Introduction
Gastric cancer (GC) ranks as the fifth most
frequently diagnosed malignancy and the third leading cause of
cancer-related mortality worldwide, with a particularly high
incidence in East Asia, where dietary exposures, chronic
inflammation and persistent Helicobacter pylori infection
are prevalent (1,2). Despite advances in surgery,
perioperative chemotherapy and targeted therapy, numerous patients
still present with advanced disease, and the 5-year survival rate
remains unsatisfactory (3,4).
Immune checkpoint blockade (ICB) targeting programmed death-1
(PD-1) or programmed death-ligand 1 (PD-L1), human epidermal factor
receptor 2 (HER2)-directed regimens and fibroblast growth factor
receptor 2b-targeted agents have improved outcomes in
biomarker-selected subgroups. However, durable benefit remains
limited to a minority of patients, indicating that conventional
clinicopathologic and genomic classifications do not fully capture
key barriers to effective antitumor immunity (5-7).
A major barrier is tumor-intrinsic metabolic
reprogramming and its impact on the tumor microenvironment (TME).
GC cells preferentially engage aerobic glycolysis, also known as
the Warburg effect, converting glucose to lactate even under
normoxic conditions. This process supports rapid adenosine
triphosphate (ATP) generation and biosynthesis while simultaneously
depleting nutrients for effector lymphocytes and acidifying the TME
(8-10). Lactate accumulation and low pH
impair dendritic cell (DC) differentiation and cross-priming and
disrupt cytotoxic T lymphocyte (CTL), natural killer (NK)-cell and
regulatory T-cell (Treg) homeostasis, collectively promoting immune
escape (11-13). This glycolytic phenotype is
reinforced by hypoxia-inducible factor-1α (HIF-1α),
phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mechanistic
target of rapamycin (mTOR) signaling, chronic Helicobacter
pylori-driven inflammation and noncoding RNA (ncRNA) networks
that upregulate glycolytic enzymes such as hexokinase 2 (HK2),
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3),
pyruvate kinase M2 (PKM2) and lactate dehydrogenase A (LDHA) and
couple inflammatory cues to PD-L1 expression (14-17).
These metabolic changes have clear clinical
associations. High LDHA and monocarboxylate transporter (MCT)4
expression are associated with deeper invasion, nodal metastasis,
immune-excluded histology and increased uptake on fluorine-18
fluorodeoxyglucose (18F-FDG) positron emission
tomography/computed tomography (PET/CT) (18,19). Diffuse-type and Epstein-Barr virus
(EBV)-positive tumors with dense stroma often display enriched
glycolytic and lactate-transport signatures, together with poor
CD8+ T-cell and DC infiltration. Glycolysis- or
lactylation-based transcriptional signatures can further stratify
patients into metabolic-immune subgroups with distinct prognosis
and differential likelihood of responding to ICB (9,20,21). The surrounding stroma further
amplifies this axis: Cancer-associated fibroblasts (CAFs) and
mesenchymal stem/stromal cells (MSCs) adopt aerobic glycolysis,
export lactate and secrete cytokines such as interleukin (IL)-6 and
IL-8. In parallel, ncRNAs, including long noncoding RNAs (lncRNAs),
microRNAs (miRNAs or miRs) and circular RNAs (circRNAs), fine-tune
glycolytic enzymes and immune checkpoints, linking glucose
metabolism to stemness, drug resistance and immune escape (22-25).
From a translational perspective, the
glycolysis-lactate axis presents both vulnerabilities and
challenges. Lactate is now recognized as a bioactive metabolite
that drives histone lactylation, M2-like macrophage polarization,
tolerogenic DC differentiation and PD-L1 upregulation through
LDHA-dependent flux and MCT-mediated export. High stromal MCT4 or
tumor LDHA/glucose transporter (GLUT)3 expression was shown to be
associated with adverse outcomes and poor response to PD-1-based
immunotherapy (11,12,26,27). Metabolic imaging with
18F-FDG PET/CT and radiomics-derived metrics, such as
metabolic tumor volume and total lesion glycolysis, can
noninvasively capture glycolytic burden, reflect immune-cold
phenotypes and support response prediction in immunotherapy trials
(1,7,28,29).
These findings support a glycolysis-centered but
metabolically flexible framework for understanding immune
suppression and therapeutic resistance in GC in which glycolytic
reprogramming in tumor and stromal compartments establishes a
lactate-rich, low-pH niche that impairs DC and T-cell activation.
At the same time, ncRNA- and inflammation-dependent programs help
stabilize this state and are linked to therapy resistance (17,30,31). The present review focuses on: i)
The molecular drivers and landscape of glycolytic reprogramming in
GC; ii) how lactate-centered crosstalk reshapes stromal and immune
compartments into an immune-refractory TME; and iii) emerging
therapeutic strategies that combine metabolic intervention, ICB and
metabolic imaging for precision patient stratification.
Glycolytic reprogramming in GC: Molecular
drivers and metabolic landscape
GC maintains a highly glycolytic state through
oncogenic signaling, hypoxia- and inflammation-driven
transcriptional programs, ncRNA-mediated regulation and persistent
metabolic crosstalk with stromal cells (12,17,32). These convergent inputs increase
glucose uptake, accelerate glycolytic flux and promote lactate
export, creating a nutrient-competitive, acidified TME that
disfavors antitumor immune responses (Fig. 1) (24,33,34).
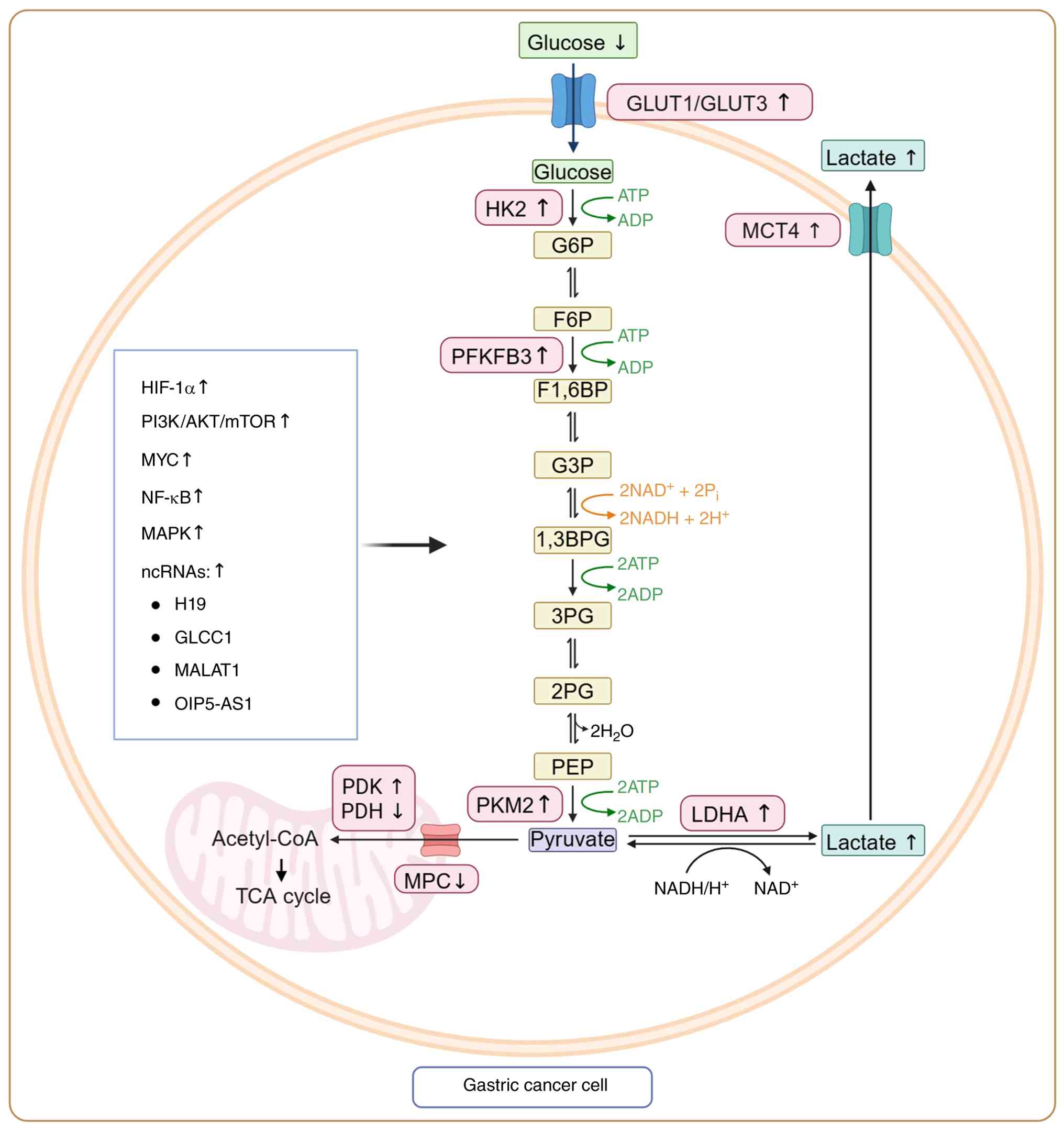 | Figure 1Glycolytic reprogramming in GC.
Tumor-intrinsic signaling pathways, including hypoxia-induced
HIF-1α, PI3K-AKT-mTOR, MAPK, NF-κB and MYC, and multiple ncRNAs
converge to activate glycolysis in GC. Upregulation of GLUT1/3
enhances glucose uptake, while HK2, PFKFB3, PKM2 and LDHA
accelerate glycolytic flux and lactate production. Mitochondrial
pyruvate oxidation is further inhibited by PDK-mediated PDH
suppression. Excess lactate is exported via MCT4, acidifying the
tumor microenvironment and initiating downstream immunosuppressive
programs. Created with BioRender.com. GC, gastic cancer; HIF-1α,
hypoxia-inducible factor-1α; PI3K, phosphoinositide 3-kinase; mTOR,
mechanistic target of rapamycin; MAPK, mitogen-activated protein
kinase; NF-κB, nuclear factor κB; ncRNAs, noncoding RNAs; GLUT1/3,
glucose transporter 1/3; HK2, hexokinase 2; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PKM2,
pyruvate kinase M2; LDHA, lactate dehydrogenase A; PDK, pyruvate
dehydrogenase kinase; PDH, pyruvate dehydrogenase kinase complex;
MCT4, monocarboxylate transporter 4; GLCC1, gastric
cancer-associated lncRNA 1; MALAT1, metastasis associated lung
adenocarcinoma transcript 1; OIP5-AS1, Opa interacting protein
5-antisense RNA1; TCA, tricarboxylic acid cycle; MPC,
mitochrondrial pyruvate carrier; ATP, adenosine triphosphate;
NAD+, oxidized nicotinamide adenine dinucleotide. |
Oncogenic, hypoxic and inflammatory
signals converging on glycolysis
Hypoxia stabilizes HIF-1α, which induces glucose
transporters and key glycolytic enzymes, including HK2, PFKFB3,
PKM2, LDHA and pyruvate dehydrogenase kinase (PDK)1, thereby
diverting pyruvate away from oxidative phosphorylation (OXPHOS)
toward lactate production (14,35-37). Coexpression of HIF-1α, GLUT1 and
lactate dehydrogenase isoforms was shown to be associated with
advanced stage, nodal metastasis and poor survival, while
hypoxia-driven mitochondrial dysfunction and reactive oxygen
species (ROS) were demonstrated to further reinforce glycolytic
dependence (38-40). Oncogenic receptor tyrosine
kinase-rat sarcoma (RAS)-mitogen-activated protein kinase (MAPK)
and PI3K/AKT/mTOR signaling converge with this hypoxic program by
enhancing GLUT trafficking, HK2-mitochondrial association and
translation of glycolytic enzymes (17,41,42). Additional regulators, including
formyl peptide receptor 3, monoamine oxidase A, mitochondrial
creatine kinase and mitochondrial topoisomerase I, promote
HK2/LDHA-dependent aerobic glycolysis, epithelialmesenchymal
transition (EMT) and peritoneal dissemination. Other
glycolysis-associated proteins, such as enolase 1 (ENO1),
hexokinase domain containing 1, PDK4 and PKM2, are also frequently
upregulated and may represent potential therapeutic targets
(43-47).
Systemic, neuroendocrine and circadian cues further
shape this metabolic circuitry. Norepinephrine enhances aerobic
glycolysis and lactate release and may predict immunotherapy
responsiveness in GC, supporting an association between sympathetic
stress signaling and glycolytic remodeling (48). Mechanistically, β-adrenergic
stimulation may activate cyclic adenosine monophosphate
(cAMP)/protein kinase A/cAMP response element-binding protein- and
HIF-1α-associated transcriptional programs, thereby increasing the
expression of glycolytic enzymes and lactate-handling molecules,
including GLUT1, HK2, LDHA and MCTs, and linking sympathetic stress
to lactate-rich immunosuppressive niches (34,48). Circadian disruption also
intersects with HER2-targeted therapy resistance. In HER2-positive
GC, trastuzumab-resistant cells exhibited circadian oscillation of
glycolysis regulated by the BMAL1-CLOCK-period circadian regulator
1 (PER1)-HK2 axis, and disruption of PER1 was shown to enhance
HK2-dependent glycolytic activity and trastuzumab resistance
(49). In this context, the
'metabolic bypass̓ refers to HK2-driven maintenance of ATP
production, biosynthetic precursor supply and redox buffering when
HER2-dependent PI3K/AKT and RAS/MAPK growth signaling is
pharmacologically inhibited. This interpretation is supported by
evidence that MACC1 promotes the Warburg effect through PI3K/AKT
pathway activation and thereby contributes to trastuzumab
resistance in HER2-positive GC (50). Lactate accumulation and
extracellular acidification may further support EMT-like
plasticity, stromal remodeling and immune escape; however, direct
evidence that lactate accumulation itself induces HER2 protein
degradation in GC remains insufficient. Therefore, lactate is
discussed here as a downstream mediator of glycolytic adaptation
and an immunosuppressive acidic niche rather than as a proven
driver of HER2 degradation. Thus, norepinephrine-related adrenergic
signaling, circadian PER1-HK2 dysregulation and HK2-dependent
glycolytic rewiring may jointly support metabolic plasticity during
HER2-targeted therapy resistance, although the precise contribution
of lactate-HER2 protein regulation requires further validation.
Helicobacter pylori remodels mitochondrial
homeostasis via ATP-dependent Lon protease, while
cytotoxin-associated gene A and Toll-like receptor 2/superoxide
dismutase 2 signaling promote mitochondrial damage, ROS
accumulation and HIF-1α activation. These changes shift tumor cells
toward a highly glycolytic and chemoresistant state (16,51-53). EBV-encoded miR-BART6-5p further
modulates transforming growth factor-β (TGF-β)/SMAD signaling and
may contribute to a virus-associated immunometabolic subtype
(16,51-53). Proinflammatory cytokines,
including IL-6 and IL-8, activate signal transducer and activator
of transcription (STAT3)-mTOR-MYC cascades, upregulating HK2, LDHA
and PD-L1 and thereby linking inflammation, glycolysis and immune
checkpoint expression (15,54,55). At the immune interface,
lactate-rich niches promote M2-like macrophage polarization through
MCT-HIF-1α signaling, while M2-derived exosomal metastasis
associated lung adenocarcinoma transcript 1 (MALAT1) and
claudin-9-mediated PD-L1 lactylation further enhance tumor
invasion, CD8+ T-cell suppression and resistance to
PD-1/PD-L1 blockade (11,56-58). Representative ncRNAs, shown in
Fig. 1, including H19, gastric
cancer-associated lncRNA 1 (GLCC1), MALAT1 and Opa interacting
protein 5-antisense RNA1 (OIP5-AS1), illustrate how ncRNA-mediated
regulation converges on GLUT1/3, HK2, PFKFB3, PKM2, LDHA and
MCT4-dependent lactate export, thereby linking glycolytic enzyme
expression to lactate production and immune escape (Fig. 1).
Mechanistically distinct ncRNA networks
in glycolytic regulation
ncRNAs, including lncRNAs, miRNAs and circRNAs,
constitute a multilayered regulatory system that fine-tunes
glycolytic reprogramming in GC. Rather than acting through a single
mechanism, these ncRNA classes regulate glucose metabolism at
different molecular levels, including chromatin remodeling,
RNA-protein interactions, mRNA stability, translational repression
and competitive endogenous RNA (ceRNA) networks (17,30,32). This mechanistic diversity is
particularly relevant in GC, where tumor cells, stromal cells and
immune cells exchange metabolic signals through soluble mediators
and exosome-associated ncRNAs, linking glycolytic flux to immune
evasion and therapeutic resistance.
lncRNAs mainly regulate glycolysis by modulating
transcriptional, epigenetic and post-transcriptional programs. For
example, deleted in lymphocytic leukemia 1 (DLEU1) was shown to
promote GC-cell proliferation and glycolysis by recruiting the
histone methyltransferase SET and MYND domain-containing protein 2
(SMYD2) to induce H3K4 trimethylation and upregulate apolipoprotein
C1, indicating that lncRNAs can remodel chromatin to sustain
metabolic gene expression (30).
GLCC1 was demonstrated to enhance tumorigenesis by strengthening
the interaction between c-Myc and insulin-like growth factor 2
mRNA-binding protein 1, thereby stabilizing glycolysis-related
oncogenic programs (59,60). The lncRNA VAL has been shown to
promote PKM2 enzymatic activity and facilitate malignant
progression, whereas m6A-modified OIP5-AS1 was revealed to enhance
glycolysis, tumorigenesis and metastasis by inhibiting
Trim21-mediated ubiquitination and degradation of heterogeneous
nuclear ribonucleoprotein A1 (32,61,62). These findings suggest that lncRNAs
often act as scaffolds or guides for chromatin modifiers and
RNA-binding proteins, enabling sustained activation of HK2-, LDHA-
and PKM2-centered glycolytic circuits.
miRNAs regulate glycolysis mainly through
sequence-specific post-transcriptional repression of metabolic
enzymes, transporters or upstream transcription factors. miR-21-5p
was shown to promote glycolytic progression by targeting pyruvate
dehydrogenase E1 subunit α1, thereby limiting pyruvate entry into
mitochondrial oxidative metabolism and favoring lactate production
(63). miR-186 was demonstrated
to inhibit aerobic glycolysis through HIF-1α regulation, whereas
miR-379 was revealed to be associated with reduced glycolytic
capacity through enhanced regulation of PKM2 (64,65). miR-148b-5p has also been linked to
metabolic remodeling of the immune microenvironment and GC
progression (66). Compared with
lncRNAs, miRNAs tend to provide more direct and rapidly adjustable
control over glycolytic enzymes and signaling nodes, thereby
shaping tumor metabolic tone, treatment sensitivity and local
immune remodeling.
circRNAs predominantly function as stable ceRNA
molecules that sequester miRNAs and derepress glycolysis-promoting
targets. circ-NRIP1, circ-0032821 and circ-ATP2B1 have been
implicated in hypoxia-induced glucose metabolism, chemoresistance,
proliferation, invasion and aerobic glycolysis by buffering
specific miRNA axes (20,67,68). Cancer-derived exosomal
circ-0038138 was shown to enhance glycolysis, growth and metastasis
through the miR-198/enhancer of zeste homolog 2 (EZH2) axis,
illustrating how circRNAs can transmit glycolytic traits between
tumor cells and the surrounding microenvironment (22). In addition, exosomal circ-ATP8A1
was demonstrated to induce M2 macrophage polarization through the
miR-1-3p/STAT6 axis, linking circRNA-mediated metabolic regulation
to myeloid immune suppression (69). Thus, circRNAs are not only
intracellular ceRNA regulators but also extracellular messengers
that disseminate glycolysis-supporting and immunosuppressive
programs across the GC TME.
The cellular and spatial context of ncRNA regulation
is also important. Tumor-cell-intrinsic ncRNAs, such as H19, DLEU1,
GLCC1, VAL and OIP5-AS1, primarily reinforce glycolytic enzyme
expression, lactate production, stemness and drug resistance. By
contrast, stromal- and immune-cell-associated ncRNAs, including
macrophage-derived exosomal MALAT1 and exosomal circ-ATP8A1,
reshape macrophage polarization, DC activation and antigen
presentation (8,58,69). This compartment-specific
regulation helps explain why glycolysis-related ncRNA signatures
may reflect not only malignant-cell metabolism but also the
metabolic state of CAFs, tumor-associated macrophages (TAMs) and
other immune populations. Overall, ncRNA networks provide a
molecular bridge between tumor glycolysis, lactate accumulation,
PD-L1 regulation, histone lactylation and immune escape.
Distinguishing lncRNA-, miRNA- and circRNA-mediated mechanisms may
therefore improve the interpretation of glycolysis-related
biomarkers and support more precise patient stratification in GC.
This cell-type-resolved interpretation is important for bulk GC
tissue analyses because glycolysis-related ncRNA signatures may
represent mixed signals from malignant epithelial cells, CAFs,
TAMs, DCs and exosome-enriched stromal or immune compartments
rather than tumor-cell-intrinsic glycolysis alone (8,58,69).
Metabolic heterogeneity and glycolytic
immunophenotypes
Despite widespread glycolytic upregulation, GC
exhibits marked intra- and intertumoral metabolic heterogeneity.
Nuclear magnetic resonance-based metabolomics has identified tumor
clusters with distinct patterns of glycolytic intermediates, amino
acids and lipid species (70-72). In orthotopic xenograft models,
glycolysis-targeted therapy produces heterogeneous reductions in
metabolic tumor volume on 18F-FDG PET/CT, underscoring
variable glycolytic dependence among lesions and its association
with Lauren classification and depth of invasion (39,73-75).
Transcriptomic and multi-omics signatures, including
nine-gene glycolysis scores, stratify GC into glycolysis-high and
glycolysis-low subgroups with distinct prognostic and immune
features (18,21,76). Glycolysis-high tumors are enriched
for EMT, angiogenesis and immunosuppressive pathways, exhibit
elevated LDHA, HK2, ENO1, PFKFB3 and MCT4, and generally contain
fewer cytotoxic T cells and DCs but more Tregs and myeloid-derived
suppressor cells (MDSCs) (26,73,77). By contrast, glycolysis-low tumors
tend to display more inflamed gene-expression patterns and may
respond better to ICB or cytotoxic chemotherapy. Imaging-derived
metrics further refine this framework: 18F-FDG PET/CT
parameters, such as metabolic tumor volume and total lesion
glycolysis, are prognostic markers and associated with HER2 status,
c-MET expression and treatment response. Radiomics models
integrating textural, metabolic and anatomical features may also
predict microvascular invasion, response to neoadjuvant
immunochemotherapy and long-term outcomes (28,29,73,78). High stromal or tumor-cell MCT4,
together with coexpression of MCT1/MCT4 and mitochondrial markers
such as mitochondrially encoded cytochrome c oxidase I,
identifies subsets with poor prognosis and strong lactate-shuttling
capacity. These findings support a continuum of glycolytic
immunophenotypes ranging from hyper-glycolytic, immune-cold tumors
to moderately glycolytic, immune-inflamed tumors (21,27,76,79).
Substrate-level heterogeneity adds another layer of
complexity. Although glucose is a dominant fuel source, some GC
cells can oxidize lactate, glutamine or fatty acids (FAs), enabling
metabolic flexibility when glycolysis is pharmacologically
inhibited (80-82). Metabolomic analyses in cell lines
and patient-derived xenografts show that tumors with robust
anaplerotic glutamine use or FA oxidation can partially escape
glycolysis blockade. Together with lactate-driven immune
suppression, this metabolic flexibility may contribute to
incomplete responses to single-agent glycolytic inhibitors
(56,70,74,83,84).
Notably, glycolysis-centered immune suppression
should be interpreted as part of a broader metabolic network rather
than as an isolated pathway. When glycolytic carbon is
preferentially diverted toward lactate production, glutamine
anaplerosis can replenish tricarboxylic acid-cycle intermediates
and support nucleotide synthesis, redox buffering and mitochondrial
function, allowing glycolysis-high tumor cells to maintain
biosynthetic capacity despite incomplete glucose oxidation
(70,74,80-84). FA uptake and FA oxidation may
provide an additional survival route under glucose-limited or
glycolysis-inhibited conditions, whereas mitochondrial rewiring
enables tumor cells to switch between glycolysis, oxidative
phosphorylation and substrate oxidation when one pathway is
therapeutically restricted (6,80-85). In parallel, stromal metabolism
reinforces this flexibility: CAFs and MSCs can export lactate,
recycle lactate-derived carbon or provide alanine, glutamine and
cytokine signals that sustain tumor anabolism and immune
suppression (24,86,87,88,89). Vascular remodeling further closes
this loop by maintaining hypoxia, HIF-1α activation, vascular
endothelial growth factor A (VEGFA) production and lactate
accumulation, thereby linking metabolic plasticity to immune
exclusion and reduced T-cell trafficking (14,37-40). Thus, glycolysis-centered
immunosuppression in GC is best viewed as a dominant but
context-dependent node within an interconnected network involving
glutamine metabolism, lipid oxidation, mitochondrial adaptation,
stromal nutrient exchange and hypoxia-driven vascular remodeling.
This interpretation avoids treating glycolysis as the sole
determinant of immune suppression and instead places glycolytic
signaling within a plastic metabolic ecosystem in which different
GC subtypes may rely on distinct combinations of glucose,
glutamine, fatty-acid and mitochondrial pathways.
Crosstalk with stromal and immune
compartments
GC frequently develops within a desmoplastic and
immunosuppressive stroma in which fibroblasts, MSCs and myeloid
cells are metabolically co-opted. GC-associated MSCs secrete IL-8
and related cytokines that activate STAT3/mTOR-c-MYC signaling in
tumor cells, thereby upregulating HK2, PD-L1 and other glycolytic
drivers; MSC-derived IL-8 has also been shown to promote EMT and
stemness (24,55). In turn, GC cells condition MSCs
and CAFs to adopt a glycolytic, lactate-exporting phenotype through
glucose-6-phosphate dehydrogenase (G6PD)-NF-κB-hepatocyte growth
factor signaling. This establishes a tumor-stroma metabolic circuit
in which stromal cells support tumor metabolism, while C-X-C motif
chemokine receptor (CXCR)2/HK2/PD-L1 signaling in MSCs further
links stromal glycolysis to checkpoint expression (86,87).
Myeloid cells integrate metabolic and inflammatory
cues from this remodeled stroma. Lactic acid promotes macrophage
polarization toward an M2-like phenotype through MCT-HIF-1α
signaling and histone lactylation. M2 TAMs secrete exosomal MALAT1
and circ-ATP8A1, which enhance GC-cell glycolysis, invasion and
immunoregulatory programming through the miR-1-3p/STAT6 axis and
related circuits (58,69,90). DCs exposed to high lactate show
impaired activation and antigen presentation, with tumor-derived
lactic acid directly modulating DC phenotype and reinforcing a
stromal-immune niche that favors tumor survival (11,91).
Endothelial cells and the vasculature also
participate in this metabolic network. Radiomics signatures
capturing angiogenic and metabolic features on 18F-FDG
PET/CT can predict microvascular invasion and survival, and
glycolysis-high tumors often exhibit abnormal vasculature and
hypoxic, immune-excluded regions that restrict immune-cell
trafficking while sustaining HIF-1α-driven glycolysis and lactate
production (14,37,38,40). Overall, tumor, stromal and immune
cells form an interconnected metabolic network in which glycolysis
is both a driver and a consequence of TME remodeling (12,24,81). Key glycolytic drivers, primary
metabolic effects, immunologic consequences and supporting
references in GC are summarized in Table I.
 | Table IKey glycolytic drivers and their
immunologic consequences in gastric cancer. |
Table I
Key glycolytic drivers and their
immunologic consequences in gastric cancer.
| Molecular
driver | Primary metabolic
effect | Immune
consequence | (Refs.) |
|---|
| HK2 | Enhances glucose
phosphorylation and glycolytic flux | Promotes
immunosuppressive metabolite accumulation | (49,81,89) |
| PFKFB3 | Increases
fructose-2,6-bisphosphate, accelerates glycolysis | May contribute to
lactate-rich metabolic conditions that limit immune-cell
function | (95) |
| PKM2 | Regulates pyruvate
conversion, supports anabolic metabolism | Supports glycolytic
remodeling associated with immune evasion | (32,42,96,127) |
| LDHA | Converts pyruvate →
lactate | Generates acidic
TME; inhibits DC/T-cell activation | (46,77,92,94) |
| MCT4 | Exports
lactate | Establishes
CD8+-poor niches in glycolysis-high tumors | (79,104) |
| HIF-1α | Transcriptionally
activates glycolytic enzymes | Upregulates PD-L1;
suppresses CTL infiltration | (35,37,38,40,99) |
| mTOR | Integrates nutrient
signaling to glycolysis | Affects T-cell
exhaustion and DC differentiation | (35,42) |
Lactate as a central mediator of
immunosuppression
GC is shaped by converging oncogenic, hypoxic and
inflammatory signals, reinforced by ncRNAs and stromal-immune
crosstalk, which together maintain a high-glycolysis,
lactate-producing state and a spectrum of glycolytic
immunophenotypes ranging from hyper-glycolytic, immune-excluded
tumors to more immune-inflamed lesions (14,21,32). Within this context, lactate has
emerged as a multifunctional metabolite. Through MCT1/4-mediated
transport, hydroxycarboxylic acid receptor 1 (HCAR1, also known as
GPR81) signaling and induction of histone lactylation, lactate
creates spatially organized niches in desmoplastic and poorly
perfused regions that are acidic, MCT4-high and depleted of
effector lymphocytes (Fig. 2)
(11,13,77). The major immune-cell consequences
of lactate accumulation in the GC TME, including impaired DC
activation and migration, reduced CD8+ T-cell
interferon-γ (IFN-γ) production and Ca2+ flux, weakened
B-cell mTOR complex 1 (mTORC1)/OXPHOS activity, reduced NK-cell
cytotoxicity and granzyme B/perforin expression, and
TAM/MDSC-associated immunosuppressive remodeling are summarized in
Fig. 2.
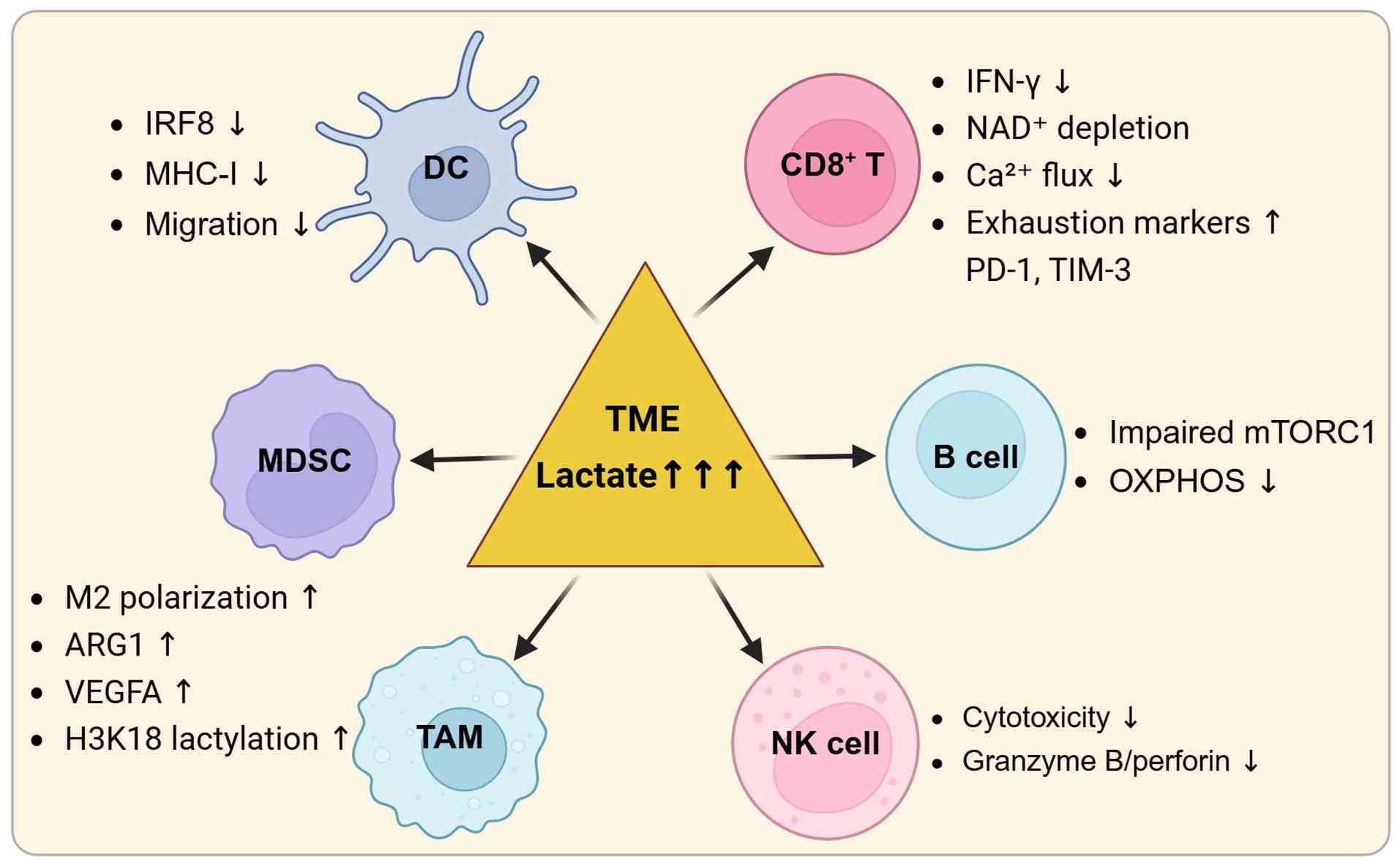 | Figure 2Immune-cell consequences of lactate
accumulation in the GC tumor microenvironment. Lactate accumulation
in the GC TME impairs DC-associated IRF8 expression, MHC-I
expression and migration, suppresses CD8+ T-cell IFN-γ
production and Ca2+ flux while increasing exhaustion
markers, weakens B-cell mTORC1 and OXPHOS activity, reduces NK-cell
cytotoxicity and granzyme B/perforin expression, and promotes
TAM/MDSC-associated immunosuppressive remodeling, including M2
polarization, ARG1 expression, VEGFA expression and H3K18
lactylation. Created with BioRender.com. GC, gastric cancer; TME, tumor
microenvironment; DC, dendritic cell; IRF8, interferon regulatory
factor 8; MHC-I, major histocompatibility complex class I; IFN-γ,
interferon-γ; mTORC1, mTOR complex 1; OXPHOS, oxidative
phosphorylation; NK, natural killer; TAM, tumor-associated
macrophage; MDSC, myeloid-derived suppressor cell; ARG1, arginase
1; VEGFA, vascular endothelial growth factor A; NAD+,
oxidized nicotinamide adenine dinucleotide; PD-1, programmed
death-1; TIM-3, T-cell immunoglobulin and mucin domain-3. |
Lactate production and export as a
metabolic circuit
GC cells display high lactate-to-pyruvate ratios
driven by LDHA activation and MCT1/4 overexpression, which are
induced by HIF-1α, MYC, NF-κB and inflammatory cytokines (10,16,18). Lactate export regenerates oxidized
nicotinamide adenine dinucleotide (NAD+), sustains
glycolytic flux and acidifies the TME, generating gradients in
which CD8+ T cells are scarce, whereas CD163+
TAMs and MDSCs accumulate (1,2,4).
To preserve biosynthetic capacity when carbons are diverted from
the tricarboxylic acid (TCA) cycle, GC cells increase glutamine
anaplerosis and FA uptake. At the same time, lactate acts as a
paracrine signal that conditions stromal and immune cells toward
immunoregulatory phenotypes (10,11,92).
Stromal cells contribute to a lactate shuttle. CAFs
and MSCs take up extracellular lactate via MCT1, oxidize it and
release alanine and glutamine that support tumor anabolism and
redox control (24,88,89). Disrupting this circuit by
inhibiting LDHA or MCT4 increases intratumoral pH, reduces M2/MDSC
abundance and restores CTL infiltration in GC models, highlighting
lactate transport as a shared metabolic and immunologic
vulnerability (80,93-95).
Direct effects on DCs and T cells
High lactate levels impair monocyte-to-DC
differentiation, downregulate CD80/CD86 and suppress IL-12 and type
I interferon production through inhibition of NF-κB and interferon
regulatory factor (IRF)1. Lactate also reduces the mitochondrial
spare respiratory capacity required for antigen processing and
cross-presentation (33,96,97). In addition, lactate skews DC
programs away from IRF8/basic leucine zipper ATF-like transcription
factor 3-dependent cross-priming toward IRF4/Kruppel-like factor
4-associated regulatory states with elevated PD-L1, IL-10 and
Treg-recruiting chemokines. Consistently,
MCT4+/lactate-rich regions in GC are almost devoid of
CD103+ conventional type 1 DCs (cDC1) (1,77,98). Activated CD8+ T cells
rely heavily on glycolysis and are therefore highly sensitive to
lactate-rich and acidic conditions. In such environments, cytosolic
acidification and NAD+ depletion impair glycolytic
enzymes, T-cell receptor-triggered Ca2+ flux,
proliferation, motility and IFN-γ/granzyme B release (12,13). LDHA silencing or lactic-acid
neutralization in GC models was shown to restore T-cell motility
and effector function. Lactate may also limit mitochondrial
biogenesis and FA oxidation, biasing differentiation toward
short-lived effector cells rather than long-lived memory subsets
(8,93,94). These findings support lactate as a
metabolic checkpoint for antitumor T-cell immunity and position
LDHA/MCT4 inhibition as an immune-adjuvant strategy rather than a
purely cytotoxic approach (Fig.
2).
Epigenetic and signaling reprogramming of
myeloid cells
Lactate directly couples metabolism to gene
expression through histone lactylation (13,90). In macrophages, lactate-driven
H3K18 lactylation activates arginase 1 (ARG1), VEGFA and mannose
receptor C-type 1 (MRC1). Together with HCAR1-mediated HIF-1α
stabilization and NF-κB dampening, these changes reinforce an
M2-like, pro-angiogenic and IL-10/TGF-β-secreting phenotype
(10,57,58). Lactate-conditioned TAMs also
upregulate chemokines that attract Tregs and C-C motif chemokine
receptor 2-positive monocytes while limiting CXCR3+
effector T-cell infiltration. Lactate-driven histone lactylation
may further cooperate with SMYD2/SET domain containing 1A, histone
lysine methyltransferase (SETD1A)/p300-dependent methylation and
acetylation programs to stabilize these transcriptional circuits
(17,30,99).
MDSCs similarly exploit lactate-rich conditions.
Lactate has been reported to enhance ARG1 and PD-L1 expression
through extracellular signal-regulated kinase (ERK) and STAT3
signaling, thereby promoting T-cell anergy and Treg expansion
(97,100). LDHA+/MCT4+
MDSC clusters are associated with immune-excluded histology and
shortened survival, and MCT4 inhibition was shown to reduce MDSC
infiltration and restore DC maturation in GC models (3,54,94). These lactate-driven changes in
TAMs and MDSCs constitute the myeloid-cell component of the
immune-cell remodeling summarized in Fig. 2.
Impact on B cells and NK cells
Lactate also dampens humoral and innate immunity.
In B cells, low pH and altered NAD+/NADH ratios impair
mTORC1 activity and OXPHOS, reducing major histocompatibility
complex (MHC) class II expression and class-switch recombination.
These changes may weaken antibody-dependent effector mechanisms in
the GC TME (8,100). In NK cells, lactate suppresses
ERK/mTORC1 signaling, cytotoxic granule exocytosis, IFN-γ
production and immunological synapse formation. Consistently,
glycolysis-high, MCT4-high GC specimens often show reduced
CD56+ NK-cell infiltration and attenuated B-cell
activation signatures (4,19,33,78). Together, these effects on B-cell
metabolic activity and NK-cell cytotoxicity are included in the
immune-cell consequences of lactate accumulation summarized in
Fig. 2.
Lactate and immune checkpoint
activation
Lactate provides a mechanistic bridge between
metabolic stress and checkpoint dominance. PD-L1 expression in GC
was shown to be associated with LDHA/MCT4 and could be induced by
histone lactylation at the CD274 promoter, with further
amplification through hypoxia-driven HIF-1α binding to PD-L1
regulatory elements (18,35,92). Lactate was also demonstrated to
stabilize MYC and increase acetyl-coenzyme A availability, favoring
acetylation events that may upregulate additional inhibitory
receptors, including T-cell immunoglobulin and mucin domain-3 and
lymphocyte activation gene-3 (LAG-3) (3,26).
CAF-derived wingless-type MMTV integration site family member 5A
(WNT5A) and MSC-derived IL-8 were reported to converge through
STAT3/mTOR-MYC signaling to further augment PD-L1, integrating
lactate, hypoxia and cytokine signaling into a broader
immunosuppressive network (54,55,89).
Functionally, this creates a metabolic-checkpoint
interface in which effector T cells encounter both high PD-L1
density and a bioenergetically hostile microenvironment. Even when
PD-1/PD-L1 interactions are blocked, residual lactate signaling may
sustain other inhibitory receptors and maintain epigenetically
imprinted exhaustion programs (3,101). Glycolysis-high GC subtypes often
show coelevation of PD-L1, LAG-3, MCT4 and myeloid-suppressive
signatures, providing a rationale for combining modulation of
lactate metabolism, HCAR1 blockade or targeting of histone
lactylation with checkpoint inhibition (21,27). This checkpoint-associated
remodeling overlaps with the lactate-driven suppression of T cells
and myeloid-cell polarization illustrated in Fig. 2.
Tumor-stroma-immune crosstalk in metabolic
immunosuppression
In GC, lactate functions not only as a metabolic
fuel but also as an epigenetic and signaling mediator. It shapes
acidic and hypoxic lactate-rich niches that suppress DCs and T
cells while skewing macrophages and MDSCs toward tolerogenic,
checkpoint-high states (Fig. 2)
(1,11,92). Tumor, stromal and immune cells
form a metabolically interdependent network that maintains
immune-silent metabolic neighborhoods characterized by high MCT4
expression, low pH and enrichment of suppressive myeloid subsets
(19,21,73). Within this framework, the
LDHA/MCT4/HCAR1 axis and stromal-immune crosstalk emerge as core
organizers of glycolysis-driven immune escape and potential targets
for immunometabolic intervention (Fig. 3) (20,24,80,95). The stromal, vascular and immune
components of this network, including CAF-derived MCT4-dependent
lactate export and extracellular matrix (ECM) remodeling,
MSC-derived IL-6/IL-8-STAT3/MYC signaling, exosome-mediated ncRNA
transfer, M2 TAM-associated lactylation and VEGFA induction,
abnormal angiogenesis and CD8+ T-cell exclusion are
integrated in Fig. 3.
Accordingly, the following section focuses on multicellular
feedback loops, stromal-vascular remodeling and spatial immune
exclusion, rather than reiterating the direct DC- and
T-cell-intrinsic effects of lactate aforementioned.
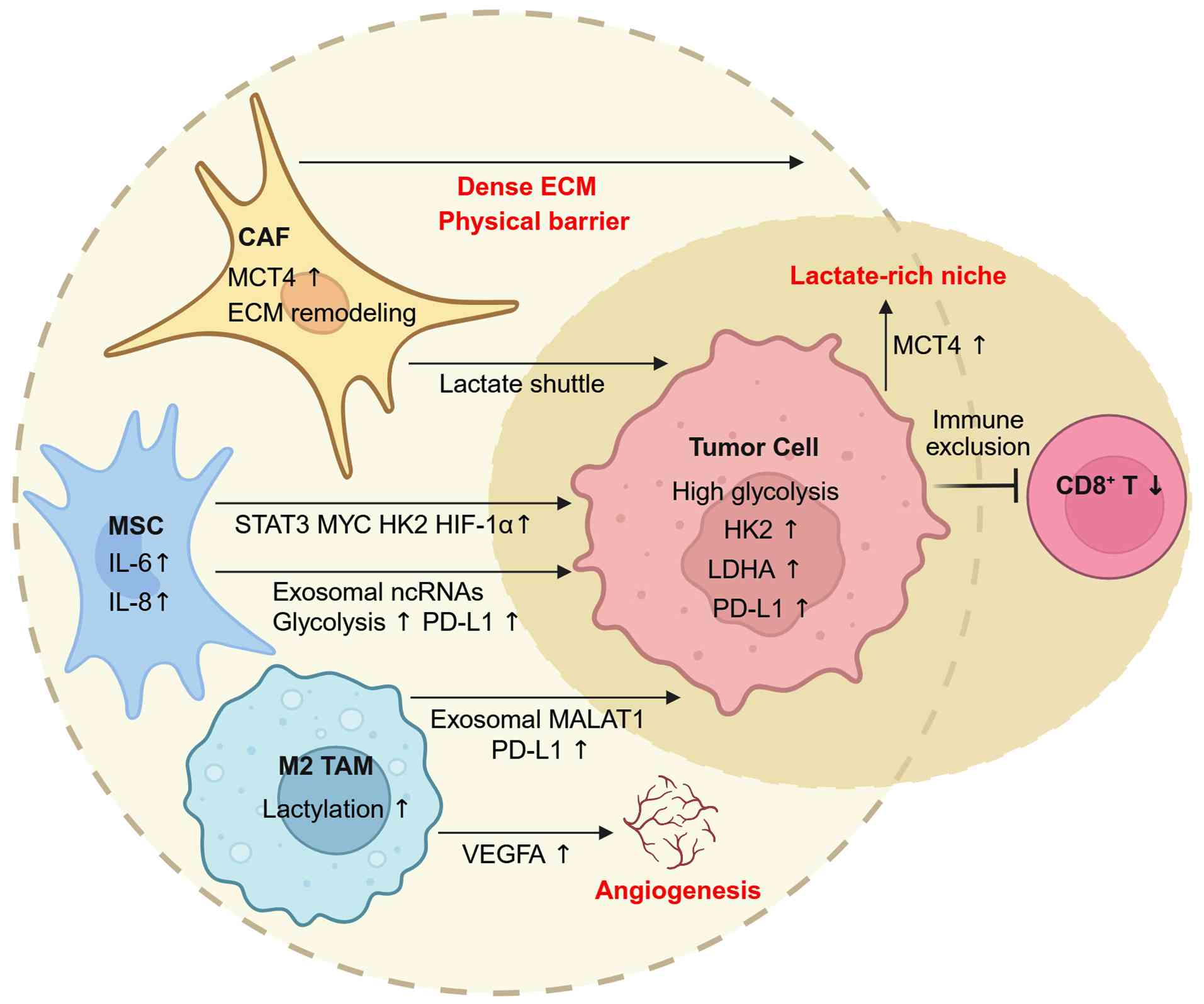 | Figure 3Metabolic and inflammatory crosstalk
between tumor, stromal and immune compartments in gastric cancer.
CAFs sustain a high-lactate niche through MCT4-dependent export and
ECM remodeling, contributing to CD8+ T-cell exclusion.
MSCs amplify tumor glycolysis via IL-6/IL-8-STAT3-MYC signaling and
deliver glycolysis-promoting or PD-L1-inducing ncRNAs through
exosomes. M2-like macrophages reinforce immune suppression by
releasing MALAT1-rich vesicles and promoting VEGFA through histone
lactylation. These reciprocal interactions generate spatially
organized metabolic-immune neighborhoods characteristic of
glycolysis-high gastric cancer. Created with BioRender.com. CAFs, cancer-associated fibroblasts;
MCT4, monocarboxylate transporter 4; ECM, extracellular matrix;
MSCs, MSC, mesenchymal stem/stromal cells; IL, interleukin; STAT3,
signal transducer and activator of transcription 3; PD-L1,
programmed death-ligand 1; ncRNAs, noncoding RNAs; MALAT1,
metastasis associated lung adenocarcinoma transcript 1; VEGFA,
vascular endothelial growth factor A; HK2, hexokinase 2; HIF-1α,
hypoxia-inducible factor-1α; LDHA, lactate dehydrogenase A; TAM,
tumor-associated macrophage. |
MSCs and CAFs as metabolic
amplifiers
MSCs and CAFs are key stromal amplifiers of
metabolic immunosuppression. MSC-derived IL-6 and IL-8 have been
shown to activate STAT3/mTOR-MYC signaling in neighboring tumor
cells, inducing HK2, PD-L1 and lactate production. Tumor-derived
lactate can in turn fuel MSC OXPHOS through MCT1 and be recycled
into alanine and glutamine to support tumor anabolism and redox
control (25,54,89,102). Single-cell analyses have
identified
CXCR2high/HK2high/PD-L1high MSC
subsets with strong immunosuppressive potential in glycolysis-high,
immune-excluded lesions (24,86,87).
CAFs, driven by TGF-β, WNT5A, hypoxia and
mechanical stress, undergo aerobic glycolysis and export lactate
through MCT4, thereby supporting oxidative tumor subclones and
dampening CTL motility and DC activation (103,104). Hypoxia and lactate further
reprogram CAFs into α-smooth muscle actin-positive myofibroblasts
expressing C-X-C motif chemokine ligand 12 (CXCL12), VEGFA and
TGF-β, which recruit MDSCs and Tregs and promote abnormal
angiogenesis (77,105). As discussed below, matrix
stiffening further amplifies this process by linking
mechanotransduction to CAF glycolytic reprogramming and immune
exclusion. Clinically, MCT4-high CAFs and IL-8-rich MSCs are
associated with immune exclusion, elevated PD-L1 and reduced
response to PD-1 blockade, supporting the role of stromal cells as
metabolic amplifiers of glycolysis-driven immunosuppression.
Exosome-mediated metabolic
communication
Exosomes provide a major route for long-range
transfer of metabolic and immunoregulatory signals (13,22,88). GC-derived exosomes carry
glycolysis-enhancing circRNAs and lncRNAs, such as circ-0038138,
circ-ATP8A1 and MALAT1, which activate EZH2-, STAT6- or
STAT3-dependent pathways in recipient macrophages and fibroblasts,
thereby promoting M2 polarization, VEGFA expression and ECM
remodeling (99,106). M2 macrophage-derived exosomes
enriched in MALAT1 and miR-21 feed back to tumor cells to increase
LDHA, HK2 and PD-L1 expression, completing a feed-forward
glycolytic loop (58,102).
CAFs and MSCs also release exosomes enriched in
PD-L1, glycolytic enzymes and immunoregulatory miRNAs, including
miR-155 and miR-23a, which suppress DC antigen presentation and CTL
granzyme B secretion (10,55,102).
Acidic pH can further imprint exosomal cargo with PD-L1, HIF-1α,
LDHA and lactylation-related signatures, facilitating dissemination
of metabolic and immunologic messages within the TME (93,99,105). Proteomic and RNA profiling of
plasma exosomes has identified stromal-associated proteins, such as
integrin-linked kinase and CD14, and ncRNAs as candidate
circulating markers of metabolic immunosuppression and ICB response
(7,85,107).
Glycolysis, angiogenesis and immune
exclusion
Glycolytic reprogramming also contributes to immune
exclusion by reshaping the vascular architecture of GC. Hypoxia and
oncogenic signaling stabilize HIF-1α, which transcriptionally
induces both glycolytic genes and pro-angiogenic mediators,
particularly VEGFA. In this setting, HIF-1α links glucose uptake,
LDHA-dependent lactate production and MCT-mediated lactate export
with VEGFA-driven angiogenesis, thereby coupling lactate-producing
metabolism to abnormal vascular remodeling (14,35-38). Although angiogenesis may increase
vessel density, the resulting vasculature is often tortuous, leaky
and poorly perfused. This leads to heterogeneous oxygen delivery,
persistent hypoxia and further activation of HIF-1α-dependent
glycolysis, forming a feed-forward loop between hypoxia,
glycolysis, lactate accumulation and vascular dysfunction.
At the immune interface, lactate and VEGFA
cooperate through several mechanisms. Tumor-derived lactic acid can
stabilize HIF-1α in macrophages and induce VEGF and ARG1
expression, thereby polarizing TAMs toward a tumor-promoting,
pro-angiogenic and immunosuppressive phenotype (108). Lactate-rich regions may also
reinforce HCAR1/GPR81- and MCT-dependent signaling in macrophages
and DCs, promoting ARG1, IL-10, TGF-β and VEGFA-associated programs
that suppress antigen presentation and favor MDSC/Treg-rich niches
(11,13,57,58,90,92,99). In parallel, VEGFA can impair
T-cell entry into tumors by inhibiting NF-κB-dependent endothelial
activation and reducing leukocyte-endothelial adhesion programs,
including intercellular adhesion molecule 1- and VCAM-1-associated
trafficking signals (109).
Thus, the HIF-1α/VEGFA/lactate network simultaneously sustains
hypoxic glycolysis, abnormal angiogenesis, suppressive myeloid
polarization and defective T-cell homing.
This metabolic-vascular loop has direct immunologic
consequences. Disorganized tumor vessels impair T-cell homing by
reducing effective perfusion, limiting endothelial activation and
generating hypoxic and acidic barriers that restrict lymphocyte
trafficking. At the same time, VEGFA and lactate promote
immunosuppressive myeloid-cell recruitment, inhibit DC maturation
and favor Treg and MDSC accumulation, thereby converting
metabolically active vascular regions into immune-excluded niches.
In GC, glycolysis-high tumors frequently display angiogenic
signatures, MCT4-rich stromal regions and poor CD8+
T-cell infiltration, supporting the concept that vascular
dysfunction is not merely a consequence of tumor growth but also an
active mediator of glycolysis-driven immune escape (21,26,73,77). This
metabolism-angiogenesis-immunity axis is incorporated into the
integrated tumor-stroma-immune network shown in Fig. 3.
Matrix stiffness, yes-associated
protein/transcriptional coactivator with PDZ-binding motif
(YAP/TAZ) signaling and CAF glycolysis
The physical properties of the GC stroma provide
another layer of metabolic regulation. In desmoplastic tumors,
excessive ECM deposition and collagen crosslinking increase stromal
stiffness, which is sensed by CAFs through integrins and focal
adhesion complexes (110).
Activation of integrin-focal adhesion kinase (FAK)/Src signaling
promotes nuclear translocation of YAP/TAZ, allowing
mechanotransduction to converge with TGF-β, WNT and
hypoxia-dependent pathways (111,112). In this context, YAP/TAZ
activation may cooperate with these pathways to enhance glycolytic
and immunoregulatory CAF phenotypes, thereby converting mechanical
stress into CAF metabolic reprogramming (112). More specifically, YAP/TAZ-TEA
domain transcription factor-associated transcriptional programs can
promote glycolysis-related gene expression, including glucose
transporters and enzymes involved in hexokinase activity,
fructose-2,6-bisphosphate production and lactate generation,
thereby providing a plausible molecular route by which matrix
stiffness may increase CAF glycolytic output and lactate release
(113).
This stiffness-driven metabolic state has important
consequences for immune exclusion. Glycolytic CAFs release lactate,
CXCL12, TGF-β and VEGFA, which reinforce ECM remodeling, abnormal
angiogenesis and myeloid-cell recruitment (77,89,105). At the same time, the stiff
matrix acts as a physical barrier to T-cell infiltration, while
CAF-derived lactate and cytokines create metabolic and immunologic
barriers that suppress DC activation and cytotoxic T-cell function
(112,114). Thus, stromal stiffness and CAF
glycolysis form a self-reinforcing circuit: Increased matrix
rigidity activates mechanotransduction, mechanotransduction
promotes glycolytic and immunoregulatory CAF phenotypes, and these
CAFs further remodel the matrix and maintain immune exclusion.
This mechanism is particularly relevant for
diffuse-type and stroma-rich GC, in which dense ECM, hypoxia,
MCT4-high CAFs and poor CD8+ T-cell infiltration often
coexist (19,21,27,77). From a therapeutic perspective,
disrupting this physical-metabolic coupling may require strategies
beyond direct glycolytic inhibition. Agents targeting integrin
signaling, FAK/Src activation, YAP/TAZ-dependent transcription,
TGF-β signaling or collagen crosslinking may reduce CAF-mediated
matrix remodeling and immune exclusion, especially when combined
with metabolic intervention and ICB (110,112,114). However, because
mechanotransduction pathways also participate in tissue repair and
immune-cell trafficking, such strategies require careful dose,
timing and patient selection.
Lactate-driven feedback loop and immune
remodeling
Central to this ecosystem is a self-reinforcing
lactate feedback circuit. Lactate released from tumor and stromal
cells engages HCAR1/GPR81 on macrophages and DCs, stabilizing
HIF-1α and inducing VEGFA, ARG1, IL-10 and TGF-β, which
collectively suppress CTL recruitment and effector function
(11,58,90,105). These cytokines, together with
prostaglandin E2 and adenosine, feed back through STAT3/mTOR-MYC
signaling to enhance glycolytic gene expression and PD-L1
expression in tumor cells (15,25,54,55). Lactate- and CXCL12-activated MDSCs
further secrete nitric oxide and ROS that inhibit DC maturation and
T-cell proliferation, while CAF-derived WNT5A and MSC-derived IL-8
reinforce this multicellular positive-feedback loop (24,33,89,97,100,103).
Epigenetic decoding of lactate adds durability to
these suppressive circuits. Histone lactylation at loci such as
ARG1 and MRC1 can lock in M2-like macrophage programs, while
similar modifications at immune checkpoint loci may help maintain
PD-L1 and LAG-3 expression in tumor cells (13,17,30,99). Lactylation-related gene-expression
models show that high lactate signaling coincides with increased
M2/MDSC infiltration and reduced cytotoxic T-cell signatures
(9,26). Spatial analyses further
demonstrate that regions populated by MCT4+ CAFs and
CD163+ TAMs are lactate-rich but CD8+-poor,
whereas regions with lower lactate burden show partial immune
infiltration (1,2,77).
In these metabolic dead zones, glucose is largely monopolized by
tumor and stromal cells, driving infiltrating lymphocytes toward
bioenergetic stress and exhausted or regulatory states (8,12,93). In preclinical GC models,
inhibition of LDHA or MCT4 was shown to normalize pH, reduce
M2/MDSC signatures and restore T-cell trafficking, indicating that
lactate stabilizes both the functional and spatial architecture of
the immunosuppressive TME (27,80,95,115).
Integrated therapeutic perspective
Viewing GC as a stromal-metabolic network reframes
therapy from single-target inhibition to multi-node modulation.
Combinatorial strategies that co-target tumor glycolysis and
stromal signaling, such as LDHA/MCT4 inhibition together with IL-8,
WNT5A or TGF-β blockade, were revealed to restore CD8+
T-cell infiltration, dismantle M2/MDSC-rich niches and weaken
CAF-mediated barriers in preclinical models (18,80,94,104). Re-educating CAFs through
inhibition of metabolic epigenetic writers, such as SMYD2 and
SETD1A, or through vitamin D analogues may further reduce lactate
output and normalize stromal architecture (17,30,99). In parallel, targeting exosome
biogenesis, such as through Ras-related protein Rab-27A knockdown
or GW4869, or targeting exosomal cargos such as MALAT1 and
circ-ATP8A1 may limit long-range dissemination of metabolic and
immunoregulatory cues (22,88,102). Co-targeting glycolysis, lactate
transport, exosome signaling and stromal reprogramming may
therefore help disrupt the lactate-driven feedback loop that
maintains the TME in a low-pH, high-PD-L1 and
immunotherapy-resistant state, providing a rational route to more
durable antitumor immunity in GC (9,19).
Therapeutic implications: Targeting the
glycolyticimmune axis
A central translational objective in GC is to
convert a metabolically cold, immune-excluded TME into one that
supports effective T-cell responses. Because glycolysis and lactate
accumulation suppress multiple components of antitumor immunity,
current strategies emphasize coordinated modulation of metabolism
and immunity through enzyme and transporter inhibition, upstream
and epigenetic targeting, repurposed metabolic drugs and rational
integration with ICB (12,33,116).
The goal is not to abolish glycolysis, which would also impair
effector lymphocytes, but to reduce glycolytic pressure in tumor
and stromal compartments and rebalance nutrients and pH in favor of
DC and T-cell function. Such approaches should be guided by
glycolysis/lactate-based signatures, 18F-FDG PET-derived
metabolic burden and MCT4-high stroma for patient selection
(3,117). An overview of this translational
framework, linking metabolic targets, metabolic-ICB combination
therapy, safety monitoring and biomarker-guided patient
stratification, including 18F-FDG PET/CT,
radiomics-derived glycolytic features, lactylation-related gene
signatures and exosomal ncRNA biomarkers is provided in Fig. 4. Therapeutic strategies,
representative agents, proposed mechanisms, expected immunologic
benefits, current evidence levels and supporting references are
summarized in Table II.
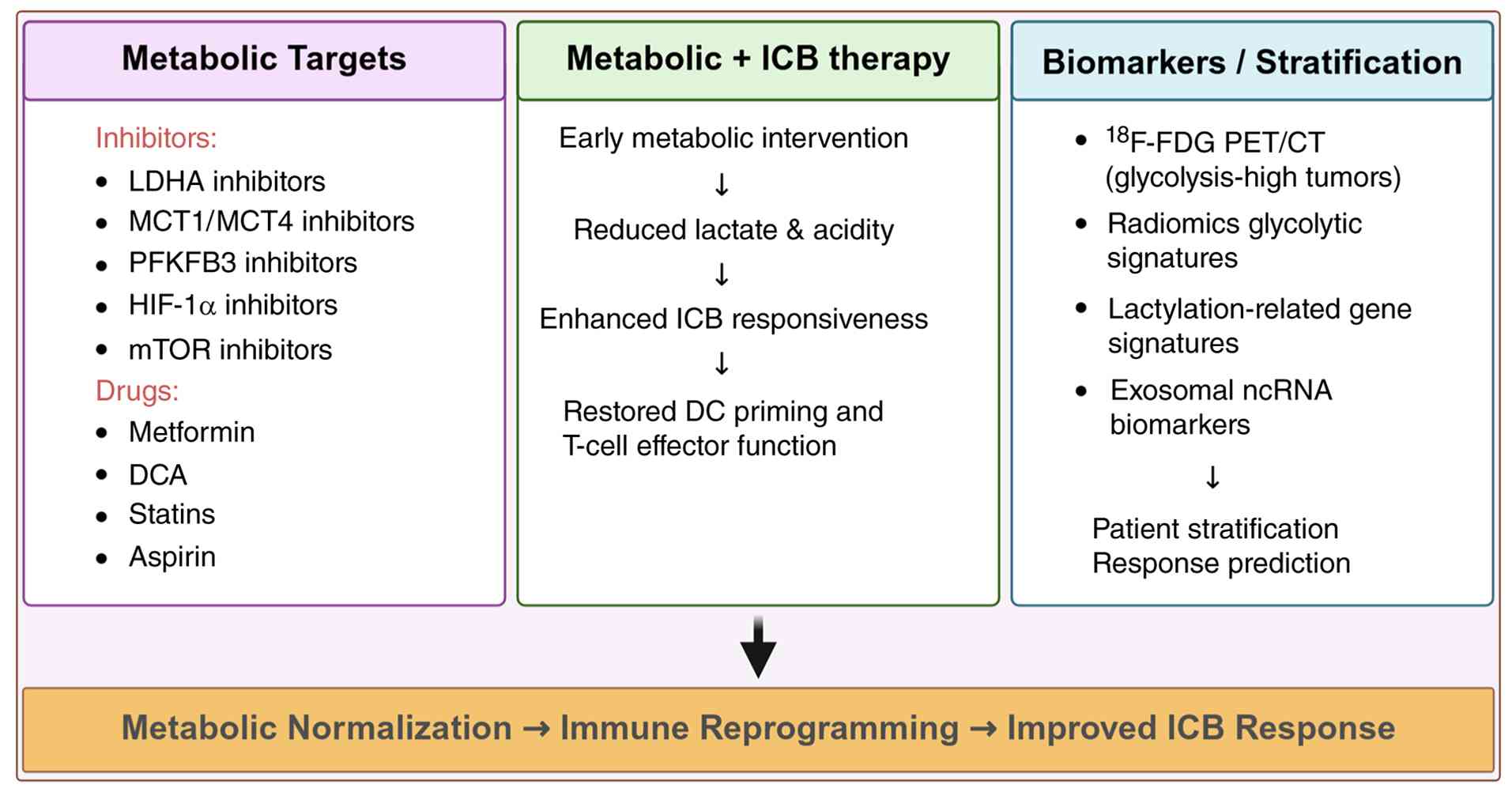 | Figure 4Therapeutic and biomarker-guided
strategies targeting glycolytic immunosuppression in gastric
cancer. Inhibitors of LDHA, MCT1/4, PFKFB3, HIF-1α and mTOR, as
well as repurposed metabolic agents such as metformin, DCA, statins
and aspirin, can normalize glycolytic pressure and lactate
accumulation. These interventions may enhance dendritic-cell
priming, restore T-cell effector function and improve
responsiveness to PD-1/PD-L1 blockade in selected metabolic
contexts. Metabolic imaging (18F-FDG PET/CT), radiomic
glycolysis signatures and lactylation-based transcriptomic
classifiers provide complementary tools for selecting
glycolysis-high tumors most likely to benefit from immunometabolic
combination strategies. Created with BioRender.com. LDHA, lactate dehydrogenase A; MCT1/4,
monocarboxylate transporter 1/4; PFKFB3,
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; HIF-1α,
hypoxia-inducible factor-1α; mTOR, mechanistic target of rapamycin;
DCA, dichloroacetate; PD-1, programmed death-1; PD-L1, programmed
death-ligand 1; 18F-FDG, fluorine-18 fluorodeoxyglucose;
ICB, immune checkpoint blockade; DC, dendritic cell; ncRNA,
noncoding RNA. |
| Table IIMetabolic-immune therapeutic
strategies and clinical relevance in GC. |
Table II
Metabolic-immune therapeutic
strategies and clinical relevance in GC.
| Strategy | Target/Drug | Mechanism | Expected
immunologic benefit | Clinical
relevance | (Refs.) |
|---|
| LDHA
inhibition | FX11, GNE-140 | Reduces lactate
production | Restores DC/T-cell
activation | Preclinical
evidence | (77,94,148) |
| MCT1/4
blockade | AZD3965,
syrosingopine | Blocks lactate
shuttle | Disrupts metabolic
immune exclusion | Early clinical
evidence for MCT1; MCT4 mainly preclinical | (79,104,146,147) |
| PFKFB3
modulation | PFKFB3-targeting
approaches | Reduces glycolytic
flux | May reduce
glycolysis-high metabolic pressure | Preclinical
evidence; GC-specific therapeutic validation remains limited | (95) |
| HIF-1α/AKT/mTORaxis
targeting | HIF-1α-, AKT- or
mTOR-related approaches | Suppresses hypoxia-
and growth-factor-driven glycolysis | May reduce
PD-L1-linked metabolic remodeling | Preclinical and
translational evidence; GC metabolic-ICB efficacy remains
unproven | (35,37,38,121,122) |
|
Dichloroacetate | PDH activation | Reduces
lactate | Restores T-cell
metabolism | Early
investigation | (36,125) |
| Chemoimmunotherapy
backbone | PD-1 blockade plus
chemotherapy | Provides an
established clinical backbone for future metabolic
combinations | Supports antitumor
immune responses in selected patients | Established
first-line clinical evidence in advanced GC/GEJ adenocarcinoma;
metabolic add-on remains unproven | (134,143-145) |
Inhibiting glycolytic enzymes and lactate
transporters
Several agents directly reduce glycolytic flux.
2-Deoxy-D-glucose (2-DG) competitively inhibits HK2, lowers ATP
levels and can downregulate PD-L1. In GC models, 2-DG combined with
pyrolyzed deketene curcumin attenuated tumor glycolysis and limited
Treg generation (15,115,118). LDHA inhibitors, such as FX11,
oxamate and related analogues, were shown to restore pyruvate
oxidation, increase intratumoral pH, enhance CTL/DC activity and
reduce invasion and EMT (77,94,95). PFKFB3 inhibitors, including PF-3,
3PO and KAN0438757, were demonstrated to suppress glycolytic flux
and normalize tumor vasculature, thereby improving T-cell
trafficking and drug delivery (4,80).
Additional targets, including PDK1/4, G6PD, transketolase-like
protein 1 and PKM2, weaken the Warburg phenotype, increase radio-
and chemosensitivity and reduce redox buffering (96,119).
Lactate transport is a complementary target. MCT1
inhibitors, including AZD3965 and AR-C155858, limit lactate uptake
by oxidative tumor cells, whereas MCT4 inhibitors, such as
syrosingopine and α-cyano-4-hydroxycinnamate, trap lactate
intracellularly, inducing metabolic stress and reducing
extracellular acidification (4,18,93). High stromal MCT4, particularly in
CAFs and TAMs, has been shown to be associated with poor survival,
immune exclusion and peritoneal carcinomatosis (27,79,104). In vivo, MCT4 blockade was
reported to normalize pH, reduce M2/MDSC signatures and restore
T-cell trafficking (80,94,115). Because systemic inhibition of
glycolysis or lactate shuttling may also affect highly glycolytic
normal tissues and activated lymphocytes, tumor-targeted
formulations, including liposomes, nanoparticles and antibody-drug
conjugates, as well as intermittent dosing schedules, are being
explored to exploit the stronger glycolytic dependence of GC cells
while preserving immune-cell fitness (6,9,12,95,100,118).
Targeting upstream signaling and
epigenetic regulators
Master regulators link oncogenic signaling,
glycolysis and immune evasion. HIF-1α, PI3K/AKT/mTOR and MYC
collectively induce GLUT1, HK2, LDHA, PD-L1 as well as other
targets (16,35,42,120). Inhibition of HIF-1α, such as
with PT2385 or BAY 87-2243, or mTOR, such as with everolimus or
rapamycin, was shown to reduce lactate output in GC models,
partially normalize metabolic competition between tumor cells and T
cells, and improve responses to anti-PD-1 therapy (37,105,121,122). Epigenetic writers such as SMYD2
and SETD1A, together with m6A-modified lncRNAs including OIP5-AS1,
were reported to stabilize glycolytic gene expression and
immune-cold phenotypes; inhibiting these pathways may lower PD-L1
expression and resensitize tumors to ICB (26,61,123,124). In myeloid cells, modulation of
histone lactylation, for example through p300 inhibition or histone
deacetylase activation, was shown to partially re-educate M2
macrophages and reverse lactate-imprinted tolerogenic memory,
providing another potential route to restore antitumor immunity
(11,13,58,90). Given the pleiotropic roles of
these pathways, most strategies favor short metabolic-priming
windows around ICB, with dosing guided by real-time metabolic and
immune readouts (1,21,28,82).
Repurposing clinically available
metabolic modulators
Several licensed agents exert immunometabolic
effects that may be therapeutically useful. Metformin, an
AMP-activated protein kinase (AMPK) activator and OXPHOS enhancer,
reduces LDHA and MCT4, limits HIF-1α stabilization and can reverse
PD-L1 upregulation by restoring the NAD+/NADH balance
(4,9,93).
Observational studies in diabetic GC suggest improved outcomes,
consistent with combined tumor-intrinsic and immune-related
benefits (6,48). Dichloroacetate (DCA) inhibits
PDKs, promotes pyruvate entry into the TCA cycle and may improve
antigen presentation. In GC models, DCA was shown to enhance
5-fluorouracil efficacy and reverse hypoxia-related resistance
through HIF-1α downregulation (36,54,64,125). Aspirin and other nonsteroidal
anti-inflammatory drugs can dampen HIF-1α/mTOR signaling, limit HK2
transcription and suppress COX-2-mediated immunosuppressive
prostaglandins (33,88,96).
Additional candidates include β-blockers, such as
propranolol, which may attenuate sympathetic HIF-1α stabilization;
statins, which target mevalonate pathways involved in membrane
organization and immune regulation; AMPK agonists such as
5-aminoimidazole-4-carboxamide ribonucleotide; proton pump
inhibitors such as pantoprazole, which can inhibit PKM2; FA
synthase inhibitors such as orlistat; and ketogenic-like diets
enriched with omega-3 FAs and medium-chain triglycerides. These
interventions can shift metabolic states, increase vulnerability to
glycolytic stress and slow GC xenograft growth in experimental
settings (126-128). They are best viewed as
background modulators that reduce glycolytic pressure, acidosis or
redox stress and create a more permissive niche for DCs and CTLs,
making them potential partners for PD-1/PD-L1 inhibitors in
biomarker-defined GC subsets (Fig.
4) (48,101,118,119,129). The therapeutic strategies
targeting glycolytic enzymes, lactate transport, upstream
signaling, repurposed metabolic modulators and immunometabolic
combination approaches are summarized in Fig. 4.
Risk factor-guided refinement may further improve
the rational use of these repurposed agents. Helicobacter
pylori-positive and chronic inflammation-enriched GC should be
regarded as etiologically high-risk and inflammation-associated
contexts rather than as established β-blocker-sensitive subtypes.
Chronic Helicobacter pylori infection induces long-standing
gastritis, disrupts local immune responses and increases the risk
of gastric adenocarcinoma, particularly in high-incidence regions,
supporting its classification as a major risk context for GC
(1,2,16,51-53). In this inflammatory background,
β-blockers may be most biologically relevant when tumors show high
sympathetic stress-related signaling, β-adrenergic pathway
activation or inflammation-associated glycolytic remodeling. This
hypothesis is supported by evidence that norepinephrine enhances
aerobic glycolysis and lactate release and may act as a predictive
factor for immunotherapy in GC (48). However, direct evidence that
Helicobacter pylori-positive GC is preferentially sensitive
to β-adrenergic blockade remains unavailable. Therefore, β-blockers
should be considered exploratory metabolic-neuroimmune modulators
for adrenergic or inflammation-linked glycolysis-high contexts
rather than universal adjuvants for all patients with
Helicobacter pylori-positive GC.
For statins, the most plausible risk-factor-guided
context may involve metabolic syndrome, dysregulated lipid
metabolism or cholesterol/mevalonate pathway activation. Metabolic
syndrome has been associated with increased GC risk in a large
prospective Korean cohort, and hypertriglyceridemia, low
high-density lipoprotein cholesterol and hyperglycemia were
independently associated with GC risk (130). In parallel, epidemiologic
studies suggest that statin exposure is associated with a reduced
risk of GC, particularly in selected metabolic backgrounds such as
elderly patients with hyperglycemia, although these findings do not
prove an immunometabolic treatment effect in established GC
(131). Mechanistically, statins
inhibit 3-hydroxy-3 methylglutaryl coenzyme A reductase and
suppress the mevalonate pathway, which can influence membrane
cholesterol availability, lipid raft organization, oncogenic
signaling and immune regulation (132). In GC models, disruption of the
mevalonate pathway has also been linked to altered tumor growth and
progression, supporting the biological plausibility of
statin-related metabolic intervention (133). Overall, β-blockers and statins
should not be considered universal metabolic adjuvants. Future
studies should incorporate Helicobacter pylori status,
chronic inflammatory background, metabolic syndrome, lipid
profiles, adrenergic-pathway activity,
cholesterol/mevalonate-pathway activation, glycolysis/lactate
signatures and immune contexture to define candidate populations
more precisely.
Combination strategies with ICB
Because metabolic inhibition alone rarely yields
durable responses, most preclinical studies now evaluate
combinations with ICB. In murine GC and colorectal models, dual
LDHA/MCT4 blockade plus anti-PD-1 was shown to increase T-cell
infiltration, reduce Tregs and prolong survival (27,80,104,115). Co-targeting HIF-1α and PD-1, or
combining metformin with anti-PD-1, achieved superior control of
hypoxia-driven tumors by lowering lactate, normalizing pH,
improving CTL metabolic fitness and attenuating STAT3/MYC signaling
and histone lactylation (26,100,105). Early-phase trials, including
NCT03721944, NCT05101237 and NCT04772633, are testing MCT1
inhibitors and other metabolic modulators with anti-PD-1/PD-L1
therapy in solid tumors, including GC or gastroesophageal cancers.
These studies are expected to clarify whether metabolic modulation
can enhance immune activation and whether such combinations have
acceptable safety profiles in biomarker-selected populations
(Fig. 4) (3,21,134).
Large randomized trials such as CheckMate 649 have
established PD-1 plus chemotherapy as a first-line standard in
advanced GC (7,135), providing a clinical backbone
onto which metabolic agents could theoretically be layered.
However, because chemoimmunotherapy already carries substantial
immune-related and hematologic toxicity, metabolic-ICB combinations
should prioritize glycolysis-high, immune-cold patients, use
temporal staggering such as short metabolic priming before or
briefly overlapping with ICB, and incorporate de-escalation
strategies guided by metabolic imaging and immune biomarkers
(5,129,136). The specific safety
considerations of these combinations are discussed below as part of
the biomarker-guided translational framework shown in Fig. 4.
Emerging biomarkers and dynamic imaging
tools
Robust biomarkers are essential for deploying
immunometabolic strategies in GC (9,18,33). MCT4 expression, LDHA activity and
lactate levels measured by magnetic resonance spectroscopy,
together with gene-expression-derived glycolysis scores
incorporating HK2, PFKFB3, LDHA, ENO1 and MCT4, are inversely
associated with CD8+ T-cell infiltration and ICB benefit
and may identify tumors more likely to benefit from metabolic
intervention (3,27,137). Circulating exosomal lncRNAs such
as H19 and MALAT1, glycolysis-associated circRNAs and serum IL-8
provide minimally invasive indicators of metabolic
immunosuppression (55,85,99,138). Integrating tissue biomarkers,
exosomal signals and peripheral immune-cell profiles may therefore
help define glycolysis-high, lactate-rich and immune-excluded GC
subsets before treatment, corresponding to the biomarker-guided
stratification module in Fig.
4.
Imaging biomarkers provide complementary,
noninvasive information on whole-tumor metabolic burden. Baseline
18F-FDG PET/CT parameters, including maximum
standardized uptake value (SUVmax), metabolic tumor volume (MTV)
and total lesion glycolysis (TLG), can capture glycolytic activity,
viable tumor burden and spatial heterogeneity beyond anatomic size
alone (1,2,4,28,29). PET/CT-derived volumetric
parameters and radiomics features have been associated with
survival, c-MET/HER2 status, lymphovascular invasion, response to
chemotherapy or ramucirumab-based regimens and outcomes after
neoadjuvant immunochemotherapy (4,28,29,73,78). These parameters may therefore
serve as baseline tools for selecting patients with FDG-avid,
glycolysis-high tumors who are more likely to require metabolic
modulation in addition to ICB, as summarized in Fig. 4.
Beyond baseline stratification, dynamic changes in
metabolic imaging during treatment may be particularly informative.
Early reductions in MTV or TLG after one or two treatment cycles
may reflect decreased viable glycolytic tumor burden before
conventional CT-based tumor shrinkage becomes apparent. Conversely,
persistent or increasing MTV/TLG despite therapy may indicate
primary metabolic resistance, inadequate immune infiltration or
continued stromal lactate production. In the setting of ICB or
chemoimmunotherapy, longitudinal PET/CT may also help distinguish
true metabolic response from mixed response, pseudoprogression,
hyperprogression or inflammatory immune-cell infiltration, although
standardized interpretation criteria for GC immunotherapy remain
insufficiently established.
Although GC-specific dynamic PET/CT data during ICB
remain limited, primary studies in other solid tumors provide
clinically relevant examples supporting this concept. In previously
treated non-small cell lung cancer (NSCLC), Kaira et al
(139) performed
18F-FDG PET/CT before and 1 month after nivolumab
therapy and calculated SUVmax, MTV and TLG; metabolic response at 1
month predicted early therapeutic efficacy and survival more
effectively than CT alone. In another prospective NSCLC study,
Umeda et al (140)
performed integrated 18F-FDG PET/magnetic resonance
imaging (MRI) before and 2 weeks after nivolumab therapy; early
changes in TLG combined with ADC changes distinguished
non-progressive disease from progressive disease and were
associated with progression-free survival (PFS) and overall
survival (OS). In the neoadjuvant setting, Tao et al
(141) evaluated baseline and
preoperative 18F-FDG PET/CT in resectable NSCLC treated
with two cycles of sintilimab; percentage changes in maximum and
peak standardized uptake values normalized by lean body mass
(SULmax and SULpeak), MTV and TLG, and PET Response Criteria in
Solid Tumors-based metabolic response were significantly associated
with major pathological response, with all partial metabolic
responders achieving major pathological response (141). These primary studies support the
rationale for prospectively testing serial PET/CT-derived MTV, TLG
and metabolic response criteria in GC immunotherapy or
chemoimmunotherapy trials, while acknowledging that
disease-specific validation in GC remains necessary.
Dynamic metabolic imaging should ideally be
interpreted together with immune and liquid-biopsy markers. For
example, early decreases in MTV/TLG accompanied by increased
peripheral effector T-cell signatures, reduced serum IL-8,
declining exosomal glycolytic ncRNAs or reduced lactate-related
gene signatures would support effective immunometabolic remodeling.
By contrast, persistently high FDG uptake together with MCT4-high
stroma, elevated IL-8 or myeloid-suppressive signatures may suggest
ongoing lactate-driven immune exclusion and the need for
therapeutic adaptation. Artificial-intelligence-assisted radiomics,
hyperpolarized 13C MRI, spatial transcriptomics and
multiplex immunohistochemistry could further refine this framework
by linking whole-body metabolic changes to regional immune
architecture (19,20,75,76,142).
Thus, PET/CT and radiomics should not be viewed
only as static prognostic tools. In future trials of metabolic
intervention plus ICB, serial imaging at baseline, early
on-treatment time points and progression should be incorporated to
evaluate whether changes in MTV, TLG, SUV-derived heterogeneity and
radiomics features can predict response, immune remodeling and
resistance earlier than anatomic criteria. Safety and toxicity
should be assessed together with clinical symptoms, laboratory
parameters and treatment exposure. Such dynamic monitoring may
support adaptive treatment strategies, including continuation,
escalation, de-escalation or switching of immunometabolic
combinations.
Challenges in clinical translation
Despite the strong biologic rationale for combining
metabolic intervention with ICB, clinical translation remains
challenging. A central concern is therapeutic selectivity.
Glycolysis and lactate transport are not unique to malignant cells;
activated T cells, DCs, intestinal epithelial cells, hematopoietic
progenitors and other proliferating normal tissues also depend on
glucose metabolism or lactate handling under stress conditions
(12,33,100). Therefore, systemic inhibition of
LDHA, MCT1/4, PFKFB3 or upstream glycolytic regulators may impair
not only tumor glycolysis but also immune-cell fitness, epithelial
repair and bone marrow recovery. Accordingly, early-phase trials
should prospectively monitor immune-cell fitness, blood counts,
mucosal toxicity, nutritional status and gastrointestinal tolerance
when glycolysis- or lactate-transport-targeted agents are combined
with chemotherapy and ICB.
Toxicity is especially important when metabolic
agents are layered onto chemotherapy plus ICB. Published
chemoimmunotherapy trials in advanced GC and gastroesophageal
junction (GEJ) adenocarcinoma provide concrete examples of how
therapeutic selectivity, patient selection and toxicity monitoring
have already been incorporated into clinical study design. In
CheckMate 649, nivolumab was combined with fluoropyrimidine- and
platinum-based chemotherapy as first-line treatment for previously
untreated, unresectable, non-HER2-positive gastric, GEJ or
esophageal adenocarcinoma. Therapeutic selectivity was implemented
through prespecified PD-L1 combined positive score (CPS) subgroups,
with OS and PFS tested primarily in patients with PD-L1 CPS ≥5.
This trial demonstrated improved OS and PFS in the CPS ≥5
population, but also showed higher grade 3-4 treatment-related
adverse events with nivolumab plus chemotherapy than with
chemotherapy alone (59% vs. 44%), most commonly nausea, diarrhea
and peripheral neuropathy, although no new safety signals were
identified (134).
ATTRACTION-4 provides an Asian population-specific
example. This randomized, double-blind phase 3 trial enrolled
patients from Japan, South Korea and Taiwan with previously
untreated, HER2-negative, unresectable advanced or recurrent GC/GEJ
cancer and Eastern Cooperative Oncology Group (ECOG) performance
status 0-1, regardless of PD-L1 expression. Nivolumab plus
oxaliplatin-based chemotherapy significantly improved PFS but not
OS. The most common treatment-related grade 3-4 adverse events were
decreased neutrophil count and decreased platelet count, and
treatment-related serious adverse events were more frequent with
nivolumab plus chemotherapy than with placebo plus chemotherapy
(143). In KEYNOTE-859,
pembrolizumab plus chemotherapy was evaluated in previously
untreated, locally advanced or metastatic HER2-negative gastric/GEJ
adenocarcinoma. Patient selection included HER2-negative status and
ECOG performance status 0-1, and randomization was stratified by
geographical region, PD-L1 status and chemotherapy backbone.
Pembrolizumab plus chemotherapy improved OS in the
intention-to-treat population and in PD-L1 CPS ≥1 and CPS ≥10
subgroups, while grade 3-5 adverse events included anemia and
decreased neutrophil count, and serious treatment-related adverse
events occurred in 23% vs. 19% of patients (144). ORIENT-16 further illustrates
population- and biomarker-informed selection in Chinese patients
with unresectable locally advanced or metastatic gastric/GEJ
adenocarcinoma. Sintilimab plus capecitabine and oxaliplatin
improved OS both in all randomized patients and in the PD-L1 CPS ≥5
subgroup; grade 3 or higher treatment-related adverse events
included decreased platelet count, decreased neutrophil count and
anemia (145).
These published trials indicate that
chemoimmunotherapy development in GC has already relied on
clinically applicable selection variables, including HER2 status,
PD-L1 CPS, ECOG performance status, disease setting, geographic or
regional population and chemotherapy backbone. They also show that
hematologic, gastrointestinal, neurologic and immune-related
toxicities must be actively monitored even before adding metabolic
inhibitors. Therefore, future metabolic-ICB combinations should not
be developed as uniform add-on strategies. Instead, they should
build on trial-tested selection variables and add experimental
metabolic biomarkers, such as LDHA/MCT4 expression, FDG avidity,
MTV/TLG, glycolysis-related gene signatures and exosomal ncRNA
markers, only after prospective validation (Fig. 4). By contrast, published clinical
data specifically evaluating chemotherapy plus ICB plus LDHA or
MCT4 inhibition in GC are not yet available. Therefore, the
possibility that LDHA/MCT4 inhibitors could increase
myelosuppression, gastrointestinal toxicity or immune-related
toxicity should currently be considered a biologically plausible
concern rather than an established clinical observation.
Available evidence on lactate-transport or
glycolysis-targeted agents further supports cautious clinical
translation. The MCT1 inhibitor AZD3965 has entered first-in-human
phase I testing in patients with advanced solid tumors or lymphoma,
providing initial human safety information for pharmacologic
lactate-transport inhibition (146). However, AZD3965 selectively
targets MCT1 rather than MCT4, and these data do not establish the
safety of combining lactate-transport inhibition with chemotherapy
plus ICB in GC. MCT4-targeting approaches remain predominantly
preclinical; for example, syrosingopine has been evaluated as a
dual MCT1/MCT4 inhibitor that modulates tumor metabolism and
extracellular acidification in experimental models (147). Similarly, LDHA inhibitors such
as FX11 and oxamate have shown antitumor activity mainly in
preclinical studies, and mature clinical toxicity data for LDHA
inhibition in GC are lacking (148). Thus, the toxicity of
chemotherapy plus ICB plus LDHA/MCT4 inhibition should be
prospectively evaluated in early-phase trials rather than inferred
from preclinical efficacy alone.
Several strategies may improve the therapeutic
window. First, metabolic intervention should be guided by patient
selection rather than applied uniformly; patients with
glycolysis-high, MCT4-high, FDG-avid and immune-excluded tumors are
more rational candidates than those with metabolically inflamed or
glycolysis-low tumors (18,21,27,28,29,73,78). Second, intermittent dosing or
short metabolic-priming windows before ICB may reduce chronic
suppression of effector lymphocytes while transiently lowering
lactate burden and improving T-cell infiltration. Third,
tumor-targeted delivery systems, pH-responsive formulations and
stromal-targeted approaches may help limit systemic exposure.
Finally, early-phase trials should prospectively monitor not only
tumor response but also neutrophil and lymphocyte dynamics, anemia,
mucosal toxicity, nutritional status, liver function, inflammatory
cytokines and immune-related adverse events.
Biomarker-integrated toxicity assessment will be
essential. Dynamic 18F-FDG PET/CT, circulating
lactate-related signatures, exosomal ncRNAs, peripheral immune-cell
profiling and serum cytokine panels may help distinguish beneficial
metabolic reprogramming from excessive systemic metabolic stress.
In this context, the goal of metabolic-ICB combination therapy
should not be maximal glycolytic inhibition, but carefully timed
and biomarker-guided reduction of tumor- and stroma-derived
metabolic pressure while preserving antitumor immune-cell function,
as summarized in Fig. 4.
Conclusions
Glycolytic reprogramming is a major organizer of
immune suppression in selected GC subtypes. Tumor-intrinsic
oncogenic signaling, hypoxia, inflammation and ncRNA-mediated
regulation enhance glucose uptake, glycolytic flux and lactate
export, while stromal cells, CAFs, MSCs and immunosuppressive
myeloid populations further amplify this metabolic state. The
resulting lactate-rich and acidic TME impairs DC maturation, T-cell
effector function and NK-cell activity, promotes M2-like
macrophages and MDSCs, and reinforces immune checkpoint activation
through signaling and epigenetic mechanisms, including HCAR1/GPR81
signaling and histone lactylation. Together, these processes
establish spatially organized immunometabolic niches in which
glycolytic stress, abnormal vasculature, stromal stiffness and
immune exclusion limit durable responses to ICB.
At the same time, GC should not be viewed as a
uniformly glycolysis-dependent disease. Glycolysis-centered immune
suppression represents an important but heterogeneous framework
that interacts with glutamine metabolism, FA oxidation,
mitochondrial rewiring, stromal metabolism and vascular remodeling.
Therefore, the concept of glycolysis-driven immunosuppression
should be interpreted as a working model for selected
glycolysis-dominant, lactate-rich and immune-excluded GC subtypes
rather than as a universal explanation for all GC biology.
Recognizing this metabolic heterogeneity is essential for
translating glycolysis-targeted strategies into rational,
biomarker-guided therapeutic combinations.
Future perspectives: Roadmap for clinical
translation
Future studies should first validate glycolysis-
and lactate-centered biomarkers in prospective GC cohorts.
Candidate markers include tumor or stromal LDHA and MCT4
expression, glycolysis-related gene signatures, lactylation-related
models, exosomal glycolytic ncRNAs, serum inflammatory mediators
such as IL-8, and baseline 18F-FDG PET/CT parameters
such as SUVmax, MTV and TLG. These markers should be integrated
with immune contexture, including CD8+ T-cell
infiltration, cDC1 abundance, TAM and MDSC signatures, PD-L1 status
and spatial immune exclusion.
Second, clinical development of immunometabolic
combinations should move from empiric combinations toward
subtype-guided trial design. Patients with FDG-avid,
LDHA/MCT4-high, lactate-rich and immune-excluded tumors may be the
most rational candidates for metabolic priming combined with ICB.
By contrast, tumors with lower glycolytic burden or stronger
dependence on glutamine, lipid or mitochondrial metabolism may
require alternative strategies. Risk-factor annotation, including
Helicobacter pylori status, EBV status, inflammatory
background, metabolic comorbidities and sympathetic stress-related
signaling, may further refine the use of repurposed agents such as
β-blockers, statins, metformin or DCA.
Third, safety and therapeutic window should remain
central considerations. Because glycolysis and lactate handling are
also required by activated immune cells, intestinal epithelium and
hematopoietic progenitors, future trials should avoid
indiscriminate metabolic inhibition and instead evaluate
intermittent dosing, short metabolic-priming windows,
tumor-targeted or pH-responsive delivery systems, and prospective
monitoring of myelosuppression, gastrointestinal toxicity,
immune-related adverse events, nutritional status and peripheral
immune-cell dynamics.
Finally, dynamic monitoring should guide adaptive
treatment. Serial 18F-FDG PET/CT, radiomics, circulating
exosomal ncRNAs, cytokine profiling and peripheral immune
monitoring may help determine whether metabolic intervention
reduces tumor glycolytic pressure while preserving immune-cell
function. Early changes in MTV, TLG, SUV-derived heterogeneity or
glycolysis-related liquid-biopsy markers may provide earlier
evidence of response or resistance than anatomic imaging alone.
Ultimately, the goal is not maximal suppression of glycolysis, but
precise remodeling of tumor- and stroma-derived metabolic barriers
to restore immune control in appropriately selected patients.
Availability of data and materials
Not applicable.
Authors' contributions
BZ and LS contributed equally to this work. BZ and
LS designed the scope and structure of the review, performed
structured literature searches, organized the figures and tables,
and drafted the initial manuscript. ZK, CW and BS contributed to
structured literature searches, critical synthesis and
interpretation of relevant findings, and revision of major sections
of the manuscript. FK conceived the review topic, supervised the
overall project, critically revised major sections of the
manuscript and approved the final version. All authors read and
approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
2-DG
|
2-deoxy-D-glucose
|
|
AMPK
|
AMP-activated protein kinase
|
|
ARG1
|
arginase 1
|
|
ATP
|
adenosine triphosphate
|
|
CAF
|
cancer-associated fibroblast
|
|
cDC1
|
conventional type 1 dendritic
cell
|
|
ceRNA
|
competitive endogenous RNA
|
|
circRNA
|
circular RNA
|
|
CTL
|
cytotoxic T lymphocyte
|
|
DC
|
dendritic cell
|
|
DCA
|
dichloroacetate
|
|
EBV
|
Epstein-Barr virus
|
|
ECM
|
extracellular matrix
|
|
EMT
|
epithelial-mesenchymal transition
|
|
FA
|
fatty acid
|
|
FAK
|
focal adhesion kinase
|
|
FDG
|
fluorodeoxyglucose
|
|
GC
|
gastric cancer
|
|
GLUT1/3
|
glucose transporter 1/3
|
|
HCAR1/GPR81
|
hydroxycarboxylic acid receptor 1/G
protein-coupled receptor 81
|
|
HER2
|
human epidermal growth factor
receptor 2
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
HK2
|
hexokinase 2
|
|
ICB
|
immune checkpoint blockade
|
|
IFN-γ
|
interferon-γ
|
|
IL
|
interleukin
|
|
IRF
|
interferon regulatory factor
|
|
LAG-3
|
lymphocyte activation gene-3
|
|
LDHA
|
lactate dehydrogenase A
|
|
lncRNA
|
long noncoding RNA
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MCT1/4
|
monocarboxylate transporter 1/4
|
|
MDSC
|
myeloid-derived suppressor cell
|
|
MHC
|
major histocompatibility complex
|
|
miRNA
|
microRNA
|
|
MRC1
|
mannose receptor C-type 1
|
|
MSC
|
mesenchymal stem/stromal cell
|
|
MTV
|
metabolic tumor volume
|
|
mTOR
|
mechanistic target of rapamycin
|
|
mTORC1
|
mTOR complex 1
|
|
NAD+
|
oxidized nicotinamide adenine
dinucleotide
|
|
ncRNA
|
noncoding RNA
|
|
NF-κB
|
nuclear factor κB
|
|
NK
|
natural killer
|
|
OXPHOS
|
oxidative phosphorylation
|
|
PD-1
|
programmed death-1
|
|
PD-L1
|
programmed death-ligand 1
|
|
PDK
|
pyruvate dehydrogenase kinase
|
|
PER1
|
period circadian regulator 1
|
|
PET/CT
|
positron emission tomography/computed
tomography
|
|
PFKFB3
|
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
PKM2
|
pyruvate kinase M2
|
|
ROS
|
reactive oxygen species
|
|
SMYD2
|
SET and MYND domain-containing
protein 2
|
|
STAT
|
signal transducer and activator of
transcription
|
|
SUVmax
|
maximum standardized uptake value
|
|
TAZ
|
transcriptional coactivator with
PDZ-binding motif
|
|
TCA
|
tricarboxylic acid
|
|
TGF-β
|
transforming growth factor-β
|
|
TLG
|
total lesion glycolysis
|
|
TME
|
tumor microenvironment
|
|
Treg
|
regulatory T cell
|
|
VEGFA
|
vascular endothelial growth factor
A
|
|
WNT5A
|
wingless-type MMTV integration site
family member 5A
|
|
YAP
|
yes-associated protein
|
|
SULmax
|
maximum standardized uptake value
normalized by lean body mass
|
|
SULpeak
|
peak standardized uptake value
normalized by lean body mass
|
Acknowledgments
Not applicable.
Funding
This work was supported by the Noncommunicable Chronic
Diseases-National Science and Technology Major Project (grant no.
2024ZD0521103); the Tianjin Public Health Science and Technology
Major Youth Project (grant no. 24ZXGQSY00090); the Science and
Technology Project of Haihe Laboratory of Modern Chinese Medicine;
National Clinical Research Center for Chinese Medicine Acupuncture
and Moxibustion Open Funding Project (grant no. NCRCOP2023007); the
Tianjin Key Research Projects in Traditional Chinese Medicine
(grant no. 2025011); the Hebei Provincial Administration of
Traditional Chinese Medicine Research Project (grant nos. T2025083
and T2025059); the Special Fund for Clinical Research of Wu Jieping
Medical Foundation (grant nos. 320.6750.2023-5-16,
320.6750.2025-6-87 and 320.6750.2023-10-5); the Pilot Demonstration
Project for the Inheritance and Innovative Development of
Traditional Chinese Medicine in Nankai District, Tianjin (grant no.
20240204019); and the Tianjin Municipal Key Disciplines and Key
Specialties Construction Program in Medicine (grant no.
TJYXZDXK-010A).
References
|
1
|
Sun G, Cheng C, Li X, Wang T, Yang J and
Li D: Metabolic tumor burden on postsurgical PET/CT predicts
survival of patients with gastric cancer. Cancer Imaging.
19:182019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Song J, Li Z, Chen P, Yu J, Wang F, Yang Z
and Wang X: A 18FDG PET/CT-based volume parameter is a predictor of
overall survival in patients with local advanced gastric cancer.
Chin J Cancer Res. 31:632–640. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang R, Mao G, Tang Y, Li C, Gao Y, Nie
W, Song T, Liu S, Zhang P, Tao K and Li W: Inhibition of glycolysis
enhances the efficacy of immunotherapy via PDK-mediated
upregulation of PD-L1. Cancer Immunol Immunother. 73:1512024.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu G, Hu Y, Cheng X, Wang Y, Gu Y, Liu T
and Shi H: Volumetric parameters on 18F-FDG PET/CT predict the
survival of patients with gastric cancer associated with their
expression status of c-MET. BMC Cancer. 19:7902019. View Article : Google Scholar
|
|
5
|
Davern M and Lysaght J: Cooperation
between chemotherapy and immunotherapy in gastroesophageal cancers.
Cancer Lett. 495:89–99. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rong Y, Teng Y and Zhou X: Advances in the
study of metabolic reprogramming in gastric cancer. Cancer Med.
14:e709482025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
An M, Mehta A, Min BH, Heo YJ, Wright SJ,
Parikh M, Bi L, Lee H, Kim TJ, Lee SY, et al: Early immune
remodeling steers clinical response to first-line
chemoimmunotherapy in advanced gastric cancer. Cancer Discov.
14:766–785. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun L, Li J, Yan W, Yao Z, Wang R, Zhou X,
Wu H, Zhang G, Shi T and Chen W: H19 promotes aerobic glycolysis,
proliferation and immune escape of gastric cancer cells through the
microRNA-519d-3p/lactate dehydrogenase A axis. Cancer Sci.
112:2245–2259. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang H, Zou X, Yang S, Zhang A, Li N and
Ma Z: Identification of lactylation related model to predict
prognostic, tumor infiltrating immunocytes and response of
immunotherapy in gastric cancer. Front Immunol. 14:11499892023.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lv M, Zhang S, Dong Y, Cao L and Guo S:
PolG inhibits gastric cancer glycolysis and viability by
suppressing PKM2 phosphorylation. Cancer Manag Res. 13:1559–1570.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang L and Li S: Lactic acid promotes
macrophage polarization through MCT-HIF1α signaling in gastric
cancer. Exp Cell Res. 388:1118462020. View Article : Google Scholar
|
|
12
|
Kooshan Z, Cárdenas-Piedra L, Clements J
and Batra J: Glycolysis, the sweet appetite of the tumor
microenvironment. Cancer Lett. 600:2171562024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu X, Ouyang W, Chen H, Liu Z, Lai Z and
Yao H: Claudin-9 (CLDN9) promotes gastric cancer progression by
enhancing the glycolysis pathway and facilitating PD-L1 lactylation
to suppress CD8+ T cell anti-tumor immunity. Cancer Pathog Ther.
3:253–266. 2024. View Article : Google Scholar
|
|
14
|
Rohwer N, Welzel M, Daskalow K, Pfander D,
Wiedenmann B, Detjen K and Cramer T: Hypoxia-inducible factor 1α
mediates anoikis resistance via suppression of α5 integrin. Cancer
Res. 68:10113–10120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang Z, Li L, Tang Y, Xie D, Wu K, Wei W
and Xiao Q: CDK2 positively regulates aerobic glycolysis by
suppressing SIRT5 in gastric cancer. Cancer Sci. 109:2590–2598.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Shen H, Xie P, Zhang Z, Wang L, Yang
Y, Yu Z, Cheng Z and Zhou J: Role of long intergenic non-protein
coding RNA 00152 in pancreatic cancer glycolysis via the
manipulation of the microRNA-185-5p/Krüppel-like factor 7 axis. J
Cancer. 12:6330–6343. 2021. View Article : Google Scholar
|
|
17
|
Wu J, Chai H, Xu X, Yu J and Gu Y: Histone
methyltransferase SETD1A interacts with HIF1α to enhance glycolysis
and promote cancer progression in gastric cancer. Mol Oncol.
14:1397–1409. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luo T, Du Y, Duan J, Liang C, Chen G,
Jiang K and Chen Y and Chen Y: Development and validation of a
scoring system based on 9 glycolysis-related genes for prognosis
prediction in gastric cancer. Technol Cancer Res Treat.
19:15330338209716702020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nam S and Lee Y: Genome-scale metabolic
model analysis of metabolic differences between Lauren diffuse and
intestinal subtypes in gastric cancer. Cancers (Basel).
14:23402022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu G, Li M, Wu J, Qin C, Tao Y and He H:
Circular RNA circ-NRIP1 sponges microRNA-138-5p to maintain
hypoxia-induced resistance to 5-fluorouracil through
HIF-1α-dependent glucose metabolism in gastric carcinoma. Cancer
Manag Res. 12:2789–2802. 2020. View Article : Google Scholar
|
|
21
|
Xu L, Liu J, An Y, Zhou L, Sun H, Xu Z,
Wang D, Liang Z, Xu C, Wang B and Li W: Glycolysis-related genes
predict prognosis and indicate immune microenvironment features in
gastric cancer. BMC Cancer. 24:9792024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zheng Y, Li P, Ma J, Yang C, Dai S and
Zhao C: Cancer-derived exosomal circ_0038138 enhances glycolysis,
growth, and metastasis of gastric adenocarcinoma via the
miR-198/EZH2 axis. Transl Oncol. 25:1014792022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cai ZR, Hu Y, Liao K, Li H, Chen DL and Ju
HQ: Circular RNAs: Emerging regulators of glucose metabolism in
cancer. Cancer Lett. 552:2159782023. View Article : Google Scholar
|
|
24
|
Chen B, Cai T, Huang C, Zang X, Sun L, Guo
S, Wang Q, Chen Z, Zhao Y, Han Z, et al: G6PD-NF-κB-HGF signal in
gastric cancer-associated mesenchymal stem cells promotes the
proliferation and metastasis of gastric cancer cells by
upregulating the expression of HK2. Front Oncol. 11:6487062021.
View Article : Google Scholar
|
|
25
|
Gao S, Song D, Liu Y, Yan H and Chen X:
Helicobacter pylori CagA protein attenuates 5-Fu sensitivity of
gastric cancer cells through upregulating cellular glucose
metabolism. Onco Targets Ther. 13:6339–6349. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng S, Li H, Li Y, Chen X, Shen J, Chen
M, Zhang C, Wu J and Sun Q: The emerging role of glycolysis and
immune evasion in gastric cancer. Cancer Cell Int. 23:3172023.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan P, Li YH, Tang ZJ, Shu X and Liu X:
High monocarboxylate transporter 4 protein expression in stromal
cells predicts adverse survival in gastric cancer. Asian Pac J
Cancer Prev. 15:8923–8929. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang L, Chu W, Li M, Xu P, Wang M, Peng M,
Wang K and Zhang L: Radiomics in gastric cancer: First clinical
investigation to predict lymph vascular invasion and survival
outcome using 18F-FDG PET/CT images. Front Oncol. 12:8360982022.
View Article : Google Scholar
|
|
29
|
Ruzzo A, Graziano F, Bagaloni I, Di
Bartolomeo M, Prisciandaro M, Aprile G, Ongaro E, Vincenzi B,
Perrone G, Santini D, et al: Glycolytic competence in gastric
adenocarcinomas negatively impacts survival outcomes of patients
treated with salvage paclitaxel-ramucirumab. Gastric Cancer.
23:1064–1074. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu H, Ba Z, Liu C and Yu X: Long noncoding
RNA DLEU1 promotes proliferation and glycolysis of gastric cancer
cells via APOC1 upregulation by recruiting SMYD2 to induce
trimethylation of H3K4 modification. Transl Oncol. 36:1017312023.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bin YL, Hu HS, Tian F, Wen ZH, Yang MF, Wu
BH, Wang LS, Yao J and Li DF: Metabolic reprogramming in gastric
cancer: Trojan horse effect. Front Oncol. 11:7452092022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dai T, Zhang X, Zhou X, Hu X, Huang X,
Xing F, Tian H and Li Y: Long non-coding RNA VAL facilitates PKM2
enzymatic activity to promote glycolysis and malignancy of gastric
cancer. Clin Transl Med. 12:e10882022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Wu M, Cao J, Zhu Y, Ma Y, Pu Y, Huo
X and Wang J: Identification and verification of a
glycolysis-related gene signature for gastric cancer. Ann Transl
Med. 10:10102022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tao L, Yu H, Liang R, Jia R, Wang J, Jiang
K and Wang Z: Rev-erbα inhibits proliferation by reducing
glycolytic flux and pentose phosphate pathway in human gastric
cancer cells. Oncogenesis. 8:572019. View Article : Google Scholar
|
|
35
|
Pan C, Liu Q and Wu X:
HIF1α/miR-520a-3p/AKT1/mTOR feedback promotes the proliferation and
glycolysis of gastric cancer cells. Cancer Manag Res.
11:10145–10156. 2019. View Article : Google Scholar
|
|
36
|
Hur H, Xuan Y, Kim YB, Lee G, Shim W, Yun
J, Ham IH and Han SU: Expression of pyruvate dehydrogenase kinase-1
in gastric cancer as a potential therapeutic target. Int J Oncol.
42:44–54. 2013. View Article : Google Scholar :
|
|
37
|
Hao LS, Liu Q, Tian C, Zhang DX, Wang B,
Zhou DX, Li ZP and Yuan ZX: Correlation and expression analysis of
hypoxia-inducible factor 1α, glucose transporter 1 and lactate
dehydrogenase 5 in human gastric cancer. Oncol Lett. 18:1431–1441.
2019.PubMed/NCBI
|
|
38
|
Xiao Y, Liu X, Xie K, Luo J, Zhang Y,
Huang X, Luo J and Tan S: Mitochondrial dysfunction induced by
HIF-1α under hypoxia contributes to the development of gastric
mucosal lesions. Clin Transl Med. 14:e16532024. View Article : Google Scholar
|
|
39
|
Ma YT, Xing XF, Dong B, Cheng XJ, Guo T,
Du H, Wen XZ and Ji JF: Higher autocrine motility
factor/glucose-6-phosphate isomerase expression is associated with
tumorigenesis and poorer prognosis in gastric cancer. Cancer Manag
Res. 10:4969–4980. 2018. View Article : Google Scholar :
|
|
40
|
Jiang J, Jiang Y, Zhang YG, Zhang T, Li
JH, Huang DL, Hou J, Tian MY, Sun L, Su XM, et al: The effects of
hypoxia on mitochondrial function and metabolism in gastric cancer
cells. Transl Cancer Res. 10:817–826. 2021. View Article : Google Scholar
|
|
41
|
Guo T, Bai YH, Cheng XJ, Han HB, Du H, Hu
Y, Jia SQ, Xing XF and Ji JF: Insulin gene enhancer protein 1
mediates glycolysis and tumorigenesis of gastric cancer through
regulating glucose transporter 4. Cancer Commun (Lond). 41:258–272.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lu J, Chen M, Gao S, Yuan J, Zhu Z and Zou
X: LY294002 inhibits the Warburg effect in gastric cancer cells by
downregulating pyruvate kinase M2. Oncol Lett. 15:4358–4364.
2018.PubMed/NCBI
|
|
43
|
Wang L, Mao X, Yu X, Su J, Li Z, Chen Z,
Ren Y, Huang H, Wang W, Zhao C and Hu Y: FPR3 reprograms glycolytic
metabolism and stemness in gastric cancer via calcium-NFATc1
pathway. Cancer Lett. 593:2168412024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Qiao H, Wang YF, Yuan WZ, Zhu BD, Jiang L
and Guan QL: Silencing of ENO1 by shRNA inhibits the proliferation
of gastric cancer cells. Technol Cancer Res Treat.
17:15330338187844112018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang MQ, Chen YR, Xu HW, Zhan JR, Suo DQ,
Wang JJ, Ma YZ, Guan XY, Li Y and Zhu SL: HKDC1 upregulation
promotes glycolysis and disease progression, and confers
chemoresistance onto gastric cancer. Cancer Sci. 114:1365–1377.
2023. View Article : Google Scholar :
|
|
46
|
Wang H, Zhou R, Sun L, Xia J, Yang X, Pan
C, Huang N, Shi M, Bin J, Liao Y and Liao W: TOP1MT deficiency
promotes gastric cancer invasion and migration via the enhancements
of LDHA expression and aerobic glycolysis. Endocr Relat Cancer.
24:565–578. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang B, Wu J, Cai Y, Luo B, Wang M and Gu
Y: AAED1 modulates proliferation and glycolysis in gastric cancer.
Oncol Rep. 40:1156–1164. 2018.PubMed/NCBI
|
|
48
|
Wang Y, Wang S, Yang Q, Li J, Yu F and
Zhao E: Norepinephrine enhances aerobic glycolysis and may act as a
predictive factor for immunotherapy in gastric cancer. J Immunol
Res. 2021:55806722021.PubMed/NCBI
|
|
49
|
Wang J, Huang Q, Hu X, Zhang S, Jiang Y,
Yao G, Hu K, Xu X, Liang B, Wu Q, et al: Disrupting circadian
rhythm via the PER1-HK2 axis reverses trastuzumab resistance in
gastric cancer. Cancer Res. 82:1503–1517. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu J, Pan C, Guo L, Wu M, Guo J, Peng S,
Wu Q and Zuo Q: A new mechanism of trastuzumab resistance in
gastric cancer: MACC1 promotes the Warburg effect via activation of
the PI3K/AKT signaling pathway. J Hematol Oncol. 9:762016.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Luo B, Wang M, Hou N, Hu X, Jia G, Qin X,
Zuo X, Liu Y, Luo K, Song W, et al: ATP-dependent Lon protease
contributes to Helicobacter pylori-induced gastric carcinogenesis.
Neoplasia. 18:242–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen L, Guo L, Sun Z, Yang G, Guo J, Chen
K, Xiao R, Yang X and Sheng L: Monoamine oxidase A is a major
mediator of mitochondrial homeostasis and glycolysis in gastric
cancer progression. Cancer Manag Res. 12:8023–8035. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu YD, Yu L, Ying L, Balic J, Gao H, Deng
NT, West A, Yan F, Ji CB, Gough D, et al: Toll-like receptor 2
regulates metabolic reprogramming in gastric cancer via superoxide
dismutase 2. Int J Cancer. 144:3056–3069. 2019. View Article : Google Scholar :
|
|
54
|
Yang Y, Chen Z, Zhou L, Wu G, Ma X, Zheng
Y, Liu M, Wang Y, Ji R, Guo Q and Zhou Y: In silico development and
validation of a novel glucose and lipid metabolism-related gene
signature in gastric cancer. Transl Cancer Res. 11:1977–1993. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sun L, Wang Q, Chen B, Zhao Y, Shen B,
Wang H, Xu J, Zhu M, Zhao X, Xu C, et al: Gastric cancer
mesenchymal stem cells derived IL-8 induces PD-L1 expression in
gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death
Dis. 9:9282018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xian SL, Cao W, Zhang XD and Lu YF:
Inhibitory effects of 3-bromopyruvate on human gastric cancer
implant tumors in nude mice. Asian Pac J Cancer Prev. 15:3175–3178.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yao X, He Z, Qin C, Deng X, Bai L, Li G
and Shi J: SLC2A3 promotes macrophage infiltration by glycolysis
reprogramming in gastric cancer. Cancer Cell Int. 20:5032020.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang Y, Zhang J, Shi H, Wang M, Yu D, Fu
M, Qian Y, Zhang X, Ji R, Wang S, et al: M2 tumor-associated
macrophages-derived exosomal MALAT1 promotes glycolysis and gastric
cancer progression. Adv Sci (Weinh). 11:e23092982024. View Article : Google Scholar :
|
|
59
|
Yang DL, Dong LF, Qiu YB and Luo GY: An
oncogenic lncRNA, GLCC1, promotes tumorigenesis in gastric
carcinoma by enhancing the c-Myc/IGF2BP1 interaction. Neoplasma.
68:1052–1062. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jiang SF and Li RR: hsa_circ_0067514
suppresses gastric cancer progression and glycolysis via
miR-654-3p/LATS2 axis. Neoplasma. 69:1079–1091. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xie R, Liu L, Lu X, He C, Yao H and Li G:
N6-methyladenosine modification of OIP5-AS1 promotes glycolysis,
tumorigenesis, and metastasis of gastric cancer by inhibiting
Trim21-mediated hnRNPA1 ubiquitination and degradation. Gastric
Cancer. 27:49–71. 2024. View Article : Google Scholar :
|
|
62
|
Yuan W, Wu S, Guo J and Chen Z, Ge J, Yang
P, Hu B and Chen Z: Silencing of TKTL1 by siRNA inhibits
proliferation of human gastric cancer cells in vitro and in vivo.
Cancer Biol Ther. 9:710–716. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu Z, Yu M, Fei B, Fang X, Ma T and Wang
D: miR-21-5p targets PDHA1 to regulate glycolysis and cancer
progression in gastric cancer. Oncol Rep. 40:2955–2963.
2018.PubMed/NCBI
|
|
64
|
Liu L, Wang Y, Bai R, Yang K and Tian Z:
miR-186 inhibited aerobic glycolysis in gastric cancer via HIF-1α
regulation. Oncogenesis. 5:e2242016. View Article : Google Scholar
|
|
65
|
Cao N, Li M, Han J, Wang Y and Wang X:
rs61991156 in miR-379 is associated with low capability of
glycolysis of gastric cancer by enhanced regulation of PKM2. Cancer
Cell Int. 18:922018. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zhang Y, Huo W, Sun L, Wu J, Zhang C, Wang
H, Wang B, Wei J, Qu C, Cao H and Jiang X: Targeting miR-148b-5p
inhibits immunity microenvironment and gastric cancer progression.
Front Immunol. 12:5904472021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen L, Chi K, Xiang H and Yang Y:
Circ_0032821 facilitates gastric cancer cell proliferation,
migration, invasion and glycolysis by regulating miR-1236-3p/HMGB1
axis. Cancer Manag Res. 12:9965–9976. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhao X, Tian Z and Liu L: circATP2B1
promotes aerobic glycolysis in gastric cancer cells through
regulation of the miR-326 gene cluster. Front Oncol. 11:6286242021.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Deng C, Huo M, Chu H, Zhuang X, Deng G, Li
W, Wei H, Zeng L, He Y, Liu H, et al: Exosome circATP8A1 induces
macrophage M2 polarization by regulating the miR-1-3p/STAT6 axis to
promote gastric cancer progression. Mol Cancer. 23:492024.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gu J, Huang C, Hu X, Xia J, Shao W and Lin
D: Nuclear magnetic resonance-based tissue metabolomic analysis
clarifies molecular mechanisms of gastric carcinogenesis. Cancer
Sci. 111:3195–3209. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Song H, Wang L, Liu HL, Wu XB, Wang HS,
Liu ZH, Li Y, Diao DC, Chen HL and Peng JS: Tissue metabolomic
fingerprinting reveals metabolic disorders associated with human
gastric cancer morbidity. Oncol Rep. 26:431–438. 2011.PubMed/NCBI
|
|
72
|
Wang H, Zhang H, Deng P, Liu C, Li D, Jie
H, Zhang H, Zhou Z and Zhao YL: Tissue metabolic profiling of human
gastric cancer assessed by 1H NMR. BMC Cancer. 16:3712016.
View Article : Google Scholar
|
|
73
|
Yuan Q, Deng D, Pan C, Ren J, Wei T, Wu Z,
Zhang B, Li S, Yin P and Shang D: Integration of transcriptomics,
proteomics, and metabolomics data to reveal HER2-associated
metabolic heterogeneity in gastric cancer with response to
immunotherapy and neoadjuvant chemotherapy. Front Immunol.
13:9511372022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Gu J, Hu X, Shao W, Ji T, Yang W, Zhuo H,
Jin Z, Huang H, Chen J, Huang C and Lin D: Metabolomic analysis
reveals altered metabolic pathways in a rat model of gastric
carcinogenesis. Oncotarget. 7:60053–60073. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang TA, Xian SL, Guo XY, Zhang XD and Lu
YF: Combined 18F-FDG PET/CT imaging and a gastric orthotopic
xenograft model in nude mice are used to evaluate the efficacy of
glycolysis-targeted therapy. Oncol Rep. 39:271–279. 2018.
|
|
76
|
Zeng J, Li M, Dai K, Zuo B, Guo J and Zang
L: A novel glycolysis-related long noncoding RNA signature for
predicting overall survival in gastric cancer. Pathol Oncol Res.
28:16106432022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang Y, Lin S, Chen Y, Yang F and Liu S:
LDH-A promotes epithelial-mesenchymal transition by upregulating
ZEB2 in intestinal-type gastric cancer. Onco Targets Ther.
11:2363–2373. 2018. View Article : Google Scholar
|
|
78
|
Xu M, Zhang Y, Zhao K, Jiang H, Wang G, Wu
Y, Wang Y, Liu N and Su X: Prediction of pathological response to
neoadjuvant immunochemotherapy with baseline and post-treatment
18F-FDG PET imaging biomarkers in patients with locally advanced
gastric cancer. BMC Cancer. 25:3782025. View Article : Google Scholar
|
|
79
|
Eskuri M, Kemi N and Kauppila JH:
Monocarboxylate transporters 1 and 4 and MTCO1 in gastric cancer.
Cancers (Basel). 13:21422021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
De Oliveira T, Goldhardt T, Edelmann M,
Rogge T, Rauch K, Kyuchukov ND, Menck K, Bleckmann A, Kalucka J,
Khan S, et al: Effects of the novel PFKFB3 inhibitor KAN0438757 on
colorectal cancer cells and its systemic toxicity evaluation in
vivo. Cancers (Basel). 13:10112021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Mi Y, Li Q, Liu B, Wang D, Liu Z, Wang T,
Wang Y, Zang Y, Zhou Y, Wen Y and Ding Y: Ubiquitous mitochondrial
creatine kinase promotes the progression of gastric cancer through
a JNK-MAPK/JUN/HK2 axis regulated glycolysis. Gastric Cancer.
26:69–81. 2023. View Article : Google Scholar :
|
|
82
|
Yang H, Li Y and Hu B: Potential role of
mitochondria in gastric cancer detection: Fission and glycolysis.
Oncol Lett. 21:4392021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Takizawa K, Muramatsu K, Maruyama K,
Urakami K, Sugino T, Kusuhara M, Yamaguchi K, Ono H and Kitagawa Y:
Metabolic profiling of human gastric cancer cells treated with
salazosulfapyridine. Technol Cancer Res Treat.
19:15330338209286212020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu C, Jin Y and Fan Z: The mechanism of
Warburg effect-induced chemoresistance in cancer. Front Oncol.
11:6980232021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhao Y, Liu Y, Lin L, Huang Q, He W, Zhang
S, Dong S, Wen Z, Rao J, Liao WJ and Shi M: The lncRNA MACC1-AS1
promotes gastric cancer cell metabolic plasticity via AMPK/Lin28
mediated mRNA stability of MACC1. Mol Cancer. 17:692018. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sun L and Yao Y: Mesenchymal stem/stromal
cells-A principal element for tumour microenvironment
heterogeneity. Front Immunol. 14:12743792023. View Article : Google Scholar
|
|
87
|
Huang C, Chen B, Wang X, Xu J, Sun L, Wang
D, Zhao Y, Zhou C, Gao Q, Wang Q, et al: Gastric cancer mesenchymal
stem cells via the CXCR2/HK2/PD-L1 pathway mediate
immunosuppression. Gastric Cancer. 26:691–707. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Szatanek R, Weglarczyk K, Stec M, Baran J,
Parlinska-Wojtan M, Siedlar M and Baj-Krzyworzeka M: Autologous
tumor-derived microvesicles influence gene expression profiles and
enhance protumorigenic chemotactic potential, signal transduction
and cellular respiration in gastric cancer cells. Int J Oncol.
56:359–367. 2020.
|
|
89
|
Xu Y, Ren Z, Zeng F, Yang H and Hu C:
Cancer-associated fibroblast-derived WNT5A promotes cell
proliferation, metastasis, stemness and glycolysis in gastric
cancer via regulating HK2. World J Surg Oncol. 22:1932024.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Shi W, Cassmann TJ, Bhagwate AV, Hitosugi
T and Ip WKE: Lactic acid induces transcriptional repression of
macrophage inflammatory response via histone acetylation. Cell Rep.
43:1137462024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gottfried E, Kunz-Schughart LA, Ebner S,
Mueller-Klieser W, Hoves S, Andreesen R, Mackensen A and Kreutz M:
Tumor-derived lactic acid modulates dendritic cell activation and
antigen expression. Blood. 107:2013–2021. 2006. View Article : Google Scholar
|
|
92
|
Yang H, Yang S, He J, Li W, Zhang A, Li N,
Zhou G and Sun B: Glucose transporter 3 (GLUT3) promotes
lactylation modifications by regulating lactate dehydrogenase A
(LDHA) in gastric cancer. Cancer Cell Int. 23:3032023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Jia L, Zhang D, Zeng X, Wu L, Tian X and
Xing N: Targeting RNA N6-methyladenosine modification-A novel
therapeutic target for HER2-positive gastric cancer. Front Oncol.
14:13874442024. View Article : Google Scholar
|
|
94
|
Liu X, Yang Z, Chen Z, Chen R, Zhao D,
Zhou Y and Qiao L: Effects of the suppression of lactate
dehydrogenase A on the growth and invasion of human gastric cancer
cells. Oncol Rep. 33:157–162. 2015. View Article : Google Scholar
|
|
95
|
Wang X, Zhang J, Wu Y, Zhang Y, Zhang S,
Li A, Wang J and Wang Z: RORα inhibits gastric cancer proliferation
through attenuating G6PD and PFKFB3 induced glycolytic activity.
Cancer Cell Int. 24:122024. View Article : Google Scholar
|
|
96
|
Shiroki T, Yokoyama M, Tanuma N, Maejima
R, Tamai K, Yamaguchi K, Oikawa T, Noguchi T, Miura K, Fujiya T, et
al: Enhanced expression of the M2 isoform of pyruvate kinase is
involved in gastric cancer development by regulating
cancer-specific metabolism. Cancer Sci. 108:931–940. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Zhao R, Cao B, Li H, Li T, Xu X, Cui H,
Deng H and Wei B: Glucose starvation suppresses gastric cancer
through targeting miR-216a-5p/Farnesyl-Diphosphate
Farnesyltransferase 1 axis. Cancer Cell Int. 21:7042021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhang B, Ohuchida K, Tsutsumi C, Shimada
Y, Mochida Y, Oyama K, Iwamoto C, Sheng N, Fei S, Shindo K, et al:
Dynamic glycolytic reprogramming effects on dendritic cells in
pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res.
43:2712024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Liu J, Liu H, Zeng Q, Xu P, Liu M and Yang
N: Circular RNA circ-MAT2B facilitates glycolysis and growth of
gastric cancer through regulating the miR-515-5p/HIF-1α axis.
Cancer Cell Int. 20:1712020. View Article : Google Scholar
|
|
100
|
Cao K, Li J, Chen J, Qian L, Wang A, Chen
X, Xiong W, Tang J, Tang S, Chen Y, et al: microRNA-33a-5p
increases radiosensitivity by inhibiting glycolysis in melanoma.
Oncotarget. 8:83660–83672. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xia R, Tang H, Shen J, Xu S, Liang Y,
Zhang Y, Gong X, Min Y, Zhang D, Tao C, et al: Prognostic value of
a novel glycolysis-related gene expression signature for
gastrointestinal cancer in the Asian population. Cancer Cell Int.
21:1542021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wang N, Qiao H, Hao J, Deng C, Zhou N,
Yang L, Zeng M and Guan Q: RNA-binding protein ENO1 promotes the
tumor progression of gastric cancer by binding to and regulating
gastric cancer-related genes. J Gastrointest Oncol. 14:585–598.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wu G, Zhang A, Yang Y and Wu D:
Circ-RNF111 aggravates the malignancy of gastric cancer through
miR-876-3p-dependent regulation of KLF12. World J Surg Oncol.
19:2592021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lee JY, Lee I, Chang WJ, Ahn SM, Lim SH,
Kim HS, Yoo KH, Jung KS, Song HN, Cho JH, et al: MCT4 as a
potential therapeutic target for metastatic gastric cancer with
peritoneal carcinomatosis. Oncotarget. 7:43492–43503. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Liang J, Liu Q, Xia L, Lin J, Oyang L, Tan
S, Peng Q, Jiang X, Xu X, Wu N, et al: Rac1 promotes the
reprogramming of glucose metabolism and the growth of colon cancer
cells through upregulating SOX9. Cancer Sci. 114:822–836. 2023.
View Article : Google Scholar :
|
|
106
|
Wainberg ZA, Kang YK, Lee KW, Qin S,
Yamaguchi K, Kim IH, Saeed A, Oh SC, Li J, Turk HM, et al:
Bemarituzumab as first-line treatment for locally advanced or
metastatic gastric/gastroesophageal junction adenocarcinoma: Final
analysis of the randomized phase 2 FIGHT trial. Gastric Cancer.
27:558–570. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Zhou C, Qiao C, Ji J, Xi W, Jiang J, Guo
L, Wu J, Qi F, Cai Q, Damink SWMO and Zhang J: Plasma exosome
proteins ILK1 and CD14 correlated with organ-specific metastasis in
advanced gastric cancer patients. Cancers (Basel). 15:39862023.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Colegio OR, Chu NQ, Szabo AL, Chu T,
Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC,
Phillips GM, et al: Functional polarization of tumour-associated
macrophages by tumour-derived lactic acid. Nature. 513:559–563.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Huang H, Langenkamp E, Georganaki M,
Loskog A, Fuchs PF, Dieterich LC, Kreuger J and Dimberg A: VEGF
suppresses T-lymphocyte infiltration in the tumor microenvironment
through inhibition of NF-κB-induced endothelial activation. FASEB
J. 29:227–238. 2015. View Article : Google Scholar
|
|
110
|
Levental KR, Yu H, Kass L, Lakins JN,
Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et
al: Matrix crosslinking forces tumor progression by enhancing
integrin signaling. Cell. 139:891–906. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Dupont S, Morsut L, Aragona M, Enzo E,
Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M,
Bicciato S, et al: Role of YAP/TAZ in mechanotransduction. Nature.
474:179–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Calvo F, Ege N, Grande-Garcia A, Hooper S,
Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary
E, Charras G and Sahai E: Mechanotransduction and YAP-dependent
matrix remodelling is required for the generation and maintenance
of cancer-associated fibroblasts. Nat Cell Biol. 15:637–646. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu QP, Luo Q, Deng B, Ju Y and Song GB:
Stiffer matrix accelerates migration of hepatocellular carcinoma
cells through enhanced aerobic glycolysis via the MAPK-YAP
signaling. Cancers (Basel). 12:4902020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Jiang H, Hegde S, Knolhoff BL, Zhu Y,
Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver
DT, et al: Targeting focal adhesion kinase renders pancreatic
cancers responsive to checkpoint immunotherapy. Nat Med.
22:851–860. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang TA, Zhang XD, Guo XY, Xian SL and Liu
YF: 3-Bromopyruvate and sodium citrate target glycolysis, suppress
survivin, and induce mitochondrial-mediated apoptosis in gastric
cancer cells and inhibit gastric orthotopic transplantation tumor
growth. Oncol Rep. 35:1287–1296. 2016. View Article : Google Scholar :
|
|
116
|
Burz C, Pop V, Silaghi C, Lupan L and
Samasca G: Prognosis and treatment of gastric cancer: A 2024
update. Cancers (Basel). 16:17082024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Dong J, Li W, Ma L, Yang J, Gan D, Xue H,
Pu L, Zhang L, Zhang K, Jia Y and Ma Q: Advances in glycolysis
research in gastric cancer: Molecular mechanisms, regulatory
networks, and therapeutic potential. Front Oncol. 15:16786812025.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
MaruYama T, Miyazaki H, Lim YJ, Gu J,
Ishikawa M, Yoshida T, Chen W, Owada Y and Shibata H: Pyrolyzed
deketene curcumin controls regulatory T cell generation and gastric
cancer metabolism cooperate with 2-deoxy-D-glucose. Front Immunol.
14:10497132023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sun Q, Yuan M, Wang H, Zhang X, Zhang R,
Wang H, Chen X, Zhu M, Liu S and Wu J: PKM2 is the target of a
multi-herb-combined decoction during the inhibition of gastric
cancer progression. Front Oncol. 11:7671162021. View Article : Google Scholar :
|
|
120
|
Xu DH, Li Q, Hu H, Ni B, Liu X, Huang C,
Zhang ZZ and Zhao G: Transmembrane protein GRINA modulates aerobic
glycolysis and promotes tumor progression in gastric cancer. J Exp
Clin Cancer Res. 37:3082018. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhao X, Huang X, Dang C, Wang X, Qi Y and
Li H: Epstein-Barr virus miRNA BART6-5p regulates the TGF-β/SMAD4
pathway to induce glycolysis and enhance proliferation and
metastasis of gastric cancer cells. Oncol Res. 32:999–1009. 2024.
View Article : Google Scholar
|
|
122
|
Wu T, Yang Y, Zhang B, Zhang ZS, Zhou S,
Jia GZ, Liu SQ, He XL, He JX and Wang N: EDDM3A drives gastric
cancer progression by promoting HIF-1α-dependent aerobic
glycolysis. Oncogenesis. 11:32022. View Article : Google Scholar
|
|
123
|
Luo X, Peng Y, Fan X, Xie X, Jin Z and
Zhang X: The crosstalk and clinical implications of circRNAs and
glucose metabolism in gastrointestinal cancers. Cancers (Basel).
15:22292023. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Huang P, Zhu S, Liang X, Zhang Q, Luo X,
Liu C and Song L: Regulatory mechanisms of lncRNAs in cancer
glycolysis: Facts and perspectives. Cancer Manag Res. 13:5317–5336.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Chen F, Zhuang M, Zhong C, Peng J, Wang X,
Li J, Chen Z and Huang Y: Baicalein reverses hypoxia-induced 5-FU
resistance in gastric cancer AGS cells through suppression of
glycolysis and the PTEN/Akt/HIF-1α signaling pathway. Oncol Rep.
33:457–463. 2015. View Article : Google Scholar
|
|
126
|
Otto C, Kaemmerer U, Illert B, Muehling B,
Pfetzer N, Wittig R, Voelker HU, Thiede A and Coy JF: Growth of
human gastric cancer cells in nude mice is delayed by a ketogenic
diet supplemented with omega-3 fatty acids and medium-chain
triglycerides. BMC Cancer. 8:1222008. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Shen Y, Chen M, Huang S and Zou X:
Pantoprazole inhibits human gastric adenocarcinoma SGC-7901 cells
by downregulating pyruvate kinase M2 expression. Oncol Lett.
11:717–722. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Cao B, Deng H, Cui H, Zhao R, Li H, Wei B
and Chen L: Knockdown of PGM1 enhances anticancer effects of
orlistat in gastric cancer under glucose deprivation. Cancer Cell
Int. 21:4812021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Chong X, Madeti Y, Cai J, Li W, Cong L, Lu
J, Mo L, Liu H, He S, Yu C, et al: Recent developments in
immunotherapy for gastrointestinal tract cancers. J Hematol Oncol.
17:382024. View Article : Google Scholar
|
|
130
|
Huang D, Shin WK, De la Torre K, Lee HW,
Min S, Shin A, Lee JK and Kang D: Association between metabolic
syndrome and gastric cancer risk: Results from the health examinees
study. Gastric Cancer. 26:481–492. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Kwon MJ, Kang HS, Kim JH, Kim JH, Kim SH,
Kim NY, Nam ES, Min KW and Choi HG: Association between statin use
and gastric cancer: A nested case-control study using a national
health screening cohort in Korea. Pharmaceuticals (Basel).
14:12832021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
King RJ, Singh PK and Mehla K: The
cholesterol pathway: Impact on immunity and cancer. Trends Immunol.
43:78–92. 2022. View Article : Google Scholar
|
|
133
|
Wang IH, Huang TT, Chen JL, Chu LW, Ping
YH, Hsu KW, Huang KH, Fang WL, Lee HC, Chen CF, et al: Mevalonate
pathway enzyme HMGCS1 contributes to gastric cancer progression.
Cancers (Basel). 12:10882020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Janjigian YY, Ajani JA, Moehler M, Shen L,
Garrido M, Gallardo C, Wyrwicz L, Yamaguchi K, Cleary JM, Elimova
E, et al: First-line nivolumab plus chemotherapy for advanced
gastric, gastroesophageal junction, and esophageal adenocarcinoma:
3-year follow-up of the phase III CheckMate 649 trial. J Clin
Oncol. 42:2012–2020. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Shitara K, Fleitas T, Kawakami H,
Curigliano G, Narita Y, Wang F, Wardhani SO, Basade M, Rha SY, Wan
Zamaniah WI, et al: Pan-Asian adapted ESMO Clinical Practice
Guidelines for the diagnosis, treatment and follow-up of patients
with gastric cancer. ESMO Open. 9:1022262024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Rached L, Laparra A, Sakkal M, Danlos FX,
Barlesi F, Carbonnel F, De Martin E, Ducreux M, Even C, Le Pavec J,
et al: Toxicity of immunotherapy combinations with chemotherapy
across tumor indications: Current knowledge and practical
recommendations. Cancer Treat Rev. 127:1027512024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wu Z, Tan J, Zhuang Y, Zhong M, Xiong Y,
Ma J, Yang Y, Gao Z, Zhao J, Ye Z, et al: Identification of crucial
genes of pyrimidine metabolism as biomarkers for gastric cancer
prognosis. Cancer Cell Int. 21:6682021. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liu Y, Jiang Y, Xu L, Qu C, Zhang L, Xiao
X, Chen W, Li K, Liang Q and Wu H: circ-NRIP1 promotes glycolysis
and tumor progression by regulating the miR-186-5p/MYH9 axis in
gastric cancer. Cancer Manag Res. 12:5945–5956. 2020. View Article : Google Scholar
|
|
139
|
Kaira K, Higuchi T, Naruse I, Arisaka Y,
Tokue A, Altan B, Suda S, Mogi A, Shimizu K, Sunaga N, et al:
Metabolic activity by 18F-FDG-PET/CT is predictive of early
response after nivolumab in previously treated NSCLC. Eur J Nucl
Med Mol Imaging. 45:56–66. 2018. View Article : Google Scholar
|
|
140
|
Umeda Y, Morikawa M, Anzai M, Ameshima S,
Kadowaki M, Waseda Y, Shigemi H, Tsujikawa T, Kiyono Y, Okazawa H
and Ishizuka T: Predictive value of integrated 18F-FDG PET/MRI in
the early response to nivolumab in patients with previously treated
non-small cell lung cancer. J Immunother Cancer. 8:e0003492020.
View Article : Google Scholar
|
|
141
|
Tao X, Li N, Wu N, He J, Ying J, Gao S,
Wang S, Wang J, Wang Z, Ling Y, et al: The efficiency of 18F-FDG
PET-CT for predicting the major pathologic response to the
neoadjuvant PD-1 blockade in resectable non-small cell lung cancer.
Eur J Nucl Med Mol Imaging. 47:1209–1219. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Park JS, Lee N, Beom SH, Kim HS, Lee CK,
Rha SY, Chung HC, Yun M, Cho A and Jung M: The Prognostic value of
volume-based parameters using 18F-FDG PET/CT in gastric cancer
according to HER2 status. Gastric Cancer. 21:213–224. 2018.
View Article : Google Scholar
|
|
143
|
Kang YK, Chen LT, Ryu MH, Oh DY, Oh SC,
Chung HC, Lee KW, Omori T, Shitara K, Sakuramoto S, et al:
Nivolumab plus chemotherapy versus placebo plus chemotherapy in
patients with HER2-negative, untreated, unresectable advanced or
recurrent gastric or gastro-oesophageal junction cancer
(ATTRACTION-4): A randomised, multicentre, double-blind,
placebo-controlled, phase 3 trial. Lancet Oncol. 23:234–247. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Rha SY, Oh DY, Yañez P, Bai Y, Ryu MH, Lee
J, Rivera F, Alves GV, Garrido M, Shiu KK, et al: Pembrolizumab
plus chemotherapy versus placebo plus chemotherapy for
HER2-negative advanced gastric cancer (KEYNOTE-859): A multicentre,
randomised, double-blind, phase 3 trial. Lancet Oncol.
24:1181–1195. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Xu J, Jiang H, Pan Y, Gu K, Cang S, Han L,
Shu Y, Li J, Zhao J, Pan H, et al: Sintilimab plus chemotherapy for
unresectable gastric or gastroesophageal junction cancer: The
ORIENT-16 randomized clinical trial. JAMA. 330:2064–2074. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Halford S, Veal GJ, Wedge SR, Payne GS,
Bacon CM, Sloan P, Dragoni I, Heinzmann K, Potter S, Salisbury BM,
et al: A phase I dose-escalation study of AZD3965, an oral
monocarboxylate transporter 1 inhibitor, in patients with advanced
cancer. Clin Cancer Res. 29:1429–1439. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Buyse C, Joudiou N, Warscotte A,
Richiardone E, Mignion L, Corbet C and Gallez B: Evaluation of
syrosingopine, an MCT inhibitor, as potential anticancer drug in
human breast cancer. Metabolites. 12:5572022. View Article : Google Scholar
|
|
148
|
Gao S, Tu DN, Li H, Jiang JX, Cao X, You
JB and Zhou XQ: Pharmacological or genetic inhibition of LDHA
reverses tumor progression of pediatric osteosarcoma. Biomed
Pharmacother. 81:388–393. 2016. View Article : Google Scholar : PubMed/NCBI
|














