colonization), EMT also plays a pivotal role in mediating therapy
resistance in PDAC; this mechanism is closely linked to EMT-induced
stemness acquisition and phenotypic plasticity, independent of
metastatic localization.
PDAC exhibits extreme resistance to current
chemotherapy (such as gemcitabine and oxaliplatin) and
radiotherapy, which contributes to its high mortality. Accumulating
evidence indicates that extracellular vesicles derived from CAFs
transmit molecules such as miR-146a (which activates Snail) and
circFARP1 [which activates the LIF/signal transducer and activator
of transcription 3 (STAT3) axis], mediating a mutually reinforcing
vicious cycle among the EMT program, acquisition of CSC properties
and treatment resistance, collectively forming the biological basis
for PDAC treatment failure (33,56,57).
The EMT program endows cancer cells with
stem-like properties
Cancer stem cells (CSCs) are subpopulation of tumor
cells with self-renewal capacity and multi-lineage differentiation
potential. They maintain stemness by activating signaling pathways
such as WNT/β-Catenin, Notch and TGF-β and serve as core drivers of
tumor initiation, recurrence and metastasis (58,59). In PDAC, cell subpopulations
expressing CD44+/CD24+/EpCAM+,
CD133+, or high aldehyde dehydrogenase 1 (ALDH1)
activity have been confirmed to be enriched in CSCs. Among these
markers, CD133 expression is regulated by hypoxia-inducible factor
1α (HIF-1α), CD44 maintains stemness via the WNT/β-Catenin pathway
and high ALDH1 activity is associated with the antioxidant
phenotype and drug resistance of CSCs (60,61). A breakthrough finding is that in
PDAC cells, TGF-β induces EMT via the Smad pathway, activating
transcription factors such as Snail and ZEB1 and enabling
differentiated non-stem cancer cells to acquire CSC
characteristics; conversely, knockout of these EMT-TFs markedly
impairs the self-renewal capacity and in vivo tumorigenicity
of CSCs by inhibiting stemness-maintaining pathways such as
WNT/β-Catenin (58,62).
Among EMT-TFs, ZEB1 plays a core integrative role:
the bidirectional negative feedback loop formed between ZEB1 and
the miR-200 family not only drives EMT phenotypic switching but
also maintains cell stemness by stabilizing the expression of the
stem cell factor Sox2; meanwhile, the direct interaction between
ZEB1 and YAP1 can respond to TGF-β signals to activate downstream
target genes, thereby coordinately regulating stem cell
pluripotency (63). The key
mechanism lies in the transcriptional inhibition of
stemness-suppressing microRNAs (such as the miR-200 family) by
ZEB1/2; in turn, the miR-200 family forms a mutually inhibitory
feedback loop by targeting the 3' untranslated region (3'UTR) of
ZEB1'2 and further indirectly regulates CSC properties by
modulating these EMT-TFs (64).
Specifically, the miR-200 family and ZEB1/2 form a mutually
inhibitory negative feedback loop; ZEB1/2 transcriptionally inhibit
miR-200 expression, while miR-200 inhibits ZEB1/2 function by
targeting their 3'UTR; simultaneously, miR-200 can directly target
and inhibit the stemness factor BMI1, thereby regulating CSC
properties (65). Thus, high ZEB1
expression in PDAC enhances the activated phenotype of stromal
myofibroblasts, activates KRAS and its downstream PI3K/AKT pathway
in cancer cells via paracrine signals, disrupts the balance of the
tumor microenvironment, accelerates the progression of PDAC from
pre-cancerous lesions to malignant tumors and markedly enhances its
in vivo tumor-initiating capacity (66).
EMT-mediated resistance to chemo- and
radiotherapy
The close association between EMT and CSCs is a key
reason for the broad-spectrum resistance of tumors to treatment.
EMT can induce cells to acquire CSC-like properties by activating
stem cell-related signaling pathways [such as Notch and Hedgehog
(Hh)] and downregulating epithelial markers (such as E-cadherin)
while upregulating mesenchymal markers (such as Id-1, α-SMA), which
specifically includes temporary entry into a dormant state (cell
cycle arrest). Most chemotherapeutic drugs, such as gemcitabine,
primarily target rapidly proliferating cells, allowing these
dormant cells to evade drug attack and become the source of
recurrence after treatment (67).
Secondly, the EMT program can enhance cell survival signals by
activating pro-survival signaling pathways such as PI3K/AKT and
increase the apoptotic threshold by upregulating anti-apoptotic
molecules (such as Bcl-xL) and inhibiting pro-apoptotic factors
(such as BOK), making cells more likely to survive DNA damage
induced by drugs or radiation (68).
In pancreatic cancer, EMT is a key mechanism
mediating resistance to chemotherapy and radiotherapy and it
affects the response of tumor cells to treatment through a
multi-dimensional regulatory network. EMT-TFs such as ZEB1 play a
central role: High ZEB1 expression is closely associated with
gemcitabine resistance and ZEB1 can maintain the EMT phenotype and
reduce drug sensitivity by inhibiting the miR-200 family to form a
negative feedback loop; conversely, ZEB1 knockdown can markedly
reverse this process (69,70).
Additionally, EMT-induced metabolic reprogramming provides an
energy basis for drug resistance: PDAC cells with ZEB1 deficiency
cannot compensatorily enhance glycolysis when oxidative
phosphorylation is inhibited, making them sensitive to metabolic
stress; by contrast, EMT-activated cells maintain energy supply by
upregulating glucose transporters [such as Glucose Transporter 3
(GLUT3)] and the Warburg effect, thereby tolerating metabolic
stress induced by chemotherapeutic drugs (69). In terms of radioresistance, EMT
achieves resistance by enhancing DNA damage repair capacity: ZEB1
can directly bind to the Ataxia Telangiectasia Mutated Protein
(ATM) promoter and form a complex with p300/PCAF to promote ATM
expression; meanwhile, ZEB1 stabilizes CHK1 by sequestering the
deubiquitinase USP7, accelerating DNA damage repair to reduce
radiation-induced cell death (69); furthermore, radiotherapy itself
can upregulate EMT-TFs such as ZEB1 and Snail by activating
pathways such as TGF-β and NF-κB, further exacerbating resistance
(71). The involvement of the TME
is another important aspect: Cytokines such as TGF-β and IL-6
secreted by CAFs can induce EMT, promoting resistance of PDAC cells
to chemotherapeutic drugs; targeting paracrine signals between CAFs
and tumor cells can partially reverse this resistance (69,71). Notably, while EMT is not necessary
for metastasis, it clearly induces chemotherapy resistance in
pancreatic cancer, this property provides a theoretical basis for
EMT-targeted therapeutic strategies (72). Meanwhile, EMT is closely
associated with the CSC phenotype: EMT-TFs can maintain the
stemness of CSCs, making them less sensitive to radiotherapy and
chemotherapy and forming a drug-resistant cell population (73).
The EMT-stemness-therapy resistance axis is central
to PDAC malignancy. TGF-β-driven ZEB1 suppresses miR-200 and
activates WNT/β-Catenin, conferring stemness (such as
CD44+/CD133+) and cell-cycle arrest to evade
chemotherapy (such as gemcitabine) and immune targeting.
Concurrently, ZEB1 enhances DNA repair (ATM/CHK1) and metabolic
reprogramming (Warburg effect) for broad resistance. Critically,
therapeutic targeting faces major limitations: EMT-related
molecules (such as ZEB1) have essential physiological roles in
normal tissues, risking off-target toxicity; and discrepancies
between preclinical models and human responses hinder clinical
translation, necessitating more precise intervention strategies
(Fig. 3).
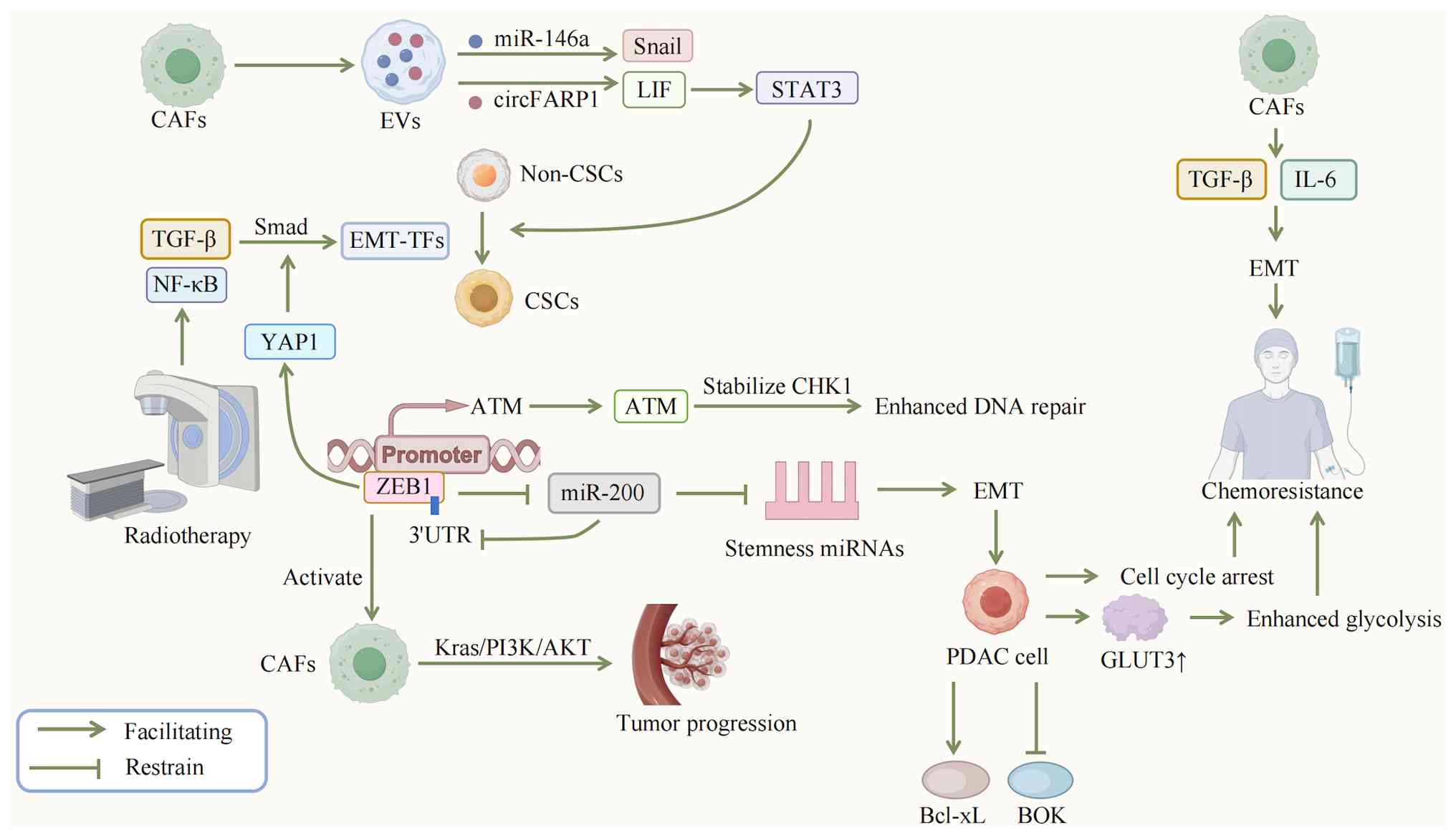 | Figure 3Therapeutic resistance mechanisms in
PDAC. The extreme resistance of PDAC to chemotherapy and
radiotherapy stems from its unique EMT-stemness-therapy resistance
axis. The initiation of this vicious cycle critically depends on
EVs secreted by CAFs. These EVs deliver molecules such as miR-146a
(activating Snail) and circFARP1 (activating the LIF/STAT3 axis),
which collaboratively induce the EMT program and the acquisition of
CSC traits. TGF-β signaling further reinforces EMT via the Smad
pathway and activates EMT-TFs such as ZEB1 and Snail, enabling
differentiated non-stem-like cancer cells to gain CSC properties.
Among these, ZEB1 directly interacts with YAP1 in response to TGF-β
signaling, coregulating stem cell pluripotency, while ZEB1/2
maintains the EMT phenotype and reduces drug sensitivity by
transcriptionally inhibiting the miR-200 family (microRNAs that
themselves form a mutual inhibitory feedback by targeting the
3'UTRs of ZEB1'2). The EMT program also enhances cell survival
signals by activating pathways such as PI3K/AKT and increases the
apoptotic threshold through upregulation of the anti-apoptotic
molecule Bcl-xL and suppression of the pro-apoptotic factor BOK,
allowing cells to improved survive DNA damage induced by drugs or
radiation. Simultaneously, EMT promotes CSC-like characteristics,
including a transient entry into a dormant, cell cycle-arrested
state, helping cells evade proliferation-targeting drugs.
Metabolically, EMT-activated cells sustain energy supply by
upregulating glucose transporter GLUT3 and enhancing the Warburg
effect, thereby tolerating metabolic stress induced by
chemotherapy. Additionally, ZEB1 directly binds to the ATM promoter
to increase ATM expression and stabilizes CHK1, accelerating DNA
damage repair and reducing radiation-induced cell death.
Radiotherapy itself can further upregulate EMT-TFs such as ZEB1 and
Snail by activating pathways such as TGF-β and NF-κB, forming a
positive feedback loop that exacerbates therapy resistance. In
summary, this multidimensional mechanism, encompassing EV-mediated
signaling initiation, EMT-stemness coupling, survival signal
enhancement, metabolic reprogramming and DNA repair activation,
collectively underlies PDAC treatment failure. PDAC, pancreatic
ductal adenocarcinoma; EMT, epithelial-mesenchymal transition; EVs
CAFs, cancer-associated fibroblasts; CSC, cancer stem cell; TGF-β,
transforming growth factor-β; EMT-TFs, EMT-transcription factors;
LIF, leukemia inhibitory factor; STAT3, signal transducer and
activator of transcription 3; ZEB1, Zinc Finger E-Box Binding
Homeobox 1; YAP1, Yes-Associated Protein 1; GLUT3, glucose
transporter 3; ATM, ataxia-telangiectasia mutated. |
Dynamic regulatory network of EMT:
Transcriptional, signaling and epigenetic hierarchies
EMT is not an isolated event but is driven by an
extraordinarily complex, multi-level and highly integrated
molecular regulatory network (2).
In the context of pancreatic cancer, dysregulation of this network
is the core of tumor malignant progression. It begins with the
perception of extracellular signals, which are transmitted through
classical signal transduction pathways and ultimately converge on a
small group of core transcription factors (EMT-TFs). The
mesenchymal cell state is then consolidated and maintained through
epigenetic remodeling. Meanwhile, dynamic changes in the TME and
adaptive reprogramming of cellular metabolism provide continuous
momentum and material basis for this process (74). Below, the key components of this
complex network and their specific mechanisms of action in
pancreatic cancer are introduced.
Core EMT transcription factor hubs:
mechanisms, synergy and feedback loops of Snail/ZEB/Twist
families
EMT-TFs are the core hubs of the regulatory network.
After integrating upstream signals (such as pathways mediated by
TGF-β and IL-6), they act as terminal effectors to directly execute
gene expression switching, binding to the promoters of target genes
to inhibit epithelial programs (such as the CDH1 gene encoding
E-cadherin) and activate mesenchymal programs (75). In pancreatic cancer, the Snail,
ZEB and Twist families are key executors of this process, playing
indispensable roles.
EMT-TFs [Snail, ZEB, Twist and Paired Related
Homeobox (PRRX) families] rely on conserved domains (zinc finger,
bHLH and homeodomain) for DNA binding and transcriptional
regulation. Their core commonality is regulating EMT via E-box
binding, suppressing epithelial markers (such as CDH1) and
activating mesenchymal genes, alongside forming miRNA negative
feedback loops. Functionally divergent due to domain differences:
Snail recruits co-repressors via SNAG domain, ZEB interacts with
YAP/AP-1, Twist requires dimerization and PRRX synergizes with
Twist to enhance invasiveness (76,77). Downstream, they form a bipolar
target network: CDH1 is directly repressed by Snail/ZEB/Twist (a
key EMT-initiating event), while mesenchymal targets (VIM, CDH2,
MMP9) and fibroblast-related genes (activated by PRRX1-TWIST1)
collectively drive PDAC progression (78,79).
Mechanisms of action of Snail, ZEB1/2 and
Twist families
As a core EMT transcription factor, Snail1 drives
the malignant progression of pancreatic cancer through a
multi-level regulatory network; its abnormal function is closely
associated with the acquisition of the EMT phenotype and enhanced
invasive/metastatic capacity. At the signaling pathway level, the
TGF-β1-Smad2/3 pathway upregulates Snail1 via Numb-PRRL activation
and synergizes with Notch1 to enhance the EMT phenotype,
characterized by downregulation of E-cadherin and upregulation of
N-cadherin and Vimentin (80).
The WNT/β-catenin pathway forms a positive feedback loop with
Snail1:STMN2 activates the WNT/β-catenin pathway (promoting
β-catenin nuclear translocation to bind TCF transcription complex)
to upregulate Snail1 expression, while the nuclear β-catenin/TCF
complex in turn transactivates STMN2 gene expression; their mutual
promotion collectively reinforces EMT and proliferation (81). At the metabolic level, RHOF
enhances glycolysis via the c-Myc-PKM2 axis and lactic acid induces
lactylation and nuclear translocation of Snail1 to drive EMT
(82); epigenetically, miR-34a
directly targets Snail1 mRNA to inhibit its expression, weakening
the invasive capacity of cancer cells (83).
The protein stability of Snail1 is dynamically
regulated by the ubiquitin-proteasome system: F-box/LRR-repeat
protein 7 inhibits EMT by ubiquitinating and degrading Snail1
(84), while TGF-β-induced
ubiquitin-specific peptidase 27X (USP27X) stabilizes Snail1 via
deubiquitination, enhancing its pro-EMT and chemoresistant
functions (85). Further studies
have shown that 17 E3 ubiquitin ligases (such as FBXL14 and βTrCP1,
which mediate Snail1 degradation via ubiquitination) and 23
deubiquitinating enzymes (such as USP47 and DUB3, which stabilize
Snail1 via deubiquitination) together form a regulatory network for
Snail1 protein stability; in addition, proteins such as COP9
signalosome subunit 2 (CSN2, not β-casein) can indirectly maintain
Snail1 stability by inhibiting the activity of E3 ubiquitin ligases
(86).
Phosphorylation regulation exhibits spatiotemporal
specificity: Glycogen synthase kinase 3 beta (GSK3β) phosphorylates
the Ser-rich domain (SRD) in the nucleus to promote Snail1 nuclear
export and synergizes with CK1ε in the cytoplasm to phosphorylate
Snail1 and form a degradation motif; by contrast, kinases such as
ATM and ERK2 phosphorylate specific sites in the nucleus, which can
recruit Heat shock protein 90 to stabilize Snail1, reflecting the
effect of subcellular localization on function (86). Modifications such as acetylation
(for instance, CBP-mediated acetylation of Snail1 at Lys146/187,
which enhances its transcriptional activation function and inhibits
degradation) and glycosylation (such as O-GlcNAc modification at
Ser112, which blocks GSK3β-mediated phosphorylation and
degradation) further regulate Snail1's transcriptional activity and
stability; its N-terminal intrinsically disordered region enables
it to flexibly bind various regulatory factors (such as E3 ligases,
deubiquitinating enzymes), making it a hub for integrating EMT
signals (86).
During the occurrence and development of pancreatic
cancer, Snail1 acts as a core driver in the progression of
precursor lesions. Inhibition of Snail1 via gene knockout or drugs
(such as GN25) can effectively delay the occurrence and development
of pancreatic intraepithelial neoplasia (PanINs) and reduce
acinar-ductal metaplasia (ADM) after pancreatic injury, suggesting
its significant potential for early intervention in pancreatic
cancer (87). From the
perspective of downstream effects of signal transduction, the
execution of the EMT phenotype mediated by Snail1 depends on the
classical BMP signaling pathway, which regulates the expression of
downstream target genes through synergy with SMAD4, ultimately
achieving EMT-related invasive and metastatic phenotypes (88). At the clinical level, the
metastasis suppressor Raf kinase inhibitor protein (RKIP) is
markedly negatively correlated with Snail1 expression: high RKIP
expression is often associated with a favorable prognosis in
pancreatic cancer patients, while high Snail1 expression predicts
disease progression and poor outcomes, this association provides an
important molecular marker for prognosis evaluation of pancreatic
cancer (39).
The ZEB family, especially ZEB1, serves as a core
driver of EMT and stem cell properties in pancreatic cancer. It
participates in multiple key links of tumor malignant progression
through a complex regulatory network and its abnormal activation is
driven by the synergy of multi-level upstream signals, non-coding
(nc)RNA networks, inflammatory pathways and cell plasticity
regulation. In the early stage of tumorigenesis, ZEB1 is a key
driver of the progression of pancreatic precursor lesions: in the
KRAS and p53 mutant pancreatic cancer model (KPC) mouse model
(carrying KRAS and p53 mutations), deletion of ZEB1 can markedly
reduce the number and grade of ADM and PanINs; in the KRAS mutant
pancreatic cancer model (KC) model (carrying KRAS mutation but no
p53 mutation), deletion of ZEB1 more markedly inhibits the
formation of KRAS-driven early lesions, highlighting its importance
in the initiation stage of pancreatic cancer (63). In terms of regulatory mechanisms:
at the epigenetic level, enhancer of zeste homolog 2 (EZH2)
inhibits miR-139-5p via lysine 27 trimethylation on histone H3
(H3K27me3), thereby relieving the targeted inhibition of ZEB1/2 by
miR-139-5p (89); in the
inflammatory microenvironment, macrophage migration inhibitory
factor (MIF) downregulates miR-200b to enhance ZEB1/2 expression
(90), while NF-κB directly binds
to the ZEB1 promoter to promote its transcription (91). In signaling pathways, vasohibin 2
(VASH2) activates the Hh pathway to upregulate ZEB1/2 (92),TGF-β-induced EMT is dependent on
ZEB1; ZEB1 deletion renders PDAC cells unable to undergo phenotypic
switching in response to TGF-β stimulation, as 91% of
TGF-β-regulated genes require ZEB1 involvement (63). ZEB1 forms a negative feedback loop
with the miR-200 family: ZEB1 deletion leads to high miR-200c
expression, which in turn inhibits stem cell markers such as Sox2
and markedly reduces sphere formation capacity and tumor-initiating
potential (63).
Functionally, ZEB1 endows pancreatic cancer with a
malignant phenotype by maintaining cell plasticity: ZEB1 deletion
fixes cells in the epithelial phenotype, losing the ability to
switch between epithelial and mesenchymal phenotypes and resulting
in the inability of the invasive front to undergo
dedifferentiation. Metabolically, ZEB1 deletion markedly reduces
oxidative phosphorylation and glycolytic reserves, rendering cells
unable to adapt to changes in TME energy demands. Unlike Snail and
Twist, ZEB1 is necessary for metastasis in the KPC model, ZEB1
deletion almost completely abolishes lung colonization capacity,
while Snail deletion has no such effect, reflecting the
non-redundancy of EMT-TFs. Clinically, high ZEB1 expression is
associated with the 'quasi-mesenchymal' subtype of pancreatic
cancer, while ZEB1 deletion enriches features of the 'classical'
subtype (which has an improved prognosis) (63). The malignant function of ZEB1 can
be counter regulated by SCAND1, which forms a complex with myeloid
zinc finger 1 (MZF1) to inhibit ZEB1/2 expression (93). By regulating precursor lesion
progression, cell plasticity, stem cell properties and metabolic
adaptation, ZEB1 serves as a core node of EMT in pancreatic cancer
and its unique role provides a specific target for targeted
therapy.
The Twist family (Twist1 and Twist2), as core
members of basic helix-loop-helix (bHLH) transcription factors,
play key roles in EMT of pancreatic cancer through
multi-dimensional regulation. Their functions are not only related
to cell plasticity in embryonic development but also involved in
malignant progression driven by the tumor microenvironment
(94,95). Twist1 and Twist2 both bind to the
promoters of epithelial marker genes such as E-cadherin to inhibit
their expression, while upregulating mesenchymal markers such as
Vimentin and N-cadherin, promoting the transformation of epithelial
cells to an invasive phenotype (95). In terms of regulatory mechanisms,
the Twist family is precisely regulated by tumor microenvironment
signals: Hypoxia activates Twist1 transcription by stabilizing
HIF-1α and this pathway is associated with lymph node metastasis
and poor prognosis of pancreatic cancer (95,96); TGF-β upregulates SOX5 via a
Smad-dependent pathway to enhance Twist1 expression and also
synergizes with RAS signals via STAT3 and ETS1/2 to strengthen its
expression (94,95); Aurora kinase A (AURKA)
phosphorylates Twist1 at sites S123, T148 and S184 to inhibit its
ubiquitination-mediated degradation and enhance its activity;
meanwhile, Twist1 maintains AURKA protein levels by inhibiting its
ubiquitination degradation and the two form a positive feedback
loop to amplify the EMT effect (97). In downstream effects, Twist1
promotes invasion and cisplatin resistance by inducing MMP2 and
GDF15 (98) and interacts with
Ring1B and EZH2 to downregulate tumor suppressor genes and enhance
proliferation (96). Twist2 is
regulated by HIF-2α, specifically binds to the E-cadherin promoter
to inhibit its expression and is negatively correlated with
E-cadherin expression (99).
Additionally, arginine deprivation (that is, reducing extracellular
arginine levels via arginine deiminase or other means to inhibit
arginine-dependent pancreatic cancer cells from acquiring this
essential amino acid) can downregulate Twist expression, thereby
inhibiting EMT (100) and
miR-539 directly targets Twist1 mRNA to inhibit its translation
(101). In metastasis biology,
Twist1-mediated EMT endows pancreatic cancer cells with the
migratory ability to detach from the primary tumor, while
colonization of metastatic lesions depends on the downregulation of
Twist1 and activation of MET. This dynamic switching reflects the
spatiotemporal specificity of the Twist family in different stages
of metastasis (95).
Interactions and feedback loops among
transcription factors
As the core EMT initiator, Snail1's stability is
regulated by TGF-β-activated deubiquitinase USP27X; TGF-β
upregulates USP27X, which then stabilizes Snail1 via
deubiquitination. Notably, Snail1 positively feeds back on TGF-β
(directly promoting TGF-β transcription or indirectly enhancing its
signaling), forming a 'TGF-β-USP27X-Snail1' loop that continuously
strengthens E-cadherin inhibition and mesenchymal phenotype
acquisition to sustain tumor progression (85). ZEB1/2 inhibits the epithelial
splicing regulatory proteins ESRP1/2, promoting the expression of
the fibroblast growth factor receptor-3 IIIc subtype; the latter
activates the MEK-ERK-ETS1/2 pathway to feedback and maintain high
expression of ZEB1/2, forming an independent self-sustaining loop.
Meanwhile, ZEB1/2 is functionally complementary to Snail1 (rather
than co-expressed), jointly mediating EMT plasticity (63). In some pancreatic cancer cells,
Slug and Snail1 exhibit a synchronized regulatory pattern in their
expression; specifically, both are often regulated by upstream
regulatory factors (such as RAB11FIP1) to be simultaneously
upregulated or downregulated; and these two factors can
synergistically respond to TGF-β signals, thereby inducing the
epithelial-mesenchymal transition (EMT) process (102).
Cross-regulation between the Twist family and other
EMT transcription factors further amplifies network effects: in a
hypoxic environment, HIF-1α can simultaneously induce the
expression of Twist1 and Snail1; among them, Twist1 enhances
chromatin modification by binding to Ring1B and EZH2, synergizing
with Snail1 to strengthen the transcriptional inhibition of
E-cadherin (96); ZEB1 and Twist1
exhibit functional differentiation: ZEB1 mainly regulates the
phenotypic plasticity and metastatic colonization capacity of tumor
cells, while Twist1 focuses more on driving invasive capacity and
chemotherapy resistance (63,72). A key feedback loop also includes
the mutual stabilization between Twist1 and AURKA: AURKA
phosphorylates Twist1 to inhibit its ubiquitination and
degradation, while Twist1 in turn prevents AURKA degradation,
forming a positive feedback loop that amplifies the EMT effect
(102). Additionally, the
miR-200 family acts as a core negative regulator that can
simultaneously target ZEB1/2 and Twist1; the tumor suppressor Par-4
constructs a 'Par-4-miR-200c-ZEB1'Twist1/ negative feedback loop by
upregulating miR-200c, limiting excessive EMT activation (103). The synergy of this network is
also reflected in the commonality of upstream regulation:arginine
deprivation can synchronously downregulate the expression of the
Snail, Slug and Twist families (98); overexpression of Dual-specificity
tyrosine-phosphorylation-regulated kinase 2 indirectly affects the
Snail/Slug-mediated EMT process by promoting the ubiquitination and
degradation of Twist (104).
These findings further confirm the close associations of various
transcription factors within the network.
The regulatory roles of Snail, ZEB and Twist
families, core EMT-TF hubs, in pancreatic cancer (PC) are analyzed
in depth, with multi-dimensional mechanisms clearly elaborated. For
Snail1, its activity is modulated by TGF-β/WNT signaling pathways,
metabolic lactylation and post-translational modifications
including ubiquitination and phosphorylation. ZEB1 exhibits
non-redundant functions, particularly in metastasis (a unique trait
validated in KPC models) and PC subtype switching. The Twist
family, dependent on hypoxia/HIF-1α signaling, regulates cell
invasion and exhibits spatiotemporal specificity in MET during
metastasis.
Critical findings cover crosstalk between EMT-TFs,
such as the TGF-β-USP27X-Snail1 positive feedback loop and
functional differentiation between ZEB1 and Twist1. Negative
regulatory networks, mediated by miR-200 and Par-4, are also
highlighted. Clinically, the associations of these TFs with PanIN
progression, prognostic evaluation (such as RKIP-Snail1
correlation) and therapeutic potential (such as GN25, arginine
deprivation) provide practical value. This comprehensive network
analysis enhances understanding of PC EMT plasticity and lays a
theoretical foundation for developing targeted therapies (Fig. 4).
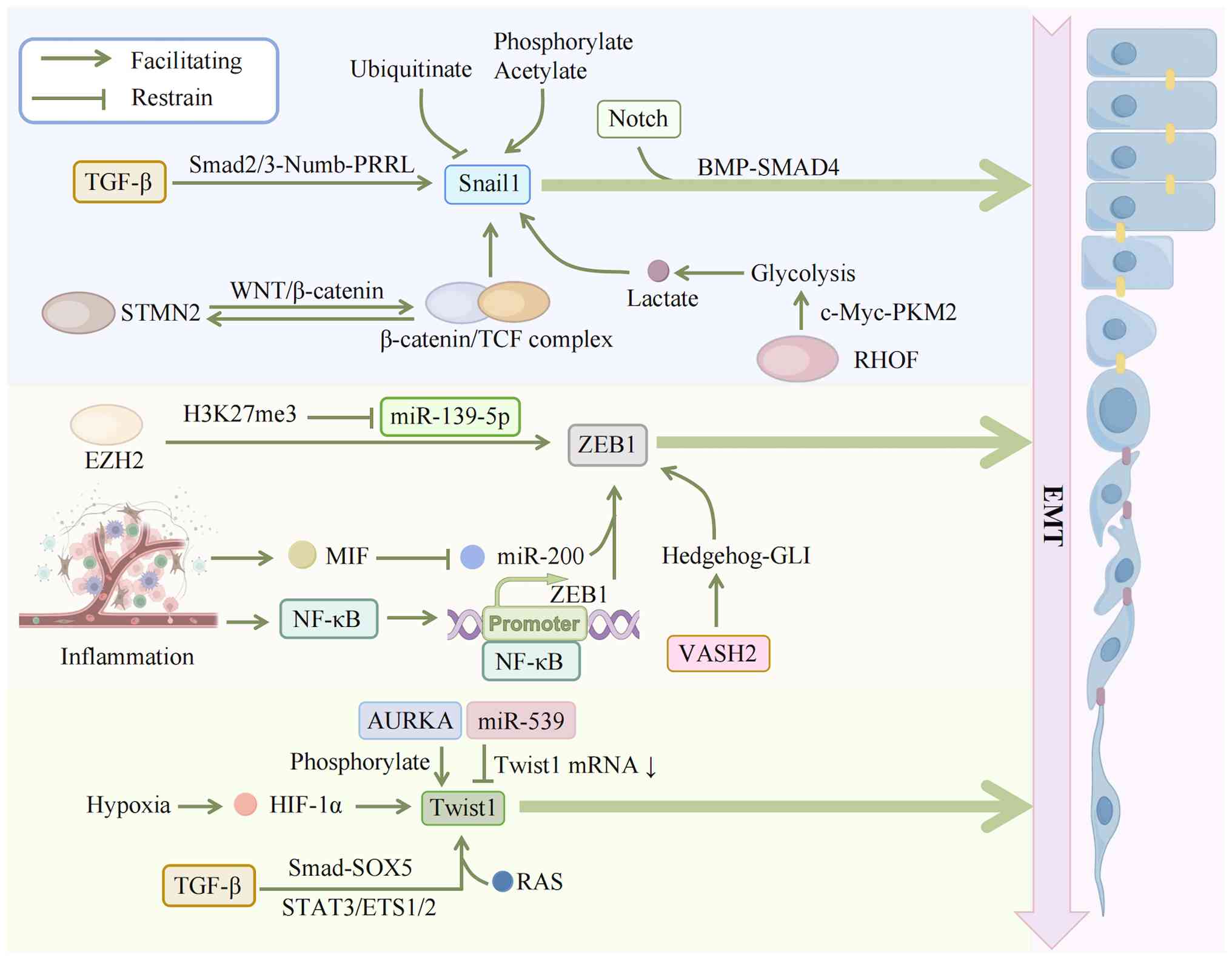 | Figure 4EMT regulatory network: Multi-layer
control of transcription factors. TGF-β1 upregulates Snail1 via the
Smad2/3-Numb-PRRL pathway and synergizes with Notch1 signaling to
enhance EMT. The WNT/β-catenin pathway forms a positive feedback
loop with Snail1: STMN2 activates the WNT pathway to upregulate
Snail1 and nuclear β-catenin/TCF complexes transactivate STMN2.
RHOF enhances glycolysis through the c-Myc-PKM2 axis and the
resulting lactate induces Snail1 lactylation and nuclear
translocation. Snail1 stability is controlled by ubiquitin-mediated
degradation, while its transcriptional activity is enhanced by
phosphorylation and acetylation. EZH2 suppresses miR-139-5p via
H3K27me3 modification, relieving its targeted inhibition of ZEB1/2.
In the inflammatory microenvironment, MIF downregulates miR-200b
and NF-κB directly binds to the ZEB1 promoter to enhance its
expression, while VASH2 upregulates ZEB1/2 by activating the Hh
pathway; moreover, TGF-β-induced EMT requires ZEB1 participation.
Upstream hypoxia stabilizes HIF-1α to activate Twist1
transcription; TGF-β upregulates Twist1 through the SMAD-SOX5
pathway and synergizes with STAT3/ETS1/2 and RAS signaling; AURKA
phosphorylates Twist1 to inhibit its ubiquitin-mediated
degradation, while miR-539 targets Twist1 mRNA to inhibit its
translation. EMT, epithelial-mesenchymal transition; TGF-β,
transforming growth factor-β; STMN2, stathmin 2; TCF, T Cell
Factor; RHOF, Ras Homolog Family Member F; c-Myc, cellular Myc;
PKM2, pyruvate kinase M2; EZH2, enhancer of Zeste Homolog 2; miR,
microRNA; ZEB1, Zinc Finger E-Box Binding Homeobox 1; MIF,
macrophage migration inhibitory factor; NF-κB, nuclear factor-kappa
B; HIF-1α, hypoxia-inducible factor 1 alpha; SMAD, Sma- and
Mad-related Protein; SOX5, SRY-Box transcription factor 5; RAS, rat
sarcoma viral oncogene homolog; AURKA, aurora kinase A. |
Tumor microenvironment-signal
integration: Spatiotemporal regulation of TGF-β/Wnt/Notch/Hh
pathways
TGF-β pathway: Dual roles, signal
crosstalk and CAF-tumor cell feedback
The TGF-β signaling pathway exerts dual effects in
EMT of PDAC The switch of its function from early tumor suppression
to late metastasis promotion depends on multi-level regulation. In
PDAC cells with retained SMAD4, TGF-β activates the SMAD2/3-SMAD4
complex to regulate transcription factors such as Snail and Twist;
meanwhile, it maintains the activity of super-enhancers of genes
such as SNAI1 and SOX9 with the help of the epigenetic regulator
PHF13, ensuring the continuous expression of EMT-related genes
(105,106); deletion of SMAD4 disrupts this
regulation, converting SOX4 from a pro-tumor factor to a
pro-apoptotic molecule, highlighting the decisive effect of SMAD4
on pathway function (105,107). Activation of the TGF-β pathway
is regulated by a 'promotion-inhibition' dynamic balance: circEIF3I
(CircBase ID: hsa_circ_0011385) promotes pathway activation by
enhancing the binding of SMAD3 to TGFβRI (108). Under TGF-β1 stimulation,
Numb-PRRL not only activates the SMAD2/3-Snail pathway downstream
of the TGF-β signaling pathway to enhance EMT but also forms a
cross-activation loop with Notch1 to synergistically strengthen the
EMT-promoting effect of this pathway. Notably, Notch inhibitors
(such as RO4929097) can block this cross-activation loop, thereby
reversing the enhancing effect of Numb-PRRL on the SMAD2/3-Snail
pathway (80). Conversely,
abnormal expression of TGF-β receptors (TGFβRI/II) disrupts the
balance, weakening tumor suppressor function and amplifying
pro-metastatic signals (109).
Signal crosstalk further expands the pro-cancer
effects of TGF-β: Numb-PRRL simultaneously participates in the
activation of the EGFR-ERK/MAPK pathway induced by EGF, enabling
TGF-β and growth factor signals to synergistically enhance tumor
migration and invasion (80);
moreover, TGF-β maintains the expression of cancer stem cell
markers such as CD24 and CXCR4 by regulating Snail and SLUG and
silencing these transcription factors can reduce the sphere
formation ability of PDAC cells (107). In the tumor microenvironment,
TGF-β activates CAFs via the TGF-β1/Smad pathway; thrombospondin 2
and MMP11 secreted by CAFs activate the MAPK and PI3K/AKT pathways,
respectively, forming a 'tumor cell-CAF' positive feedback loop
that exacerbates stromal fibrosis (110-112).
Phase Ib/II clinical trial data show that the
combination of the TGFβRI inhibitor vactosertib and gemcitabine can
reduce extracellular matrix components by inhibiting the
TGF-β/Smad2 pathway, thereby enhancing the penetration of the
chemotherapeutic drug (113);
the combination of TGF-β inhibitors and PD-1/PD-L1 blockers can
reverse T-cell exclusion and restore anti-tumor immunity (114,115). However, treatment response needs
to be combined with patient molecular subtypes (such as SMAD4
status may affect efficacy), suggesting that multi-target
combination is required to achieve comprehensive regulation of EMT,
CSC and the microenvironment (105-107).
The Wnt/β-catenin pathway: Link to
PDAC stemness
In the EMT process of PDAC, the Wnt/β-catenin
pathway is a core upstream driver. Its dysregulation connects
intracellular regulation of cancer cells, paracrine signals of
cancer-associated fibroblasts CAFs and molecular subtype
characteristics of PDAC, forming a multi-dimensional regulatory
network (116-119). In cancer cells: FAM83A disrupts
the β-catenin degradation complex by binding to β-catenin and BLK
phosphorylation can strengthen this effect to promote β-catenin
nuclear translocation by inhibiting the β-catenin cytoplasmic
destruction complex; serine/threonine/tyrosine kinase 1 (STYK1)
sequesters GSK3β into multivesicular bodies, maintaining pathway
activity to support EMT (116,117). Among ncRNAs circRREB1 promotes
WNT7B transcription and glycolysis adaptation to EMT by binding to
the RRM domain of YAP1; circPHF14 stabilizes WNT7A mRNA to induce
EMT transcription factors; exosomal miR-146a and lncRNA H19 derived
from CAFs further potentiate Wnt/β-catenin pathway activity
(118,120,121). Pathway activation shows
significant subtype specificity (119): The squamous subtype is enriched
with TP53/KDM6A mutations, highly expresses TP63∆N and has
hypermethylation of endodermal genes, thereby relieving the
inhibition of pathways by these endodermal genes; the pancreatic
progenitor subtype highly expresses developmental factors such as
PDX1 and transforming growth factor beta receptor 2 (TGFBR2)
mutations weaken TGF-β pathway inhibition and also enhance the
interaction between ECM and β-catenin through mucins; the ADEX
subtype relies on oncogenic KRAS to synergize with Wnt ligands to
drive EMT (119). At the
metabolic level: Frizzled5 binds to cholesterol to maintain
Wnt/β-catenin pathway activation (more common in the squamous
subtype); methyltransferase-like 3 mediates m6A modification of APC
mRNA, which further recruits YTHDF proteins to promote APC mRNA
degradation; this degradation reduces APC protein expression,
thereby relieving APC's inhibitory effect on the Wnt'β-catenin
pathway, activating downstream effectors including β-catenin,
Cyclin D1, c-Myc and PKM2 and ultimately enhancing aerobic
glycolysis (to support energy supply) and abnormal cell
proliferation to drive tumor growth; a regulatory cascade that is
more significant in the pancreatic progenitor subtype (122,123). In CAFs, myofibroblastic
cancer-associated fibroblasts (myCAFs) secrete Wnt2 to create the
immune microenvironment required for EMT; the squamous subtype
additionally features a 'TP63∆N-CAF-ECM-Wnt'positive feedback loop,
whereby TP63∆N induces CAF activation, these activated CAFs remodel
the ECM; the altered ECM then activates Wnt signaling and this Wnt
activation in turn reinforces the loop to strengthen the pathway
(118,119).
Treatment needs to be adapted to subtypes: Fisetin
inhibits β-catenin nuclear accumulation (suitable for the
pancreatic progenitor subtype); riluzole directly blocks the
Wnt-β-catenin pathway (targeting metabolism-related EMT in the
squamous subtype); inhibiting CAF-derived Wnt2 can enhance the
efficacy of anti-PD-1 in the immunogenic subtype; the squamous
subtype can be combined with epigenetic drugs and Wnt inhibitors to
restore endodermal gene expression (118,119,124,125).
Notch and Hh pathways: regulatory
roles in the pancreatic cancer microenvironment
In the pancreatic cancer microenvironment, the Notch
and Hh pathways regulate EMT through dynamic interactions with
stromal cells, serving as key upstream signals driving tumor
invasion and treatment resistance. In pancreatic cancer, the Notch
pathway is activated by membrane-bound ligands (such as DLL, Jagged
families) from adjacent cells. Upon ligand binding, the Notch
receptor is cleaved, releasing its intracellular domain (NICD) into
the nucleus to form a complex with transcription factors such as
CSL and drive target gene expression. The Hh pathway is initiated
by secreted ligands [such as Sonic Hedgehog (SHH)] binding to the
PTCH receptor, which relieves its inhibition of SMO and ultimately
leads to the activation of GLI family transcription factors.
Together, these pathways co-regulate the EMT process (126).
EGFR/ERBB2 signals synergize with the Notch/Hh
pathway to drive EMT in myCAFs induced by TGF-β (107). Specifically, TGF-β induces
myCAFs to secrete autocrine amphiregulin (AREG), activating
EGFR/ERBB2 heterodimer signals; IL-6 secreted by these
CD90− myCAFs (CD90−) can upregulate the
expression of Snail and Twist in cancer cells via the STAT3
pathway; meanwhile, EGFR/ERBB2 signals enhance the γ-secretase
activity of the Notch pathway to promote NICD nuclear
translocation, forming a 'TGF-β-AREG-EGFR'Notch/cascade effect
(107); Hh-highly active myCAFs
remodel collagen structure via the PTCH-SMO-GLI1 axis, forming
functional complementarity of 'structural remodeling-soluble factor
regulation'with EGFR⁺myCAFs, reflecting the selective activation of
pathways by CAF subtypes (107,118).
At the molecular level the long non-coding (lnc)RNA
TRPM2-AS binds to miR-31-5p/miR-146a-5p via a ceRNA mechanism,
relieving their inhibition of NUMB, thereby releasing the
inhibition of Notch1 ubiquitination and degradation and maintaining
continuous activation of the Notch pathway (107); meanwhile, GLI1 downstream of Hh
can form an intranuclear complex with EGFR phosphorylation
products, synergistically binding to the ZEB1 promoter;
Notch-activated cMYC, which acts as an evolutionarily conserved
proto-oncogenic transcription factor, strengthens EGFR signals by
upregulating AREG transcription, constructing a multi-pathway
positive feedback loop (107,127).
Clinical translation studies have shown that the
combination of EGFR/ERBB2 inhibitors and Notch inhibitors (such as
DAPT) can markedly reduce the expression of EMT markers induced by
myCAFs and reverse CAF-mediated gemcitabine resistance in PDAC
organoid models (107). This
strategy avoids the problem of CAF subtype switching caused by the
single use of Hh inhibitors (such as vismodegib) (118). in addition, dietary
phytochemicals (such as curcumin) can indirectly inhibit the
synergistic activation of EGFR/Notch by downregulating SP1,
providing a supplement for combination therapy (107,128). These findings further support
the multi-target therapeutic value of the pathway
crosstalk-CAFs-EMT axis.
The paradigm of CAF-secreted TGF-β inducing EMT has
been challenged by single-cell spatial transcriptomics; novel
inflammatory CAFs (iCAFs) independently activate EMT via the
IL-1β/JAK/STAT axis and this subpopulation markedly expands in the
microenvironment after chemotherapy. More paradoxically, while
stroma-depleting therapies targeting CAFs enhance drug delivery,
they may accelerate tumor spread (the 'FA paradox'); although
combining with CXCR4 inhibitors inhibits metastasis in KPC mice,
differences in human stromal density may weaken efficacy. These
findings require a re-evaluation of the clinical translation path
of stroma targeting (Fig. 5).
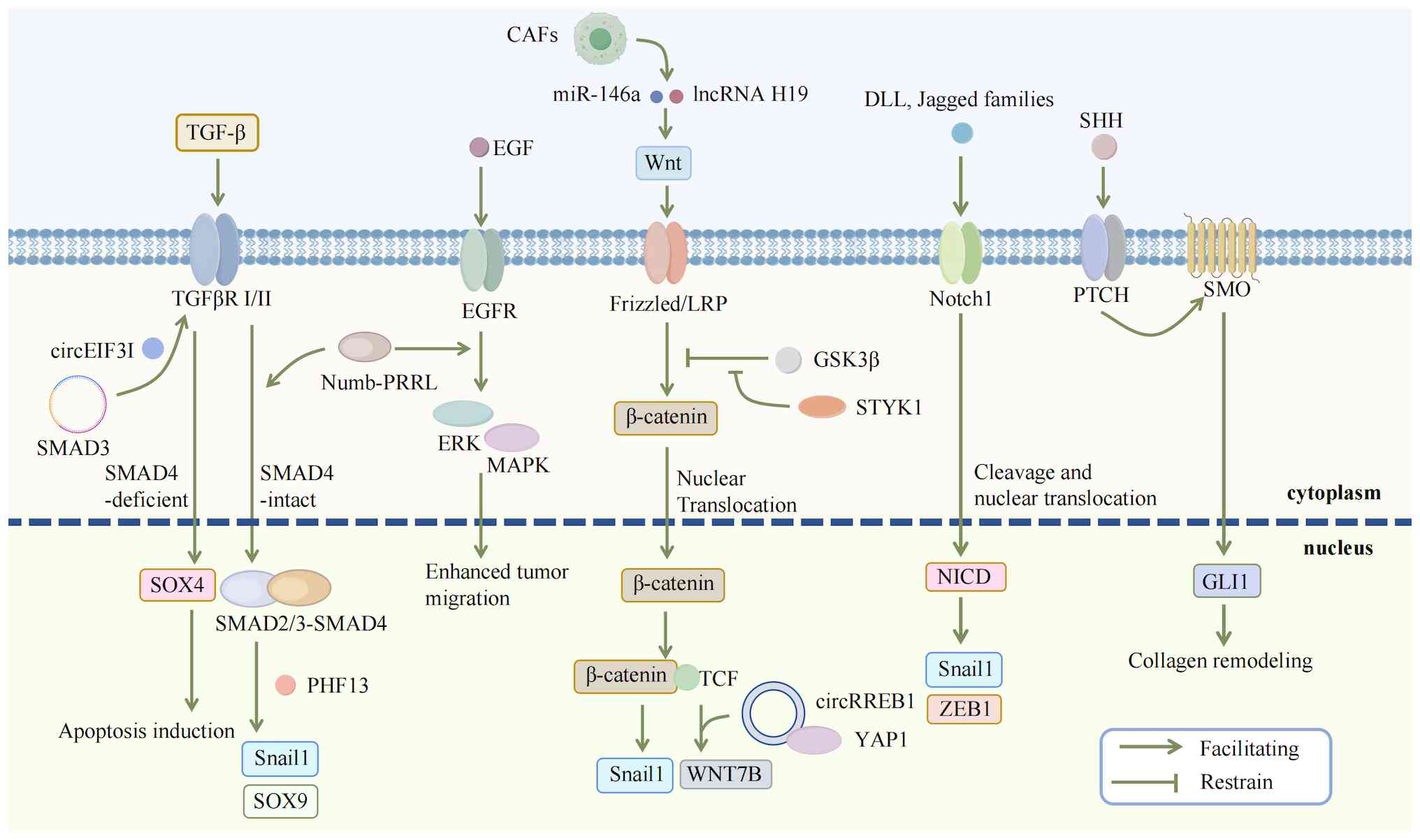 | Figure 5The dual role of signaling and
pathway crosstalk in pancreatic cancer EMT. The role of the TGF-β
pathway in EMT of pancreatic cancer is context-dependent. In the
presence of SMAD4, TGF-β not only activates the SMAD2/3-SMAD4
complex to regulate key transcription factors such as Snail and
Twist but also, with the assistance of PHF13, maintains the
super-enhancer activity of SNAI1 and SOX9, ensuring sustained
expression of EMT genes. Conversely, SMAD4 loss switches SOX4 from
an oncogenic to a pro-apoptotic function. CircEIF3I enhances
pathway activation by strengthening the interaction between SMAD3
and TGFβRI. Under TGF-β1 stimulation, Numb-PRRL activates the
SMAD2/3-Snail pathway and forms a cross-activation loop with
Notch1, synergistically promoting EMT. TGF-β also exhibits
crosstalk with the EGF-EGFR-ERK/MAPK pathway to collectively
enhance cancer cell migration and invasion. Intracellularly, FAM83A
binds to β-catenin to disrupt the degradation complex, an effect
potentiated by BLK phosphorylation, jointly promoting β-catenin
nuclear translocation and Snail transcription; STYK1 maintains
pathway activity by sequestering GSK3β into multivesicular bodies.
Non-coding RNAs contribute to this regulation, such as circRREB1
binds to YAP1 to promote WNT7B transcription and glycolysis.
Furthermore, the Notch signaling pathway, activated by
membrane-bound ligands (such as DLL and Jagged) from neighboring
cells, and the Hh pathway, initiated by secreted ligands (such as
SHH) binding to the PTCH receptor, also participate in the
fine-tuned regulation of EMT. EMT, epithelial-mesenchymal
transition; TGF-β, transforming growth factor-β; SMAD4, Sma- and
Mad-related Protein 4; PHF13, PHD Finger Protein 13; SOX, SRY-Box
Transcription Factor; TGFβRI, Transforming Growth Factor-beta
Receptor I; EGF, Epidermal Growth Factor; EGFR, Epidermal Growth
Factor Receptor; ERK, Extracellular Signal-Regulated Kinase; MAPK,
Mitogen-Activated Protein Kinase; FAM83A, Family With Sequence
Similarity 83 Member A; BLK, B Lymphoid Tyrosine Kinase; STYK1,
Serine/Threonine/Tyrosine Kinase 1; GSK3β, Glycogen Synthase Kinase
3 Beta; YAP1, Yes-Associated Protein 1; DLL, Delta-like ProteinHh,
Hedgehog; SHH, Sonic Hedgehog; PTCH, Patched. |
Epigenetic programming: epigenetic
memory for stabilizing EMT states
DNA methylation plays a core role in the EMT of
PDAC by regulating the tumor microenvironment, epigenetic landscape
and signaling pathway networks. In the tumor microenvironment, PDAC
cells with organ-specific metastatic potential can induce genomic
DNA methylation changes in CAFs, for example, hypermethylation of
metabolic genes NQO1 and ALDH1a3 leads to downregulation of their
mRNA expression. This metabolic reprogramming provides a metabolic
basis for CAFs to support the EMT process and the heterogeneity of
CAFs further affects the transmission of EMT-related signals
through epigenetic regulation (129,130).
Subtype-specific differences in the epigenetic
landscape are another key aspect of EMT regulation: In the basal
subtype of PDAC, key EMT pathways such as TGF-β are abnormally
activated due to epigenetic modifications and the methylation
imbalance of pathway genes enhances signal activity, directly
triggering the transition of epithelial cells to a mesenchymal
phenotype; the classical subtype indirectly affects the EMT process
by regulating the methylation status of pancreatic
development-related transcription factors (131). At the molecular mechanism level,
high expression of DNA methyltransferase 1 (DNMT1) is a core
driving factor: It targets and silences the promoters of EMT
suppressors such as Krüppel-like factor 4, thereby relieving the
inhibition that these suppressors originally exert on EMT
transcription factors such as Snail and Twist (132,133). Meanwhile, DNMT1 forms a negative
feedback regulatory axis with miR-148a, whereby DNMT1
overexpression in pancreatic cancer drives hypermethylation of the
miR-148a promoter to suppress miR-148a expression, while miR-148a
directly targets the 3'UTR of DNMT1 mRNA to inhibit its own
upstream regulator. Disruption of this inhibitory loop (that is,
the loss of miR-148a-mediated restraint on DNMT1) exacerbates the
methylation-dependent silencing of EMT-related genes, thereby
further promoting EMT (134).
At the signaling pathway level, abnormal
methylation of DNA methylation-driven genes (such as GPRC5A) can
activate the PI3K-AKT pathway; methylation silencing of HIP1R
enhances the migration and invasion ability of tumor cells via this
pathway and this process has crosstalk with downstream signals of
KRAS specifically, during pancreatic acinar-ductal metaplasia
(ADM), a pre-neoplastic lesion of pancreatic cancer. This crosstalk
between KRAS downstream pathways is achieved through the
coordinated activation of the PI3K pathway (which regulates cell
proliferation and survival) and the Rho/Rac/Cdc42 GTPase pathway
(which modulates cytoskeletal remodeling and morphological
transformation) (130,135,136). Notably, the DNA methylation
'memory' formed during pancreatic acinar-ductal metaplasia can
continuously affect PI3K and Rho GTPase signals even in the absence
of KRAS mutations, providing an epigenetic basis for the continuous
activation of EMT (108). In
addition, changes in global DNA methylation levels also participate
in cytoskeletal reorganization by affecting the β-sheet structure
of proteins, ultimately supporting EMT-mediated tumor invasion and
metastasis (137).
Histone modifications play a core role in the EMT
of pancreatic cancer by constructing a multi-level epigenetic
regulatory network, with mechanisms involving the synergy and
crosstalk of multiple modification types such as methylation,
acetylation and ubiquitination. At the level of histone
methylation, the protein arginine methyltransferase (PRMT) family
exhibits significant functions; PRMT1 activates the Wnt pathway by
binding to the β-catenin promoter, or enhances the transcriptional
activity of Gli1 via methylation to induce the expression of
EMT-related genes (such as ZEB1) (138,139); PRMT5 upregulates β-catenin via
the EGFR/AKT/β-catenin axis, promoting the migration and invasion
of pancreatic cancer cells (140). Among lysine methyltransferases
(KMTs) loss of SETD2 accelerates KRAS-driven acinar-ductal
metaplasia and EMT by continuously activating AKT and
downregulating α-catenin (141);
KMT5A upregulates stem cell and EMT-related genes by inducing ROR1
expression (142), KMT5C
indirectly inhibits EMT by regulating epithelial transcription
factors such as FOXA1 (143).
The functional differentiation of histone demethylases (KDMs) is
also critical: KDM2B activates the Hippo signal by inhibiting MOB1,
or inhibits epithelial markers (such as CDH1) by regulating H1A
ubiquitination, promoting migration and invasion (144,145). KDM3A is activated by HIF1α under
hypoxic conditions, initiating EMT by upregulating DCLK1 (146). KDM4B directly activates ZEB1
transcription in response to TGF-β-induced EMT (147). KDM5A is regulated by NOX4 under
hypoxic conditions, promoting invasion and metastasis by activating
SNAIL1 (148). Loss of KDM6A
enhances the mesenchymal phenotype by activating the activin A-p38
pathway (149).
The dynamic balance of histone acetylation is
another important node in EMT regulation. p300/CBP activates GATA6
expression via H3K27ac, which directly or indirectly inhibits EMT
(150,151). PCAF forms a complex with p300,
activating miR200c transcription by acetylating ZEB1 and
downregulation of the miR200 family is key to maintaining the
mesenchymal phenotype (152).
Among histone deacetylases (HDACs): HDAC1 and HDAC2 form a
Snail/HDAC1/HDAC2 inhibitory complex to downregulate E-cadherin,
enhancing tumor invasiveness (153,154); SIRT1 regulates the Wnt pathway
by deacetylating β-catenin, participating in the process of
pancreatic acinar-ductal metaplasia (155).
Histone ubiquitination affects EMT by regulating
chromatin structure and transcription factor activity.
Ring1A/Ring1B of the PRC1 complex is recruited by Snail with the
assistance of EZH2 to inhibit E-cadherin transcription (156). Overexpression of Bmi1 directly
promotes EMT by downregulating E-cadherin (157). Among deubiquitinases: USP22
upregulates ZEB1 and Snail by activating FAK signals, reducing
E-cadherin expression (158) and
USP28 activates the Wnt/β-catenin pathway by stabilizing FOXM1,
inducing the expression of EMT-related genes (159).
These mechanisms collectively constitute a complex
network of histone modification regulating EMT. Microenvironmental
factors such as hypoxia and inflammation further amplify regulatory
effects by affecting the activity of modification enzymes (such as
activation of KDM3A and KDM5A under hypoxia) (146,148). Such regulation is not only
involved in tumor invasion and metastasis but also associated with
chemotherapy resistance and stem cell properties, providing
multi-dimensional targets for the development of EMT intervention
strategies targeting epigenetic modifications.
NcRNAs regulatory networks: Modulators
of signaling pathways
NcRNAs construct a complex regulatory system for
pancreatic cancer EMT through multi-dimensional interaction
networks with proteins, covering aspects such as direct regulation
of core EMT factors, activation of key signaling pathways,
remodeling of the metabolic microenvironment and regulation of
immune-related EMT paracrine signals.
At the level of circular RNAs (circRNAs) circRTN4
maintains the protein stability of RAB11FIP1 by masking the
ubiquitination site at Lys578 of RAB11FIP1, continuously activating
downstream EMT genes (102).
circEIF3I promotes the aggregation of TGFβ receptor I in early
endosomes and the phosphorylation of SMAD3 by simultaneously
binding to AP2A1 and SMAD3, driving the expression of
invasion-related genes such as MMP2/9 (108). The newly identified circRREB1
promotes the nuclear localization of YAP1 by binding to the RRM
domain of YAP1, activating WNT7B transcription and the
Wnt/β-catenin pathway and upregulating EMT transcription factors
such as Snail and Twist (120).
These circRNAs form functional complexes with proteins through
specific domains, precisely regulating key signal nodes of EMT.
lncRNAs exhibit bidirectional regulatory
characteristics: in positive regulation, ZFAS1 maintains EMT
activity through an AMPK-ZEB1 positive feedback loop under
metabolic stress (160).
FGD5-AS1, upregulated by the IL-6/STAT3 pathway, enhances the
acetylation of STAT3 by bridging p300 and STAT3, activating the
STAT3/NF-κB pathway and inducing M2 macrophage polarization, which
reinforces EMT via paracrine TGF-β (161). LINC00842 binds to the inhibitory
domain of PGC-1α in a high-glucose environment, maintaining its
activity to support EMT-related fatty acid synthesis metabolism
(162). In negative regulation,
MTSS1-AS accelerates the degradation of MZF1 by promoting the
interaction between MZF1 and STUB1, upregulating the EMT suppressor
MTSS1 (163). KCNK15-AS1
activates PTEN by mediating REST degradation, inhibiting the
expression of Vimentin driven by the PI3K/Akt pathway (164). The newly identified MEG3, as a
tumor-suppressive lncRNA, upregulates E-cadherin expression by
sponging miR-421 and can also regulate SLFN5 expression by
targeting miR-146b-5p, dualistically inhibiting the EMT process
(165,166).
MicroRNAs (miRNAs) play a significant role in
hypoxic adaptation regulation: miR-301a is transcriptionally
activated by HIF-2α under hypoxic conditions, inhibiting the
tumor-suppressive branch of the TGF-β pathway by targeting SMAD4,
while downregulating PTEN and TP63 to enhance Akt activity and
upregulate mesenchymal markers, respectively, forming a
'hypoxia-HIF-2α-miR-301a-EMT' regulatory axis (167). Exosomal miRNAs spread the EMT
phenotype through intercellular communication; for example, the
absence of exosomal miR-128-3p leads to overexpression of its
target gene Bmi1, which not only promotes EMT but also enhances
drug efflux capacity, being associated with gemcitabine resistance
in pancreatic cancer (168).
The transfer (t)RNA-derived stress-induced RNAs
(tiRNAs) RNA-Val-CAC-2 prevents the ubiquitination and degradation
of FUBP1 by binding to the RRM domain of FUBP1; stabilized FUBP1
accumulates in the FUSE region of the c-Myc promoter, activating
its transcription to drive the expression of EMT effector genes
such as N-cadherin and Twist1 (169).
These mechanisms collectively reveal the molecular
basis of ncRNAs regulating pancreatic cancer EMT through a cascade
mode of 'protein stability regulation-signaling pathway
activation-metabolic adaptation-phenotype spread.'
Microenvironmental factors such as hypoxia and metabolic stress
further amplify regulatory effects by selectively upregulating
specific ncRNAs (such as ZFAS1, miR-301a under hypoxia). Such
regulatory networks are not only involved in tumor invasion and
metastasis but also closely associated with chemotherapy
resistance, providing diverse targets for the development of EMT
intervention strategies targeting ncRNAs.
Targeted therapeutic strategies for EMT:
From basic research to clinical translation
Given the core role of EMT in PC invasion,
metastasis, maintenance of stem cell properties and treatment
resistance, direct targeting of EMT-related pathways has become a
highly attractive therapeutic direction. This strategy aims to
reverse the invasive phenotype of tumor cells, restore their
sensitivity to conventional treatments and inhibit metastatic
spread by interfering with upstream EMT-inducing signals, core
transcriptional regulatory networks, or downstream effector
molecules. Currently, numerous preclinical studies and several
clinical trials are exploring multiple EMT-targeted strategies,
covering small-molecule inhibitors, natural compounds, gene therapy
and innovative combination therapeutic regimens.
Inhibiting upstream signaling pathways
that drive EMT
The initiation and maintenance of EMT are driven by
the continuous activation of multiple upstream signaling pathways,
forming a complex regulatory network. Directly targeting the source
of these signals represents a fundamental strategy to inhibit EMT
and its associated malignant phenotypes in pancreatic cancer.
Current research has focused on several key pathways, with the
TGF-β pathway being one of the most intensively studied. The TGF-β
signaling pathway is a potent inducer of EMT and its aberrant
activation is closely linked to disease progression and poor
prognosis (57). Clinical
translation efforts are exploring combinations, such as TGFβRI
inhibitor with nal-IRI/5-FU/LV chemotherapy, in phase Ib/II trials.
Preclinical studies showed that this combination could inhibit
tumor invasion and prolong survival in mouse models by inducing the
tumor suppressor gene CCDC80 and blocking EMT (170). Repurposed drugs have also shown
unexpected efficacy; the antifungal agent itraconazole suppresses
the TGF-β/SMAD2/3 pathway to inhibit EMT, invasion and migration
(171), while the NSAID
indomethacin blocks TGF-β-mediated EMT by upregulating E-cadherin
and downregulating N-cadherin and Snail (172). The regulatory network is highly
complex, involving molecules such as Menin, which enhances
TGF-β-mediated EMT by inhibiting C/EBPβ and autocrine TGF-β1, which
can form a self-sustaining feed-forward loop via the ALK5-MEK-ERK
signal, antagonizing the effects of exogenous TGF-β1 (173,174). Furthermore, super-enhancers can
regulate TGFBR2 expression, offering an epigenetic target (175). Natural products provide
additional resources; extracts from Cynanchum paniculatum
inhibit EMT by suppressing the TGF-β1/Smad2/3 pathway (176) and baicalein disrupts the
TGF-β/FTO/ZEB1 axis by downregulating the m6A demethylase FTO,
reducing ZEB1 mRNA stability (177). However, the dual role of TGF-β
as both a tumor suppressor and promoter necessitates careful
consideration, as some agents such as calycosin can inhibit
proliferation but paradoxically promote EMT by upregulating TGF-β1
(178). A multi-faceted
intervention system has thus been established, encompassing TGFβRI
inhibitors, approved drugs, natural compounds and combination
therapies with chemotherapy to improve drug penetration and
clinical translation potential.
Beyond TGF-β, the Wnt/β-catenin and Hh pathways are
critical targets. The Wnt/β-catenin pathway is inhibited by various
natural products: oridonin suppresses the pathway and reduces
β-catenin levels to inhibit migration and EMT (179). Trametes robiniophila
(Huaier) extract inhibits proliferation, migration and EMT by
downregulating β-catenin (180)
and high-dose vitamin C blocks invasion and metastasis by
suppressing Wnt/β-catenin-mediated EMT (181). At the molecular level, restoring
the expression of the Wnt antagonist DKK3 blocks hypoxia-induced
nuclear translocation of β-catenin and reverses EMT, enhancing
gemcitabine's effect (182).
Silencing molecules such as LRRFIP1 enhances β-catenin
phosphorylation and reduces its nuclear localization, while
silencing GPX2 downregulates key Wnt pathway molecules, both
effectively reversing the EMT phenotype (183,184). Similarly, CHRNB2 exerts
anti-tumor effects by downregulating β-catenin pathway activity via
a non-acetylcholine-dependent mechanism (185). Epigenetic regulation also plays
a role, as TET1 catalyzes DNA hydroxymethylation in the SFRP2
promoter, activating this Wnt inhibitor and blocking both canonical
and non-canonical Wnt signaling (186). The Hh pathway maintains cancer
stem cell properties and promotes EMT by regulating transcription
factors such as Gli. Curcumin exerts multi-target effects,
reversing hypoxia-induced EMT by suppressing Hh pathway activation
(downregulating SHH, SMO, GLI1) and inhibiting crosstalk between
CAFs and tumor cells (107,128,187). The resveratrol derivative
triacetyl resveratrol (TCRV) inhibits EMT transcription factors
such as Zeb1 by upregulating the miR-200 family and simultaneously
inhibits the SHH pathway (188).
Another natural product, α-mangostin, targets Hh pathway molecules
(Gli1 and Smoothened) and core EMT-TFs (Snail and Slug); when
encapsulated in PLGA nanoparticles, it effectively targets Shh
pathway-related genes in vivo, reducing cancer stem cell
markers and inhibiting tumorigenesis (189,190). Sedum sarmentosum Bunge
extract also downregulates Hh pathway activity (191). A significant challenge is the
compensatory activation of other pathways; for instance, Hh pathway
inhibitors (SHhi) can improve drug delivery by enhancing vascular
patency but may induce an FGF-driven EMT phenotype. This can be
addressed by a dual inhibition strategy combining SHhi with FGFR
inhibitors such as infigratinib, which reverses EMT while retaining
improved vascular permeability (192).
Interventions targeting other key pathways, such as
PI3K/Akt/mTOR and MAPK/ERK, further enrich the therapeutic arsenal.
The PI3K/Akt/mTOR pathway is inhibited by agents such as the Aurora
kinase A inhibitor alisertib, which reduces N-cadherin and
upregulates E-cadherin to block EMT while inducing cell cycle
arrest and autophagy (193).
Natural products such as betulinic acid and irisin inhibit EMT and
stemness by activating AMPK to suppress mTOR signaling (194,195) and plumbagin inhibits the
PI3K/Akt/mTOR pathway while synergistically regulating p38 MAPK and
AMPK activation (196).
Inhibiting KIN17 downregulates this pathway, upregulating
E-cadherin and reducing vimentin and N-cadherin (197), while arsenic trioxide reverses
EMT and enhances chemosensitivity by inhibiting TIMP1-mediated
PI3K/Akt/mTOR activation (198).
The MAPK/ERK pathway is another key target. Curcumin inhibits
IL-6-mediated ERK and NF-κB phosphorylation by suppressing PSC
activation and IL-6 secretion under hypoxia, thereby disrupting
tumor-stroma crosstalk (199).
HNRNPA2B1 promotes EMT by activating the ERK/Snail axis (200), whereas miR-338-5p inhibits the
EMT process by targeting EGFR and blocking EGF-induced MAPK/ERK
signaling (201). The TAp73
protein inhibits basal and TGF-β-induced ERK activation in a
SMAD4-dependent manner, upregulating E-cadherin and downregulating
Snail to inhibit migration (202). Targeting receptor tyrosine
kinases such as MET and AXL is also promising. The inhibitor
NPS-1034 targets the MET/PI3K/AKT axis to suppress EMT and
migration, shows synergistic effects with chemotherapy and exhibits
immunomodulatory potential by inducing type I interferon and TNF
(203). Similarly, a novel
AXL/triple angiokinase inhibitor effectively suppresses AXL and
lung metastasis, with its mechanism involving EMT inhibition
(204). Furthermore, inhibiting
the HGF/c-MET pathway not only directly inhibits tumor growth but
also enhances cytotoxic T-cell infiltration by reducing TGF-β
secretion, linking EMT inhibition to immune regulation (205).
In summary, a comprehensive intervention system
targeting upstream signaling pathways of EMT has been established
(Table I). This system includes
diverse strategies: Blocking the TGF-β pathway with inhibitors and
repurposed drugs, targeting the Wnt/β-catenin and Hh pathways with
natural products, molecular interventions and combination therapies
and inhibiting pathways such as PI3K/Akt/mTOR and HGF/c-MET with
specific inhibitors that can synergize with chemotherapy. These
multi-dimensional approaches, validated in preclinical studies, aim
to reverse the invasive phenotype, restore treatment sensitivity
and inhibit metastasis, providing a strong rationale for their
clinical translation to address therapeutic resistance in
pancreatic cancer.
 | Table ICore strategies targeting EMT
signaling pathways. |
Table I
Core strategies targeting EMT
signaling pathways.
| First author/s,
year | Targeted
pathway | Key target(s) | Reresentative
intervention(s) | Mechanism and
effects | Research model | (Refs.) |
|---|
| Hong et al,
2020 | TGF-β Signaling
Pathway | TGFβRI | Vactosertib | Blocks receptor
kinase activity; inhibits SMAD2/3 phosphorylation and Snail/ZEB1
expression; reverses EMT. | Preclinical models,
pancreatic tumor mouse models | (170) |
| Zhao et al,
2024 | | TGFβRI | Riboflavin (Vitamin
B2) | Directly binds and
inhibits TGFβRI, blocking TGF-β signaling and inhibiting EMT and
metastasis. | In vitro and
in vivo models | (206) |
| Chen et al,
2018 | | TGF-β/SMAD
axis | Itraconazole | Suppresses the
TGF-β/SMAD2/3 signaling pathway, inhibiting EMT, invasion and
migration. | Pancreatic cancer
cells, animal models | (171) |
| Sezer et al,
2022 | | TGF-β-mediated
EMT | Indomethacin | Upregulates
E-cadherin, downregulates N-cadherin and Snail, blocking
TGF-β-induced EMT. | Pancreatic cancer
cells models | (172) |
| Cheng et al,
2019 | | Menin/C/EBPβ
axis | Menin inhibition
(mechanism study) | Menin enhances
TGF-β signal-mediated EMT by inhibiting C/EBPβ expression; exhibits
prometastatic effects when C/EBPβ is absent. | Pancreatic cancer
cells models | (173) |
| Cheng et al,
2019 | | TGF-β signaling
feedback | -(Mechanism
study) | Autocrine TGF-β1
forms a self-sustaining feed-forward loop via ALK5-MEK-ERK,
antagonizing exogenous TGF-β1 effects. | Pancreatic cancer
cells models | (173) |
| Zhu et al,
2020 | | TGFBR2 epigenetic
regulation | -(Mechanism
study) | Super-enhancers
regulate TGFBR2 expression, affecting TGF-β signaling activity and
pancreatic cancer progression. | Pancreatic cancer
cells models | (175) |
| Cheng et al,
2024 | | TGF-β/Smad2/3
axis | Cynanchum
paniculatum extract | Suppresses the
TGF-β1/Smad2/3 signaling pathway, exerting EMT-inhibiting effects
at non-cytotoxic doses. | In vitro
cell models and in vivo animal models | (176) |
| Ungefroren et
al, 2021 | | TGF-β/FTO/ZEB1
axis | Baicalein | Downregulates m6A
demethylase FTO, enhancing m6A modification of ZEB1 mRNA and
reducing its stability, disrupting EMT. | Pancreatic cancer
cells models | (174) |
| Zhao et al,
2025 | Wnt/β-catenin
Signaling Pathway | β-catenin nuclear
translocation | Oridonin | Inhibits
Wnt/β-catenin pathway, reduces cytoplasmic and nuclear β-catenin
levels, suppressing migration and EMT. | Pancreatic cancer
cells models and in vivo animal models | (177) |
| Zhou et al,
2020 | | Wnt/β-catenin
pathway | Trametes
robiniophila (Huaier) extract | Suppresses
Wnt/β-catenin pathway (downregulates β-catenin), inhibiting
proliferation, migration, invasion and EMT. | Pancreatic cancer
cells and animal models | (180) |
| Kim et al,
2022 | |
Wnt/β-catenin-mediated EMT | High-dose Vitamin
C | Inhibits glycolysis
and Wnt/β-catenin-mediated EMT, blocking cancer cell growth and
metastasis. | Pancreatic cancer
cells models | (181) |
| Guo et al,
2015 | | DKK3 | DKK3 gene
restoration | Antagonizes Wnt
signaling, blocks hypoxia-induced β-catenin nuclear translocation,
reverses EMT, enhances gemcitabine efficacy. | Pancreatic cancer
Bxpc-3 cells models, clinical sample analysis and animal
models | (182) |
| Douchi et
al, 2015 | | β-catenin
degradation complex | Silencing
LRRFIP1 | Enhances β-catenin
phosphorylation and reduces its nuclear localization by targeting
the degradation complex, reversing EMT phenotype. | Pancreatic cancer
cells models, animal models | (183) |
| Li et al,
2020 | | Wnt pathway key
molecules | Silencing GPX2 | Downregulates key
Wnt pathway molecules, inhibiting Wnt/β-catenin pathway and
reversing EMT. | Pancreatic cancer
cells models, clinical research | (184) |
| Qin et al,
2022 | | β-catenin
pathway | CHRNB2 | Downregulates
β-catenin pathway activity via a non-acetylcholine-dependent
mechanism, inhibiting migration and invasion; associated with EMT
inhibition. | Pancreatic cancer
cells models, clinical research | (185) |
| Wu et al,
2019 | | SFRP2 epigenetic
activation | TET1 | Catalyzes DNA
hydroxymethylation in the SFRP2 promoter, initiating its
transcriptional activation, inhibiting canonical/noncanonical Wnt
pathways and blocking EMT. | Clinical sample
analysis, cell and animal models | (186) |
| Cao et al,
2017 | Hedgehog Signaling
Pathway | SMO/Gli | Curcumin | Reverses
hypoxia-induced EMT by downregulating SHH, SMO, GLI1 expression;
also targets other pathways (EGFR/Notch, PSC activation). | Pancreatic cancer
cells models | (187) |
| Ma et al,
2019 | | Hh pathway and
EMT-TFs | α-Mangostin | Targets Hh pathway
molecules (Gli1, Smoothened) and core EMT-TFs (Snail, Slug),
weakening CSC properties and blocking EMT. | Pancreatic cancer
cells models | (190) |
| Verma et al,
2016 | | Shh pathway-related
genes | α-Mangostin-PLGA
nanoparticles | Specifically
targets Shh pathway-related genes in KPC mice, reducing CSC marker
expression and inhibiting tumorigenesis and liver metastasis. | KPC transgenic
mouse model, cell model | (189) |
| Bai et al,
2016 | | Hh pathway | Sedum
sarmentosum extract (SSBE) | Downregulates Hh
pathway activity, inhibits cancer cell proliferation, weakens stem
cell properties, blocks EMT. | Pancreatic cancer
cells model, animal models | (191) |
| Chaudhuri et
al, 2024 | | SHHi-induced FGF
compensation | SHHi + FGFR
inhibitor (such as Infigratinib) | SHHi improves drug
delivery but induces FGF-driven EMT; combination reverses EMT while
retaining improved vascular permeability. | Preclinical
studies | (192) |
| Fu et al,
2019 | | Shh pathway and
miR-200 | Triacetyl
resveratrol (TCRV) | Upregulates miR-200
family to inhibit Zeb1, inhibits Shh pathway activity reducing
Cyclin D1 and Bcl-2, synergistically inhibiting EMT. | Pancreatic cancer
cells model, animal model | (188) |
| Wang et al,
2015 | Other key signaling
pathways (PI3K/Akt/mTOR, MAPK/ERK, HGF/c-MET) | PI3K/Akt/mTOR | Alisertib (Aurora
kinase A inhibitor) | Inhibits
PI3K/Akt/mTOR pathway, reduces N-cadherin, upregulates E-cadherin,
blocks EMT; induces G2/M arrest and autophagy. | Pancreatic cancer
cells model | (193) |
| Sun et al,
2019 | | AMPK/mTOR | Betulinic acid | Activates AMPK,
suppressing downstream mTOR signaling, inhibiting EMT and stem cell
properties. | Pancreatic cancer
cells model | (194) |
| Wang et al,
2015 | | PI3K/Akt/mTOR and
p38 MAPK | Plumbagin | Inhibits
PI3K/Akt/mTOR, synergistically regulates p38 MAPK and AMPK
activation, upregulates E-cadherin, downregulates N-cadherin,
blocks EMT, induces autophagy. | Pancreatic cancer
cells model, animal model | (196) |
| Li et al,
2020 | | MAPK/ERK (IL-6
mediated) | Curcumin | Suppresses hypoxic
PSC activation and IL-6 secretion, inhibiting IL-6-mediated ERK and
NF-κB phosphorylation, interfering with tumor-stroma interactions
and EMT. | Pancreatic cancer
cells models | (199) |
| Sun et al,
2021 | | MAPK/ERK (EGF
induced) | miR-338-5p | Directly targets
EGFR, blocking EGF-induced MAPK/ERK pathway activation, inhibiting
EMT and metastasis. | Pancreatic cancer
cells models, clinical research | (201) |
| Luan et al,
2024 | | HGF/c-MET
pathway | NPS-1034 (MET/AXL
inhibitor) | Inhibits
MET/PI3K/AKT axis-induced EMT, suppresses migration; synergizes
with chemotherapy; shows immunomodulatory potential. | Pancreatic cancer
cells models | (203) |
| Mekapogu et
al, 2023 | | HGF/c-MET pathway
and Immunity | c-MET
inhibitors/antibodies | Inhibiting
HGF/c-MET directly inhibits tumor growth/metastasis and increases
cytotoxic T-cell infiltration by reducing TGF-β secretion. | Animal models and
cells models | (205) |
Targeting core EMT regulators
In addition to blocking upstream signals, directly
targeting the core molecules that execute the EMT program,
specifically EMT-transcription factors (EMT-TFs) and the epigenetic
mechanisms governing their expression, represents a more precise
therapeutic strategy.
A primary approach involves inhibiting key EMT-TFs
such as Snail, Slug, ZEB and Twist. This can be achieved through
diverse mechanisms. For instance, miRNAs offer precise regulation:
miR-34a directly targets Snail1 and Notch1 (83), miR-539 inhibits TWIST1 (101) and miR-200b-3p suppresses ZEB1
(204). Nutrient deprivation
strategies, such as arginine deprivation, can simultaneously
downregulate multiple EMT-TF families (Snail, Slug and Twist) while
upregulating E-cadherin and inhibiting invasion (100). Natural products also exhibit
multi-target effects; α-mangostin inhibits Snail and Slug (190), while the resveratrol derivative
TCRV targets Snail, Slug and Zeb1 (188). Furthermore, the protein DACH1
can directly bind to SNAI1 to inhibit its transcriptional activity,
thereby blocking EMT and promoting apoptosis (207). These strategies collectively
reverse the EMT phenotype by upregulating epithelial markers and
downregulating mesenchymal markers.
Beyond direct TF targeting, interventions against
epigenetic and post-transcriptional mechanisms have emerged as
powerful tools. ncRNAs function as critical regulators: lncRNA GAS5
acts as a ceRNA for miR-221 to upregulate SOCS3, reversing EMT and
gemcitabine resistance (208),
whereas LINC00958 promotes EMT by sponging miR-330-5p (209). Conversely, lncRNA GATA6-AS1
inhibits EMT by reducing SNAI1 mRNA stability in an m6A-dependent
manner by inhibiting FTO (210).
The m6A modification itself is a key regulatory layer; the 'eraser'
FTO promotes EMT and can be targeted for intervention (212) and the 'demethylase' ALKBH5
exerts anti-tumor effects by demethylating mRNAs of iron metabolism
regulators, indirectly leading to SNAI1 degradation (211). Histone modifications are another
major target. HDAC inhibitors, such as a novel dual HDAC2/6
inhibitor and the pan-HDAC inhibitor LAQ824, can reverse
TGF-β-induced EMT, induce apoptosis and enhance antigen
presentation (212,213). Similarly, the histone
methyltransferase inhibitor DZNep reshapes miRNA expression to
block the TGF-β signaling pathway and associated EMT (214,215).
In summary, a two-dimensional intervention system
has been established, encompassing the direct inhibition of EMT-TFs
and the regulation of their epigenetic landscape (Table II). Preclinical studies confirm
that these strategies can effectively reverse the EMT phenotype,
weaken tumor stemness and invasive capacity and provide precise
molecular targets for overcoming therapeutic resistance in
pancreatic cancer.
| Table IITargeting core EMT regulators and
microenvironment. |
Table II
Targeting core EMT regulators and
microenvironment.
| First author/s,
year | Target
category | Specific
target/molecule | Representative
intervention(s) | Mechanism of
action | Research model
(cell/animal) | (Refs.) |
|---|
| Tang et al,
2017 | Inhibiting EMT
transcription factors (EMT-TFs) | Snail1/Notch1 | miR-34a | Directly targets
and inhibits Snail1 and Notch1 mRNA. | Pancreatic cancer
cells | (83) |
| Yu et al,
2019 | | TWIST1 | miR-539 | Exerts
tumor-suppressive function by targeting TWIST1 mRNA. | Pancreatic cancer
cells | (101) |
| Gui et al,
2017 | | ZEB1 | miR-200b-3p | Inhibits EMT by
directly targeting ZEB1 mRNA. | Pancreatic cancer
cells Animal model | (216) |
| Wang et al,
2020 | | Snail, Slug,
Twist | Arginine
deprivation | Downregulates
multiple EMT-TF families (Snail, Slug, Twist), upregulates
E-cadherin, inhibits MMP-1/9, weakens PI3K/Akt pathway. | Pancreatic cancer
cells | (100) |
| Ma et al,
2019 | | Snail/Slug | α-Mangostin | Exerts multi-target
effects by inhibiting Snail and Slug. | Pancreatic cancer
cells | (190) |
| Fu et al,
2019 | | Snail, Slug,
Zeb1 | Triacetyl
resveratrol | Inhibits multiple
EMT-TFs (Snail, Slug, Zeb1) by upregulating the miR-200 family | Pancreatic cancer
cells | (188) |
| Bu et al,
2016 | | SNAI1 | DACH1
overexpression | Directly binds to
SNAI1 to inhibit its transcriptional activity, upregulates
E-cadherin and promotes apoptosis via Bcl-2 axis. | Pancreatic cancer
cells | (207) |
| Garg et al,
2022 | | FTO | FTO inhibitors | Targeting the m6A
'eraser' FTO reverses EMT and inhibits tumor growth. | Pancreatic cancer
cells | (217) |
| Kim et al,
2023 | | Snail (protein
stability) | ERK3 inhibition
(mechanism study) | Targeting ERK3 may
inhibit Snail activity by preventing ERK3 from enhancing Snail
stability (via inhibiting FBXO11 binding). | Clinical Sample
Analysis, Cell Model | (218) |
| Liu et al,
2018 | Epigenetic and
post-transcriptional regulation | miR-221/SOCS3 | lncRNA GAS5 | Acts as a ceRNA to
'sponge' miR-221, upregulating SOCS3 expression, thereby reversing
EMT and gemcitabine resistance. | Pancreatic cancer
cells, animal model | (208) |
| Chen et al,
2019 | |
miR-330-5p/PAX8 | Silencing
LINC00958 | Silencing this
lncRNA inhibits progression by sponging miR-330-5p to downregulate
PAX8. | Pancreatic cancer
cells, animal model | (209) |
| Zhou et al,
2023 | | FTO/SNAI1 mRNA
stability | lncRNA
GATA6-AS1 | Reduces SNAI1 mRNA
stability in an m6A-dependent manner by inhibiting the m6A
demethylase FTO. | Animal model cells,
model clinical research | (210) |
| Hu et al,
2018 | | NLRP3 promoter | lncRNA XLOC_
000647 | Downregulates
adjacent gene NLRP3 by inhibiting its promoter activity, inhibiting
EMT and invasion. | Pancreatic cancer
cells, animal models, clinical research | (219) |
| Li et al,
2023 | |
miR-140-3p/TRAM2 | circ-STK39 | Acts as a ceRNA for
miR-140-3p, relieving its inhibition of TRAM2 to promote EMT. | Pancreatic cancer
cells, animal models, clinical research | (220) |
| Cao et al,
2024 | | miR-147b/SOCS1 | circTMEM59 | Sponges miR-147b to
upregulate SOCS1, exerting an inhibitory effect on EMT. | Pancreatic cancer
cells, clinical research | (221) |
| Huang et al,
2021 | |
ALKBH5/FBXL5/SNAI1 | ALKBH5 (mechanism
study) | Demethylates mRNA
of iron metabolism regulators (such as FBXL5), indirectly leading
to SNAI1 degradation, inhibiting migration/invasion. | Pancreatic cancer
cells | (211) |
| Su et al,
2023 | | METTL3/AREG mRNA
stability | miR-33a-3p | Targets METTL3 to
inhibit its-mediated m6A modification and stabilization of AREG
mRNA, inhibiting invasion/metastasis. | Pancreatic cancer
cells. animal model | (222) |
| Schiedlauske et
al, 2024 | | HDAC2/HDAC6 | Novel dual HDAC2/6
inhibitor | Upregulates
E-cadherin, reverses TGF-β-induced EMT, induces apoptosis and cell
cycle arrest. | Pancreatic cancer
cells | (212) |
| Jia et al,
2025 | | Pan-HDAC/MHC-I | Pan-HDAC inhibitor
LAQ824 | Inhibits HDAC
activity, blocks EMT, induces apoptosis; enhances chromatin
accessibility of MHC-I genes to promote antigen presentation. | Pancreatic cancer
cells, animal model | (213) |
| Edderkaoui et
al, 2018 | | GSK3B/HDACs | Metavert (dual
inhibitor) | Weakens EMT and
inhibits tumorgrowth/metastasis. | KPC mouse models,
cells model | (223) |
Mody et al,
2016
Mody et al, 2017 | | EZH2/miRNAs
(miR-202, etc.) | 3-deazaneplanocin A
(DZNep) | Reshapes miRNA
expression (such as miR-202 targets TGFBR1/2; miR-663a/miR-4787-5p
target ligands), blocking TGF-β signaling and EMT. | Pancreatic cancer
cells, animal model | (214, 215) |
Disrupting the mesenchymal phenotype and
TME crosstalk
EMT not only alters the characteristics of cancer
cells themselves but also reshapes their interactions with the TME,
particularly with CAFs and the ECM. Disrupting this crosstalk is
therefore a critical strategy to inhibit metastasis.
Targeting tumor stroma and ECM: The dense fibrotic
stroma of PC acts as a physical barrier to drug penetration and
actively drives EMT and immune suppression through the secretion of
various factors (224).
Therapeutic strategies targeting stromal components have shown
promising results. For instance, the oncolytic adenovirus
HEmT-DCN/sLRP6 co-expresses decorin (to degrade ECM) and a Wnt
decoy receptor (sLRP6), thereby improving drug penetration and
directly inhibiting the Wnt/β-catenin pathway to block EMT
(225). In clinical translation,
RNA oligonucleotide STNM01, which targets carbohydrate
sulfotransferase 15 (CHST15), has been shown in phase II trials to
increase tumor-infiltrating T cells and improve patient survival
(226). Additionally, inhibiting
YAP expression can reduce connective tissue growth factor
secretion, suppressing CAF activation and tumor-stroma interactions
(227).
Blocking key tumor-stroma signaling molecules:
Beyond the physical stroma, blocking key signaling molecules
between tumor and stromal cells is equally important.
Pharmacological inhibition of the Gas6/AXL axis, primarily produced
by tumor-associated macrophages and CAFs, can partially reverse EMT
and activate NK cells to inhibit metastasis (228). Similarly, a
nanotechnology-delivered CXCL13 'trap' can counteract the dual
effects of this chemokine, which recruits immunosuppressive
regulatory B cells (Bregs) and directly stimulates EMT, thereby
inhibiting tumor growth (229).
Furthermore, targeted inhibition of the HGF/c-MET pathway has
demonstrated stronger anti-metastatic activity than standard
chemotherapy in animal models (230). These strategies highlight the
potential of simultaneously reversing the EMT phenotype and
activating anti-tumor immunity.
Combination strategies and clinical
translation
Given the complexity of pancreatic cancer,
combining EMT-targeted strategies with conventional therapies is
essential to overcome resistance and achieve synergistic effects
(Table III).
| Table IIICombination strategies and clinical
translation. |
Table III
Combination strategies and clinical
translation.
| First author/s,
year | Therapeutic
strategy | Target/pathway | Representative
intervention(s) | Mechanism of
action | Research model
(cell/animal) | (Refs.) |
|---|
| Li et al,
2019 | Targeting tumor
stroma and ECM | ECM barrier and Wnt
pathway | Oncolytic
adenovirus HEmT-DCN/sLRP6 | Co-expresses
decorin (degrades ECM) and sLRP6 (Wnt decoy); enhances drug
penetration and blocks EMT. | Animal model, cells
model | (225) |
| Fujisawa et
al, 2022 | | CHST15 | STMN01 (RNA
oligonucleotide) | Targets
carbohydrate sulfotransferase 15; increases tumor-infiltrating T
cells and improves survival. | Clinical trial
(patients with unresectable PDAC) | (226) |
| Jiang et al,
2018 | | YAP/CTGF | YAP inhibition | Reduces Connective
Tissue Growth Factor (CTGF) secretion; inhibits CAF activation and
tumor-stroma interactions. | Animal model, cells
model | (227) |
| Ireland et
al L, 2020 | Blocking
Tumor-Stroma Signals | Gas6/AXL axis | Pharmacological
Gas6 inhibitor | Partially reverses
EMT and activates NK cells to inhibit metastasis. | Animal model, cells
model | (228) |
| Shen et al,
2022 | | CXCL13 | CXCL13 'trap'
(nanotechnology) | Blocks
CXCL13-mediated Breg recruitment and direct EMT stimulation;
inhibits tumor growth. | Animal model | (229) |
| Pothula et
al, 2016 | | HGF/c-MET
pathway | c-MET
inhibitors | Inhibits tumor
growth, metastasis and enhances anti-metastatic activity compared
to gemcitabine. | Animal model, cells
model | (230) |
| Mondal et
al, 2017 | Sensitization to
Chemo-/Radiotherapy | EGFR and
miR-205 | EGFR-targeted
micelles (co-deliver Gemcitabine + miR-205) | Reverses EMT and
inhibits growth of drug-resistant tumors. | Animal model, cells
model | (231) |
| Cao et al,
2019 | | AKT/GSK-3β
pathway | β-sitosterol +
Gemcitabine | Inhibits EMT
markers; synergistically enhances inhibition of growth, migration
and invasion. | Animal model, cells
model | (232) |
| Wei et al,
2019 | | Akt pathway | EGCG +
Gemcitabine | Regulates
EMT-related phenotypic switching (such as cadherin switching, ZEB1
downregulation). | Animal model, cells
model | (233) |
| Li et al,
2024 | | TGM2 | GK921 (TGM2
inhibitor) + Cisplatin | Inhibits
TGM2-induced EMT; enhances cisplatin-induced apoptosis and cell
cycle arrest. | Animal model, cells
model | (234) |
| Jin et al,
2018 | | EMT reversal | Hyperthermia +
Gemcitabine | Reverses EMT to
restore sensitivity in gemcitabine-resistant cells. | Pancreatic cancer
cells (such as PANC-1) | (235) |
| Momeny et
al, 2020 | | VEGF receptors | Cediranib
(pan-VEGFR inhibitor) + Radiotherapy | Directly inhibits
EMT and enhances radiosensitivity. | Pancreatic cancer
cells | (236) |
| Mondal et
al, 2016 | | Glucocorticoid
Receptor and Hsp90 | GR-targeted
liposomes (codeliver drug + anti-Hsp90 gene) | Reverses EMT and
induces drug sensitivity. | Animal model, cells
model | (237) |
| Gui et al,
2017 | Combination with
Immunotherapy | HDAC/MHC-I | LAQ824 (pan-HDAC
inhibitor) + ICIs | Upregulates MHC-I
molecules (enhances antigen presentation) while inhibiting
EMT. | Animal model, cells
model | (216) |
| Mekapogu et
al, 2023 | |
HGF/c-MET/TGF-β | c-MET inhibitors +
ICIs | Reduces TGF-β
secretion, relieving immune suppression and increasing cytotoxic
T-cell infiltration. | Animal model, cells
model | (205) |
| Li et al,
2024 | | PD-L1/CXCR4 | Bispecific nanobody
(anti-PD-L1/CXCR4) | Delays EMT by
inhibiting SDF-1/CXCR4 pathway; disrupts TME and promotes T-cell
infiltration. | Cells model | (224) |
Sensitization to chemotherapy/radiotherapy:
Reversing EMT can resensitize tumors to chemotherapy and
radiotherapy. Nanotechnology platforms enable effective
co-delivery; for instance, EGFR-targeted micelles co-delivering
gemcitabine and miR-205 can inhibit the growth of resistant tumors
and reduce EMT (231). Natural
products also show promise in combination regimens. β-sitosterol
and epigallocatechin-3-gallate (EGCG) both synergize with
gemcitabine by inhibiting the AKT/GSK-3β pathway and regulating
EMT-related phenotypic switching, respectively (232,233). The TGM2 inhibitor GK921 enhances
cisplatin-induced apoptosis by inhibiting EMT (234), while hyperthermia can restore
gemcitabine sensitivity in resistant cells by reversing EMT
(235). In radiotherapy, the
pan-VEGFR inhibitor cediranib can directly inhibit EMT and enhance
radiosensitivity (236).
Combination with immunotherapy to remodel the
immune microenvironment: Combining EMT inhibition with
immunotherapy represents another promising direction. The pan-HDAC
inhibitor LAQ824 not only blocks EMT but also upregulates MHC-I
molecules to enhance antigen presentation and tumor immunogenicity
(213). Inhibiting the HGF/c-MET
pathway can reduce TGF-β secretion, thereby relieving
immunosuppression and increasing cytotoxic T-cell infiltration in
the TME (205). Furthermore, a
bispecific nanobody targeting PD-L1 and CXCR4 can delay EMT and
disrupt the immunosuppressive TME, promoting T cell infiltration
(224). These approaches aim to
transform 'cold' tumors into immunoresponsive ones, improving the
efficacy of immune checkpoint inhibitors.
In summary, a multi-pronged therapeutic approach
targeting EMT has emerged, ranging from disrupting the TME to
designing rational combination therapies (Table III). The future challenge lies
in the precise clinical translation of these strategies, leveraging
biomarkers to identify patient populations most likely to
benefit.
Challenges in clinical translation of
EMT-targeted therapies
Despite promising preclinical data, EMT-targeted
therapies for PDAC face substantial translational barriers.
Reliable EMT-specific biomarkers are lacking; traditional markers
fail to capture EMT heterogeneity, single-cell
sequencing-identified hybrid E/M states are hard to translate into
routine assays (237) and CA19-9
reflects tumor burden but not EMT activity (238,239). Systemic toxicity and off-target
effects further narrow the therapeutic window; EMT pathways (such
as TGF-β, Wnt) have critical physiological roles, leading to severe
side effects such as cardiovascular toxicity that halts anti-TGF-β
therapies clinically (240),
while core EMT-TFs (such as Snail and Twist) are 'undruggable' and
upstream inhibition disrupts basal physiology (241).
Tumor cells exhibit robust compensatory mechanisms:
blocking one EMT pathway activates parallel signaling networks,
with combined PI3K/Akt and MAPK inhibition more effective than
monotherapy and EMT-TME crosstalk offsets targeted effects via
CAF-derived pro-EMT signals (242). Additionally, preclinical models
(such as KPC mice) differ from human PDAC in TME, immune context
and pharmacokinetics (243),
while patient-specific factors (genetic heterogeneity and
comorbidities) are underrepresented, exacerbating the translational
gap. Addressing these challenges requires integrated strategies for
biomarker development, improved drug specificity, compensatory
network disruption and model refinement.
Conclusions
EMT serves as an indispensable driver of PDAC
malignant progression, being tightly linked to PDAC's most
devastating clinical characteristics; early metastasis,
chemoresistance and a dismal ~9% 5-year survival rate. As detailed
in the present review, EMT exerts its pathological effects not
through a simple epithelial-mesenchymal binary switch, but via a
spectrum of stable intermediate (hybrid E/M) states that are
well-adapted to PDAC's unique pathophysiology. Specifically, PDAC's
extremely dense desmoplastic microenvironment (fibroproliferative
reaction) acts both as a physical barrier and a key signaling hub:
CAFs and PSCs secrete TGF-β and HGF to activate EMT-TFs, such as
ZEB1 and Snail1). These TFs orchestrate a cascade of molecular
events; they disrupt epithelial adhesion by suppressing E-cadherin
and switching to N-cadherin (enabling cancer cells to detach from
ductal structures); upregulate MMP-14 and MMP-2 to degrade type
I/III collagen (creating invasive channels through the
extracellular matrix) and form a reciprocal 'EMT-stemness-therapy
resistance' axis (via crosstalk between ZEB1 and YAP1 or
miR-200/ZEB1 feedback loops) that sustains CD44+/CD133+ CSCs and
helps evade gemcitabine or radiotherapy.
At the regulatory level, EMT is governed by a
hierarchical, context-dependent network; upstream signals (TGF-β,
Wnt and Notch) converge on core TFs, which are further stabilized
by epigenetic modifications (DNMT1-mediated CDH1 silencing,
HDAC1/2-driven E-cadherin suppression) and ncRNAs (circEIF3I
promoting SMAD3 phosphorylation, lncRNA H19 sponging miR-200b).
Importantly, translational studies confirm that targeting this
network, whether using TGFβR1 inhibitors (vactosertib) to reverse
EMT-driven metastasis, HDAC inhibitors (LAQ824) to restore MHC-I
expression and overcome immune escape, or CAF-targeted agents
(STNM01, a CHST15-targeting RNA oligonucleotide) to reduce stromal
TGF-β, can suppress PDAC progression in preclinical models.
However, these advances also highlight a core challenge: EMT's
phenotypic plasticity allows tumor cells to compensate for
single-target inhibition (for example, Snail1 silencing leads to
Slug upregulation), emphasizing the need for combinatorial
strategies that disrupt the entire EMT network rather than
individual nodes. Collectively, these findings confirm EMT as a
central therapeutic target for PDAC and provide a mechanism-based
framework to guide the design of next-generation interventions.
Among the EMT-targeted strategies summarized, three
approaches show the highest promise for inhibiting PDAC metastasis:
Disrupting CAF-TME crosstalk (such as STNM01 targeting CHST15)
addresses the root of EMT activation in the desmoplastic
microenvironment; combined pathway inhibition (such as Hh + FGFR
inhibitors and TGFβRI inhibitors + chemotherapy) overcomes
compensatory signaling; and epigenetic modulation (such as pan-HDAC
inhibitors) synergistically targets EMT and immunosuppression.
Direct EMT-TF targeting is less feasible due to off-target
risks.
Given EMT's early activation in PanIN lesions,
early intervention relies on three practical directions: using EMT
biomarkers (miR-200/ZEB1 ratio, Vimentin+/CD44v6+ CTCs) to stratify
high-risk populations; repurposing low-toxicity agents (metformin,
aspirin) to block risk factor-driven EMT; and inhibiting upstream
signals (TGF-β and HGF) to halt EMT initiation before invasion.
This strategy intercepts metastasis at its origin, complementing
late-stage therapies.
Future perspectives
While mechanistic insights into PDAC-related EMT
have advanced rapidly, translating these discoveries into clinical
benefits remains limited by unresolved gaps; gaps that must be
addressed to meet the rigor and innovation standards of top-tier
oncology journals. Below is outlined priority research directions
based on the unmet needs highlighted in the present review:
Define molecular signatures of EMT
spectrum states for subtype-specific targeting
The present review emphasized that hybrid E/M
states (rather than full EMT) confer the highest metastatic
potential and chemoresistance in PDAC. However, the molecular
markers that distinguish these states (such as specific EMT-TF,
miRNA, or protein post-translational modification signatures)
remain poorly defined. Current PDAC subtype classifications
(classical compared with quasi-mesenchymal) also lack clear links
to EMT trajectories; for example, high ZEB1 expression correlates
with quasi-mesenchymal subtypes, but how this translates to
therapeutic vulnerability is unclear. Future studies must use
single-cell spatial multi-omics technologies (single-cell RNA
sequencing combined with spatial proteomics) on patient-derived
samples (primary tumors, CTCs, liver metastases) to map EMT
phenotypic landscapes. This will enable identification of
subtype-specific EMT drivers (such as Twist1 for classical PDAC,
ZEB1 for quasi-mesenchymal PDAC) and development of companion
diagnostics (such as circulating miR-200/ZEB1 ratio) to stratify
patients for targeted therapy, avoiding the failure of unselected
trials seen with earlier EMT inhibitors.
Dissect bidirectional TME-EMT crosstalk
to overcome stromal compensation
PDAC's desmoplastic stroma is not just a passive
barrier but an active driver of EMT: CAFs secrete AREG to activate
EGFR/Notch signaling in tumor cells, while PSCs deposit hyaluronan
to amplify mechanical tension-driven EMT. Yet, the field still
lacks a clear understanding of CAF subtype-specific regulation of
EMT (myCAFs compared with inflammatory CAFs) and how EMT
reciprocally reshapes stromal function (for example, tumor-derived
PDGF activating PSCs). Current strategies targeting the stroma
(such as Hh inhibitors) often trigger compensatory EMT via FGF
signaling, highlighting the need for co-targeting approaches.
Future work should use exosome tracing and cell-cell communication
modeling to dissect paracrine loops (such as CAF-derived miR-146a
activating Snail1) and develop rational combinations; for example,
pairing CAF subtype-specific inhibitors (CHST15 antisense
oligonucleotides) with EMT blockers (TGFβR1 inhibitors) to disrupt
the 'EMT-stroma' positive feedback loop without destabilizing
beneficial stromal functions (such as immune cell recruitment).
Address translational bottlenecks in
EMT-targeted therapy
Two critical barriers limit clinical translation,
as highlighted in the present review: The absence of dynamic EMT
monitoring tools and poor in vivo efficacy of EMT
inhibitors. For biomarkers, while CTCs with hybrid E/M phenotypes
correlate with poor prognosis, current CTC detection methods (based
on EpCAM enrichment) miss mesenchymal CTCs with low EpCAM
expression. Future efforts should validate EMT-specific CTC markers
(such as Vimentin/CD44v6) or circulating extracellular vesicle (EV)
cargo (circFARP1, miR-146a) as surrogate endpoints for therapy
response. For drug delivery, EMT inhibitors (such as HDAC
inhibitors, TGF-β antagonists) suffer from low tumor penetration
and off-target toxicity; potential solutions include ECM-targeted
nanoparticles (decorin-functionalized carriers that bind collagen)
or CAF-homing liposomes to deliver EMT-TF siRNAs (such as ZEB1
siRNA) directly to the stroma-tumor interface. Additionally,
clinical trials must incorporate molecular stratification (such as
TGF-β pathway activity, ZEB1 expression) to enrich for responsive
patients, as seen in the phase I/II trial of STNM01 (a CHST15
inhibitor) which improved survival only in PDAC patients with high
CAF levels.
Leverage early EMT intervention to target
modifiable risk factors
A less emphasized finding in the present review is
that modifiable risk factors (smoking, obesity, diabetes) account
for 65.6% of PDAC cases in patients ≤60 years, yet how these
factors drive EMT (such as chronic inflammation activating
NF-κB/Snail1, hyperglycemia upregulating LINC00842/PGC-1α) remains
understudied. Early PDAC lesions (PanIN) already exhibit EMT
features, presenting a window for chemoprevention. Future research
should explore repurposed agents targeting risk factor-driven EMT
(such as metformin inhibiting mTOR/EMT in diabetic patients,
aspirin blocking NF-κB/Snail1 in obese patients) in high-risk
cohorts. Combining these agents with EMT monitoring tools (such as
plasma EMT-related ncRNAs) could enable early intervention to halt
PanIN progression to invasive PDAC, addressing the 'late diagnosis'
dilemma that is the root cause of PDAC's poor prognosis.
Modifiable risk factors account for 65.6% of PDAC
cases in patients ≤60 years, driving EMT via chronic inflammation
or hyperglycemia. Early PanIN lesions already exhibit EMT features,
offering a chemoprevention window. Future research should
prioritize: i) Validating non-invasive EMT biomarkers for high-risk
stratification; ii) evaluating repurposed agents (metformin,
aspirin) in chemoprevention trials; iii) dissecting risk factor-EMT
crosstalk to identify precise early targets. Combining these with
dynamic EMT monitoring can halt PanIN progression to invasive PDAC,
addressing late diagnosis.
In summary, advancing PDAC-EMT research requires a
shift from 'broad EMT inhibition' to 'precision EMT modulation,'
integrating insights into phenotypic heterogeneity, stromal
crosstalk and early intervention. Only by addressing these gaps can
EMT-targeted strategies fulfill their potential to transform PDAC
from a lethal disease to a manageable one.
Abbreviations:
|
ADM
|
acinar-ductal metaplasia
|
|
CAFs
|
cancer-associated fibroblasts
|
|
CDH2
|
n-cadherin
|
|
CHST15
|
carbohydrate sulfotransferase 15
|
|
circRNAs
|
circular RNAs
|
|
CSC
|
cancer stem cell
|
|
CSCs
|
cancer stem cells
|
|
CTCs
|
circulating tumor cells
|
|
DKK3
|
Dickkopf-related protein 3
|
|
DNMTs
|
DNA methyltransferases
|
|
ECM
|
extracellular matrix
|
|
EGCG
|
epigallocatechin-3-gallate
|
|
EMT
|
epithelial-mesenchymal transition
|
|
EMT-TFs
|
EMT transcription factors
|
|
FAK
|
focal adhesion kinase
|
|
Gas6
|
growth arrest-specific protein 6
|
|
HDACs
|
histone deacetylases
|
|
HGF
|
hepatocyte growth factor
|
|
ICIs
|
immune checkpoint inhibitors
|
|
LIF
|
leukemia inhibitory factor
|
|
MET
|
mesenchymal-epithelial transition
|
|
MMPs
|
matrix metalloproteinases
|
|
myCAFs
|
myofibroblastic CAFs
|
|
PanINs
|
pancreatic intraepithelial
neoplasia
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
PDGF
|
platelet-derived growth factor
|
|
PSCs
|
pancreatic stellate cells
|
|
TCRV
|
triacetyl resveratrol
|
|
TGF-β
|
transforming growth factor-β
|
|
TME
|
tumor microenvironment
|
Availability of data and materials
Not applicable.
Authors' contributions
GZ, YW, MW and SH were co-first authors, JW was
correspnding author. JW, GZ, SH, MW, QW and YW designed the study.
JW, GZ and SH wrote the paper. ZX and SL contributed to manuscript
revision and academic quality control. Data authentication is not
applicable. All authors reviewed and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National
Natural Scientific Foundation of China (grant no. 82201694),
Guangxi Youth Science Foundation Project (grant no.2023JJB140089),
Guangxi university young and middle-aged teachers' basic ability
improvement project (grant nos.2022KY0327), Guangxi Science and
technology base and talent project (grant no. 2022AC21033),
Development, PhD project, Guangxi University of Science and
Technology (grant no. 22Z12, 19Z26).
References
|
1
|
Klein AP: Pancreatic cancer epidemiology:
Understanding the role of lifestyle and inherited risk factors. Nat
Rev Gastroenterol Hepatol. 18:493–502. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nieto MA, Huang RY, Jackson RA and Thiery
JP and Thiery JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yuan C, Kim J, Wang QL, Lee AA and Babic
A: PanScan/PanC4 I-III Consortium; Amundadottir LT, Klein AP, Li D,
McCullough ML, et al: The age-dependent association of risk factors
with pancreatic cancer. Ann Oncol. 33:693–701. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yeo TP: Demographics, epidemiology, and
inheritance of pancreatic ductal adenocarcinoma. Semin Oncol.
42:8–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Martini G, Dienstmann R, Ros J, Baraibar
I, Cuadra-Urteaga JL, Salva F, Ciardiello D, Mulet N, Argiles G,
Tabernero J and Elez E: Molecular subtypes and the evolution of
treatment management in metastatic colorectal cancer. Ther Adv Med
Oncol. 12:17588359209360892020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Whatcott CJ, Han H and Von Hoff DD: The
role of the tumor microenvironment in resistance to therapy in
pancreatic cancer. Br J Cancer. 112(Suppl 1): S9–S15. 2015.
|
|
8
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar
|
|
9
|
Gaianigo N, Melisi D and Carbone C: EMT
and treatment resistance in pancreatic cancer. Cancers (Basel). 9.
pp. 1222017, View Article : Google Scholar
|
|
10
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lamouille S, Xu J and Derynck R: Molecular
mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 15:178–196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao W, Zhou J, Deng Z, Gao Y and Cheng Y:
SPOP promotes tumor progression via activation of β-catenin/TCF4
complex in clear cell renal cell carcinoma. Int J Oncol.
49:1001–1008. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aiello NM, Maddipati R, Norgard RJ, Balli
D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, et al:
EMT subtype influences epithelial plasticity and mode of cell
migration. Dev Cell. 45681–695. (e4)2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou P, Li B, Liu F, Zhang M, Wang Q, Liu
Y, Yao Y and Li D: The epithelial to mesenchymal transition (EMT)
and cancer stem cells: Implication for treatment resistance in
pancreatic cancer. Mol Cancer. 16:522017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Venhuizen JH, Jacobs FJC, Span PN and
Zegers MM: P120 and E-cadherin: Double-edged swords in tumor
metastasis. Semin Cancer Biol. 60:107–120. 2020. View Article : Google Scholar
|
|
16
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat Rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sánchez-Tilló E, Lázaro A, Torrent R,
Cuatrecasas M, Vaquero EC, Castells A, Engel P and Postigo A: ZEB1
represses E-cadherin and induces an EMT by recruiting the SWI/SNF
chromatin-remodeling protein BRG1. Oncogene. 29:3490–3500. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nowak E and Bednarek I: Aspects of the
epigenetic regulation of EMT related to cancer metastasis. Cells.
10:34352021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hazan RB, Phillips GR, Qiao RF, Norton L
and Aaronson SA: Exogenous expression of N-cadherin in breast
cancer cells induces cell migration, invasion, and metastasis. J
Cell Biol. 148:779–790. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang YE and Stuelten CH: Alternative
splicing in EMT and TGF-β signaling during cancer progression.
Semin Cancer Biol. 101:1–11. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chastney MR, Kaivola J, Leppänen VM and
Ivaska J: The role and regulation of integrins in cell migration
and invasion. Nat Rev Mol Cell Biol. 26:147–167. 2025. View Article : Google Scholar
|
|
23
|
Hosseini K, Taubenberger A, Werner C and
Fischer-Friedrich E: EMT-induced Cell-mechanical changes enhance
mitotic rounding strength. Adv Sci (Weinh). 7. pp. 20012762020,
View Article : Google Scholar
|
|
24
|
Zhu Y, Herndon JM, Sojka DK, Kim KW,
Knolhoff BL, Zuo C, Cullinan DR, Luo J, Bearden AR, Lavine KJ, et
al: Tissue-resident macrophages in pancreatic ductal adenocarcinoma
originate from embryonic hematopoiesis and promote tumor
progression. Immunity. 47323–338. (e6)2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quemerais C, Jean C, Brunel A, Decaup E,
Labrousse G, Audureau H, Raffenne J, Belhabib I, Cros J, Perraud A,
et al: Unveiling FKBP7 as an early endoplasmic reticulum sentinel
in pancreatic stellate cell activation, collagen remodeling and
tumor progression. Cancer Lett. 614:2175382025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Akhmetkaliyev A, Alibrahim N, Shafiee D
and Tulchinsky E: EMT/MET plasticity in cancer and Go-or-Grow
decisions in quiescence: The two sides of the same coin? Mol
Cancer. 22:902023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Niland S, Riscanevo AX and Eble JA: Matrix
metalloproteinases shape the tumor microenvironment in cancer
progression. Int J Mol Sci. 23:1462021. View Article : Google Scholar
|
|
28
|
Strouhalova K, Tolde O, Rosel D and Brábek
J: Cytoplasmic Tail of MT1-MMP: A Hub of MT1-MMP Regulation and
Function. Int J Mol Sci. 24:50682023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tao X, Xiang H, Pan Y, Shang D, Guo J, Gao
G and Xiao GG: Pancreatitis initiated pancreatic ductal
adenocarcinoma: Pathophysiology explaining clinical evidence.
Pharmacol Res. 168:1055952021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sarkar R, Xu Z, Perera CJ and Apte MV:
Emerging role of pancreatic stellate cell-derived extracellular
vesicles in pancreatic cancer. Semin Cancer Biol. 93:114–122. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng D, Fu M, Wang M, Wei Y and Wei X:
Targeting TGF-β signal transduction for fibrosis and cancer
therapy. Mol Cancer. 21:1042022. View Article : Google Scholar
|
|
32
|
Li J, Chen X, Kang R, Zeh H, Klionsky DJ
and Tang D: Regulation and function of autophagy in pancreatic
cancer. Autophagy. 17:3275–3296. 2021. View Article : Google Scholar :
|
|
33
|
Lin D, Shen L, Luo M, Zhang K, Li J, Yang
Q, Zhu F, Zhou D, Zheng S, Chen Y and Zhou J: Circulating tumor
cells: Biology and clinical significance. Signal Transduct Target
Ther. 6:4042021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Haber DA and Velculescu VE: Blood-based
analyses of cancer: Circulating tumor cells and circulating tumor
DNA. Cancer Discov. 4:650–661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lawrence R, Watters M, Davies CR, Pantel K
and Lu YJ: Circulating tumour cells for early detection of
clinically relevant cancer. Nat Rev Clin Oncol. 20:487–500. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mullins RDZ, Pal A, Barrett TF, Heft Neal
ME and Puram SV: Epithelial-mesenchymal plasticity in tumor immune
evasion. Cancer Res. 82:2329–2343. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hariri A, Mirian M, Khosravi A, Zarepour
A, Iravani S and Zarrabi A: Intersecting pathways: The role of
hybrid E/M cells and circulating tumor cells in cancer metastasis
and drug resistance. Drug Resist Updat. 76:1011192024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jolly MK, Somarelli JA, Sheth M, Biddle A,
Tripathi SC, Armstrong AJ, Hanash SM, Bapat SA, Rangarajan A and
Levine H: Hybrid epithelial/mesenchymal phenotypes promote
metastasis and therapy resistance across carcinomas. Pharmacol
Ther. 194:161–184. 2019. View Article : Google Scholar
|
|
39
|
Bustamante A, Baritaki S, Zaravinos A and
Bonavida B: Relationship of signaling pathways between RKIP
expression and the inhibition of EMT-inducing transcription factors
SNAIL1/2, TWIST1/2 and ZEB1/2. Cancers (Basel). 16:31802024.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vidlarova M, Rehulkova A, Stejskal P,
Prokopova A, Slavik H, Hajduch M and Srovnal J: Recent advances in
methods for circulating tumor cell detection. Int J Mol Sci.
24:39022023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Padmanaban V, Krol I, Suhail Y, Szczerba
BM, Aceto N, Bader JS and Ewald AJ: E-cadherin is required for
metastasis in multiple models of breast cancer. Nature.
573:439–444. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ward MP, E Kane L, A Norris L, Mohamed BM,
Kelly T, Bates M, Clarke A, Brady N, Martin CM, Brooks RD, et al:
Platelets, immune cells and the coagulation cascade; friend or foe
of the circulating tumour cell? Mol Cancer. 20:592021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Leblanc R and Peyruchaud O: Metastasis:
New functional implications of platelets and megakaryocytes. Blood.
128:24–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tao J, Zhu L, Yakoub M, Reißfelder C,
Loges S and Schölch S: Cell-Cell interactions drive metastasis of
circulating tumor microemboli. Cancer Res. 82:2661–2671. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou J, Yang S, Zhu D, Li H, Miao X, Gu M,
Xu W, Zhang Y, Tang W, Shen R, et al: The crosstalk between anoikis
and epithelial-mesenchymal transition and their synergistic roles
in predicting prognosis in colon adenocarcinoma. Front Oncol.
13:11842152023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cao Z, Livas T and Kyprianou N: Anoikis
and EMT: Lethal 'Liaisons' during Cancer Progression. Crit Rev
Oncog. 21:155–168. 2016. View Article : Google Scholar
|
|
47
|
Schuster E, Taftaf R, Reduzzi C, Albert
MK, Romero-Calvo I and Liu H: Better together: Circulating tumor
cell clustering in metastatic cancer. Trends Cancer. 7:1020–1032.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Costa C, Muinelo-Romay L, Cebey-López V,
Pereira-Veiga T, Martínez-Pena I, Abreu M, Abalo A, Lago-Lestón RM,
Abuín C, Palacios P, et al: Analysis of a Real-world cohort of
metastatic breast cancer patients shows circulating tumor cell
clusters (CTC-clusters) as predictors of patient outcomes. Cancers
(Basel). 12:11112020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou W, Ye XL, Xu J, Cao MG, Fang ZY, Li
LY, Guan GH, Liu Q, Qian YH and Xie D: The lncRNA H19 mediates
breast cancer cell plasticity during EMT and MET plasticity by
differentially sponging miR-200b/c and let-7b. Sci Signal.
10:eaak95572017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Alečković M, McAllister SS and Polyak K:
Metastasis as a systemic disease: Molecular insights and clinical
implications. Biochim Biophys Acta Rev Cancer. 1872:89–102. 2019.
View Article : Google Scholar
|
|
51
|
Shen S, Vagner S and Robert C: Persistent
cancer cells: The deadly survivors. Cell. 183:860–874. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gerstberger S, Jiang Q and Ganesh K:
Metastasis. Cell. 186:1564–1579. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Aguirre-Ghiso JA: Models, mechanisms and
clinical evidence for cancer dormancy. Nat Rev Cancer. 7:834–846.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Massagué J and Obenauf AC: Metastatic
colonization by circulating tumour cells. Nature. 529:298–306.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Costa-Silva B, Aiello NM, Ocean AJ, Singh
S, Zhang H, Thakur BK, Becker A, Hoshino A, Mark MT, Molina H, et
al: Pancreatic cancer exosomes initiate pre-metastatic niche
formation in the liver. Nat Cell Biol. 17:816–826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lin Z, Li G, Jiang K, Li Z and Liu T:
Cancer therapy resistance mediated by cancer-associated
fibroblast-derived extracellular vesicles: Biological mechanisms to
clinical significance and implications. Mol Cancer. 23:1912024.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang X, Eichhorn PJA and Thiery JP: TGF-β,
EMT, and resistance to anti-cancer treatment. Semin Cancer Biol.
97:1–11. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chu X, Tian W, Ning J, Xiao G, Zhou Y,
Wang Z, Zhai Z, Tanzhu G, Yang J and Zhou R: Cancer stem cells:
Advances in knowledge and implications for cancer therapy. Signal
Transduct Target Ther. 9:1702024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Bayik D and Lathia JD: Cancer stem
cell-immune cell crosstalk in tumour progression. Nat Rev Cancer.
21:526–536. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chang CH and Pauklin S: ROS and TGFβ: From
pancreatic tumour growth to metastasis. J Exp Clin Cancer Res.
40:1522021. View Article : Google Scholar
|
|
61
|
Stoica AF, Chang CH and Pauklin S:
Molecular therapeutics of pancreatic ductal adenocarcinoma:
Targeted pathways and the role of cancer stem cells. Trends
Pharmacol Sci. 41:977–993. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Modi SJ and Kulkarni VM: Discovery of
VEGFR-2 inhibitors exerting significant anticancer activity against
CD44+ and CD133+ cancer stem cells (CSCs): Reversal of TGF-β
induced epithelial-mesenchymal transition (EMT) in hepatocellular
carcinoma. Eur J Med Chem. 207:1128512020. View Article : Google Scholar
|
|
63
|
Krebs AM, Mitschke J, Lasierra Losada M,
Schmalhofer O, Boerries M, Busch H, Boettcher M, Mougiakakos D,
Reichardt W, Bronsert P, et al: The EMT-activator Zeb1 is a key
factor for cell plasticity and promotes metastasis in pancreatic
cancer. Nat Cell Biol. 19:518–529. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Feng J, Hu S, Liu K, Sun G and Zhang Y:
The role of MicroRNA in the regulation of tumor
Epithelial-mesenchymal transition. Cells. 11:19812022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bracken CP, Goodall GJ and Gregory PA: RNA
regulatory mechanisms controlling TGF-β signaling and EMT in
cancer. Semin Cancer Biol. 102-103:4–16. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Sangrador I, Molero X, Campbell F,
Franch-Expósito S, Rovira-Rigau M, Samper E, Domínguez-Fraile M,
Fillat C, Castells A and Vaquero EC: Zeb1 in stromal myofibroblasts
promotes Kras-driven development of pancreatic cancer. Cancer Res.
78:2624–2637. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Duan H, Liu Y, Gao Z and Huang W: Recent
advances in drug delivery systems for targeting cancer stem cells.
Acta Pharm Sin B. 11:55–70. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pan G, Liu Y, Shang L, Zhou F and Yang S:
EMT-associated microRNAs and their roles in cancer stemness and
drug resistance. Cancer Commun (Lond). 41:199–217. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Brabletz S, Schuhwerk H, Brabletz T and
Stemmler MP: Dynamic EMT: A multi-tool for tumor progression. EMBO
J. 4:e1086472021. View Article : Google Scholar
|
|
70
|
Funamizu N, Honjo M, Tamura K, Sakamoto K,
Ogawa K and Takada Y: microRNAs associated with gemcitabine
resistance via EMT, TME, and drug metabolism in pancreatic cancer.
Cancers (Basel). 15. pp. 12302023, View Article : Google Scholar
|
|
71
|
Redfern AD, Spalding LJ and Thompson EW:
The Kraken Wakes: Induced EMT as a driver of tumour aggression and
poor outcome. Clin Exp Metastasis. 35:285–308. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zheng X, Carstens JL, Kim J, Scheible M,
Kaye J, Sugimoto H, Wu CC, LeBleu VS and Kalluri R:
Epithelial-to-mesenchymal transition is dispensable for metastasis
but induces chemoresistance in pancreatic cancer. Nature.
527:525–530. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Singh A and Settleman J: EMT, Cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Bulle A and Lim KH: Beyond just a tight
fortress: Contribution of stroma to epithelial-mesenchymal
transition in pancreatic cancer. Signal Transduct Target Ther.
5:2492020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Joshi VB, Gutierrez Ruiz OL and Razidlo
GL: The cell biology of metastatic invasion in pancreatic cancer:
Updates and mechanistic insights. Cancers (Basel). 15. pp.
21692023, View Article : Google Scholar
|
|
76
|
Youssef KK and Nieto MA:
Epithelial-mesenchymal transition in tissue repair and
degeneration. Nat Rev Mol Cell Biol. 25:720–739. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bracken CP and Goodall GJ: The many
regulators of epithelial-mesenchymal transition. Nat Rev Mol Cell
Biol. 23:89–90. 2022. View Article : Google Scholar
|
|
78
|
Mittal V: Epithelial mesenchymal
transition in tumor metastasis. Annu Rev Pathol. 13:395–412. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Debnath P, Huirem RS, Dutta P and
Palchaudhuri S: Epithelial-mesenchymal transition and its
transcription factors. Biosci Rep. 42:BSR202117542022. View Article : Google Scholar :
|
|
80
|
Sheng W, Tang J, Cao R, Shi X, Ma Y and
Dong M: Numb-PRRL promotes TGF-β1- and EGF-induced
epithelial-to-mesenchymal transition in pancreatic cancer. Cell
Death Dis. 13:1732022. View Article : Google Scholar
|
|
81
|
Shao M, Wang L, Zhang Q, Wang T and Wang
S: STMN2 overexpression promotes cell proliferation and EMT in
pancreatic cancer mediated by WNT/β-catenin signaling. Cancer Gene
Ther. 30:472–480. 2023.
|
|
82
|
Zhao R, Yi Y, Liu H, Xu J, Chen S, Wu D,
Wang L and Li F: RHOF promotes Snail1 lactylation by enhancing
PKM2-mediated glycolysis to induce pancreatic cancer cell
endothelial-mesenchymal transition. Cancer Metab. 12:322024.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Tang Y, Tang Y and Cheng YS: miR-34a
inhibits pancreatic cancer progression through Snail1-mediated
epithelial-mesenchymal transition and the Notch signaling pathway.
Sci Rep. 7:382322017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tang L, Ji M, Liang X, Chen D, Liu A, Yang
G, Shi L, Fu Z and Shao C: Downregulated F-Box/LRR-Repeat protein 7
facilitates pancreatic cancer metastasis by regulating snail1 for
proteasomal degradation. Front Genet. 12:6500902021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Lambies G, Miceli M, Martínez-Guillamon C,
Olivera-Salguero R, Peña R, Frías CP, Calderó I, Atanassov BS, Dent
SYR, Arribas J, et al: TGFβ-Activated USP27X Deubiquitinase
regulates cell migration and chemoresistance via stabilization of
snail1. Cancer Res. 79:33–46. 2019. View Article : Google Scholar
|
|
86
|
García de Herreros A: Control of snail1
protein stability by post-translational modifications: The basis
for a complex regulation of Snail1 function. Int J Biol Sci.
21:3183–3196. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fendrich V, Jendryschek F, Beeck S, Albers
M, Lauth M, Esni F, Heeger K, Dengler J, Slater EP, Holler JPN, et
al: Genetic and pharmacologic abrogation of Snail1 inhibits
acinar-to-ductal metaplasia in precursor lesions of pancreatic
ductal adenocarcinoma and pancreatic injury. Oncogene.
37:1845–1856. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Frey P, Devisme A, Schrempp M, Andrieux G,
Boerries M and Hecht A: Canonical BMP signaling executes
epithelial-mesenchymal transition downstream of SNAIL1. Cancers
(Basel). 12. pp. 10192020, View Article : Google Scholar
|
|
89
|
Ma J, Zhang J, Weng YC and Wang JC:
EZH2-Mediated microRNA-139-5p Regulates Epithelial-mesenchymal
transition and lymph node metastasis of pancreatic cancer. Mol
Cells. 41:868–880. 2018.PubMed/NCBI
|
|
90
|
Funamizu N, Hu C, Lacy C, Schetter A,
Zhang G, He P, Gaedcke J, Ghadimi MB and Ried T: Macrophage
migration inhibitory factor induces epithelial to mesenchymal
transition, enhances tumor aggressiveness and predicts clinical
outcome in resected pancreatic ductal adenocarcinoma. Int J Cancer.
132:785–794. 2013. View Article : Google Scholar
|
|
91
|
Mirzaei S, Saghari S, Bassiri F, Raesi R,
Zarrabi A, Hushmandi K, Sethi G and Tergaonkar V: NF-κB as a
regulator of cancer metastasis and therapy response: A focus on
epithelial-mesenchymal transition. J Cell Physiol. 237:2770–2795.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhang Y, Xue X, Zhao X, Qin L, Shen Y, Dou
H, Sun J, Wang T and Yang DQ: Vasohibin 2 promotes malignant
behaviors of pancreatic cancer cells by inducing
epithelial-mesenchymal transition via Hedgehog signaling pathway.
Cancer Med. 7:5567–5576. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Eguchi T, Csizmadia E, Kawai H, Sheta M,
Yoshida K, Prince TL, Wegiel B and Calderwood SK: SCAND1 reverses
Epithelial-to-mesenchymal transition (EMT) and suppresses prostate
cancer growth and migration. Cells. 11:39932022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Saitoh M: Transcriptional regulation of
EMT transcription factors in cancer. Semin Cancer Biol. 97:21–29.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ghafoor S, Garcia E, Jay DJ and Persad S:
Molecular mechanisms regulating epithelial mesenchymal transition
(EMT) to promote cancer progression. Int J Mol Sci. 26:43642025.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chen S, Chen JZ, Zhang JQ, Chen HX, Yan
ML, Huang L, Tian YF, Chen YL and Wang YD: Hypoxia induces
TWIST-activated epithelial-mesenchymal transition and proliferation
of pancreatic cancer cells in vitro and in nude mice. Cancer Lett.
383:73–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wang J, Nikhil K, Viccaro K, Chang L,
Jacobsen M, Sandusky G and Shah K: The Aurora-A-Twist1 axis
promotes highly aggressive phenotypes in pancreatic carcinoma. J
Cell Sci. 130:1078–1093. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ji H, Lu HW, Li YM, Lu L, Wang JL, Zhang
YF and Shang H: Twist promotes invasion and cisplatin resistance in
pancreatic cancer cells through growth differentiation factor 15.
Mol Med Rep. 12:3841–3848. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yang J, Zhang X, Zhang Y, Zhu D, Zhang L,
Li Y, Zhu Y, Li D and Zhou J: HIF-2α promotes
epithelial-mesenchymal transition through regulating Twist2 binding
to the promoter of E-cadherin in pancreatic cancer. J Exp Clin
Cancer Res. 35:262016. View Article : Google Scholar
|
|
100
|
Wang H, Li QF, Chow HY, Choi SC and Leung
YC: Arginine deprivation inhibits pancreatic cancer cell migration,
invasion and EMT via the down regulation of Snail, Slug, Twist, and
MMP1/9. J Physiol Biochem. 76:73–83. 2020. View Article : Google Scholar
|
|
101
|
Yu H, Gao G, Cai J, Song H, Ma Z, Jin X,
Ji W and Pan B: MiR-539 functions as a tumor suppressor in
pancreatic cancer by targeting TWIST1. Exp Mol Pathol. 108:143–149.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wong CH, Lou UK, Fung FK, Tong JHM, Zhang
CH, To KF, Chan SL and Chen Y: CircRTN4 promotes pancreatic cancer
progression through a novel CircRNA-miRNA-lncRNA pathway and
stabilizing epithelial-mesenchymal transition protein. Mol Cancer.
21:102022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Katoch A, Jamwal VL, Faheem MM, Kumar S,
Senapati S, Yadav G, Gandhi SG and Goswami A: Overlapping targets
exist between the Par-4 and miR-200c axis which regulate EMT and
proliferation of pancreatic cancer cells. Transl Oncol.
14:1008792021. View Article : Google Scholar
|
|
104
|
Pan H, Liu Y, Bao K, Wang Y, Zhang Y and
Zhou L: DYRK2 regulates epithelial-mesenchymal transition
restriction in pancreatic cancer liver metastasis by inhibiting
Twist. Digestion. Nov;19:2024.Epub ahead of print. View Article : Google Scholar
|
|
105
|
Gabitova-Cornell L, Surumbayeva A, Peri S,
Franco-Barraza J, Restifo D, Weitz N, Ogier C, Goldman AR, Hartman
TR, Francescone R, et al: Cholesterol pathway inhibition induces
TGF-β Signaling to promote basal differentiation in pancreatic
cancer. Cancer Cell. 38:567–583.e11. 2020. View Article : Google Scholar
|
|
106
|
Sun Y, Li D, Liu H, Huang Y, Meng F, Tang
J, Li Z and Xie W: PHF13 epigenetically activates TGFβ driven
epithelial to mesenchymal transition. Cell Death Dis. 13:4872022.
View Article : Google Scholar
|
|
107
|
Uddin MH, Al-Hallak MN, Philip PA, Chen H,
El-Rayes B and Azmi AS: Aberrant transcription factors in the
cancers of the pancreas. Semin Cancer Biol. 86:28–45. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhao Z, Yang W, Kong R, Zhang Y, Li L,
Song Z, Chen H, Luo Y, Zhang T, Cheng C, et al: circEIF3I
facilitates the recruitment of SMAD3 to early endosomes to promote
TGF-β signalling pathway-mediated activation of MMPs in pancreatic
cancer. Mol Cancer. 22:1522023. View Article : Google Scholar
|
|
109
|
Zhou K, Liu Y, Tang C and Zhu H:
Pancreatic cancer: Pathogenesis and clinical studies. MedComm
(2020). 6:e701622025. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Nan P, Dong X, Bai X, Lu H, Liu F, Sun Y
and Zhao X: Tumor-stroma TGF-β1-THBS2 feedback circuit drives
pancreatic ductal adenocarcinoma progression via integrin
αvβ3/CD36-mediated activation of the MAPK pathway. Cancer Lett.
528:59–75. 2022. View Article : Google Scholar
|
|
111
|
Wang Z, Guo X, Li X, Wang J, Zhang N, Amin
B, Xu G and Zhu B: Cancer-associated fibroblast-derived MMP11
promotes tumor progression in pancreatic cancer. Cancer Sci.
116:643–655. 2025. View Article : Google Scholar :
|
|
112
|
Guo H, Zhao Z and Liu L: HIF-1α modulates
pancreatic cancer ECM proteins via the TGF-β1/Smad signaling
pathway introduction. Front Oncol. 15:15646552025. View Article : Google Scholar
|
|
113
|
Lee JE, Lee P, Yoon YC, Han BS, Ko S, Park
MS, Lee YJ, Kim SE, Cho YJ, Lim JH, et al: Vactosertib, TGF-β
receptor I inhibitor, augments the sensitization of the anti-cancer
activity of gemcitabine in pancreatic cancer. Biomed Pharmacother.
162:1147162023. View Article : Google Scholar
|
|
114
|
TGFβ promotes immune evasion to limit the
efficacy of Anti-PD-1/PD-L1. Cancer Discov. 8:OF102018. View Article : Google Scholar
|
|
115
|
Oronsky B, Cabrales P, Alizadeh B, Caroen
S, Stirn M, Williams J and Reid TR: TGF-β TGF-β: The apex predator
of immune checkpoints. Future Oncol. 19:2013–2015. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhou C, Zhu X, Liu N, Dong X, Zhang X,
Huang H, Tang Y, Liu S and Hu M: B-lymphoid tyrosine
kinase-mediated FAM83A phosphorylation elevates pancreatic
tumorigenesis through interacting with β-catenin. Signal Transduct
Target Ther. 8:662023. View Article : Google Scholar
|
|
117
|
Zhou C, Dong X, Li S, Xi Y, Liu Y, Qian X,
Song Z, Zhou L, Zhang R, Lyu H, et al: Serine/threonine/tyrosine
Kinase 1 drives pancreatic carcinogenesis via GSK3β
sequestration-mediated Wnt/β-catenin pathway hyperactivation.
Signal Transduct Target Ther. 10:2052025. View Article : Google Scholar
|
|
118
|
Fang Z, Meng Q, Xu J, Wang W, Zhang B, Liu
J, Liang C, Hua J, Zhao Y, Yu X and Shi S: Signaling pathways in
cancer-associated fibroblasts: Recent advances and future
perspectives. Cancer Commun (Lond). 43:3–41. 2023. View Article : Google Scholar :
|
|
119
|
Bailey P, Chang DK, Nones K, Johns AL,
Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC,
et al: Genomic analyses identify molecular subtypes of pancreatic
cancer. Nature. 531:47–52. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Rong Z, Xu J, Yang J, Wang W, Tang R,
Zhang Z, Tan Z, Meng Q, Hua J, Liu J, et al: CircRREB1 mediates
metabolic reprogramming and stemness maintenance to facilitate
pancreatic ductal adenocarcinoma progression. Cancer Res.
84:4246–4263. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Fang Z, Wu Z, Yu C, Xie Q, Zeng L and Chen
R: EIF4E-mediated biogenesis of circPHF14 promotes the growth and
metastasis of pancreatic ductal adenocarcinoma via Wnt/β-catenin
pathway. Mol Cancer. 24:562025. View Article : Google Scholar
|
|
122
|
Zheng S, Lin J, Pang Z, Zhang H, Wang Y,
Ma L, Zhang H, Zhang X, Chen M, Zhang X, et al: Aberrant
cholesterol metabolism and Wnt/β-catenin signaling coalesce via
Frizzled5 in supporting cancer growth. Adv Sci (Weinh).
9:e22007502022. View Article : Google Scholar
|
|
123
|
Wang W, Shao F, Yang X, Wang J, Zhu R,
Yang Y, Zhao G, Guo D, Sun Y, Wang J, et al: METTL3 promotes tumour
development by decreasing APC expression mediated by APC mRNA
N6-methyladenosine-dependent YTHDF binding. Nat Commun.
12:38032021. View Article : Google Scholar
|
|
124
|
Fu X, Ma J, Ma F, Guo S, Wang X, Li Y,
Tang Y, Qi J, Zhang W and Ye L: MISP-mediated enhancement of
pancreatic cancer growth through the Wnt/β-catenin signaling
pathway is suppressed by Fisetin. Biochim Biophys Acta Mol Basis
Dis. 1871:1675152025. View Article : Google Scholar
|
|
125
|
Roy SK, Ma Y, Lam BQ, Shrivastava A,
Srivastav S, Shankar S and Srivastava RK: Riluzole regulates
pancreatic cancer cell metabolism by suppressing the Wnt-β-catenin
pathway. Sci Rep. 12:110622022. View Article : Google Scholar
|
|
126
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Sivakumar S, de Santiago I, Chlon L and
Markowetz F: Master regulators of oncogenic KRAS response in
pancreatic cancer: An integrative network biology analysis. PLoS
Med. 14:e10022232017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Kumar G, Farooqui M and Rao CV: Role of
dietary cancer-preventive phytochemicals in pancreatic cancer stem
cells. Curr Pharmacol Rep. 4:326–335. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Pan X, Zhou J, Xiao Q, Fujiwara K, Zhang
M, Mo G, Gong W and Zheng L: Cancer-associated fibroblast
heterogeneity is associated with organ-specific metastasis in
pancreatic ductal adenocarcinoma. J Hematol Oncol. 14:1842021.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Zhu S, Xu H, Chen R, Shen Q, Yang D, Peng
H, Tong J and Fu Q: DNA methylation and miR-92a-3p-mediated
repression of HIP1R promotes pancreatic cancer progression by
activating the PI3K/AKT pathway. J Cell Mol Med. 27:788–802. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Lomberk G, Blum Y, Nicolle R, Nair A,
Gaonkar KS, Marisa L, Mathison A, Sun Z, Yan H, Elarouci N, et al:
Distinct epigenetic landscapes underlie the pathobiology of
pancreatic cancer subtypes. Nat Commun. 9:19782018. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Silverman BR and Shi J: Alterations of
epigenetic regulators in pancreatic cancer and their clinical
implications. Int J Mol Sci. 17:21382016. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Xie VK, Li Z, Yan Y, Jia Z, Zuo X, Ju Z,
Wang J, Du J, Xie D, Xie K and Wei D: DNA-Methyltransferase 1
induces dedifferentiation of pancreatic cancer cells through
silencing of Krüppel-like factor 4 expression. Clin Cancer Res.
23:5585–5597. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Hong L, Sun G, Peng L, Tu Y, Wan Z, Xiong
H, Li Y and Xiao W: The interaction between miR-148a and DNMT1
suppresses cell migration and invasion by reactivating tumor
suppressor genes in pancreatic cancer. Oncol Rep. 40:2916–2925.
2018.PubMed/NCBI
|
|
135
|
Xiao M, Liang X, Yan Z, Chen J, Zhu Y, Xie
Y, Li Y, Li X, Gao Q, Feng F, et al: A DNA-methylation-driven genes
based prognostic signature reveals immune microenvironment in
pancreatic cancer. Front Immunol. 13:8039622022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Lo EKW, Idrizi A, Tryggvadottir R, Zhou W,
Hou W, Ji H, Cahan P and Feinberg AP: DNA methylation memory of
pancreatic acinar-ductal metaplasia transition state altering
Kras-downstream PI3K and Rho GTPase signaling in the absence of
Kras mutation. Genome Med. 17:322025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Szymoński K, Lipiec E, Sofińska K,
Skirlińska-Nosek K, Czaja M, Seweryn S, Wilkosz N, Birarda G,
Piccirilli F, Vaccari L, et al: Variabilities in global DNA
methylation and β-sheet richness establish spectroscopic landscapes
among subtypes of pancreatic cancer. Eur J Nucl Med Mol Imaging.
50:1792–1810. 2023. View Article : Google Scholar
|
|
138
|
Lin Z, Chen Y, Lin Z, Chen C and Dong Y:
Overexpressing PRMT1 inhibits proliferation and invasion in
pancreatic cancer by inverse correlation of ZEB1. IUBMB Life.
70:1032–1039. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Wang Y, Hsu JM, Kang Y, Wei Y, Lee PC,
Chang SJ, Hsu YH, Hsu JL, Wang HL, Chang WC, et al: Oncogenic
functions of gli1 in pancreatic adenocarcinoma are supported by Its
PRMT1-mediated methylation. Cancer Res. 76:7049–7058. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Ge L, Wang H, Xu X, Zhou Z, He J, Peng W,
Du F, Zhang Y, Gong A and Xu M: PRMT5 promotes
epithelial-mesenchymal transition via EGFR-β-catenin axis in
pancreatic cancer cells. J Cell Mol Med. 24:1969–1979. 2020.
View Article : Google Scholar :
|
|
141
|
Niu N, Lu P, Yang Y, He R, Zhang L, Shi J,
Wu J, Yang M, Zhang ZG, Wang LW, et al: Loss of Setd2 promotes
Kras-induced acinar-to-ductal metaplasia and epithelia-mesenchymal
transition during pancreatic carcinogenesis. Gut. 69:715–726. 2020.
View Article : Google Scholar
|
|
142
|
Liu M, Shi Y, Hu Q, Qin Y, Ji S, Liu W,
Zhuo Q, Fan G, Ye Z, Song C, et al: SETD8 induces stemness and
epithelial-mesenchymal transition of pancreatic cancer cells by
regulating ROR1 expression. Acta Biochim Biophys Sin (Shanghai).
53:1614–1624. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Viotti M, Wilson C, McCleland M, Koeppen
H, Haley B, Jhunjhunwala S, Klijn C, Modrusan Z, Arnott D, Classon
M, et al: SUV420H2 is an epigenetic regulator of
epithelial/mesenchymal states in pancreatic cancer. J Cell Biol.
217:763–777. 2018. View Article : Google Scholar :
|
|
144
|
Wanna-Udom S, Terashima M, Suphakhong K,
Ishimura A, Takino T and Suzuki T: KDM2B is involved in the
epigenetic regulation of TGF-β-induced epithelial-mesenchymal
transition in lung and pancreatic cancer cell lines. J Biol Chem.
296:1002132021. View Article : Google Scholar
|
|
145
|
Quan M, Chen Z, Jiao F, Xiao X, Xia Q,
Chen J, Chao Q, Li Y, Gao Y, Yang H, et al: Lysine demethylase 2
(KDM2B) regulates hippo pathway via MOB1 to promote pancreatic
ductal adenocarcinoma (PDAC) progression. J Exp Clin Cancer Res.
39:132020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Dandawate P, Ghosh C, Palaniyandi K, Paul
S, Rawal S, Pradhan R, Sayed AAA, Choudhury S, Standing D,
Subramaniam D, et al: The histone demethylase KDM3A, increased in
human pancreatic tumors, regulates expression of DCLK1 and promotes
tumorigenesis in mice. Gastroenterology. 1571646–1659. (e11)2019.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Li S, Wu L, Wang Q, Li Y and Wang X: KDM4B
promotes epithelial-mesenchymal transition through up-regulation of
ZEB1 in pancreatic cancer. Acta Biochim Biophys Sin (Shanghai).
47:997–1004. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Li H, Peng C, Zhu C, Nie S, Qian X, Shi Z,
Shi M, Liang Y, Ding X, Zhang S, et al: Hypoxia promotes the
metastasis of pancreatic cancer through regulating
NOX4/KDM5A-mediated histone methylation modification changes in a
HIF1A-independent manner. Clin Epigenetics. 13:182021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Yi Z, Wei S, Jin L, Jeyarajan S, Yang J,
Gu Y, Kim HS, Schechter S, Lu S, Paulsen MT, et al: KDM6A regulates
cell plasticity and pancreatic cancer progression by noncanonical
activin pathway. Cell Mol Gastroenterol Hepatol. 13:643–667. 2022.
View Article : Google Scholar :
|
|
150
|
Zhong Z, Harmston N, Wood KC, Madan B and
Virshup DM: A p300/GATA6 axis determines differentiation and Wnt
dependency in pancreatic cancer models. J Clin Invest.
132:e1563052022. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Martinelli P, Carrillo-de Santa Pau E, Cox
T, Sainz B Jr, Dusetti N, Greenhalf W, Rinaldi L, Costello E,
Ghaneh P, Malats N, et al: GATA6 regulates EMT and tumour
dissemination, and is a marker of response to adjuvant chemotherapy
in pancreatic cancer. Gut. 66:1665–1676. 2017. View Article : Google Scholar :
|
|
152
|
Mizuguchi Y, Specht S, Lunz JG III, Isse
K, Corbitt N, Takizawa T and Demetris AJ: Cooperation of p300 and
PCAF in the control of microRNA 200c/141 transcription and
epithelial characteristics. PLoS One. 7:e324492012. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
von Burstin J, Eser S, Paul MC, Seidler B,
Brandl M, Messer M, von Werder A, Schmidt A, Mages J, Pagel P, et
al: E-cadherin regulates metastasis of pancreatic cancer in vivo
and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex.
Gastroenterology. 137:361–371. e1–e5. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Aghdassi A, Sendler M, Guenther A, Mayerle
J, Behn CO, Heidecke CD, Friess H, Büchler M, Evert M, Lerch MM and
Weiss FU: Recruitment of histone deacetylases HDAC1 and HDAC2 by
the transcriptional repressor ZEB1 downregulates E-cadherin
expression in pancreatic cancer. Gut. 61:439–448. 2012. View Article : Google Scholar
|
|
155
|
Wauters E, Sanchez-Arévalo Lobo VJ, Pinho
AV, Mawson A, Herranz D, Wu J, Cowley MJ, Colvin EK, Njicop EN,
Sutherland RL, et al: Sirtuin-1 regulates acinar-to-ductal
metaplasia and supports cancer cell viability in pancreatic cancer.
Cancer Res. 73:2357–2367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Chen J, Xu H, Zou X, Wang J, Zhu Y, Chen
H, Shen B, Deng X, Zhou A, Chin YE, et al: Snail recruits Ring1B to
mediate transcriptional repression and cell migration in pancreatic
cancer cells. Cancer Res. 74:4353–4363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Guo S, Xu X, Tang Y, Zhang C, Li J, Ouyang
Y, Ju J, Bie P and Wang H: miR-15a inhibits cell proliferation and
epithelial to mesenchymal transition in pancreatic ductal
adenocarcinoma by down-regulating Bmi-1 expression. Cancer Lett.
344:40–46. 2014. View Article : Google Scholar
|
|
158
|
Ning Z, Wang A, Liang J, Xie Y, Liu J, Yan
Q and Wang Z: USP22 promotes epithelial-mesenchymal transition via
the FAK pathway in pancreatic cancer cells. Oncol Rep.
32:1451–1458. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Chen L, Xu Z, Li Q, Feng Q, Zheng C, Du Y,
Yuan R and Peng X: USP28 facilitates pancreatic cancer progression
through activation of Wnt/β-catenin pathway via stabilising FOXM1.
Cell Death Dis. 12:8872021. View Article : Google Scholar
|
|
160
|
Zhuo W, Zeng Z, Hu Y, Hu P, Han S, Wang D,
Wang F, Zhao Y, Huang Y, Wang J, et al: Metabolic stress-induced
reciprocal loop of long noncoding RNA ZFAS1 and ZEB1 promotes
epithelial-mesenchymal transition and metastasis of pancreatic
cancer cells. Cancer Sci. 114:3623–3635. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
He Z, Wang J, Zhu C, Xu J, Chen P, Jiang
X, Chen Y, Jiang J and Sun C: Exosome-derived FGD5-AS1 promotes
tumor-associated macrophage M2 polarization-mediated pancreatic
cancer cell proliferation and metastasis. Cancer Lett.
548:2157512022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Huang X, Pan L, Zuo Z, Li M, Zeng L, Li R,
Ye Y, Zhang J, Wu G, Bai R, et al: LINC00842 inactivates
transcription co-regulator PGC-1α to promote pancreatic cancer
malignancy through metabolic remodelling. Nat Commun. 12:38302021.
View Article : Google Scholar
|
|
163
|
Hu Y, Wang F, Xu F, Fang K, Fang Z, Shuai
X, Cai K, Chen J, Hu P, Chen D, et al: A reciprocal feedback of Myc
and lncRNA MTSS1-AS contributes to extracellular acidity-promoted
metastasis of pancreatic cancer. Theranostics. 10:10120–10140.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
He Y, Yue H, Cheng Y, Ding Z, Xu Z, Lv C,
Wang Z, Wang J, Yin C, Hao H and Chen C: ALKBH5-mediated
m6A demethylation of KCNK15-AS1 inhibits pancreatic
cancer progression via regulating KCNK15 and PTEN/AKT signaling.
Cell Death Dis. 12:11212021. View Article : Google Scholar
|
|
165
|
Ji Y, Feng G, Hou Y, Yu Y, Wang R and Yuan
H: Long noncoding RNA MEG3 decreases the growth of head and neck
squamous cell carcinoma by regulating the expression of miR-421 and
E-cadherin. Cancer Med. 9:3954–3963. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Gu X, Li J, Zuo X, Chen K, Wan G, Deng LL,
Zhao W and Lu C: The long noncoding RNA MEG3 retains
Epithelial-mesenchymal transition by sponging miR-146b-5p to
regulate SLFN5 expression in breast cancer cells. J Immunol Res.
2022:18241662022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Zhang KD, Hu B, Cen G, Yang YH, Chen WW,
Guo ZY, Wang XF, Zhao Q and Qiu ZJ: MiR-301a transcriptionally
activated by HIF-2α promotes hypoxia-induced epithelial-mesenchymal
transition by targeting TP63 in pancreatic cancer. World J
Gastroenterol. 26:2349–2373. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Guo C, Liu J, Zhou Q, Song J, Zhang Z, Li
Z, Wang G, Yuan W and Sun Z: Exosomal noncoding RNAs and tumor drug
resistance. Cancer Res. 80:4307–4313. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Xiong Q, Zhang Y, Xu Y, Yang Y, Zhang Z,
Zhou Y, Zhang S, Zhou L, Wan X, Yang X, et al: tiRNA-Val-CAC-2
interacts with FUBP1 to promote pancreatic cancer metastasis by
activating c-MYC transcription. Oncogene. 43:1274–1287. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Hong E, Park S, Ooshima A, Hong CP, Park
J, Heo JS, Lee S, An H, Kang JM, Park SH, et al: Inhibition of
TGF-β signalling in combination with nal-IRI plus
5-Fluorouracil/Leucovorin suppresses invasion and prolongs survival
in pancreatic tumour mouse models. Sci Rep. 10:29352020. View Article : Google Scholar
|
|
171
|
Chen K, Cheng L, Qian W, Jiang Z, Sun L,
Zhao Y, Zhou Y, Zhao L, Wang P, Duan W, et al: Itraconazole
inhibits invasion and migration of pancreatic cancer cells by
suppressing TGF-β/SMAD2/3 signaling. Oncol Rep. 39:1573–1582.
2018.PubMed/NCBI
|
|
172
|
Sezer G, Onses MS, Sakir M, Sahin F,
Çamdal A, Sezer Z, Inal A and Ciftci Z: Indomethacin prevents
TGF-β-induced epithelial-to-mesenchymal transition in pancreatic
cancer cells; evidence by Raman spectroscopy. Spectrochim Acta A
Mol Biomol Spectrosc. 280:1214932022. View Article : Google Scholar
|
|
173
|
Cheng P, Chen Y, He TL, Wang C, Guo SW, Hu
H, Ni CM, Jin G and Zhang YJ: Menin coordinates C/EBPβ-mediated
TGF-β signaling for epithelial-mesenchymal transition and growth
inhibition in pancreatic cancer. Mol Ther Nucleic Acids.
18:155–165. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Ungefroren H, Christl J, Eiden C, Wellner
UF, Lehnert H and Marquardt JU: Autocrine TGFβ1 opposes exogenous
TGFβ1-induced cell migration and growth arrest through sustainment
of a feed-forward loop involving MEK-ERK signaling. Cancers
(Basel). 13. pp. 13572021, View Article : Google Scholar
|
|
175
|
Zhu X, Zhang T, Zhang Y, Chen H, Shen J,
Jin X, Wei J, Zhang E, Xiao M, Fan Y, et al: A super-enhancer
controls TGF-β signaling in pancreatic cancer through
downregulation of TGFBR2. Cell Signal. 66:1094702020. View Article : Google Scholar
|
|
176
|
Cheng CS, Wu Y, Jin JB, Xu JY, Yang PW,
Zhu WH, Zheng L and Chen JX: Cynanchum paniculatum(Bunge) Kitag. ex
H.Hara inhibits pancreatic cancer progression by inducing
caspase-dependent apoptosis and suppressing TGF-β-mediated
epithelial-mesenchymal transition. Front Pharmacol. 15:12843712024.
View Article : Google Scholar
|
|
177
|
Zhao L, Chen G, Li D, Wang K, Schaefer M,
Herr I and Yan B: Baicalein disrupts TGF-β-induced EMT in
pancreatic cancer by FTO-dependent m6A demethylation of ZEB1.
Biochim Biophys Acta Mol Cell Res. 1872:1199692025. View Article : Google Scholar
|
|
178
|
Zhang Z, Auyeung KK, Sze SC, Zhang S, Yung
KK and Ko JK: The dual roles of calycosin in growth inhibition and
metastatic progression during pancreatic cancer development: A
'TGF-β paradox'. Phytomedicine. 68:1531772020. View Article : Google Scholar
|
|
179
|
Liu QQ, Chen K, Ye Q, Jiang XH and Sun YW:
Oridonin inhibits pancreatic cancer cell migration and
epithelial-mesenchymal transition by suppressing Wnt/β-catenin
signaling pathway. Cancer Cell Int. 16:572016. View Article : Google Scholar
|
|
180
|
Zhou C, Li J, Qian W, Yue Y, Xiao Y, Qin
T, Ma Q and Li X: Huaier extract restrains pancreatic cancer by
suppressing Wnt/β-catenin pathway. Biomed Pharmacother.
127:1101262020. View Article : Google Scholar
|
|
181
|
Kim JH, Hwang S, Lee JH, Im SS and Son J:
Vitamin C suppresses pancreatic carcinogenesis through the
inhibition of both glucose metabolism and Wnt signaling. Int J Mol
Sci. 23:122492022. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Guo Q and Qin W: DKK3 blocked
translocation of β-catenin/EMT induced by hypoxia and improved
gemcitabine therapeutic effect in pancreatic cancer Bxpc-3 cell. J
Cell Mol Med. 19:2832–2841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Douchi D, Ohtsuka H, Ariake K, Masuda K,
Kawasaki S, Kawaguchi K, Fukase K, Oikawa M, Motoi F, Naitoh T, et
al: Silencing of LRRFIP1 reverses the epithelial-mesenchymal
transition via inhibition of the Wnt/β-catenin signaling pathway.
Cancer Lett. 365:132–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Li F, Dai L and Niu J: GPX2 silencing
relieves epithelial-mesenchymal transition, invasion, and
metastasis in pancreatic cancer by downregulating Wnt pathway. J
Cell Physiol. 235:7780–7790. 2020. View Article : Google Scholar
|
|
185
|
Qin C, Li T, Wang Y, Zhao B, Li Z, Li T,
Yang X, Zhao Y and Wang W: CHRNB2 represses pancreatic cancer
migration and invasion via inhibiting β-catenin pathway. Cancer
Cell Int. 22:3402022. View Article : Google Scholar
|
|
186
|
Wu J, Li H, Shi M, Zhu Y, Ma Y, Zhong Y,
Xiong C, Chen H and Peng C: TET1-mediated DNA hydroxymethylation
activates inhibitors of the Wnt/β-catenin signaling pathway to
suppress EMT in pancreatic tumor cells. J Exp Clin Cancer Res.
38:3482019. View Article : Google Scholar
|
|
187
|
Cao L, Xiao X, Lei J, Duan W, Ma Q and Li
W: Curcumin inhibits hypoxia-induced epithelial-mesenchymal
transition in pancreatic cancer cells via suppression of the
hedgehog signaling pathway. Oncol Rep. 35:3728–3734. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Fu J, Shrivastava A, Shrivastava SK,
Srivastava RK and Shankar S: Triacetyl resveratrol upregulates
miRNA-200 and suppresses the Shh pathway in pancreatic cancer: A
potential therapeutic agent. Int J Oncol. 54:1306–1316.
2019.PubMed/NCBI
|
|
189
|
Verma RK, Yu W, Shrivastava A, Shankar S
and Srivastava RK: α-Mangostin-encapsulated PLGA nanoparticles
inhibit pancreatic carcinogenesis by targeting cancer stem cells in
human, and transgenic (Kras(G12D), and Kras(G12D)/tp53R270H) mice.
Sci Rep. 6:327432016. View Article : Google Scholar
|
|
190
|
Ma Y, Yu W, Shrivastava A, Srivastava RK
and Shankar S: Inhibition of pancreatic cancer stem cell
characteristics by α-Mangostin: Molecular mechanisms involving
Sonic hedgehog and Nanog. J Cell Mol Med. 23:2719–2730. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Bai Y, Chen B, Hong W, Liang Y, Zhou M and
Zhou L: Sedum sarmentosum Bunge extract induces apoptosis and
inhibits proliferation in pancreatic cancer cells via the hedgehog
signaling pathway. Oncol Rep. 35:2775–2784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Roy Chaudhuri T, Lin Q, Stachowiak EK,
Rosario SR, Spernyak JA, Ma WW, Stachowiak MK, Greene MK, Quinn GP,
McDade SS, et al: Dual-hit strategy for therapeutic targeting of
pancreatic cancer in patient-derived xenograft tumors. Clin Cancer
Res. 30:1367–1381. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He
ZX, Pan ST, Yang YX, Wang ZZ, Zhang X, et al: Alisertib induces
cell cycle arrest and autophagy and suppresses
epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and
sirtuin 1-mediated signaling pathways in human pancreatic cancer
cells. Drug Des Devel Ther. 9:575–601. 2015.PubMed/NCBI
|
|
194
|
Sun L, Cao J, Chen K, Cheng L, Zhou C, Yan
B, Qian W, Li J, Duan W, Ma J, et al: Betulinic acid inhibits
stemness and EMT of pancreatic cancer cells via activation of AMPK
signaling. Int J Oncol. 54:98–110. 2019.
|
|
195
|
Liu J, Song N, Huang Y and Chen Y: Irisin
inhibits pancreatic cancer cell growth via the AMPK-mTOR pathway.
Sci Rep. 8:152472018. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Wang F, Wang Q, Zhou ZW, Yu SN, Pan ST, He
ZX, Zhang X, Wang D, Yang YX, Yang T, et al: Plumbagin induces cell
cycle arrest and autophagy and suppresses epithelial to mesenchymal
transition involving PI3K/Akt/mTOR-mediated pathway in human
pancreatic cancer cells. Drug Des Devel Ther. 9:537–560.
2015.PubMed/NCBI
|
|
197
|
Li Q, Yang Y, Lin X, Chu LT, Chen H, Chen
L, Tang J and Zeng T: Regulation of pancreatic cancer cells by
suppressing KIN17 through the PI3K/AKT/mTOR signaling pathway.
Oncol Rep. 53:312025. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Tian Z, Tan Y, Lin X, Su M, Pan L, Lin L,
Ou G and Chen Y: Arsenic trioxide sensitizes pancreatic cancer
cells to gemcitabine through downregulation of the
TIMP1/PI3K/AKT/mTOR axis. Transl Res. 255:66–76. 2023. View Article : Google Scholar
|
|
199
|
Li W, Sun L, Lei J, Wu Z, Ma Q and Wang Z:
Curcumin inhibits pancreatic cancer cell invasion and EMT by
interfering with tumor-stromal crosstalk under hypoxic conditions
via the IL-6/ERK/NF-κB axis. Oncol Rep. 44:382–392. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Dai S, Zhang J, Huang S, Lou B, Fang B, Ye
T, Huang X, Chen B and Zhou M: HNRNPA2B1 regulates the
epithelial-mesenchymal transition in pancreatic cancer cells
through the ERK/snail signalling pathway. Cancer Cell Int.
17:122017. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Sun J, Chen L and Dong M: MiR-338-5p
inhibits EGF-induced EMT in pancreatic cancer cells by targeting
EGFR/ERK signaling. Front Oncol. 11:6164812021. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Ungefroren H, Konukiewitz B, Braun R,
Wellner UF, Lehnert H and Marquardt JU: TAp73 inhibits EMT and cell
migration in pancreatic cancer cells through promoting SMAD4
expression and SMAD4-dependent inhibition of ERK activation.
Cancers (Basel). 15. pp. 37912023, View Article : Google Scholar
|
|
203
|
Luan YZ, Wang CC, Yu CY, Chang YC, Sung WW
and Tsai MC: The therapeutic role of NPS-1034 in pancreatic ductal
adenocarcinoma as monotherapy and in combination with chemotherapy.
Int J Mol Sci. 25:69192024. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Yao H, Ren Y, Wu F, Cao L, Liu J, Yan M
and Li X: The discovery of a Novel AXL/triple angiokinase inhibitor
based on 6-chloro-substituted indolinone and side chain Methyl
substitution inhibiting pancreatic cancer growth and metastasis. J
Med Chem. 68:465–490. 2025. View Article : Google Scholar
|
|
205
|
Mekapogu AR, Xu Z, Pothula S, Perera C,
Pang T, Hosen SMZ, Damalanka V, Janetka J, Goldstein D, Pirola R,
et al: HGF/c-Met pathway inhibition combined with chemotherapy
increases cytotoxic T-cell infiltration and inhibits pancreatic
tumour growth and metastasis. Cancer Lett. 568:2162862023.
View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Zhao J, Liu X, Jin X, Dong T, Gao X, Wang
J, Li Y and Ma E: Riboflavin protects against pancreatic cancer
metastasis by targeting TGF-β receptor 1. Bioorg Chem.
146:1072742024. View Article : Google Scholar
|
|
207
|
Bu XN, Qiu C, Wang C and Jiang Z:
Inhibition of DACH1 activity by short hairpin RNA represses cell
proliferation and tumor invasion in pancreatic cancer. Oncol Rep.
36:745–754. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Liu B, Wu S, Ma J, Yan S, Xiao Z, Wan L,
Zhang F, Shang M and Mao A: lncRNA GAS5 reverses EMT and tumor stem
cell-mediated gemcitabine resistance and metastasis by targeting
miR-221/SOCS3 in pancreatic cancer. Mol Ther Nucleic Acids.
13:472–482. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Chen S, Chen JZ, Zhang JQ, Chen HX, Qiu
FN, Yan ML, Tian YF, Peng CH, Shen BY, Chen YL, et al: Silencing of
long noncoding RNA LINC00958 prevents tumor initiation of
pancreatic cancer by acting as a sponge of microRNA-330-5p to
down-regulate PAX8. Cancer Lett. 446:49–61. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Zhou Y, Zhou X, Ben Q, Liu N, Wang J, Zhai
Y, Bao Y and Zhou L: GATA6-AS1 suppresses epithelial-mesenchymal
transition of pancreatic cancer under hypoxia through regulating
SNAI1 mRNA stability. J Transl Med. 21:8822023. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Huang R, Yang L, Zhang Z, Liu X, Fei Y,
Tong WM, Niu Y and Liang Z: RNA m6A Demethylase ALKBH5
protects against pancreatic ductal adenocarcinoma via targeting
regulators of iron metabolism. Front Cell Dev Biol. 9:7242822021.
View Article : Google Scholar
|
|
212
|
Schiedlauske K, Deipenbrock A, Pflieger M,
Hamacher A, Hänsel J, Kassack MU, Kurz T and Teusch NE: Novel
histone deacetylase (HDAC) inhibitor induces apoptosis and
suppresses invasion via E-cadherin upregulation in pancreatic
ductal adenocarcinoma (PDAC). Pharmaceuticals (Basel). 17. pp.
7522024, View Article : Google Scholar
|
|
213
|
Jia Y, Li J, Mei W, Zhang H, Wang Z, Xie
X, Gao C, Xu X and Li F: Pan-HDAC inhibitor LAQ824 inhibits the
progression of pancreatic ductal adenocarcinoma and suppresses
immune escape by promoting antigen presentation. Int
Immunopharmacol. 154:1145282025. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Mody HR, Hung SW, AlSaggar M, Griffin J
and Govindarajan R: I n h ibit ion of S -Adenosyl met h ion i ne -D
ep endent Methyltransferase Attenuates TGFβ1-induced EMT and
metastasis in pancreatic cancer: Putative roles of miR-663a and
miR-4787-5p. Mol Cancer Res. 14:1124–1135. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Mody HR, Hung SW, Pathak RK, Griffin J,
Cruz-Monserrate Z and Govindarajan R: miR-202 diminishes TGFβ
receptors and attenuates TGFβ1-induced EMT in pancreatic cancer.
Mol Cancer Res. 15:1029–1039. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Gui Z, Luo F, Yang Y, Shen C, Li S and Xu
J: Oridonin inhibition and miR-200b-3p /ZEB1 axis in human
pancreatic cancer. Int J Oncol. 50:111–120. 2017. View Article : Google Scholar
|
|
217
|
Garg R, Melstrom L, Chen J, He C and Goel
A: Targeting FTO suppresses pancreatic carcinogenesis via
regulating stem cell maintenance and EMT pathway. Cancers (Basel).
14. pp. 59192022, View Article : Google Scholar
|
|
218
|
Kim SH, Ryu KJ, Hong KS, Kim H, Han H, Kim
M, Kim T, Ok DW, Yang JW, Hwangbo C, et al: ERK3 increases snail
protein stability by inhibiting FBXO11-mediated snail
ubiquitination. Cancers (Basel). 16. pp. 1052023, View Article : Google Scholar
|
|
219
|
Hu H, Wang Y, Ding X, He Y, Lu Z, Wu P,
Tian L, Yuan H, Liu D, Shi G, et al: Long non-coding RNA
XLOC_000647 suppresses progression of pancreatic cancer and
decreases epithelial-mesenchymal transition-induced cell invasion
by down-regulating NLRP3. Mol Cancer. 17:182018. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Li C, Cai J, Liu W, Gao Z and Li G:
Downregulation of circ-STK39 suppresses pancreatic cancer
progression by sponging mir-140-3p and regulating TRAM2-mediated
epithelial-mesenchymal transition. Apoptosis. 28:1024–1034. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Cao T, Hong L, Yu D, Shen J, Jiang L, Hu N
and He S: Circular RNA circTMEM59 inhibits progression of
pancreatic ductal adenocarcinoma by targeting miR-147b/SOCS1: An in
vitro study. Heliyon. 10:e244022024. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Su X, Lai T, Tao Y, Zhang Y, Zhao C, Zhou
J, Chen E, Zhu M, Zhang S, Wang B, et al: miR-33a-3p regulates
METTL3-mediated AREG stability and alters EMT to inhibit pancreatic
cancer invasion and metastasis. Sci Rep. 13:135872023. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Edderkaoui M, Chheda C, Soufi B, Zayou F,
Hu RW, Ramanujan VK, Pan X, Boros LG, Tajbakhsh J, Madhav A, et al:
An inhibitor of GSK3B and HDACs kills pancreatic cancer cells and
slows pancreatic tumor growth and metastasis in mice.
Gastroenterology. 1551985–1998. (e5)2018. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Li Y, Zheng Y, Xu S, Hu H, Peng L, Zhu J
and Wu M: The nanobody targeting PD-L1 and CXCR4 counteracts
pancreatic stellate cell-mediated tumour progression by disrupting
tumour microenvironment. Int Immunopharmacol. 132:1119442024.
View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Li Y, Hong J, Jung BK, Oh E and Yun CO:
Oncolytic Ad co-expressing decorin and Wnt decoy receptor overcomes
chemoresistance of desmoplastic tumor through degradation of ECM
and inhibition of EMT. Cancer Lett. 459:15–29. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Fujisawa T, Tsuchiya T, Kato M, Mizuide M,
Takakura K, Nishimura M, Kutsumi H, Matsuda Y, Arai T, Ryozawa S,
et al: STNM01, the RNA oligonucleotide targeting carbohydrate
sulfotransferase 15, as second-line therapy for
chemotherapy-refractory patients with unresectable pancreatic
cancer: An open label, phase I/IIa trial. EClinicalMedicine.
55:1017312022. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Jiang Z, Zhou C, Cheng L, Yan B, Chen K,
Chen X, Zong L, Lei J, Duan W, Xu Q, et al: Inhibiting YAP
expression suppresses pancreatic cancer progression by disrupting
tumor-stromal interactions. J Exp Clin Cancer Res. 37:692018.
View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Ireland L, Luckett T, Schmid MC and Mielgo
A: Blockade of stromal Gas6 alters cancer cell plasticity,
activates NK Cells, and inhibits pancreatic cancer metastasis.
Front Immunol. 11:2972020. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Shen L, Li J, Liu Q, Das M, Song W, Zhang
X, Tiruthani K, Dorosheva O, Hu H, Lai SK, et al: Nano-trapping
CXCL13 reduces regulatory B cells in tumor microenvironment and
inhibits tumor growth. J Control Release. 343:303–313. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Pothula SP, Xu Z, Goldstein D, Biankin AV,
Pirola RC, Wilson JS and Apte MV: Hepatocyte growth factor
inhibition: A novel therapeutic approach in pancreatic cancer. Br J
Cancer. 114:269–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Mondal G, Almawash S, Chaudhary AK and
Mahato RI: EGFR-Targeted cationic polymeric mixed micelles for
codelivery of gemcitabine and miR-205 for treating advanced
pancreatic cancer. Mol Pharm. 14:3121–3133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Cao ZQ, Wang XX, Lu L, Xu JW, Li XB, Zhang
GR, Ma ZJ, Shi AC, Wang Y and Song YJ: β-Sitosterol and gemcitabine
exhibit synergistic Anti-pancreatic cancer activity by modulating
apoptosis and inhibiting Epithelial-mesenchymal transition by
deactivating Akt/GSK-3β signaling. Front Pharmacol. 9:15252019.
View Article : Google Scholar
|
|
233
|
Wei R, Penso NEC, Hackman RM, Wang Y and
Mackenzie GG: Epigallocatechin-3-Gallate (EGCG) suppresses
pancreatic cancer cell growth, invasion, and migration partly
through the inhibition of akt pathway and Epithelial-mesenchymal
transition: Enhanced efficacy when combined with gemcitabine.
Nutrients. 11:18562019. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Li M, Wang X, Chen X, Hong J, Du Y and
Song D: GK921, a transglutaminase inhibitor, strengthens the
antitumor effect of cisplatin on pancreatic cancer cells by
inhibiting epithelial-to-mesenchymal transition. Biochim Biophys
Acta Mol Basis Dis. 1870:1669252024. View Article : Google Scholar
|
|
235
|
Jin H, Zhao Y, Zhang S, Yang J, Zhang X
and Ma S: Hyperthermia inhibits the motility of
gemcitabine-resistant pancreatic cancer PANC-1 cells through the
inhibition of epithelial-mesenchymal transition. Mol Med Rep.
17:7274–7280. 2018.PubMed/NCBI
|
|
236
|
Momeny M, Alishahi Z, Eyvani H, Esmaeili
F, Zaghal A, Ghaffari P, Tavakkoly-Bazzaz J, Alimoghaddam K,
Ghavamzadeh A and Ghaffari SH: Anti-tumor activity of cediranib, a
pan-vascular endothelial growth factor receptor inhibitor, in
pancreatic ductal adenocarcinoma cells. Cell Oncol (Dordr).
43:81–93. 2020. View Article : Google Scholar
|
|
237
|
Mondal SK, Jinka S, Pal K, Nelli S, Dutta
SK, Wang E, Ahmad A, AlKharfy KM, Mukhopadhyay D and Banerjee R:
Glucocorticoid Receptor-targeted liposomal codelivery of lipophilic
drug and Anti-Hsp90 gene: Strategy to Induce Drug-sensitivity,
EMT-Reversal, and reduced malignancy in aggressive tumors. Mol
Pharm. 13:2507–2523. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Guo D, Sheng K, Zhang Q, Li P, Sun H, Wang
Y, Lyu X, Jia Y, Wang C, Wu J, et al: Single-cell transcriptomic
analysis reveals the landscape of epithelial-mesenchymal transition
molecular heterogeneity in esophageal squamous cell carcinoma.
Cancer Lett. 587:2167232024. View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Buchholz M, Lausser L, Schenk M, Earl J,
Lawlor RT, Scarpa A, Sanjuanbenito A, Carrato A, Malats N, Tjaden
C, et al: Combined analysis of a serum mRNA/miRNA marker signature
and CA 19-9 for timely and accurate diagnosis of recurrence after
resection of pancreatic ductal adenocarcinoma: A prospective
multicenter cohort study. United European Gastroenterol J.
13:353–363. 2025. View Article : Google Scholar :
|
|
240
|
Teixeira AF, Ten Dijke P and Zhu HJ:
On-Target Anti-TGF-β therapies are not succeeding in clinical
cancer treatments: What are remaining challenges? Front Cell Dev
Biol. 8:6052020. View Article : Google Scholar
|
|
241
|
Chen Q, Wang H, Liu Q and Luo C: CTHRC1: A
key player in colorectal cancer progression and immune evasion.
Front Immunol. 16:15796612025. View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Gu M, Liu Y, Xin P, Guo W, Zhao Z, Yang X,
Ma R, Jiao T and Zheng W: Fundamental insights and molecular
interactions in pancreatic cancer: Pathways to therapeutic
approaches. Cancer Lett. 588:2167382024. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Zhu SL, Qi M, Chen MT, Lin JP, Huang HF,
Deng LJ and Zhou XW: A novel DDIT3 activator dehydroevodiamine
effectively inhibits tumor growth and tumor cell stemness in
pancreatic cancer. Phytomedicine. 128:1553772024. View Article : Google Scholar : PubMed/NCBI
|














