Introduction
Among all risk factors for cancer, smoking is the
most important and controllable one; notably, it is frequently
observed that smoking cessation can result in a reduction of this
risk (1,2). Cigarette smoke has been reported to
contain carcinogenic compounds, including nitrosamines, polycyclic
aromatic hydrocarbons and volatile organic compounds, which lead to
mutations in critical cancer genes, such as KRAS and TP53 (3,4).
Colorectal cancer (CRC) is the third most prevalent type of cancer
worldwide (5), and globally,
smoking is a common risk factor for CRC, contributing to ~13.3% of
CRC cases (6). Previous studies
have confirmed the association between smoking and CRC by
performing correlation and Mendelian randomization (MR) analyses
(7,8). Animal experiments have also revealed
that cigarette smoke increases the incidence of CRC and promotes
cell proliferation by inducing gut microbiota dysbiosis (9). However, there is still a lack of
research on critical genes that regulate the progression of
smoking-associated CRC.
The National Health and Nutrition Examination Survey
(NHANES) database (https://wwwn.cdc.gov/nchs/nhanes/) contains abundant
survey data and has been widely used in observation studies
(10-13). MR analysis is a robust method for
causality assessment (14);
notably, genome-wide association studies (GWAS) provide ideal
instrumental variables (IVs) and have been widely used in MR
analyses (11). In the present
study, observational studies and MR analyses were employed to
systematically assess the link between different smoking
characteristics and CRC.
Bioinformatics analyses have been widely used to
identify critical molecules of diseases (15). In the current study, the critical
genes implicated in the smoking-enhanced progression of CRC were
identified by integrative bioinformatics analyses. By overlapping
CRC-related genes and smoking-related genes, the current study
aimed to filter out the key genes involved in smoking-enhanced CRC.
Cytoskeleton-associated protein 2-like (CKAP2L) is a component of
the cell centrosome, which serves a critical role in spindle
formation (16,17). As reported in bladder cancer,
esophageal squamous cell carcinoma and lung cancer, CKAP2L mediates
the cell cycle and promotes the progression and dissemination of
tumors (18-20). A recent study reported that
regulatory factor X5-regulated CKAP2L can stimulate the
proliferation, migration and invasion of CRC cells (21). Amphiregulin (AREG) is a ligand of
epidermal growth factor receptor (EGFR) and promotes cancer
progression via activating EGFR signaling (22). The present study aimed to identify
the key genes involved in smoking-enhanced CRC and reveal the
potential mechanisms by which key genes promote the progression of
CRC.
Materials and methods
Investigation of the association between
smoking and CRC
The clinical association between smoking features
(smoking status, cotinine and age started smoking) and CRC was
analyzed based on 1999-2018 NHANES survey datasets (https://wwwn.cdc.gov/nchs/nhanes/default.aspx). After
excluding participants under the age of 20 years, those who were
pregnant or those with missing variables, 37,091 participants were
finally included in the cross-sectional study.
Smoking status was defined by two questions: 'Smoked
at least 100 cigarettes in life?' and 'Do you now smoke
cigarettes?' Cotinine is a major metabolite of nicotine and can be
used as a marker for smokers and an indicator of secondhand smoke
exposure. Due to the recall bias in self-reported secondhand smoke
exposure (exposure source or duration), for individuals who have
never smoked, a serum cotinine level of ≥0.015 ng/ml (lower
detection limit) was considered secondhand smoke exposure according
to previous studies (23,24). The participants were subsequently
divided into four different smoking statuses: No smoking, past
smoking, current smoking and secondhand smoking. Previous studies
have reported that 3 ng/ml is a recommended cut-off point for
cotinine levels (25-27), whereas ≥10 ng/ml is always
observed in active smokers (28).
Therefore, serum cotinine levels were divided into <0.015,
0.015-3, 3-10 and ≥10 ng/ml. Age at which individuals started
smoking cigarettes regularly was divided into three groups: Never
regularly smoked, <20 and ≥20 years. CRC was defined according
to the first reported cancer type. The following covariates were
adjusted in the subsequent analyses: Age, sex, ethnicity, marital
status, education, ratio of family income to poverty, body mass
index and alcohol consumption. Those who had consumed ≥12 alcohol
drinks/1 year or lifetime were defined as having alcohol
consumption. The baseline characteristics revealed the differences
between the CRC and non-CRC groups (Table SI). Continuous data were compared
using survey-weighted linear regression, whereas categorical data
were compared using survey-weighted χ2 test.
Due to the complex sampling design of the NHANES
survey, sample weights, strata and primary sampling units were
considered in all analyses. The association between CRC and smoking
was assessed by three logistic regression models: Model 1,
non-adjusted; model 2, adjusted for age, sex and ethnicity [these
characteristics show notable differences in smoking habits and CRC
occurrence (29-31)]; and model 3, fully adjusted. All
analyses were performed using EmpowerStats software (version 4.1;
X&Y Solutions, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
The causality of smoking and CRC was measured by
two-sample MR analyses. GWAS data for CRC (n=293,646) were
downloaded from FinnGen (https://www.finngen.fi/en/access_results). Pooled GWAS
data for age of initiation of regular smoking (n=341,427),
cigarettes per day (n=337,334), smoking cessation (n=547,219) and
smoking initiation (n=1,232,091) were extracted from a
meta-analysis (32). GWAS data
for pack years of adult smoking as a proportion of life span
exposed to smoking (n=142,387) were downloaded from the IEU
database (https://gwas.mrcieu.ac.uk/datasets/ukb-b-7460/). All
participants in these GWAS datasets were European and the details
of the GWAS data are listed in Table
SII.
The identification of IVs conformed to three
assumptions: Relevance, independence and exclusion restriction.
Single-nucleotide polymorphisms (SNPs) with P<5×10−8
and a linkage disequilibrium of r2<0.001 within
10,000-kb windows were selected as IVs for smoking. Palindromic
SNPs were harmonized. Finally, 13 SNPs were identified as IVs for
pack years of adult smoking. R2 was calculated to reveal
the proportion of exposure variation explained by SNPs. The
F-statistic was calculated to assess the strength of the
association between the SNP and exposure. SNPs with F≥10 were
considered as strong genetic instruments and were included in the
subsequent analysis.
The inverse-variance weighted (IVW), weighted
median, simple mode and weighted mode methods in 'TwoSampleMR'
package (version 0.6.19; https://github.com/MRCIEU/TwoSampleMR) in R software
4.2.2 (https://www.r-project.org/) were used
for MR analysis, in which the IVW method was the main method.
Selection of the IVW fixed effect (IVW_FE) or IVW multiplicative
random effect (IVW_MRE) method was dependent on heterogeneity,
which was evaluated by Cochran's Q test. If heterogeneity existed,
IVW_MRE was the main method. MR-Egger intercept analysis was
performed to assess directional pleiotropy. MR-PRESSO, funnel plot
and leave-one-out analyses were conducted to detect outliers.
Outliers were excluded by MR-PRESSO and the remaining IVs were
included in the final MR analysis. MR analyses were conducted with
the 'TwoSampleMR' package in R software 4.2.2 (https://www.r-project.org/). Bonferroni-corrected
P<0.01 (0.05/5=0.01) was defined as the significance criterion
for MR estimation, whereas P<0.05 was considered to indicate a
statistically significant difference in other tests. Both
cross-sectional and MR analyses were supervised by a trained
statistician and a bioinformatician.
Bioinformatics analyses
To identify hub genes of CRC, The Cancer Genome
Atlas (TCGA) datasets (https://portal.gdc.cancer.gov/) for CRC (colon
adenocarcinoma and rectum adenocarcinoma), including 650 tumors and
51 healthy controls, were downloaded. In addition, bulk RNA
sequencing [RNA-seq; GSE200130 (33), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200130;
12 tumor samples and 13 healthy control samples] and single-cell
RNA-seq [scRNA-seq; GSE200997 (34), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200997;
16 tumors and 7 adjacent normal tissues] datasets were downloaded
from the Gene Expression Omnibus database. These datasets were used
in the bioinformatics analysis.
Differentially expressed genes (DEGs) in epithelial
cells between CRC and normal samples were filtered out based on
scRNA-seq, using 'Seurat' (v5; https://satijalab.org/seurat/) and 'harmony' (release
0.1; https://github.com/immunogenomics/harmony) packages.
The upregulated DEGs were identified with log2FoldChange
(FC)>0.585 and adjusted P<0.05. For GSE200130 and TCGA
datasets, upregulated DEGs with logFC>1 and adjusted P<0.05
were identified with the 'DESeq2' (35) package (version 1.44.0; https://www.bioconductor.org/packages/release/bioc/html/DESeq2.html).
Moreover, weighted gene co-expression network analysis (WGCNA) was
performed on TCGA datasets to further select genes positively
linked to CRC using the 'WGCNA' (36) package (version 1.73). By
overlapping the aforementioned upregulated DEGs and genes
positively related to CRC, the CRC-related genes were
identified.
The key genes involved in smoking-enhanced CRC were
selected by overlapping the CRC-related genes and smoking-related
genes. All analyses were conducted using R software 4.2.2.
Cell lines and inhibitor
To investigate the hypothesis that key genes promote
the progression of smoking-enhanced CRC in different subtypes of
CRC, and ensure the reliability and universality of the research
results, four human CRC cell lines were used. The human CRC cell
lines SW480, Caco-2, HCT116 and RKO, the mouse CRC cell line CT26,
the human normal colonic cell line NCM460 and 293T cells were
purchased from Ubigene Biosciences. All cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (FBS; Pricella; Elabscience Bionovation Inc.) at 37°C
and 5% CO2. The signal transducer and activator of
transcription 3 (STAT3) phosphorylation inhibitor Stattic (cat. no.
HY-13818; MedChemExpress) was dissolved in DMSO for cell culture.
HCT116 cells were cultured with Stattic at a final concentration of
2 μM for 24 h at 37°C and 5% CO2.
Construction of cell models treated with
CSE
According to previous studies (18,37,38), CSE was freshly prepared and used
to treat cells within 30 min. The details of CSE preparation and
usage were published in our previous study (18). Briefly, a cigarette (brand:
Hongtashan) was burned for ~5 min and its mainstream smoke was
absorbed in 10 ml DMEM. After filtering through a 0.22-μm
filter, the solution was regarded as a 100% CSE solution. Upon
preparation, CSE was diluted to the concentrations of 0.0625,
0.125, 0.25, 0.5, 1, 2 and 4% CSE using DMEM and was then used to
treat the CRC cell lines at 37°C for 4 days, including Caco-2, RKO
and HCT116 cells. The cells were then incubated with the Cell
Counting Kit 8 (CCK8; cat. no. BS350C; Biosharp Life Sciences)
reagent for 1 h at 37°C to evaluate the impact of varying
concentrations of CSE on cell viability. Subsequently, according to
the results of short-time CSE treatment, CRC cells (RKO and HCT116)
were cultured with 0.125 and 0.25% CSE for 2 months to reveal the
influence of chronic smoke exposure on proliferation, migration and
invasion.
Cell transfection and transduction
All plasmids and small interfering RNA (siRNA) were
synthetized by Beijing Tsingke Biotech Co., Ltd., and the details
of the short hairpin RNA (shRNA; Beijing Tsingke Biotech Co., Ltd.)
and siRNA sequences are shown in Table SIII. A total of four shCKAP2L
plasmids were synthesized and used, and the shRNA-negative control
(non-targeting) plasmid was set as the control. For the CKAP2L
overexpression (CKAP2L-OE) plasmid, the corresponding empty plasmid
without the CKAP2L gene insert was used as the control. First, all
of the OE and shRNA plasmids (PLVX-puro; Beijing Tsingke Biotech
Co., Ltd.) were transfected into 293T cells to generate lentiviral
particles, and these lentiviral particles were then transduced into
CRC cells. Briefly, to generate lentiviral particles, 293T cells
were seeded in a 10-cm culture dish and incubated overnight until
the cell density reached 70-80%. A total of 10 μg plasmids
(5 μg target plasmid, 3.75 μg psPAX2 and 1.25
μg pMD2.G) were co-transfected into 293T cells with 20
μl Lipofectamine® 2000 (cat. no. 11668027;
Invitrogen; Thermo Fisher Scientific, Inc.). The viruses were then
collected 48 and 72 h after transfection, and were mixed together
for cell transduction according to previous studies (39-41) in CRC cell lines (RKO, Caco-2,
HCT116 and CT26). Cells were transduced with 500 μl
collected supernatant containing lentiviral particles for 18 h at
37°C and 5% CO2. A total of 48 h post-transduction,
stable cells were filtered with puromycin (selection, 2
μg/ml; maintenance, 1 μg/ml).
For siAREG and siRNA-negative control transfection,
HCT116 cells were pre-plated in a 6-well plate and incubated
overnight until they reached 70-80% confluence. Subsequently, 50
μM siRNA was transfected into the cells with 5 μl
Lipofectamine 2000. After incubation at 37°C for 4-6 h, the
transfection complex was replaced with fresh culture medium. A
total of 24 h post-transfection, the mRNA expression was measured,
and after 48 h, the protein expression, and cell migration and
proliferation were detected.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA was extracted from cells using
TRIzol® reagent (cat. no. 15596026; Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. RNA was then reverse transcribed into cDNA using the
qPCR RT Master Mix (cat. no. FSQ-201; Toyobo Co., Ltd.) (RNA: 1
μg; 5X RT Master Mix: 2 μl; nuclease-free water: ≤10
μl; steps: 37°C for 15 min, 50°C for 5 min, 98°C for 5 min
and maintained at 4°C). qPCR was performed using the
SYBR® Green Realtime PCR Master Mix (cat. no. QPK-201;
Toyobo Co., Ltd.) according to the manufacturer's instructions
[nuclease-free water: 3.2 μl; forward primer (10 μM):
0.4 μl; reverse primer (10 μM): 0.4 μl; cDNA:
1 μl; SYBR Green Realtime PCR Master Mix: 5 μl;
steps: 95°C for 60 sec, followed by 40 cycles at 95°C for 5 sec,
60°C for 15 sec and 72°C for 45 sec, and melting curve analysis].
The mRNA expression levels were calculated using the
2−ΔΔCq method (42).
The primer sequences are listed in Table SIV.
RNA-seq
HCT116 cells were treated with CSE for 8 weeks and
underwent RNA extraction. RKO cells stably transduced with shCKAP2L
#1 and HCT116 cells stably transduced with CKAP2L-OE also underwent
RNA extraction using TRIzol reagent, according to the
manufacturer's instructions. Subsequently, the extracted RNA was
sent to Sangon Biotech Co., Ltd. for further processing and RNA
sequencing. The integrity of the RNA and DNA contamination were
detected by agarose gel electrophoresis on 1% gels. Based on the
Illumina HiSeq 2500 platform (Illumina, Inc.), 150-bp paired-end
sequencing (direction: Forward and reverse) was conducted using the
HiSeq Rapid SBS Kit v2 (cat. no. FC-402-4022; Illumina, Inc.). The
loading concentration of the final library was 6 pM. The sequencing
data were analyzed using the 'DESeq2' package and were visualized
in volcano plots. For self-conducted RNA-seq, the upregulated genes
(logFC>0.263 and adjusted P<0.05) in the CSE-treated group
were identified as smoking-related genes using the 'DESeq2'
package.
Western blotting
RIPA buffer (cat. no. P0013B; Beyotime
Biotechnology) was employed to extract total proteins from cells
and a BCA kit (cat. no. P0010S; Beyotime Biotechnology) was used to
detect protein concentration. A total of 40 μg protein/lane
was separated by SDS-PAGE (cat. no. P0012AC; Beyotime
Biotechnology) on 10-12% gels, and the proteins were then
transferred to PVDF membranes. Subsequently, the membranes were
incubated in 5% skim milk at room temperature for 1 h, and the
washed membranes were immersed in primary antibodies (Table SV) at 4°C overnight. β-actin was
used as the loading control. After incubation with the
corresponding secondary antibodies (HRP-conjugated Goat Anti-Rabbit
IgG, 1:5,000, cat. no. SA00001-2, Proteintech Group, Inc.;
HRP-conjugated Goat Anti-Mouse IgG, 1:5,000, cat. no. SA00001-1,
Proteintech Group, Inc.) at room temperature for 1 h, an ECL kit
(cat. no. P0018S; Beyotime Biotechnology) and Fusion software
(Fusion FX; Vilber Lourmat) were employed to measure protein
signals. Subsequently, the semi-quantification of images was
performed by ImageJ (version 1.54 g; National Institutes of
Health).
Enzyme-linked immunosorbent assay
(ELISA)
HCT116 cells were maintained with complete medium
supplemented with 2% FBS for 24 h and the medium was collected. The
concentration of secreted AREG in the culture medium was then
measured using the Human AREG ELISA Kit (cat. no. EK0304; Wuhan
Boster Biological Technology, Ltd.) according to the manufacturer's
instructions. Briefly, samples were added to the plate that was
precoated with antibody and were incubated at 37°C for 90 min.
After discarding the liquid, the biotin-labeled anti-human AREG
antibody was added to the plate and incubated at 37°C for 60 min.
After washing the plate, ABC solution was added and incubated at
37°C for 30 min. Finally, the plate was washed again and TMB
solution was added for color generation. The OD value at 450 nm was
detected using a microplate reader (Tecan Group, Ltd.).
Chromatin immunoprecipitation (ChIP)
The transcriptional regulatory effect of STAT3 on
AREG was evaluated using the Cistrome Data Browser (http://cistrome.org/db/) (43) and JASPAR database (https://jaspar.elixir.no/) (44). This effect was confirmed using a
ChIP assay kit (cat. no. P2080S; Beyotime Biotechnology), which was
performed according to the manufacturer's instructions. Briefly,
RKO cells were cultured in a 10-cm culture dish. Cross-linking was
carried out by incubation with 1% formaldehyde at 37°C for 10 min,
after which, SDS lysis buffer was added to the cells and incubated
on ice for 10 min. Subsequently, chromatin was fragmented to
400-800 bp using ultrasonication at 0°C (20 kHz; power: 38W;
ultrasonic treatment for 10 sec, pause for 10 sec; 20 cycles.).
After fragmentation, the lysate was incubated with 2 μg
anti-STAT3 (cat. no. 60199-1-Ig; Proteintech Group, Inc.) or 2
μg IgG (cat. no. 30000-0-AP; Proteintech Group, Inc.) at 4°C
overnight. Subsequently, 80 μl protein A/G beads
(MedChemExpress) were incubated with the aforementioned antibody
and lysate complex at 4°C for 1 h and chromatin was eluted with
elution buffer (1% SDS and 0.1 M NaHCO3) and was
incubated at 65°C for 4 h to reverse crosslinking. The purified DNA
was subjected to qPCR analysis as aforementioned.
Cell proliferation assays
The CCK8 reagent (cat. no. BS350C; Biosharp Life
Sciences) was employed to measure cell proliferation. Transduced
and transfected cells (RKO, Caco-2 and HCT116) were cultured at a
density of 2,000 cells/well for 0, 24, 48 and 72 h. Subsequently,
10 μl CCK8 reagent was added to 100 μl medium and
incubated with the cells for 1-2 h. Subsequently, a microplate
reader (Tecan Group, Ltd.) was employed to detect the absorbance at
450 nm.
The 5-ethynyl-2'-deoxyuridine (EdU) assay was also
conducted to assess cell proliferation. EdU reagent was added to
medium and incubated with the CSE-treated or transduced RKO, Caco-2
and HCT116 cells (60-70% confluence) for 2 h. Subsequently, the
cells were incubated with 4% paraformaldehyde at room temperature
for 15 min, treated with Triton X-100 at room temperature for 15
min, and stained with an EdU kit (cat. no. BL917A; Biosharp Life
Sciences) and Hoechst. Finally, images were obtained under a
fluorescence microscope (Leica Microsystems GmbH).
A colony formation assay was also conducted using
6-well plates. In each well, 1,500 transfected cells (RKO, Caco-2
and HCT116) were seeded and cultured for 14 days. Subsequently, the
colonies were fixed with 4% paraformaldehyde at room temperature
for 15 min and stained with 0.1% crystal violet at room temperature
for 15 min, images were captured and the colonies consisting of
>50 cells were counted under a light microscope (Leica
Microsystems GmbH).
Cell cycle analysis
Flow cytometry was employed to detect cell cycle
progression. Transduced RKO, Caco-2 and HCT116 cells at 80%
confluence were harvested and fixed with 75% ethanol for >24 h
at 4°C. The fixed cells were then stained with PI/RNase Staining
Buffer (cat. no. 550825; BD Biosciences) at room temperature for 15
min, and immediately submitted to cell cycle detection by flow
cytometry using a CytoFLEX flow cytometer (Beckman Coulter, Inc.)
and CytExpert software (Version 2.6; Beckman Coulter, Inc.).
Assessment of cell migration and
invasion
Transwell assay is a classic method used to assess
cell migration and invasion. To measure migration, 5×104
cells (RKO, Caco-2 and HCT116) were resuspended in 200 μl
serum-free medium and seeded in the upper chamber of a Transwell
plate (cat. no. 3422; Corning, Inc.). Subsequently, complete medium
was added to the lower chamber. After 48 h at 37°C, the migrated
cells were incubated with 4% paraformaldehyde at room temperature
for 15 min and 0.1% crystal violet at room temperature for 15 min,
successively. Under a light microscope, images of migrated cells
were captured. For invasion, after pre-coating the Transwell plate
with Matrigel (cat. no. 356234; Corning, Inc.), 5×104
cells were resuspended in 100 μl serum-free medium and
seeded in the upper chamber. The subsequent steps were identical to
those performed in the migration assay.
The wound healing assay is also a common method used
to measure cell migration. Briefly, RKO and HCT116 cells were
cultured to reach 100% confluence. Subsequently, the cell monolayer
was scraped and the cells were maintained in serum-free medium for
0 and 48 h at 37°C. At each time point, images were captured at the
same location under a light microscope (Leica Microsystems
GmbH).
Subcutaneous tumor model
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Chongqing Medical
University (approval no. IACUC-CQMU-2025-0457; Chongqing, China).
In the present study, 4-6-week-old female BALB/c mice (weight,
14-18 g) were obtained from and raised at the Experimental Animal
Center of Chongqing Medical University at a room temperature of
20-24°C and 45-55% humidity under a 12-h light/dark cycle, with
free access to water and food. Using the random number method, a
total of eight mice were randomly divided into two groups: Vector
and CKAP2L groups (n=4 mice/group). Briefly, CT26 cells
(2×106) were stably infected with CKAP2L-OE and vector
lentiviruses as aforementioned and then suspended in 100 μl
PBS. This mix was subcutaneously injected into the right flank of
each mouse, after which, tumor growth was independently and blindly
measured every 3 days by two experimenters. The mice were
continuously fed for 21 days, and the tumor size, health and
behavior of the mice were checked every 1-2 days. The maximum
allowable tumor diameter was 15 mm. When the tumor size reached 15
mm, when ulceration occurred on the body surface, or when the mice
exhibited weakness, poor appetite and symptoms such as convulsions,
tremors and paralysis, the experiment was terminated and euthanasia
was performed. In the current study, no mice reached the
aforementioned humane endpoints. Euthanasia was performed by
intraperitoneal injection of an overdose of sodium pentobarbital
(150 mg/kg), followed by confirmation of respiratory cessation,
cardiac arrest and loss of vital reflexes. Subsequently, the tumors
were removed and weighed.
Statistical analysis
GraphPad Prism 10 (Dotmatics) and SPSS 27 (IBM
Corp.) software were used for statistical analyses. Each experiment
was independently repeated three times and all data are presented
as the mean ± standard deviation. Unpaired Student's t-test was
used to analyze the differences between two groups, whereas one-way
ANOVA followed by Dunnett's test was used to analyze the
differences among multiple groups. For comparisons among multiple
groups with two treatment factors, two-way ANOVA followed by Tukey
test was adopted. Adjusted P<0.05 was considered to indicate a
statistically significant difference.
Results
Association between smoking and CRC
Among the participants with CRC, 60.5% of them had
reported a history of smoking and 24.1% had reported secondhand
smoke exposure (Table SI). A
positive link between smoking and CRC was determined by logistic
regression analyses (Table I).
After fully adjusting for covariates, the risk of CRC in the past
smoking [odds ratio (OR), 1.595; 95% confidence interval (CI),
1.020-2.493] and current smoking (OR, 1.738; 95% CI, 1.033-2.923)
groups was higher than that reported in the no smoking history
group. No difference in the risk of CRC was observed between the
secondhand smoking and no smoking groups (OR, 1.184; 95% CI,
0.706-1.984).
 | Table IAssociation between smoking and
colorectal cancer. |
Table I
Association between smoking and
colorectal cancer.
| Characteristic | Non-adjusted model
| Adjusted model I
| Adjusted model II
|
|---|
| OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value |
|---|
| Smoking | | | | | | |
| No smoking | Ref | | Ref | | Ref | |
| Past smoking | 2.208 (1.489,
3.273) | 0.0001 | 1.684 (1.111,
2.552) | 0.0154 | 1.595 (1.020,
2.493) | 0.0429 |
| Current
smoking | 0.824 (0.506,
1.342) | 0.4377 | 1.786 (1.106,
2.885) | 0.0191 | 1.738 (1.033,
2.923) | 0.0394 |
| Secondhand
smoking | 0.860 (0.516,
1.431) | 0.5619 | 1.179 (0.708,
1.962) | 0.5285 | 1.184 (0.706,
1.984) | 0.5237 |
| Cotinine,
ng/ml | | | | | | |
| <0.015 | Ref | | Ref | | Ref | |
| 0.015-2.99 | 0.612 (0.169,
2.210) | 0.4545 | 0.634 (0.175,
2.301) | 0.4900 | 0.626 (0.173,
2.258) | 0.4753 |
| 3-10 | 1.517 (1.043,
2.208) | 0.0310 | 0.823 (0.557,
1.215) | 0.3277 | 0.847 (0.575,
1.248) | 0.4035 |
| ≥10 | 1.786 (1.168,
2.729) | 0.0083 | 0.732 (0.475,
1.127) | 0.1589 | 0.752 (0.478,
1.184) | 0.2209 |
| Age at which
started smoking cigarettes regularly | | | | | | |
| Never regularly
smoked | Ref | | Ref | | Ref | |
| <20 years | 0.999 (0.360,
2.776) | 0.9986 | 1.042 (0.361,
3.005) | 0.9400 | 1.059 (0.365,
3.074) | 0.9169 |
| ≥20 years | 1.335 (0.460,
3.872) | 0.5963 | 0.986 (0.327,
2.972) | 0.9794 | 1.006 (0.335,
3.025) | 0.9916 |
Serum cotinine, a major metabolite of nicotine, was
used to assess the short-term smoke exposure levels, and was
classified as <0.015, 0.015-2.99, 3-10 and ≥10 ng/ml. In all
models, no difference was observed among patients with different
cotinine levels (Table I). The
effect of age at the start of smoking cigarettes regularly on the
risk of CRC was also explored; notably, no difference among
patients in the different age groups was observed in all models
(Table I).
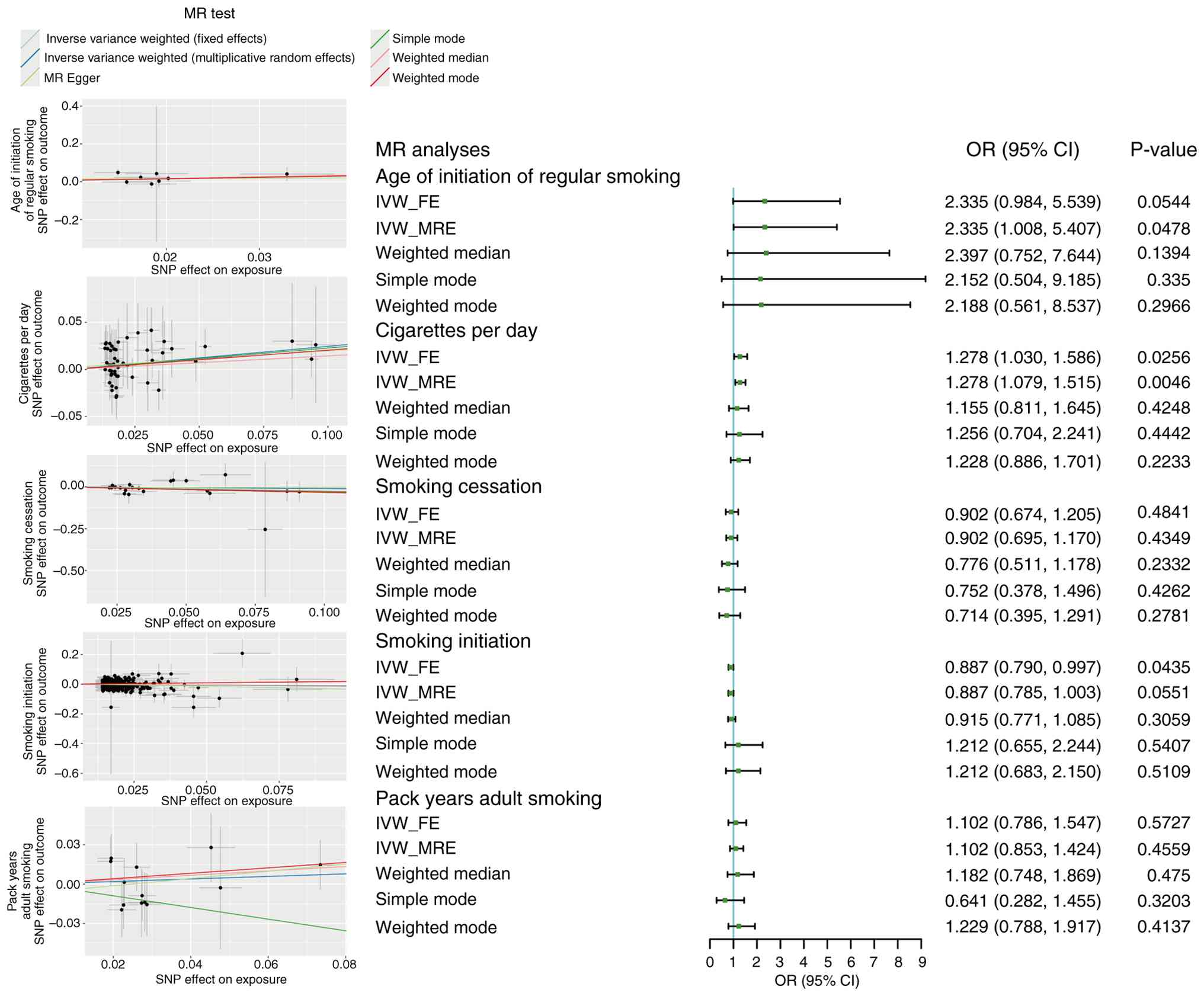
Causality of smoking and CRC
A total of 52 IVs were included in MR analysis of
cigarettes per day. MR analysis confirmed that cigarettes per day
had a causal effect on CRC (IVW_FE: OR, 1.278; 95% CI, 1.030-1.586;
IVW_MRE: OR, 1.278; 95% CI, 1.079-1.515) (Fig. 1). No heterogeneity (Cochran's Q
test, P=0.982), pleiotropy (MR-Egger intercept, P=0.676) and
outliers (MR-PRESSO, P=0.977) were observed, which confirmed the
reliability of the results (Table
SII).
A total of 8, 21, 331 and 13 IVs were included for
the age of initiation of regular smoking, smoking cessation,
smoking initiation and pack years of adult smoking, respectively.
No causality was observed between these factors and CRC (all
P>0.05; Fig. 1). No
heterogeneity, pleiotropy and outliers were observed in these MR
analyses. The details of Cochran's Q test, MR-Egger intercept and
MR-PRESSO are listed in Table
SII.
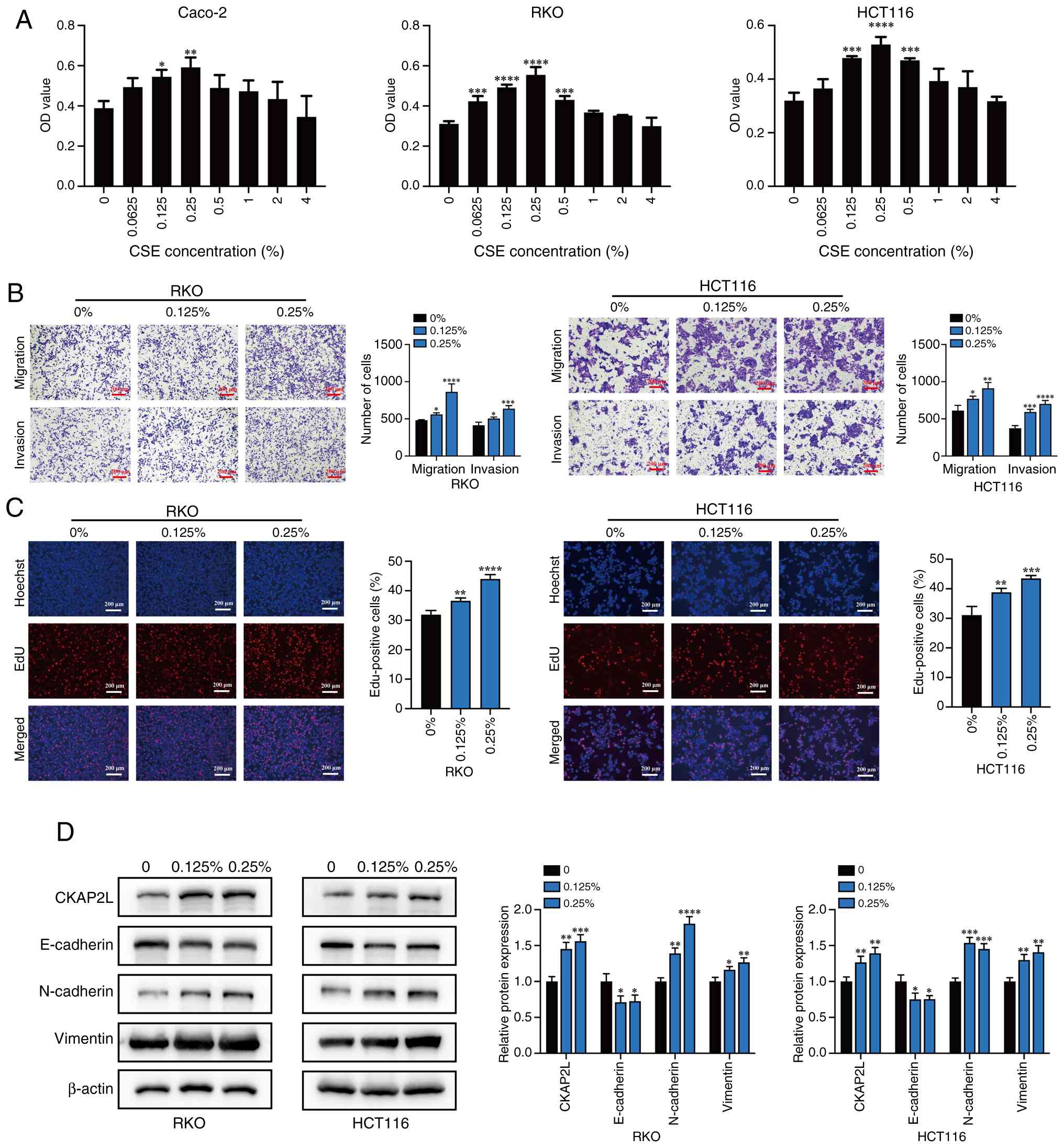
CSE promotes CRC progression
After 4 days of treatment with CSE, compared with
that in the control group, treatment with CSE increased cell
viability; among all groups, CSE concentrations of 0.125 and 0.25%
had the highest capacity to promote cell viability in all three
cell lines, as confirmed by CCK8 (Fig. 2A). In groups treated with 4% CSE,
cell viability was not significantly promoted. Therefore, in the
subsequent experiments, CSE concentrations of 0.125 and 0.25% were
selected to establish the chronic smoke exposure model, and were
used for functional experiments on RKO and HCT116 cells. Among the
three cell lines, RKO and HCT116 cells showed a more significant
increase in cell proliferation after short-term CSE treatment, and
were selected for subsequent experiments.
Transwell assays revealed that smoke exposure
increased the number of migratory and invasive RKO and HCT116 cells
(Fig. 2B). The EdU assay also
showed that smoke exposure significantly stimulated the
proliferation of CRC cells (Fig.
2C). Moreover, smoke exposure promoted the protein expression
levels of N-cadherin and Vimentin, but inhibited the expression
levels of E-cadherin, indicating that CSE promoted
epithelial-mesenchymal transition (EMT) (Fig. 2D). These results suggested that
CSE promotes CRC cell progression and increases the expression of
CKAP2L.
Identifying key genes related to both
smoking and CRC
The scRNA-seq analysis identified 2,450 DEGs in the
epithelial cells of CRC. Subsequently, 677 upregulated DEGs were
screened from the bulk RNA-seq and 2,973 upregulated DEGs were
filtered out from TCGA dataset. To further identify genes related
to CRC, 245 genes were identified using the WGCNA method based on
TCGA dataset. Based on TCGA, scRNA-seq and bulk RNA-seq datasets, a
total of 12 CRC-related genes were screened out by overlapping the
aforementioned DEGs (Fig.
3A).
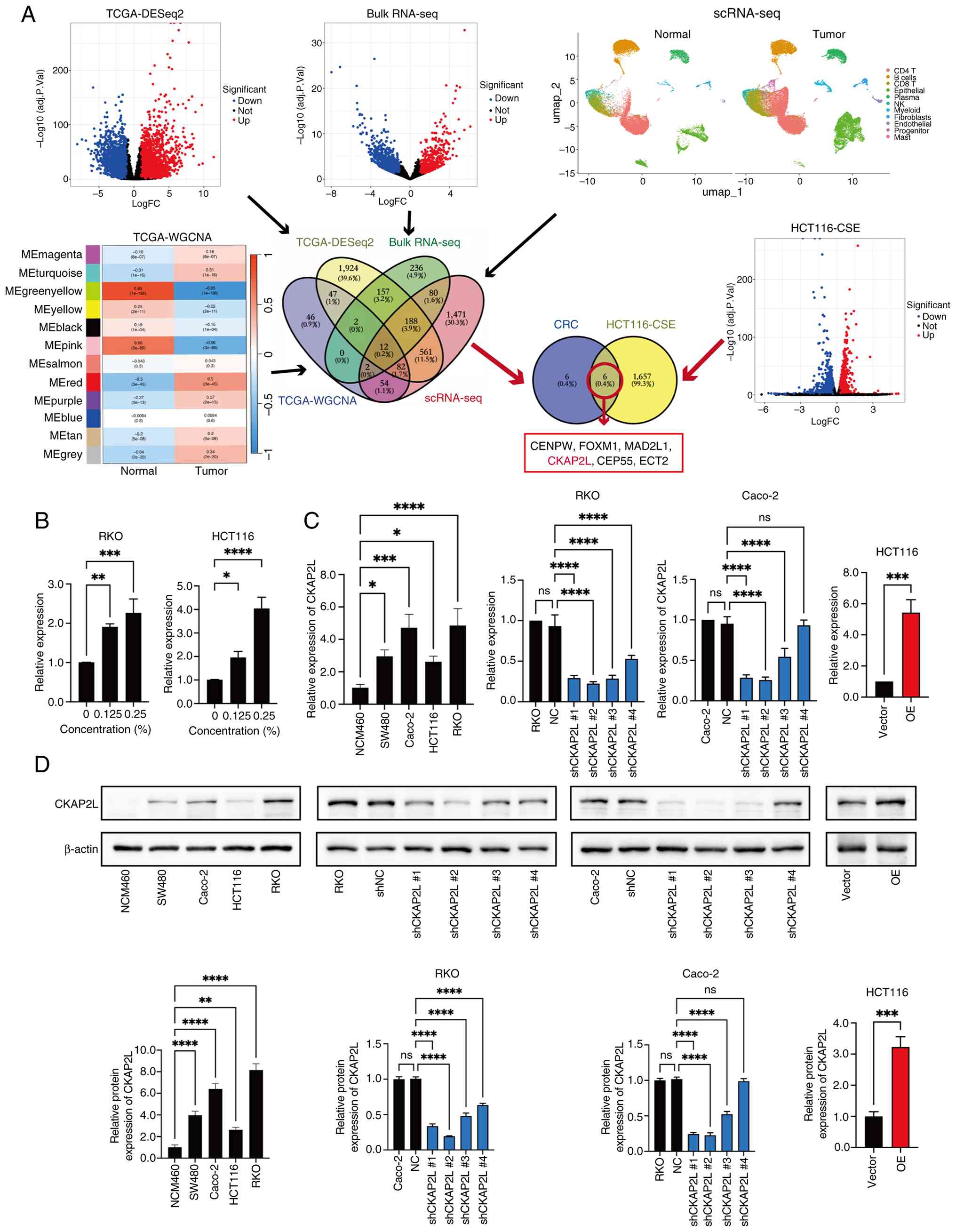 | Figure 3Bioinformatics analyses and
investigation of CKAP2L expression. (A) Identification of key genes
involved in smoking-enhanced CRC progression. (B) Relative
expression of CKAP2L in CRC cells treated with CSE for 2 months
detected by reverse transcription-quantitative PCR and normalized
against β-actin. (C) Expression of CKAP2L was measured by reverse
transcription-quantitative PCR in colorectal cell lines and
transduced CRC cells. (D) CKAP2L expression was measured by western
blotting in colorectal cell lines and transduced CRC cells. Data
were analyzed using one-way ANOVA followed by Dunnett's test for
multiple groups, or unpaired Student's t-test for two groups.
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. CKAP2L,
cytoskeleton-associated protein 2-like; CRC, colorectal cancer;
CSE, cigarette smoke extract; FC, fold change; NC, negative
control; OE, over expression; RNA-seq, RNA sequencing; scRNA-seq,
single-cell RNA-seq; sh, short hairpin; TCGA, The Cancer Genome
Atlas. |
By performing RNA-seq in CSE-treated HCT116 cells,
1,663 upregulated genes were identified in the CSE-treated group as
smoking-related genes. Finally, six key genes involved in
smoking-enhanced CRC progression were selected by overlapping
CRC-related genes and smoking-related genes, including CENPW,
FOXM1, MAD2L1, CKAP2L, CEP55 and ECT2 (Fig. 3A).
Among these six key genes, CKAP2L is an emerging
cell cycle-related gene (19).
Moreover, both the mRNA and protein expression levels of CKAP2L
were upregulated by CSE treatment (P<0.05; Figs. 2D and 3B). These results suggested that CKAP2L
may be a critical molecule in smoking-enhanced CRC progression.
Verification of CKAP2L expression
As shown in Fig. 3B
and C, both the mRNA and protein expression levels of CKAP2L
were higher in CRC cells than in normal colonic cells. Furthermore,
CKAP2L expression was higher in Caco-2 and RKO cells, but lower in
HCT116 cells. Therefore, knockdown of CKAP2L was performed in
Caco-2 and RKO cells, and overexpression of CKAP2L was performed in
HCT116 cells. The most effective shRNA plasmids (shCKAP2L #1 and
#2) were determined by RT-qPCR and western blotting, and were used
in subsequent experiments. The expression of CKAP2L was
significantly suppressed and upregulated following transduction
with shCKAP2L #1 and #2, and the CKAP2L-OE plasmid, respectively
(Fig. 3B and C).
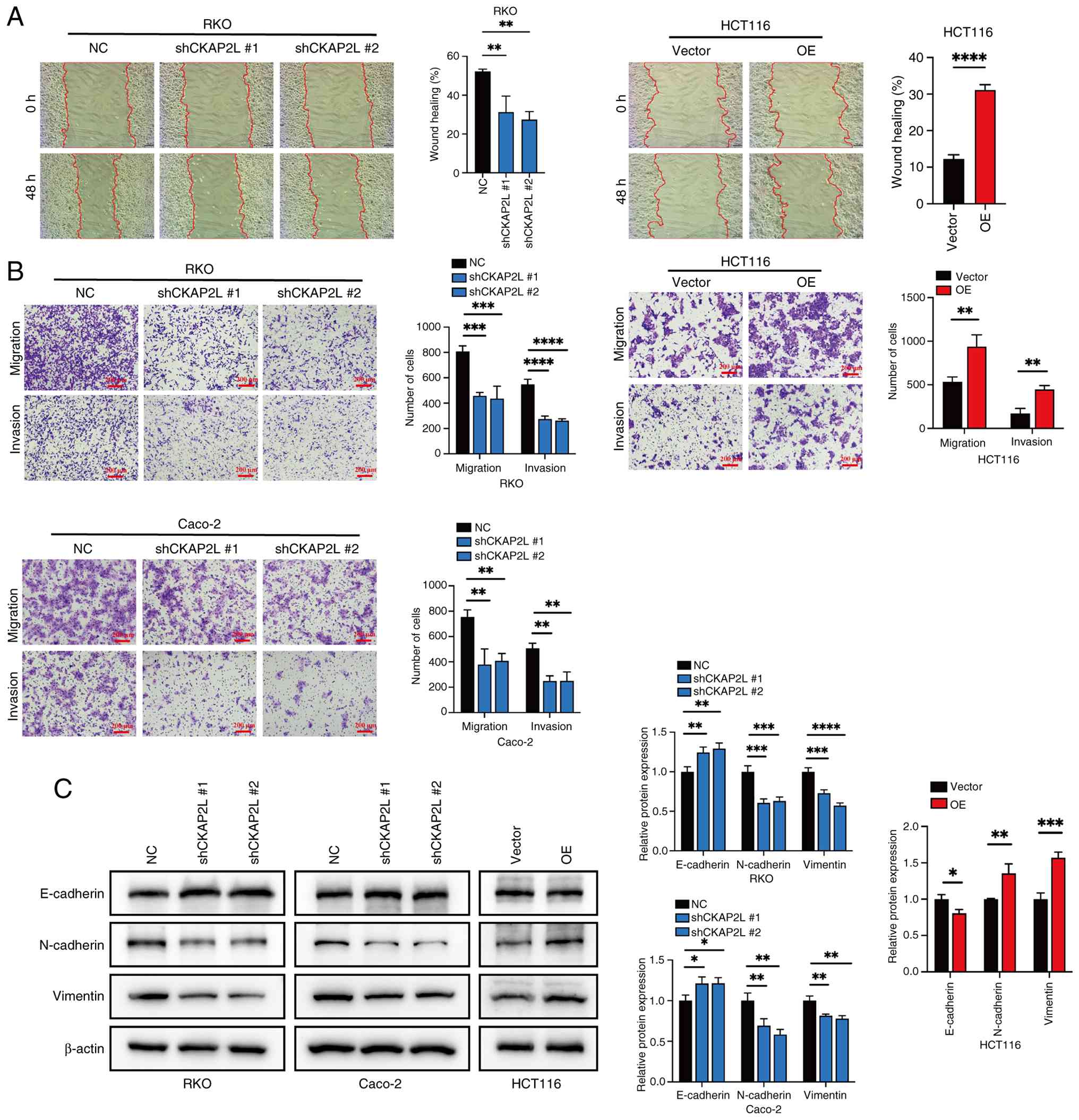
CKAP2L promotes cell migration and
invasion
Wound healing assay confirmed that knockdown of
CKAP2L inhibited the migratory ability of RKO cells (P<0.05;
Fig. 4A). By contrast, promotion
of migration was observed in HCT116 cells after CKAP2L
overexpression (P<0.05; Fig.
4A). The same trend in migration and invasion was confirmed by
Transwell assay (Fig. 4B).
Western blotting revealed that knockdown of CKAP2L enhanced
E-cadherin expression, and suppressed N-cadherin and Vimentin
expression (Fig. 4C), whereas
overexpression of CKAP2L showed the opposite trend. These results
revealed that CKAP2L may enhance the migration and invasion of CRC
cells by promoting EMT.
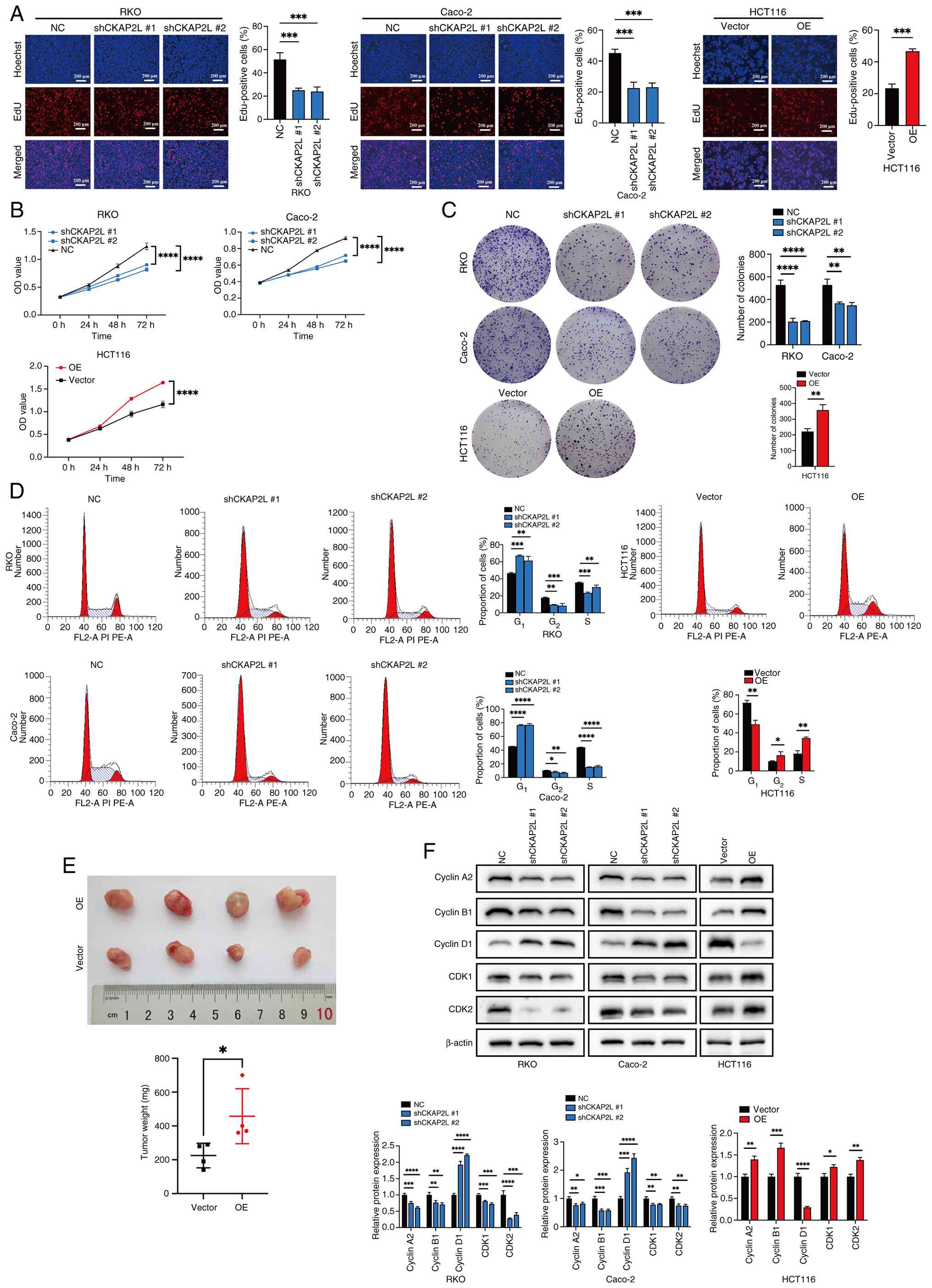
CKAP2L promotes cell proliferation
The proliferation of cells in vitro was
confirmed by EdU, CCK8 and colony formation assays. Notably, EdU
(Fig. 5A), CCK8 (Fig. 5B) and colony formation (Fig. 5C) assays showed that knockdown of
CKAP2L significantly suppressed the proliferation of Caco-2 and RKO
cells. By contrast, overexpression of CKAP2L promoted the
proliferation of HCT116 cells.
Flow cytometry confirmed that knockdown of CKAP2L
significantly reduced the proportion of cells in G2 and
S phases, whereas the opposite trend was shown after CKAP2L
overexpression (Fig. 5D).
Cell proliferation in vivo was confirmed by
animal experiments. After stable transduction of CT26 cells with
the CKAP2L-OE plasmid (Fig.
S1A), these cells were used to establish a subcutaneous tumor
model. Among all groups, the measured diameter of the largest tumor
was 12 mm, and the maximum volume was 486 mm3. The
results showed that overexpression of CKAP2L increased formed tumor
weight (mean, 225.0 vs. 457.5 mg; 95% CI, 14.81-450.2; P=0.0399;
Fig. 5E). These results supported
that CKAP2L can promote the proliferation of CRC cells in
vivo.
Further western blotting results revealed that
knockdown of CKAP2L inhibited the expression of cyclin A2, cyclin
B1, CDK1 and CDK2, and promoted the expression of cyclin D1,
whereas overexpression of CKAP2L generated the opposite results
(Fig. 5F). These results
suggested that CKAP2L promotes CRC cell proliferation by regulating
the cell cycle.
CKAP2L promotes CRC progression by
upregulating AREG expression
To explore the molecular mechanism by which CKAP2L
promotes the progression of CRC, RNA-seq was performed on CRC cells
that had been transfected with shCKAP2L and CKAP2L-OE plasmids
(Fig. 6A). Subsequently, 20 key
genes were filtered out by overlapping upregulated DEGs in
CKAP2L-OE cells, downregulated DEGs in shCKAP2L cells and
upregulated DEGs in HCT116-CSE cells (Fig. 6B). Among these 20 key genes
regulated by CKAP2L, AREG is a biomarker for cancer progression
(45), indicating that CKAP2L can
regulate the expression of AREG to promote CRC progression. The
effect of CSE on AREG expression was then assessed; the mRNA levels
of AREG were upregulated by CSE treatment (Fig. 6C) and CKAP2L overexpression
(Fig. 6D). However, after
successfully inhibiting AREG expression via transfection of cells
with siAREG (Fig. S1B), the
expression of CKAP2L did not change (Fig. 6D). ELISA confirmed that
overexpression of CKAP2L also promoted the secretory protein levels
of AREG (Fig. 6E). These results
reveled that smoking may upregulate AREG expression by increasing
the expression levels of CKAP2L.
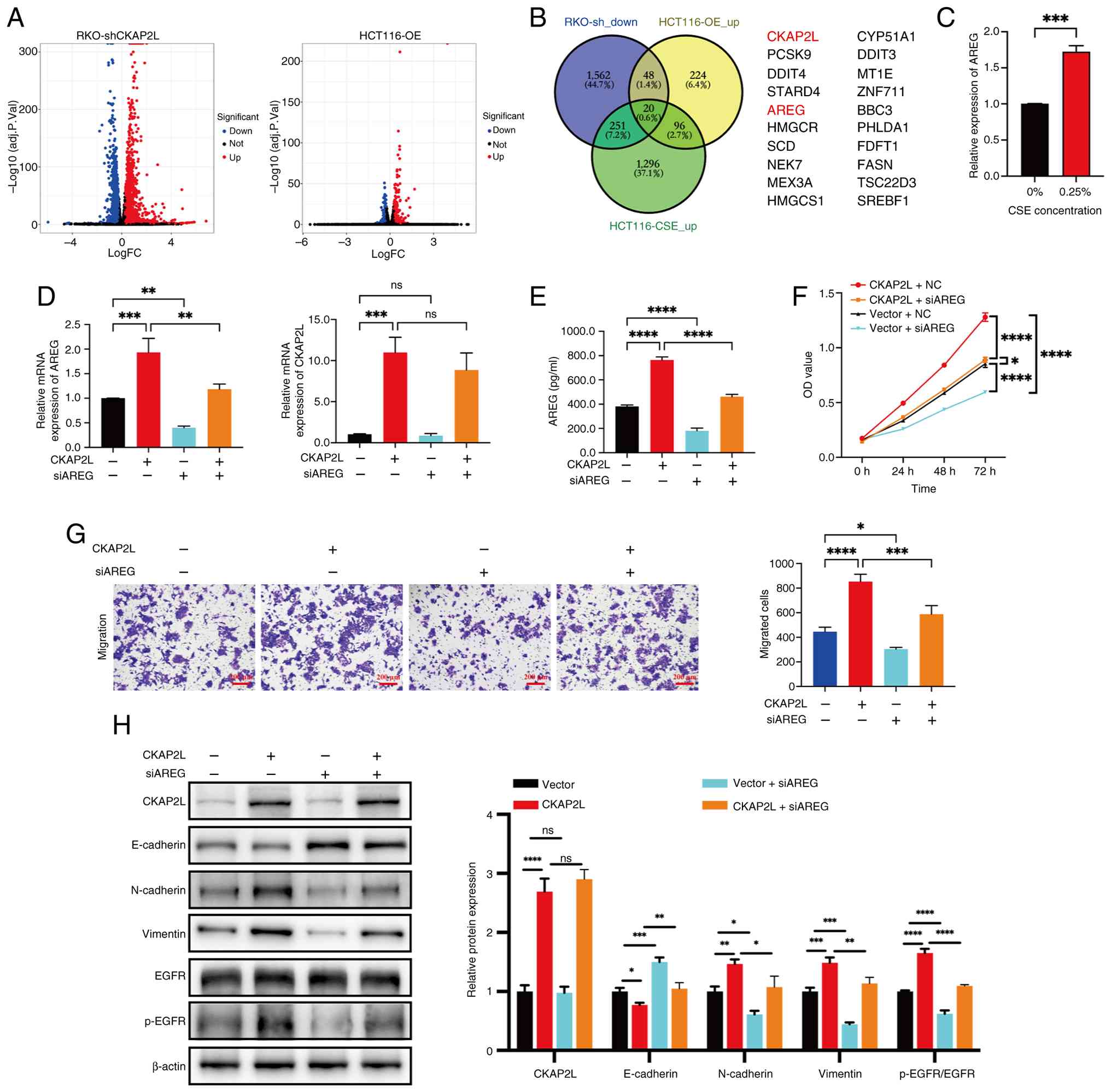 | Figure 6CKAP2L promotes proliferation and
migration of CRC cells by promoting AREG expression. (A) RNA
sequencing results for transduced CRC cells. (B) Venn diagram shows
the overlapping genes between transduced cells and CSE-treated
HCT116 cells. (C) Relative expression of AREG in CRC cells treated
with CSE detected by RT-qPCR and normalized against β-actin. (D)
Relative expression levels of AREG and CKAP2L were detected by
RT-qPCR and normalized against β-actin. (E) Secreted protein levels
of AREG were detected by enzyme-linked immunosorbent assay. (F)
Proliferation of transduced/transfected cells was measured by Cell
Counting Kit 8. (G) Migration of transduced/transfected cells was
measured by Transwell assay (×100 magnification). (H) Expression
levels of proteins were measured by western blotting. Data were
analyzed using two-way ANOVA followed by Tukey test, or unpaired
Student's t-test for two groups. *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001. AREG, amphiregulin; CKAP2L,
cytoskeleton-associated protein 2-like; CRC, colorectal cancer;
CSE, cigarette smoke extract; EGFR, epidermal growth factor
receptor; NC, negative control; OE, overexpression; p-,
phosphorylated; RT-qPCR, reverse transcription-quantitative PCR;
sh, short hairpin; si, small interfering. |
To evaluate whether CKAP2L promotes CRC progression
by upregulating the expression of AREG, CCK8 and Transwell assays
were conducted. As shown in Fig.
6F, AREG knockdown partially suppressed the enhanced
proliferative ratio of CRC cells caused by overexpression of
CKAP2L. Transwell assay results confirmed that knockdown of AREG
expression decreased the number of migratory cells induced by
CKAP2L overexpression (Fig. 6G).
Western blotting further confirmed that inhibiting the expression
of AREG partially restored the upregulated protein levels of
N-cadherin and Vimentin and phosphorylation level of EGFR, and
downregulated the protein levels of E-cadherin caused by
overexpression of CKAP2L (Fig.
6H). These results suggested that CKAP2L may promote CRC
progression by activating the AREG/EGFR pathway.
CKAP2L increases STAT3 phosphorylation to
promote the expression of AREG
After overexpression of CKAP2L, the phosphorylation
level of STAT3 increased, whereas the opposite occurred when CKAP2L
was knocked down, as determined by western blotting (Fig. 7A). To explore whether CKAP2L
promotes CRC progression by enhancing STAT3 phosphorylation, the
STAT3 phosphorylation inhibitor Stattic was used. After culturing
HCT116 cells in medium containing Stattic for 24 h, cell
proliferation was measured using CCK8 to calculate the
IC50; the results showed that the IC50 of
Stattic was 6.106 μM (Fig.
7B). Both RT-qPCR and ELISA confirmed that Stattic inhibited
AREG levels enhanced by CKAP2L overexpression (Fig. 7C). Furthermore, Transwell and CCK8
assays indicated that treatment with Stattic inhibited the
migration and proliferation of HCT116 cells enhanced by
overexpression of CKAP2L (Fig. 7D and
E). Western blotting revealed that suppression of STAT3
phosphorylation inhibited the protein expression levels of
N-cadherin and Vimentin and the phosphorylation level of EGFR
enhanced by CKAP2L overexpression, and restored the expression
level of E-cadherin downregulated by CKAP2L overexpression
(Fig. 7F). These results
indicated that CKAP2L may promote AREG expression by increasing the
phosphorylation of STAT3.
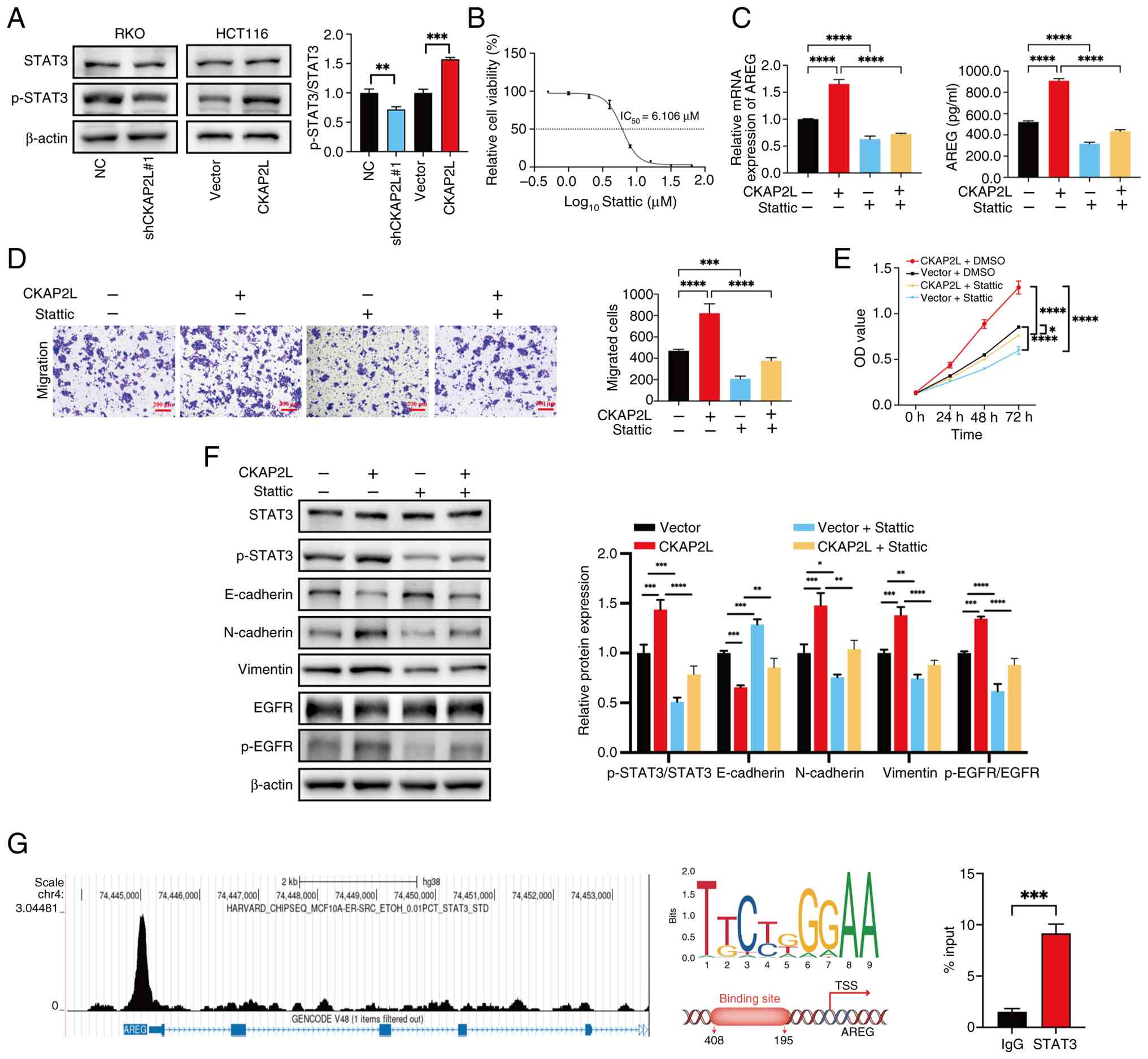 | Figure 7CKAP2L promotes proliferation and
migration of colorectal cancer cells through the STAT3/AREG/EGFR
axis. (A) Expression levels of STAT3 and p-STAT3 proteins measured
by western blotting. (B) Calculation of IC50 in HCT116
cells treated with Stattic for 24 h. (C) Levels of AREG were
measured by reverse transcription-quantitative PCR and
enzyme-linked immunosorbent assay. (D) Migration of cells was
detected by Transwell assay (×100 magnification). (E) Proliferation
of cells was measured by Cell Counting Kit 8. (F) Expression levels
of proteins measured by western blotting. (G) Binding peak of STAT3
on the promoter region of AREG was detected by Cistrome Data
Browser, the binding motif was predicted using the JASPAR database,
and the binding of STAT3 to the AREG promoter was evaluated by
chromatin immunoprecipitation assay. Data were analyzed using (A
and G) Unpaired Student's t-test, and (C-F) two-way ANOVA followed
by Tukey test. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. AREG,
amphiregulin; CKAP2L, cytoskeleton-associated protein 2-like; EGFR,
epidermal growth factor receptor; NC, negative control; p-,
phosphorylated; sh, short hairpin; STAT3, signal transducer and
activator of transcription 3; TSS, transcription start site. |
The current study further investigated the
interaction between STAT3 and AREG. The Cistrome Data Browser
showed that STAT3 has a significant binding peak in the promoter
region of AREG, suggesting that STAT3 is a transcription factor of
AREG (Fig. 7G). Subsequently, the
binding motif of STAT3 and the potential binding site was predicted
by JASPAR database. ChIP-qPCR assay confirmed the significant
enrichment of STAT3 on the promoter region of AREG (Fig. 7G). These results confirmed that
STAT3 is a transcription factor of AREG.
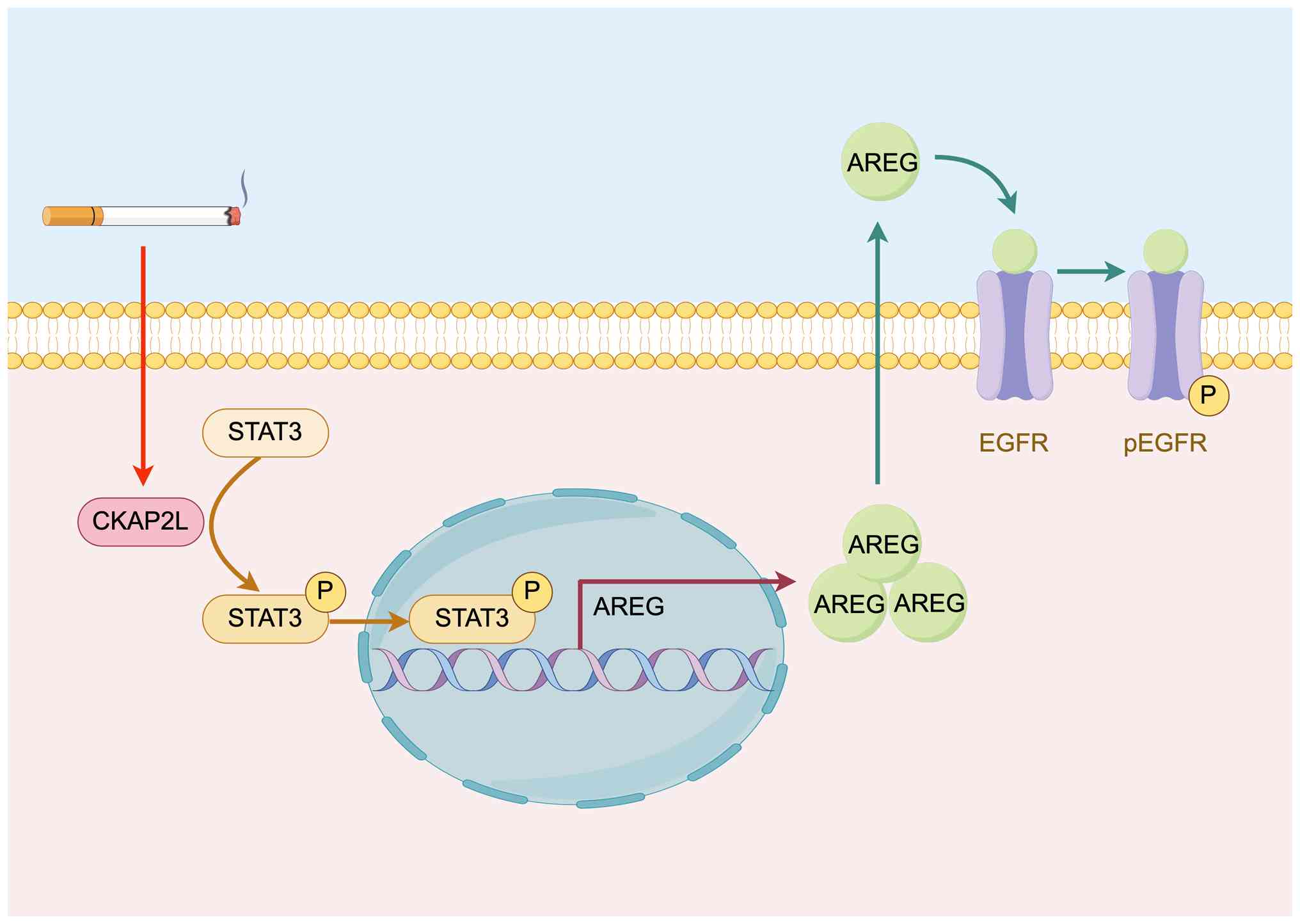
These results revealed that CKAP2L may promote the
phosphorylation level of STAT3. Subsequently, STAT3 promotes AREG
transcription and activates the AREG/EGFR pathway, resulting in CRC
progression (Fig. 8).
Discussion
Smoking is a major risk factor for CRC that also
affects the survival of patients with CRC (46-49). In a previous study, a cigarette
smoke-exposed C57BL/6 mice model demonstrated that cigarette smoke
promotes CRC progression by modulating the gut microbiota and
related metabolites (9). Tumor
immunity serves a crucial role in the progression of cancer and
some inflammatory markers, such as albumin-to-globulin ratio and
neutrophil percentage to albumin ratio, are notably associated with
the prognosis of CRC (50). It
has been shown that smoking is markedly associated with reduced
macrophage densities and T-lymphocyte response in patients with
CRC, which contributes to CRC carcinogenesis (51,52). Smoking is also positively
associated with high CpG island methylation, BRAF mutations and
high microsatellite instability, which increases the risk of CRC
(53). In the present study, the
cross-sectional study confirmed that past smoking and current
smoking were significantly linked with a higher CRC risk compared
with not smoking. In addition, the ORs of the current smoking group
were higher than those of the past smoking group, indicating a
reduced risk of CRC after smoking cessation. However, no difference
in CRC risk was observed between secondhand smoking and no smoking.
A previous study also reported that harmful associations between
secondhand smoking and cancer were supported by weak evidence
(54). Moreover, no significant
relationship between CRC and cotinine levels was observed,
suggesting that short-term smoking exposure levels had no effect on
CRC risk. Analysis of the age at which individuals started smoking
cigarettes regularly also showed no significant relationship with
CRC risk. Previous MR analyses have reported the causality between
smoking and CRC (7,55). In the current study, the causality
between cigarettes per day and CRC was also confirmed by MR
analysis. Consistent with previous studies, these results confirmed
that smoking is positively linked to CRC risk, and a causality
exists between the two, whereas quitting smoking can reduce this
risk. These results revealed the positive association between
smoking and CRC.
To investigate the hypothesis in various subtypes of
CRC, four CRC cell lines were selected for use in the subsequent
experiments. SW480 cells were selected as a model of primary
colorectal adenocarcinoma; this cell line harbors a KRAS G12D
mutation and a p53 R273H mutation, representing common oncogenic
drivers in CRC. HCT116 cells were selected as a model of mismatch
repair-deficient/microsatellite instability-high (MSI-H) CRC; this
cell line carries an activating KRAS G12D mutation. RKO cells were
selected as another model of MSI-H CRC, which carries a wild-type
APC gene that is distinct from numerous other CRC cell lines.
Caco-2 cells were selected due to their unique ability to
spontaneously differentiate into enterocyte-like cells. By using
these cell lines, the current study aimed to ensure that the
findings were robust and not limited to a single genetic subtype of
CRC.
The CSE treatment cell model was constructed to
assess the direct effect of cigarette smoke on CRC cells. In the
present study, CSE concentrations of 0.125 and 0.25% were selected
by CCK8 (cells treated with 0.125 and 0.25% CSE showed peak
proliferation) to establish the chronic CSE treatment model
according to previous studies (18,37,56). Assuming that the blood volume of
an adult is 5,000 ml, one cigarette dissolved in the blood is equal
to 0.20% CSE. Therefore, CSE concentrations of 0.125 and 0.25% are
approximately equal to the physiological exposure concentration and
were selected to establish the chronic CSE treatment model. The EdU
assay was also employed to detect the cytotoxicity of CSE, rather
than only the short-term CCK8 assay. The results confirmed an
increase in viable cell number in the CSE treatment groups without
causing notable cell death. In the chronic CSE treatment model,
smoke exposure enhanced CRC cell proliferation, migration and
invasion, and increased the expression of proliferation-related
proteins (cyclin A2, cyclin B1, CDK1 and CDK2) and EMT-associated
proteins (N-cadherin and Vimentin). These results suggested that
smoking may enhance CRC cell progression.
RNA-seq was conducted on CSE-treated HCT116 cells
and it was revealed that CKAP2L may be a critical gene involved in
smoking-enhanced CRC. Radmis, the mouse ortholog of CKAP2L, has
been identified as a novel microtubule-associated protein by Yumoto
et al (57). Notably,
scaffold proteins are involved in promoting tumor progression and
metabolic adaptation (58).
Previous bioinformatics analyses have identified CKAP2L as a hub
gene in multiple types of cancer (such as CRC and clear cell renal
cell carcinoma) (59,60), and the expression of CKAP2L has
been shown to be upregulated in tumor tissues, leading to a poor
prognosis (61,62). Mechanistically, CKAP2L serves a
vital role in regulating the cell cycle and the tumor immune
microenvironment, thereby promoting the progression of cancer cells
(63,64). The present study reported that
CKAP2L expression in CRC cells was significantly upregulated by CSE
treatment. Furthermore, overexpression of CKAP2L enhanced the
proliferation, migration and invasion of CRC cells. Cell cycle
analysis also revealed that CKAP2L was involved in regulation of S
and G2/M phases. Moreover, CKAP2L increased the levels
of EMT-, and S and G2/M phase-related proteins (for
example, N-cadherin, Vimentin, cyclin A2, cyclin B1, CDK1 and
CDK2). These results confirmed that CKAP2L is upregulated by
smoking, promoting CRC progression through its regulation of the
cell cycle.
By regulating CKAP2L expression, both the mRNA and
protein expression levels of AREG were affected. AREG is an EGFR
ligand, that mainly serves its role by interacting with EGFR
(65). EGFR has an important role
in CRC progression and is a fundamental therapeutic target
(66). The AREG/EGFR axis is
involved in regulating proliferation, metastasis, tumor
microenvironment and tumor immune tolerance in multiple tumors,
such as melanoma and esophageal squamous cell carcinoma (45,67,68). The present study revealed that
CKAP2L could upregulate AREG expression, leading to the promotion
of EGFR phosphorylation and activation of the AREG/EGFR signaling
pathway. Further experiments are needed to explore the downstream
EGFR pathways that are involved in smoking-enhanced CRC
progression. Furthermore, the knockdown of AREG only partially
inhibited the cell proliferation and migration induced by CKAP2L,
indicating that there are other downstream pathways involved. Other
mechanisms still require further investigation.
To explain how CKAP2L promote the expression of
AREG, a ChIP assay was performed. The results revealed that CKAP2L
increased STAT3 phosphorylation, leading to the progression of CRC
cells. STAT3 is a classic transcription factor that participates in
the regulation of almost all malignant characteristics of tumors
(69-71) and is also a therapeutic target for
patients with CRC (72).
Inhibiting the phosphorylation of STAT3 was shown to suppress the
expression of AREG, which verified the regulatory effect of STAT3
on AREG. Moreover, the results of the ChIP assay confirmed that
STAT3 promoted AREG transcription by binding to its promoter
region.
The present study confirmed the positive
association between smoking and CRC. In addition, the study
explored the influence of smoking features on CRC. Further
experiments also revealed that smoke exposure may stimulate CRC
progression through the CKAP2L/STAT3/AREG/EGFR axis.
Notably, there are several limitations in the
present study. Self-reported NHANES data may introduce recall bias.
Moreover, there was a lack of detailed clinicopathological
information about cancer, such as stage, therapies, drug resistance
and prognosis, which limits comprehensive evaluation of the impact
of smoking on CRC. Furthermore, some confounders not included in
this cross-sectional analysis may affect the link between smoking
and CRC. Due to the lack of information about electronic cigarettes
during the period of 1999-2014, the present study did not collect
or analyze data related to electronic cigarettes. For MR analysis,
GWAS data from Europe limits the generalizability of these MR
analysis results to other populations, and the brands and
formulations of cigarettes may lead to differences in the
experimental results. Moreover, CSE cannot fully replicate human
smoking exposure conditions. Further experiments are needed to
explore the downstream EGFR pathways and other mechanisms involved
in smoking-enhanced CRC.
In conclusion, the current study demonstrated that
both past smoking and current smoking are positively related to
CRC, and identified a causality between smoking and CRC. CSE
promoted the expression of CKAP2L, which regulated the cell cycle,
and promoted the proliferation, migration and invasion of CRC
cells. These findings revealed that smoke exposure may enhance CRC
progression through CKAP2L/AREG signaling.
Supplementary Data
Availability of data and materials
The RNA-seq data generated in the present study may
be found in the Gene Expression Omnibus database under accession
numbers GSE305020 and GSE305039, or at the following URLs:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE305020,
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE305039.
Other data generated in the present study are available from the
corresponding author.
Authors' contributions
SW contributed to study design, carried out the
cross-section and Mendelian randomization analyses, performed cell
experiments and wrote the main manuscript. FW performed the
bioinformatics analysis and animal experiment, and was a major
contributor in writing the manuscript. XL, ZhiJ and FL performed
cell experiments, data collection and analysis. ZheJ designed the
study and was involved in revising the manuscript. SW and ZheJ
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the
Institutional Animal Care and Use Committee of Chongqing Medical
University (approval no. IACUC-CQMU-2025-0457).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
MR
|
Mendelian randomization
|
|
ChIP
|
chromatin immunoprecipitation
|
|
CKAP2L
|
cytoskeleton-associated protein
2-like
|
|
AREG
|
amphiregulin
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
GWAS
|
genome-wide association studies
|
|
IV
|
instrumental variable
|
|
EGFR
|
epidermal growth factor receptor
|
|
SNP
|
single-nucleotide polymorphism
|
|
IVW
|
inverse-variance weighted
|
|
FE
|
fixed effect
|
|
MRE
|
multiplicative random effect
|
|
RNA-seq
|
RNA sequencing
|
|
scRNA-seq
|
single-cell RNA-seq
|
|
DEG
|
differentially expressed gene
|
|
CSE
|
cigarette smoke extract
|
|
CCK8
|
cell Counting Kit 8
|
|
shRNA
|
short hairpin RNA
|
|
OE
|
overexpression
|
|
siRNA
|
small interfering RNA
|
|
ELISA
|
enzyme-linked immunosorbent assay
|
|
EdU
|
5-ethynyl-2'-deoxyuridine
|
|
EMT
|
epithelial-mesenchymal transition
|
Acknowledgements
Not applicable.
Funding
The present study was supported by the China Early
Gastrointestinal Cancer Physician Common Growth Program (grant no.
GTCZ-2024-08).
References
|
1
|
Harris E: Cancer risk decreased 10 years
after quitting smoking. JAMA. 331:8222024.PubMed/NCBI
|
|
2
|
Park E, Kang HY, Lim MK, Kim B and Oh JK:
Cancer risk following smoking cessation in Korea. JAMA Netw Open.
7:e23549582024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hecht SS and Hatsukami DK: Smokeless
tobacco and cigarette smoking: Chemical mechanisms and cancer
prevention. Nat Rev Cancer. 22:143–155. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jiang M, Han J, Ma Q, Chen X, Xu R, Wang
Q, Zheng J, Wang W, Song J, Huang Y and Chen Y: Nicotine-derived
NNK promotes CRC progression through activating TMUB1/AKT pathway
in METTL14/YTHDF2-mediated m6A manner. J Hazard Mater.
467:1336922024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siegel RL, Kratzer TB, Giaquinto AN, Sung
H and Jemal A: Cancer statistics, 2025. CA Cancer J Clin. 75:10–45.
2025.PubMed/NCBI
|
|
6
|
GBD 2019 Colorectal Cancer Collaborators:
Global, regional, and national burden of colorectal cancer and its
risk factors, 1990-2019: A systematic analysis for the global
burden of disease study 2019. Lancet Gastroenterol Hepatol.
7:627–647. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou X, Xiao Q, Jiang F, Sun J, Wang L, Yu
L, Zhou Y, Zhao J, Zhang H, Yuan S, et al: Dissecting the
pathogenic effects of smoking and its hallmarks in blood DNA
methylation on colorectal cancer risk. Br J Cancer. 129:1306–1313.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li H, Chen X, Hoffmeister M and Brenner H:
Associations of smoking with early- and late-onset colorectal
cancer. JNCI Cancer Spectr. 7:pkad0042023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai X, Wei H, Liu W, Coker OO, Gou H, Liu
C, Zhao L, Li C, Zhou Y, Wang G, et al: Cigarette smoke promotes
colorectal cancer through modulation of gut microbiota and related
metabolites. Gut. 71:2439–2450. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu S and Wu F: Association of urinary
incontinence with depression among men: A cross-sectional study.
BMC Public Health. 23:9442023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu S, Yuan G, Wu L, Zou L and Wu F:
Identifying the association between depression and constipation: An
observational study and Mendelian randomization analysis. J Affect
Disord. 359:394–402. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hsiao CC, Yang AM, Wang C and Lin CY:
Association between glyphosate exposure and cognitive function,
depression, and neurological diseases in a representative sample of
US adults: NHANES 2013-2014 analysis. Environ Res. 237:1168602023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie Z, Wang L, Sun M, Wang R, Li J, Wang
X, Guo R, Dong Y, Wang Y and Li B: Mediation of 10-year
cardiovascular disease risk between inflammatory diet and handgrip
strength: Base on NHANES 2011-2014. Nutrients. 15:9182023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang H, Wu Z, Chen Q, Yu G, Chen L, Ma Y
and Chen Y: Subtype-specific causal effects of hypothyroidism on
obstructive sleep apnea: A bidirectional Mendelian randomization
study. Medicine (Baltimore). 104:e432662025. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu S, Wu F and Jiang Z: Identification of
hub genes, key miRNAs and potential molecular mechanisms of
colorectal cancer. Oncol Rep. 38:2043–2050. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hussain MS, Battaglia A, Szczepanski S,
Kaygusuz E, Toliat MR, Sakakibara S, Altmüller J, Thiele H,
Nürnberg G, Moosa S, et al: Mutations in CKAP2L, the human homolog
of the mouse Radmis gene, cause Filippi syndrome. Am J Hum Genet.
95:622–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jakobsen L, Vanselow K, Skogs M, Toyoda Y,
Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg
EA, et al: Novel asymmetrically localizing components of human
centrosomes identified by complementary proteomics methods. EMBO J.
30:1520–1535. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu F, Wu S, Huang Y, Xiao X, Yu H, Bai X,
Zhang C, Feng Z, Li L, Mei Y, et al: Smoking promotes the
progression of bladder cancer through FOXM1/CKAP2L axis. J Transl
Med. 23:7852025. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen W, Wang Y, Wang L, Zhao H and Li X:
CKAP2L promotes esophageal squamous cell carcinoma progression and
drug-resistance by modulating cell cycle. J Oncol.
2022:23782532022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Monteverde T, Sahoo S, La Montagna M,
Magee P, Shi L, Lee D, Sellers R, Baker AR, Leong HS, Fassan M and
Garofalo M: CKAP2L promotes non-small cell lung cancer progression
through regulation of transcription elongation. Cancer Res.
81:1719–1731. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Luo Q, Zhu B, Wang C and Wang Y: CKAP2L
plays a pivotal role in colorectal cancer progression via the dual
regulation of cell cycle and epithelial-mesenchymal transition.
Discov Med. 37:182–192. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim TR, Son B, Lee CG and Park HO:
Amphiregulin in fibrotic diseases and cancer. Int J Mol Sci.
26:69452025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Z and Li Z, Zhang X, Ye W, Chen J,
Wang L, Lin Z, Li J and Li Z: Association between secondhand smoke
and cancers in adults in the US population. J Cancer Res Clin
Oncol. 149:3447–3455. 2023. View Article : Google Scholar
|
|
24
|
Dove MS, Dockery DW and Connolly GN:
Smoke-free air laws and secondhand smoke exposure among nonsmoking
youth. Pediatrics. 126:80–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Benowitz NL, Bernert JT, Caraballo RS,
Holiday DB and Wang J: Optimal serum cotinine levels for
distinguishing cigarette smokers and nonsmokers within different
racial/ethnic groups in the United States between 1999 and 2004. Am
J Epidemiol. 169:236–248. 2009. View Article : Google Scholar
|
|
26
|
Hou W, Chen S, Zhu C, Gu Y, Zhu L and Zhou
Z: Associations between smoke exposure and osteoporosis or
osteopenia in a US NHANES population of elderly individuals. Front
Endocrinol (Lausanne). 14:10745742023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du X, Peng T, Ma L and Cheng G: Serum
cotinine levels and adolescents' sleep health outcomes from NHANES
2005 to 2018. Sci Rep. 14:210762024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hukkanen J, Jacob P III and Benowitz NL:
Metabolism and disposition kinetics of nicotine. Pharmacol Rev.
57:79–115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen FF, Klemperer EM, Erath TG, DeSarno
M, Leventhal AM and Higgins ST: Age, cumulative psychosocial risk
factors, and health disparities in U.S. adult cigarette smoking:
2002-2019. BMC Med. 23:6642025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reitsma MB, Flor LS, Mullany EC, Gupta V,
Hay SI and Gakidou E: Spatial, temporal, and demographic patterns
in prevalence of smoking tobacco use and initiation among young
people in 204 countries and territories, 1990-2019. Lancet Public
Health. 6:e472–e481. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Taylor E, Tattan-Birch H, Oldham M, East
K, Walsh H and Jackson S: Smoking and drinking among the Gypsy and
Traveller communities: A population study in England. Addiction.
February 17–2026.Epub ahead of print. View Article : Google Scholar
|
|
32
|
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM,
Chen F, Datta G, Davila-Velderrain J, McGuire D, Tian C, et al:
Association studies of up to 1.2 million individuals yield new
insights into the genetic etiology of tobacco and alcohol use. Nat
Genet. 51:237–244. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Potenza A, Balestrieri C, Spiga M,
Albarello L, Pedica F, Manfredi F, Cianciotti BC, De Lalla C,
Botrugno OA, Faccani C, et al: Revealing and harnessing CD39 for
the treatment of colorectal cancer and liver metastases by
engineered T cells. Gut. 72:1887–1903. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khaliq AM, Erdogan C, Kurt Z, Turgut SS,
Grunvald MW, Rand T, Khare S, Borgia JA, Hayden DM, Pappas SG, et
al: Refining colorectal cancer classification and clinical
stratification through a single-cell atlas. Genome Biol.
23:1132022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun X, Deng Q, Liang Z, Liu Z, Geng H,
Zhao L, Zhou Q, Liu J, Ma J, Wang D, et al: Cigarette smoke extract
induces epithelial-mesenchymal transition of human bladder cancer
T24 cells through activation of ERK1/2 pathway. Biomed
Pharmacother. 86:457–465. 2017. View Article : Google Scholar
|
|
38
|
Liang Z, Lu L, Mao J, Li X, Qian H and Xu
W: Curcumin reversed chronic tobacco smoke exposure induced
urocystic EMT and acquisition of cancer stem cells properties via
Wnt/β-catenin. Cell Death Dis. 8:e30662017. View Article : Google Scholar
|
|
39
|
Qian Y, Liu Z, Liu Q, Tian X, Mo J, Leng
L, Wang C, Xu G, Zhang S and Xie J: Transduction of lentiviral
vectors and ADORA3 in HEK293T cells modulated in gene expression
and alternative splicing. Int J Mol Sci. 26:44312025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Escrivá-Fernández J, Cueto-Ureña C,
Solana-Orts A, Lledó E, Ballester-Lurbe B and Poch E: A CRISPR
interference strategy for gene expression silencing in multiple
myeloma cell lines. J Biol Eng. 17:342023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Elegheert J, Behiels E, Bishop B, Scott S,
Woolley RE, Griffiths SC, Byrne EFX, Chang VT, Stuart DI, Jones EY,
et al: Lentiviral transduction of mammalian cells for fast,
scalable and high-level production of soluble and membrane
proteins. Nat Protoc. 13:2991–3017. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
43
|
Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang
C, Zhu M, Wu J, Shi X, Taing L, et al: Cistrome data browser: A
data portal for ChIP-Seq and chromatin accessibility data in human
and mouse. Nucleic Acids Res. 45(D1): D658–D662. 2017. View Article : Google Scholar :
|
|
44
|
Rauluseviciute I, Riudavets-Puig R,
Blanc-Mathieu R, Castro-Mondragon JA, Ferenc K, Kumar V, Lemma RB,
Lucas J, Chèneby J, Baranasic D, et al: JASPAR 2024: 20th
anniversary of the open-access database of transcription factor
binding profiles. Nucleic Acids Res. 52(D1): D174–D182. 2024.
View Article : Google Scholar :
|
|
45
|
Nakanishi T, Koma YI, Miyako S, Torigoe R,
Yokoo H, Omori M, Yamanaka K, Ishihara N, Tsukamoto S, Kodama T, et
al: AREG upregulation in cancer cells via direct interaction with
cancer-associated fibroblasts promotes esophageal squamous cell
carcinoma progression through EGFR-Erk/p38 MAPK signaling. Cells.
13:17332024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Keum N and Giovannucci E: Global burden of
colorectal cancer: Emerging trends, risk factors and prevention
strategies. Nat Rev Gastroenterol Hepatol. 16:713–732. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Roshandel G, Ghasemi-Kebria F and
Malekzadeh R: Colorectal cancer: Epidemiology, risk factors, and
prevention. Cancers (Basel). 16:15302024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gausman V, Dornblaser D, Anand S, Hayes
RB, O'Connell K, Du M and Liang PS: Risk factors associated with
early-onset colorectal cancer. Clin Gastroenterol Hepatol.
18:2752–2759.e2. 2020. View Article : Google Scholar
|
|
49
|
Islami F, Marlow EC, Thomson B, McCullough
ML, Rumgay H, Gapstur SM, Patel AV, Soerjomataram I and Jemal A:
Proportion and number of cancer cases and deaths attributable to
potentially modifiable risk factors in the United States, 2019. CA
Cancer J Clin. 74:405–432. 2024.PubMed/NCBI
|
|
50
|
Li K, Zhang R, Zhang J, Zhang Z, Wang K,
Lu Y, Zhao Z, Chen Y and Ma S: The prognostic role of
inflammation-based hematologic markers in stage I-III colorectal
cancer: A retrospective analysis. BMC Cancer. 25:18222025.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ugai T, Väyrynen JP, Haruki K, Akimoto N,
Lau MC, Zhong R, Kishikawa J, Väyrynen SA, Zhao M, Fujiyoshi K, et
al: Smoking and incidence of colorectal cancer subclassified by
tumor-associated macrophage infiltrates. J Natl Cancer Inst.
114:68–77. 2022. View Article : Google Scholar :
|
|
52
|
Hamada T, Nowak JA, Masugi Y, Drew DA,
Song M, Cao Y, Kosumi K, Mima K, Twombly TS, Liu L, et al: Smoking
and risk of colorectal cancer sub-classified by tumor-infiltrating
T cells. J Natl Cancer Inst. 111:42–51. 2019. View Article : Google Scholar :
|
|
53
|
Botteri E, Borroni E, Sloan EK, Bagnardi
V, Bosetti C, Peveri G, Santucci C, Specchia C, van den Brandt P,
Gallus S and Lugo A: Smoking and colorectal cancer risk, overall
and by molecular subtypes: A meta-analysis. Am J Gastroenterol.
115:1940–1949. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Flor LS, Anderson JA, Ahmad N, Aravkin A,
Carr S, Dai X, Gil GF, Hay SI, Malloy MJ, McLaughlin SA, et al:
Health effects associated with exposure to secondhand smoke: A
burden of proof study. Nat Med. 30:149–167. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dimou N, Yarmolinsky J, Bouras E, Tsilidis
KK, Martin RM, Lewis SJ, Gram IT, Bakker MF, Brenner H, Figueiredo
JC, et al: Causal effects of lifetime smoking on breast and
colorectal cancer risk: Mendelian randomization study. Cancer
Epidemiol Biomarkers Prev. 30:953–964. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ferraro M, Di Vincenzo S, Lazzara V, Pinto
P, Patella B, Inguanta R, Bruno A and Pace E: Formoterol exerts
anti-cancer effects modulating oxidative stress and
epithelial-mesenchymal transition processes in cigarette smoke
extract exposed lung adenocarcinoma cells. Int J Mol Sci.
24:160882023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yumoto T, Nakadate K, Nakamura Y, Sugitani
Y, Sugitani-Yoshida R, Ueda S and Sakakibara S: Radmis, a novel
mitotic spindle protein that functions in cell division of neural
progenitors. PLoS One. 8:e798952013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu Y, Chen Y, Wang F, Lin J, Tan X, Chen
C, Wu LL, Zhang X, Wang Y, Shi Y, et al: Caveolin-1 promotes glioma
progression and maintains its mitochondrial inhibition resistance.
Discov Oncol. 14:1612023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang Y, Luo J, Liu Z, Liu X, Ma Y, Zhang
B, Chen Y, Li X, Feng Z, Yang N, et al: Identification of hub genes
in colorectal cancer based on weighted gene co-expression network
analysis and clinical data from the cancer genome atlas. Biosci
Rep. 41:BSR202112802021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu Z, Zhang J, Shen D, Hu X, Ke Z, Ehrich
Lister IN and Sihombing B: Prognostic significance of CKAP2L
expression in patients with clear cell renal cell carcinoma. Front
Genet. 13:8738842023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xiong G, Li L, Chen X, Song S, Zhao Y, Cai
W and Peng J: Up-regulation of CKAP2L expression promotes lung
adenocarcinoma invasion and is associated with poor prognosis. Onco
Targets Ther. 12:1171–1180. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li Q, Yan M, Wang C, Wang K and Bao G:
CKAP2L, a crucial target of miR-326, promotes prostate cancer
progression. BMC Cancer. 22:6662022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yi B, Fu Q, Zheng Z, Zhang M, Liu D, Liang
Z, Xu S and Zhang Z: Pan-cancer analysis reveals the prognostic and
immunotherapeutic value of cytoskeleton-associated protein 2-like.
Sci Rep. 13:83682023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chi F, Chen L, Jin X, He G, Liu Z and Han
S: CKAP2L, transcriptionally inhibited by FOXP3, promotes breast
carcinogenesis through the AKT/mTOR pathway. Exp Cell Res.
412:1130352022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Berasain C and Avila MA: Amphiregulin.
Semin Cell Dev Biol. 28:31–41. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Napolitano S, Martini G, Ciardiello D, Del
Tufo S, Martinelli E, Troiani T and Ciardiello F: Targeting the
EGFR signalling pathway in metastatic colorectal cancer. Lancet
Gastroenterol Hepatol. 9:664–676. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Zhang W, Zhang W, Tang C, Hu Y, Yi K, Xu X
and Chen Z: Silencing AREG enhances sensitivity to irradiation by
suppressing the PI3K/AKT signaling pathway in colorectal cancer
cells. Biologics. 18:273–284. 2024.PubMed/NCBI
|
|
68
|
Sun R, Zhao H, Gao DS, Ni A, Li H, Chen L,
Lu X, Chen K and Lu B: Amphiregulin couples IL1RL1+
regulatory T cells and cancer-associated fibroblasts to impede
antitumor immunity. Sci Adv. 9:eadd73992023. View Article : Google Scholar
|
|
69
|
Hu Y, Dong Z and Liu K: Unraveling the
complexity of STAT3 in cancer: Molecular understanding and drug
discovery. J Exp Clin Cancer Res. 43:232024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zou S, Tong Q, Liu B, Huang W, Tian Y and
Fu X: Targeting STAT3 in cancer immunotherapy. Mol Cancer.
19:1452020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Samad MA, Ahmad I, Hasan A, Alhashmi MH,
Ayub A, Al-Abbasi FA, Kumer A and Tabrez S: STAT3 signaling pathway
in health and disease. MedComm (2020). 6:e701522025. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Pennel KAF, Hatthakarnkul P, Wood CS, Lian
GY, Al-Badran SSF, Quinn JA, Legrini A, Inthagard J, Alexander PG,
van Wyk H, et al: JAK/STAT3 represents a therapeutic target for
colorectal cancer patients with stromal-rich tumors. J Exp Clin
Cancer Res. 43:642024. View Article : Google Scholar : PubMed/NCBI
|