Introduction
Cancer cachexia is a complicated syndrome associated
with tissue damage caused by multiple factors and is fatal in ~20%
of patients with cancer (1,2).
Patients with gastrointestinal types of cancer (GIC), such as
pancreatic, gastroesophageal and colorectal cancer, frequently
experience weight loss at diagnosis (3,4). A
major feature of cancer cachexia is the loss of skeletal muscle
mass (5). To date, previous
studies have highlighted increased muscle protein catabolism and
decreased protein synthesis as key mechanisms underlying cancer
cachexia (6,7). In particular, cancer cachexia has
been considered to be the degradation of muscle proteins. This
process is accelerated via the ubiquitin-proteasome system (UPS) or
autophagy-lysosome system (ALS) mediated by inflammation (cytokine
and downstream IL1β/TNFα-NFκB and IL-6-JAK-STAT3 pathways) or TGF-β
(myostatin/activinA-SMAD2/3) pathway (8). However, the majority of these
studies were based on rodent models using mouse colon 26 (C26) and
Lewis lung carcinoma (LLC) (8-11).
Comparative studies have reported that cachexia induced by C26 and
LLC cells did not fully reflect the cachectic features observed in
patients with cancer (8,12-17). Therefore, the identification of
alternative mechanisms that are distinct from those in rodent
models may provide novel insights into the pathogenesis of
cancer-induced cachexia in patients with cancer.
Impaired skeletal muscle differentiation and
regeneration have also attracted attention as alternative inducers
of cancer cachexia (18). A
previous study has shown that defective myoblast differentiation
and fusion result in the accumulation of muscle precursor cells in
cancer-cachectic mouse muscles (18). Another study reported that
patients with cancer display reduced expression of key myogenic
factors in the cachectic muscles (16,18-20).
During the known process of postnatal myogenic
differentiation and regeneration, Pax7-positive myogenic stem
cells, called satellite cells, are first activated and express
myogenic regulatory factors (MRFs), such as MyoD and/or Myf5. These
activated cells proliferate to produce muscle precursor cells,
known as myoblasts. These cells subsequently decrease Pax7, and
instead elevate the expression of other MRFs, such as myogenin and
MRF4, which drive further differentiation and promote myocyte
fusion by upregulating downstream target genes associated with
myogenesis, eventually increasing muscle fiber size (21-25).
Previous studies have reported the key role of bone
morphogenetic protein (BMP) signaling in myogenic differentiation
(26-31). The BMP family (comprising BMP
1-15), a member of TGF-β, binds to the BMP receptor and activates
intracellular signaling pathways. This interaction phosphorylates
the BMP receptor-regulated Smad (R-Smad), including Smad1, Smad5
and Smad8 (32-34). Phosphorylated (p) Smad1/5/8
further induces heteromeric assembly with common-partner Smad
(co-Smad; Smad4) and translocates into the nucleus, upregulating
the expression of target genes (35-38). Previously, the Smad1/5/8-Smad4
complex was reported to directly bind to the Smad-responsive DNA
element within the inhibitor of DNA binding (Id)1 and Id3 gene
promoters to upregulate their expression (39,40). BMP-Smad-induced Id has been
reported to suppress myogenic differentiation by directly
inhibiting the transcriptional activity of MRFs, especially MyoD
(28,29). Other studies have demonstrated
that hyperactivation of BMP signaling during muscle injury causes
delayed muscle regeneration (26,27). By contrast, the proper activation
of the BMP-Smad-Id signaling pathway is critically committed to
muscle development and adult muscle regeneration (30,31). However, to the best of our
knowledge, few studies have investigated whether cancer-derived
BMPs suppress myoblast differentiation exogenously.
Therefore, the present study aimed to identify
cancer-derived factors that impair myogenic differentiation using
conditioned medium (CM) from 20 human GIC cell lines. In addition,
we sought to explore the potential signaling pathways through which
these factors may affect myogenic differentiation in C2C12
myoblasts.
Materials and methods
Cell culture and differentiation
Human colorectal cancer cell lines HT29 (cat. no.
JCRB1383) and DLD1 (cat. no. JCRB1382) were purchased from the
Japanese Collection of Research Bioresources Cell Bank, gastric
cancer cell line KATO III (cat. no. RCB2088) was purchased from the
RIKEN BioResource Center, 44As3 was obtained from Dr Kazuyoshi
Yanagihara (National Cancer Center Hospital, Kashiwa, Japan), and
pancreatic cancer cell line BxPC3 from Dr. Kenoki Ohuchida (Kyusyu
University, Fukuoka, Japan). These cells were cultured in RPMI 1640
medium (Nacalai Tesque, Inc.) supplemented with 10% FBS (Nichirei
Biosciences, Inc.) and 1% penicillin-streptomycin (Fujifilm Wako
Pure Chemical Corporation). The details of the other human cancer
cell lines used in the present study are shown in Table I. The murine colon cancer cell
line C26 (cat. no. RCB2657) was obtained from the RIKEN Cell Bank
and cultured in RPMI1640 medium supplemented with 10% FBS and 1%
penicillin-streptomycin. Murine myoblasts C2C12 (cat. no.
CRL-1772), obtained from the American Type Culture Collection and
were grown in growth medium (GM) consisting of DMEM (Nacarai
Tesque, Inc.) supplemented with 10% FBS and 1%
penicillin-streptomycin. Myoblast differentiation was induced by
replacing GM with differentiation medium (DM) consisting of DMEM
supplemented with 2% horse serum (MillporeSigma) and 1%
penicillin-streptomycin or conditioned medium (CM) prepared as
described in CM preparation. All cell lines were incubated
under standard culture conditions in a humidified 5% CO2
incubator at 37°C. The cell lines were confirmed to be free of
Mycoplasma contamination for at least 6 months.
 | Table ISummary of human cancer cell lines
used in the present study. |
Table I
Summary of human cancer cell lines
used in the present study.
| Tissue | Cell line | Medium | Provided by |
|---|
| Esophagus | KYSE30 | RPMI1640 | JCRB |
| KYSE150 | RPMI1640 | JCRB |
| TE-1 | RPMI1640 | RIKEN BioResource
Center |
| TE-5 | RPMI1640 | RIKEN BioResource
Center |
| TE-6 | RPMI1640 | RIKEN BioResource
Center |
| Stomac | 44As3 | RPMI1640 | Dr. K.
Yanagihara |
| 58As9 | RPMI1640 | Dr. K.
Yanagihara |
| KATO III | RPMI1640 | RIKEN BioResource
Center |
| MKN45 | RPMI1640 | RIKEN BioResource
Center |
| MKN74 | RPMI1640 | RIKEN BioResource
Center |
| Colon | SW480 | RPMI1640 | American Type
Culture Collection |
| HT29 | RPMI1640 | JCRB |
| DLD1 | RPMI1640 | JCRB |
| HCT116 | DMEM | RIKEN BioResource
Center |
| LoVo | HamF12 | RIKEN BioResource
Center |
| Pancreas | BxPC3 | RPMI1640 | Dr. K.
Ohuchida |
| SUIT-2 | RPMI1640 | Dr. K.
Ohuchida |
| AsPC-1 | RPMI1640 | Dr. K.
Ohuchida |
| Panc-1 | RPMI1640 | RIKEN BioResource
Center |
| MIA Paca2 | DMEM | RIKEN BioResource
Center |
CM preparation
Cancer cells were seeded at a density of
1.5-2.0×106 cells in 100-mm culture dishes (Corning,
Inc.) and cultured in growth medium for 2-3 days. When the cancer
cells reached 50-60% confluence, they were washed with PBS and 10
ml of fresh DM was added. After 48 h, when the cells reached 80-90%
confluence, the culture medium was collected and centrifuged at
1,000 × g for 10 min at room temperature, and stored at −80°C until
use. Finally, a CM consisting of 33% cancer cell culture medium and
66% fresh DM was prepared.
Treatment with dorsomorphin
C2C12 cells were treated with 5 μM
dorsomorphin (cat. no. S7840; Selleck Chemicals) at the time of
inducing differentiation with DM or CM. After 1 h of treatment, the
medium containing dorsomorphin was removed and fresh DM or CM was
added. DMSO was used as a vehicle control at a final concentration
of 0.05%.
Gene knockdown of BMP4 by small
interfering RNA (siRNA)
Transient transfection was performed in HT29 cells
using 15 nM control siRNA (ON-TARGETplus Non-targeting siRNA; cat.
no. D-001810-01-05; Revvity) or 15 nM BMP4 siRNA (ON-TARGETplus
Human BMP4 siRNA SMARTpool; cat. no. L-11221-00-0005; Revvity). The
BMP4 siRNA SMARTpool consists of four siRNA duplexes targeting
human BMP4, with the following sequences (5' to 3'):
GAGCCAUGCUAGUUUGAUA, UAGCAAGAGUGCCGUCAUU, CGACACUUCUGCAGAUGUU, and
CAGGAUUAGCCGAUCGUUA. The non-targeting control siRNA, designed not
to target any known human gene, has the following sequence (5' to
3'): UGGUUUACAUGUCGACUAA. Lipofectamine® RNAiMAX (Thermo
Fisher Scientific, Inc.) was used as the transfection reagent
according to the manufacturer's protocol. After 24 h of
transfection at 37°C in a humidified 5% CO2 incubator,
cells were washed with PBS and the medium was changed to DM. The
culture medium was collected for CM and ELISA after 48 h of
incubation.
Assessment of cell growth
C2C12 myoblasts were cultured in DM or CM for 5
days, and the number of viable cells was determined daily using the
trypan blue exclusion test as described previously (41). Briefly, the cells were detached
using 0.05% trypsin-EDTA, collected after neutralization with
growth medium, and resuspended to single-cell suspension for
counting. An aliquot (10 μl) of the cell suspension was
mixed with 0.4% trypan blue (cat. no. T8154; Merck KGaA) and the
number of living cells was determined using a TC20 cell counter
(Bio-Rad Laboratories, Inc.). The number of cells was counted every
day from day 0 to day 5.
Immunofluorescence staining (IFS)
C2C12 myotubes on sterile glass coverslips were
washed in PBS and fixed with 4% paraformaldehyde (cat. no
163-20145; FUJIFILM Wako Pure Chemical Corporation) for 15 min at
room temperature followed by permeabilization with 0.2% Triton
X-100 in PBS for 10 min at room temperature. Samples were blocked
with 5% donkey serum (cat. no SIG-D9663; MilliporeSigma) and 1% BSA
(cat. no 01-2030-2; MilliporeSigma) in PBS for 60 min at room
temperature, and then incubated with primary MYH antibody (1:100;
cat. no. sc376157; Santa Cruz Biotechnology, Inc.) overnight at
4°C, followed by incubation with Donkey Anti-Mouse IgG H&L
(Alexa Fluor 488) secondary antibody (1:200; cat. no. ab150105;
Abcam) for 1 h at room temperature. Nuclei were stained with DAPI
(cat. no. ab-104139; Abcam) for 5 min at room temperature. Images
were captured using a Zeiss LSM 880 Fast Airyscan Confocal and
analyzed using IMARIS (Oxford Instruments). The differentiation
index was calculated as the percentage of nuclei expressing MYH
cells relative to the total nuclei. The fusion index was calculated
as the percentage of nuclei in multinucleated cells with two or
more nuclei relative to the total number of nuclei. These indices
were determined by randomly analyzing at least 10 images from each
sample.
Western bolt analysis
Whole-cell lysates were extracted using lysis buffer
(150 mM NaCl; 50 mM Tris-HCl; pH 7.5; 2 mM EDTA; 1% Triton X-100;
1% sodium deoxycholate and 2% sodium dodecyl sulfate) containing
protease inhibitors (Roche Diagnostics) and phenylmethylsulfonyl
fluoride (Roche Diagnostics). The total protein concentration was
determined using Protein Assay Dye Reagent (Bio-Rad Laboratories,
Inc.) according to the manufacturer's protocol. The samples were
dissolved in NuPage LDS sample buffer (Thermo Fisher Scientific
Inc.) and 1 M dithiothreitol, and then heated for 5 min at 95°C.
Proteins (20-30 μg) were separated on 5-20% Bis-Tris gels
(International Techno Center Co., Ltd.) and transferred to
Hybond-ECL membranes (Cytiva). Membranes were blocked with 5% skim
milk at room temperature for 60 min and then incubated overnight at
4°C with the following primary antibodies: MYH (1:20,000; cat. no.
sc-376157; Santa Cruz Biotechnology, Inc.), MyoD (1:500; cat. no.
sc-377460; Santa Cruz Biotechnology, Inc.), myogenin (1:1,000;
cat.no. sc-12732; Santa Cruz Biotechnology, Inc.), myomaker
(1:1,000; cat. no. NBP2-34175; Novus Biologicals, LLC), myomixer
(1:2,000; cat. no. AF4580; R&D systems, Inc.), Pax7 (1:1,000;
cat. no. AB_528428; Developmental Studies Hybridoma Bank), Id1
(1:1,000; cat. no. 18475-1-AP, Proteintech Group Inc.), Id3
(1:1,000; cat. no. 10389-1-AP, Proteintech Group, Inc.),
p-Smad1/5/8 (1:500; cat. no. 13820, Cell Signaling technology,
Inc.), Smad1/5/8 (1:1,000; cat. no. NB600-962, Novus Biologicals,
LLC), Smad4 (1:1,000; cat. no. 38454, Cell Signaling Technology,
Inc.), BMP4 (1:2,000; cat. no. ab39973; Abcam), MuRF-1 (1:100; cat.
no. sc-398608, Santa Cruz Biotechnology, Inc.), LC3B (1:5,000; cat.
no. 2775; Cell Signaling Technology, Inc.), GAPDH (1:20,000; cat.
no. 60004-1-ig; Proteintech Group Inc.), ACTB (1:1,000; cat. no
A1978; Merck KGaA). Membranes were then washed and incubated with
the corresponding HRP-conjugated secondary antibodies [goat
anti-rabbit IgG (1:3,000; cat. no. 4050-05; SouthernBiotech), goat
anti-mouse IgG (1:3,000; cat. no. 1031-05; SouthernBiotech) and
donkey anti-sheep IgG (1:1,000; cat. no. HAF016, R&D Systems,
Inc.)] for 60 min at room temperature. The signals were detected
using the ECL Prime Western Blotting Detection Reagent (Cytiva) and
images were acquired using a FUSION-FX7 imaging system
(Vilber-Lourmat).
Isolation of RNA and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from cells using Isogen II
(Nippon Gene Co., Ltd.,) and 1 μg aliquots were
reverse-transcribed to cDNA using ReverTra Ace qPCR RT Master Mix
(cat. no. FSQ-201; Toyobo Co., Ltd.). qPCR was performed using the
CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories,
Inc.) with TB Green Premix Ex Taq™ II Fast qPCR (cat. no. RR830A;
Takara Bio, Inc.) according to the manufacturer's protocol. After
performing a denaturation step at 95°C for 3 min, PCR amplification
was conducted using 50 cycles of 15 sec of denaturation at 95°C, 5
sec, annealing at 60°C and 10 sec of extension at 72°C.
Quantitative values were calculated using the 2−ΔΔCq
(42) method and normalized to
the expression of ACTB and GAPDH. RT-qPCR primers were designed for
either human or mouse genes depending on the experimental system.
Primers for MyoD, myogenin, myomaker, myomixer, Id1, Id3, Pax7 and
GAPDH were specific for mouse genes and used for C2C12 cells,
whereas primers for BMP family genes and IL-6 were designed for
human genes. The primers are listed in Table II.
| Table IISequences of primer used for reverse
transcription-quantitative PCR in the present study. |
Table II
Sequences of primer used for reverse
transcription-quantitative PCR in the present study.
| Gene name | Species | Forward primer (5'
to 3') | Reverse primer (5'
to 3') |
|---|
| Pax7 | Mouse |
GTGCCCTCAGTGAGTTCGAT |
CGGGTTCTGATTCCACATCT |
| MyoD | Mouse |
AGTGAATGAGGCCTTCGAGA |
GCATCTGAGTCGCCACTGTA |
|
myogenin | Mouse |
CTACAGGCCTTGCTCAGCTC |
ATGGACGTAAGGGAGTGCAG |
|
myomaker | Mouse |
GGCCTTTACCACCTTCTCC |
AAGCACAGCACAGACAAACC |
|
myomixier | Mouse |
AGTGAACTCCTTAACCAGCTTTC |
CACCTCTGTACTCCCCAGTTT |
| BMP4 | Human |
GGAAGCTAGGTGAGTGTGGC |
CTACGGAATGGCTCCATAGGTC |
| BMP2 | Human |
AGAATGCAAGCAGGTGGGAA |
CCACTTCCACCACGAATCCA |
| BMP6 | Human |
TCAACCGCAAGAGCCTTC |
TTGTCGTACTCCACCAGGTT |
| BMP7 | Human |
CTCTGGCCAGCCTGCAAGATA |
CCGGAACTCTCGATGGTGGA |
| Id1 | Mouse |
GGTACTTGGTCTGTCGGAGC |
GCAGGTCCCTGATGTAGTCG |
| Id3 | Mouse |
ACTTACCCTGAACTCAACGCC |
CAGGCCACCCAAGTTCAGTC |
| IL-6 | Human |
TACCCCCAGGAGAAGATTCC |
TTTTCTGCCAGTGCCTCTTT |
| GAPDH | Mouse |
CAGGGCAAATTCAACGGCACAGTCAA |
GTTCACACCCATCACAAACATGG |
| ACTB | Human |
ACGCCTCTGGCCGTACCACT |
TAATGTCACGCACGATTTCCC |
ELISA
BMP4 concentration in the culture supernatant from
cancer cells was determined in triplicate using a Human BMP4
Quantikine ELISA kit (cat. no DBP400; R&D Systems, Inc.)
according to the manufacturer's protocol.
Statistical analysis
The data were analyzed using JMP Pro 14 (SAS
Institute, Inc.). To compare two groups, differences in mean values
were evaluated using a two-tailed unpaired Student's t-test. For
comparisons of three or more groups, ANOVA followed by Dunnett's or
Tukey's post hoc tests was performed. P<0.05 was considered to
indicate a statistically significant difference and all results
were expressed as the means ± SD.
Results
CM from several human GIC cells inhibit
myoblast differentiation in C2C12
The present study first investigated the
morphological changes in C2C12 cells cultured in DM or CM from
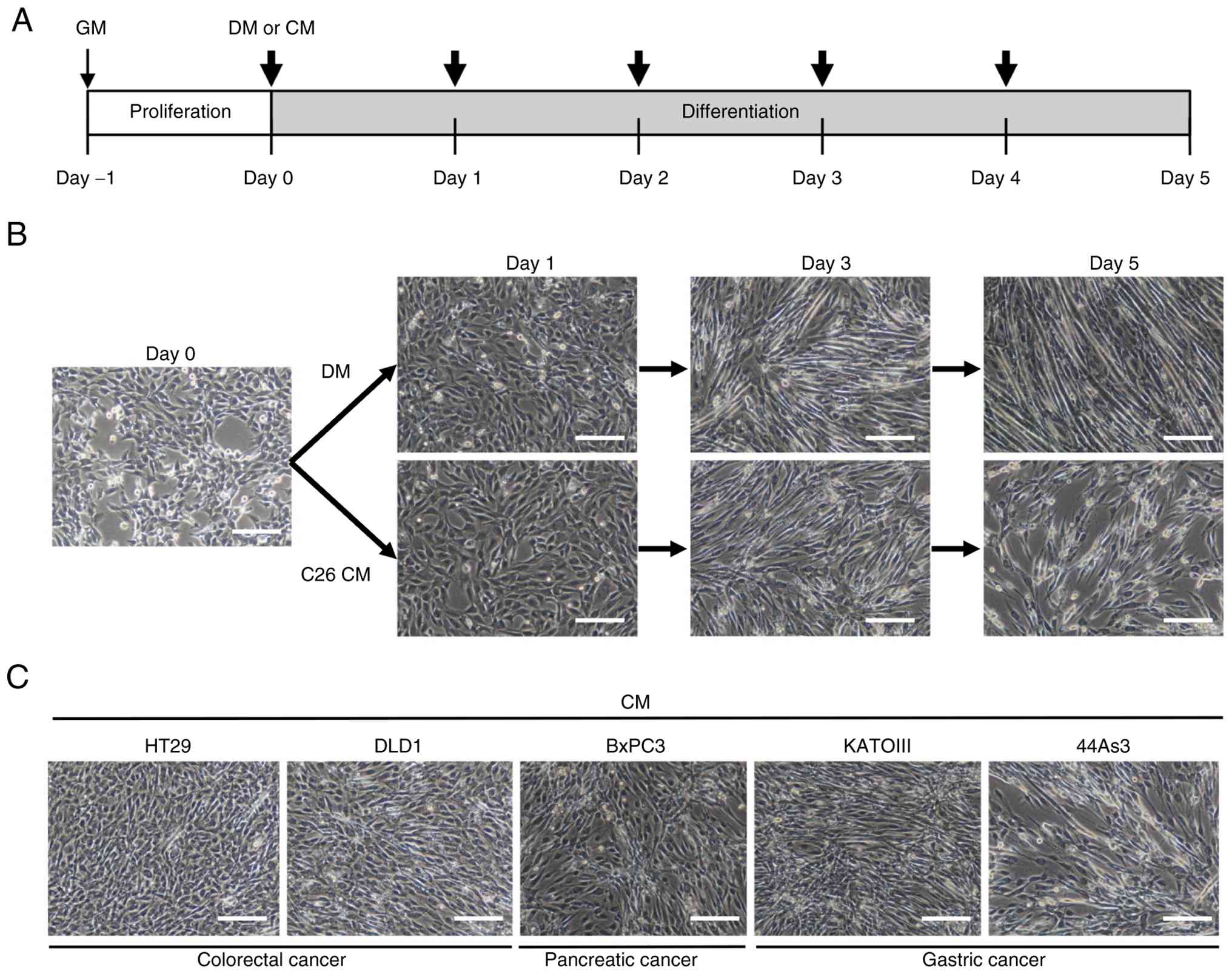
cancer cells for 5 days. From the beginning of C2C12
differentiation induction in DM or CM (designated as day 0), the
medium was changed every 24 h (Fig.
1A). C2C12 myoblasts cultured in DM fused with each other and
transformed into myotubes with multiple nuclei on days 3 and 5
(Fig. 1B). By contrast, C2C12
cells cultured with CM from C26, formed fewer myotubes on day 5
(Fig. 1B). Next, the inhibitory
effects of CM from 20 human GIC cell lines on myotube formation in
C2C12 cells was explored. As shown in Fig. 1C, CM from the colorectal cancer
cell lines HT29 and DLD1, the pancreatic cancer cell lines BxPC3
and the gastric cancer cell lines KATOIII and 44As3, inhibited the
myogenic differentiation of C2C12 cells. By contrast, CMs from the
remaining GIC cell lines exhibited either minimal or weak
inhibitory effects on myoblast differentiation (Fig. S1).
GIC CM inhibits myoblast differentiation
into myotubes and suppresses the expression of myogenic factors in
C2C12
To investigate the mechanism underlying the
inhibitory effect of CM from GIC on myoblast differentiation,
subsequent analysis focused on HT29 and DLD1 cell lines, which
exhibited the most pronounced inhibitory effects on C2C12
morphology based on visual assessment in the initial screening
(Figs. 1C and S1). The 58As9 cell line, which did not
inhibit the differentiation, was used as a negative control.
In C2C12 cells cultured with DM and 58As9 CM, cell
growth was markedly suppressed, and mature myotubes with
MYH-positive staining appeared on day 5 (Fig. 2A and B). Differentiation and
fusion indices were estimated to be >50% (Fig. 2C). By contrast, C2C12 cells
cultured with HT29 and DLD1 CMs proliferated with time dependency
by day 5 (Fig. 2A). The formation
of MYH-positive myotubes (Fig.
2B), and differentiation and fusion indices were significantly
suppressed compared with cells cultured in DM and 58As9 CM
(Fig. 2C).
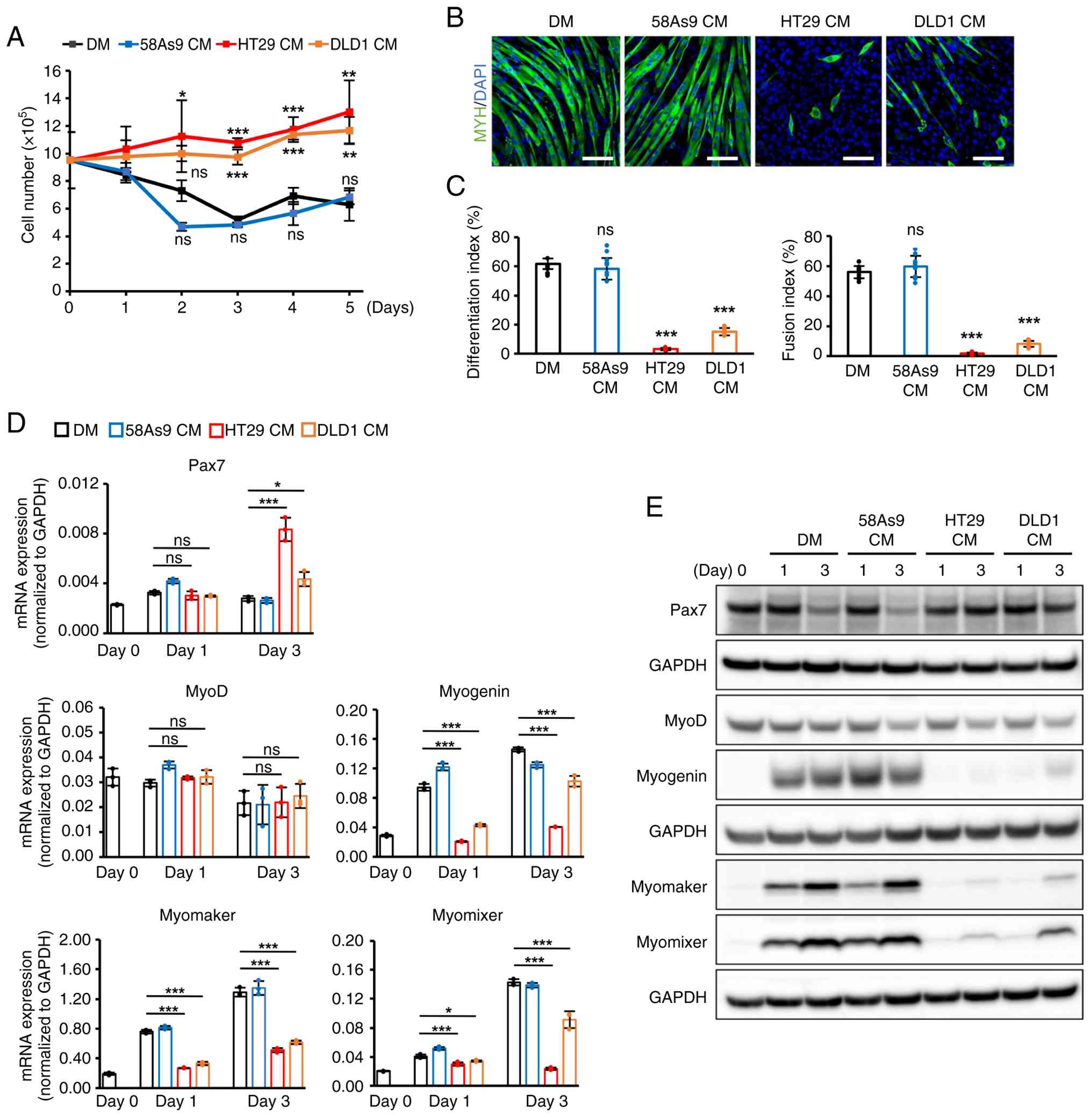 | Figure 2Analysis of gene expression
associated with myogenic differentiation in C2C12 cells which
cultured with DM or CM from control 58As9, HT29 and DLD1 cells. (A)
Cell proliferation of C2C12 myoblast cultured with DM or three GIC
CMs for 5 days (n=3). (B) Immunofluorescence staining of MYH in
C2C12 cells with DM or CMs from three GIC cells on day 5.
Representative images are shown (green, MYH; blue, DAPI). Scale
bar, 100 μm. (C) Differentiation and the fusion indices that
were quantitatively estimated in C2C12 cells with DM or CM from
three GIC cells (n=10). (D) Reverse transcription-quantitative PCR
(n=3) and (E) western blot analysis of Pax7, MyoD, myogenin,
myomaker and myomixer in C2C12 cells with DM or three GIC CMs (on
day 0, day 1 and day 3). All experiments were independently
repeated at least three times. Data are presented as mean ± SD.
Statistical significance was determined by comparison with control
(DM) on each day. ns not significant, *P<0.05,
**P<0.01, ***P<0.001. DM,
differentiation medium; CM, conditioned medium. |
Next, the present study evaluated the expression of
myogenic genes related to differentiation and cell fusion. In C2C12
cells with controls, the protein expression level of Pax7, which is
a key marker for satellite cells and myoblasts, visibly decreased
on day 3, whereas the expression of the MRF member MyoD showed a
slight reduction. Conversely, the mRNA and protein expression
levels of another MRF member, myogenin and its downstream targets,
myomaker and myomixer, increased over time (days 1-3) under control
conditions (Fig. 2D and E). By
contrast, in C2C12 cells cultured with HT29 and DLD1 CM, Pax7 mRNA
expression significantly increased, and its protein expression
level was preserved on day 3. The expression of MyoD did not show a
significant difference when compared with cells cultured in DM and
58As9 CM. However, the expression of myogenin and its downstream
targets was remarkably suppressed compared with cells cultured in
DM and 58As9 CM (Fig. 2D and E).
These results suggest that CMs from HT29 and DLD1 inhibited C2C12
differentiation from myoblasts to myotubes by decreasing the
expression of myogenin and its downstream factors. At this point,
we hypothesized that some secreted factor from HT29 and DLD1 may
exogenously inhibit myoblast differentiation in C2C12 cells by
activating the intrinsic signaling pathway.
HT29 and DLD1 cells secrete BMP4, which
activates Smad-Id signaling in C2C12 myoblasts
The present study focused on BMP signaling as a
possible mechanism underlying impaired differentiation in C2C12
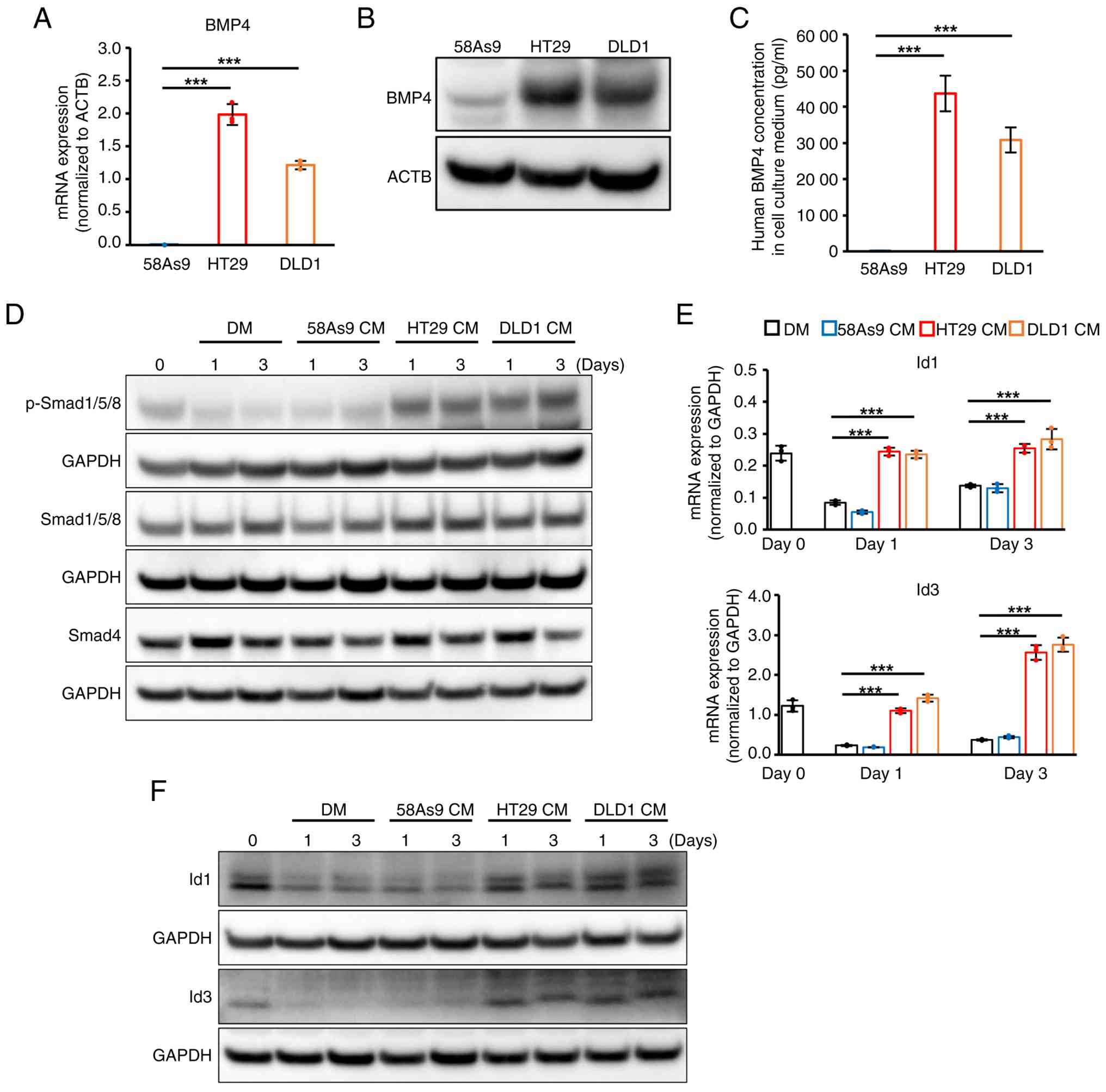
cells treated with HT29 and DLD1 CMs. First, the mRNA expression
levels of BMP family members BMP2, BMP4, BMP6 and BMP7 in control
58As9, HT29 and DLD1 cells were investigated. BMP4 mRNA expression
was significantly higher in HT29 and DLD1 when compared with that
in 58As9 cells (Fig. 3A). Higher
expression of BMP4 protein expression was also observed in cell
lysates and culture supernatants from HT29 and DLD1 cells when
compared with that in 58As9 cells (Fig. 3B and C). In addition, mRNA
expression of the other BMPs was not commonly expressed in HT29 and
DLD1 cells (Fig. S2). Next, the
present study analyzed whether BMP downstream Smad-Id signaling was
activated in C2C12 cells treated with HT29 and DLD1 CM. Expression
of p-Smad1/5/8 (pSmad1/5/8), which is the activated form of
Smad1/5/8, was visibly higher in C2C12 cells treated with HT29 and
DLD1 CMs when compared with the other two controls during
differentiation (Fig. 3D). There
was no apparent difference in the expression levels of total
Smad1/5/8 or Smad4 among all four groups (Fig. 3D). With respect to the Id family,
mRNA expression of Id1 and Id3 was observed in C2C12 cells on day
0. Expression of these mRNAs declined in C2C12 cells treated with
DM and 58As9 CM on day 1 and 3; however, high expression of Id1 and
Id3 was sustained in C2C12 cells treated with HT29 and DLD1 CMs
(Fig. 3E). Furthermore, western
blotting analysis demonstrated findings consistent with RT-qPCR
results for the protein levels of Id1 and Id3 (Fig. 3F). These results indicate that
exogenous BMP4, which is secreted by HT29 and DLD1, may inhibit
C2C12 differentiation by activating the Smad-Id pathway.
Dorsomorphin ameliorates myogenic
differentiation by suppressing Smad-Id signaling in C2C12 with HT29
and DLD1 CMs
To clarify whether the inhibitory effect of HT29 and
DLD1 CMs on C2C12 differentiation is caused by the activation of
BMP-Smad signaling, the present study analyzed the reverse effect
of an inhibitor of the BMP type I receptor, dorsomorphin. C2C12
cells cultured in HT29 and DLD1 CMs were treated with or without 5
μM dorsomorphin. IFS analysis showed that dorsomorphin
markedly increased the number of MYH-positive myotubes on day 5
(Fig. 4A). The differentiation
and fusion indices were also increased by this treatment (Fig. 4B). Moreover, the expression levels
of myogenin, myomaker and myomixer were markedly restored by
dorsomorphin, whereas MyoD expression was slightly decreased
(Fig. 4C). The present study
further confirmed that the drug treatment decreased the expression
of p-Smad1/5/8, Id1 and Id3 in C2C12 cells treated with both HT29
and DLD1 CMs (Fig. 4D-F). These
results demonstrated that attenuation of BMP-Smad signaling by
dorsomorphin reversed the inhibitory effect of HT29 and DLD1 CM on
C2C12 differentiation.
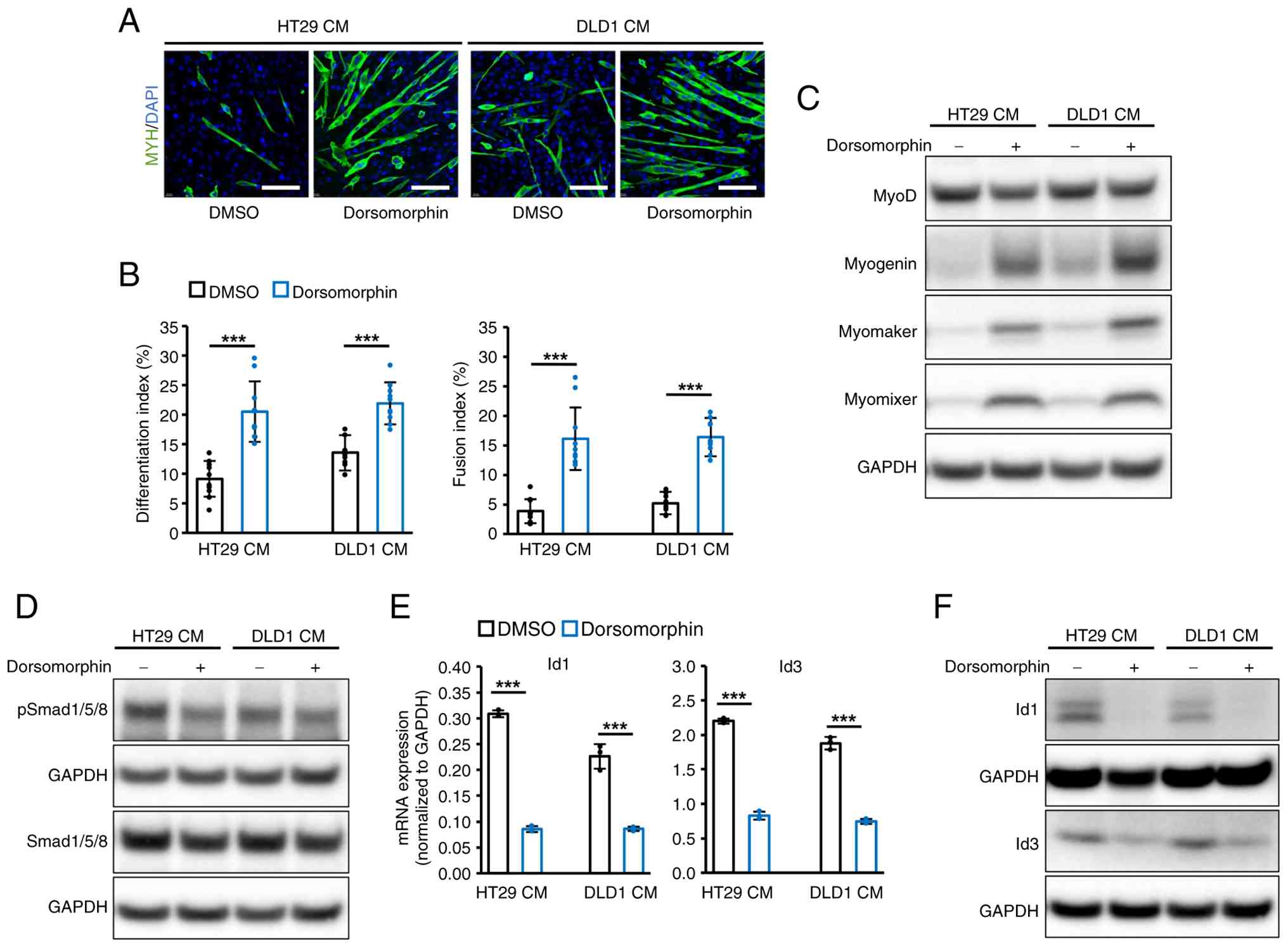 | Figure 4Dorsomorphin ameliorated the
inhibitory effect of HT29 and DLD1 CMs on myogenic differentiation
in C2C12 cells. (A) Immunofluorescence staining of MYH in C2C12
cells cultured with CM from HT29 and DLD1 on day 5 with or without
Dorsomorphin treatment. Representative images were shown (green,
MYH; blue, DAPI). Scale bar, 100 μm. (B) Differentiation and
fusion indices were quantitatively assessed in C2C12 cells with
HT29 and DLD1 CMs with or without Dorsomorphin (n=10). (C) The
protein expression of MyoD, myogenin, myomaker and myomixer in
C2C12 cells with HT29 and DLD1 CMs with or without Dorsomorphin on
day 1. (D) WB analysis of Smad1/5/8 and p-Smad1/5/8 in C2C12 cells
with HT29 and DLD1 CMs with or without Dorsomorphin on day 1. (E)
Reverse transcription-quantitative PCR (n=3) and (F) WB analysis of
Id1 and Id3 in C2C12 cells with HT29 and DLD1 CMs with or without
Dorsomorphin on day 1. For panels (D) Smad1/5/8 and (F) Id3, the
corresponding GAPDH loading controls were derived from the same
membrane processed on the same experimental day; different exposure
times were used for visualization. All experiments were
independently repeated at least three times, and data are presented
as mean ± SD. Statistical significance was determined by
comparisons between the dorsomorphin-treated and -untreated C2C12
cells with HT29 and DLD1 CMs. ***P<0.001. WB, western
blotting; DM, differentiation medium; CM, conditioned medium. |
Assessment of BMP-Smad signal in other
GIC cell lines exhibiting the inhibitory effect on C2C12
differentiation
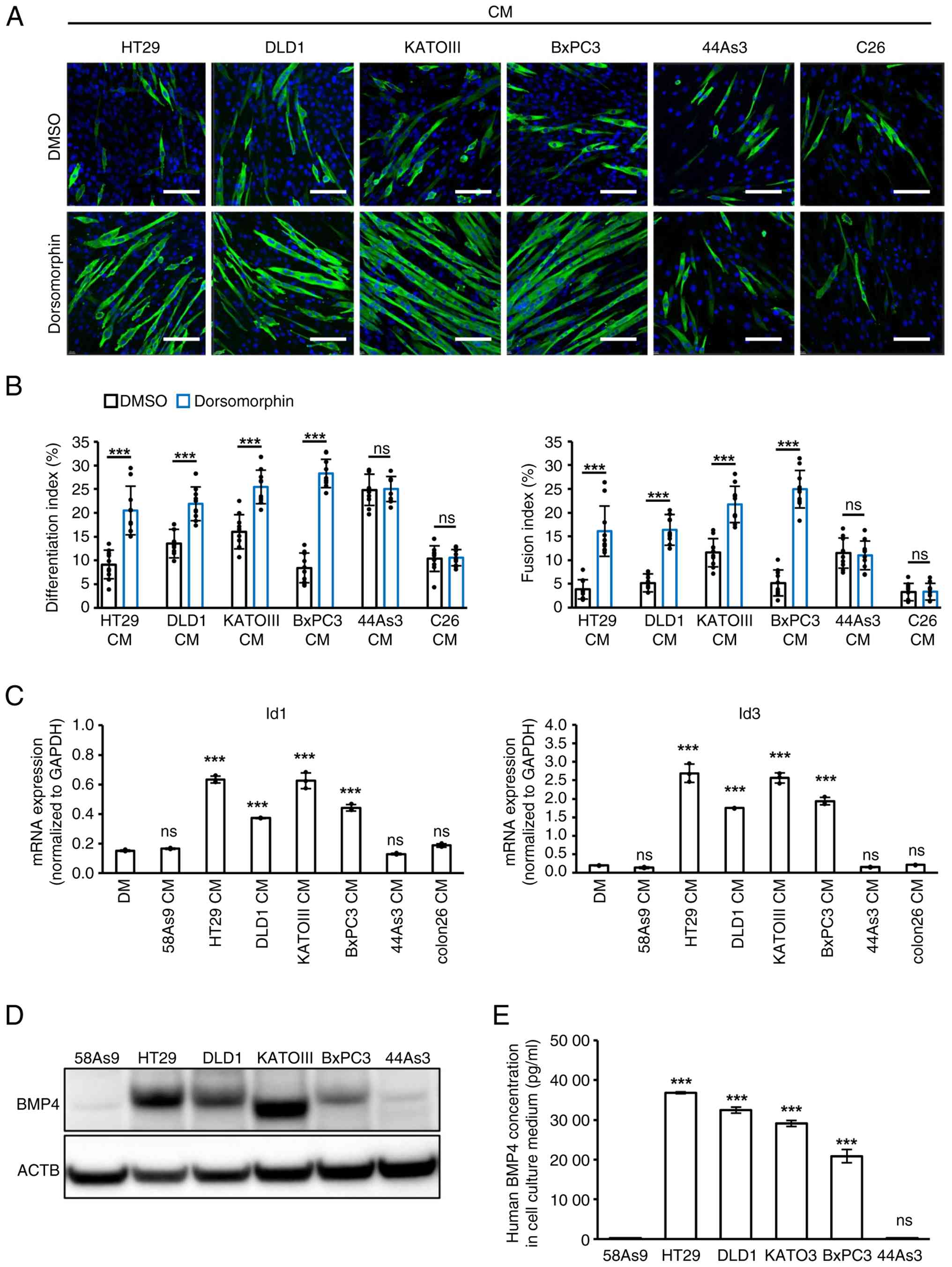
Whether the inhibition of C2C12 differentiation by
CM from other human GIC cells (KATOIII, 44As3 and BxPC3) or mouse
C26 cells is mediated by the BMP-Smad-Id signaling pathway was next
investigated. The morphological changes with or without
dorsomorphin in C2C12 cells cultured with CMs from these cells were
first analyzed. Dorsomorphin treatment significantly restored
MYH-positive myotube formation, along with increased
differentiation and fusion indices, in C2C12 cells treated with
KATOIII and BxPC3 CMs, as observed in HT29 and DLD1 CMs (Fig. 5A and B). However, this treatment
did not affect the inhibitory effects of 44As3 or C26 CMs (Fig. 5A and B). Furthermore, CM from
KATOIII and BxPC3, in addition to HT29 and DLD1, significantly
increased the expression of Id1 and Id3 mRNAs in C2C12 cells
compared to DM or CMs from other remaining cells (Fig. 5C). Finally, BMP4 was highly
expressed and secreted not only in HT29 and DLD1 but also in
KATOIII and BxPC3 cells, compared with control 58As9 cells. By
contrast, 44As3 cells did not express or secrete BMP4 (Fig. 5D and E). These results suggest
that the inhibitory effect of KATOIII and BxPC3 CMs on C2C12
differentiation was induced via the BMP4-Smad-Id signaling
pathway.
By contrast, C26 cells are known to induce muscle
atrophy via UPS and ALS, which are activated by proinflammatory
cytokines, including IL-6 and TNFα (8-11).
Thus, an experiment to analyze whether CM from 44As3 and C26 caused
atrophy in myotubes differentiated from C2C12 cells was conducted
(Fig. S3A). Analysis revealed
that CM from 44As3 and C26, but not HT29 or DLD1, induced visible
atrophy in myotubes, with a significant decrease in myotube
diameter (Fig. S3B and C).
Higher expression levels of UPS (associated with MuRF-1) and ALS
(associated with LC3B-II) were observed in myotubes cultured with
44As3 and C26 CMs than with HT29 and DLD1 CMs (Fig. S3D). In addition, 44As3 cells
expressed higher levels of IL-6 mRNA compared with 4
BMP4-expressing GIC (Fig. S3E).
These findings suggest that 44As3- and C26-derived IL-6 not only
inhibited myogenic differentiation of C2C12 cells but also
accelerated UPS- and ALS-dependent atrophy in myotubes.
BMP4 gene silencing by siRNA in HT29
cells rescues myogenic differentiation
The present study attempted to confirm whether
cancer-derived BMP4 inhibits myogenic differentiation by activating
Smad-Id signaling in C2C12 cells. A knockdown analysis was
performed in HT29 cells using siBMP4. After siBMP4 transfection,
BMP4 expression and secretion were effectively inhibited in
siBMP4-HT29 cells, compared with siCtl (Fig. 6A-C). When C2C12 cells were
cultured with CM from siBMP4-HT29, myotube formation with
multinuclei was remarkably restored relative to siCtl-HT29, and
significantly higher indices of differentiation and fusion were
observed (Fig. 6D and E).
Moreover, the expression levels of myogenin, myomaker and myomixer
were apparently increased in C2C12 cells with CM from siBMP4-HT29
(Fig. 6F). Finally, siBMP4-HT29
CM did not elevate the expression of p-Smad1/5/8, Id1 and Id3 in
C2C12 cells relative to siCtl-HT29 CM (Fig. 6G and H). Taken together, these
results suggest that HT29-derived BMP4 itself inhibited C2C12
differentiation into myotubes by activating the Smad1/5/8-Id1 and
-Id3 signaling pathways.
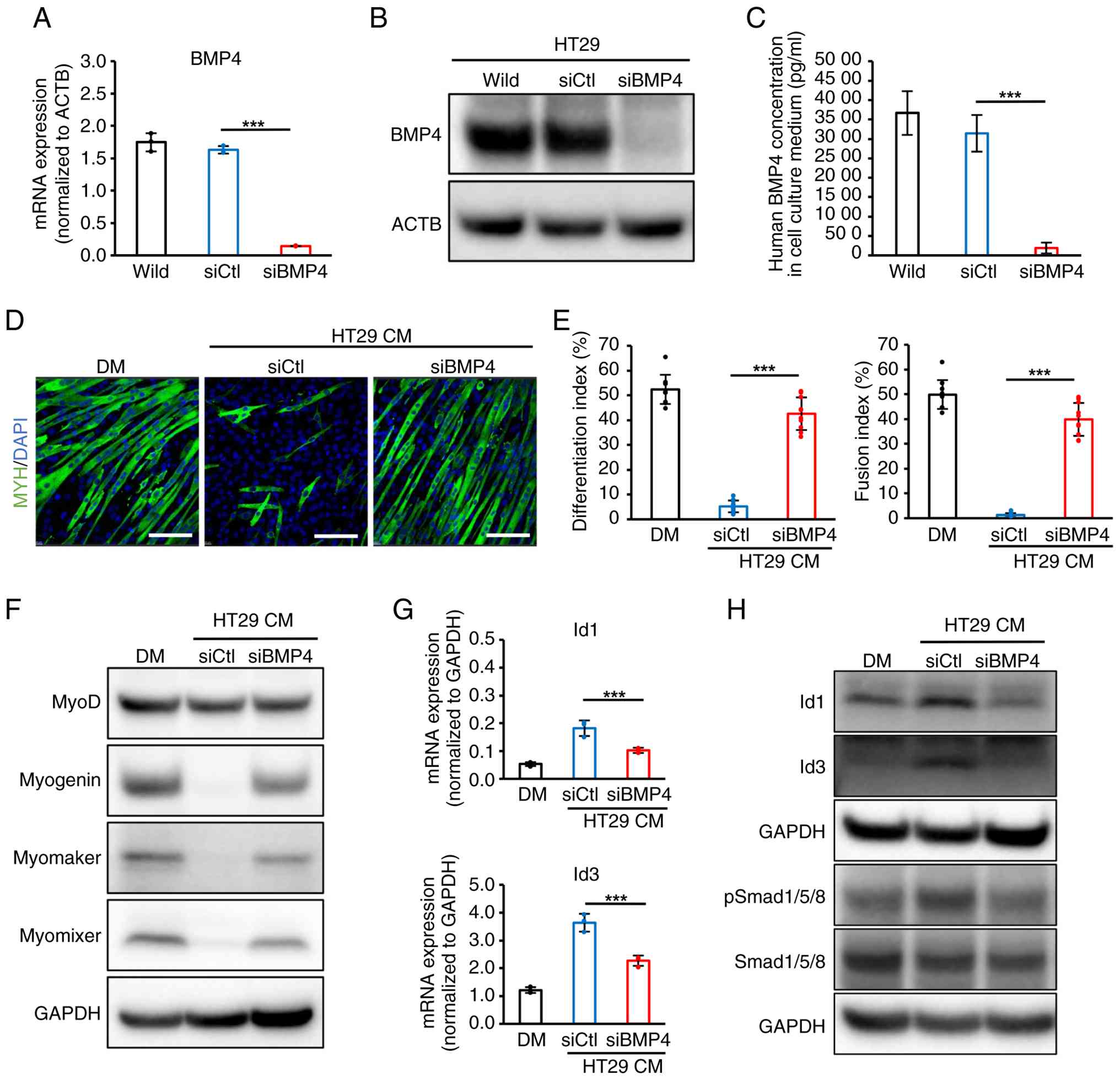 | Figure 6Abrogation of BMP4 by siRNA reversed
the inhibitory effect of HT29 CM on myogenic differentiation in
C2C12 cells. (A) The efficiency of BMP4 knockdown in HT29 cells was
evaluated by (A) RT-qPCR (n=3) and (B) WB analysis. (C) BMP4
secretion from HT29 cell was evaluated by ELISA in triplicate. A
total of two independent experiments were performed. (D)
Representative images and (E) quantification of the effects of
siBMP4 transfection to HT29 cells on C2C12 differentiation.
Immunofluorescence staining of MYH in C2C12 cells on day 5, which
was cultured with DM or CMs from siCtl- and siBMP4-transfected HT29
cells. (D) Scale bar, 100 μm. (E) Differentiation and fusion
indices were estimated and plotted on graph (n=10). (F) WB analysis
of MyoD, myogenin, Myomaker and Myomixer in C2C12 cells on day 1
with DM or CMs from siCtl- and siBMP4-transfected HT29 cells. (G)
RT-qPCR of Id1and Id3 in C2C12 cells on day 1 (n=3). (H) WB
analysis of Id1, Id3 and Smad signaling proteins in C2C12 cells on
day 1 with DM or CMs from siCtl- and siBMP4-HT29 cells. These
experiments were independently repeated at least three times, and
data are presented as mean ± SD. Statistical significance was
determined by comparison between siBMP4 and siCtl.
***P<0.001. si, small interfering; WB, western
blotting; RT-qPCR, reverse transcription-quantitative PCR; ctl,
control; DM, differentiation medium; CM, conditioned medium; p,
phosphorylation. |
Discussion
The present study found that CM treatment from the
five human GIC cell lines and mouse C26 cells morphologically
inhibited myotube formation in C2C12 cells. Among the five GIC cell
lines, HT29 and DLD1 cells were subjected to subsequent analyses.
C2C12 cells cultured with CMs from these cells proliferated with
time dependency and failed to fuse with each other. Furthermore,
the expression of myogenin and its downstream targets was markedly
suppressed, whereas Pax7 expression was sustained. In
differentiating fetal myoblasts, Pax7 is co-expressed with MyoD,
but is absent in myogenin-expressing myotubes (43). Thus, we hypothesized that a factor
secreted by HT29 and DLD1 cells may inhibit the switching of gene
expression from Pax7 to myogenin and drive C2C12 myoblasts out of
the normal process of myogenic differentiation.
Previous studies have demonstrated that the number
of muscle precursor cells increases under various muscle atrophy
conditions, including cancer cachexia (16,44,45). This accumulation of muscle
precursor cells in atrophying muscles may be the result of a fusion
defect that inhibits myoblast differentiation and regeneration
(16). At this point, both
sustained proliferation with Pax7 expression and unsuccessful cell
fusion, which were observed in C2C12 cells cultured with HT29 and
DLD1, may be consistent with the accumulation of muscle precursor
cells as reported in cancer-induced muscle atrophy (16,44,45).
Next, the BMP signaling pathway was investigated. We
analyzed the mRNA expression of BMP-2, 4, 6 and 7, as studies have
reported that these BMP members are expressed in human cancer cells
and tissues (46-50). HT29 and DLD1 cells commonly
expressed BMP4 mRNA, but not the mRNAs of the other three BMPs. The
secretion of BMP4 protein was also confirmed. Furthermore, the
present study showed that CMs from these cells inhibited C2C12
differentiation through activation of the Smad-Id signaling and
blocked BMP4-Smad1/5/8 signaling with dorsomorphin, restoring
myoblast differentiation. Finally, abrogation of BMP4 by siRNA
verified that HT29-derived BMP4 was key for inhibiting myogenic
differentiation in C2C12. Taken together, these results suggest
that HT29- and DLD1-derived BMP4 triggered the impairment of C2C12
differentiation by activating the Smad1/5/8-Id1 and -Id3 signaling
axis.
The present study also revealed that CMs from HT29
and DLD1 suppressed the expression of MRF myogenin and its
downstream targets, but not that of MRF MyoD in C2C12 cells. MRFs
are muscle-specific basic helix-loop-helix (bHLH) transcriptional
factors that function as transcriptional activators via
heterodimerization with a subfamily of bHLH member E-protein
(51,52). In particular, the MyoD/E-protein
complex transactivates another bHLH gene, myogenin, by binding to
the E-box DNA element within the myogenin promoter, which
cooperatively interacts with homeodomain transcription factors,
such as Pbx and Meis (22,53-57).
These proteins form a higher-order transcriptional complex that
facilitates chromatin remodeling at the myogenin locus and allows
the recruitment of additional transcriptional regulators (56,57). By contrast, Id proteins prevent
MyoD activity by forming antagonistic dimers with E-protein
(28,29). This sequestration of E-proteins by
Id proteins prevents the formation of the MyoD/E-protein complex,
thereby inhibiting the recruitment of MyoD to the myogenin promoter
and impairing chromatin remodeling required for transcriptional
activation (28,29). Given these results, the induction
of Id1 and Id3 via BMP4-Smad signaling may suppress myogenin
transcription by forming E-protein/Id1 and/or Id3 complexes,
instead of E-protein/MyoD. A proposed scheme of the inhibitory
effect of human GIC-derived BMP4 on myoblast differentiation is
shown in Fig. 7.
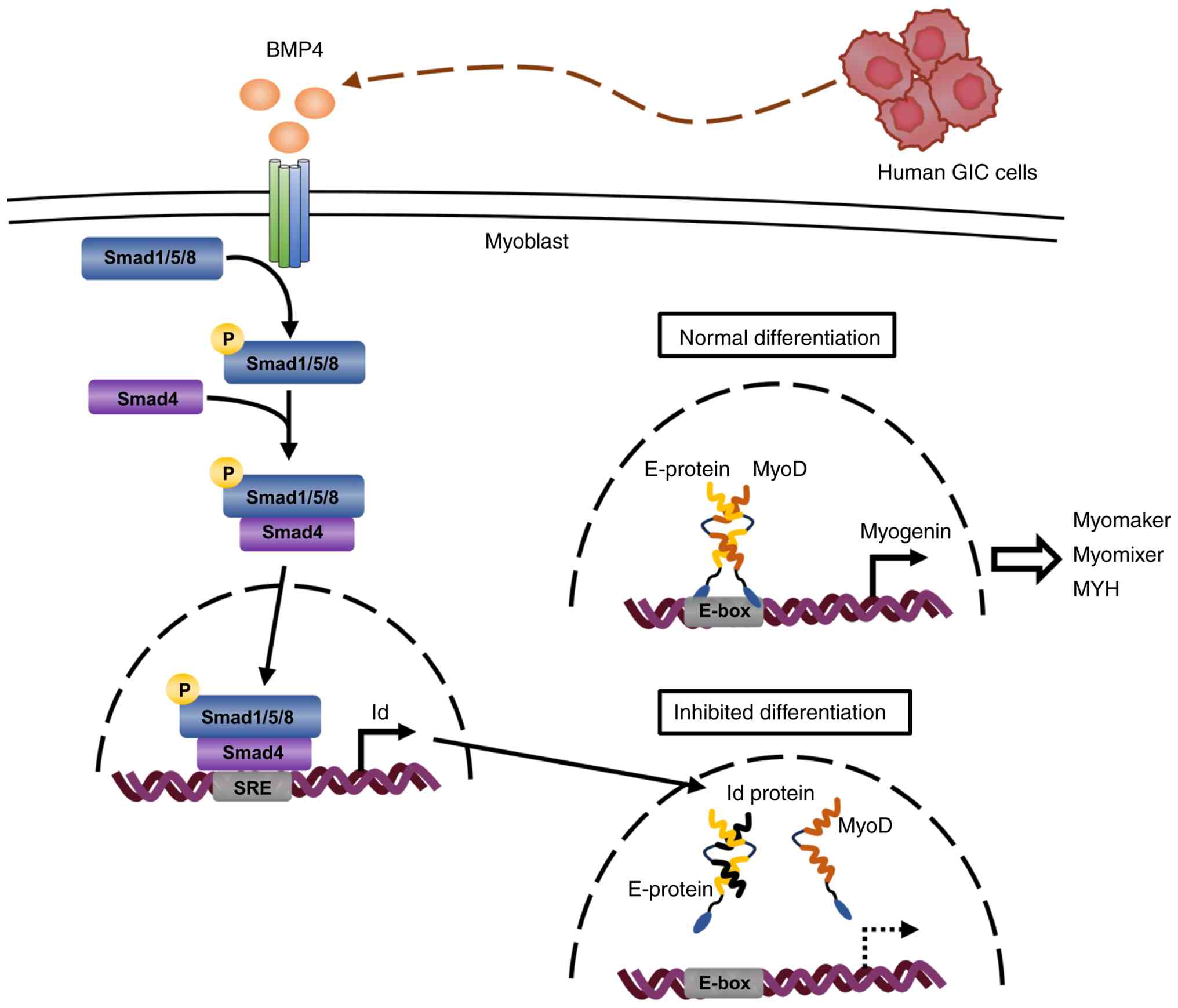 | Figure 7A possible mechanism of inhibitory
effect of human GIC-derived BMP4 on myoblast differentiation
through Smad-Id signaling. During the normal process of myogenic
differentiation, a heterodimer complex MyoD/E-protein is formed
through each HLH domain and activates the transcription of myogenin
gene via binding to E-box DNA element within its promoter.
Subsequently, myogenin protein transactivates the expression of the
downstream genes, and eventually completes the terminal
differentiation to myotube. By contrast, human GIC cells-derived
BMP4 binds to BMP receptors, which in turn phosphorylates
Smad1/5/8. The p-Smad1/5/8 forms a complex with Smad4 and
translocates to the nucleus. The p-Smad1/5/8-Smad4 complex binds to
the Smad responsive DNA element (SRE) in the Id gene promoter to
upregulate mRNA expression. As Id protein contains an HLH domain,
but it lacks a basic DNA binding region, Id prevents MyoD activity
by forming antagonistic dimers with E-protein through each HLH.
GIC, gastrointestinal cancer; p, phosphorylated; HLH,
helix-loop-helix; BMP, bone morphogenetic protein. |
Previous studies have reported that BMP signaling
modulates myogenesis-related microRNAs, including miR-1, miR-133
and miR-206, which regulate the balance between myoblast
proliferation and differentiation (58,59). BMP-Smad signaling may also
influence the expression of these post-transcriptional regulators,
which are essential for myogenic differentiation. Moreover,
epigenetic mechanisms, such as chromatin remodeling and histone
modifications, may contribute to the regulation of myogenic gene
expression downstream of BMP signaling (60). These alternative mechanisms may
cooperate with the Id-mediated inhibition of the MyoD activity to
suppress the transcriptional activation of myogenic genes, such as
myogenin.
Previously, Ono et al (61) demonstrated that in satellite cells
isolated from mouse skeletal muscle, myogenic differentiation was
inhibited by the addition of recombinant BMP4 protein. Conversely,
blocking the BMP4-Smad1/5/8 signaling axis with the BMP antagonist
Noggin or Dorsomorphin induced precocious differentiation (61). Furthermore, the study speculated
that myogenic cells per se may secrete BMP4 and act on
satellite cells in vivo, and concluded that during muscle
regeneration, BMP4 signaling may be initially required to allow the
expansion of the satellite cell pool by stimulating proliferation
and preventing precocious differentiation (61). Given this previous study and the
present observations, autocrine BMP4 secretion from satellite cells
may be temporarily essential for myoblast proliferation during the
early phase. However, secretion from cancer cells may continuously
activate Smad1/5/8-Id signaling to prevent myoblasts from
undergoing myogenic differentiation and may eventually induce
muscle wasting.
The present study determined that KATOIII and BxPC3,
consistent with HT29 and DLD1, could inhibit differentiation via
BMP4-Smad1/5/8-Id signaling. These results suggest that the
activation of BMP4-Smad1/5/8-Id signaling may be a central
mechanism underlying myogenic differentiation inhibition in human
GIC cells, because four (including HT29, DLD1, KATOIII and BxPC3)
of the five GIC cell types inhibited C2C12 differentiation via this
signaling pathway. Additionally, the present study investigated the
cross-species interaction between human GIC cells and mouse
myoblasts. Future validation using human primary myoblasts would
improve the contextualization of the findings for human
pathophysiology. However, the present study suggests that BMP4
derived from human GIC cells inhibited myogenic differentiation in
murine C2C12 cells using pharmacological inhibition and genetic
knockdown. Recombinant human BMP4 is well established to be
biologically active in C2C12 cells, where it induces Smad1/5/8
phosphorylation and inhibits myogenic differentiation, indicating
effective activation of murine BMP receptors (62). Furthermore, BMP signaling and
receptor activation mechanisms are highly conserved across species
(63-65). Notably, the amino acid sequence
identity of the BMP receptor BMPR-II between humans and mice is
~96.6%, indicating structural conservation (66). Taken together, these findings
suggest that potential species-specific differences in receptor
affinity are unlikely to affect downstream signaling or confound
the interpretation of the results.
Meanwhile, previous studies have reported that
cancer-secreted BMPs promote proliferation, invasion and
epithelial-mesenchymal transition in an autocrine manner (46-50). Notably, autocrine
BMP4-Smad1/5/8-Id signaling was activated in HT29 and DLD1 cells
per se, and contributed to accelerating tumor growth of the
HT29 xenograft in nude mice (49). Therefore, BMP4-expressing GIC
cancer, such as HT29 and DLD1, may carry out dual roles in
promoting tumor growth and cancer cachexia. Furthermore, an in
vitro study demonstrated that muscle differentiation in human
skeletal muscle myoblasts was inhibited by sera from
cancer-cachectic patients, including patients with colorectal
cancer (67). Moreover, previous
studies have reported that circulating BMP4 has been detected at
~230 pg/ml by ELISA in human serum, and its levels are associated
with disease status in patients with cancer, suggesting that BMP4
may function as a systemic factor in cancer progression (68,69). In the future, analyzing the
association between the serum BMP4 levels and the cachexia grade in
patients with GIC may be important to verify these in vitro
findings.
In conclusion, the present study identified
cancer-derived BMP4 as an essential factor that inhibits the
myogenic differentiation of C2C12 in human GIC cells. As the
present study was limited to in vitro experiments, further
in vivo studies using mouse xenografts or clinical samples
from patients are needed to clarify whether cancer-derived BMP4
inhibits skeletal muscle differentiation and eventually causes
cachexia. However, this novel insight may provide clues for the
elucidation of the complicated mechanisms underlying cancer-induced
cachexia in humans.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
KH designed and performed the experiments, analyzed
and interpreted the data, and drafted the manuscript. YK designed
and supervised the study, analyzed and interpreted the data, and
drafted and reviewed the manuscript. NK, SI and SM performed the
experiments. TT interpreted the data and reviewed the manuscript.
HN contributed to the conception and design of the study, provided
critical interpretation of the data, and supervised the overall
research direction. KH and YK confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
GIC
|
gastrointestinal cancer
|
|
UPS
|
ubiquitin-protease system
|
|
ALS
|
autophagy-lysosome system
|
|
GM
|
growth medium
|
|
DM
|
differentiation medium
|
|
CM
|
conditioned medium
|
|
IFS
|
immunofluorescence staining
|
|
siRNA
|
small interfering RNA
|
|
siCtl
|
control siRNA
|
|
siBMP4
|
BMP4 siRNA
|
Acknowledgements
Not applicable.
Funding
No funding was received.
References
|
1
|
Tisdale MJ: Biology of cachexia. J Natl
Cancer Inst. 89:1763–1773. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Argilés JM, Busquets S, Stemmler B and
Lopez-Soriano FJ: Cancer cachexia: Understanding the molecular
basis. Nat Rev Cancer. 14:754–762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gannavarapu BS, Lau SKM, Carter K, Cannon
NA, Gao A, Ahn C, Meyer JJ, Sher DJ, Jatoi A, Infante R and Iyengar
P: Prevalence and survival impact of pretreatment cancer-associated
weight loss: A tool for guiding early palliative care. J Oncol
Pract. 14:e238–e250. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gilmore LA, Olaechea S, Gilmore BW,
Gannavarapu BS, Alvarez CM, Ahn C, Iyengar P and Infante RE: A
preponderance of gastrointestinal cancer patients transition into
cachexia syndrome. J Cahexia Sarcopenia Muscle. 13:2920–2931. 2022.
View Article : Google Scholar
|
|
5
|
Fearon K, Strasser F, Anker SD, Bosaeus I,
Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N,
Mantovani G, et al: Definition and classification of cancer
cachexia: An international consensus. Lancet Oncol. 12:489–495.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Norton JA, Shamberger R, Stein TP, Milne
GWA and Brennan MF: The influence of tumor-bearing on protein
metabolism in the rat. J Surg Res. 30:456–462. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Smith KL and Tisdale MJ: Increased protein
degradation and decreased protein synthesis in skeletal muscle
during cancer cachexia. Br J Cancer. 67:680–685. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Martin A, Gallot YS and Freyssenet D:
Molecular mechanisms of cancer cachexia-related loss of skeletal
muscle mass: Data analysis from preclinical and clinical studies. J
Cachexia Sarcopenia Muscle. 14:1150–1167. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Acharyya S, Ladner KJ, Nelson LL, Damrauer
J, Reiser PJ, Swoap S and Guttridge DC: Cancer cachexia is
regulated by selective targeting of skeletal muscle gene products.
J Clin Invest. 114:370–378. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cai D, Frantz JD, Tawa NE Jr, Melendez PA,
Oh BC, Lidov HGW, Hasselgren PO, Frontera WR, Lee J, Glass DJ and
Shoelson SE: IKKbeta/NF-kappaB activation causes severe muscle
wasting in mice. Cell. 119:285–298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Paul PK, Gupta SK, Bhatnagar S, Panguluri
SK, Darnay BG, Choi Y and Kumar A: Targeted ablation of TRAF6
inhibits skeletal muscle wasting in mice. J Cell Biol.
191:1395–1411. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cao Z, Zhao K, Jose I, Hoogenraad NJ and
Osellame LD: Biomarkers for cancer cachexia: A mini review. Int J
Mol Sci. 22:45012021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gallagher IJ, Stephens NA, MacDonald AJ,
Skipworth RJE, Husi H, Greig CA, Ross JA, Timmons JA and Fearon
KCH: Suppression of skeletal muscle turnover in cancer cachexia:
Evidence from the transcriptome in sequential human muscle
biopsies. Clin Cancer Res. 18:2817–2827. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonneto A, Penna F, Aversa Z, Mercantini
P, Baccino FM, Costelli P, Ziparo V, Lucia S, Fanelli FR and
Muscaritoli M: Early changes of muscle insulin-like growth factor-1
and myostatin gene expression in gastric cancer patients. Muscle
Nerve. 48:387–392. 2013. View Article : Google Scholar
|
|
15
|
D'orland C, Marzetti E, François S,
Lorenzi M, Conti V, Stasio ED, Rosa F, Brunelli S, Doglietto GB,
Pacelli F and Bossola M: Gastric cancer does not affect the
expression of atrophy-related genes in human skeletal muscle.
Muscle Nerve. 49:528–533. 2014. View Article : Google Scholar
|
|
16
|
Talbert EE and Guttridge DC: Impaired
regeneration: A role for the muscle microenvironment in cancer
cachexia. Semin Cell Dev Biol. 54:82–91. 2016. View Article : Google Scholar
|
|
17
|
Talbert EE, Cuitino MC, Landner KJ,
Rajasekerea PV, Siebert M, Shakya R, Leone GW, Ostrowski MC, Paleo
B, Weisleder N, et al: Modeling human cancer-induced cachexia. Cell
Rep. 28:1612–1622.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arneson PC and Doles JD: Impaired muscle
regeneration in cancer-associated cachexia. Trends in Cancer.
5:579–582. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Penna F, Costamagna D, Fanzani A, Bonelli
G, Baccino FM and Costelli P: Muscle wasting and impaired
myogenesis in tumor bearing mice are prevented by ERK inhibition.
PLoS One. 5:e136042010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramamoorthy S, Donohue M and Buck M:
Decreased Jun-D expression in muscle wasting of human cachexia. Am
J Physiol Endocrinol Metab. 297:E392–E401. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hawke TJ and Garry DJ: Myogenic satellite
cells: Physiology to molecular biology. J Appl Physiol (1985).
91:534–551. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hollenberg SM, Cheng PF and Weintraub H:
Use of a conditional MyoD transcription factor in studies of MyoD
trans-activation and muscle determination. Proc Natl Acad Sci USA.
90:8028–8032. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berkes CA and Tapscott SJ: MyoD and the
transcriptional control of myogenesis. Semin Cell Dev Biol.
16:585–595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Q, Vashisht AA, O'Rourke J, Corbel
SY, Moran R, Romero A, Miraglia L, Zhang J, Durrant E, Schmedt C,
et al: The microprotein Minion controls cell fusion and muscle
formation. Nat Commun. 8:156642017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ganassi M, Badodi S, Quiroga HPO, Zammit
PS, Hinits Y and Hughes SM: Myogenin promotes myocyte fusion to
balance fibre number and size. Nat Commun. 9:42322018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dey BK, Gagan J, Yan Z and Dutta A:
miR-26a is required for skeletal muscle differentiation and
regeneration in mice. Genes Dev. 26:2180–2191. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Agarwal S, Cholok D, Loder S, Li J,
Breuler C, Chung MT, Sung HH, Ranganathan K, Habbouche J, Drake J,
et al: mTOR inhibition and BMP signaling act synergistically to
reduce muscle fibrosis and improve myofiber regeneration. JCI
Insight. 1:e898052016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Benezra R, Davis RL, Lockshon D, Turner DL
and Weintraud H: The protein Id: A negative regulator of
helix-loop-helix DNA binding proteins. Cell. 61:49–59. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jen Y, Weintraub H and Benezra R:
Overexpression of Id protein inhibits the muscle differentiation
program: In vivo association of Id with E2A proteins. Genes Dev.
6:1466–1479. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clever JL, Sakai Y, Wang RA and Schneider
DB: Inefficient skeletal muscle repair in inhibitor of
differentiation knockout mice suggests a crucial role for BMP
signaling during adult muscle regeneration. Am J Cell Physiol.
298:C1087–C1099. 2010. View Article : Google Scholar
|
|
31
|
Winbanks CE, Chen JL, Qian H, Liu Y,
Bernardo B, Beyer C, Watt KI, Thomson RE, Connor T, Turner BJ, et
al: The bone morphogenetic protein axis is a positive regulator of
skeletal muscle mass. J Cell Biol. 203:345–357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hogan BL: Bone morphogenetic proteins:
Multifunctional regulators of vertebrate development. Genes Dev.
10:1580–1594. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Walsh DW, Godson C, Brazil DP and Martin
F: Extracellular BMP-antagonist regulation in development and
disease: Tied up in knots. Trends Cell Biol. 20:244–256. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koosha E and Eames BF: Two modulators of
skeletal development: BMPs and Proteoglycans. J Dev Biol.
10:152022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miyazono K, Maeda S and Imamura T: BMP
receptor signaling: Transcriptional targets, regulation of signals,
and signaling cross-talk. Cytokine Growth Factor Rev. 16:251–263.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ogata T, Wozney JM, Benezra R and Noda M:
Bone morphogenetic protein 2 transiently enhances expression of a
gene, Id (inhibitor of differentiation), encoding a
helix-loop-helix molecule in osteoblast-like cells. Proc Natl Acad
Sci USA. 90:9219–9222. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hollnagel A, Oehlmann V, Heymer J, Rüther
U and Nordheim A: Id genes are direct targets of bone morphogenetic
protein induction in embryonic stem cells. J Biol Chem.
274:19838–19845. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Borok MJ, Mademtzoglou D and Relaix F:
Bu-M-P-ing iron: How BMP signaling regulates muscle growth and
regeneration. J Dev Biol. 8:42020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Korchynskyi O and Dijke PT: Identification
and functional characterization of distinct critically important
bone morphogenetic protein-specific response elements in the Id1
promoter. J Biol Chem. 277:4883–4891. 2002. View Article : Google Scholar
|
|
40
|
Shepherd TG, Thériault BL and Nachtigal
MW: Autocrine BMP4 signalling regulates ID3 proto-oncogene
expression in human ovarian cancer cells. Gene. 414:95–105. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsufuji S, Kitajima Y, Higure K, Kimura
N, Maeda S, Yamada K, Ito K, Tanaka T, Kai K and Noshiro H: A
HIF-1α inhibitor combined with palmitic acid and L-carnitine
treatment can prevent the fat metabolic reprogramming under hypoxia
and induce apoptosis in hepatocellular carcinoma cells. Cancer
Metab. 11:252023. View Article : Google Scholar
|
|
42
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
43
|
Reimann J, Brimah K, Schroder R, Wering A,
Beauchamp JR and Partridge TA: Pax7 distribution in human skeletal
muscle biopsies and myogenic tissue cultures. Cell Tissue Res.
315:233–242. 2004. View Article : Google Scholar
|
|
44
|
Borisov AB, Debkov EI and Carlson BM:
Differentiation of activated satellite cells in denervated muscle
following single fusions in situ and in cell culture. Histochem
Cell Biol. 124:13–23. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
He WA, Berardi E, Cardillo VM, Acharyya S,
Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P,
et al: NF-κB-mediated Pax7 dysregulation in the muscle
microenvironment promotes cancer cachexia. J Clin Invest.
123:4821–4835. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kleeff J, Maruyama H, Ishiwata T, Sawhney
H, Friess H, Büchler MW and Korc M: Bone morphogenetic protein 2
exerts diverse effects on cell growth in vitro and is expressed in
human pancreatic cancer in vivo. Gastroenterology. 116:1202–1216.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Aoki M, Ishigami S, Uenosono Y, Arigami T,
Uchikado Y, Kita Y, Kurahara H, Matsumoto M, Ueno S and Natsugoe S:
Expression of BMP-7 in human gastric cancer and its clinical
significance. B J Cancer. 104:714–718. 2011. View Article : Google Scholar
|
|
48
|
Guo X, Xiong L, Zou L and Zhao J:
Upregulation of bone morphogenetic protein 4 is associated with
poor prognosis in patients with hepatocellular carcinoma. Pathol
Oncol Res. 18:635–640. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yokoyama Y, Watanabe T, Tamura Y,
Hashizume Y, Miyazono K and Ehata S: Autocrine BMP-4 signaling is a
therapeutic target in colorectal cancer. Cancer Res. 77:4026–4038.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Davis H, Raja E, Miyazono K, Thubakihara Y
and Moustakas A: Mechanism of action of bone morphogenetic proteins
in cancer. Cytokine Growth Factor Rev. 27:81–92. 2016. View Article : Google Scholar
|
|
51
|
Murre C, McCaw PS and Baltimore D: A new
DNA binding and dimerization motif in immunoglobulin enhancer
binding, daughterless, MyoD, and myc proteins. Cell. 56:777–783.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Davis RL, Cheng PF, Lassar AB and
Weintraub H: The MyoD DNA binding domain contains a recognition
code for muscle-specific gene activation. Cell. 60:733–746. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ishibashi J, Perry RL, Asakura A and
Rudnicki MA: MyoD induces myogenic differentiation through
cooperation of its NH2- and COOH-terminal regions. J Cell Biol.
171:471–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Parker MH, Perry RLS, Fauteux MC, Berkes
CA and Rudnicki MA: MyoD synergizes with the E-protein HEB beta to
induce myogenic differentiation. Mol Cell Biol. 26:5771–5783. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Faralli H and Dilworth J: Turning on
myogenin in muscle: A paradigm for understanding mechanisms of
tissue-specific gene expression. Comp Funct Genomics.
2012:8363742012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Berkes CA, Bergstrom DA, Penn BH, Seaver
KJ, Knoepfler PS and Tapscott SJ: Pbx marks genes for activation by
MyoD indicating a role for a homeodomain protein in establishing
myogenic potential. Mol Cell. 14:465–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gerber AN, Klesert TR, Bergstrom DA and
Tapscott SJ: Two domains of MyoD mediate transcriptional activation
of genes in repressive chromatin: A mechanism for lineage
determination in myogenesis. Genes Dev. 11:436–450. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lopez MA, Si Y, Hu X, Williams V, Qushair
F, Carlyle J, Alesce L, Conklin M, Gilbert S, Bamman MM, et al:
Smad8 is increased in duchenne muscular dystrophy and suppresses
miR-1, miR-133a, and miR-133b. Int J Mol Sci. 23:75152022.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pasero M, Giovarelli M, Bucci G, Gherzi R
and Briata P: Bone morphogenetic protein/SMAD signaling orients
cell fate decision by impairing KSRP-dependent microRNA maturation.
Cell Rep. 2:1159–1168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jin W, Peng J and Jiang S: The epigenetic
regulation of embryonic myogenesis and adult muscle regeneration by
histone methylation modification. Biochem Biophys Rep. 6:209–219.
2016.PubMed/NCBI
|
|
61
|
Ono Y, Calhabeu F, Morgan JE, Katagiri T,
Amthor H and Zammit PS: BMP signalling permits population expansion
by preventing premature myogenic differentiation in muscle
satellite cells. Cell Death Differ. 18:222–234. 2011. View Article : Google Scholar :
|
|
62
|
Terada K, Misao S, Katase N, Nishimatsu S
and Nohno T: Interaction of Wnt signaling with BMP/Smad signaling
during the transition from cell proliferation to myogenic
differentiation in mouse myoblast-derived cells. Int J Cell Biol.
2013:6162942013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Abrams KL, Xu J, Nativelle-Serpentini C,
Dabirshahsahebi S and Rogers MB: An Evolutionary and molecular
analysis of Bmp2 expression. J Biol Chem. 279:15916–15928. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Miyazono K, Kamiya Y and Morikawa M: Bone
morphogenetic protein receptors and signal transduction. J Biochem.
147:35–51. 2010. View Article : Google Scholar
|
|
65
|
Mueller TD and Nickel J: Promiscuity and
specificity in BMP receptor activation. FEBS Lett. 586:1846–1859.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Beppu H, Minowa O, Miyazono K and Kawabata
M: cDNA cloning and genomic organization of the mouse BMP type II
receptor. Biochem Biophys Res Commun. 235:499–504. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nakane A, Nakagawa H and Nagata H:
Advanced high-content phenotypic screening to identify drugs that
ameliorate the inhibition of skeletal muscle cell differentiation
induced by cancer cachexia serum. Pharmaceuticals (Basel).
18:4452025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kosacka M, Dyła T, Chaszczewska-Markowska
M, Bogunia-Kubik K and Brzecka A: Decreased thrombospondin-1 and
bone morphogenetic protein-4 serum levels as potential indices of
advanced stage lung cancer. J Clin Med. 10:38592021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Shi YJ and Pan XT: BMP6 and BMP4
expression in patients with cancer-related anemia and its
relationship with hepcidin and s-HJV. Gen Mol Res.
15:gmr.150171302015.
|