Introduction
Pancreatic ductal adenocarcinoma (PDAC) is highly
lethal, contributing to >400,000 mortalities across the globe
each year. Epidemiological projections from North America and
Europe suggest that PDAC is poised to become the second leading
cause of cancer-related mortality within the next decade. This
trend is the lack of reliable early diagnostic tools, limited
therapeutic options and the increasing incidence of the disease
across all age groups and sexes (1,2).
In United States alone, ~50,000 individuals are diagnosed annually
and >45,000 of them succumb to the disease (3). The overall five-year survival rate
for PDAC remains <10%, largely attributed to delayed diagnosis
in >80% of cases (4). Despite
advances in adjuvant therapies, the five-year disease-free survival
rate remains low, at just 19-26% (5). Although the median overall survival
of these patients has doubled over the past 15 years, from ~6 to 12
months, it remains unacceptably low. Long-term survival rate
remains poor, with less than 3% of patients surviving beyond five
years (6,7).
Pancreatic cancer has a significant genetic basis,
with mutations in the Kirsten rat sarcoma viral oncogene homolog
(KRAS) found in >80% of patients. These alterations are
consistently associated with reduced overall survival and
independent of stage of the disease (8,9).
KRAS mutation testing is already part of clinical practice for
several epithelial malignancies, including lung and colorectal
cancers, where it guides therapeutic decision-making and
eligibility for targeted therapy trials (10,11). Due to its clinical significance,
KRAS mutation analysis is increasingly incorporated into PDAC
diagnostic workflows, particularly through endoscopic
ultrasound-guided fine-needle aspiration (EUS-FNA) in specialized
settings (12).
Current treatment strategies for PDAC encompass
surgical resection, chemotherapy, radiation therapy and emerging
targeted or immunotherapeutic approaches. Translational research
has revealed that PDAC is characterized by profound molecular and
cellular heterogeneity, which likely contributes to its pronounced
resistance to conventional therapies (13). Consequently, molecular biomarkers
are increasingly recognized as predictive and prognostic tools for
guiding personalized management of PDAC. However, the molecular of
tumor progression and chemoresistance remain poorly elucidated,
underscoring the urgent need for additional mechanistic and
translational investigations.
Beyond identifying gene expression signatures in
pancreatic cancer, elucidating the genetic and epigenetic
mechanisms that regulate transcription and translation may unveil
novel prognostic biomarkers. In recent years, microRNAs (miRNAs)
have been recognized as key post-transcriptional modulators of gene
expression, controlling the stability and translation of multiple
target mRNAs. This has established miRNAs as promising biomarkers
for predicting disease prognosis and therapeutic response (14,15).
Notably, despite the prevalence of KRAS activation
through oncogenic mutations in the vast majority of PDAC cases,
several miRNAs are capable of directly targeting KRAS, including
miR-96, miR-217 and miR-126 and are consistently downregulated
(16). Enforced expression of
miR-96 and miR-217 have been shown to reduce KRAS protein levels
and consequent attenuation of downstream v-Akt murine thymoma viral
oncogene homolog (AKT) signaling (17). As reduced expression of these
miRNAs correlates with increased KRAS activity, their dysregulation
likely contributes to the hyperactivation of RAS-driven signaling
pathways. Furthermore, several miRNAs that normally suppress the
expression of KRAS oncoproteins are usually downregulated in PDAC,
promoting sustained RAS pathway activation irrespective of
activating KRAS mutations (18).
Despite recent advances in diagnostic techniques,
including high-resolution imaging and EUS-guided FNA and advances
in therapy with regimens such as gemcitabine, nab-paclitaxel plus
gemcitabine and folinic acid, 5-fluorouracil, irinotecan and
oxaliplatin (FOLFIRINOX), the overall prognosis of PDAC remains
poor (19,20). The PRODIGE 24 analysis showed that
FOLFIRINOX therapy, patient age, tumor grade, disease stage and
care at high-volume centers, markedly influenced overall survival
(7). However, to date, no
specific molecular determinant has been conclusively linked to
prognosis in patients undergoing surgical resection for PDAC
(21).
The present review outlined the multifaceted role of
oncogenic KRAS in pathobiology, diagnosis, prognosis and
therapeutic management of pancreatic cancer. It also explored the
spectrum of RAS mutations across malignancies, with a particular
emphasis on KRAS aberrations in PDAC and elucidated the molecular
mechanisms by which miRNAs modulate KRAS-mediated signaling.
Furthermore, the present review focused on some major modes of
'crosstalk' between KRAS and miRNAs in PDAC, including: i) Direct
targeting of KRAS by specific miRNAs (e.g., miR-217, miR-216a-3p),
ii) feedback effects whereby mutant KRAS alters the expression of
miRNAs and iii) co-regulation of shared downstream effectors,
forming feedback loops that shape cell signaling and tumor
behavior.
KRAS-driven signaling in pancreatic ductal
cells
Pancreatic cells rely on an intricate network of
signaling cascades to coordinate their highly specialized exocrine
and endocrine functions. Key regulatory pathways include Hippo,
Wnt/β-catenin, phosphoinositide 3-kinase/protein kinase
B/mechanistic target of rapamycin (PI3K-Akt-mTOR), nuclear
factor-κB (NF-κB) and Janus kinase-signal transducer and activator
of transcription (JAK-STAT). These signaling pathways control key
cellular processes such as tissue development, differentiation,
proliferation and maintenance of homeostatic (22-24). Perturbations in these signaling
axes, whether through aberrant activation or dysregulation,
profoundly affect pancreatic physiology (25). Oncogenic mutations in KRAS disrupt
these pathways, driving pathological outcomes including
uncontrolled cellular proliferation, dedifferentiation,
inflammation and the eventual development of chronic pancreatitis
and progression to PDAC (9).
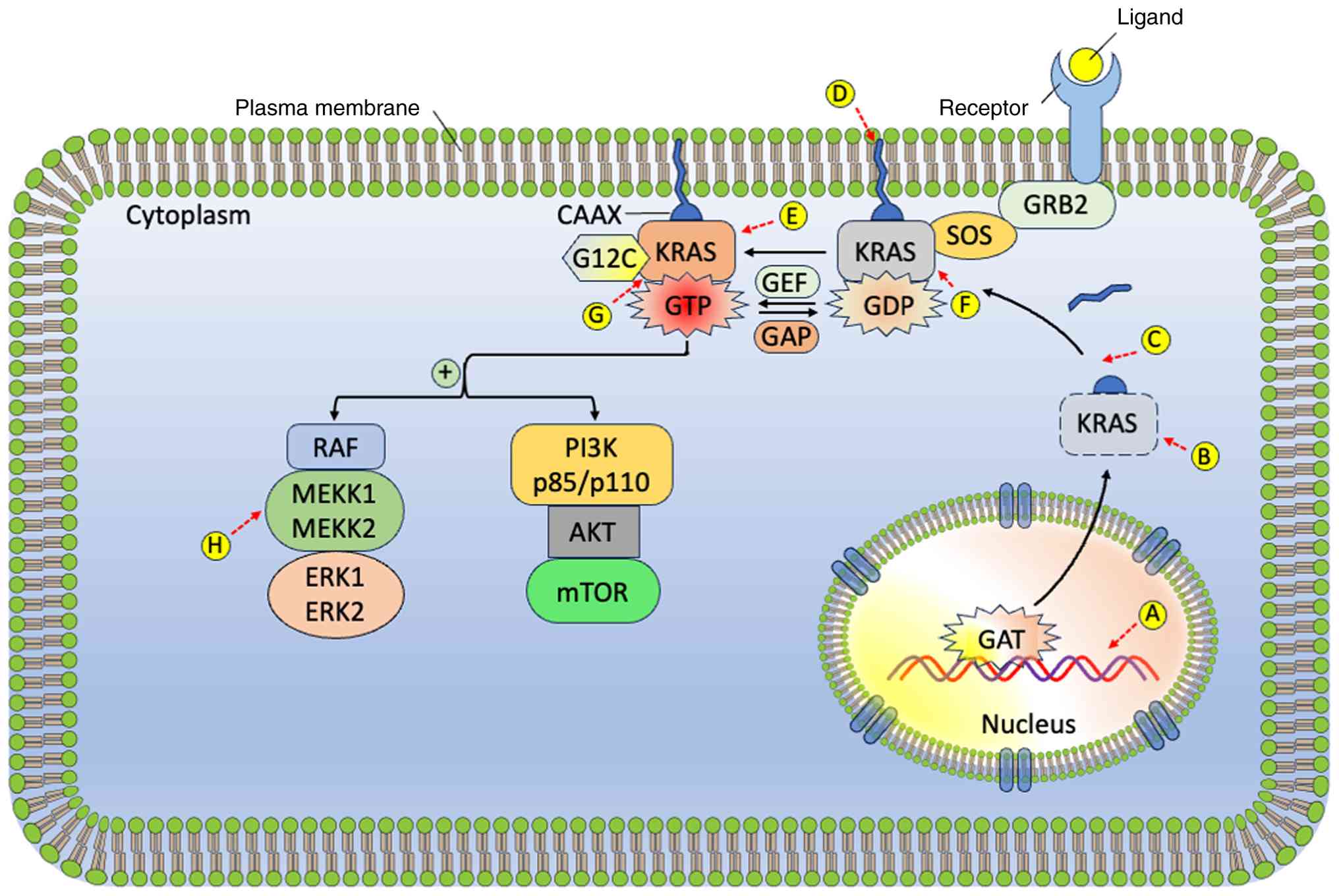
The RAS superfamily comprises a set of small
GTP-binding proteins that act as molecular switches within
pancreatic ductal epithelial cells, orchestrating signal
transduction from membrane-bound receptors to nuclear effectors.
Among these, KRAS plays a pivotal role within the RAS-MAPK
signaling cascade, a central regulatory network governing cell
growth, survival and differentiation. Under physiological
conditions, KRAS cycles between inactive GDP-bound and active
GTP-bound states, thereby transmitting extracellular cues to
intracellular effectors that maintain normal cellular behavior
(26) (Fig. 1).
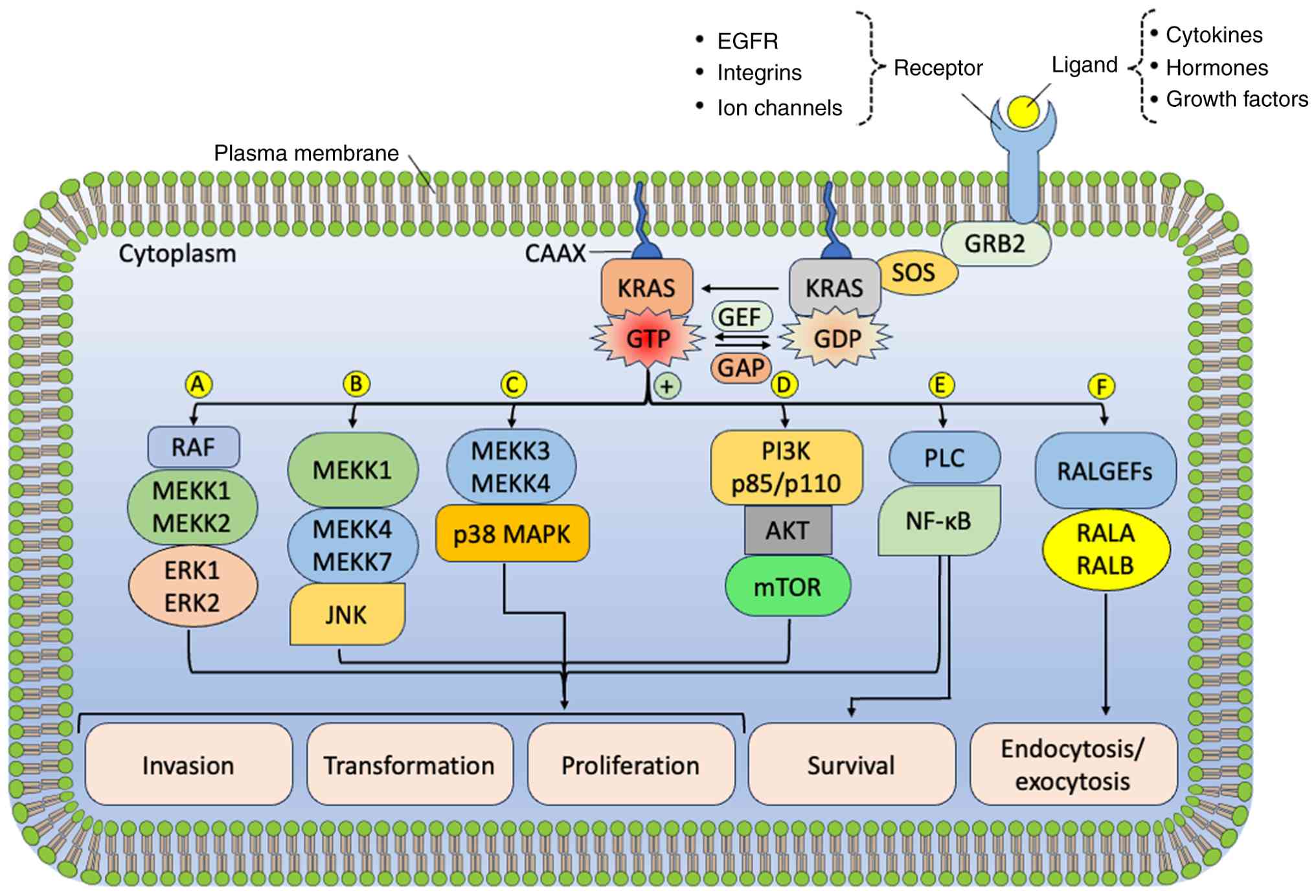 | Figure 1Stimulation of KRAS and the
subsequent activation of intracellular key signaling networks. The
major signaling pathways triggered by activated KRAS include (A)
the MAPK/ERK pathway, (B) the JNK pathway, (C) the p38 MAPK
pathway, (D) the PI3K pathway, (E) the NF-κB pathway and (F) The
RALA-RALB pathway. JNK, c-Jun N-terminal Kinase; KRAS, Kirsten rat
sarcoma viral oncogene homolog; MAPK/ERK, mitogen-activated protein
kinase/extracellular signal-regulated kinase; NF-κB, nuclear
factor-κB; PI3K, phosphoinositide 3-kinase; RALA, RAS-like
proto-oncogene A; RALB, RAS-like proto-oncogene B. |
However, activating mutations in KRAS, which occur
in the vast majority of PDAC cases, convert this tightly regulated
molecular switch into a constitutively active oncoprotein. Mutant
KRAS chronically stimulates a range of downstream effector
pathways, thereby orchestrating oncogenic signaling that sustains
tumor cell proliferation, metabolic reprogramming, metastatic
dissemination and therapeutic resistance. The principal downstream
cascades driven by mutant KRAS include: i) The RAF-MEK-ERK (MAPK)
pathway, the canonical and most extensively characterized effector
route, which promotes cellular proliferation and survival (27); ii) MEKK1-4→MEKK4/MEKK7→JNK
pathway, a pathway that is a part of the stress-activated MAP
kinase (SAPK) network, also known as the JNK pathway; iii)
MEKK3/MEKK4→p38 MAPK pathway, part of the SAPK system, parallel to
JNK pathway; iv) the PI3K-Akt-mTOR pathway, which regulates
metabolism, growth and anti-apoptotic signaling (28); v) PLC→NFκB pathway, this pathway
links membrane receptor activation to inflammatory, immune and
survival gene expression and vi) the Ras-like (RAL) Guanine
Nucleotide Exchange Factor pathway (GEF) pathway (29), involved in cytoskeletal
reorganization, vesicle trafficking and cell migration (Fig. 1). Besides these pathways, there
can occur some other aberrantly activated pathways such as
JAK-STAT, Wnt/β-catenin pathway, Hippo-YAP/TAZ Pathway, Notch
pathway and FAK-SRC pathway. The Hedgehog (Hh) signaling pathway is
aberrantly activated in the tumor microenvironment by oncogenic
KRAS (30), facilitating
reciprocal communication between cancer cells and the surrounding
stroma to promote tumor progression. Collectively, these
KRAS-mediated signaling networks highlight the pivotal role of this
oncogene in orchestrating pancreatic cellular reprograming,
positioning it as a master regulator of PDAC initiation and
evolution (9).
KRAS and its interaction with associated
proteins
The KRAS gene encrypts the KRAS protein, a small
GTP-binding enzyme composed of ~189 amino acid residues having a
molecular weight of ~21.6 kDa. Functionally, KRAS belongs to the
RAS family of small GTPases, which act as molecular switches
linking cell-surface growth factor receptors to a variety of
intracellular signaling cascades and transcriptional networks that
regulate cell survival, proliferation and differentiation (31). The human RAS protein family,
comprising HRAS, NRAS and KRAS (KRAS4A and KRAS4B splice variants),
shares a conserved structural organization with two major domains:
The G domain (GTPase domain) and the hypervariable region, the
membrane-targeting domain. The G domain (residues 1-164)
encompasses the nucleotide-binding pocket responsible for GDP/GTP
binding and hydrolysis, as well as regions essential for effector
interactions. The C-terminal membrane-targeting domain (residues
165-188/189) contains the CAAX motif, where C denotes cysteine, A
denotes aliphatic residues and X specifies the terminal amino acid
(26). Post-translational
modifications of this motif are critical for proper membrane
localization and function. Specifically, a 15-carbon farnesyl
isoprenoid group is attached covalently to the cysteine residue by
farnesyltransferase enzymes, enabling the protein to anchor to the
inner leaflet of the plasma membrane, an essential step for its
biological activity (32,33).
The dynamic cycle of active GTP and inactive GDP is
tightly controlled by two classes of regulatory proteins: guanine
nucleotide exchange factors (GEFs), which catalyze the exchange of
GDP for GTP to activate KRAS and GTPase-activating proteins (GAPs),
which accelerate GTP hydrolysis, thereby returning KRAS to its
inactive state (34) (Fig. 1). Mutations in KRAS, particularly
at codons 12, 13 and 61, impair intrinsic GTPase activity or
GAP-mediated hydrolysis, effectively locking KRAS in its active,
GTP-bound conformation and leading to constitutive signal
transduction (35). Once
activated, KRAS engages with a broad spectrum of downstream
effectors, >80 proteins have been identified to date, initiating
a multitude of signaling cascades. Upon activation of growth factor
receptors, such as receptor tyrosine kinases or G-protein-coupled
receptors, growth factor receptor-bound protein 2 associates with
the guanine nucleotide exchange factor Son of Sevenless (SOS) and
subsequently engages the KRAS protein (36).
For KRAS to become functionally active, it must
localize to the plasma membrane. This membrane anchoring is
mediated by the covalent attachment of a farnesyl isoprenoid group
to a cysteine residue within the C-terminal CAAX motif of KRAS, a
reaction catalyzed by farnesyltransferases. Once properly
localized, KRAS attains its active configuration upon binding GTP.
Mutations in KRAS typically diminish its GTPase activity, rendering
it resistant to GAP-mediated inactivation and thus locking the
protein in its GTP-bound, constitutively active state. This
persistent activation drives multiple downstream signaling cascades
and nuclear transcription programs that promote cell proliferation,
survival and oncogenic transformation (37).
The prominent signal transduction pathways include
RAF-MEK-ERK (MAPK) pathway, which governs cellular proliferation
and differentiation; the PI3K-AKT-mTOR axis, which regulates cell
metabolism, growth and survival; and the RAL-GEF and Tiam1-RAC
pathways, which influence cytoskeletal remodeling, motility and
vesicular trafficking (38).
Through these signaling routes, activated KRAS orchestrates the
transcriptional activation of several nuclear factors such as
ETS-like gene 1 (ELK1), JUN and myelocytomatosis (MYC), which
collectively promote cell cycle progression, transformation and
resistance to apoptosis (39).
The precise localization and post-translational
modifications of KRAS at the plasma membrane not only dictates its
functional activation but also influences its selective engagement
with specific effectors, thereby conferring signaling specificity.
This precise spatial and temporal control of KRAS activity
highlights its role as a master integrator of extracellular signals
and intracellular responses, accounting for the potent oncogenicity
and therapeutic intractability of mutant KRAS in PDAC. The
stimulation of KRAS activity and its downstream molecular signaling
routes is shown in Fig. 1.
KRAS driven signaling and the role of
miRNAs
KRAS functions as a membrane-associated molecular
integrator that transduces extracellular cues, such as growth
factors, cytokines, or cellular stress signals, received through
receptor tyrosine kinases, ion channels or integrins into
intracellular signaling cascades, primarily the KRAS-ERK and
PI3K-Akt pathways (40) (Fig. 1). In non-malignant cells with
wild-type KRAS, this signaling axis is stringently synchronized
through different layers of control. First, the equilibrium between
active and inactive KRAS is governed by the opposing activities of
GAPs and GEFs. Second, scaffold and adaptor proteins fine-tune the
spatial and temporal fidelity of KRAS-MAPK signaling by ensuring
the appropriate localization and assembly of pathway components.
Proteins such as Sprouty Related EVH1 Domain Containing 1 (SPRED1)
are essential for targeting GAPs to the membrane (41). Third, microRNAs (miRNAs) introduce
an additional post-transcriptional regulatory layer by modulating
the expression of RAS pathway constituents, scaffolds and
modulators (42).
miRNAs have emerged as crucial regulators of
oncogenic signaling networks, including the KRAS pathway and are
being actively explored as therapeutic targets across multiple
cancer types (43). In pancreatic
cancer, oncogenic KRAS mutations result in constitutive activation
of downstream pathways that drive malignant proliferation,
metabolic adaptation and therapeutic resistance. Concurrently,
several miRNAs function either as tumor suppressors, by directly
repressing KRAS or its downstream effectors, or as oncogenic
drivers that silence tumor-suppressive genes. The downregulation of
tumor-suppressive miRNAs such as miR-96, miR-217 and the Let-7
family leads to enhanced KRAS expression and activity, thereby
promoting tumor growth, invasiveness and survival. Conversely,
certain miRNAs, including miR-31, are upregulated by oncogenic KRAS
itself, further amplifying pro-metastatic signaling (17).
Extensive preclinical investigations have
demonstrated that miRNA dysregulation profoundly influences PDAC
initiation, progression and chemoresistance, highlighting the
potential of miRNA-based therapeutics as a novel strategy to
counteract KRAS-driven oncogenesis. By restoring tumor-suppressive
miRNAs or inhibiting oncogenic ones, miRNA-directed interventions
could redefine therapeutic paradigms in pancreatic cancer
management (14).
The Let-7 family of miRNAs directly targets all
major RAS isoforms, including KRAS, HRAS and NRAS. Negative
regulators of the RAS-ERK cascade, such as RAS p21 protein
activator 1 (RASA1) and Sprouty-related EVH1 domain-containing
protein 1 (SPRED1), are jointly suppressed by miR-206 and miR-21.
The GAP protein neurofibromin 1 (NF1) has been identified as a
probable catalytic partner of SPRED1. Another inhibitor of the same
pathway, Sprouty RTK signaling antagonist 1 (SPRY1), along with the
PI3K-AKT pathway suppressor phosphatase and tensin homolog (PTEN),
are uniquely targeted by miR-21. Furthermore, the downstream
effector and tumor suppressor programmed cell death 4 (PDCD4) is
co-regulated by miR-21 and miR-206 (44). The interplay between oncogenic RAS
signaling and microRNAs targeting key components of the RAS pathway
in cancer is diagrammatically represented in Fig. 2.
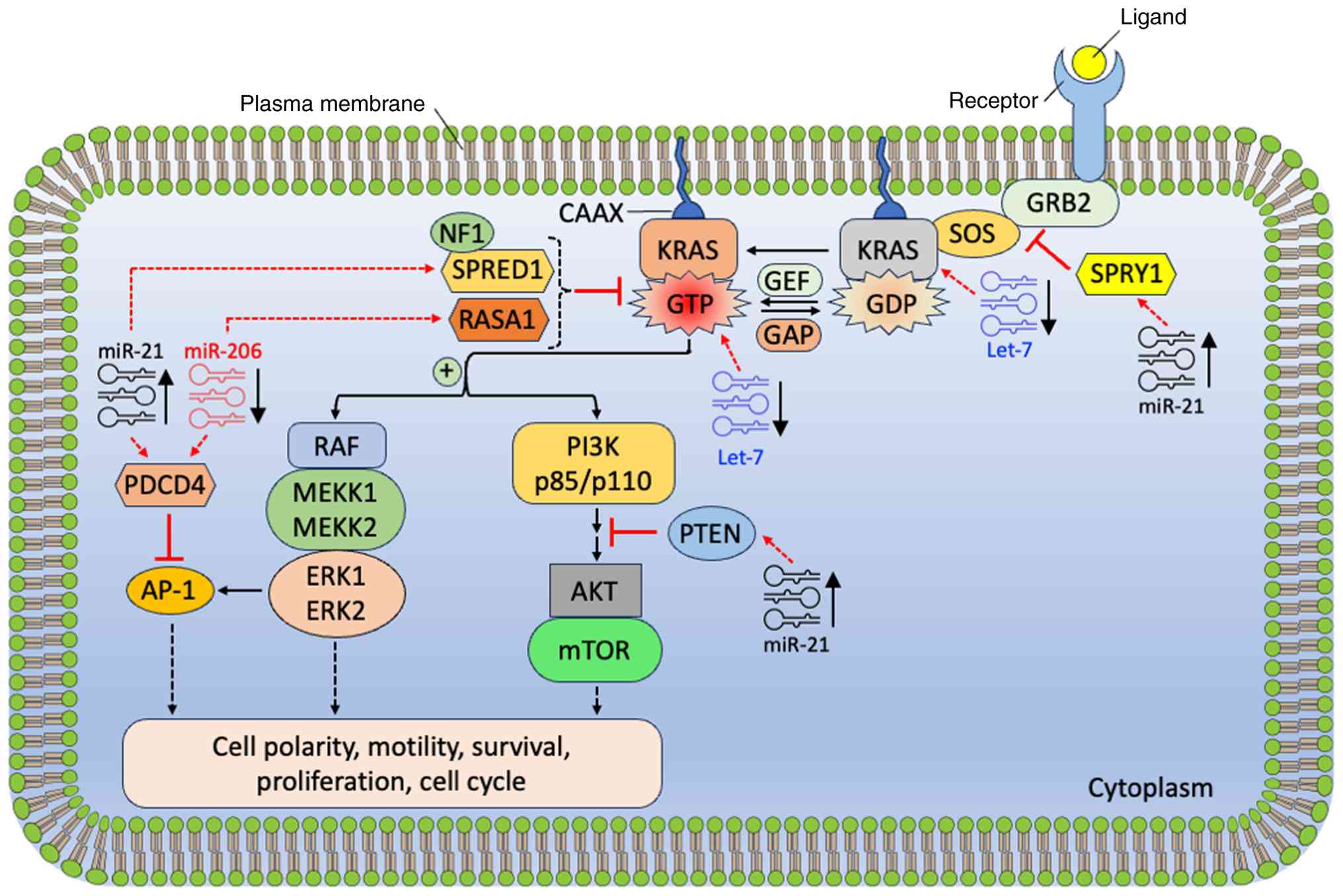 | Figure 2Interactions between oncogenic RAS
signaling and miRNAs in cancer. RAS GTPases function as molecular
switches, cycling between an inactive GDP-bound conformation and an
active GTP-bound form. In cancer, RAS signaling frequently becomes
constitutively active due to elevated RAS-GTP levels, which promote
enhanced cell survival, proliferation and migration. miRNAs, such
as Let-7 family and miR-206 are down regulated (↓), while miR-21 is
upregulated (↑), capable of modulating multiple components of the
RAS signaling network. GTPase, guanosine triphosphatase; miRNAs,
microRNAs; RAS, rat sarcoma. |
Feedback loops and reciprocal regulation
between KRAS and miRNAs
Accumulating evidence indicates that the interaction
between oncogenic KRAS signaling and miRNAs in PDAC is not
unidirectional but instead organized into feedback loops and
reciprocal regulatory circuits. These mechanisms contribute to the
stabilization and amplification of KRAS-driven oncogenic programs
and have important implications for tumor progression, therapeutic
resistance and biomarker development (45,46).
One of the best-characterized examples is the
KRAS-RREB1-miR-143/145 regulatory loop. Oncogenic KRAS activates
the transcription factor Ras-responsive element binding protein 1
(RREB1), which directly represses the transcription of the
tumor-suppressive miR-143/145 cluster. In turn, miR-143 and miR-145
directly target KRAS and RREB1, respectively. Loss of miR-143/145
expression in PDAC therefore leads to de-repression of KRAS
signaling, creating a feed-forward loop that reinforces oncogenic
KRAS activity. Disruption of this circuit has been shown to enhance
cell proliferation, invasion and tumorigenicity, highlighting its
functional importance in pancreatic cancer progression (47).
In addition to the miR-143/145 axis, several other
KRAS-targeting miRNAs participate in reciprocal suppression loops.
Members of the Let-7 family, as well as miR-127, miR-193a and
miR-27b, directly bind the KRAS 3'-UTR and suppress its expression.
However, sustained KRAS activation downregulates these miRNAs
through downstream MAPK and MYC-dependent transcriptional programs,
thereby relieving inhibitory pressure on KRAS itself. Such
reciprocal regulation ensures persistent KRAS pathway activation
and contributes to the addiction of PDAC cells to KRAS signaling
(42,48).
Conversely, oncogenic KRAS can induce the expression
of certain oncogenic miRNAs that further enhance KRAS pathway
output by targeting its negative regulators. For example,
KRAS-driven up-regulation of miR-21, miR-31 and miR-155 has been
reported to suppress tumor suppressors such as PTEN, RASA1 and NF1,
thereby strengthening downstream MAPK and PI3K signaling. These
miRNAs form positive feedback loops that indirectly amplify KRAS
signaling and promote tumor growth, survival and chemoresistance
(46).
Collectively, these findings underscore that
KRAS-miRNA crosstalk in PDAC is governed by a network of
interconnected feedback loops, rather than isolated regulatory
events. A conceptual overview of these reciprocal mechanisms,
including the KRAS→RREB1→miR-143/145 suppression→KRAS de-repression
loop and additional KRAS-miRNA feedback circuits, is presented in
Fig. 3. Understanding these
regulatory architectures provides a framework for the rational
design of therapeutic strategies aimed at restoring
tumor-suppressive miRNAs or disrupting KRAS-reinforcing feedback
loops. These approaches hold promises for improving diagnostic,
prognostic and therapeutic strategies in pancreatic cancer.
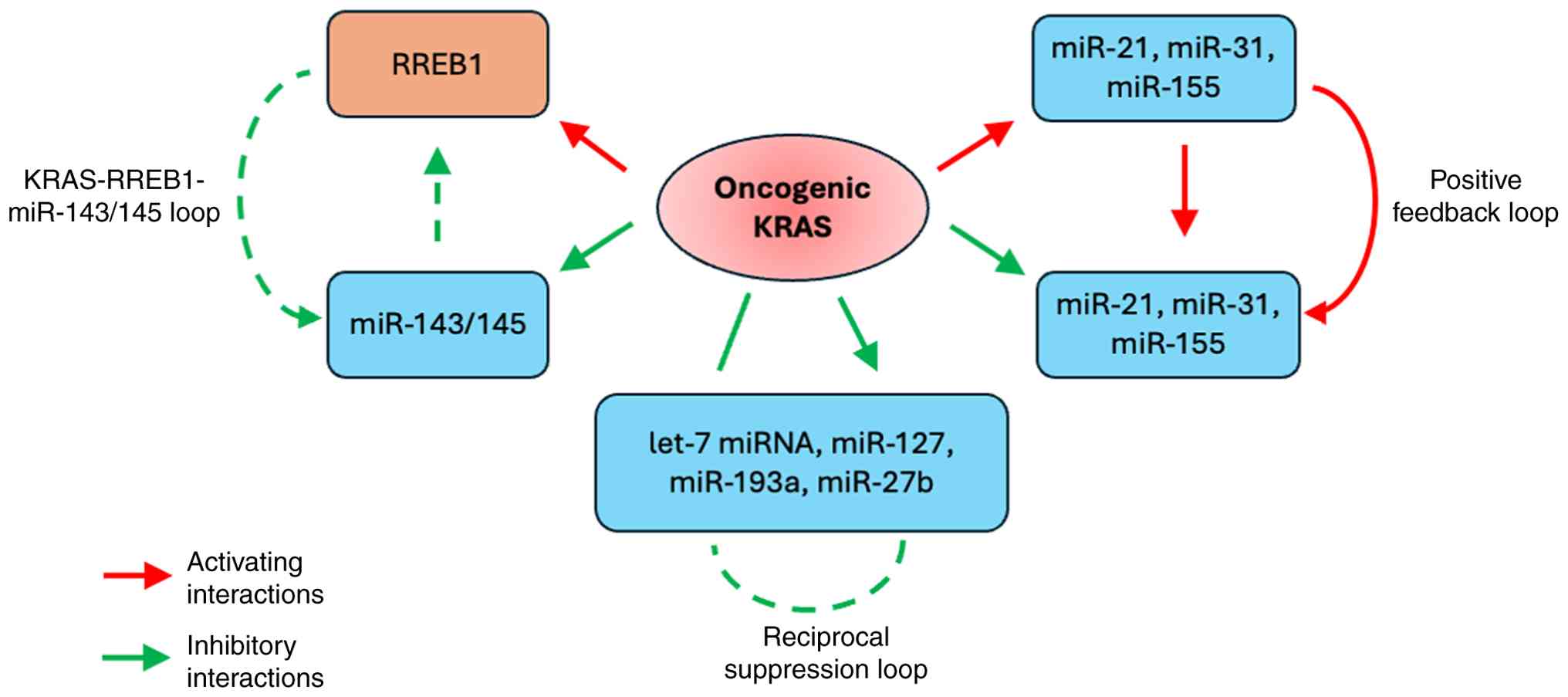 | Figure 3Feedback loops and reciprocal
regulation between KRAS and miRNAs in PDAC. The diagram illustrates
interconnected regulatory circuits between oncogenic KRAS signaling
and miRNAs. KRAS → RREB1 → miR-143/145 suppression → KRAS
de-repression forms a feed-forward loop that reinforces KRAS
activity. Tumor-suppressive miRNAs (e.g., Let-7 family,
miR-143/145) inhibit KRAS expression, but their levels are
downregulated by KRAS-driven transcriptional programs. Oncogenic
miRNAs (miR-21, miR-31, miR-155) are upregulated by KRAS and target
negative regulators (PTEN, RASA1, NF1), amplifying downstream MAPK
and PI3K signaling pathways. Collectively, these reciprocal
circuits stabilize KRAS-driven oncogenic programs, promoting tumor
growth, survival and chemoresistance. KRAS, Kirsten rat sarcoma
viral oncogene homolog; KRAS, Kirsten rat sarcoma viral oncogene
homolog; MAPK, mitogen activated protein kinase; miRNAs, microRNAs;
NF1, neurofibromin 1; PDAC, pancreatic ductal adenocarcinoma; PI3K,
phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog;
RASA1, RAS p21 protein activator 1; RREB1, Ras-responsive
element-binding protein 1. |
Role of KRAS in pancreatic cancer
KRAS is the principal oncogenic driver in PDAC,
harboring activating mutations, most notably KRASG12D
and KRASG12V, in >90% of tumors (49,50). Insights from genetically
engineered mouse models, such as the Pdx1-Cre;
LSL-KRASG12D system, have been pivotal in delineating
the stepwise transformation from normal pancreatic epithelium to
pancreatic intraepithelial neoplasia (PanIN) and ultimately to
invasive carcinoma. These models have underscored that sustained
KRAS activation is sufficient to initiate neoplastic lesions and,
in cooperation with additional genetic or epigenetic alterations,
to drive full malignant progression (51).
Oncogenic KRAS engages a wide array of downstream
effectors and signaling cascades, including the MAPK-ERK,
PI3K-AKT-mTOR and RAL-GDS pathways, which collectively drive
enhanced proliferation, altered metabolic programming, resistance
to apoptosis and immune evasion. The magnitude and duration of KRAS
signaling is directly associated with disease aggressiveness,
metastatic potential and poor clinical outcomes, positioning KRAS
mutation profiling as a valuable tool for early detection,
prognosis and patient stratification (52).
From a therapeutic perspective, targeting KRAS
remains a central focus of translational research. Emerging
therapeutic modalities include direct pharmacological inhibition of
KRAS (such as allele-specific inhibitors targeting
KRASG12C), RNA interference-based silencing approaches
and RAS-directed peptide or mRNA-based vaccines designed to elicit
anti-tumor immunity (53,54). Moreover, mutant KRAS expression
was markedly reduced by using CRISPR/Cas13a-mediated mRNA knockdown
(55). In addition, inhibitors of
downstream signaling nodes, including MEK, ERK and PI3K are being
explored either as monotherapies or in rational combination
regimens (Fig. 4). Although these
approaches are still evolving, they represent promising avenues for
overcoming the long-standing challenge of therapeutically targeting
KRAS-driven pancreatic cancer (56).
 | Figure 4Various therapeutic approaches
directed at the KRAS gene and KRAS protein: (A) RNAi strategies
designed to suppress the expression of mutant KRAS, (B) FTIs that
block the farnesylation of the KRAS protein, (C) Inhibition of
PDEδ, (D) Disruption of RAS signaling by detaching farnesylated
KRAS from the cell membrane, (E) KRAS-targeted vaccination, (F)
Small-molecule inhibitors that bind to the RAS-GDP complex, (G)
Blocking the KRAS-GTP interaction, (H) Targeting downstream
signaling cascades, specifically the RAF-MEK-ERK and PI3K-AKT-mTOR
pathways. FTIs, farnesyltransferase inhibitors; KRAS, Kirsten rat
sarcoma viral oncogene homolog; RNAi, PDEδ, phosphodiesterase 6
delta; PI3K-AKT-mTOR, phosphoinositide 3-kinase-protein kinase
B-mechanistic target of rapamycin; RAF-MEK-ERK, rapidly accelerated
fibrosarcoma-mitogen-activated protein kinase-extracellular
signal-regulated kinase; RAS, rat sarcoma; RNA interference. |
KRAS mutations in PDAC and associated
biological processes
A point mutation in KRAS oncogene, most commonly at
codon 12, represents the initiating genetic alteration in most of
the PDAC cases, occurring in 70-95% of cases (8,57).
This single-nucleotide substitution alters the wild-type GGT codon
(encoding glycine) to GAT or GTT, [encoding aspartic acid (G12D),
or valine (G12V), GCT (alanine; G12A), or CGT (arginine; G12R)]
(52). Less frequent mutations
are also detected at codons 11, 13, 61 and 146, each conferring
distinct biochemical properties and oncogenic potential (8,52,58).
These activating point mutations compromise the
intrinsic GTPase activity of KRAS, rendering it resistant to
regulation by GAPs. As a result, mutant KRAS remains constitutively
locked in its GTP-bound, active state, persistently transmitting
proliferative and pro-survival signals through downstream pathways
such as RAF-MEK-ERK, PI3K-AKT-mTOR and RAL-GDS (38) (Fig.
1). Importantly, this aberrant activation of KRAS is frequently
accompanied by the genetic inactivation of key tumor suppressor
pathways, including inhibitor of cyclin-dependent kinase
4a/alternate reading frame (INK4a/ARF), tumor protein 53 (TP53) and
deleted in pancreatic carcinoma locus 4/SMAD family member 4
(DPC4/SMAD4). Together, these alterations synergistically promote
malignant transformation and drive tumor progression (8). Notably, KRAS mutations represent one
of the earliest molecular events in pancreatic tumorigenesis and
are detectable in preneoplastic lesions, including PanINs and
intraductal papillary mucinous neoplasms (IPMNs), highlighting
their central role in initiating the neoplastic cascade (59).
Extensive evidence highlights the critical role of
KRAS mutations in driving multiple pancreatic cancer cell
behaviors, including enhanced proliferation, survival, migration
and invasion. Oncogenic KRAS extensively reprograms cellular
metabolism, promoting increased glucose uptake, the Warburg effect
and increased lactate and reactive oxygen species (ROS) production
(38,60). Additionally, mutant KRAS
stimulates macropino-cytosis and autophagy, supporting nutrient
scavenging and tumor growth. Collectively, these metabolic and
catabolic adaptations facilitate PDAC expansion and metastatic
progression.
Beyond its cell-intrinsic functions, KRAS also
coordinates dynamic interactions between tumor cells and the
surrounding tumor microenvironment (61). These interactions are mediated
largely through paracrine signaling. KRAS-mutant tumor cells
secrete range of chemokines, including interleukin-6 (IL-6) and
granulocyte-macrophage colony-stimulating factor (GM-CSF). These
factors recruit and activate T cells, myeloid-derived suppressor
cells, B cells and macrophages, thereby enhancing inflammation and
tumor progression. Furthermore, KRAS is essential for communication
with cancer-associated fibroblasts (CAFs), which are activated via
tumor growth factor-β (TGF-β) and sonic hedgehog pathways. CAFs
modulate the extracellular matrix, including hyaluronic acid and
collagen, creating a supportive niche that enhances tumor cell
proliferation (62).
Tumor aggressiveness associated with
specific KRAS mutants
Although several studies have investigated the
impact of distinct KRAS mutant alleles on tumor aggressiveness and
phenotype across various cancer types, no definitive consensus has
been reached. This uncertainty may arise from the lack of a single
dominant signaling pathway that is consistently altered across
different mutations. To date, no studies have directly compared the
transforming efficiency of G12R, G12D and G12V alleles in
pancreatic tumors. However, research in lung and colorectal cancer
models, where KRAS is also frequently mutated, suggests that most
KRAS variants have broadly similar effects on tumor growth and
dissemination (63).
Subtle differences in downstream effector engagement
are likely to influence the ultimate tumor phenotype. For example,
in non-small cell lung carcinoma (NSCLC) cell lines,
KRASG12D preferentially activates PI3K and MEK pathways,
whereas KRASG12V predominantly engages RAL-GEFs and
shows reduced AKT activation. Additionally, the G12R, G12D and G12V
mutants exhibit lower affinity for RAF compared to wild-type KRAS
(64). Molecular dynamics
simulations further indicate that the conformational flexibility of
KRAS mutants differs from wild type, with significant variation
among mutants themselves, highlighting the complexity of
allele-specific effects (65).
Notably, the switch-I region, which mediates effector binding, is
more accessible in mutant forms, particularly G12D bound to GDP,
providing mechanistic insight into differential effector
interactions (65).
Prognosis of KRAS mutations
Several studies have investigated whether the
occurrence of KRAS mutations affects the PDAC prognosis (66). Analyses have been conducted on
both resected tumor specimens and EUS-FNA-derived samples, in
studies including more than 50 patients. The KRAS mutation were
consistently associated with poorer survival, independent of
surgical intervention (66,67). It should be noted that some study
cohorts were heterogeneous, including resected PDAC, ampullary
carcinomas, non-resectable tumors and recurrent lesions. Despite
these variations, most investigations concluded that KRAS mutations
adversely affect survival irrespective of curative surgery.
Emerging evidence suggests that specific KRAS
mutational subtypes may further influence prognosis (52,57). For example, PDAC patients
harboring KRASG12D mutations exhibited considerably
reduced overall survival (~6 months) compared with those with
wild-type KRAS (~9 months), KRASG12V (~9 months), or
KRASG12R (~14 months), independent of chemotherapy
(57). Other similar studies
indicate that KRASG12D, KRASG12R, or
combinations of mutant alleles are linked to associated with poorer
clinical outcomes (68). These
differences may reflect allele-specific engagement of downstream
signaling pathways (69).
To establish definitive prognostic correlations,
multi-center studies with larger, homogeneous cohorts (>200
patients) are required. Nevertheless, KRAS mutation testing already
provides a robust prognostic biomarker for PDAC. Integrating KRAS
status with additional molecular markers, such as S100A2
expression, immune infiltration metrics, or microRNAs, may further
refine risk stratification (70).
Targeting KRAS for treatment
In pancreatic cancer, therapeutic targeting of KRAS
has been extensively investigated; however, two major challenges
remain; its historical designation as the 'undruggable' and the
emergence of bypass pathways despite KRAS inhibition. KRAS mutants
exhibit high GTP-binding affinity and a sterically constrained
active site and their signaling relies on protein-protein
interactions that lack well-defined binding pockets, complicating
direct inhibition. Conceptually, KRAS blockade should suppress the
cancer growth and induce regression in KRAS-dependent PDAC. Indeed,
KRAS inactivation in mouse models causes tumor shrinkage; however,
spontaneous recurrence occurs, with ~1/3 of recurrent tumors
lacking KRAS expression (71). In
these KRAS-independent tumors, Yes associated protein 1 (YAP1)
amplification collaborates with E2F for cell cycle activation and
DNA repair, driving aggressive, quasi-mesenchymal phenotypes
(72).
Despite these challenges, multiple approaches have
been advanced for targeting KRAS mutants, including genetic
approaches and small-molecule inhibitors identified through in
silico and in vitro screening platforms (73). Various therapeutic approaches have
been tried that direct at the KRAS gene and its protein. These
approaches include RNA interference (RNAi) strategies designed to
overwhelm the expression of KRAS mutant (predominantly the
KRASG12D variant) (74,75). The other approach involved the use
of farnesyltransferase inhibitors (FTIs) (such as lonafarnib or
tipifarnib) that block the farnesylation of the KRAS protein at the
C-terminal CAAX motif. This approach led to the prevention of its
localization to the endoplasmic reticulum and Golgi apparatus
(32,76). Moreover, inhibition of
phosphodiesterase 6δ (PDEδ) with deltarasin was also employed, as
it facilitates the transport of farnesylated KRAS to the plasma
membrane (32,77).
In addition, disruption of RAS signaling by
detaching farnesylated KRAS from the cell membrane (such as
farnesyl thiosalicylic acid, also known as Salirasib) has also been
attempted (32). Furthermore,
KRAS-targeted vaccination has also been employed, utilizing mutant
peptide antigens that contain amino acid substitutions
characteristic of various KRAS mutations (78). In addition, small-molecule
inhibitors that bind to the RAS-GDP complex, obstructing the
interaction between KRAS and the SOS, thereby preventing
SOS-mediated nucleotide exchange has been worked out (79-81).
Moreover, blocking the KRAS-GTP interaction prevents
KRAS binding with RAF. This strategy includes covalent small
molecules that selectively recognize and irreversibly bind to the
KRASG12C mutant, neutralizing its downstream signaling.
Furthermore, targeting downstream signaling cascades, specifically
the RAF-MEK-ERK and PI3K-AKT-mTOR pathways, using small-molecule
inhibitors has also been routinely worked out (82,83). These approaches and their
mechanisms of action are summarized in Fig. 3.
KRAS inhibition by RNAi
RNAi has emerged as a novel approach to suppress
KRAS expression in mutant-driven cancers, including pancreatic
cancer (75). In vivo and
in vitro studies validate that RNAi-mediated knockdown of
mutant KRAS in PDAC cells reduces proliferation,
anchorage-independent growth and tumorigenicity (32). These findings suggest that
KRAS-targeted small interfering RNAs (siRNAs) may represent a
viable therapeutic approach (74). However, systemic delivery of
siRNAs is limited by enzymatic degradation, renal clearance and
challenges in achieving tumor-specific targeting. To overcome these
barriers, siRNAs have been incorporated into Local Drug Eluter
(LODER) implants, a decomposable polymeric matrix that protects
siRNAs and enables its sustained, localized release within tumors
over several months. In preclinical models, LODER-mediated delivery
inhibited pancreatic tumor growth and improved survival (84,85). This approach was translated into a
phase I-IIa clinical trial in combination with FOLFIRINOX
chemotherapy, demonstrating encouraging efficacy, with a median
survival of almost15 months and the survival rate of 18-months
(86).
Another innovative strategy employs human
fibroblast-derived inhibitory exosomes (iExosomes) loaded with
KRAS-specific siRNAs. These vesicles, expressing CD47 to evade
uptake by the reticuloendothelial system, efficiently deliver siRNA
via macropinocytosis and show robust antitumor activity in
preclinical PDAC models (87).
Clinical trials are currently underway to evaluate their safety and
efficacy in metastatic PDAC. Additionally, systemic delivery of
KRAS-targeting siRNAs using dioleoyl-phosphatidylcholine-based
nanoliposomal platforms represents a promising approach for in
vivo therapy (88).
KRAS-binding pocket targeting
Some small molecules have been found to directly
bind KRAS hydrophobic pocket on the inactive KRAS-GDP complex,
disrupting the interaction between KRAS and SOS, inhibiting
nucleotide exchange (79,80). A second strategy involved
compounds that obstruct the interface of GTP-bound KRAS and RAF or
other downstream effectors, effectively blocking signal
transduction. A third approach targets the KRAS-SOS complex,
preventing activation of KRAS by small-molecule binding. While
these approaches remain largely preclinical and have yet to be
fully validated in vivo, they challenge the long-standing
view of KRAS as an 'undruggable' oncogenic target.
Anti-KRAS vaccination
An alternative strategy to inhibit KRAS involves
peptide-based vaccination using RAS peptides harboring specific
amino acid mutations. To date, KRAS-targeted vaccination approaches
in have not demonstrated clear clinical benefit, although novel
peptide formulations are currently under clinical investigation
(78,80). The first human trial of GI-4000, a
recombinant heat-inactivated Saccharomyces cerevisiae
vaccine expressing mutant KRAS, demonstrated a favorable safety
profile and elicited measurable immune responses in most patients
with pancreatic cancer (89).
KRAS membrane localization
disruption
Another strategy to target KRAS focuses on
disrupting its membrane localization, a critical step for its
activation. KRAS undergoes farnesylation and is trafficked from the
endoplasmic reticulum to the plasma membrane, where it attaches GTP
and becomes active. Farnesyltransferase inhibitors (such as
tipifarnib) were designed to block this farnesylation. However,
clinical results have been largely disappointing as KRAS can
undergo alternative prenylation via geranylgeranyl transferase 1,
bypassing the blockade (32,76).
An alternative approach targets KRAS translocation
by inhibiting phosphodiesterase 6δ (PDEδ), a chaperone that
facilitates the membrane trafficking of farnesylated KRAS.
Deltarasin, a high-affinity PDEδ inhibitor, disrupts KRAS membrane
association and effectively reduces the proliferation in
KRAS-dependent PDAC cell lines (32,90). A related strategy involves
farnesyl-cysteine mimetics that compete with KRAS for membrane
anchoring. Farnesyl thiosalicylic acid (FTS, Salirasib) displaces
farnesylated KRAS from the membrane, induces its proteolytic
degradation and inhibits downstream signaling (32). FTS has demonstrated preclinical
and early clinical potential as a KRAS-targeted therapy in PDAC
(91).
RAF-MEK-ERK pathway targeting
Beyond post-translational modifications and membrane
localization of KRAS, its mutant-driven signaling networks
constitute critical therapeutic targets (Fig. 1). Among these pathways, the
RAF-MEK-ERK cascade is a major effector pathway, prompting the
development of multiple MEK inhibitors. For instance, trametinib,
an allosteric inhibitor of MEK1/2 blocks both its activation and
kinase potential (92).
Similarly, selumetinib, another oral MEK1/2 inhibitor, demonstrated
limited clinical benefit, showing no significant survival advantage
over capecitabine in gemcitabine-refractory PDAC (93,94). In addition, some trials are
ongoing, evaluating other inhibitors of MEK such as refametinib and
pimasertib, often in addition with gemcitabine, to enhance the
therapeutic efficacy (80,82).
PI3K-AKT-mTOR targeting
A variety of inhibitors targeting PI3K, RAF, AKT and
mTOR are under preclinical and clinical investigation (80). While early phase I trials
occasionally observed partial tumor responses, most approaches
targeting AKT, PI3K, or mTOR have yielded limited efficacy in
KRAS-mutant tumors (95).
Preclinical studies, however, demonstrate synergistic antitumor
effects when PI3K pathway inhibitors are combined with RAF-MEK-ERK
inhibitors in mouse models (79,96). Several clinical trials (phase
I-III) have evaluated combinatorial approaches, including AKT
inhibitors with MEK inhibitors (such as selumetinib), mTOR
inhibitors (everolimus and temsirolimus), PI3K inhibitors
(rigosertib), or multi-kinase inhibitors (sorafenib), with or
without gemcitabine. Despite these efforts, most combinations have
failed to achieve meaningful clinical benefit, often resulting in
enhanced toxicity or treatment-related adverse effects (57).
Other inhibitors and targets
The downstream members of KRAS, the GTPases, Ras
like proto-oncogene A (RALA) and RALB play crucial roles in PDAC
cell transformation and invasion (66). Once activated, RALA and RALB
regulate key cellular processes, including autophagy, cytokine
signaling, endocytosis and transcriptional control. Dysregulation
of these activities can promote cell propagation, apoptosis
resistance and metastatic spread (32,38). Autophagy can be therapeutically
targeted using inhibitors such as hydroxychloroquine, often in
combination with MEK-ERK pathway inhibitors (97). Clinical trials evaluating the
combination of trametinib (MEK1/2 inhibitor) and hydroxychloroquine
are ongoing, highlighting the potential of such combinatorial
strategies in PDAC.
Additional KRAS-pathway targets under investigation
include ruxolitinib (a JAK1/2 inhibitor) and NF-κB inhibitors.
Other approaches focus on agents affecting synthetic lethal
interactions, transcriptional programs, survival signaling, cell
cycle regulation, protein kinases, apoptosis and senescence
(79,80). Beyond canonical effectors,
KRAS-regulated metabolic pathways such as glucose and glutamine
metabolism, macropinocytosis and autophagy present promising
therapeutic targets (98).
Challenges and future directions of
targeting the KRAS-miRNA axis in PDAC
Despite increasing evidence supporting miRNAs as
regulators of KRAS signaling in PDAC, several challenges hinder
their clinical translation (99,100). Efficient and tumor-specific
delivery of miRNA-based therapeutics remains a major obstacle,
largely due to the dense desmoplastic stroma and poor
vascularization characteristic of PDAC (101). Although emerging delivery
platforms, including lipid nanoparticles and exosome-based systems,
have improved miRNA stability and uptake, further refinement is
required to enhance tissue specificity and reduce systemic toxicity
(102,103).
An additional challenge is the context-dependent
nature of miRNA function (104).
Individual miRNAs may exert divergent roles depending on tumor
type, genetic background and oncogenic signaling, complicating
therapeutic development (105,106). This highlights the need for
PDAC-specific functional validation using relevant preclinical
models (107). Moreover, because
miRNAs regulate multiple downstream targets, off-target effects and
unintended pathway modulation remain concerns, necessitating
improved target prediction and transcriptomic profiling approaches
(108).
Given these limitations, combination strategies are
likely to be essential (109).
Modulating KRAS-associated miRNAs may enhance responses to
inhibitors of the MEK-ERK pathway, autophagy, or inflammatory
signaling, thereby overcoming adaptive resistance to single-agent
therapies (110). Importantly,
the role of specific miRNAs must be interpreted within the PDAC
context; for example, miR-96 promotes KRAS-driven tumor progression
in PDAC but has been reported to function as a tumor suppressor in
other cancers (111).
Collectively, these considerations underscore the importance of
context-aware and combinatorial approaches for translating
KRAS-miRNA-targeted therapies into clinical applications (112).
Paradigm shift towards understanding the
role of miRNA in PDAC
Recent innovations in high-throughput miRNA
profiling have revolutionized our understanding of PDAC, uncovering
robust miRNA signatures with clinical utility in diagnosis,
prognosis and therapy. Recent computational studies have identified
several dysregulated miRNAs inversely associated with KRAS
expression, suggesting their potential as biomarkers and regulators
of KRAS-driven oncogenesis in PDAC (113). A multicenter study reported a
five-miRNA serum signature (hsa-miR-1343-5p, -4632-5p, -4665-5p,
-665 and -6803-5p) capable of detecting early-stage
pancreatobiliary tumors with >80% sensitivity and specificity,
markedly outperforming CA19-9 (114).
A recent meta-analysis confirmed the diagnostic
accuracy of miRNAs in pancreatic cancer, notably, miR-320,
miR-1290, miR-93, miR-25, miR-451, miR-20, miR-21, miR-223 and
miR-122. In addition, prognostically, miR-10, miR-21 and miR-221
associated strongly with survival (115). Furthermore, circulating and
exosomal miRNAs (e.g., miR-21, miR-10b, miR-205-5p, miR-1246,
miR-191-5p) have demonstrated utility in distinguishing PDAC from
benign conditions such as pancreatitis, with diagnostic accuracy
reaching 94% (116,117). These findings substantiate the
paradigm shift: miRNAs are not peripheral players but central
regulators of PDAC's molecular architecture, influencing
KRAS-driven signaling, tumor progression and therapeutic
response.
Therapeutic modulation of miRNAs is gaining
traction. For instance, miR-665 has shown inhibitory effects on
cancer cell growth, highlighting the potential of miRNA mimics and
anti-miRNAs as precision therapeutics targeting tumor-specific
molecular profiles (114).
Integration of miRNA-based strategies into personalized medicine,
tailoring interventions to individual tumor signatures. This may
offer a promising avenue for improving outcomes in this aggressive
disease.
Overview of miRNA structure, biogenesis and
mechanism of action
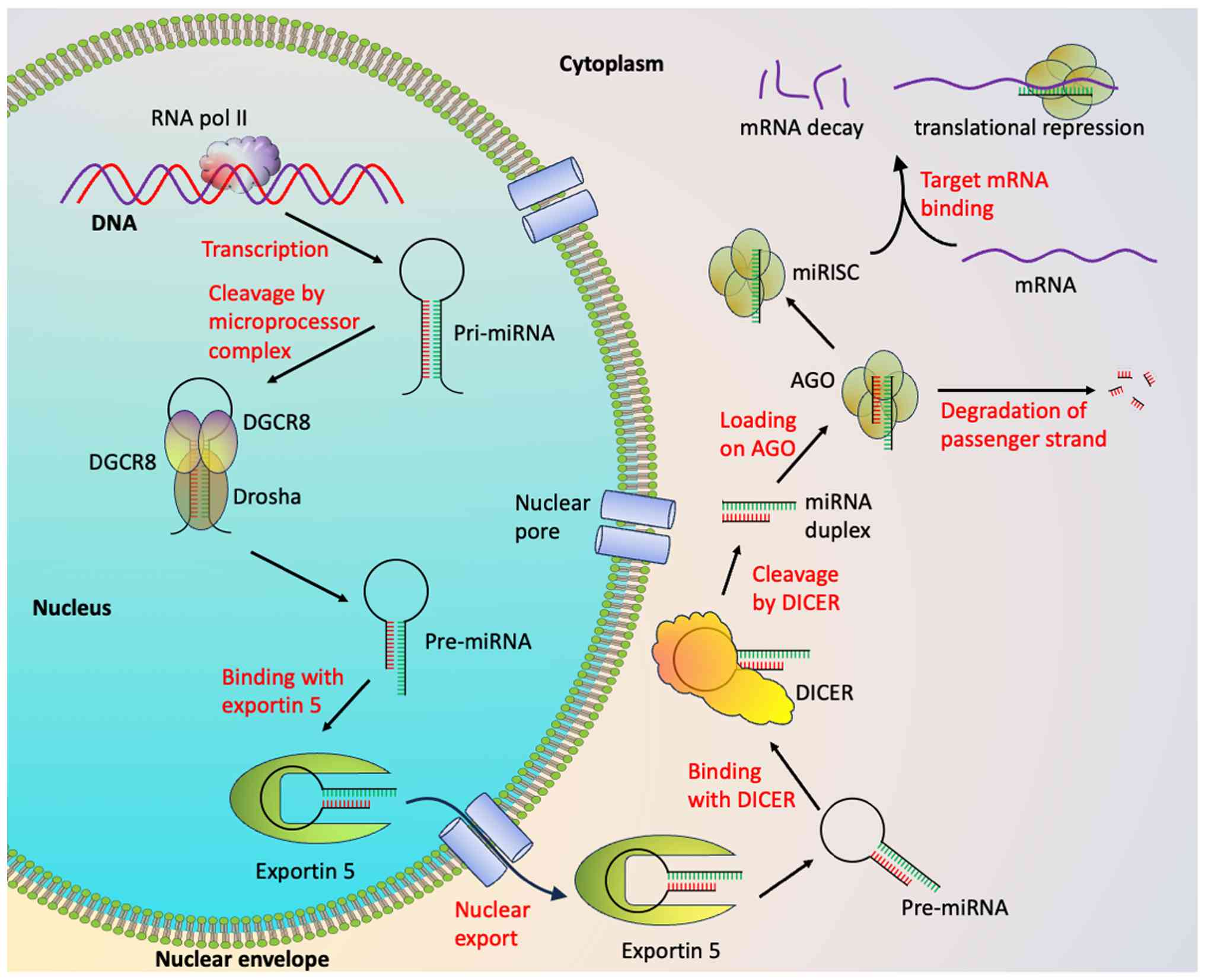
miRNAs are small, single stranded (20-25
nucleotides), non-coding type RNAs that constitute one of the
largest gene regulatory families, modulating gene expression via
mRNA degradation or translational repression (14,118,119). Of miRNAs, ~50% are transcribed
from non-coding genomic regions, while the rest are located within
introns of protein-coding genes. The biogenesis of miRNA begins
with transcription by RNA polymerase II, generating hairpin-shaped
primary transcripts (pri-miRNAs) (14) (Fig.
5). These are processed by the Drosha-DGCR8 complex into ~60-70
nucleotide precursor (pre-)miRNAs. The pre-miRNAs are exported out
of the nucleus by binding with Exportin-5 to the cytoplasm in a
GTP-dependent manner (120,121). In the cytoplasm, Dicer cleaves
pre-miRNAs into miRNA duplexes. One strand, known as the passenger
strand, is degraded, while the other strand known as guide strand,
is incorporated into the RNA-induced silencing complex (RISC)
(122) (Fig. 5).
Argonaute (Ago) proteins, especially Ago2, are core
RISC components, with Piwi-Argonaute-Zwille and P-element-Induced
Wimpy testis domains facilitating binding to mature miRNAs and
interaction with target mRNAs (123,124). miRNA binding typically occurs
via the seed sequence (nucleotides 2-8) within the 3' untranslated
region (3' UTR) of target mRNAs. Perfect complementarity usually
triggers mRNA cleavage, while partial pairing results in
translational repression, allowing a single miRNA to regulate
multiple targets and vice versa (125).
Therapeutic applications include the use of miRNA
mimics for replacement therapy and antisense oligonucleotides
(ASOs) to suppress overexpressed miRNAs. ASOs can target
pri-miRNAs, pre-miRNAs, or mature miRNAs, providing flexibility to
modulate miRNA activity in diseases (126,127). Studies also reveal alternative
mechanisms of miRNA action, highlighting the versatility and
complexity of miRNA-mediated gene regulation (128,129).
Role of miRNAs in pancreatic cancer
In PDAC, miRNAs can function as either tumor
suppressors or oncogenes and therapeutic strategies are accordingly
categorized into miRNA replacement therapy and miRNA inhibition
therapy (55). Profiling studies
have shown that dysregulation of specific miRNAs correlates with
poor prognosis, chemoresistance, invasion, metastasis and
epithelial-mesenchymal transition (EMT). Several miRNAs implicated
in PDAC development and progression include miR-21, miR-10b,
miR-218, miR-205, miR-200c and Let-7 (130,131).
miRNA replacement therapy restores downregulated
tumor-suppressive miRNAs, thereby reactivating normal cellular
programs and suppressing oncogenic processes. For instance, the
miR-200 family reduces cancer stemness by suppressing
sex-determining region Y-box 2, Smad interacting protein 1 and zinc
finger E-box binding homeobox 1, while the Let-7 family acts as a
tumor suppressor across multiple cancers, including pancreatic,
lung, ovarian and breast (132,133).
Conversely, oncogenic miRNAs such as miR-21,
miR-210, miR-155 and miR-221 are often upregulated in PDAC,
promoting tumor progression and correlating with reduced survival.
These can be targeted through miRNA inhibition therapy approaches,
using antisense oligonucleotides (AMOs) to degrade or block miRNA
function (134). miRNAs are
detectable via non-invasive samples including blood, saliva, urine
and feces (135). For example, a
study by Abue et al showed markedly elevated plasma levels
of miR-483-3p and miR-21 in PDAC patients compared to healthy
controls, highlighting their potential as diagnostic biomarkers
(136).
Interactions between miRNAs and KRAS in
PDAC
miRNAs serve as key regulators of KRAS expression.
Several miRNAs, including the members of Let-7 and miR-143/145
families, bind directly to the 3'-UTR of KRAS mRNA, resulting in
its degradation or translational repression. Loss or dysregulation
of these miRNAs leads to aberrant KRAS activation, promoting
uncontrolled proliferation, enhanced invasiveness and resistance to
apoptosis. Consequently, the miRNA-KRAS network serves as a key
regulatory axis in pancreatic tumor development and represents a
promising avenue for targeted therapies.
Regulation of signaling via miRNAs
The coordinated interplay of miRNAs can modulate
communication among multiple pathway components, thereby
influencing the key signaling networks such as the RAS-RAF-MAPK
cascade (42,137). Key pathway members, including
HRAS, KRAS and NRAS, are directly targeted by Let-7 (42) (Fig.
2). miRNAs also modulate essential regulatory proteins,
including RASA1, SPRED1, SPRY1 and the tumor suppressor PTEN. For
instance, miR-21 and miR-206 jointly target SPRED1, RASA1 and
SPRY1, whereas miR-21 alone regulates PTEN (138,139).
Beyond direct pathway components, miRNAs can also
regulate upstream activators and downstream effectors; miR-9-3p
targets the integrin subunit β1 (140) and miR-206 and miR-21 targets
tumor suppressor PDCD4 (141,142). Notably, a number of miRNAs
exhibit dynamic expression patterns during the progression of
pancreatic cancer, modulating signaling networks and contributing
to PDAC pathogenesis (143).
miRNAs targeting KRAS
The first evidence of miRNA-mediated regulation of
KRAS proteins was demonstrated by Johnson et al (144), who demonstrated that the Let-7
family directly targets KRAS mRNA in lung cancer. Subsequent
studies have identified multiple miRNAs that modulate RAS
expression across different malignancies. Given that oncogenic KRAS
is a principal driver and lineage-survival gene in PDAC,
understanding miRNA-mediated regulation of KRAS is of biological
and therapeutic relevance (145,146).
Although the mutational activation of KRAS is
nearly universal in PDAC, accumulating evidence indicates that
post-transcriptional regulation by miRNAs markedly influences KRAS
protein output and downstream signaling strength. Importantly,
these regulatory relationships can be either unidirectional, where
miRNAs suppress KRAS expression, or bidirectional, where KRAS
signaling feeds back to repress miRNA expression, forming
self-reinforcing oncogenic loops.
To date, miRNAs reported to directly target KRAS in
PDAC, include miR-126, miR-96, miR-145, miR-143, miR-193b, miR-217,
miR-206, miR-3923, Let-7a and Let-7b (Fig. 2; Table I) (147,148). Most of these miRNAs function as
tumor suppressors and their downregulation contributes to sustained
KRAS signaling, tumor progression and therapeutic resistance. Some
of these miRNAs include:
 | Table IDifferentially expressed miRNAs and
their target proteins in pancreatic cancer. |
Table I
Differentially expressed miRNAs and
their target proteins in pancreatic cancer.
| First author/s,
year | miRNAs | Roles in pancreatic
cancer |
Role/expression |
Context-dependency | Targets | (Refs.) |
|---|
| A, miRNAs directly
regulating KRAS |
|
| Dai et al,
2015 | Let-7 | Drug resistance,
proliferation | Tumor
suppressor/down regulated | Depends on KRAS
mutation burden and LIN28 expression; loss is particularly
oncogenic in KRAS-driven PDAC | KRAS, MAPK, IGF
family, c-Myc, STAT3, HMGA1, IGF2BP1/3, RRM2, N-cadherin/ZEB1, NF2,
LIN28 | (151) |
| Lavacchi et
al, 2023 | miR-143 | Proliferation,
invasion, metastasis | Tumor
suppressor/down regulated | Context-dependent
via stromal-epithelial interactions and KRAS signaling
activity | KRAS, COX2, GET1,
GET2, TAK1, RREB1 | (215) |
| Jin et al,
2015; Khalilian et al, 2026 | miR-193b | Inhibit cell growth
and malignant transformation | Tumor
suppressor/down regulated | Suppressive effects
depend on KRAS dependency and early tumorigenic stage | KRAS | (161,216) |
| Hara et al,
2014 | miR-126 | Proliferation | Tumor
suppressor/down regulated | Function varies
with angiogenic signaling and growth-factor-rich
microenvironment | KRAS, c-Myc, MAPK,
E2F2, STAT3, ADAM9 | (217) |
| Liu et al,
2021 | miR-96 | Tumor growth,
invasion | Tumor
suppressor/down regulated | Highly
context-dependent; tumor-suppressive in pancreatic cancer but
oncogenic in other tissues | KRAS, AKT | (218) |
| Singh et al,
2021 | miR-206 | Inhibits
angiogenesis, inflammation | Tumor
suppressor/down regulated | Depends on
inflammatory signaling and NF-κB activation status | KRAS, KRAS-induced
NF-κB | (219) |
| Zhao et al,
2010 | miR-217 | Decreases
proliferation and survival | Tumor
suppressor/down regulated | Context-dependent
on KRAS addiction and metabolic rewiring | KRAS, AKT,
ATAD2 | (164) |
| Li et al,
2016 | miR-3923 | Emerging
regulator | Tumor
suppressor | Function influenced
by lncRNA-miRNA-mRNA networks and ceRNA competition | KRAS via
lncRNA/miRNA network | (165) |
|
| B, miRNAs
regulating downstream or parallel components of the KRAS signaling
pathway |
|
| Hu et al,
2025 | miR-203 | EMT, cell cycle,
apoptosis | Tumor
suppressor/down regulated | Depends on EMT
status and epigenetic silencing in advanced tumors | Bmi-1, CAV1, CKAP2,
WASF1, ASAP1, SNAI1/2, RUNX2, LASP1, ZEB1/2, AKT2 | (220) |
| Guo et al,
2020 | miR-34a/b | DNA repair,
angiogenesis, apoptosis | Tumor
suppressor/down regulated | Strongly dependent
on TP53 mutational status | TP53, c-Myc,
Notch1/2/3, Snail1, E2F1/3, Bcl-2, CDK6, SIRT1, SMAD3, CCND1 | (221) |
| Jin et al,
2023 | miR-124 | Proliferation,
invasion, metastasis | Tumor
suppressor/down regulated | Context-dependent
via epigenetic regulation and invasive tumor phenotype | RAC1 | (222) |
| Zhang et al,
2021 | miR-146a | Invasion | Tumor
suppressor/down regulated | Function varies
with EGFR and inflammatory pathway activation | EGFR, IRAK1 | (223) |
| Diaz-Riascos et
al, 2019 | miR-200a | EMT | Tumor
suppressor/down regulated | Dependent on
epithelial-mesenchymal plasticity and tumor stage | ZEB1/2, Vimentin,
PTEN, MMPs, SOX2, E-cadherin | (224) |
| Chen et al,
2024 | miR-21 | Cell division,
proliferation |
Oncogene/upregulated | Oncogenic role
amplified by TGF-β signaling and fibrotic tumor
microenvironment | PTEN, EGFR, CDK6,
PDCD4, BCL2, TIMP2/3, SOCS5, FAS | (117) |
| Wang et al,
2015 | miR-155 | Apoptosis |
Oncogene/upregulated | Context-dependent
via immune-cell infiltration and inflammatory cytokines | SOCS1, SOCS3,
TP53INP1 | (225) |
| Ghafouri-Fard et
al, 2023 | miR-424 | Migration,
proliferation |
Oncogene/upregulated | Depends on
cell-cycle dependency and SOCS6 repression | SOCS6 | (226) |
| Yang et al,
2025 | miR-192 | Cell cycle,
proliferation |
Oncogene/upregulated | Context-dependent
via p53 signaling and DNA damage response | Cell cycle
regulators, SIP1 | (227) |
| Ouyang et
al, 2017 | miR-10 | Invasion,
metastasis |
Oncogene/upregulated | Dependent on HOX
gene dysregulation and metastatic potential | HOXB8, TFAP2C,
HOXA1, HOXA3 | (228) |
| Ji et al,
2025 | miR-208a | EMT |
Oncogene/upregulated | Context-dependent
through EMT signaling activation in late-stage tumors | CDH1 | (229) |
| Yang et al,
2016 | miR-375 | Apoptosis,
proliferation |
Oncogene/upregulated | Dual behavior
depending on metabolic state and cancer subtype | 14-3-3ζ, PDK1 | (230) |
| Ding et al,
2020 | miR-16-1/2 | Angiogenesis,
apoptosis |
Oncogene/upregulated | Oncogenic in PDAC
despite suppressive roles in other cancers | BCL2L1, MYBL2,
FGFR2, NAIP5 | (231) |
| Kanno et al,
2017 |
miR-196a-2/miR-196 | - |
Oncogene/upregulated | Depends on HOX gene
regulation and gastrointestinal lineage | HOXB8, HMGA2,
ANXA1 | (232) |
| Shopit et
al, 2020 | miR-421 | Colony formation,
proliferation |
Oncogene/upregulated | Oncogenic effects
enhanced in SMAD4-deficient tumors | SMAD4 | (233) |
| Di Martino et
al, 2022 | miR-221/222 | Cell cycle
progression |
Oncogene/upregulated | Context-dependent
via PTEN loss and checkpoint dysregulation | PTEN, MMP-2/9,
CDKN1B, PUMA, BIM | (234) |
| Pancratov et
al, 2013 | miR-310a | Proliferation,
metastasis |
Oncogene/upregulated | Depends on
apoptotic threshold and BIM repression | NKRF, BIM | (235) |
| Ghafouri-Fard et
al, 2022 | miR-15a/b | Angiogenesis,
apoptosis |
Oncogene/upregulated | Oncogenic in
pancreatic cancer despite tumor-suppressive roles elsewhere | BCL2L1, MYBL2,
FGFR2, WNT3A, BMI-1 | (236) |
| Liu et al,
2020 | miR-210 | Tumor-stroma
interaction |
Oncogene/upregulated | Strongly dependent
on hypoxia and stromal remodeling | HOXA9, RAD52, E2F3,
ACVR1B | (237) |
Let-7
The human Let-7 miRNA family comprises at least 13
members, including Let-7a and Let-7b. These miRNAs are
well-established regulators of cell proliferation, differentiation
and apoptosis through the direct targeting of key oncogenes, MYC,
RAS and high mobility group AT-hook 2 (149). Let-7 is widely regarded as a
canonical tumor-suppressive miRNA family and its downregulation is
frequently observed across multiple cancer types, including PDAC
(150).
The interaction between Let-7 and KRAS in
pancreatic cancer is predominantly unidirectional, with Let-7
directly binding to complementary sites within the 3'-UTR of KRAS
mRNA and suppressing its translation. This interaction has been
validated by luciferase reporter assays and KRAS protein analyses
in PDAC cell models (151).
However, emerging evidence also supports an indirect feedback
component, whereby oncogenic KRAS signaling suppresses Let-7
biogenesis through upregulation of LIN28A/B, known inhibitors of
Let-7 maturation. This KRAS-LIN28-Let-7 axis, described in PDAC and
other KRAS-driven cancers, establishes a feed-forward oncogenic
loop that reinforces KRAS expression at the post-transcriptional
level (48,152).
Functionally, restoration of Let-7 expression in
vitro reduces KRAS protein levels, inhibits PDAC cell
proliferation and suppresses tumorigenicity. Nevertheless, in
vivo studies have produced variable results, underscoring the
importance of delivery efficiency and tumor microenvironmental
context (153). Collectively,
Let-7 functions as both a direct KRAS suppressor and a node within
a broader regulatory loop sustaining KRAS signaling.
miR-96
miR-96 belongs to the miR-183-96-182 cluster, whose
members display context-dependent oncogenic or tumor-suppressive
functions (154). While miR-96
is upregulated in several epithelial cancers, its expression is
notably reduced in PDAC (155).
Current evidence supports a strictly unidirectional regulatory
relationship between miR-96 and KRAS in PDAC. miR-96 directly binds
to the KRAS 3'-UTR, leading to translational repression and
attenuation of KRAS-driven MAPK and PI3K signaling pathways
(156). Functional studies have
not demonstrated reciprocal regulation of miR-96 by KRAS signaling,
indicating that miR-96 acts primarily as an upstream suppressor
rather than part of a feedback loop. Restoration of miR-96
expression inhibits PDAC cell growth, migration and tumor
progression, highlighting its role as a tumor-suppressive regulator
of KRAS output (155,156).
miR-126
miR-126 is encoded within intron 7 of the EGFL7
gene on chromosome 9 and plays key roles in angiogenesis,
inflammation and tumor biology (157). In PDAC, miR-126 acts as a
unidirectional suppressor of KRAS, directly targeting its 3'-UTR
and reducing KRAS protein expression (158). Loss of miR-126 enhances
downstream KRAS signaling, promoting tumor growth and invasion
(158).
While KRAS-mediated repression of miR-126 has been
observed indirectly via epigenetic silencing mechanisms in other
cancers, direct evidence of a feedback loop in pancreatic cancer
remains limited (157). Thus,
current data support miR-126 as a primarily upstream regulator of
KRAS rather than a reciprocal feedback component.
miR-143/145
The miR-143/145 cluster is one of the
best-characterized miRNA systems involved in KRAS regulation. Its
tumor-suppressive role was first identified in colorectal cancer,
where miR-143 was shown to inhibit KRAS translation via binding to
its 3'-UTR (159). In pancreatic
cancer, the miR-143/145 cluster forms a well-defined negative
feedback loop with KRAS. Kent et al (47) demonstrated that oncogenic KRAS
represses miR-143/145 transcription through activation of RREB1,
which binds the miR-143/145 promoter. In turn, both KRAS and RREB1
are direct targets of miR-143/145, creating a self-reinforcing
feed-forward loop that sustains oncogenic KRAS signaling when
miR-143/145 is lost (47). Loss
of this cluster enhances RAS-GTP loading and downstream pathway
activation, including MAPK, PI3K and JNK signaling, highlighting
miR-143/145 as a central node in KRAS-driven regulatory circuitry
rather than a simple unidirectional regulator (47,160).
miR-193b
miR-193b is frequently downregulated in multiple
types of cancer and functions as a tumor-suppressive miRNA in PDAC.
Evidence indicates a unidirectional regulatory relationship, where
miR-193b directly targets the KRAS 3'-UTR, resulting in reduced
KRAS expression and suppression of both AKT and ERK signaling
pathways (161). Functional
restoration of miR-193b inhibits PDAC cell proliferation and
invasion (161). Although
KRAS-dependent transcriptional repression of miR-193b has been
suggested in other tumor contexts, direct confirmation of a
feedback loop in pancreatic cancer remains lacking. Current data
support miR-193b primarily as an upstream regulator of KRAS rather
than a reciprocal component of a feedback loop.
miR-206
miR-206 is markedly downregulated in PDAC, as
demonstrated by gene expression omnibus-based expression profiling
and validation studies (162).
miR-206 directly targets KRAS mRNA, leading to inhibition of the
KRAS-NF-κB signaling axis. This interaction has been validated
across multiple in vitro and in vivo PDAC models
(162). Reduced KRAS-NF-κB
activity subsequently diminishes inflammatory and pro-angiogenic
gene expression, including IL-8, CXCL1, CXCL2, CCL2, GM-CSF and
VEGF-C (162). While KRAS-driven
inflammatory signaling may indirectly influence miR-206 expression,
a defined reciprocal feedback loop has not yet been conclusively
demonstrated in PDAC.
miR-216/217
The miR-216/217 cluster is highly expressed in
normal pancreatic tissue and markedly reduced in PDAC (163). This cluster exhibits features of
a bidirectional regulatory relationship. miR-217 directly targets
KRAS, suppressing tumor growth in PDAC models (164). Conversely, KRAS activation
suppresses miR-216/217 expression, as demonstrated in KRAS-mutant
mouse models, suggesting a feed-forward oncogenic loop in which
KRAS signaling reinforces its own expression by repressing
inhibitory miRNAs (163). This
reciprocal regulation positions the miR-216/217 cluster as an
important brake on KRAS signaling that is actively dismantled
during pancreatic tumorigenesis.
miR-3923
miR-3923 has been implicated in PDAC with
context-dependent roles. Although some data indicate that miR-3923
can target the 3'-UTR of KRAS mRNA, potentially reducing KRAS
expression, the predominant behavior of miR-3923 in PDAC appears
oncogenic. Li et al (165) demonstrated that hypoxia-induced
lncRNA NUTF2P3-001 acts as a competitive endogenous RNA,
derepressing KRAS by sponging miR-3923, which suggested that
miR-3923 can directly bind KRAS 3'-UTR but fails to suppress its
expression when sequestered.
Studies have consistently reported aberrant
overexpression of miR-3923 in PDAC tissues and patient serum,
associating elevated levels with increased tumor cell
proliferation, invasion and migration. These oncogenic effects are
considered to result from suppression of tumor suppressor genes and
activation of pathways related to EMT and metastasis (165,166).
Consequently, miR-3923 is emerging as a potential
diagnostic biomarker and prognostic indicator in PDAC (165). The regulation is unidirectional
as miR-3923 acts upstream, capable of targeting KRAS mRNA but
unable to initiate any feedback control over its own expression via
KRAS signaling. However, Further research is necessary to delineate
its full molecular targets and determine whether a dual regulatory
potential exists under differing cellular contexts.
miRNAs for pancreatic cancer therapy
miRNAs represent a promising class of therapeutic
agents in pancreatic cancer, owing to their capacity to
simultaneously modulate several tumor-suppressive and oncogenic
pathways. Therapeutic strategies focus on either restoring the
expression of tumor-suppressive miRNAs or inhibiting oncogenic
miRNAs, using synthetic mimics or antagomirs, respectively. These
interventions target critical molecular regulators such as KRAS,
TP53 and signaling pathways that govern cell proliferation,
apoptosis and metastasis. Although some challenges exist regarding
efficient stability, delivery and off-target effects, miRNA-based
therapies are being actively investigated in preclinical models and
early-phase clinical trials, offering a novel, personalized
approach to improving outcomes in pancreatic cancer treatment.
miRNAs as therapeutic targets for
PDAC
RAS proteins are central drivers of multiple
oncogenic signaling pathways across various cancers, making them
prime targets for therapeutic intervention (167,168). Emerging evidence indicates that
modulating KRAS-directing miRNAs can effectively suppress cancer
growth (147). For example,
miR-34 expression inhibits migration and cell proliferation in lung
cancer and markedly reduces cancer growth by targeting KRAS
(169). Similarly,
downregulation of miR-31, which directly affects RASA1, a RAS-MAPK
pathway regulator, decreases cell proliferation and tumor size in
colorectal cancer models (170).
In squamous cell cancer, miR-181a has been shown to directly target
KRAS and suppress its proliferation (171), while miR-451 overexpression
suppresses tumor growth in NSCLC by targeting RAB14 (172).
Considering the significant role of KRAS signaling
in PDAC, miRNA-based strategies targeting KRAS represent an
underexplored therapeutic opportunity. Although limited, studies in
PDAC have demonstrated promising results. For instance,
nanoparticle-mediated delivery of miR-34a, miR-143 and miR-145 in
animal models potentially inhibited the cancerous growth (173). Antisense oligonucleotides
targeting overexpressed oncogenic miRNAs such as miR-21, miR-221,
miR-132 and miR-212 enhanced gemcitabine efficacy and inhibited
tumor growth by modulating tumor suppressors like Rb1 (174,175). Suppression of miR-10a reduces
pancreatic tumor growth and metastasis.
Alternative delivery strategies, including viral
vectors and adenovirus-mediated expression, have been employed to
deliver miR-143, miR-145 and miR-150, effectively suppressing
pancreatic tumor development and metastasis (47). However, restoration of Let-7, a
well-characterized anticancer miRNA, reduces proliferation in PDAC
cells by downregulating KRAS and inhibiting MAPK signaling
(153).
P21-activated kinase 4 inhibition, together with
Nicotinamide phosphoribosyl transferase modulators, suppresses
proliferation in pancreatic cancer cells. This effect is mediated
by reduced phosphorylation and enhanced expression of
tumor-suppressive miRNAs including Let-7c/d, miR-145, miR-34c,
miR-320 and miR-100 (176).
These findings suggest that therapeutic strategies aimed at
enhancing cancer-suppressive miRNAs targeting the KRAS cascade
could offer an encouraging approach to overcome treatment-resistant
pancreatic cancer.
Strategies to target miRNAs in PDAC
Despite extensive efforts, direct targeting of KRAS
protein or its signaling pathway in pancreatic cancer has achieved
limited success. However, modulating miRNAs that regulate RAS and
other oncogenic pathways offers a promising alternative. Although
no clinical trials currently focus on KRAS-targeting miRNAs, the
rapidly expanding field of miRNA research suggests that innovative
therapeutic applications are likely to emerge in future. The
identification of dysregulated miRNAs as novel contributors in PDAC
highlights the requirement for effective tools to manipulate these
molecules.
One prominent strategy involves the use of AMOs to
inhibit oncogenic miRNAs. These molecules bind specifically to
target miRNAs, inducing miRNA silencing (177). For instance, anti-miRNA strategy
has demonstrated the ability to specifically block mutant
KRASG12D, while sparing wild-type KRAS, thereby
selectively downregulating oncogenic signaling (178).
Another approach, miRNA replacement therapy,
restores the expression of anticancer miRNAs that are downregulated
in PDAC. Synthetic miRNA mimics supplement endogenous miRNA levels
and are effective because of their smaller size, high target
affinity and minimal immunogenicity (179). For example, miR-489, an
anticancer nucleic acid, is downregulated by KRAS signaling in
pancreatic cancer, which can be restored using miRNA mimetics to
potentially inhibit metastasis (180).
Despite their therapeutic potential, miRNA-based
treatments face several challenges, including limited cell
specificity, minimal in vivo stability, uneven
biodistribution, interference with endogenous RNA system and
substantial off-target effects. All these parameters complicate the
efficient delivery and function of miRNA-based approaches (181). To overcome these limitations,
chemical modifications of oligonucleotides are engineered to
enhance their stability and binding affinity. One successful
example is Locked Nucleic Acid (LNA) technology, in which nucleic
acid analogs contain LNA nucleotide monomers possessing a furanose
unit fastened in an RNA-like conformation. This configurational
constraint enhances hybridization to complementary RNA targets and
has demonstrated higher efficacy (181).
miRNAs as PDAC biomarkers
Early detection of PDAC is critical, as surgical
intervention remains the only option for cure, yet is practicable
in only 15-20% of patients diagnosed at early stages (182,183). In addition, postoperative
complications are common and conditions such as pancreatic
tuberculosis or chronic pancreatitis are often difficult to
distinguish from malignancy (184). Currently, only carbohydrate
antigen 19-9 (CA 19-9) is FDA-approved biomarker for monitoring
treatment response in PDAC (185), but it suffers from low
sensitivity and specificity. Other markers, including CEA and
CA125, have limited utility for early detection but may still aid
in monitoring therapy (186).
In this situation, miRNAs have played a novel role
as biomarkers for early PDAC detection due to their stability in
serum, non-invasive accessibility and ease of measurement (187). For example, Lee et al
(188) profiled over 200 miRNA
precursors across normal pancreas, pancreatic cancer tissues,
paired benign tissues, pancreatitis tissues and related cell lines,
enabling detection of premalignant changes during the transition
from benign to malignant states. Key upregulated cancer-related
miRNAs included miR-100, miR-424, miR-212, miR-301 and miR-125b-1,
while miR-142-P, miR-345 and miR-139 were downregulated as compared
to normal pancreas (189).
Several miRNAs are also associated with precursor
lesions and histological progression. Notably, miR-21 and miR-155
are expressed more in pancreatic PanIN and show specificity in
pancreatic juice samples (190).
Other studies recognized miR-21, miR-196a, miR-27a, miR-146a and
miR-200a as most upregulated miRNAs via FNA, while miR-96, miR-217,
miR-141, miR-20a and miR-29c were among the most downregulated
(184). Additional upregulated
candidates confirmed by reverse transcription-quantitative PCR
include miR-15b, miR-186, miR-190, miR-196a, miR-200b, miR-221,
miR-222 and miR-95 (191), as
well as miR-21, miR-26b, miR-194, miR-200b/c, miR-320, miR-374 and
miR-429 in pancreatic cancer cell lines compared to normal
pancreatic cells (Table I).
Early KRAS mutations, frequently identified in
PanIN lesions, can alter miRNA expression (192). Using a KRASG12D mouse
model, upregulation of miR-21, miR-205 and miR-200, was identified
in early adenocarcinoma lesions and miR-21 levels associated with
the level of morphological changes in PanIN samples (193). These findings indicate that
miRNA profiling may have strong potential for early diagnosis, risk
stratification and monitoring disease progression in pancreatic
cancer.
miRNAs as PDAC prognostic factors
Beyond their roles in diagnosis and therapy, miRNAs
have emerged as important prognostic indicators in PDAC.
Dysregulated expression of specific miRNAs correlates with tumor
aggressiveness, metastatic potential and overall patient survival.
For example, enhanced levels of miR-155, miR-21 and miR-196a are
consistently designated with low prognosis, reflecting their
involvement in promoting proliferation, invasion and resistance to
apoptosis (194). Conversely,
minimal expression of tumor-suppressive miRNAs such as Let-7,
miR-34a, miR-217 and miR-96 often predicts more aggressive disease
and worse clinical outcomes, likely due to their role in
restraining oncogenic pathways such as KRAS and RAS-MAPK signaling
(195).
In addition to single miRNAs, miRNA expression
signatures or panels are proposed as more robust prognostic tools.
These signatures integrate multiple upregulated oncogenic miRNAs
and downregulated tumor suppressors for improved stratification of
patients according to risk and likely response to therapy. The
prognostic relevance of miRNAs is further supported by their
detectability in serum, plasma, or pancreatic juice, enabling
non-invasive monitoring of disease progression and response to
treatment. Overall, miRNAs serve as both molecular readouts of
tumor biology and predictive biomarkers, providing valuable insight
into patient outcomes and helping guide personalized therapeutic
strategies in PDAC (196,197).
miRNAs may also serve as valuable predictors of
chemoresistance and therapeutic responsiveness in pancreatic
cancer. For instance, miR-320c has been identified as a prognostic
marker for predicting clinical response to gemcitabine (198). Similarly, patients exhibiting
higher post-surgical levels of miR-200c demonstrated improved
survival compared to those with lower expression. This clinical
observation aligns with in vitro findings, where
upregulation of miR-200c reduced cellular invasiveness, suggesting
a functional role in modulating tumor aggressiveness (199).
Clinical trials involving KRAS and miRNAs in
pancreatic cancer
Multiple preclinical studies have demonstrated that
specific miRNAs can directly suppress KRAS expression or modulate
its downstream signaling pathways, resulting in reduced pancreatic
cancer cell proliferation, invasion and therapeutic resistance
(200). Conversely, oncogenic
KRAS signaling has been shown to reprogram the miRNA regulatory
landscape by altering miRNA transcription, processing and
maturation, such as through DROSHA, DICER and AGO2 modulation,
thereby reinforcing tumor-promoting networks in PDAC (46). These findings establish a strong
biological basis for targeting the KRAS-miRNA axis in pancreatic
cancer.
Despite this compelling preclinical rationale,
translation of miRNA-based modulation of KRAS signaling into
clinical practice remains limited and no registered clinical trial
has yet combined KRAS-targeted agents with miRNA-based therapeutics
in PDAC as of 2025. Current clinical efforts primarily focus on
either KRAS inhibition or RNA interference strategies. For example,
a phase I clinical trial (NCT03608631; iExoKrasG12D) is evaluating
mesenchymal stromal cell-derived exosomes loaded with
KRASG12D siRNA in patients with metastatic
KRASG12D-mutant pancreatic cancer (201). Recent reports indicate that this
approach is well tolerated, with no dose-limiting toxicities
observed, alongside evidence of KRASG12D DNA
downregulation, reduced phospho-ERK signaling and increased
intratumoral CD8+ T-cell infiltration in a subset of
patients (201,202).
In parallel, small-molecule KRAS inhibitors have
entered early-phase clinical testing. A phase I/Ib study of the
KRASG12D-selective inhibitor zoldonrasib (RMC-9805;
NCT06040541) is currently recruiting patients with advanced solid
tumors, including PDAC (203).
Initial clinical data reported in 2025 demonstrate promising
antitumor activity, including reductions in circulating
KRASG12D mutant ctDNA in treated PDAC patients (204).
In preclinical models, MRTX1133 has shown potent
inhibition of KRAS signaling, reduced cell viability and caused
tumor regressions in xenograft and immunocompetent tumor models,
especially in pancreatic ductal adenocarcinoma (205). Additionally, the pan-RAS
inhibitor daraxonrasib (RMC-6236; NCT05379985) is undergoing phase
I/II evaluation in pancreatic and other gastrointestinal cancers,
with phase III studies active in previously treated metastatic PDAC
(206).
ASP3082 is a KRAS G12D-selective degrader,
proteolysis targeting chimera (PROTAC), designed to induce targeted
degradation of the oncogenic KRAS G12D protein rather than just
inhibiting it. It has entered Phase I clinical trials for advanced
solid tumors harboring the KRAS G12D mutation, including studies
both as monotherapy and in combination regimens (207). BI-1823911 is a small-molecule
KRAS inhibitor developed by Boehringer Ingelheim that has also
entered a Phase I clinical trial (NCT04973163) to assess safety,
dosing and preliminary efficacy in patients with advanced cancers
that harbor KRAS mutations. While BI-1823911 was initially
characterized as targeting KRAS G12C, its clinical study enrollment
includes patients with diverse KRAS mutations and it is being
evaluated both alone and in combination with other agents.
Notably, none of these clinical strategies
integrate miRNA-based therapeutics with KRAS-targeted agents,
despite strong preclinical evidence suggesting potential synergy.
Several miRNAs, such as miR-34a, miR-143/145, Let-7 family members
and miR-217 have been shown to concurrently suppress KRAS
expression, attenuate MAPK and PI3K signaling and overcome adaptive
resistance mechanisms induced by KRAS inhibition (42,200). From a mechanistic standpoint,
combining KRAS inhibitors with tumor-suppressive miRNAs could i)
enhance pathway suppression at multiple regulatory levels, ii)
prevent or delay compensatory feedback activation and iii) target
parallel oncogenic programs driven by KRAS, including metabolic
reprogramming and immune evasion (46,200).
However, several scientific and clinical challenges
likely explain the current absence of such combination trials.
miRNAs exhibit pleiotropic effects and regulate hundreds of
targets, raising concerns regarding off-target toxicity and
unpredictable pharmacodynamic interactions when combined with KRAS
inhibitors (208). Furthermore,
KRAS-mutant PDAC is known to disrupt miRNA biogenesis machinery,
including DROSHA, DICER and AGO2, which may compromise the efficacy
of exogenously delivered miRNAs (46). From a clinical perspective,
efficient and tumor-selective delivery of miRNA therapeutics to the
dense and desmoplastic PDAC microenvironment remains a major
barrier, particularly when combined with systemic targeted agents
(208,209).
In summary, while early-phase clinical trials
targeting KRAS through siRNA or small-molecule inhibitors represent
significant progress, the lack of clinical studies combining
KRAS-directed therapies with miRNA-based approaches reflects
unresolved biological, pharmacological and delivery-related
challenges rather than a lack of scientific rationale. Future
advances in RNA delivery platforms, improved miRNA target
specificity and rational biomarker-driven patient selection may
enable the successful clinical translation of KRAS-miRNA
combination strategies in pancreatic cancer.
Future perspectives
Although compelling evidence supports
miRNA-mediated regulation of KRAS signaling in pancreatic cancer,
the translation of these findings into effective therapies has been
markedly limited, underscoring several unresolved biological and
technical challenges. While numerous miRNAs, including Let-7,
miR-143/145 and miR-217, exhibit tumor-suppressive activity through
direct or indirect targeting of KRAS, the complexity of KRAS-driven
regulatory networks and the pancreatic tumor microenvironment have
hindered therapeutic progress.
One major challenge lies in the delivery efficiency
of miRNA-based therapeutics. PDAC is characterized by an extensive
desmoplastic stroma, hypovascularity and elevated interstitial
pressure, all of which markedly impair the penetration and
distribution of nucleic acid-based drugs. Even advanced delivery
platforms, including lipid nanoparticles, polymeric carriers and
viral vectors, show limited intratumoral accumulation in PDAC
models. Studies emphasize that stromal targeting or co-delivery
strategies may be required to overcome these physical barriers, yet
such approaches increase formulation complexity and regulatory
burden (210,211).
Another critical limitation is the pleiotropic
nature of miRNAs. Unlike small-molecule inhibitors or
allele-specific KRAS inhibitors, individual miRNAs can regulate
dozens to hundreds of targets. While this multi-target capacity may
be advantageous for suppressing oncogenic networks, it also raises
concerns regarding off-target effects, unintended pathway
activation and systemic toxicity. The termination of the MRX34
clinical trial, despite its success in preclinical models,
highlighted immune-related adverse events as a significant obstacle
for systemic miRNA delivery (212). These findings underscore the
need for more precise miRNA engineering, tissue-specific delivery
and improved immunocompatibility.
Importantly, context-dependent and occasionally
contradictory roles of miRNAs further complicate therapeutic
development. Several miRNAs reported as tumor suppressors in PDAC
may exhibit oncogenic or neutral effects in other cellular contexts
or disease stages. Additionally, KRAS-driven feedback mechanisms,
such as repression of miR-143/145 suggest that restoring a single
miRNA may be insufficient to durably suppress KRAS signaling due to
compensatory network rewiring (47). Recent systems-level analyses
indicate that combinatorial miRNA strategies or integration with
KRAS pathway inhibitors may be required to achieve sustained
therapeutic benefit (110).
Despite these challenges, emerging evidence
suggests potential avenues for progress. For example,
overexpression of KRAS-targeting miRNAs such as Let-7 has been
shown to enhance radiosensitivity and chemosensitivity in PDAC
models, supporting the concept of miRNA-based adjuvant therapies
rather than standalone treatments (213). Advances in RNA chemistry,
including backbone modifications and ligand-directed nanoparticles,
have improved miRNA stability and tumor targeting in preclinical
studies (214). Furthermore, the
growing clinical success of RNA-based therapeutics, such as
patisiran (Onpattro™), demonstrates the feasibility of overcoming
delivery and safety barriers when appropriate platforms are
employed.
In the future, large-scale clinical validation,
standardized miRNA detection methodologies (including liquid biopsy
approaches) and rigorous patient stratification will be essential
for successful translation. Given the pronounced heterogeneity of
pancreatic cancer, miRNA expression profiling may enable
personalized therapeutic strategies, identifying patients most
likely to benefit from miRNA modulation in combination with
chemotherapy, radiotherapy, or emerging KRAS inhibitors.
Ultimately, integrating miRNA-based approaches into multimodal
treatment frameworks, while acknowledging and addressing their
inherent limitations, will be critical for realizing their
potential in pancreatic cancer management.
Conclusion
KRAS mutation remains a central oncogenic driver in
pancreatic cancer, orchestrating tumor initiation, progression and
therapeutic resistance. The present review underscored a critical
insight: The miRNA-KRAS negative feedback loop is a defining
feature of PDAC biology. Under normal conditions, KRAS and its
regulatory miRNAs maintain a tightly controlled equilibrium;
however, in the cancerous state, activated KRAS disrupts this
balance by downregulating tumor-suppressive miRNAs such as miR-126,
miR-96, miR-145, miR-143, miR-193b, miR-206, miR-217 and the Let-7
family, thereby fueling uncontrolled signaling and disease
progression. Recognizing this feedback loop not only deepens our
mechanistic understanding of PDAC but also opens new avenues for
precision medicine. Therapeutic strategies aimed at restoring miRNA
expression or targeting KRAS-driven pathways could overcome
resistance and improve patient outcomes. Furthermore, integrating
miRNA profiling into clinical workflows, alongside imaging and
liquid biopsy, may enable early detection, dynamic monitoring and
personalized treatment planning. Future research should prioritize
unraveling the context-specific regulation of this axis, optimizing
miRNA delivery systems and validating these approaches in
large-scale clinical trials to translate these insights into
tangible benefits for PDAC patients.
Availability of data and materials
Not applicable.
Authors' contributions
AAA and AAK conceived the study. AAA, AMA and AAK
wrote the original draft. AAA, AMA, MA, AHR and AAK reviewed and
edited the manuscript. Data authentication is not applicable. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The researchers would like to thank the Deanship of
Graduate Studies and Scientific Research at Qassim University for
financial support (QU-APC-2026).
Funding
The present study was supported by the Deanship of Graduate
Studies and Scientific Research at Qassim University
(QU-APC-2026).
References
|
1
|
Bouvier AM, Uhry Z, Jooste V, Drouillard
A, Remontet L, Launoy G and Leone N; French Network of Cancer
Registries (FRANCIM): Focus on an unusual rise in pancreatic cancer
incidence in France. Int J Epidemiol. 46:1764–1772. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Caban M and Małecka-Wojciesko E: Gaps and
opportunities in the diagnosis and treatment of pancreatic cancer.
Cancers (Basel). 15:55772023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Amundadottir LT: Pancreatic cancer
genetics. Int J Biol Sci. 12:314–325. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bengtsson A, Andersson R and Ansari D: The
actual 5-year survivors of pancreatic ductal adenocarcinoma based
on real-world data. Sci Rep. 10:164252020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Strobel O, Lorenz P, Hinz U, Gaida M,
König AK, Hank T, Niesen W, Kaiser JÖR, Al-Saeedi M, Bergmann F, et
al: Actual five-year survival after upfront resection for
pancreatic ductal adenocarcinoma. Ann Surg. 275:962–971. 2022.
View Article : Google Scholar
|
|
6
|
Neoptolemos JP, Kleeff J, Michl P,
Costello E, Greenhalf W and Palmer DH: Therapeutic developments in
pancreatic cancer: Current and future perspectives. Nat Rev
Gastroenterol Hepatol. 15:333–348. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Conroy T, Castan F, Lopez A, Turpin A, Ben
Abdelghani M, Wei AC, Mitry E, Biagi JJ, Evesque L, Artru P, et al:
Five-year outcomes of FOLFIRINOX vs gemcitabine as adjuvant therapy
for pancreatic cancer. JAMA Oncol. 8:1571–1578. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haigis KM: KRAS alleles: The devil is in
the detail. Trends Cancer. 3:686–697. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo J: KRAS mutation in pancreatic cancer.
Semin Oncol. 48:10–18. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takeda M, Yoshida S, Inoue T, Sekido Y,
Hata T, Hamabe A, Ogino T, Miyoshi N, Uemura M, Yamamoto H, et al:
The role of KRAS mutations in colorectal cancer: Biological
insights, clinical implications, and future therapeutic
perspectives. Cancers (Basel). 17:4282025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ghosh S, Bhuniya T, Dey A, Koley M, Roy P,
Bera A, Gol D, Chowdhury A, Chowdhury R and Sen S: An updated
review on KRAS mutation in lung cancer (NSCLC) and its effects on
human health. Appl Biochem Biotechnol. 196:4661–4678. 2024.
View Article : Google Scholar
|
|
12
|
Tamura T, Ashida R, Wan K, Shimokawa T and
Kitano M: K-ras gene mutation analysis to diagnosis pancreatic
adenocarcinoma from endoscopic ultrasound-guided tissue
acquisition; a systematic review and meta-analysis. Pancreatology.
24:78–87. 2024. View Article : Google Scholar
|
|
13
|
Puleo F, Nicolle R, Blum Y, Cros J, Marisa
L, Demetter P, Quertinmont E, Svrcek M, Elarouci N, Iovanna J, et
al: Stratification of pancreatic ductal adenocarcinomas based on
tumor and microenvironment features. Gastroenterology.
155:1999–2013.e3. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aloliqi AA, Alnuqaydan AM, Albutti A,
Alharbi BF, Rahmani AH and Khan AA: Current updates regarding
biogenesis, functions and dysregulation of microRNAs in cancer:
Innovative approaches for detection using CRISPR/Cas13-based
platforms (Review). Int J Mol Med. 55:902025. View Article : Google Scholar
|
|
15
|
Rana S, Valbuena GN, Curry E, Bevan CL and
Keun HC: MicroRNAs as biomarkers for prostate cancer prognosis: A
systematic review and a systematic reanalysis of public data. Br J
Cancer. 126:502–513. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rawat M, Kadian K, Gupta Y, Kumar A, Chain
PSG, Kovbasnjuk O, Kumar S and Parasher G: MicroRNA in pancreatic
cancer: from biology to therapeutic potential. Genes (Basel).
10:7522019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ibrahim H and Lim YC: KRAS-associated
microRNAs in colorectal cancer. Oncol Rev. 14:4542020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roncarati R, Lupini L, Shankaraiah RC and
Negrini M: The importance of microRNAs in RAS oncogenic activation
in human cancer. Front Oncol. 9:9882019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cecchini M, Salem RR, Robert M, Czerniak
S, Blaha O, Zelterman D, Rajaei M, Townsend JP, Cai G, Chowdhury S,
et al: Perioperative modified FOLFIRINOX for resectable pancreatic
cancer. JAMA Oncol. 10:1027–1035. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ducreux M, Cuhna AS, Caramella C,
Hollebecque A, Burtin P, Goéré D, Seufferlein T, Haustermans K, Van
Laethem JL, Conroy T, et al: Cancer of the pancreas: ESMO clinical
practice guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 26(Suppl 5): v56–v68. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Keane F, O'Connor C, Moss D, Chou JF,
Perry MA, Crowley F, Saxena P, Chan A, Schoenfeld JD, Singhal A, et
al: Adjuvant modified FOLFIRINOX for resected pancreatic
adenocarcinoma: Clinical insights and genomic features from a large
contemporary cohort. J Natl Cancer Inst. 117:496–506. 2025.
View Article : Google Scholar :
|
|
22
|
Stanciu S, Ionita-Radu F, Stefani C,
Miricescu D, Stanescu-Spinu II, Greabu M, Ripszky Totan A and Jinga
M: Targeting PI3K/AKT/mTOR signaling pathway in pancreatic cancer:
From molecular to clinical aspects. Int J Mol Sci. 23:101322022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ansari D, Ohlsson H, Althini C, Bauden M,
Zhou Q, Hu D and Andersson R: The Hippo Signaling Pathway in
Pancreatic Cancer. Anticancer Res. 39:3317–3321. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao J, Long B and Wang Z: Role of Notch
signaling pathway in pancreatic cancer. Am J Cancer Res. 7:173–186.
2017.PubMed/NCBI
|
|
25
|
Mustafa M, Abbas K, Alam M, Habib S,
Zulfareen, Hasan GM, Islam S, Shamsi A and Hassan I: Investigating
underlying molecular mechanisms, signaling pathways, emerging
therapeutic approaches in pancreatic cancer. Front Oncol.
14:14278022024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pantsar T: The current understanding of
KRAS protein structure and dynamics. Comput Struct Biotechnol J.
18:189–198. 2019. View Article : Google Scholar
|
|
27
|
Drosten M and Barbacid M: Targeting the
MAPK Pathway in KRAS-Driven Tumors. Cancer Cell. 37:543–550. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang Y, Liu JL and Wang J: KRAS gene
silencing inhibits the activation of PI3K-Akt-mTOR signaling
pathway to regulate breast cancer cell epithelial-mesenchymal
transition, proliferation and apoptosis. Eur Rev Med Pharmacol Sci.
24:3085–3096. 2020.PubMed/NCBI
|
|
29
|
Kim HJ, Lee HN, Jeong MS and Jang SB:
Oncogenic KRAS: Signaling and drug resistance. Cancers (Basel).
13:55992021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee C, Yi J, Park J, Ahn B, Won YW, Jeon
J, Lee BJ, Cho WJ and Park JW: Hedgehog signalling is involved in
acquired resistance to KRASG12C inhibitors in lung cancer cells.
Cell Death Dis. 15:562024. View Article : Google Scholar
|
|
31
|
Huang L, Guo Z, Wang F and Fu L: KRAS
mutation: from undruggable to druggable in cancer. Signal Transduct
Target Ther. 6:3862021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeitouni D, Pylayeva-Gupta Y, Der C and
Bryant K: KRAS mutant pancreatic cancer: No lone path to an
effective treatment. Cancers (Basel). 8:452016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cox AD, Der CJ and Philips MR: Targeting
RAS Membrane Association: Back to the Future for Anti-RAS Drug
Discovery? Clin Cancer Res. 21:1819–1827. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Menyhárd DK, Pálfy G, Orgován Z, Vida I,
Keserű GM and Perczel A: Structural impact of GTP binding on
downstream KRAS signaling. Chem Sci. 11:9272–9289. 2020. View Article : Google Scholar
|
|
35
|
Varshavi D, Varshavi D, McCarthy N,
Veselkov K, Keun HC and Everett JR: Metabolic characterization of
colorectal cancer cells harbouring different KRAS mutations in
codon 12, 13, 61 and 146 using human SW48 isogenic cell lines.
Metabolomics. 16:512020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bhadhadhara K, Jani V, Koulgi S, Sonavane
U and Joshi R: Studying early structural changes in SOS1 mediated
KRAS activation mechanism. Curr Res Struct Biol. 7:1001152023.
View Article : Google Scholar
|
|
37
|
Godwin I, Anto NP, Bava SV, Babu MS and
Jinesh GG: Targeting K-Ras and apoptosis-driven cellular
transformation in cancer. Cell Death Discov. 7:802021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jonckheere N, Vasseur R and Van Seuningen
I: The cornerstone K-RAS mutation in pancreatic adenocarcinoma:
From cell signaling network, target genes, biological processes to
therapeutic targeting. Crit Rev Oncol Hematol. 111:7–19. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hibshman PS, Stalnecker CA, Klomp JA,
Drizyte-Miller K, Klomp JE, Edwards AC, Pita LM, Hodge RG, Diehl
JN, Mouery RD, et al: Defining the MYC-regulated transcriptome and
kinome that support KRAS- and ERK-dependent growth of pancreatic
cancer. Sci Signal. 18:eadu71452025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mondal K, Posa MK, Shenoy RP and
Roychoudhury S: KRAS mutation subtypes and their association with
other driver mutations in oncogenic pathways. Cells. 13:12212024.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hou H, Li J, Xing J and Hou R: The role of
SPRED1 mutation in melanoma. Eur J Med Res. 30:9732025. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shui B, La Rocca G, Ventura A and Haigis
KM: Interplay between K-RAS and miRNAs. Trends Cancer. 8:384–396.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Singhal A, Li BT and O'Reilly EM:
Targeting KRAS in cancer. Nat Med. 30:969–983. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ghafouri-Fard S, Hussen BM, Mohaqiq M,
Shoorei H, Baniahmad A, Taheri M and Jamali E: Interplay between
non-coding RNAs and programmed cell death proteins. Front Oncol.
12:8084752022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mok ETY, Chitty JL and Cox TR: miRNAs in
pancreatic cancer progression and metastasis. Clin Exp Metastasis.
41:163–186. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bortoletto AS and Parchem RJ: KRAS Hijacks
the miRNA regulatory pathway in cancer. Cancer Res. 83:1563–1572.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kent OA, Chivukula RR, Mullendore M,
Wentzel EA, Feldmann G, Lee KH, Liu S, Leach SD, Maitra A and
Mendell JT: Repression of the miR-143/145 cluster by oncogenic Ras
initiates a tumor-promoting feed-forward pathway. Genes Dev.
24:2754–2759. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Balzeau J, Menezes MR, Cao S and Hagan JP:
The LIN28/let-7 pathway in cancer. Front Genet. 8:312017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ardalan B, Ciner A, Baca Y, Hinton A,
Darabi S, Kasi A, Lou E, Azqueta JI, Xiu J, Datta J, et al:
Distinct molecular and clinical features of specific variants of
KRAS Codon 12 in pancreatic adenocarcinoma. Clin Cancer Res.
31:1082–1090. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
McIntyre CA, Grimont A, Park J, Meng Y,
Sisso WJ, Seier K, Jang GH, Walch H, Aveson VG, Falvo DJ, et al:
Distinct clinical outcomes and biological features of specific KRAS
mutants in human pancreatic cancer. Cancer Cell. 42:1614–1629.e5.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Walcheck MT, Nukaya M, Ranheim EA,
Matkowskyj KA and Ronnekleiv-Kelly S: Pdx1 expression in
hematopoietic cells activates Kras -mutation to drive leukemia in
KC (Pdx1-Cre; LSL-Kras G12D/+) mice. Leuk Lymphoma.
64:1112–1122. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Buscail L, Bournet B and Cordelier P: Role
of oncogenic KRAS in the diagnosis, prognosis and treatment of
pancreatic cancer. Nat Rev Gastroenterol Hepatol. 17:153–168. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Huang X, Zhang G, Tang TY, Gao X and Liang
TB: Personalized pancreatic cancer therapy: from the perspective of
mRNA vaccine. Mil Med Res. 9:532022.PubMed/NCBI
|
|
54
|
Dragnev KH, Lubet RA, Miller MS, Sei S,
Fox JT and You M: Primary prevention and interception studies in
ras-mutated tumor models employing small molecules or vaccines.
Cancer Prev Res (Phila). 16:549–560. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Allemailem KS, Rahmani AH, Almansour NM,
Aldakheel FM, Albalawi GM, Albalawi GM and Khan AA: Current updates
on the structural and functional aspects of the CRISPR/Cas13 system
for RNA targeting and editing: A next-generation tool for cancer
management (Review). Int J Oncol. 66:422025. View Article : Google Scholar
|
|
56
|
Ma Y, Schulz B, Trakooljul N, Al Ammar M,
Sekora A, Sender S, Hadlich F, Zechner D, Weiss FU, Lerch MM, et
al: Inhibition of KRAS, MEK and PI3K demonstrate synergistic
anti-tumor effects in pancreatic ductal adenocarcinoma cell lines.
Cancers (Basel). 14:44672022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bournet B, Muscari F, Buscail C, Assenat
E, Barthet M, Hammel P, Selves J, Guimbaud R, Cordelier P and
Buscail L: KRAS G12D mutation subtype is a prognostic factor for
advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol.
7:e1572016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bijlsma MF, Sadanandam A, Tan P and
Vermeulen L: Molecular subtypes in cancers of the gastrointestinal
tract. Nat Rev Gastroenterol Hepatol. 14:333–342. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Toriyama K, Masago K, Shibata N, Haneda M,
Kuwahara T, Natsume S, Kobayashi S, Fujita Y, Sasaki E, Yamao K, et
al: Clinicopathological and molecular characterization of KRAS
wild-type pancreatic ductal adenocarcinomas reveals precursor
lesions with oncogenic mutations and fusions in RAS pathway genes.
J Pathol. 266:337–351. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Liu YH, Hu CM, Hsu YS and Lee WH:
Interplays of glucose metabolism and KRAS mutation in pancreatic
ductal adenocarcinoma. Cell Death Dis. 13:8172022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ma Q, Zhang W, Wu K and Shi L: The roles
of KRAS in cancer metabolism, tumor microenvironment and clinical
therapy. Mol Cancer. 24:142025. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bansod S, Dodhiawala PB and Lim KH:
Oncogenic KRAS-induced feedback inflammatory signaling in
pancreatic cancer: An overview and new therapeutic opportunities.
Cancers (Basel). 13:54812021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Araujo LH, Souza BM, Leite LR, Parma SAF,
Lopes NP, Malta FSV and Freire MCM: Molecular profile of KRAS
G12C-mutant colorectal and non-small-cell lung cancer. BMC Cancer.
21:1932021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hunter JC, Manandhar A, Carrasco MA,
Gurbani D, Gondi S and Westover KD: Biochemical and structural
analysis of common cancer-associated KRAS mutations. Mol Cancer
Res. 13:1325–1335. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pantsar T, Rissanen S, Dauch D, Laitinen
T, Vattulainen I and Poso A: Assessment of mutation probabilities
of KRAS G12 missense mutants and their long-timescale dynamics by
atomistic molecular simulations and Markov state modeling. PLoS
Comput Biol. 14:e10064582018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bournet B, Buscail C, Muscari F, Cordelier
P and Buscail L: Targeting KRAS for diagnosis, prognosis, and
treatment of pancreatic cancer: Hopes and realities. Eur J Cancer.
54:75–83. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Qian ZR, Rubinson DA, Nowak JA,
Morales-Oyarvide V, Dunne RF, Kozak MM, Welch MW, Brais LK, Da
Silva A, Li T, et al: Association of alterations in main driver
genes with outcomes of patients with resected pancreatic ductal
adenocarcinoma. JAMA Oncol. 4:e1734202018. View Article : Google Scholar :
|
|
68
|
Cao H, Ma Z, Huang Q, Han H, Li Y, Zhang Y
and Chen H: Clinicopathologic features, concurrent genomic
alterations, and clinical outcomes of patients with KRAS G12D
mutations in resected lung adenocarcinoma. Eur J Cancer.
202:1139852024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Strickler JH, Yoshino T, Stevinson K,
Eichinger CS, Giannopoulou C, Rehn M and Modest DP: Prevalence of
KRAS G12C mutation and co-mutations and associated clinical
outcomes in patients with colorectal cancer: A systematic
literature review. Oncologist. 28:e981–e994. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chen Q, Guo H, Jiang H, Hu Z, Yang X, Yuan
Z, Gao Y, Zhang G and Bai Y: S100A2 induces epithelial-mesenchymal
transition and metastasis in pancreatic cancer by coordinating
transforming growth factor β signaling in SMAD4-dependent manner.
Cell Death Discov. 9:3562023. View Article : Google Scholar
|
|
71
|
Ischenko I, D'Amico S, Rao M, Li J, Hayman
MJ, Powers S, Petrenko O and Reich NC: KRAS drives immune evasion
in a genetic model of pancreatic cancer. Nat Commun. 12:14822021.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kapoor A, Yao W, Ying H, Hua S, Liewen A,
Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al: Yap1 activation
enables bypass of oncogenic Kras addiction in pancreatic cancer.
Cell. 158:185–197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Ajmal A, Alkhatabi HA, Alreemi RM, Alamri
MA, Khalid A, Abdalla AN, Alotaibi BS and Wadood A: Prospective
virtual screening combined with bio-molecular simulation enabled
identification of new inhibitors for the KRAS drug target. BMC
Chem. 18:572024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shao YT, Ma L, Zhang TH, Xu TR, Ye YC and
Liu Y: The Application of the RNA interference technologies for
KRAS: Current status, future perspective and associated challenges.
Curr Top Med Chem. 19:2143–2157. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Papke B, Azam SH, Feng AY, Gutierrez-Ford
C, Huggins H, Pallan PS, Van Swearingen AED, Egli M, Cox AD, Der CJ
and Pecot CV: Silencing of oncogenic KRAS by mutant-selective small
interfering RNA. ACS Pharmacol Transl Sci. 4:703–712. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Berndt N, Hamilton AD and Sebti SM:
Targeting protein prenylation for cancer therapy. Nat Rev Cancer.
11:775–791. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kaya P, Schaffner-Reckinger E, Manoharan
GB, Vukic V, Kiriazis A, Ledda M, Burgos Renedo M, Pavic K,
Gaigneaux A, Glaab E and Abankwa DK: An improved PDE6D inhibitor
combines with sildenafil to inhibit KRAS mutant cancer cell growth.
J Med Chem. 67:8569–8584. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Quandt J, Schlude C, Bartoschek M, Will R,
Cid-Arregui A, Schölch S, Reissfelder C, Weitz J, Schneider M,
Wiemann S, et al: Long-peptide vaccination with driver gene
mutations in p53 and Kras induces cancer mutation-specific effector
as well as regulatory T cell responses. Oncoimmunology.
7:e15006712018. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wilson CY and Tolias P: Recent advances in
cancer drug discovery targeting RAS. Drug Discov Today.
21:1915–1919. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Asati V, Mahapatra DK and Bharti SK: K-Ras
and its inhibitors towards personalized cancer treatment:
Pharmacological and structural perspectives. Eur J Med Chem.
125:299–314. 2017. View Article : Google Scholar
|
|
81
|
Sabt A, Tawfik HO, Khaleel EF, Badi RM,
Ibrahim HAA, Elkaeed EB and Eldehna WM: An overview of recent
advancements in small molecules suppression of oncogenic signaling
of K-RAS: An updated review. Mol Divers. 28:4581–4608. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Van Cutsem E, Hidalgo M, Canon J,
Macarulla T, Bazin I, Poddubskaya E, Manojlovic N, Radenkovic D,
Verslype C, Raymond E, et al: Phase I/II trial of pimasertib plus
gemcitabine in patients with metastatic pancreatic cancer. Int J
Cancer. 143:2053–2064. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li Q, Li Z, Luo T and Shi H: Targeting the
PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol
Biomed. 3:472022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Mirzaei S, Gholami MH, Ang HL, Hashemi F,
Zarrabi A, Zabolian A, Hushmandi K, Delfi M, Khan H, Ashrafizadeh
M, et al: Pre-clinical and clinical applications of small
interfering RNAs (siRNA) and Co-delivery systems for pancreatic
cancer therapy. Cells. 10:33482021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Aghamiri S, Raee P, Talaei S,
Mohammadi-Yeganeh S, Bayat S, Rezaee D, Ghavidel AA, Teymouri A,
Roshanzamiri S, Farhadi S and Ghanbarian H: Nonviral siRNA delivery
systems for pancreatic cancer therapy. Biotechnol Bioeng.
118:3669–3690. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Golan T, Khvalevsky EZ, Hubert A, Gabai
RM, Hen N, Segal A, Domb A, Harari G, David EB, Raskin S, et al:
RNAi therapy targeting KRAS in combination with chemotherapy for
locally advanced pancreatic cancer patients. Oncotarget.
6:24560–24570. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Kamerkar S, LeBleu VS, Sugimoto H, Yang S,
Ruivo CF, Melo SA, Lee JJ and Kalluri R: Exosomes facilitate
therapeutic targeting of oncogenic KRAS in pancreatic cancer.
Nature. 546:498–503. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Smith JP, Chen W, Shivapurkar N, Gerber M,
Tucker RD, Kallakury B, Dasa SSK, Kularatne RN and Stern ST:
Target-specific nanoparticle polyplex down-regulates mutant kras to
prevent pancreatic carcinogenesis and halt tumor progression. Int J
Mol Sci. 24:7522023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cohn A, Morse MA, O'Neil B, Whiting S,
Coeshott C, Ferraro J, Bellgrau D, Apelian D and Rodell TC: Whole
recombinant saccharomyces cerevisiae yeast expressing ras mutations
as treatment for patients with solid tumors bearing ras mutations:
Results from a phase 1 trial. J Immunother. 41:141–150. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chandra A, Grecco HE, Pisupati V, Perera
D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M,
Venkitaraman AR, et al: The GDI-like solubilizing factor PDEδ
sustains the spatial organization and signalling of Ras family
proteins. Nat Cell Biol. 14:148–158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Du R, Sullivan DK, Azizian NG, Liu Y and
Li Y: Inhibition of ERAD synergizes with FTS to eradicate
pancreatic cancer cells. BMC Cancer. 21:2372021. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lugowska I, Kosela-Paterczyk H, Kozak K
and Rutkowski P: Trametinib: A MEK inhibitor for management of
metastatic melanoma. Onco Targets Ther. 8:2251–2259.
2015.PubMed/NCBI
|
|
93
|
Bodoky G, Timcheva C, Spigel DR, La Stella
PJ, Ciuleanu TE, Pover G and Tebbutt NC: A phase II open-label
randomized study to assess the efficacy and safety of selumetinib
(AZD6244 [ARRY-142886]) versus capecitabine in patients with
advanced or metastatic pancreatic cancer who have failed first-line
gemcitabine therapy. Invest New Drugs. 30:1216–1223. 2012.
View Article : Google Scholar
|
|
94
|
Infante JR, Somer BG, Park JO, Li CP,
Scheulen ME, Kasubhai SM, Oh DY, Liu Y, Redhu S, Steplewski K and
Le N: A randomised, double-blind, placebo-controlled trial of
trametinib, an oral MEK inhibitor, in combination with gemcitabine
for patients with untreated metastatic adenocarcinoma of the
pancreas. Eur J Cancer. 50:2072–2081. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mahapatra DK, Asati V and Bharti SK: MEK
inhibitors in oncology: A patent review (2015-Present). Expert Opin
Ther Pat. 27:887–906. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ning C, Liang M, Liu S, Wang G, Edwards H,
Xia Y, Polin L, Dyson G, Taub JW, Mohammad RM, et al: Targeting ERK
enhances the cytotoxic effect of the novel PI3K and mTOR dual
inhibitor VS-5584 in preclinical models of pancreatic cancer.
Oncotarget. 8:44295–44311. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Kinsey CG, Camolotto SA, Boespflug AM,
Guillen KP, Foth M, Truong A, Schuman SS, Shea JE, Seipp MT, Yap
JT, et al: Protective autophagy elicited by RAF MEK ERK inhibition
suggests a treatment strategy for RAS-driven cancers. Nat Med.
25:620–627. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Shodeinde A, Ginjupalli K, Lewis HD, Riaz
S and Barton BE: STAT3 inhibition induces apoptosis in cancer cells
independent of STAT1 or STAT2. J Mol Biochem. 2:18–26.
2013.PubMed/NCBI
|
|
99
|
Stickler S, Rath B and Hamilton G:
Targeting KRAS in pancreatic cancer. Oncol Res. 32:799–805. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Seyhan AA: Circulating microRNAs as
potential biomarkers in pancreatic cancer-advances and challenges.
Int J Mol Sci. 24:133402023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Chakkera M, Foote JB, Farran B and
Nagaraju GP: Breaking the stromal barrier in pancreatic cancer:
Advances and challenges. Biochim Biophys Acta Rev Cancer.
1879:1890652024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
O'Brien J, Hayder H, Zayed Y and Peng C:
Overview of MicroRNA biogenesis, mechanisms of actions, and
circulation. Front Endocrinol (Lausanne). 9:4022018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Zhang Y, Liu Q, Zhang X, Huang H, Tang S,
Chai Y, Xu Z, Li M, Chen X, Liu J and Yang C: Recent advances in
exosome-mediated nucleic acid delivery for cancer therapy. J
Nanobiotechnology. 20:2792022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Plawgo K and Raczynska KD:
Context-dependent regulation of gene expression by non-canonical
small RNAs. Noncoding RNA. 8:292022.PubMed/NCBI
|
|
105
|
Iorio MV and Croce CM: MicroRNA
dysregulation in cancer: Diagnostics, monitoring and therapeutics.
A comprehensive review. EMBO Mol Med. 4:143–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Sidorova EA, Zhernov YV, Antsupova MA,
Khadzhieva KR, Izmailova AA, Kraskevich DA, Belova EV, Simanovsky
AA, Shcherbakov DV, Zabroda NN and Mitrokhin OV: The role of
different types of microRNA in the pathogenesis of breast and
prostate cancer. Int J Mol Sci. 24:19802023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Salu P and Reindl KM: Advancements in
preclinical models of pancreatic cancer. Pancreas. 53:e205–e220.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bereczki Z, Benczik B, Balogh OM, Marton
S, Puhl E, Pétervári M, Váczy-Földi M, Papp ZT, Makkos A, Glass K,
et al: Mitigating off-target effects of small RNAs: Conventional
approaches, network theory and artificial intelligence. Br J
Pharmacol. 182:340–379. 2025. View Article : Google Scholar
|
|
109
|
Perurena N, Situ L and Cichowski K:
Combinatorial strategies to target RAS-driven cancers. Nat Rev
Cancer. 24:316–337. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Fanini F, Bandini E, Plousiou M, Carloni
S, Wise P, Neviani P, Murtadha M, Foca F, Fabbri F, Vannini I and
Fabbri M: MicroRNA-16 restores sensitivity to tyrosine kinase
inhibitors and outperforms MEK inhibitors in KRAS-mutated non-small
cell lung cancer. Int J Mol Sci. 22:133572021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Wu Y, Xie Z, Deng S, Xia Y, Lei X and Yang
X: Microrna-96 in human cancers. Comb Chem High Throughput Screen.
26:1285–1297. 2023. View Article : Google Scholar
|
|
112
|
de Jesus VHF, Mathias-Machado MC, de
Farias JPF, Aruquipa MPS, Jácome AA and Peixoto RD: Targeting KRAS
in pancreatic ductal adenocarcinoma: The long road to cure. Cancers
(Basel). 15:50152023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Garibaldi-Ríos AF, Figuera LE,
Zúñiga-González GM, Gómez-Meda BC, García-Verdín PM,
Carrillo-Dávila IA, Gutiérrez-Hurtado IA, Torres-Mendoza BM and
Gallegos-Arreola MP: In silico identification of dysregulated
miRNAs Targeting KRAS gene in pancreatic cancer. Diseases.
12:1522024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Mitsunaga S, Ueno M, Tsuda M, Kuwahara T,
Takayama Y, Hanada K, Yoshida H, Takahashi K, Nakata K, Iwama H, et
al: Abstract 6686: Tumor suppressor miRNA-665 based serum biomarker
for detecting pancreatobiliary cancer. Cancer Res. 83(Suppl 7):
66862023. View Article : Google Scholar
|
|
115
|
Hasani F, Masrour M, Khamaki S, Jazi K,
Hosseini S, Heidarpour H and Namazee M: Diagnostic and prognostic
accuracy of MiRNAs in pancreatic cancer: A systematic review and
meta-analysis. J Cell Mol Med. 29:e703372025. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Lai X, Wang M, McElyea SD, Sherman S,
House M and Korc M: A microRNA signature in circulating exosomes is
superior to exosomal glypican-1 levels for diagnosing pancreatic
cancer. Cancer Lett. 393:86–93. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Chen C, Demirkhanyan L and Gondi CS: The
multifaceted role of miR-21 in pancreatic cancers. Cells.
13:9482024. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Catalanotto C, Cogoni C and Zardo G:
MicroRNA in control of gene expression: An overview of nuclear
functions. Int J Mol Sci. 17:17122016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Mulholland EJ, Dunne N and McCarthy HO:
MicroRNA as therapeutic targets for chronic wound healing. Mol Ther
Nucleic Acids. 8:46–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Guo S, Fesler A, Wang H and Ju J: microRNA
based prognostic biomarkers in pancreatic cancer. Biomark Res.
6:182018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Chaudhary AK, Mondal G, Kumar V, Kattel K
and Mahato RI: Chemosensitization and inhibition of pancreatic
cancer stem cell proliferation by overexpression of microRNA-205.
Cancer Lett. 402:1–8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Shang R, Lee S, Senavirathne G and Lai EC:
microRNAs in action: Biogenesis, function and regulation. Nat Rev
Genet. 24:816–833. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Schultz NA, Andersen KK, Roslind A,
Willenbrock H, Wøjdemann M and Johansen JS: Prognostic MicroRNAs in
cancer tissue from patients operated for pancreatic cancer-five
MicroRNAs in a prognostic index. World J Surg. 36:2699–2707. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Li Z, Xu Q, Zhong J, Zhang Y, Zhang T,
Ying X, Lu X, Li X, Wan L, Xue J, et al: Structural insights into
RNA cleavage by PIWI Argonaute. Nature. 639:250–259. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Beg MS, Brenner A, Sachdev J, Ejadi S,
Borad M, Kang YK, Lim H, Kim TY, Bader A, Stoudemire J, et al:
Abstract C43: Safety, tolerability, and clinical activity of MRX34,
the first-in-class liposomal miR-34 mimic, in patients with
advanced solid tumors. Mol Cancer Ther. 14(12 Suppl 2): C432015.
View Article : Google Scholar
|
|
126
|
Cole G, Ali AA, McErlean E, Mulholland EJ,
Short A, McCrudden CM, McCaffrey J, Robson T, Kett VL, Coulter JA,
et al: DNA vaccination via RALA nanoparticles in a microneedle
delivery system induces a potent immune response against the
endogenous prostate cancer stem cell antigen. Acta Biomater.
96:480–490. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
McCarthy HO, McCaffrey J, McCrudden CM,
Zholobenko A, Ali AA, McBride JW, Massey AS, Pentlavalli S, Chen
KH, Cole G, et al: Development and characterization of
self-assembling nanoparticles using a bio-inspired amphipathic
peptide for gene delivery. J Control Release. 189:141–149. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Yi J, Li S, Wang C, Cao N, Qu H, Cheng C,
Wang Z, Wang L and Zhou L: Potential applications of polyphenols on
main ncRNAs regulations as novel therapeutic strategy for cancer.
Biomed Pharmacother. 113:1087032019. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Li Z, Mao K, Liu L, Xu S, Zeng M, Fu Y,
Huang J, Li T, Gao G, Teng ZQ, et al: Nuclear microRNA-mediated
transcriptional control determines adult microglial homeostasis and
brain function. Cell Rep. 43:1139642024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Sun T, Kong X, Du Y and Li Z: Aberrant
MicroRNAs in pancreatic cancer: Researches and clinical
implications. Gastroenterol Res Pract. 2014:3865612014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Senaratne M, Swami SS, Aye SL, Trivedi Y,
Bolgarina Z, Desai HN and Mohammed L: Clinical value of circulating
microRNAs in diagnosis and prognosis of pancreatic cancer: A
systematic review. Cureus. 15:e439312023.PubMed/NCBI
|
|
132
|
Cheng H, Shi S, Cai X, Long J, Xu J, Liu C
and Yu X: microRNA signature for human pancreatic cancer invasion
and metastasis. Exp Ther Med. 4:181–187. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Haselmann V, Kurz A, Bertsch U, Hübner S,
Olempska-Müller M, Fritsch J, Häsler R, Pickl A, Fritsche H,
Annewanter F, et al: Nuclear death receptor TRAIL-R2 Inhibits
Maturation of Let-7 and promotes proliferation of pancreatic and
other tumor cells. Gastroenterology. 146:278–290. 2014. View Article : Google Scholar
|
|
134
|
Schneider AO, Birsan S, Anderco P, Ichim
C, Todor SB, Dura H, Fleacă R and Boicean A: The evolving landscape
of microRNAs in cholangiocarcinoma and pancreatic cancer.
Diagnostics (Basel). 15:22852025. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Słotwiński R, Lech G and Słotwińska SM:
MicroRNAs in pancreatic cancer diagnosis and therapy. Cent Eur J
Immunol. 43:314–324. 2018. View Article : Google Scholar :
|
|
136
|
Abue M, Yokoyama M, Shibuya R, Tamai K,
Yamaguchi K, Sato I, Tanaka N, Hamada S, Shimosegawa T, Sugamura K
and Satoh K: Circulating miR-483-3p and miR-21 is highly expressed
in plasma of pancreatic cancer. Int J Oncol. 46:539–547. 2015.
View Article : Google Scholar :
|
|
137
|
Poenitzsch Strong AM, Setaluri V and
Spiegelman VS: microRNA-340 as a modulator of RAS-RAF-MAPK
signaling in melanoma. Arch Biochem Biophys. 563:118–124. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Hatley ME, Patrick DM, Garcia MR,
Richardson JA, Bassel-Duby R, van Rooij E and Olson EN: Modulation
of K-Ras-Dependent lung tumorigenesis by MicroRNA-21. Cancer Cell.
18:282–293. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Meng F, Henson R, Wehbe-Janek H, Ghoshal
K, Jacob ST and Patel T: MicroRNA-21 regulates expression of the
PTEN tumor suppressor gene in human hepatocellular cancer.
Gastroenterology. 133:647–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zawistowski JS, Nakamura K, Parker JS,
Granger DA, Golitz BT and Johnson GL: MicroRNA 9-3p Targets
β1 Integrin To sensitize claudin-low breast cancer cells
to MEK inhibition. Mol Cell Biol. 33:2260–2274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Lin CC, Sharma SB, Farrugia MK, McLaughlin
SL, Ice RJ, Loskutov YV, Pugacheva EN, Brundage KM, Chen D and
Ruppert JM: Kruppel-like factor 4 signals through microRNA-206 to
promote tumor initiation and cell survival. Oncogenesis.
4:e1552015. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Frankel LB, Christoffersen NR, Jacobsen A,
Lindow M, Krogh A and Lund AH: Programmed cell death 4 (PDCD4) is
an important functional target of the MicroRNA miR-21 in breast
cancer cells. J Biol Chem. 283:1026–1033. 2008. View Article : Google Scholar
|
|
143
|
Momi N, Kaur S, Rachagani S, Ganti AK and
Batra SK: Smoking and microRNA dysregulation: A cancerous
combination. Trends Mol Med. 20:36–47. 2014. View Article : Google Scholar
|
|
144
|
Johnson SM, Grosshans H, Shingara J, Byrom
M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D and Slack
FJ: RAS Is Regulated by the let-7 MicroRNA family. Cell.
120:635–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Mokhlis HA, Bayraktar R, Kabil NN, Caner
A, Kahraman N, Rodriguez-Aguayo C, Zambalde EP, Sheng J, Karagoz K,
Kanlikilicer P, et al: The modulatory role of MicroRNA-873 in the
Progression of KRAS-Driven cancers. Mol Ther Nucleic Acids.
14:301–317. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Yu SN, Ma YH, Zhao WG, Jin XL, Yang HY,
Liu PP and Chen J: KRAS -related noncoding RNAs in pancreatic
ductal adenocarcinoma. Chronic Dis Transl Med. 2:215–222. 2016.
|
|
147
|
Rachagani S, Macha MA, Menning MS, Dey P,
Pai P, Smith LM, Mo YY and Batra SK: Changes in microRNA (miRNA)
expression during pancreatic cancer development and progression in
a genetically engineered KrasG12D;Pdx1-Cre mouse (KC) model.
Oncotarget. 6:40295–40309. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Dong Y, Fu C, Guan H, Zhang Z, Zhou T and
Li B: Prognostic significance of miR-126 in various cancers: A
meta-analysis. Onco Targets Ther. 9:2547–2555. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Boyerinas B, Park SM, Hau A, Murmann AE
and Peter ME: The role of let-7 in cell differentiation and cancer.
Endocr Relat Cancer. 17:F19–F36. 2010. View Article : Google Scholar
|
|
150
|
Kim M and Slack FJ: MicroRNA-mediated
regulation of KRAS in cancer. J Hematol Oncol. 7:842014. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Dai X, Jiang Y and Tan C: Let-7 Sensitizes
KRAS mutant tumor cells to chemotherapy. PLoS One. 10:e01266532015.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Liu Y, Wang D, Zhou M, Chen H, Wang H, Min
J, Chen J, Wu S, Ni X, Zhang Y, et al: The KRAS/Lin28B axis
maintains stemness of pancreatic cancer cells via the let-7i/TET3
pathway. Mol Oncol. 15:262–278. 2021. View Article : Google Scholar :
|
|
153
|
Torrisani J, Bournet B, du Rieu MC,
Bouisson M, Souque A, Escourrou J, Buscail L and Cordelier P: Let-7
MicroRNA transfer in pancreatic cancer-derived cells inhibits in
vitro cell proliferation but fails to alter tumor progression. Hum
Gene Ther. 20:831–844. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Li S, Meng W, Guo Z, Liu M, He Y, Li Y and
Ma Z: The miR-183 Cluster: Biogenesis, functions, and cell
communication via exosomes in cancer. Cells. 12:13152023.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Huang X, Lv W, Zhang JH and Lu DL: miR-96
functions as a tumor suppressor gene by targeting NUAK1 in
pancreatic cancer. Int J Mol Med. 34:1599–1605. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Yu S, Lu Z, Liu C, Meng Y, Ma Y, Zhao W,
Liu J, Yu J and Chen J: miRNA-96 suppresses KRAS and functions as a
tumor suppressor gene in pancreatic cancer. Cancer Res.
70:6015–6025. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Liao L, Tang Y, Zhou Y, Meng X, Li B and
Zhang X: MicroRNA-126 (MiR-126): Key roles in related diseases. J
Physiol Biochem. 80:277–286. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Jiao LR, Frampton AE, Jacob J, Pellegrino
L, Krell J, Giamas G, Tsim N, Vlavianos P, Cohen P, Ahmad R, et al:
MicroRNAs targeting oncogenes are down-regulated in pancreatic
malignant transformation from benign tumors. PLoS One.
7:e320682012. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Chen X, Guo X, Zhang H, Xiang Y, Chen J,
Yin Y, Cai X, Wang K, Wang G, Ba Y, et al: Role of miR-143
targeting KRAS in colorectal tumorigenesis. Oncogene. 28:1385–1392.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Kent OA, Fox-Talbot K and Halushka MK:
RREB1 repressed miR-143/145 modulates KRAS signaling through
downregulation of multiple targets. Oncogene. 32:2576–2585. 2013.
View Article : Google Scholar
|
|
161
|
Jin X, Sun Y, Yang H, Li J, Yu S, Chang X,
Lu Z and Chen J: Deregulation of the MiR-193b-KRAS axis contributes
to impaired cell growth in pancreatic cancer. PLoS One.
10:e01255152015. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Keklikoglou I, Hosaka K, Bender C, Bott A,
Koerner C, Mitra D, Will R, Woerner A, Muenstermann E, Wilhelm H,
et al: MicroRNA-206 functions as a pleiotropic modulator of cell
proliferation, invasion and lymphangiogenesis in pancreatic
adenocarcinoma by targeting ANXA2 and KRAS genes. Oncogene.
34:4867–4878. 2015. View Article : Google Scholar :
|
|
163
|
Azevedo-Pouly AC, Sutaria DS, Jiang J,
Elgamal OA, Amari F, Allard D, Grippo PJ, Coppola V and Schmittgen
TD: miR-216 and miR-217 expression is reduced in transgenic mouse
models of pancreatic adenocarcinoma, knockout of miR-216/miR-217
host gene is embryonic lethal. Funct Integr Genomics. 17:203–212.
2017. View Article : Google Scholar :
|
|
164
|
Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM and
Chen J: The miR-217 microRNA functions as a potential tumor
suppressor in pancreatic ductal adenocarcinoma by targeting KRAS.
Carcinogenesis. 31:1726–1733. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Li X, Deng SJ, Zhu S, Jin Y, Cui SP, Chen
JY, Xiang C, Li QY, He C, Zhao SF, et al: Hypoxia-induced
lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic
cancer by derepressing the miR-3923/KRAS pathway. Oncotarget.
7:6000–6014. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Yang X, Zhang Z, Zhang L and Zhou L:
MicroRNA hsa-mir-3923 serves as a diagnostic and prognostic
biomarker for gastric carcinoma. Sci Rep. 10:46722020. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Oom AL, Humphries BA and Yang C:
MicroRNAs: Novel players in cancer diagnosis and therapies. Biomed
Res Int. 2014:9594612014. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Takács T, Kudlik G, Kurilla A, Szeder B,
Buday L and Vas V: The effects of mutant Ras proteins on the cell
signalome. Cancer Metastasis Rev. 39:1051–1065. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Kasinski AL and Slack FJ: miRNA-34
prevents cancer initiation and progression in a therapeutically
resistant K-ras and p53-induced mouse model of lung adenocarcinoma.
Cancer Res. 72:5576–5587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Sun D, Yu F, Ma Y, Zhao R, Chen X, Zhu J,
Zhang CY, Chen J and Zhang J: MicroRNA-31 activates the RAS pathway
and functions as an oncogenic MicroRNA in human colorectal cancer
by repressing RAS p21 GTPase activating protein 1 (RASA1). J Biol
Chem. 288:9508–9518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Shin KH, Bae SD, Hong HS, Kim RH, Kang MK
and Park NH: miR-181a shows tumor suppressive effect against oral
squamous cell carcinoma cells by downregulating K-ras. Biochem
Biophys Res Commun. 404:896–902. 2011. View Article : Google Scholar
|
|
172
|
Wang R, Wang ZX, Yang JS, Pan X, De W and
Chen LB: MicroRNA-451 functions as a tumor suppressor in human
non-small cell lung cancer by targeting ras-related protein 14
(RAB14). Oncogene. 30:2644–2658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Pramanik D, Campbell NR, Karikari C,
Chivukula R, Kent OA, Mendell JT and Maitra A: Restitution of tumor
suppressor MicroRNAs using a systemic nanovector inhibits
pancreatic cancer growth in mice. Mol Cancer Ther. 10:1470–1480.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Park JK, Lee EJ, Esau C and Schmittgen TD:
Antisense Inhibition of microRNA-21 or -221 arrests cell cycle,
induces apoptosis, and sensitizes the effects of gemcitabine in
pancreatic adenocarcinoma. Pancreas. 38:e190–e199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Park JK, Henry JC, Jiang J, Esau C, Gusev
Y, Lerner MR, Postier RG, Brackett DJ and Schmittgen TD: miR-132
and miR-212 are increased in pancreatic cancer and target the
retinoblastoma tumor suppressor. Biochem Biophys Res Commun.
406:518–523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Mohammad RM, Li Y, Muqbil I, Aboukameel A,
Senapedis W, Baloglu E, Landesman Y, Philip PA and Azmi AS:
Targeting Rho GTPase effector p21 activated kinase 4 (PAK4)
suppresses p-Bad-microRNA drug resistance axis leading to
inhibition of pancreatic ductal adenocarcinoma proliferation. Small
GTPases. 10:367–377. 2019. View Article : Google Scholar :
|
|
177
|
Ebert MS and Sharp PA: MicroRNA sponges:
Progress and possibilities. RNA. 16:2043–2050. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Acunzo M, Romano G, Nigita G, Veneziano D,
Fattore L, Laganà A, Zanesi N, Fadda P, Fassan M, Rizzotto L, et
al: Selective targeting of point-mutated KRAS through artificial
microRNAs. Proc Natl Acad Sci USA. 114:E4203–E4212. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Hosseinahli N, Aghapour M, Duijf PHG and
Baradaran B: Treating cancer with microRNA replacement therapy: A
literature review. J Cell Physiol. 233:5574–5588. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Yuan P, He XH, Rong YF, Cao J, Li Y, Hu
YP, Liu Y, Li D, Lou W and Liu MF: KRAS/NF-κB/YY1/miR-489 signaling
axis controls pancreatic cancer metastasis. Cancer Res. 77:100–111.
2017. View Article : Google Scholar
|
|
181
|
Passadouro M and Faneca H: Managing
pancreatic adenocarcinoma: A special focus in MicroRNA gene
therapy. Int J Mol Sci. 17:7182016. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Strobel O, Neoptolemos J, Jäger D and
Büchler MW: Optimizing the outcomes of pancreatic cancer surgery.
Nat Rev Clin Oncol. 16:11–26. 2019. View Article : Google Scholar
|
|
183
|
Masiak-Segit W, Rawicz-Pruszyński K,
Skórzewska M and Polkowski WP: Surgical treatment of pancreatic
cancer. Pol Przegl Chir. 90:45–53. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Hong TH and Park IY: MicroRNA expression
profiling of diagnostic needle aspirates from surgical pancreatic
cancer specimens. Ann Surg Treat Res. 87:290–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Ballehaninna UK and Chamberlain RS: Serum
CA 19-9 as a biomarker for pancreatic cancer-a comprehensive
review. Indian J Surg Oncol. 2:88–100. 2011. View Article : Google Scholar
|
|
186
|
Winter JM, Yeo CJ and Brody JR:
Diagnostic, prognostic, and predictive biomarkers in pancreatic
cancer. J Surg Oncol. 107:15–22. 2013. View Article : Google Scholar
|
|
187
|
Li Y and Sarkar FH: MicroRNA targeted
therapeutic approach for pancreatic cancer. Int J Biol Sci.
12:326–337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Lee EJ, Baek M, Gusev Y, Brackett DJ,
Nuovo GJ and Schmittgen TD: Systematic evaluation of microRNA
processing patterns in tissues, cell lines, and tumors. RNA.
14:35–42. 2008. View Article : Google Scholar :
|
|
189
|
Natesh NS, White BM, Bennett MMC, Uz M,
Kalari Kandy RR, Batra SK, Mallapragada SK and Rachagani S:
Emerging Role of miR-345 and Its effective delivery as a potential
therapeutic candidate in pancreatic cancer and other cancers.
Pharmaceutics. 13:19872021. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Habbe N, Koorstra JBM, Mendell JT,
Offerhaus GJ, Ryu JK, Feldmann G, Mullendore ME, Goggins MG, Hong
SM and Maitra A: MicroRNA miR-155 is a biomarker of early
pancreatic neoplasia. Cancer Biol Ther. 8:340–346. 2009. View Article : Google Scholar
|
|
191
|
Zhang Y, Li M, Wang H, Fisher WE, Lin PH,
Yao Q and Chen C: Profiling of 95 MicroRNAs in pancreatic cancer
cell lines and surgical specimens by real-time PCR analysis. World
J Surg. 33:698–709. 2009. View Article : Google Scholar
|
|
192
|
Humeau M, Torrisani J and Cordelier P:
miRNA in clinical practice: Pancreatic cancer. Clin Biochem.
46:933–936. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
du Rieu MC, Torrisani J, Selves J, Al
Saati T, Souque A, Dufresne M, Tsongalis GJ, Suriawinata AA,
Carrère N, Buscail L and Cordelier P: MicroRNA-21 is induced early
in pancreatic ductal adenocarcinoma precursor lesions. Clin Chem.
56:603–612. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Daoud AZ, Mulholland EJ, Cole G and
McCarthy HO: MicroRNAs in pancreatic cancer: Biomarkers,
prognostic, and therapeutic modulators. BMC Cancer. 19:11302019.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Calatayud D, Dehlendorff C, Boisen MK,
Hasselby JP, Schultz NA, Werner J, Immervoll H, Molven A, Hansen CP
and Johansen JS: Tissue MicroRNA profiles as diagnostic and
prognostic biomarkers in patients with resectable pancreatic ductal
adenocarcinoma and periampullary cancers. Biomark Res. 5:82017.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Liu J, Gao J, Du Y, Li Z, Ren Y, Gu J,
Wang X, Gong Y, Wang W and Kong X: Combination of plasma microRNAs
with serum CA19-9 for early detection of pancreatic cancer. Int J
Cancer. 131:683–691. 2012. View Article : Google Scholar
|
|
197
|
Brunetti O, Russo A, Scarpa A, Santini D,
Reni M, Bittoni A, Azzariti A, Aprile G, Delcuratolo S, Signorile
M, et al: MicroRNA in pancreatic adenocarcinoma:
predictive/prognostic biomarkers or therapeutic targets?
Oncotarget. 6:23323–23341. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Iwagami Y, Eguchi H, Nagano H, Akita H,
Hama N, Wada H, Kawamoto K, Kobayashi S, Tomokuni A, Tomimaru Y, et
al: miR-320c regulates gemcitabine-resistance in pancreatic cancer
via SMARCC1. Br J Cancer. 109:502–511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Yu J, Ohuchida K, Mizumoto K, Sato N,
Kayashima T, Fujita H, Nakata K and Tanaka M: MicroRNA,
hsa-miR-200c, is an independent prognostic factor in pancreatic
cancer and its upregulation inhibits pancreatic cancer invasion but
increases cell proliferation. Mol Cancer. 9:1692010. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Dong F, Zhou J, Wu Y, Gao Z, Li W and Song
Z: MicroRNAs in pancreatic cancer drug resistance: Mechanisms and
therapeutic potential. Front Cell Dev Biol. 12:14991112025.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Kalluri VS, Smaglo BG, Mahadevan KK,
Kirtley ML, McAndrews KM, Mendt M, Yang S, Maldonado AS, Sugimoto
H, Salvatierra ME, et al: Engineered exosomes with KrasG12D
specific siRNA in pancreatic cancer: A phase I study with
immunological correlates. Nat Commun. 16:86962025. View Article : Google Scholar
|
|
202
|
LeBleu VS, Smaglo BG, Mahadevan KK,
Kirtley ML, McAndrews KM, Mendt M, et al: KRAS G12D
-specific targeting with engineered exosomes reprograms the immune
microenvironment to enable efficacy of immune checkpoint therapy in
PDAC patients. medRxiv. [Preprint]:2025.03.03.25322827. 2025.
|
|
203
|
Shi K and Li A: Targeting KRAS G12D:
Advances in inhibitor design. Thorac Cancer. 16:e702032025.
View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Krupa K, Fudalej M, Włoszek E, Miski H,
Badowska-Kozakiewicz AM, Mękal D, Budzik MP, Czerw A and Deptała A:
Treatment of KRAS-mutated pancreatic cancer: New hope for the
patients? Cancers (Basel). 17:24532025. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Hallin J, Bowcut V, Calinisan A, Briere
DM, Hargis L, Engstrom LD, Laguer J, Medwid J, Vanderpool D, Lifset
E, et al: Anti-tumor efficacy of a potent and selective
non-covalent KRASG12D inhibitor. Nat Med. 28:2171–2182. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Navarro-Jiménez M, González B, Mulet N,
Hierro C and Alonso S: KRAS-targeted therapies in colorectal
cancer: A systematic analysis of mutations, inhibitors, and
clinical trials. NPJ Precis Oncol. 9:3802025. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Yoshinari T, Nagashima T, Ishioka H,
Inamura K, Nishizono Y, Tasaki M, Iguchi K, Suzuki A and Sato C:
Discovery of KRAS(G12D) selective degrader ASP3082. Commun Chem.
8:2542025. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Wang Z, Peng Y, Zhou H, Zhang M, Ju D and
Chen Z: MiRNA-based drugs: Challenges and delivery strategies. Appl
Microbiol Biotechnol. 109:2472025. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Liu T, Chen Z, Chen W, Evans R, Xu J,
Reeves ME, de Vera ME and Wang C: Dysregulated miRNAs modulate
tumor microenvironment associated signaling networks in pancreatic
ductal adenocarcinoma. Precis Clin Med. 6:pbad0042023. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Chattopadhyay S, Liao YP, Wang X and Nel
AE: Use of stromal intervention and exogenous neoantigen
vaccination to boost pancreatic cancer chemo-immunotherapy by
nanocarriers. Bioengineering (Basel). 10:12052023. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Lu T and Prakash J: Nanomedicine
strategies to enhance tumor drug penetration in pancreatic cancer.
Int J Nanomedicine. 16:6313–6328. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Hong DS, Kang YK, Borad M, Sachdev J,
Ejadi S, Lim HY, Brenner AJ, Park K, Lee JL, Kim TY, et al: Phase 1
study of MRX34, a liposomal miR-34a mimic, in patients with
advanced solid tumours. Br J Cancer. 122:1630–1637. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Leonhardt L and Hebrok M: KRAS degradation
averts PDAC chemoresistance. Nat Cancer. 5:375–377. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Garreau M, Weidner J, Hamilton R,
Kolosionek E, Toki N, Stavenhagen K, Paris C, Bonetti A, Czechtizky
W, Gnerlich F and Rydzik A: Chemical modification patterns for
microRNA therapeutic mimics: A structure-activity relationship
(SAR) case-study on miR-200c. Nucleic Acids Res. 52:2792–2807.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Lavacchi D, Polvani S, Taddei A, Scolari
F, Messerini L, Caliman E, Moraldi L, Guidolin A, Grazi GL, Galli
A, et al: KRAS-related miR-143 expression is associated with lymph
node involvement and correlates with outcome in pancreatic
adenocarcinoma patients. Front Oncol. 13:12959362023. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Khalilian S, Fathi M, Arezi S and
Ghafouri-Fard S: Updates on the role of miR-193b in the
pathogenesis of human cancers and other diseases. Biochem Biophys
Rep. 45:1023922025.PubMed/NCBI
|
|
217
|
Hara T, Jones MF, Subramanian M, Ling Li
X, Ou O, Zhu Y, Yang Y, Wakefield LM, Hussain SP, Gaedcke J, et al:
Selective targeting of KRAS-Mutant cells by miR-126 through
repression of multiple genes essential for the survival of
KRAS-Mutant cells. Oncotarget. 5:7635–7650. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Liu T, Zhou L, He Z, Chen Y, Jiang X, Xu J
and Jiang J: Circular RNA hsa_circ_0006117 facilitates pancreatic
cancer progression by regulating the miR-96-5p/KRAS/MAPK signaling
pathway. J Oncol. 2021:92132052021. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Singh A, Pruett N, Pahwa R, Mahajan AP,
Schrump DS and Hoang CD: MicroRNA-206 suppresses mesothelioma
progression via the Ras signaling axis. Mol Ther Nucleic Acids.
24:669–681. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Hu Y, Hu Y, Zhang S, Guo Y, Wang F, Du Y,
Wang L, Li P, Xu Y, Zhang H, et al: Tumor-derived miR-203a-3p
potentiates muscle wasting by inducing muscle ferroptosis in
pancreatic cancer. Cancer Lett. 614:2175232025. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Guo X, Zhou Q, Su D, Luo Y, Fu Z, Huang L,
Li Z, Jiang D, Kong Y, Li Z, et al: Circular RNA circBFAR promotes
the progression of pancreatic ductal adenocarcinoma via the
miR-34b-5p/MET/Akt axis. Mol Cancer. 19:832020. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Jin J, Ma M, Yan B, Qiu B, Lu S, Yang L,
Nie Y, Tai S, Yu Z and Teng CB: A SLC31A1-MEK-DNMT1-miR-124
feedback loop contributes to pancreatic cancer progression. Genes
Dis. 10:654–656. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Zhang K and Zhang X: MiR-146b-3p protects
against AR42J cell injury in cerulein-induced acute pancreatitis
model through targeting Anxa2. Open Life Sci. 16:255–265. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Diaz-Riascos ZV, Ginesta MM, Fabregat J,
Serrano T, Busquets J, Buscail L, Cordelier P and Capellá G:
Expression and Role of MicroRNAs from the miR-200 family in the
tumor formation and metastatic propensity of pancreatic cancer. Mol
Ther Nucleic Acids. 17:491–503. 2016. View Article : Google Scholar
|
|
225
|
Wang P, Zhu CF, Ma MZ, Chen G, Song M,
Zeng ZL, Lu WH, Yang J, Wen S, Chiao PJ, et al: Micro-RNA-155 is
induced by K-Ras oncogenic signal and promotes ROS stress in
pancreatic cancer. Oncotarget. 6:21148–21158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Ghafouri-Fard S, Askari A, Hussen BM,
Taheri M and Akbari Dilmaghani N: Role of miR-424 in the
carcinogenesis. Clin Transl Oncol. 26:16–38. 2024. View Article : Google Scholar :
|
|
227
|
Yang Y, Razak SRA, Ismail IS, Ma Y and
Yunus MA: Molecular mechanisms of miR-192 in cancer: A biomarker
and therapeutic target. Cancer Cell Int. 25:942025. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Ouyang H, Gore J, Deitz S and Korc M:
Erratum: microRNA-10b enhances pancreatic cancer cell invasion by
suppressing TIP30 expression and promoting EGF and TGF-β actions.
Oncogene. 36:4952. 2017. View Article : Google Scholar
|
|
229
|
Ji W, Bi Y, Zheng Q, Sun R and Dai K: The
association between radiation-induced miR-208a, cancer recurrence
and death risk in patients with non-small cell lung cancer. Int J
Gen Med. 18:6203–6209. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Yang D, Yan R and Zhang X, Zhu Z, Wang C,
Liang C and Zhang X: Deregulation of MicroRNA-375 inhibits cancer
proliferation migration and chemosensitivity in pancreatic cancer
through the association of HOXB3. Am J Transl Res. 8:1551–1559.
2016.PubMed/NCBI
|
|
231
|
Ding Z, Liu SJ, Liu XW, Ma Q and Qiao Z:
MiR-16 inhibits proliferation of cervical cancer cells by
regulating KRAS. Eur Rev Med Pharmacol Sci. 24:10419–10425.
2020.PubMed/NCBI
|
|
232
|
Kanno S, Nosho K, Ishigami K, Yamamoto I,
Koide H, Kurihara H, Mitsuhashi K, Shitani M, Motoya M, Sasaki S,
et al: MicroRNA-196b is an independent prognostic biomarker in
patients with pancreatic cancer. Carcinogenesis. 38:425–431. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Shopit A, Li X, Tang Z, Awsh M, Shobet L,
Niu M, Wang H, Mousa H, Alshwmi M, Tesfaldet T, et al: miR-421
up-regulation by the oleanolic acid derivative K73-03 regulates
epigenetically SPINK1 transcription in pancreatic cancer cells
leading to metabolic changes and enhanced apoptosis. Pharmacol Res.
161:1051302020. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Di Martino MT, Arbitrio M, Caracciolo D,
Cordua A, Cuomo O, Grillone K, Riillo C, Caridà G, Scionti F,
Labanca C, et al: miR-221/222 as biomarkers and targets for
therapeutic intervention on cancer and other diseases: A systematic
review. Mol Ther Nucleic Acids. 27:1191–1224. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Pancratov R, Peng F, Smibert P, Yang S Jr,
Olson ER, Guha-Gilford C, Kapoor AJ, Liang FX, Lai EC, Flaherty MS
and DasgGupta R: The miR-310/13 cluster antagonizes β-catenin
function in the regulation of germ and somatic cell differentiation
in the Drosophila testis. Development. 140:2904–2916. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Ghafouri-Fard S, Khoshbakht T, Hussen BM,
Jamal HH, Taheri M and Hajiesmaeili M: A comprehensive review on
function of miR-15b-5p in malignant and non-malignant disorders.
Front Oncol. 12:8709962022. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Liu G, Shao C, Li A, Zhang X, Guo X and Li
J: Diagnostic value of plasma miR-181b, miR-196a, and miR-210
combination in pancreatic cancer. Gastroenterol Res Pract.
2020:60731502020. View Article : Google Scholar : PubMed/NCBI
|