Introduction
Cervical cancer (CC) ranks among the most prevalent
gynecological malignancies, constituting a persistent and
substantial global health challenge (1). While advances in human
papillomavirus (HPV) screening and vaccination have improved
prevention and early detection, a notable number of patients still
experience delayed diagnosis, treatment resistance, recurrence and
poor clinical outcomes. Thus, elucidating the molecular mechanisms
that underlie CC initiation and progression is fundamental to
improving disease management and refining therapeutic strategies
(2).
Ubiquitination and deubiquitination are essential
processes for maintaining cellular function and homeostasis
(3). Ubiquitination is regulated
by a coordinated enzymatic cascade involving ubiquitin-activating
enzyme E1, ubiquitin-conjugating enzyme E2 and ubiquitin ligase E3
(3). This process is reversed by
deubiquitination, which is mediated by deubiquitinating enzymes
(DUBs) (3). Notably, >100 DUBs
have thus far been identified and classified into nine
superfamilies based on sequence and structural features (Fig. 1): Ubiquitin-specific peptidase
(USP), ovarian tumor-related protease (OTU), ubiquitin C-terminal
hydrolase (UCH), Machado-Joseph domain-containing protease (MJD),
JAMM/MPN domain-associated (JAMM/MPN) metallopeptidase,
motif-interacting with ubiquitin-containing novel DUB (MINDY),
monocyte chemotactic protein-induced protein (MCPIP), permuted
papain fold peptidases of dsRNA viruses and eukaryotes (PPPDE) and
zinc finger-containing ubiquitin peptidase 1 (ZUP1) families
(3). A subgroup termed
pseudo-DUBs has been proposed to describe DUB family members that
lack canonical catalytic activity because of defects in key
residues or domains (4). However,
subsequent studies have suggested that this classification is not
absolute. Several proteins previously categorized as pseudo-DUBs,
including USP39 (5), USP53
(6), USP54 (6), PSMD7 (7), eukaryotic translation initiation
factor 3 subunit F (EIF3F) (8)
and STAMBPL1 (9), retain or
exhibit deubiquitinating activity under specific conditions.
Enzymatic activity has also been confirmed for PPPDE1 (10), MCPIP1 (11) and MCPIP4 (12). These findings indicate that the
classification of pseudo-DUBs is dynamic and should be interpreted
cautiously as functional evidence continues to emerge.
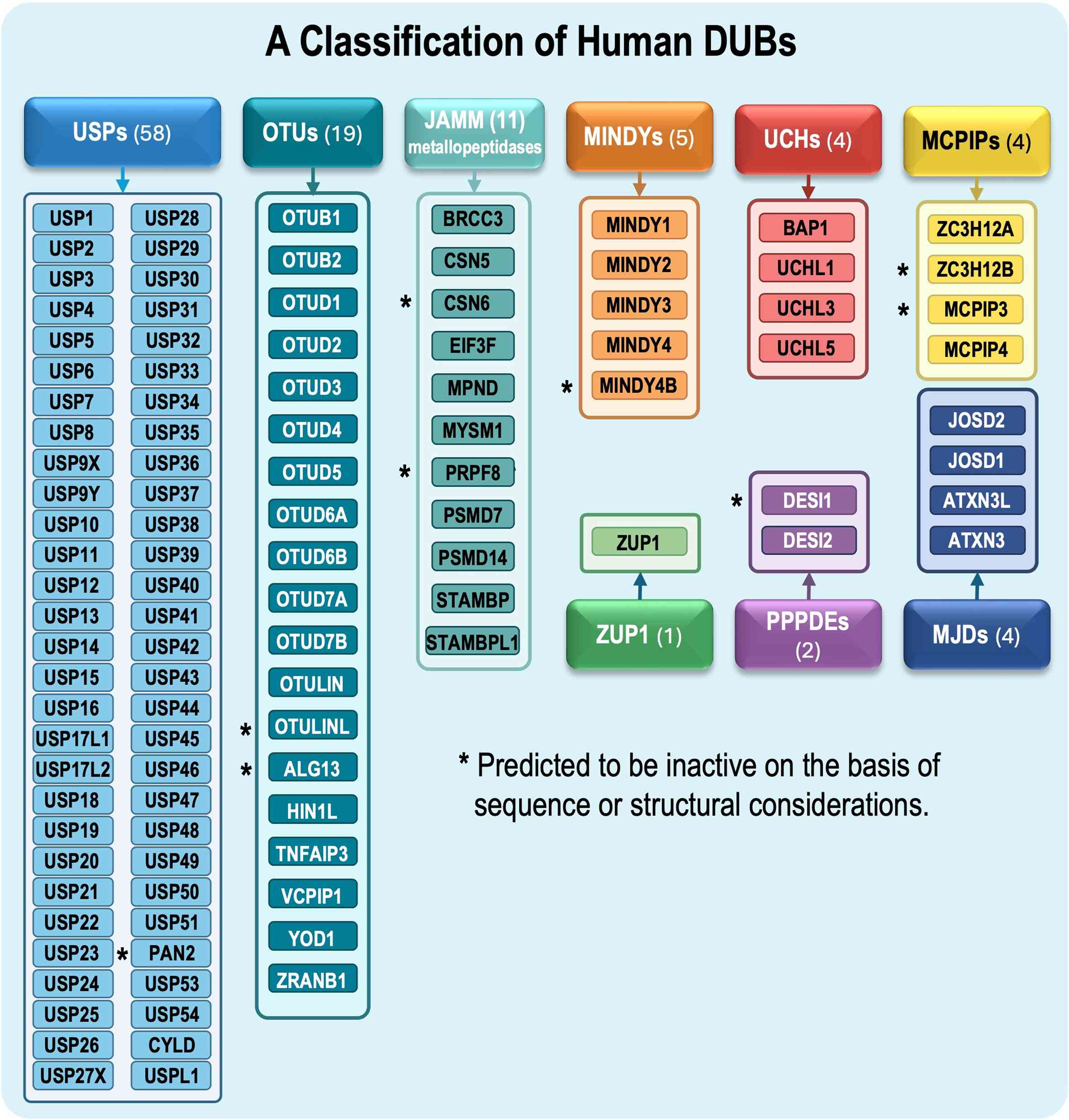 | Figure 1Classification of human DUBs. Human
DUBs are classified into cysteine protease or metallopeptidase
families based on their sequence and structural features. The
largest family comprises 58 USPs, followed by 19 OTUs and 9 JAMM
metallopeptidases. Additional cysteine protease families include 5
MINDY proteins, 4 UCHs, 4 MCPIP-related proteins, 4 MJDs, 2 PPPDEs
and ZUP1. Individual family members are listed within each colored
box. Proteins marked with an asterisk are predicted to lack
catalytic activity based on their amino-acid sequence analysis or
structural evidence. DUB, deubiquitinating enzyme; JAMM
metallopeptidase, JAMM/MPN domain-associated metallopeptidase;
MCPIP, monocyte chemotactic protein-induced protein; MINDY,
motif-interacting with ubiquitin-containing novel DUB; MJD,
Machado-Joseph domain-containing protease; OTU, ovarian
tumor-related protease; PPPDE, permuted papain fold peptidase of
dsRNA viruses and eukaryotes; UCH, ubiquitin C-terminal hydrolase;
USP, ubiquitin-specific peptidase; ZUP1, zinc finger-containing
ubiquitin peptidase 1. |
DUBs function by recognizing ubiquitin-tagged
proteins and hydrolyzing isopeptide bonds between ubiquitin
molecules, or between ubiquitin and substrate proteins. Through
these actions, DUBs regulate cell cycle progression, DNA damage
repair, apoptosis, immune regulation and signal transduction, among
other cellular events (13,14). Growing evidence has shown that
dysregulated DUB activity contributes to the development and
progression of various malignancies, including gynecologic cancer
(15-17). Although several studies have
examined DUBs in CC (13,15,18), a comprehensive review of their
molecular functions, regulatory mechanisms and therapeutic
relevance is lacking. The present review systematically summarizes
current research on DUBs in CC, with emphasis on their roles in
HPV-associated carcinogenesis, signaling pathway regulation, tumor
progression, therapy response and potential DUB-targeted
therapies.
USPs: Key regulatory factors in the
development and progression of CC
The USP family is the largest subgroup of DUBs, with
>50 members that account for ~60% of all DUBs. USPs are mainly
involved in cell cycle regulation, DNA damage repair and chromatin
repair, thereby affecting various cellular functions and leading to
disease progression. Notably, >50 USP family members have been
implicated in the onset, progression and treatment of cancer
(18-20).
USP3
USP3 is a cysteine protease and one of the few known
members of the USP family capable of efficiently cleaving the
ubiquitin-proline bond (21).
Data from The Cancer Genome Atlas (TCGA) have indicated that high
USP3 expression is associated with poor survival in adrenocortical
carcinoma, breast cancer and pancreatic adenocarcinoma, whereas
elevated USP3 expression predicts better survival in two brain
tumor types, including low-grade glioma and glioblastoma (21). No statistically significant
difference in USP3 expression has been observed in CC. Studies have
also demonstrated that USP3 is closely associated with DNA damage
response (DDR), cell cycle regulation, apoptosis and immune
regulation (22-24). Evidence has suggested that USP3
regulates the cell cycle by stabilizing the cell division cycle 25A
(Cdc25A) protein through deubiquitination, and this process is
implicated in cancer development and progression. A previous
mechanistic study indicated that USP3 expression is positively
associated with Cdc25A protein levels; knockdown of USP3 can reduce
Cdc25A protein levels, thereby inhibiting the G1-to-S
phase transition and suppressing tumor growth in xenograft models
established using USP3-depleted or -overexpressing HeLa cells
(21). Additionally, USP3 and
Cdc25A have been shown to co-localize and interact within the
nucleus, where USP3 extends the half-life of Cdc25A via
deubiquitination, ensuring its stability throughout the cell cycle
(21). Analysis of TCGA database
has revealed that USP3 and Cdc25A are upregulated in multiple
cancer types. Clinically, elevated expression of USP3 and Cdc25A
has been associated with poor prognosis in breast cancer,
supporting a pro-tumorigenic role for this axis in certain types of
cancer (21). However, TCGA data
have shown no notable difference in USP3 expression between
patients with CC (n=305) and healthy normal controls, suggesting
that its clinical relevance varies across tumor types (http://gepia.cancer-pku.cn/detail.php?gene=USP3###)
(25). Collectively, these
findings identify USP3 as a functionally important DUB in
oncogenesis and support the USP3/Cdc25A axis as a potential
therapeutic target, although its cancer-type-specific importance
requires further investigation.
USP7
USP7, also known as herpesvirus-associated
ubiquitin-specific protease, is one of the most extensively studied
DUBs (26). Recent studies have
shown that USP7 is associated with various types of cancer,
including glioblastoma (27),
breast cancer (28), colorectal
cancer (29) and ovarian cancer
(30). In CC, USP7 is
consistently upregulated and is associated with malignant
progression; notably, increased USP7 expression is associated with
advanced histological grade and poor patient prognosis (31,32).
Functionally, USP7 promotes oncogenic phenotypes in
CC, including increased proliferation, migration, invasion,
angiogenesis, immune evasion and resistance to DNA damage, through
several distinct but complementary molecular mechanisms. In the
DDR, USP7 interacts with the MRE11-RAD50-NBS1 (MRN) complex and
mediator of DNA damage checkpoint protein 1 (MDC1), stabilizing the
MRN-MDC1 complex through MDC1 deubiquitination. This stabilization
facilitates recruitment of key DDR factors, such as 53BP1 and BRCA1
to DNA double-strand breaks. Loss of USP7 disrupts this process,
impairs DDR and increases CC cell sensitivity to DNA damage
(31). USP7 also promotes tumor
progression by deubiquitinating and stabilizing metadherin (MTDH),
enhancing proliferation, migration, invasion, angiogenesis and M2
macrophage polarization. Silencing USP7 reduces MTDH levels,
inhibits tumor progression and promotes apoptosis (32). In addition, USP7 stabilizes Cdc25A
through deubiquitination, protecting it from DDR-induced
degradation and sustaining CC cell proliferation. In HeLa cells,
USP7 knockout has been shown to markedly impair Cdc25A-dependent
proliferation, migration and colony formation, reduce xenograft
tumor growth, and increase sensitivity to etoposide, hydroxyurea
and ultraviolet radiation (33).
Moreover, USP7 regulates the EZH2/TIMP2/NF-κB/PD-L1 signaling axis.
USP7-mediated upregulation of EZH2 suppresses TIMP2 through
methylation, activates NF-κB signaling and increases PD-L1
expression, thus promoting immune escape, tumor growth and
metastasis (34). Clinically,
elevated USP7 expression is associated with aggressive
clinicopathological features and poor prognosis in CC, supporting
its value as a potential biomarker.
Collectively, these findings demonstrate the
multifaceted oncogenic role of USP7 in CC and highlight several
therapeutically actionable pathways, including the USP7/MDC1,
USP7/MTDH, USP7/Cdc25A and USP7/EZH2/TIMP2/NF-κB/PD-L1 axes.
Targeting USP7 may thus represent a promising strategy to sensitize
CC cells to DNA-damaging agents and suppress tumor progression.
USP8
USP8 is a multifunctional DUB involved in mitophagy,
autophagy, cell cycle regulation, apoptosis and the DDR (35). Evidence has demonstrated that USP8
is highly expressed in CC tissues relative to normal cervical
tissues; this elevation is particularly pronounced in cervical
squamous cell carcinoma (CSCC), and has been validated at both the
mRNA and protein levels (36,37). Functionally, USP8 exerts oncogenic
effects by promoting tumor cell proliferation, migration, invasion,
survival and overall tumor growth (36,37). Mechanistically, USP8
deubiquitinates and stabilizes FLICE-like inhibitory protein
(FLIPL), thereby suppressing extrinsic apoptosis induced by Fas,
TRAIL and TNF-α, and facilitating tumor progression (37). Consistent with these findings,
USP8 inhibition has been shown to markedly reduce tumor volume and
weight in xenograft models, further supporting its tumor-promoting
role (37). Clinically, elevated
USP8 expression is closely associated with advanced tumor stage,
poor prognosis and reduced overall survival in patients with CSCC,
indicating its potential as a prognostic biomarker (36). Collectively, these findings
underscore the therapeutic potential of USP8 in CC and suggest that
targeting the USP8/FLIPL axis may effectively attenuate tumor
progression and improve patient outcomes.
USP11
USP11 is a key member of the largest subfamily of
cysteine protease DUBs (38). It
is widely distributed in the nuclei of mitotic cells, consistent
with its role in nuclear events related to cell proliferation and
genome maintenance (39,40). In CC, its oncogenic relevance is
closely linked to HPV, particularly the HPV-16 E7 oncoprotein, the
stability of which is essential for its transforming activity.
Mechanistically, USP11 stabilizes HPV-16 E7 by reducing its
ubiquitination and preventing proteasomal degradation; this effect
depends on its DUB activity, as a catalytically inactive USP11
mutant has been reported to fail to protect E7 and restore its
normal degradation rate (41).
USP11-mediated stabilization of HPV-16 E7 also affects downstream
targets, including pRb, Bcl-2 and Cdc2, thereby promoting CC cell
proliferation and transformation (41). These findings indicate that USP11
may contribute to cervical carcinogenesis by sustaining HPV-16 E7
stability and oncogenic function. Targeting USP11 could therefore
destabilize HPV-16 E7, and inhibit CC cell proliferation and
transformation.
USP14
USP14 is aberrantly expressed in several
gynecological diseases, including deep infiltrating endometriosis
(42), premature ovarian failure
(43) and endometrial cancer
(44). Evidence has suggested
that USP14 contributes to CC progression, although its expression
pattern in CC remains incompletely defined. Functionally, USP14
promotes CC cell proliferation, and regulates cell cycle
progression and survival (45).
Mechanistically, USP14 facilitates proliferation through modulating
the protein stability of murine double minute protein 2 (MDM2).
Pharmacological inhibition of USP14 by IU1 can reduce MDM2
expression, activate the autophagy-lysosome degradation pathway,
enhance ubiquitin-proteasome system activity, block the
G0/G1-to-S transition and induce apoptosis
(45). These findings suggest
that USP14 supports tumor cell proliferation and contributes to
disease progression. Targeting the USP14/MDM2 axis or developing
USP14 inhibitors may therefore represent a promising therapeutic
strategy.
USP15
USP15 contributes to disease initiation and
progression through DNA damage repair (46), regulation of tumor-related
signaling pathways (47) and
modulation of immune responses (48). In CC, evidence mainly reflects
functional regulation of USP15 rather than a clearly defined
expression pattern. Under hypoxic conditions, microRNA
(miRNA/miR)-100 is markedly upregulated in CC cells and directly
suppresses USP15 translation, reducing USP15 mRNA and protein
levels (49). Functionally, USP15
supports viral oncoprotein stability and influences
chemotherapeutic response. Mechanistically, USP15 interacts with
the HPV-16 E6 oncoprotein and prevents its degradation through
deubiquitination, thereby stabilizing E6 protein levels, enhancing
its oncogenic activity and promoting CC cell proliferation
(50). USP15 is also linked to
paclitaxel resistance. Hypoxia-induced miR-100 downregulates USP15
and contributes to induced drug responsiveness in CC cells
(49,51). Clinically, these findings suggest
that USP15 participates in both HPV-driven carcinogenesis and
chemotherapy resistance, although its clinical relevance remains
limited and evidence for paclitaxel resistance is currently
restricted to an in vitro study (49). Collectively, these observations
highlight the therapeutic potential of USP15 in CC. Targeting
USP15, its DUB activity toward HPV-16 E6 or its regulatory
interaction with miR-100 may offer promising treatment
strategies.
USP17
USP17 comprises a family of structurally related
proteins (52). Aberrant USP17
expression has been implicated in several diseases, including
preeclampsia (53), colorectal
cancer (54) and lung
adenocarcinoma (55), suggesting
a context-dependent role in human pathophysiology. With regard to
biological function, available evidence has indicated that USP17
exerts tumor-suppressive effects in CC by promoting apoptosis, and
inhibiting cell proliferation and cell cycle progression. At the
molecular level, USP17 induces apoptosis and suppresses
proliferation of cervical adenocarcinoma cells through
Lys63-specific deubiquitination of suppressor of defective
silencing 3 (SDS3), a component of histone deacetylase
(HDAC)-dependent Sin3A co-repressor complex, thereby negatively
regulating HCAC activity (56).
USP17 also inhibits the localization of H-Ras and N-Ras, but not
K-Ras, to the plasma membrane, attenuating MEK/ERK and PI3K/JNK
signaling, and leading to cell cycle arrest (57). These findings suggest that USP17
may function as a negative regulator of CC progression. Modulation
of the USP17/SDS3/HDAC and USP17/Ras axes could therefore represent
a potential therapeutic strategy, although further studies are
required to clarify its mechanisms and translational relevance.
USP18
USP18 regulates immune responses and cell death,
particularly through the type I interferon pathway, which has been
implicated in tumor development via induction of immunogenic cell
death (58,59). USP18 is markedly upregulated in CC
tissues and cell lines (60).
With regard to biological function, USP18 promotes CC cell
proliferation and suppresses apoptosis, indicating a
pro-tumorigenic role (60). At
the molecular level, USP18 is associated with the PI3K/AKT
signaling pathway. The oncogenic effects of USP18 upregulation are
markedly reduced by the PI3K/AKT inhibitor LY294002, suggesting
that USP18 promotes tumor progression at least partly through
activation of PI3K/AKT signaling (60). Elevated USP18 expression may
therefore contribute to disease progression and serve as a
biomarker of malignant behavior, and targeting USP18 alone or in
combination with PI3K/AKT pathway inhibition may represent a
promising therapeutic strategy.
USP19
USP19 serves a context-dependent role in tumor
progression, exhibiting tumor-promoting or tumor-suppressive
effects depending on cellular context (61). Dysregulated USP19 expression has
been associated with CC progression. Functionally, USP19 promotes
CC cell proliferation, migration and invasion. At the molecular
level, USP19 directly interacts with the tumor suppressor p53 and
enhances its ubiquitination and degradation, thereby reducing p53
stability (62). Because p53 is a
key regulator of the DDR (63)
and its loss is closely linked to malignant progression,
USP19-mediated suppression of p53 provides a mechanistic basis for
CC development. CRISPR/Cas9-mediated USP19 knockout has been shown
to markedly increase p53 levels and inhibit malignant behavior in
CC cells (62). Clinically, these
findings indicate that USP19 contributes to CC aggressiveness
through modulation of the p53 signaling axis. Accordingly, the
USP19/p53 axis may serve as a biomarker and therapeutic target for
precision treatment in CC.
USP21
USP21 has been implicated in tumor progression
through its roles in cell signaling, DNA damage and repair, and
histone modification (64).
Elevated USP21 expression is positively associated with
radioresistance in CC tissues (65). Functionally, USP21 promotes
radioresistance and inhibits apoptosis, whereas its knockdown
enhances radiosensitivity and induces apoptosis in CC cells
(65). At the molecular level,
USP21 regulates the stability of the transcription factor FOXM1;
silencing USP21 may reduce FOXM1 stability, inhibit YAP1 nuclear
translocation and activate Hippo signaling, ultimately increasing
CC cell sensitivity to radiotherapy (65). Activation of Hippo signaling
promotes apoptosis and enhances radiosensitivity. Consistently,
USP21 knockdown has been reported to promote apoptosis, increase
radiosensitivity and suppress xenograft tumor growth (65). These findings indicate that USP21
contributes to treatment resistance and disease progression in CC.
Accordingly, the USP21/FOXM1/Hippo axis may serve as a biomarker
and therapeutic target for improving radiosensitivity in CC.
USP22
USP22 functions as an oncogenic DUB in several
malignancies, including lung, breast, ovarian and liver cancer, by
regulating cell cycle progression, signal transduction and immune
responses (66). USP22 is also
highly expressed in CC (67).
Although its specific biological functions and molecular mechanisms
in CC remain unclear, evidence from other cancer types suggests
that USP22 contributes to tumor progression through oncogenic
activity (66,67). Clinically, elevated USP22
expression in CC is markedly associated with adverse
clinicopathological parameters, including advanced International
Federation of Gynecology and Obstetrics (FIGO) stage, high Ki67
index, lymph node metastasis and poor histological differentiation
(67). In addition, patients with
CC and high USP22 expression exhibit significantly reduced overall
survival and disease-free survival compared with those with low
expression (P<0.001) (67).
Collectively, these findings indicate that USP22 may serve as a
prognostic biomarker, and an auxiliary indicator for guiding
systemic treatment and predicting therapeutic outcomes in CC.
USP26
USP26 was initially identified in mouse
spermatogonia and linked to male infertility, but has since been
implicated in cancer progression and bone homeostasis (68). Its expression pattern in CC
remains incompletely defined, although functional evidence suggests
a tumor-suppressive role (15).
Ye et al (15)
demonstrated that USP26 can inhibit CC cell proliferation and
migration. At the molecular level, this effect is mediated through
the deubiquitination and stabilization of Krüppel-like factor 6
(KLF6), a transcription factor that regulates cell cycle
progression and promotes apoptosis, and is widely recognized as a
tumor suppressor in several malignancies, including prostate and
breast cancer (15,69). By maintaining KLF6 protein
stability, USP26 suppresses malignant behavior in CC cells
(15). These findings indicate
that USP26 expression is associated with a less aggressive CC
phenotype and the USP26/KLF6 axis may represent a potential
therapeutic target; however, further clinical studies are needed to
clarify its expression pattern and prognostic value.
USP39
USP39 is aberrantly expressed in several
malignancies, including gastric, colorectal and endometrial cancer,
and it contributes to tumor progression (70). In CC, USP39 is highly expressed in
both tissues and cell lines (71). Functionally, USP39 promotes
proliferation, survival, autophagy and oxidative stress responses
in CSCC cells (71). At the
molecular level, sirtuin7 (SIRT7) interacts with and deacetylates
USP39, enhancing its protein stability (71). USP39 then regulates alternative
splicing of FOXM1 pre-mRNA and upregulates SIRT7 expression;
subsequently, SIRT7 positively regulates USP39 and FOXM1,
establishing a positive feedback loop that sustains malignant
progression (71). Elevated USP39
expression is associated with CC progression and aggressiveness
(71). The SIRT7/USP39/FOXM1 axis
may thus represent a potential therapeutic target.
USP45
USP45 exhibits oncogenic DUB activity; however, its
precise role in tumorigenesis has not been fully elucidated
(72). USP45 is upregulated in
tumors and highly expressed in CC cells (72,73). Functionally, USP45 promotes CC
cell proliferation, stemness and drug resistance (73). At the molecular level, USP45
stabilizes Myc through deubiquitination, thereby enhancing
proliferative capacity and stem cell-like properties in CC cells
(73). Clinically, elevated USP45
expression is negatively associated with overall survival and
recurrence-free survival, indicating its potential value as a
prognostic marker (72).
Targeting the USP45/Myc axis may thus represent a promising
therapeutic strategy. Notably, α-mangostin has been identified as a
specific USP45 inhibitor that suppresses proliferation and reduces
drug resistance in CC cells (73). Collectively, these findings
support an oncogenic role for USP45 in CC and highlight its
potential as a therapeutic target.
USP53
USP53 is broadly expressed in human tissues and has
been reported to function as a tumor suppressor in several types of
cancer, including lung, colorectal and liver malignancies (74). In CC, USP53 expression is
associated with radiosensitivity; functionally, USP53 enhances the
response of CC cells to radiotherapy (75). At the molecular level, USP53
interacts with DNA damage-binding protein 2 (DDB2) and upregulates
its expression, promoting nucleotide excision repair and increasing
DNA repair capacity (75). USP53
also induces G2/M arrest, and alters CDK1 and CDK2
expression, which may further contribute to radiosensitization
(75). These findings identify
USP53 as a predictive biomarker for radiotherapy response and a
potential target for radiosensitization strategies in CC.
CYLD
CYLD is a well-established tumor suppressor that
regulates cell cycle progression, signal transduction and DDR, and
its dysregulation is frequently linked to malignant progression
(76). In CC, CYLD expression is
significantly downregulated and negatively associated with miR-501
levels (77). Reduced CYLD
expression is also associated with enhanced Aurora B activation and
increased histological invasiveness in advanced CC specimens
(78). Functionally, CYLD
inhibits CC cell proliferation, migration, invasion and colony
formation while promoting apoptosis (77,78). Mechanistically, miR-501-mediated
repression of CYLD contributes to malignant phenotypes and
apoptosis resistance, possibly through activation of NF-κB p65
signaling, upregulation of Bcl-2 and downregulation of Bax
(77). CYLD also directly
deubiquitinates Aurora B, suppressing its activation and mitotic
function, delaying mitosis and inhibiting CC development (78). Clinically, low CYLD expression is
significantly associated with large tumor size, advanced FIGO
stage, lymph node metastasis, heightened Aurora B activity and
increased histological invasiveness (77,78). These findings support CYLD as both
a prognostic biomarker and a potential therapeutic target in CC
(77,78).
The USP family serves a broad and complex role in CC
and appears to function as an integrated regulatory system
connecting viral oncogenesis, cell cycle control, DDR, aberrant
signaling and immune evasion (Fig.
2; Table I). Most USP members
reported in CC, including USP3, USP7, USP8, USP11, USP14, USP15,
USP18, USP19, USP21, USP22, USP39 and USP45, generally show
tumor-promoting functions. These proteins enhance cell
proliferation, migration, invasion, angiogenesis and immune escape,
and contribute to resistance to radiotherapy and chemotherapy.
Mechanistically, these effects are mediated through stabilization
of oncogenic substrates, suppression of tumor suppressor turnover,
activation of key signaling pathways such as PI3K/AKT,
Wnt/β-catenin, NF-κB, and RAS/MAPK, and modulation of HPV-16 E6/E7
oncogenic functions. By contrast, several members, including USP17,
USP26, USP53 and CYLD, exhibit tumor-suppressive or
radiosensitizing effects in specific contexts, highlighting the
context-dependent nature of USP-mediated regulation in CC.
Clinically, dysregulated USP expression is associated with FIGO
stage, lymph node metastasis, tumor burden, prognosis and treatment
response, supporting their potential use as biomarkers. In
addition, advances in small-molecule inhibitors and
deubiquitinase-targeting chimera (DUBTAC)-based approaches suggest
that targeting selected USPs may be a feasible strategy for
precision therapy. Taken together, the USP family represents not
only an important contributor to CC progression, but also a
potentially valuable source of diagnostic, prognostic and
therapeutic targets.
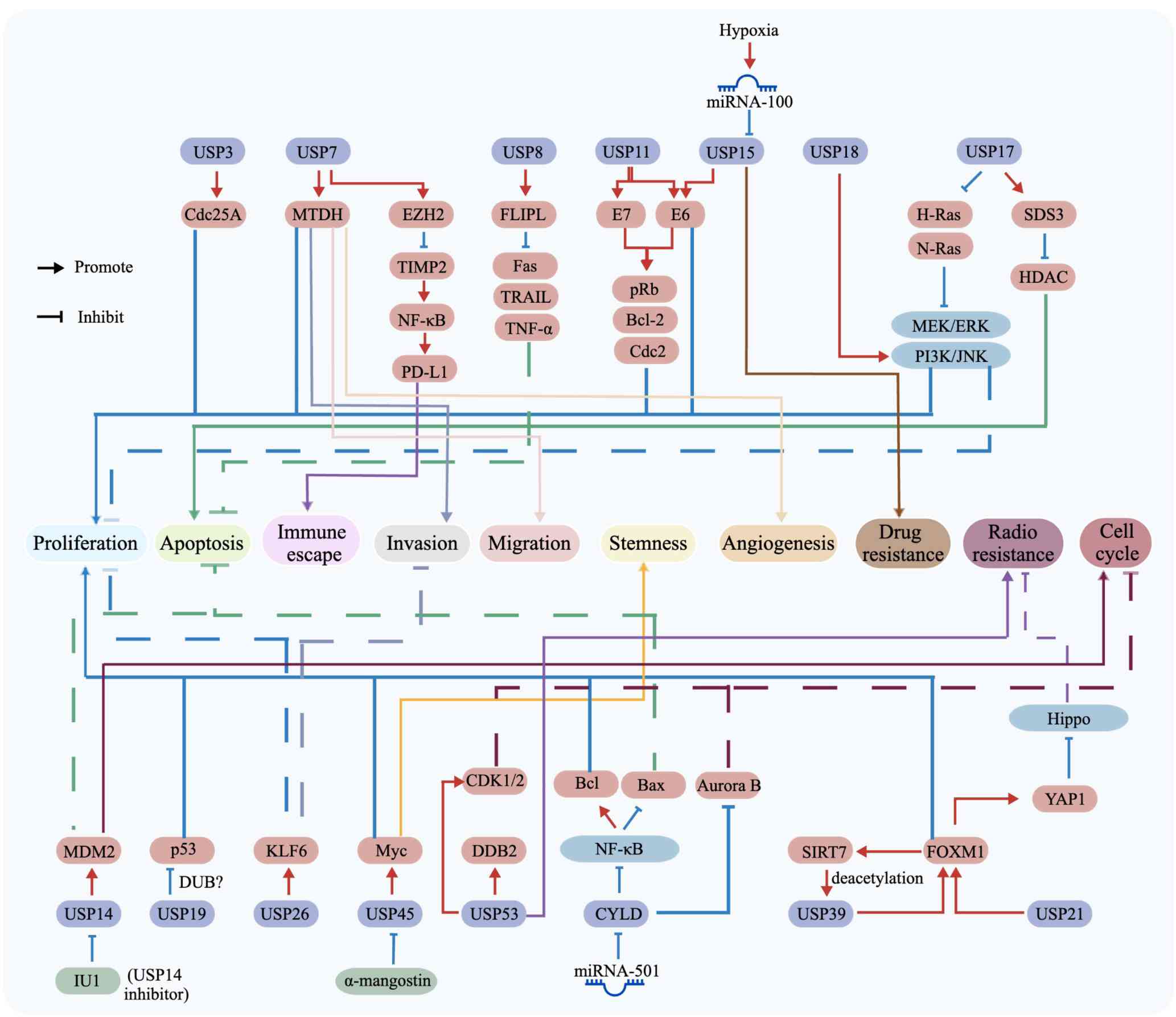 | Figure 2Regulatory network of USPs in tumor
progression and therapy resistance. Schematic diagram illustrating
the roles of multiple USPs and their downstream targets in
regulating malignant phenotypes in CC. USP3, USP7, USP8, USP11,
USP15, USP17, USP18, USP19, USP21, USP26, USP39, USP45, USP53 and
CYLD modulate diverse oncogenic or tumor-suppressive pathways
through substrates or signaling mediators including Cdc25A, MTDH,
EZH2, TIMP2, NF-κB, PD-L1, Fas, TRAIL, TNF-α, HPV E6/E7, pRb,
Bcl-2, Cdc2, Ras, HDAC, MEK/ERK, PI3K/JNK, MDM2, p53, KLF6, Myc,
CDK1/2, DDB2, Bax, Aurora B, SIRT7, FOXM1, and YAP1. These
signaling events converge on key cancer-associated processes,
including proliferation, apoptosis, immune escape, invasion,
migration, stemness, angiogenesis, drug resistance, radioresistance
and cell cycle regulation. The influence of non-coding RNAs and
microenvironmental cues, such as hypoxia/miRNA-100 and miRNA-501,
as well as small molecule modulators, including α-mangostin and
IU1, on USP-centered signaling networks are also highlighted.
Arrows indicate activation or positive regulation, whereas
blunt-ended lines indicate inhibition or negative regulation.
Overall, the diagram emphasizes the central role of USP-mediated
deubiquitination in coordinating tumor progression, cellular
adaptation and therapeutic response in CC. CC, cervical cancer;
Cdc, cell division cycle; DDB2, DNA damage-binding protein 2; DUB,
deubiquitinating enzyme; FLIPL, FLICE-like inhibitory protein;
HDAC, histone deacetylase; HPV, human papillomavirus; KLF6,
Krüppel-like factor 6; MDM2, murine double minute protein 2; miRNA,
microRNA; MTDH, metadherin; SDS3, HDAC-dependent Sin3A co-repressor
complex; USP, ubiquitin-specific protease. |
 | Table IList of DUBs, their substrates and
functions associated with CC. |
Table I
List of DUBs, their substrates and
functions associated with CC.
| First author,
year | DUB | Expression of level
in CC | Substrates or
related proteins | Role of DUB | Mechanisms and
functions | (Refs.) |
|---|
| Das, 2020 | USP3 | High
expression | Cdc25A | Stabilizes protein
structure | USP3 promotes CC
progression by regulating cell cycle progression by
deubiquitinating Cdc25A | (21) |
| Su, 2018 | USP7 | High
expression | MDC1 | Stabilizes protein
structure | USP7 enhances the
DDR by deubiquitinating MDC1, thus affecting accumulation of the
MRN-MDC1 complex and promoting CC development | (31) |
| Wang, 2024 | USP7 | High
expression | MTDH | Stabilizes protein
structure | USP7 promotes CC
progression by deubiquitinating MTDH | (32) |
| Das, 2020 | USP7 | Not mentioned | Cdc25A | Stabilizes protein
structure | USP7 promotes CC
development by deubiquitinating Cdc25A | (33) |
| Li, 2022 | USP7 | High
expression | EZH2, TIMP2 | Mediates signal
transduction | USP7 promotes
immune escape by upregulating EZH2; EZH2 inhibits TIMP2 to activate
NF-κB signaling and increase PD-L1 expression | (34) |
| Jeong, 2017 | USP8 | High
expression | FLIPL, Fas, TRAIL,
TNF-α | Stabilizes protein
structure | USP8 promotes CC
progression by deubiquitinating FLIPL and inhibiting apoptosis | (37) |
| Lin, 2008 | USP11 | Not mentioned | HPV-16 E7 | Stabilizes protein
structure | USP11 promotes CC
progression by deubiquitinating HPV-16 E7 | (41) |
| Xu, 2020 | USP14 | Not mentioned | MDM2 | Stabilizes protein
structure | USP14 promotes CC
progression by deubiquitinating MDM2 | (45) |
| Yaginuma, 2018 | USP15 | Not mentioned | HPV-16 E6 | Stabilizes protein
structure | USP15 promotes CC
progression by deubiquitinating HPV-16 E6 | (50) |
| Yu, 2024 | USP15 | Not mentioned | miR-100 | Downstream
factor | Hypoxia upregulates
miR-100 expression, reduces USP15 levels, and thereby induces
chemoresistance in CC | (51) |
| Ramakrishna,
2011 | USP17 | Not mentioned | SDS3, HDAC | Stabilizes protein
structure | USP17 inhibits cell
apoptosis by regulating the activities of SDS3 and HDAC | (56) |
| de la Vega,
2010 | USP17 | Not mentioned | Ras, N-Ras | Mediates signal
transduction | USP17 suppresses CC
progression by inhibiting the localization of H-Ras and N-Ras to
the plasma membrane, leading to the downregulation of MAPK,
MEK/ERK, and PI3K/JNK signaling pathways | (57) |
| Diao, 2020 | USP18 | High
expression | PI3K/AKT | Mediates signal
transduction | USP18 promotes CC
progression through the PI3K/AKT signaling pathway | (60) |
| Tyagi, 2024 | USP19 | Not mentioned | p53 | Stabilizes protein
structure | USP19 promotes CC
progression by deubiquitinating p53 | (62) |
| Li, 2022 | USP21 | Not mentioned | FOXM1 | Stabilizes protein
structure and mediates signal transduction | USP21
deubiquitinates FOXM1 to mediate the Hippo signaling pathway,
thereby affecting radiotherapy resistance and promoting the
progression of CC | (65) |
| Ye, 2024 | USP26 | Not mentioned | KLF6 | Stabilizes protein
structure | USP26 inhibits CC
progression by deubiquitinating KLF6 | (15) |
| Yu, 2023 | USP39 | High
expression | SIRT7, FOXM1 | Stabilizes protein
structure | USP39 promotes CC
progression through interaction with SIRT7 and FOXM1 | (71) |
| Tu, 2023 | USP45 | High
expression | Myc | Stabilizes protein
structure | USP45 promotes
chemoresistance and progression of CC by deubiquitinating Myc | (73) |
| Zhou, 2020 | USP53 | Not mentioned | DDB2 | Stabilizes protein
structure | USP53 promotes
radiosensitivity in CC by mediating DDR through interaction with
DDB2 | (75) |
| Sanches, 2018 | CYLD | Low expression | miR-501 | Downstream factor
and mediates signal transduction | miR-501 inhibits
CYLD expression, thereby activating the NF-κB signaling pathway and
promoting CC progression | (77) |
| Huang, 2023 | CYLD | Not mentioned | Aurora B | Stabilizes protein
structure | CYLD suppresses CC
progression by deubiquitinating Aurora B | (78) |
| Zhang, 2022 | UCHL1 | High
expression | PROX1 | Stabilizes protein
structure | UCHL1 promotes CC
progression and metastasis by deubiquitinating PROX1 | (83) |
| Lei, 2024 | UCHL3 | High
expression | NRF2 | Stabilizes protein
structure | UCHL3 promotes CC
progression and metastasis by deubiquitinating NRF2 | (84) |
| Bao, 2024 | UCHL5 | High
expression | Wnt/β-catenin | Mediates signal
transduction | UCHL5 promotes CC
progression by regulating the Wnt/β-catenin signaling pathway | (89) |
| Li, 2019 | | Not mentioned | β-catenin | Stabilize protein
structure | UCHL5 promotes CC
progression by deubiquitinating β-catenin | (90) |
| Wang, 2017 | BAP1 | Not mentioned | miR-31 | Downstream
factor | miR-31 inhibits
BAP1 expression and promotes CC progression | (93) |
| Cao, 2026 | CSN5 | High
expression | ENO3, PKM2, HK2,
LDHA | Stabilizes protein
structure | CSN5 stabilizes
ENO3, increasing glycolytic flux | (98) |
| Gao, 2015 | CSN6 | High
expression | E6AP | Stabilizes protein
structure | CSN6 regulates p53
activity by binding with E6AP, thereby promoting CC
progression | (96) |
| Zhang, 2018 | BRCC3 | High
expression | E-cadherin,
Vimentin, MMP-2, MMP-9, Snail-1, Snail-2 | Mediates
EMT-related signal transduction | BRCC3 promotes CC
progression by maintaining a mesenchymal-like phenotype and
activating Snail family-mediated EMT signaling, thereby enhancing
cell viability, migration and invasion | (99) |
| Lee, 2016 | EIF3F | Low expression | CLU, especially
sCLU, AKT, ERK, GSK-3β, Elk-1, Egr-1, p53, p21, Bax | Mediates
survival/apoptosis signal transduction | EIF3F suppresses CC
progression by interacting with sCLU to interfere with its
maturation/secretion, thereby inhibiting AKT/ERK signaling,
stabilizing p53, and upregulating p21 and Bax, ultimately inducing
apoptosis and inhibiting proliferation and tumor growth | (102) |
| Wu, 2019 | OTUD1 | High
expression | MCL1 | Stabilizes protein
structure | OTUD1 promotes CC
progression by deubiquitinating MCL1 | (108) |
| Bossler, 2019 | OTUD1 | Not mentioned | AKT | Mediates signal
transduction | OTUD1 inhibits the
proliferation of CC cells by inhibiting the level of AKT | (110) |
| Xiao, 2024 | OTUB2 | High
expression | FOXM1 | Stabilizes protein
structure | OTUB2 promotes CC
progression by deubiquitinating FOXM1 | (113) |
| Song, 2023 | OTUB2 | High
expression | RBM15, m6A,
AKT/mTOR | Downstream factor,
mediates signal transduction | RBM15 and m6A are
jointly involved in regulating the mRNA level of OTUB2, thereby
activating the AKT/mTOR signaling pathway and promoting the
malignant behavior of cervical cancer cells | (114) |
| Yin, 2019 | OTUD5 | Not mentioned | AKT | Stabilizes protein
structure | OTUD5 regulates the
sensitivity of CC to radiotherapy by deubiquitinating AKT | (117) |
UCHs: Potential targets and mechanisms in
the diagnosis and treatment of CC
At present, the human UCH subfamily contains only
four members, namely UCHL1, UCHL3, UCHL5 and BRCA1-associated
protein-1 (BAP1). The primary function of UCHs is to hydrolyze the
isopeptide bonds at the C-terminus of protein substrates. Research
has increasingly highlighted the role of UCHs in malignant tumors,
especially in gynecological malignancies, breast cancer and liver
cancer (79,80).
UCHL1
UCHL1 is a DUB involved in protein degradation, cell
cycle control and apoptosis, and exhibits context-dependent roles
in cancer (80,81). In CC, UCHL1 is highly expressed in
both CSCC and small cell neuroendocrine carcinoma (SCNEC) (82,83). Functionally, UCHL1 promotes
proliferation, migration and invasion in CSCC, and is associated
with increased metastatic potential in SCNEC (82,83). Mechanistically, UCHL1 reduces
prospero homeobox 1 (PROX1) ubiquitination, stabilizing this
transcription factor, and promoting migration, invasion and lymph
node metastasis, particularly in SCNEC (83). Clinically, elevated UCHL1
expression is associated with poor prognosis and reduced overall
survival in CSCC, and increased lymph node metastasis in SCNEC
(82,83). These findings suggest that UCHL1
functions as a context-dependent oncogenic regulator, and exhibits
potential as a prognostic biomarker and a therapeutic target.
UCHL3
UCHL3 regulates cell cycle progression, apoptosis
and DNA damage repair, and has tumor-promoting roles in several
cancer types, including liver, bladder and ovarian cancer (84,85). In CC, UCHL3 is highly expressed
and its expression is negatively associated with patient survival
(85). A cell-based study has
shown that UCHL3 promotes CC cell proliferation, migration and
invasion (85), whereas an in
vivo study demonstrated that UCHL3 knockdown suppresses tumor
growth and reduces lung metastasis in mouse models (85). Mechanistically, UCHL3 stabilizes
nuclear factor erythroid 2-related factor 2 (NRF2) through
deubiquitination, increasing NRF2 levels and enhancing downstream
oncogenic effects. A catalytically inactive UCHL3 (C92A) mutant
fails to stabilize NRF2, indicating that this effect depends on its
deubiquitinating activity (85).
Clinically, elevated UCHL3 expression is associated with poor
prognosis in patients with CC, highlighting its potential relevance
as a prognostic biomarker. These findings suggest that the
UCHL3/NRF2 axis serves a critical role in CC progression and may
represent a promising therapeutic target.
UCHL5
UCHL5 (also known as UCH37) is a conserved DUB that
is aberrantly expressed in multiple types of cancer and often
exhibits oncogenic activity, although its prognostic importance is
tumor type-dependent (86,87).
In CC, UCHL5 is markedly upregulated in tumor tissues, and is
closely associated with tumor stage and patient prognosis (88,89). Functionally, UCHL5 promotes CC
cell proliferation and migration while suppressing apoptosis
(89,90). Mechanistically, UCHL5 regulates
the Wnt/β-catenin signaling pathway by reducing β-catenin
ubiquitination and preventing its proteasomal degradation, thereby
maintaining c-Myc expression and promoting malignant progression
(89,90). As a proteasome-associated DUB,
UCH37 also contributes to proteasomal homeostasis by facilitating
Rpn13 recruitment and reversing the ubiquitination of key
substrates (90). Clinically,
UCHL5 expression is associated with patient prognosis and features
of the tumor immune microenvironment, suggesting a role in disease
progression and immunotherapy response (90). These findings support UCHL5 as a
prognostic biomarker and potential therapeutic target in CC,
particularly through modulation of the Wnt/β-catenin signaling
axis.
BAP1
BAP1 is an important DUB involved in cell cycle
regulation, DNA damage repair and apoptosis, and generally
functions as a tumor suppressor in multiple types of cancer
(91,92). In CC, BAP1 has been identified as
a direct target of miR-31, an oncogenic miRNA that is highly
expressed in CC cells and tissues (93). Functionally, miR-31 promotes CC
cell proliferation, epithelial-mesenchymal transition (EMT) and
tumor growth in vivo, suggesting that suppression of BAP1
contributes to CC progression (93). Mechanistically, miR-31 directly
inhibits BAP1 expression, weakening its tumor-suppressive activity
and facilitating malignant phenotypes (93). Clinically, elevated miR-31
expression is associated with lymph node metastasis, stromal
invasion, lymphovascular space invasion, advanced FIGO stage,
larger tumor size and poor prognosis (93). These findings highlight the
clinical relevance of the miR-31/BAP1 axis and suggest that it may
serve as a potential therapeutic target.
In summary, the UCH family predominantly exhibits
tumor-promoting roles in CC, although functional diversity exists
among individual members. Biologically, UCH proteins regulate
protein stability and ubiquitin-dependent signaling, influencing
malignant phenotypes such as proliferation, migration, invasion,
apoptosis, EMT, DDR and metastasis. Mechanistically, these effects
involve key substrates and pathways, including PROX1, NRF2, BAP1
and the Wnt/β-catenin/c-Myc axis (Fig. 3; Table I). Clinically, aberrant expression
of UCH family members is strongly associated with tumor
progression, lymph node metastasis and poor patient survival across
various CC subtypes. These associations underscore their potential
as biomarkers for prognostic stratification and metastatic risk
assessment. UCH proteins may also represent promising therapeutic
targets. However, existing data are limited, necessitating further
studies to clarify the context-dependent functions of individual
UCH members and to validate their translational utility in the
clinical management of CC.
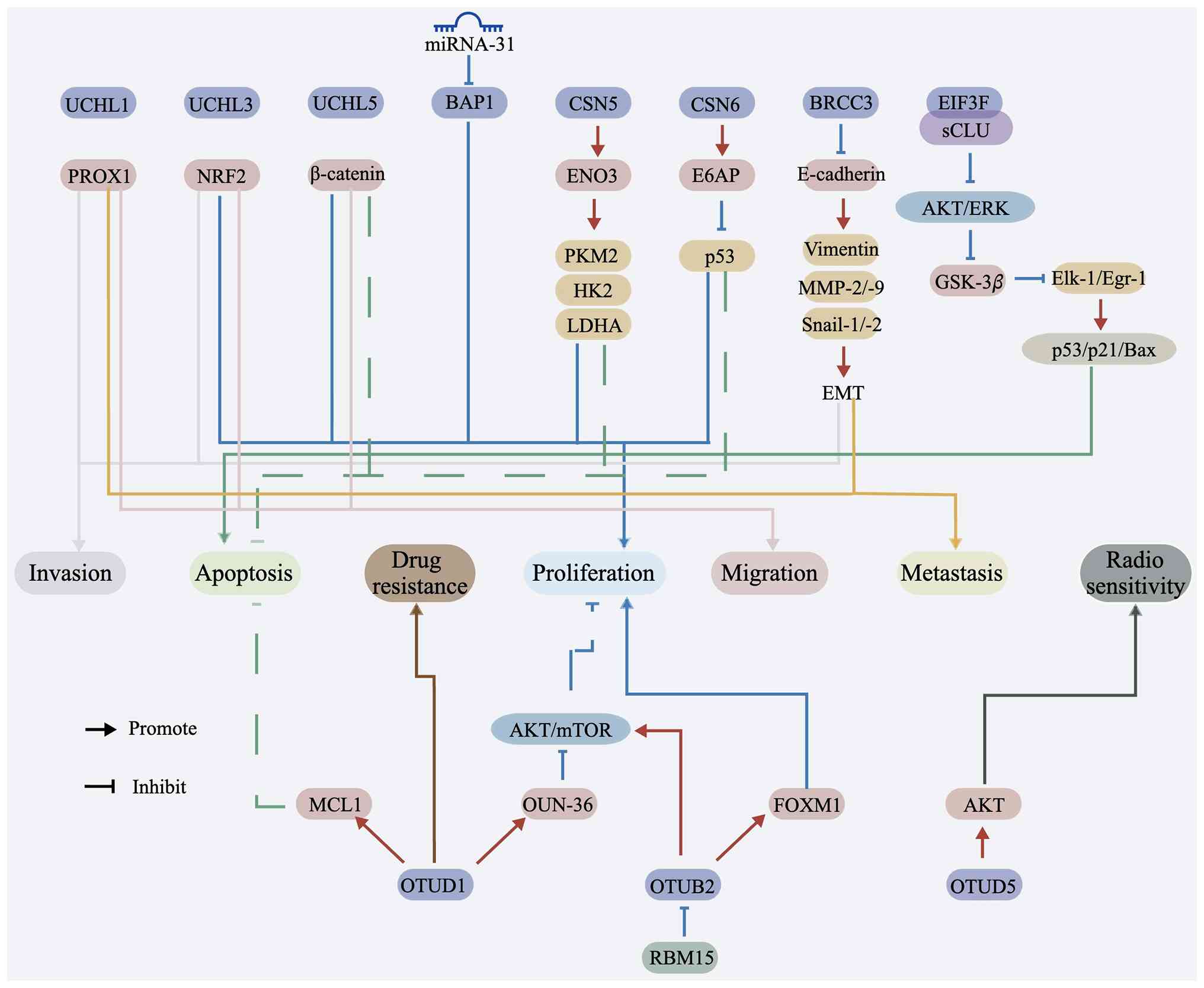 | Figure 3Regulatory roles of DUBs and
associated factors in tumor progression. Schematic diagram
illustrating how multiple DUBs and related regulatory proteins
contribute to malignant phenotypes in CC through distinct
downstream signaling pathways. Members of the UCH family, including
UCHL1, UCHL3 and UCHL5; as well as BAP1, CSN5, CSN6, BRCC3, EIF3F,
OTUD1 and OTUB2, modulate the stability or activity of key
effectors involved in tumor progression. Their downstream targets
include PROX1, NRF2, β-catenin, ENO3, E6AP, p53, E-cadherin,
vimentin, MMP-2/-9, Snail-1/-2, GSK-3β, Elk-1/Egr-1, p53/p21/Bax,
MCL1, OUN-36, AKT/mTOR, FOXM1 and RBM15. Through these regulatory
interactions, DUBs influence major cancer-related processes,
including proliferation, apoptosis, invasion, migration, metastasis
and drug resistance. Upstream regulation by miRNA-31 targeting BAP1
is also shown, and pathway crosstalk involving AKT/ERK and AKT/mTOR
signaling is highlighted. Arrows indicate activation or positive
regulation, whereas lines indicate inhibition or negative
regulation. Overall, the diagram emphasizes the central role of
deubiquitination-dependent signaling in coordinating tumor growth,
dissemination and therapeutic response in CC. BAP1,
BRCA1-associated protein-1; CC, cervical cancer; DUB,
deubiquitinating enzyme; EMT, epithelial-mesenchymal transition;
ENO3, enolase 3; HK2, hexokinase 2; LDHA, lactate dehydrogenase A;
miRNA, microRNA; NRF2, nuclear factor erythroid 2-related factor 2;
OTU, ovarian tumor-related protease; PKM2, pyruvate kinase M2;
PROX1, prospero homeobox 1; UCH, ubiquitin C-terminal hydrolase;
USP, ubiquitin-specific protease. |
JAMM/MPN metallopeptidases: New advances and
challenges in CC research
CSN5 (COPS5) and CSN6 (COPS6)
As the only metallopeptidase DUBs, members of the
JAMM/MPN metallopeptidase family are defined by their
Zn2+-dependent catalytic activity (94,95). CSN5 and CSN6 are upregulated in
CC, and high CSN5 expression is associated with advanced stage,
poor differentiation and lymph node involvement (96,97). Functionally, these proteins
promote proliferation, tumor growth, glycolysis and survival while
inhibiting apoptosis (96-98).
Mechanistically, CSN6 interacts with E6AP and modulates p53
activity, thereby influencing proliferation and apoptosis (96). CSN5 stabilizes enolase 3 (ENO3) by
preventing its ubiquitin-dependent degradation, which increases
glycolytic flux, and upregulates pyruvate kinase M2, hexokinase 2,
lactate dehydrogenase A and lactate production (98). Inhibition of glycolysis with
2-deoxy-D-glucose or ENO3 silencing reverses the oncogenic effects
of CSN5 upregulation (98).
Together, these findings indicate that JAMM/MPN proteins promote CC
progression through coordinated regulation of apoptosis and
metabolic reprogramming. The CSN6/E6AP/p53 axis and the CSN5/ENO3
pathway may therefore serve as biomarkers and therapeutic
targets.
BRCC3
BRCC3 (also known as BRCC36) is a JAMM/MPN DUB
involved in DNA damage signaling, cell cycle control and
inflammatory pathways (99). In
CC, it acts as an oncogenic regulator; BRCC3 is upregulated in CC
tissues, and in HeLa, SiHa and C33A cells compared with in normal
cervical controls (99). High
expression is associated with advanced FIGO stage, poor
differentiation and shorter overall survival, indicating aggressive
disease (99). Functionally,
BRCC3 promotes cell viability, migration and invasion, whereas its
knockdown suppresses these phenotypes in vitro (99). Mechanistically, BRCC3 drives EMT.
Silencing BRCC3 increases E-cadherin and reduces Vimentin, MMP-2,
MMP-9, Snail-1 and Snail-2, indicating loss of a mesenchymal state
and reduced invasive capacity (99). These findings support BRCC3 as a
prognostic biomarker and potential therapeutic target, particularly
for strategies aimed at limiting invasion and metastasis.
EIF3F
EIF3F is a component of the eukaryotic translation
initiation factor 3 complex, which also functions as a
non-canonical DUB with tumor-suppressive properties (100,101). In CC, EIF3F is markedly
downregulated in HeLa and CaSki cells, whereas clusterin (CLU),
particularly the secretory form (sCLU), is highly expressed
(102). Restoring EIF3F
suppresses proliferation, reduces colony formation, induces
apoptosis in vitro and inhibits tumor growth in xenograft
models (102). Mechanistically,
EIF3F binds the α-chain of sCLU in the cytoplasm and disrupts its
maturation and secretion, reducing intracellular precursor CLU and
mature α/β-CLU (102). This
inhibition suppresses AKT and ERK signaling, reduces GSK-3β
phosphorylation, downregulates EST-like-1 (Elk-1) and early growth
response 1 (Egr-1), stabilizes p53 and increases p21 and Bax
expression, partly in a p53-independent manner (102). Rescue experiments show that
EIF3F silencing restores CLU expression and downstream survival
signaling (102). Although
prognostic data remain limited, the combined loss of EIF3F and
upregulation of CLU suggest that EIF3F deficiency contributes to
apoptosis resistance and tumor progression (102). Restoring EIF3F activity or
targeting CLU-dependent survival pathways may therefore provide
therapeutic strategies.
Collectively, current evidence indicates that
JAMM/MPN metallopeptidase family DUBs largely promote tumor
progression in CC, although some members, such as EIF3F, act as
tumor suppressors in specific contexts. Overall, this family has
been implicated in the regulation of CC progression through protein
stability, apoptosis, EMT and metabolic reprogramming. Oncogenic
members, including CSN5 and BRCC3, enhance tumor growth, survival,
glycolysis and invasion through pathways such as CSN5/ENO3-mediated
metabolic activation and BRCC3-driven EMT signaling. By contrast,
EIF3F suppresses malignant progression by disrupting the
anti-apoptotic CLU pathway and restoring pro-death signaling
(Fig. 3; Table I). Clinically, dysregulated
expression of JAMM/MPN metallopeptidase family members is
associated with advanced stage, poor differentiation, metastasis
and reduced survival, supporting their potential utility as
biomarkers of disease progression. Together, these findings suggest
that JAMM/MPN proteins contribute to CC pathogenesis in a
context-dependent but mechanistically convergent manner. Targeting
oncogenic members while restoring tumor-suppressive ones may
represent a promising strategy for CC therapy.
OTUs: Association with CC and implications
for clinical treatment
OTUs are a vital subset of the DUB family. Initially
discovered as genes essential for Drosophila egg formation,
they share homology with ovarian cancer genes. This group contains
19 proteases and can be primarily classified into four subfamilies:
The Otubain subfamily, the OTUD subfamily, the A20 subfamily and
the OTULIN subfamily (103,104).
OTUD1
OTUD1 is a conserved OTUD family DUB involved in
cell signaling, protein homeostasis, cell cycle control and immune
regulation (103,105,106). Although it generally acts as a
tumor suppressor in a number of types of cancer (107), OTUD1 functions in a
context-dependent oncogenic role in CC. Bioinformatics analyses
have shown that OTUD1 is highly expressed in CC and is associated
with poor prognosis (108).
Functionally, OTUD1 exerts opposite effects on CC cells through
different mechanisms (108).
Mechanistically, OTUD1 interacts with MCL1 and enhances its
deubiquitination and stability, thereby antagonizing BH3
mimetic-induced apoptosis. Accordingly, OTUD1 knockdown sensitizes
CC cells to ABT-263 (Navitoclax; Bcl-2 family protein inhibitor),
whereas OTUD1 upregulation increases resistance (108,109). In addition, the OTUD1-derived
peptide OUN-36 inhibits AKT signaling by binding the RH domain of
AKT and blocking its PIP3-binding site, thereby preventing membrane
localization, reducing phosphorylation and suppressing CC cell
proliferation (110,111). These findings suggest that OTUD1
may serve as both a prognostic marker and a therapeutic target in
CC. The OTUD1/MCL1 axis and OUN-36-mediated AKT inhibition
represent promising strategies for drug development.
OTUB2
OTUB2, a member of the OTU DUB family, promotes
tumor progression by regulating DNA damage repair, immune escape
and metastasis-related signaling pathways (112). In CC, OTUB2 is highly expressed
in tissues and cell lines, including CSCC, and its expression is
associated with poor prognosis (113,114). Functionally, OTUB2 promotes CC
cell proliferation, tumor growth and malignant progression
(113,114). Mechanistically, OTUB2 stabilizes
FOXM1 through deubiquitination, thereby enhancing proliferation
(113). OTUB2 expression is also
upregulated by RBM15-mediated m6A modification, and elevated OTUB2
activates the AKT/mTOR signaling pathway to drive malignant
phenotypes in CSCC cells (114).
Clinically, these findings support OTUB2 as a marker of poor
prognosis in CC. The OTUB2/FOXM1 axis and the
RBM15/m6A/OTUB2/AKT/mTOR pathway represent potential therapeutic
targets.
OTUD5
OTUD5 is a deubiquitinating cysteine protease
involved in cell cycle regulation, DNA damage repair and signal
transduction, and its dysregulation has been linked to tumor
development (115). In CC, OTUD5
is downregulated and associated with poor prognosis (116). Its expression is negatively
associated with FIGO stage, lymph node metastasis and tumor type,
suggesting a tumor-suppressive role (116). Functionally, OTUD5 inhibits CC
progression and increases radiosensitivity (116,117). Mechanistically, bioinformatics
analyses have identified regulatory networks involving co-expressed
genes, miRNAs and interacting proteins associated with OTUD5
(116). OTUD5 upregulation
reduces AKT ubiquitination and enhances radiosensitivity in CC
cells, indicating that the OTUD5/AKT axis serves an important role
in radiotherapy response (117).
These findings indicate that OTUD5 may serve as a prognostic and
predictive biomarker for radiotherapy response, as well as a
potential therapeutic target in CC (116,117).
In summary, the OTU family serves diverse and
context-dependent roles in CC by regulating the stability and
activity of key substrates involved in apoptosis, cell cycle
control, DDR, signal transduction and therapeutic sensitivity
(Fig. 3; Table I). Only a limited number of OTU
family members have been investigated in CC, but current evidence
indicates that they contribute to tumor progression through both
tumor-promoting and tumor-suppressive mechanisms. OTUD1 and OTUB2
generally exert oncogenic effects by stabilizing proteins such as
MCL1 and FOXM1, or by activating signaling pathways including AKT
and AKT/mTOR, thereby promoting proliferation, survival and
treatment resistance. By contrast, OTUD5 may function as a tumor
suppressor or radiosensitizer in specific contexts, highlighting
the functional heterogeneity of this family. Notably, the
biological effects of OTU proteins extend beyond individual
enzyme-substrate interactions and instead reflect a broader
regulatory network in which multiple members converge on shared
pathways, particularly those involved in AKT signaling, cellular
survival and stress responses. Clinically, aberrant expression of
OTU family members is strongly associated with prognosis, FIGO
stage, lymph node metastasis and radiotherapy response, indicating
their potential value as biomarkers for disease stratification and
treatment prediction. Emerging approaches, including peptide-based
interventions, targeted inhibitors and DUBTAC-related strategies,
further support their translational potential. A deeper
understanding of OTU molecular functions and regulatory
interactions may therefore enable improved biomarker development
and precision therapy in CC.
Crosstalk and pathway convergence of DUBs in
CC
Although the roles of individual DUBs in CC have
been increasingly characterized, growing evidence suggests that
their biological importance extends beyond isolated
enzyme-substrate interactions. Instead, multiple DUBs participate
in interconnected regulatory networks through shared substrates,
overlapping signaling pathways and common biological processes.
Therefore, a comprehensive understanding of DUB function in CC
requires both substrate-level mapping and network-level analysis of
crosstalk.
One notable feature is the convergence of multiple
DUBs on proteins involved in cell cycle progression and checkpoint
control. For example, both USP3 and USP7 have been reported to
stabilize Cdc25A, suggesting that maintenance of Cdc25A is a shared
requirement for CC cell proliferation. This observation raises the
possibility of redundant, cooperative or context-dependent
regulation of the same substrate by distinct DUBs. In addition,
regulation of MDM2 by USP14 and p53 by USP19 further supports
convergence on the cell cycle checkpoint network. Together, these
findings suggest that DUBs collectively promote cell cycle
progression by sustaining cell cycle transition and weakening
stress-induced checkpoint responses.
Beyond shared substrates, multiple DUBs converge on
major oncogenic signaling pathways. FOXM1 emerges as a central
node, as USP21, USP39 and OTUB2 enhance FOXM1-associated oncogenic
activity through distinct mechanisms, including protein
stabilization, alternative splicing regulation and pathway
reinforcement. This suggests that FOXM1 may function as a
convergence hub for DUB-mediated regulation in CC, contributing to
proliferation, survival and radioresistance. Similarly, the
Wnt/β-catenin pathway is regulated by several DUBs. UCHL5/UCH37
stabilize β-catenin, while USP45 enhances Myc stability, a key
downstream effector of Wnt signaling. Together, these effects
amplify pathway output and promote malignant progression.
A comparable pattern is observed in the PI3K/AKT
pathway. USP18 and OTUB2 promote pathway activation, whereas OTUD1
and OTUD5 regulate AKT signaling in a context-dependent manner.
This observation suggests that DUBs influence not only protein
stability but also signaling dynamics and cellular stress
responses. Accordingly, DUB function in CC should not be strictly
categorized as oncogenic or tumor-suppressive, but instead
interpreted within specific molecular and signaling contexts.
DUB-mediated regulation must also be considered in
the context of HPV-driven carcinogenesis, a defining feature of CC.
Several DUBs directly modulate viral oncoproteins or their
downstream pathways. USP11 and USP15 stabilize HPV-16 E7 and E6,
respectively, and CSN6 regulates E6AP-mediated p53 degradation.
USP7 further contributes by modulating the DDR, potentially
supporting survival of HPV-transformed cells under replication
stress. Collectively, these findings support a model in which DUBs
function as components of an integrated regulatory network rather
than isolated enzymes. A deeper understanding of DUB crosstalk and
pathway convergence will be essential for identifying key
regulatory nodes and developing effective DUB-targeted therapies in
CC.
DUB-based therapeutic strategies for CC
Small-molecule DUB inhibitors: Current
progress and translational challenges
Despite strong biological rationale for targeting
DUBs, the clinical translation of DUB inhibitors remains in its
early stages. Most DUB-targeting compounds are still confined to
preclinical development, including inhibitors of PSMD14, UCHL1,
USP1, USP2, USP4, USP7, USP8, USP9X, USP10/USP13, USP11, USP14,
USP20 and USP30 (118). These
agents have shown promising antitumor, anti-inflammatory or
neuroprotective effects in biochemical assays, cell-based systems
and animal models, supporting the druggability of multiple DUB
family members; however, most have not progressed beyond
preclinical proof-of-concept. VLX1570, a dual inhibitor of UCHL5
and USP14, is one of the few to have entered early-phase clinical
trials in oncology (118).
However, its development was later suspended owing to safety
concerns, highlighting key translational challenges in this field.
Future work should therefore extend beyond mechanistic validation
in CC models, and prioritize the development of inhibitors with
improved selectivity, safety and pharmacokinetic properties. In
addition, rational patient stratification and biomarker-guided
approaches will be essential to support successful translation into
phase I/II clinical trials.
DUBTACs: An emerging strategy for
targeted protein stabilization in CC
DUBTACs represent an emerging strategy for targeted
protein stabilization. In contrast to proteolysis-targeting
chimeras (PROTACs), which induce degradation of target proteins
through recruitment of E3 ubiquitin ligases, DUBTACs recruit DUBs
to remove degradation-associated ubiquitin chains from proteins of
interest, thereby preventing proteasomal degradation and increasing
protein stability (119). A
typical DUBTAC consists of three components: A ligand for the
target protein, a ligand for the recruited DUB and a chemical
linker (120). DUBTACs induce
ternary complex formation by bringing the target protein and a DUB
into close proximity; this process facilitates the cleavage of
K48-linked polyubiquitin chains, extending the half-life of the
target protein (121). This
approach offers a strategy to stabilize proteins that are
beneficial for disease control but difficult to modulate using
conventional small-molecule activators or inhibitors.
The feasibility of this approach, although still at
an early stage of development, has been demonstrated in several
studies. Early work has shown that OTUB1 can be chemically
recruited to stabilize otherwise unstable proteins, establishing
proof of concept for pharmacological redirection of DUB activity
(122). This strategy has been
expanded to transcription factors and other traditionally
'undruggable' proteins in subsequent studies, suggesting the broad
application potential of DUBTACs (123). Specifically, a well-established
study (123) has developed three
series of transcription factor-targeting DUBTACs (TF-DUBTACs),
namely FOXO-DUBTAC, p53-DUBTAC and interferon regulatory factor
(IRF)-DUBTAC (123). These
TF-DUBTACs selectively stabilize the tumor suppressor transcription
factors FOXO3A, p53 and IRF3 in cells, respectively, in an
OTUB1-dependent manner (123).
Recently, USP7-based DUBTACs have been reported to stabilize AMPK
and activate AMPK signaling in HeLa cells, providing direct
evidence that this strategy can function in a CC-derived cellular
context and produce measurable downstream biological effects
(124). These findings indicate
that DUBTAC technology is not only conceptually attractive but also
experimentally feasible. However, compared with PROTACs and other
targeted protein regulation technologies, DUBTACs remain less
developed, with limited available DUB recruiters, an incomplete
understanding of ternary complex formation and no clinical-stage
agents reported to date (119).
In CC, DUBTACs may offer several potential
applications. First, they may stabilize tumor suppressors or
negative regulators of oncogenic pathways. For example, p53 is
often functionally impaired in CC because of HPV E6/E6AP-mediated,
ubiquitin-dependent degradation, which suggests that restoring p53
stability helps recover its cell cycle arrest and pro-apoptotic
functions (125). Similarly,
KLF6 (15) and CYLD (78) are potential candidates, as both
exhibit tumor-suppressive effects in CC. Second, DUBTACs may
enhance radiosensitivity. The USP53/DDB2 and OTUD5/AKT axes
regulate the radiation response in CC; thus, stabilization of
radiosensitizing proteins serves to improve the susceptibility of
tumor cells to radiation-induced DNA damage (75,117). Third, DUBTACs may also be useful
for modulating oncogenic and immune-related signaling. Pathways
such as FOXM1 (113), AKT/mTOR
(126), NF-κB (127) and Wnt/β-catenin (128) are frequently dysregulated in CC,
and some DUBs, including USP7 (129) and CYLD (130), are involved in PD-L1 expression
and tumor immune regulation.
Despite these prospects, several limitations should
be noted. Some recruitable DUBs, such as USP7, also have oncogenic
roles in CC, raising the risk of stabilizing oncogenic substrates.
The effects of protein stabilization may be context dependent,
particularly for DNA repair-related proteins (31,75). In addition, DUBTACs face
pharmacological challenges, including large molecular size, poor
cell permeability and limited CC-specific preclinical validation
(131).
In summary, DUBTAC technology offers a novel
strategy for targeted protein stabilization and expands the
therapeutic scope of ubiquitin-related interventions in CC. It may
be particularly promising for restoring tumor suppressors,
enhancing radiosensitivity and stabilizing key negative regulators
of oncogenic pathways. However, DUBTACs remain at an early stage of
development, with challenges in recruiter selection, target
prioritization, ternary complex optimization and safety evaluation.
Future studies integrating chemoproteomics, structural biology and
functional screening in CC models will be essential to identify
suitable substrates and DUB recruiters, and to advance DUBTAC-based
precision therapies for CC.
Conclusions and future prospects
In conclusion, current evidence indicates that DUBs
are biologically important and potentially actionable regulators in
CC, although the depth and maturity of available evidence vary
substantially among individual family members. Therefore, future
research should focus less on simply expanding the list of
CC-associated DUBs, and more on prioritizing those with the
strongest clinical and translational potential.
Among the DUBs reviewed, USP7 appears to be the
most promising near-term candidate because it is supported by
relatively consistent evidence linking dysregulated expression,
clinicopathological relevance, functional importance and
therapeutic tractability. USP8 has also emerged as a strong
candidate based on its prognostic associations and in vivo
functional validation. In addition, USP21, USP53 and OTUD5 warrant
particular attention in the context of radiotherapy, where
DUB-based patient stratification and radiosensitization strategies
may offer practical clinical benefits.
From a therapeutic perspective, small molecule
inhibition of oncogenic DUBs currently represents the most feasible
route for clinical translation in CC. By contrast, DUBTAC-based
approaches remain highly promising but are still at an early
developmental stage and will require substantial optimization
before clinical application in CC becomes realistic.
Subsequent research should prioritize three key
directions: i) Validation of leading DUB candidates in clinically
annotated patient cohorts; ii) evaluation in treatment-oriented
preclinical models; and iii) biomarker-guided development of
DUB-targeted strategies. Ultimately, the greatest near-term
progress is likely to come from concentrated work on a limited
number of well-supported DUBs rather than from broad but
superficial expansion of candidate lists.
Availability of data and materials
Not applicable.
Authors' contributions
CZP collected literature and drafted the
manuscript. XXS helped to draft and modify the manuscript. HX
conceived the topic of the present review and revised the
manuscript. KHB offered significant guidance and critically
reviewed this manuscript. Data authentication is not applicable.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural Science
Foundation of China (grant no. 82460310) awarded to CZP and the
Ministry of Health & Welfare, Republic of Korea (grant no.
RS-2025-02215666) awarded to KHB. The funding agencies had no role
in the study design, literature collection, analysis,
interpretation of data, manuscript preparation or the decision to
submit the article for publication.
References
|
1
|
Li T, Zhang H, Lian M, He Q, Lv M, Zhai L,
Zhou J, Wu K and Yi M: Global status and attributable risk factors
of breast, cervical, ovarian, and uterine cancers from 1990 to
2021. J Hematol Oncol. 18:52025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bohlin KS, Brännström M and Dahm-Kähler P:
Gynecological cancer during pregnancy-from a gyne-oncological
perspective. Acta Obstet Gynecol Scand. 103:761–766. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim SH and Baek KH: Regulation of cancer
metabolism by deubiquitinating enzymes: The warburg effect. Int J
Mol Sci. 22:61732021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clague MJ, Urbe S and Komander D: Breaking
the chains: Deubiquitylating enzyme specificity begets function.
Nat Rev Mol Cell Biol. 20:338–352. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen X, Tian L, Zhang L, Gao W, Yu M, Li Z
and Zhang W: Deubiquitinase USP39 promotes SARS-CoV-2 replication
by deubiquitinating and stabilizing the envelope protein. Antiviral
Res. 221:1057902024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wendrich K, Gallant K, Recknagel S,
Petroulia S, Kazi NH, Hane JA, Fuhrer S, Bezstarosti K, O'Dea R,
Demmers J and Gersch M: Discovery and mechanism of
K63-linkage-directed deubiquitinase activity in USP53. Nat Chem
Biol. 21:746–757. 2025. View Article : Google Scholar :
|
|
7
|
Luo C, Yu Y, Zhu J, Chen L, Li D, Peng X,
Liu Z, Li Q, Cao Q, Huang K and Yuan R: Deubiquitinase PSMD7
facilitates pancreatic cancer progression through activating nocth1
pathway via modifying SOX2 degradation. Cell Biosci. 14:352024.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pan Q, Yu F, Jin H, Zhang P, Huang X, Peng
J, Xie X, Li X, Ma N, Wei Y, et al: eIF3f mediates SGOC pathway
reprogramming by enhancing deubiquitinating activity in colorectal
cancer. Adv Sci (Weinh). 10:e23007592023. View Article : Google Scholar
|
|
9
|
Tang X, Qiu J, Chen C, Zhang J, Tan R, Li
W, Huang J, Chen H and Yang Z: STAMBPL1 promotes the progression of
gastric cancer via deubiquitinating IQGAP1 to activate the
JAK2/STAT3 pathway. J Gastroenterol. 61:250–265. 2026. View Article : Google Scholar
|
|
10
|
Xie X, Wang X, Jiang D, Wang J, Fei R,
Cong X, Wei L, Wang Y and Chen H: PPPDE1 is a novel deubiquitinase
belonging to a cysteine isopeptidase family. Biochem Biophys Res
Commun. 488:291–296. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suk FM, Chang CC, Sun PC, Ke WT, Chung CC,
Lee KL, Chan TS and Liang YC: MCPIP1 enhances TNF-α-mediated
apoptosis through downregulation of the NF-κB/cFLIP axis. Biology
(Basel). 10:6552021.
|
|
12
|
Huang S, Qi D, Liang J, Miao R, Minagawa
K, Quinn T, Matsui T, Fan D, Liu J and Fu M: The putative tumor
suppressor Zc3h12d modulates toll-like receptor signaling in
macrophages. Cell Signal. 24:569–576. 2012. View Article : Google Scholar
|
|
13
|
Ren J, Yu P, Liu S, Li R, Niu X, Chen Y,
Zhang Z, Zhou F and Zhang L: Deubiquitylating enzymes in cancer and
immunity. Adv Sci (Weinh). 10:e23038072023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu X, Wu Y, Yao M, Chen Z and Li Q: The
other side of the coin: Protein deubiquitination by
ubiquitin-specific protease 1 in cancer progression and therapy.
Future Med Chem. 17:329–345. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ye Y, Li M, Pan Q, Fang X, Yang H, Dong B,
Yang J, Zheng Y, Zhang R and Liao Z: Machine learning-based
classification of deubiquitinase USP26 and its cell proliferation
inhibition through stabilizing KLF6 in cervical cancer. Comput Biol
Med. 168:1077452024. View Article : Google Scholar
|
|
16
|
Zhou T, Li X, Zhao F, Zhou J and Sun B:
Lactamase β reprograms lipid metabolism to inhibit the progression
of endometrial cancer through attenuating MDM2-mediated p53
ubiquitination and degradation. Arch Biochem Biophys.
764:1102872025. View Article : Google Scholar
|
|
17
|
Li M, Tian Y, Si L, Fu H, Lai T and Guo R:
OTUD4-mediated inhibition of YAP1 signaling pathway in ovarian
cancer: Implications for macrophage polarization and recruitment.
Int Immunopharmacol. 147:1140112025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao H, Xi Z, Dai J, Xue J, Guan X, Zhao L,
Chen Z and Xing F: Drug resistance mechanisms and treatment
strategies mediated by ubiquitin-specific proteases (USPs) in
cancers: New directions and therapeutic options. Mol Cancer.
23:882024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cockram PE, Kist M, Prakash S, Chen SH,
Wertz IE and Vucic D: Ubiquitination in the regulation of
inflammatory cell death and cancer. Cell Death Differ. 28:591–605.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roberts JZ, Crawford N and Longley DB: The
role of ubiquitination in apoptosis and necroptosis. Cell Death
Differ. 29:272–284. 2022. View Article : Google Scholar :
|
|
21
|
Das S, Chandrasekaran AP, Suresh B, Haq S,
Kang JH, Lee SJ, Kim J, Kim J, Lee S, Kim HH, et al: Genome-scale
screening of deubiquitinase subfamily identifies USP3 as a
stabilizer of Cdc25A regulating cell cycle in cancer. Cell Death
Differ. 27:3004–3020. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang H, Liu W, Wu Y and Chen C: USP3: Key
deubiquitylation enzyme in human diseases. Cancer Sci.
115:2094–2106. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Y, Shi Y, Niu K, Yang R, Lv Q, Zhang
W, Feng K and Zhang Y: Ubiquitin specific peptidase 3: An emerging
deubiquitinase that regulates physiology and diseases. Cell Death
Discov. 10:2432024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou JM, Dai WX, Wang RJ, Xu WQ, Xiang Z,
Wang YX, Zhang T, Zhao YM, Wang L and Mao AR: Organoid modeling
identifies USP3-AS1 as a novel promoter in colorectal cancer liver
metastasis through increasing glucose-driven histone lactylation.
Acta Pharmacol Sin. 46:1404–1418. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: Gepia: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45:W98–W102.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
He Y, Jiang S, Zhong Y, Wang X, Cui Y,
Liang J, Sun Y, Zhu Z, Huang Z and Mao X: Usp7 promotes
non-small-cell lung cancer cell glycolysis and survival by
stabilizing and activating c-Abl. Clin Transl Med. 13:e15092023.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu T, Sun T, Chen X, Wu J, Sun X, Liu X,
Yan H, Fu Q, Fan Z, Wang X, et al: Targeting ARPC1B overcomes
immune checkpoint inhibitor resistance in glioblastoma by reversing
pro-tumorigenic macrophage polarization. Cancer Res. 85:1236–1252.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yi J, Li H, Chu B, Kon N, Hu X, Hu J,
Xiong Y, Kaniskan HU, Jin J and Gu W: Inhibition of USP7 induces
p53-independent tumor growth suppression in triple-negative breast
cancers by destabilizing FOXM1. Cell Death Differ. 30:1799–1810.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Sun X and Huang Z: USP7 facilitates
deubiquitination of LRRC42 in colorectal cancer to accelerate
tumorigenesis and augment wnt/β-catenin signaling. Biochim Biophys
Acta Mol Cell Res. 1872:1198592025. View Article : Google Scholar
|
|
30
|
Yin Y, Li Y, Ma B, Ren C, Zhao S and Li J,
Gong Y, Yang H and Li J: Mitochondrial-derived peptide MOTS-c
suppresses ovarian cancer progression by attenuating USP7-mediated
LARS1 deubiquitination. Adv Sci (Weinh). 11:e24056202024.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Su D, Ma S, Shan L, Wang Y, Wang Y, Cao C,
Liu B, Yang C, Wang L, Tian S, et al: Ubiquitin-specific protease 7
sustains DNA damage response and promotes cervical carcinogenesis.
J Clin Invest. 128:4280–4296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang N, Xu J, Wang Y, Zhang X and Zhang H:
USP7 promotes cervical cancer progression by stabilizing mtdh
expression through deubiquitination. J Cancer Res Clin Oncol.
150:1962024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Das S, Chandrasekaran AP, Jo KS, Ko NR, Oh
SJ, Kim KS and Ramakrishna S: HAUSP stabilizes Cdc25A and protects
cervical cancer cells from DNA damage response. Biochim Biophys
Acta Mol Cell Res. 1867:1188352020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li N, Geng F, Liang SM and Qin X: USP7
inhibits TIMP2 by up-regulating the expression of EZH2 to activate
the NF-κB/PD-L1 axis to promote the development of cervical cancer.
Cell Signal. 96:1103512022. View Article : Google Scholar
|
|
35
|
Dufner A and Knobeloch KP:
Ubiquitin-specific protease 8 (USP8/UBPy): A prototypic multidomain
deubiquitinating enzyme with pleiotropic functions. Biochem Soc
Trans. 47:1867–1879. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yan M, Zhao C, Wei N, Wu X, Cui J and Xing
Y: High expression of ubiquitin-specific protease 8 (USP8) is
associated with poor prognosis in patients with cervical squamous
cell carcinoma. Med Sci Monit. 24:4934–4943. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jeong M, Lee EW, Seong D, Seo J, Kim JH,
Grootjans S, Kim SY, Vandenabeele P and Song J: USP8 suppresses
death receptor-mediated apoptosis by enhancing FLIPL
stability. Oncogene. 36:458–470. 2017. View Article : Google Scholar
|
|
38
|
Xu Y, Zeng J, Liu K, Li D, Huang S, Fu S,
Ye M, Tao S and Wu J: USP11 promotes lipogenesis and tumorigenesis
by regulating SREBF1 stability in hepatocellular carcinoma. Cell
Commun Signal. 22:5502024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cui L, Yang L, Lai B, Luo L, Deng H, Chen
Z and Wang Z: Integrative and comprehensive pan-cancer analysis of
ubiquitin specific peptidase 11 (USP11) as a prognostic and
immunological biomarker. Heliyon. 10:e345232024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liao Y, Zhou D, Wang P, Yang M and Jiang
N: Ubiquitin specific peptidase 11 as a novel therapeutic target
for cancer management. Cell Death Discov. 8:2922022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lin CH, Chang HS and Yu WC: USP11
stabilizes HPV-16E7 and further modulates the E7 biological
activity. J Biol Chem. 283:15681–15688. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shi S, Huang C, Tang X, Liu H, Feng W and
Chen C: Identification and verification of diagnostic biomarkers
for deep infiltrating endometriosis based on machine learning
algorithms. J Biol Eng. 18:702024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma LZ, Wang A, Lai YH, Zhang J, Zhang XF,
Chen SL and Zhou XY: USP14 inhibition promotes DNA damage repair
and represses ovarian granulosa cell senescence in premature
ovarian insufficiency. J Transl Med. 22:8342024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gong X, Jia L, Zhou L and Hu T: USP14
predicts poorer survival outcomes and promotes tumor progression in
endometrial carcinoma by activating NF-κB signaling. Aging (Albany
NY). 15:12120–12135. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xu L, Wang J, Yuan X, Yang S, Xu X, Li K,
He Y, Wei L, Zhang J and Tian Y: IU1 suppresses proliferation of
cervical cancer cells through MDM2 degradation. Int J Biol Sci.
16:2951–2963. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhu R, Li M, Wang D, Liu C, Xie L, Yang Y,
Gu X, Zhao K, Tian Y and Cai S: USP15 regulates radiation-induced
DNA damage and intestinal injury through K48-linked
deubiquitination and stabilisation of ATM. Mol Med. 30:2052024.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fang R, Jia Z, Xin Y, Zhao K, Qin W, Lu H,
Zhou Y, Yang Y and Fang H: N6-methyladenosine-modification of USP15
regulates chemotherapy resistance by inhibiting LGALS3
ubiquitin-mediated degradation via akt/mtor signaling activation
pathway in hepatocellular carcinoma. Cell Death Discov. 11:32025.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang J, Li C, Hou Y, Liu D, Li Q, Wang Z,
Tang R, Zheng K, Guo H and Wang W: Mir-26a exerts broad-spectrum
antiviral effects via the enhancement of RIG-I-mediated type I
interferon response by targeting USP15. Microbiol Spectr.
12:e03124232024. View Article : Google Scholar :
|
|
49
|
Nishi H, Ono M, Ohno S, Yamanaka Z, Sasaki
T, Ohyashiki K, Ohyashiki JH and Kuroda M: Hypoxia-induced
paclitaxel resistance in cervical cancer modulated by miR-100
targeting of USP15. Gynecol Oncol Rep. 45:1011382023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yaginuma Y, Yoshimoto M and Tokuda A:
USP15 inhibits HPV16 E6 degradation and catalytically inactive
USP15 has reduced inhibitory activity. Acta Virol. 62:147–156.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yu L, Chen Z, Wu Y, Xu M, Zhong D, Xu H
and Zhu W: Unraveling role of ubiquitination in drug resistance of
gynecological cancer. Am J Cancer Res. 14:2523–2537. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ducker C and Shaw PE: USP17-mediated
de-ubiquitination and cancer: Clients cluster around the cell
cycle. Int J Biochem Cell Biol. 130:1058862021. View Article : Google Scholar
|
|
53
|
Li A, Wang T, Zhou S, Han J and Wu W:
USP17 regulates preeclampsia by modulating the NF-κB signaling
pathway via deubiquitinating HDAC2. Placenta. 145:9–18. 2024.
View Article : Google Scholar
|
|
54
|
Huang S, Li J, Wu S, Zheng Z, Wang C, Li
H, Zhao L, Zhang X, Huang H, Huang C and Xie Q: C4orf19 inhibits
colorectal cancer cell proliferation by competitively binding to
Keap1 with TRIM25 via the USP17/Elk-1/CDK6 axis. Oncogene.
42:1333–1346. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang L, Ma Y, Zhang S, Yang Y and Huang B:
NFATC2 promotes lactate and M2 macrophage polarization through
USP17 in lung adenocarcinoma. Anticancer Drugs. 35:385–396. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ramakrishna S, Suresh B, Lee EJ, Lee HJ,
Ahn WS and Baek KH: Lys-63-specific deubiquitination of SDS3 by
USP17 regulates HDAC activity. J Biol Chem. 286:10505–10514. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
de la Vega M, Burrows JF, McFarlane C,
Govender U, Scott CJ and Johnston JA: The deubiquitinating enzyme
USP17 blocks N-Ras membrane trafficking and activation but leaves
K-Ras unaffected. J Biol Chem. 285:12028–12036. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Holicek P, Guilbaud E, Klapp V, Truxova I,
Spisek R, Galluzzi L and Fucikova J: Type I interferon and cancer.
Immunol Rev. 321:115–127. 2024. View Article : Google Scholar
|
|
59
|
Arimoto KI, Miyauchi S, Troutman TD, Zhang
Y, Liu M, Stoner SA, Davis AG, Fan JB, Huang YJ, Yan M, et al:
Expansion of interferon inducible gene pool via USP18 inhibition
promotes cancer cell pyroptosis. Nat Commun. 14:2512023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Diao W, Guo Q, Zhu C, Song Y, Feng H, Cao
Y, Du M and Chen H: USP18 promotes cell proliferation and
suppressed apoptosis in cervical cancer cells via activating AKT
signaling pathway. BMC Cancer. 20:7412020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chandrasekaran AP, Tyagi A, Poondla N,
Sarodaya N, Karapurkar JK, Kaushal K, Park CH, Hong SH, Kim KS and
Ramakrishna S: Dual role of deubiquitinating enzyme USP19 regulates
mitotic progression and tumorigenesis by stabilizing survivin. Mol
Ther. 30:3414–3429. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tyagi A, Karapurkar JK, Colaco JC,
Sarodaya N, Antao AM, Kaushal K, Haq S, Chandrasekaran AP, Das S,
Singh V, et al: USP19 negatively regulates p53 and promotes
cervical cancer progression. Mol Biotechnol. 66:2032–2045. 2024.
View Article : Google Scholar
|
|
63
|
Dyson HJ and Wright PE: How does p53 work?
Regulation by the intrinsically disordered domains. Trends Biochem
Sci. 50:9–17. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu Z, Dong C, Yang Y, Zheng Z and Ge X:
USP21 stabilizes immune checkpoint of CD276 and serves as an
immunological and tumor prognostic biomarker. Biochem Biophys Res
Commun. 745:1512212025. View Article : Google Scholar
|
|
65
|
Li Z, Liu X, Yu H, Wang S, Zhao S and
Jiang G: USP21 regulates hippo signaling to promote radioresistance
by deubiquitinating FOXM1 in cervical cancer. Hum Cell. 35:333–347.
2022. View Article : Google Scholar
|
|
66
|
Feng T, Ling S, Xu C, Ying L, Su D and Xu
X: Ubiquitin-specific peptidase 22 in cancer. Cancer Lett.
514:30–37. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yang M, Liu YD, Wang YY, Liu TB, Ge TT and
Lou G: Ubiquitin-specific protease 22: A novel molecular biomarker
in cervical cancer prognosis and therapeutics. Tumour Biol.
35:929–934. 2014. View Article : Google Scholar
|
|
68
|
Sheng CL, Jiang BD, Zhang CQ, Huang JH,
Wang Z and Xu C: USP26 suppresses type I interferon signaling by
targeting TRAF3 for deubiquitination. PLoS One. 19:e03077762024.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li L and Zhang X, Li Y, Xiao B, Pei S,
Jiang H and Zhang X: Transcription factor KLF16 activates MAGT1 to
regulate the tumorigenesis and progression of breast cancer. Int J
Mol Med. 50:1152022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lu C, Cai Y, Wu S, Wang Y, Li JB, Xu G and
Ma J: Deubiquitinating enzyme USP39 promotes the growth and
metastasis of gastric cancer cells by modulating the degradation of
rna-binding protein RBM39. J Biol Chem. 300:1077512024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yu J, Yuan S, Song J and Yu S: USP39
interacts with SIRT7 to promote cervical squamous cell carcinoma by
modulating autophagy and oxidative stress via FOXM1. J Transl Med.
21:8072023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li K, Wang Q and Bian H: USP45 acts as an
oncogene to regulate the proliferation of esophageal cancer cells.
Genes Dis. 11:1011352024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tu X, Li C, Sun W, Tian X, Li Q, Wang S,
Ding X and Huang Z: Suppression of cancer cell stemness and drug
resistance via myc destabilization by deubiquitinase USP45
inhibition with a natural small molecule. Cancers (Basel).
15:9302023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Xia G, Guo Y, Zhang J, Han M, Meng X and
Lv J: An overview of the deubiquitinase USP53: A promising
diagnostic marker and therapeutic target. Curr Protein Pept Sci.
25:708–718. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhou Q, Yao X, Wu C, Chen S and Fan D:
Knockdown of ubiquitin-specific protease 53 enhances the
radiosensitivity of human cervical squamous cell carcinoma by
regulating DNA damage-binding protein 2. Technol Cancer Res Treat.
19:15330338209297922020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li Y, Yang C, Xie L, Shi F, Tang M, Luo X,
Liu N, Hu X, Zhu Y, Bode AM, et al: CYLD induces high oxidative
stress and DNA damage through class I HDACS to promote
radiosensitivity in nasopharyngeal carcinoma. Cell Death Dis.
15:952024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Sanches JGP, Xu Y, Yabasin IB, Li M, Lu Y,
Xiu X, Wang L, Mao L, Shen J, Wang B, et al: miR-501 is upregulated
in cervical cancer and promotes cell proliferation, migration and
invasion by targeting CYLD. Chem Biol Interact. 285:85–95. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Huang Y, Zhang W, Yu Z, Su H, Zeng B, Piao
J, Wang J and Wu J: A tumor suppressive role of CYLD as a novel
potential DUB of aurora B in cervical cancer. Clin Med Insights
Oncol. 17:117955492311808322023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Xu Z, Zhang N and Shi L: Potential roles
of uch family deubiquitinases in tumorigenesis and chemical
inhibitors developed against them. Am J Cancer Res. 14:2666–2694.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fang Y and Shen X: Ubiquitin
carboxyl-terminal hydrolases: Involvement in cancer progression and
clinical implications. Cancer Metastasis Rev. 36:669–682. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu S, Garcia-Marques FJ, Shen M, Bermudez
A, Pitteri SJ and Stoyanova T: Ubiquitin C-terminal hydrolase L1 is
a regulator of tumor growth and metastasis in double-negative
prostate cancer. Am J Clin Exp Urol. 12:306–322. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Jia Q, Wang H, Xiao X, Sun Y, Tan X, Chai
J, Yang Y, Yin Z, Li M, Wang K and Liu J: UCHL1 acts as a
prognostic factor and promotes cancer stemness in cervical squamous
cell carcinoma. Pathol Res Pract. 247:1545742023. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang Y, Ding J, Zhang X and Hua K:
Ubiquitin C-terminal hydrolase l1 promotes lymph node metastasis in
small cell neuroendocrine carcinomas of the cervix. J Int Med Res.
50:30006052210876202022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lei H, Xu H and Wu Y: Role of UCHL3 in
health and disease. Biochem Biophys Res Commun. 734:1506262024.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Liu T, Fan MQ, Xie XX, Shu QP, Du XH, Qi
LZ, Zhang XD, Zhang MH, Shan G, Du RL and Li SZ: Activation of
CTNNB1 by deubiquitinase UCHL3-mediated stabilization facilitates
bladder cancer progression. J Transl Med. 21:6562023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wan B, Cheng M, He T and Zhang L: UCHL5
promotes hepatocellular carcinoma progression by promoting
glycolysis through activating wnt/β-catenin pathway. BMC Cancer.
24:6182024. View Article : Google Scholar
|
|
87
|
Song A, Hazlett Z, Abeykoon D, Dortch J,
Dillon A, Curtiss J, Martinez SB, Hill CP, Yu C, Huang L, et al:
Branched ubiquitin chain binding and deubiquitination by UCH37
facilitate proteasome clearance of stress-induced inclusions.
Elife. 10:e727982021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Rolen U, Kobzeva V, Gasparjan N, Ovaa H,
Winberg G, Kisseljov F and Masucci MG: Activity profiling of
deubiquitinating enzymes in cervical carcinoma biopsies and cell
lines. Mol Carcinog. 45:260–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Bao L, Wu Y, Ren Z, Huang Y, Jiang Y, Li
K, Xu X, Ye Y and Gui Z: Comprehensive pan-cancer analysis
indicates UCHL5 as a novel cancer biomarker and promotes cervical
cancer progression through the wnt signaling pathway. Biol Direct.
19:1392024. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Li Z, Zhou L, Jiang T, Fan L, Liu X and
Qiu X: Proteasomal deubiquitinase UCH37 inhibits degradation of
β-catenin and promotes cell proliferation and motility. Acta
Biochim Biophys Sin (Shanghai). 51:277–284. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sturgill IR, Raab JR and Hoadley KA:
Expanded detection and impact of BAP1 alterations in cancer. NAR
Cancer. 6:zcae0452024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Puri S, Chen SN, Chiu YH, Draczkowski P,
Ko KT, Yang TJ, Wang YS, Uchiyama S and Hsu SD: Impacts of
cancer-associated mutations on the structure-activity relationship
of BAP1. J Mol Biol. 434:1675532022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Wang N, Li Y and Zhou J: miR-31 functions
as an oncomir which promotes epithelial-mesenchymal transition via
regulating BAP1 in cervical cancer. Biomed Res Int.
2017:63614202017.PubMed/NCBI
|
|
94
|
Zou S, Qin B, Yang Z, Wang W, Zhang J,
Zhang Y, Meng M, Feng J, Xie Y, Fang L, et al: CSN6 mediates
nucleotide metabolism to promote tumor development and
chemoresistance in colorectal cancer. Cancer Res. 83:414–427. 2023.
View Article : Google Scholar
|
|
95
|
Du W, Zhang R, Muhammad B and Pei D:
Targeting the COP9 signalosome for cancer therapy. Cancer Biol Med.
19:573–590. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gao S, Fang L, Phan LM, Qdaisat A, Yeung
SC and Lee MH: COP9 signalosome subunit 6 (CSN6) regulates
E6AP/UBE3A in cervical cancer. Oncotarget. 6:28026–28041. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Wang L, Zeng X, Yang G, Liu G and Pan Y:
Pan-cancer analyses of Jab1/COPS5 reveal oncogenic role and
clinical outcome in human cancer. Heliyon. 8:e125532022. View Article : Google Scholar
|
|
98
|
Cao Z, Yang S, Zou J, Jin Y, Zhang Y, Chen
T and Shen Y: CSN5 overexpression promotes the integral progression
of cervical cancer by enhancing ENO3-mediated glycolysis.
Apoptosis. 31:412026. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang F and Zhou Q: Knockdown of BRCC3
exerts an anti-tumor effect on cervical cancer in vitro. Mol Med
Rep. 18:4886–4894. 2018.PubMed/NCBI
|
|
100
|
Hershey JW: Regulation of protein
synthesis and the role of EIF3 in cancer. Braz J Med Biol Res.
43:920–930. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Shi J, Kahle A, Hershey JW, Honchak BM,
Warneke JA, Leong SP and Nelson MA: Decreased expression of
eukaryotic initiation factor 3f deregulates translation and
apoptosis in tumor cells. Oncogene. 25:4923–4936. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lee JY, Kim HJ, Rho SB and Lee SH: eIF3f
reduces tumor growth by directly interrupting clusterin with
anti-apoptotic property in cancer cells. Oncotarget. 7:18541–18557.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Oikawa D, Shimizu K and Tokunaga F:
Pleiotropic roles of a KEAP1-associated deubiquitinase, OTUD1.
Antioxidants (Basel). 12:3502023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Du J, Fu L, Sui Y and Zhang L: The
function and regulation of otu deubiquitinases. Front Med.
14:542–563. 2020. View Article : Google Scholar
|
|
105
|
Liu Q, Mallette E, Zheng H and Zhang W:
Development of an OTUD1 ubiquitin variant inhibitor. Biochem J.
480:1317–1330. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zhou X, Xing Y, Wang Y, Lv M, Zhang P, Zhu
S, Ge J, Liu L, Zhao M, Gong H, et al: OTUD1 regulates cytokine
expression and related pathways in goose fatty liver by promoting
deubiquitination of its target proteins. Poult Sci. 103:1043822024.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zheng S, Li Y, Song X, Wu M, Yu L, Huang
G, Liu T, Zhang L, Shang M, Zhu Q, et al: OTUD1 ameliorates
cerebral ischemic injury through inhibiting inflammation by
disrupting K63-linked deubiquitination of RIP2. J
Neuroinflammation. 20:2812023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Wu L, Lin Y, Feng J, Qi Y, Wang X, Lin Q,
Shi W, Zheng E, Wang W, Hou Z, et al: The deubiquitinating enzyme
OTUD1 antagonizes BH3-mimetic inhibitor induced cell death through
regulating the stability of the MCL1 protein. Cancer Cell Int.
19:2222019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Deng H, Han Y, Liu L, Zhang H, Liu D, Wen
J, Huang M and Zhao L: Targeting myeloid leukemia-1 in cancer
therapy: Advances and directions. J Med Chem. 67:5963–5998. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Bossler F, Hoppe-Seyler K and Hoppe-Seyler
F: PI3K/AKT/mTOR signaling regulates the Virus/Host cell crosstalk
in HPV-positive cervical cancer cells. Int J Mol Sci. 20:21882019.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fan G, Wang F, Chen Y, Zheng Q, Xiong J,
Lv Q, Wu K, Xiong J, Wei L, Li D, et al: The deubiquitinase OTUD1
noncanonically suppresses Akt activation through its N-terminal
intrinsically disordered region. Cell Rep. 42:1119162023.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Li J, Zhang N, Li M, Hong T, Meng W and
Ouyang T: The emerging role of OTUB2 in diseases: From cell
signaling pathway to physiological function. Front Cell Dev Biol.
10:8207812022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Xiao J, Wang L, Zhuang Y, Zhu Q, Li W,
Liao H, Chen X and Liu Z: The deubiquitinase OTUB2 promotes
cervical cancer growth through stabilizing FOXM1. Am J Transl Res.
16:75–84. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Song Y and Wu Q: RBM15 m6 a
modification-mediated OTUB2 upregulation promotes cervical cancer
progression via the AKT/mTOR signaling. Environ Toxicol.
38:2155–2164. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fu L, Lu K, Jiao Q, Chen X and Jia F: The
regulation and double-edged roles of the deubiquitinase OTUD5.
Cells. 12:11612023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Bai M, Che Y, Lu K and Fu L: Analysis of
deubiquitinase OTUD5 as a biomarker and therapeutic target for
cervical cancer by bioinformatic analysis. PeerJ. 8:e91462020.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yin F, He H, Zhang B, Zheng J, Wang M,
Zhang M and Cui H: Effect of deubiquitinase ovarian tumor
domain-containing protein 5 (OTUD5) on radiosensitivity of cervical
cancer by regulating the ubiquitination of akt and its mechanism.
Med Sci Monit. 25:3469–3475. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Harrigan JA, Jacq X, Martin NM and Jackson
SP: Deubiquitylating enzymes and drug discovery: Emerging
opportunities. Nat Rev Drug Discov. 17:57–78. 2018. View Article : Google Scholar
|
|
119
|
Wang D, Min W, Jiang B, Sun H, Sun C and
Yang P: From concept to application: Exploring the evolution and
potential of DUBTAC technology. Acta Pharm Sin B. 16:770–787. 2026.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Chen Y, Xue H and Jin J: Applications of
protein ubiquitylation and deubiquitylation in drug discovery. J
Biol Chem. 300:1072642024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Noblejas-Lopez MDM, Tebar-Garcia D,
Lopez-Rosa R, Alcaraz-Sanabria A, Cristobal-Cueto P, Pinedo-Serrano
A, Rivas-Garcia L and Galan-Moya EM: TACkling cancer by targeting
selective protein degradation. Pharmaceutics. 15:24422023.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Henning NJ, Boike L, Spradlin JN, Ward CC,
Liu G, Zhang E, Belcher BP, Brittain SM, Hesse MJ, Dovala D, et al:
Deubiquitinase-targeting chimeras for targeted protein
stabilization. Nat Chem Biol. 18:412–421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Liu J, Yu X, Chen H, Kaniskan HU, Xie L,
Chen X, Jin J and Wei W: TF-DUBTACs stabilize tumor suppressor
transcription factors. J Am Chem Soc. 144:12934–12941. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Liu J, Hu X, Luo K, Xiong Y, Chen L, Wang
Z, Inuzuka H, Qian C, Yu X, Xie L, et al: USP7-based
deubiquitinase-targeting chimeras stabilize AMPK. J Am Chem Soc.
Apr 10–2024. View Article : Google Scholar : Epub ahead of
print.
|
|
125
|
Li S, Hong X, Wei Z, Xie M, Li W, Liu G,
Guo H, Yang J, Wei W and Zhang S: Ubiquitination of the HPV
oncoprotein E6 is critical for E6/E6AP-mediated p53 Degradation.
Front Microbiol. 10:24832019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Gao M, Cui Z, Song H, Li Y, Li A, Zheng J,
Liu L, Lin X, Fu W and Sun Y: IRF4 enhances radiosensitivity of
cervical cancer by inhibiting the PI3K/Akt/mTOR pathway to regulate
autophagy. Cancer Sci. 117:958–971. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Deng S, Yuan P and Sun J: The role of
NF-κB in carcinogenesis of cervical cancer: Opportunities and
challenges. Mol Biol Rep. 51:5382024. View Article : Google Scholar
|
|
128
|
Zhang X, Wang M, Zhang Y, Yang J and Duan
W: Knockdown of CENPU inhibits cervical cancer cell migration and
stemness through the FOXM1/Wnt/β-catenin pathway. Tissue Cell.
81:1020092023. View Article : Google Scholar
|
|
129
|
Li B and Wang B: USP7 enables immune
escape of glioma cells by regulating PD-L1 expression. Immunol
Invest. 51:1921–1937. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Umemura S, Zhu J, Chahine JJ, Kallakury B,
Chen V, Kim IK, Zhang YW, Goto K, He Y and Giaccone G:
Downregulation of CYLD promotes IFN-γ mediated PD-L1 expression in
thymic epithelial tumors. Lung Cancer. 147:221–228. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Ma Z, Zhou M, Chen H, Shen Q and Zhou J:
Deubiquitinase-targeting chimeras (DUBTACs) as a potential
paradigm-shifting drug discovery approach. J Med Chem.
68:6897–6915. 2025. View Article : Google Scholar : PubMed/NCBI
|














