Introduction
The implication of constitutive and often
exacerbated enzymatic activity of casein kinase 2 (CK2) in the
maintenance of the malignant phenotype has been firmly established
by in vitro and in vivo preclinical experimentation
and epidemiological findings in several types of tumors (1,2).
Based on such knowledge, different groups are currently engaged in
the quest for highly potent and specific CK2 inhibitors, which may
overcome the limitations of first-generation anti-CK2 compounds,
such as 5,6-dichloro-1-(β-D-ribofuranosyl)-1H-benzimidazole (DRB),
4,5,6,7-tetrabromo-1H-benzimidazole,
4,5,6,7-tetrabromo-1H-benzotriazole (TBB) and
2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (3–8).
Such new-generation anti-CK2 compounds are ultimately represented
by CX-4945, a small-molecule adenosine triphosphate (ATP)
competitor currently in clinical cancer trials (9,10).
Focusing on the same tumor-specific CK2 signaling but aiming to
abrogate such events by binding to the phosphoaceptor domain on its
substrates, the CIGB-300 peptide was discovered by screening a
phage display library against a model CK2 substrate (11).
In different experimental settings and preclinical
cancer models, CIGB-300 was shown to inhibit the CK2-mediated
phosphorylation of the validated CK2 substrate B23/nucleophosmin,
leading to a fast cell death by apoptosis (12,13).
However, considering the high degree of conservation for the CK2
phosphoaceptor domain, a multitarget effect may be anticipated
(14). Such a multitarget effect
may better explain the diverse arrays of proteins and processes
that appear to be modulated by CIGB-300 and its already established
antiangiogenic effect (15,16).
Of note, CIGB-300 exerts a broad antiproliferative effect on cell
lines derived from breast, cervical, lung, colon and prostate
cancer, while a robust antitumor effect was also observed in
vivo in mouse models of cervical and lung cancer (13,17,18).
In the clinical setting, CIGB-300 was investigated in a phase I
clinical trial on high-grade squamous intraepithelial lesions
(HSILs), establishing its safety and tolerability by local
injection (19). More recently,
another phase I clinical study on patients with stage 1B2/II
cervical cancer allowed us to estimate the maximum tolerated dose
(MTD) and the pharmacokinetics/biodistribution profiles for
CIGB-300 following local administration (20). Moreover, a phase I study on lung
and haematological cancers further demonstrated that CIGB-300 may
also be administered by intravenous injection without significant
toxicity (unpublished data). Of note, the first evidence regarding
the antitumor effect of CIGB-300 in humans was recently collected
from case studies (21).
Despite these promising preclinical and clinical
findings using the CIGB-300 peptide, the clinical oncology setting
and cumulative knowledge on cancer biology suggest that drug
combinations are more likely to cope with tumor complexity compared
to single agents (22). Of note,
previous data from proteomics studies demonstrated that CIGB-300
modulates a group of proteins directly involved in anticancer drug
resistance (15). Therefore, in
this study, we evaluated the antiproliferative effect of CIGB-300
when combined with four different chemotherapeutics drugs in two
model cell lines derived from lung and cervical cancer. With the
aim to select the optimal combination in these two clinically
relevant tumor types, the peptide was combined in a pairwise manner
with anticancer drugs, such as antimitotic (paclitaxel), alkylating
(cisplatin), antitopoisomerase II (doxorubicin) or DNA/RNA
antimetabolite (5-fluorouracil) agents. Based on the estimation of
the combination index (CI) (23),
all the interactions were classified as synergistic, additive or
antagonistic and the direct effect on cell viability and
proliferation was analyzed. Finally, the potential benefit of one
selected drug combination over each monotherapy was corroborated
in vivo in a mouse model of cervical cancer.
Materials and methods
Cell lines and chemotherapeutic
agents
The NCI-H125 non-small-lung cancer cell line and the
SiHa cervix-derived squamous carcinoma cell line were originally
acquired from American Type Culture Collection (Manassas, VA, USA)
and cultured in RPMI or Dulbecco’s modified Eagle’s medium
supplemented with 10% fetal bovine serum, unless otherwise stated.
The clinical grade chemotherapeutic drugs paclitaxel, cisplatin,
doxorubicin and 5-fluorouracil (Drug Research and Development
Center, Havana, Cuba) were kindly provided by the Oncology Service
of the National Institute of Oncology and Radiobiology (Havana,
Cuba) or from the Hermanos Ameijeiras Hospital (Havana, Cuba).
CIGB-300
The CIGB-300 peptide was synthesized on solid phase,
purified by reverse-phase high-performance liquid chromatography to
>98% purity on acetonitrile/H2O-trifluoroacetic acid
gradient and confirmed by ion-spray mass spectrometry (Micromass,
Manchester, UK).
Drug combination assays
The 48-h sulforhodamine B (SRB)-based assay was
adopted from the National Cancer Institute to measure the cytotoxic
effect of each drug combination (24). Sulforhodamine B sodium salt was
purchased from Sigma-Aldrich (St. Louis, MO, USA). NCI-H125 or SiHa
cell suspensions (6×104 cells/ml) were seeded on 96-well
plates (Corning Costar; Sigma-Aldrich, ) and on the next day, a
curve of serial dilutions for each compound (1:2 or 1:10 on RPMI)
were added and incubated for 48 h. Following plate reading, the
data were analyzed by the CalcuSyn software, which is based on the
median effect equation (23,25).
The type of interaction was scored as synergistic, additive or
antagonistic, according to the obtained CI. The interaction maps
were represented by color-coded surfaces rendering a 3D visual
effect within a 2D plot. The maps were built by superimposing
filled contours (CI) and isoline plots (Fa, fraction affected),
partially scripted with Matlab® R2012a software
(7.14.0.0739; http://www.mathworks.com/).
Cytotoxicity by propidium iodide (PI)
exclusion
NCI-H125 or SiHa cells were seeded at
6×104 cells/ml on 24-well culture plates (Corning
Costar) and incubated for 24 h. On the next day, selected
concentrations of CIGB-300, paclitaxel, cisplatin, or their
combinations, were added to the plates and incubated for 0.5, 2, 5,
8, 24 and 36 h. The cells were subsequently washed with cold
phosphate-buffered saline (PBS) and stained for 10 min with 5 μg/ml
PI solution. Finally, the stained cells were analyzed on the FL2
channel using the Partec PAS III particle analyzing system flow
cytometer and their proprietary FloMax v2.4f software (Partec GmbH,
Münster, Germany).
Cell cycle analysis
The cell cultures were incubated with selected
concentrations of CIGB-300, paclitaxel, cisplatin, or their
combinations for 24 h. Subsequently, the cells were collected by
trypsinization, washed and fixed with ice-cold methanol/acetone
(4:1) at 4°C for 1 h. The cells were then stained by incubation for
20 min at 37°C with a solution of 100 mg/ml PI and 10 mg/ml of
DNase-free RNase. Finally, the stained cells were analyzed on the
abovementioned PAS III flow cytometer using the cell cycle analyzer
from the FloMax software. Prior to fitting the DNA distribution to
a diploid DNA content for cell cycle profiling, cellular debris and
doublets were properly excluded by gating out in FL3-A vs. FL3-W
two-parameter dot plots.
In vivo experiments
Scheme A
A total of 28, 8-week-old male nude mice (Harlan
Laboratories GmbH, Eystrup, Germany/National Atomic Energy
Commission, Buenos Aires, Argentina) were injected subcutaneously
in the right flank with 4×106 SiHa cells in 300 μl PBS.
When the tumors reached 30 mm3, the mice were randomly
assigned into 7 groups (n=4 per group) and were administered
placebo, CIGB-300 (days 1–5, 50 or 200 μg), cisplatin (days 1, 3
and 5, 1 or 4 mg/kg), or their combinations, by intraperitoneal
(cisplatin) or intratumoral (CIGB-300) routes.
Scheme B
A total of 35 8-week-old female nude mice (Harlan
Laboratories GmbH/National Atomic Energy Commission) were injected
with 3×106 SiHa cells as described for scheme A. When
the tumors reached 30 mm3, the mice were randomly
assigned into 7 groups (n=5 per group) and injected with placebo,
CIGB-300 (days 1–5, 100 or 200 μg), cisplatin (days 1, 3 and 5, 3
or 6 mg/kg), or their combinations, by the routes indicated in
scheme A. The tumors were measured every other day with a caliper
and their volumes were calculated as follows: volume = length ×
width2/2. Survival was daily registered during the
experimentation, unless the tumor volumes reached 2,000
mm3, in which case the animals were sacrificed due to
ethical considerations. The mice were maintained under
pathogen-free conditions and all the procedures were performed in
accordance with the recommendations for the proper use and care of
laboratory animals at the Center for Genetic Engineering and
Biotechnology (Havana, Cuba).
Statistical analysis
The tumor volumes among the different groups were
compared using one-way analysis of variance (ANOVA) and Tukey’s
post hoc test. The log-rank test was applied for the survival
analysis of the Kaplan-Meier curves. All the statistical analyses
were performed using GraphPad Prism version 4.00 for Windows
(GraphPad Software, San Diego CA, USA).
Results
Antiproliferative effect of CIGB-300 and
chemotherapeutic drugs as single agents
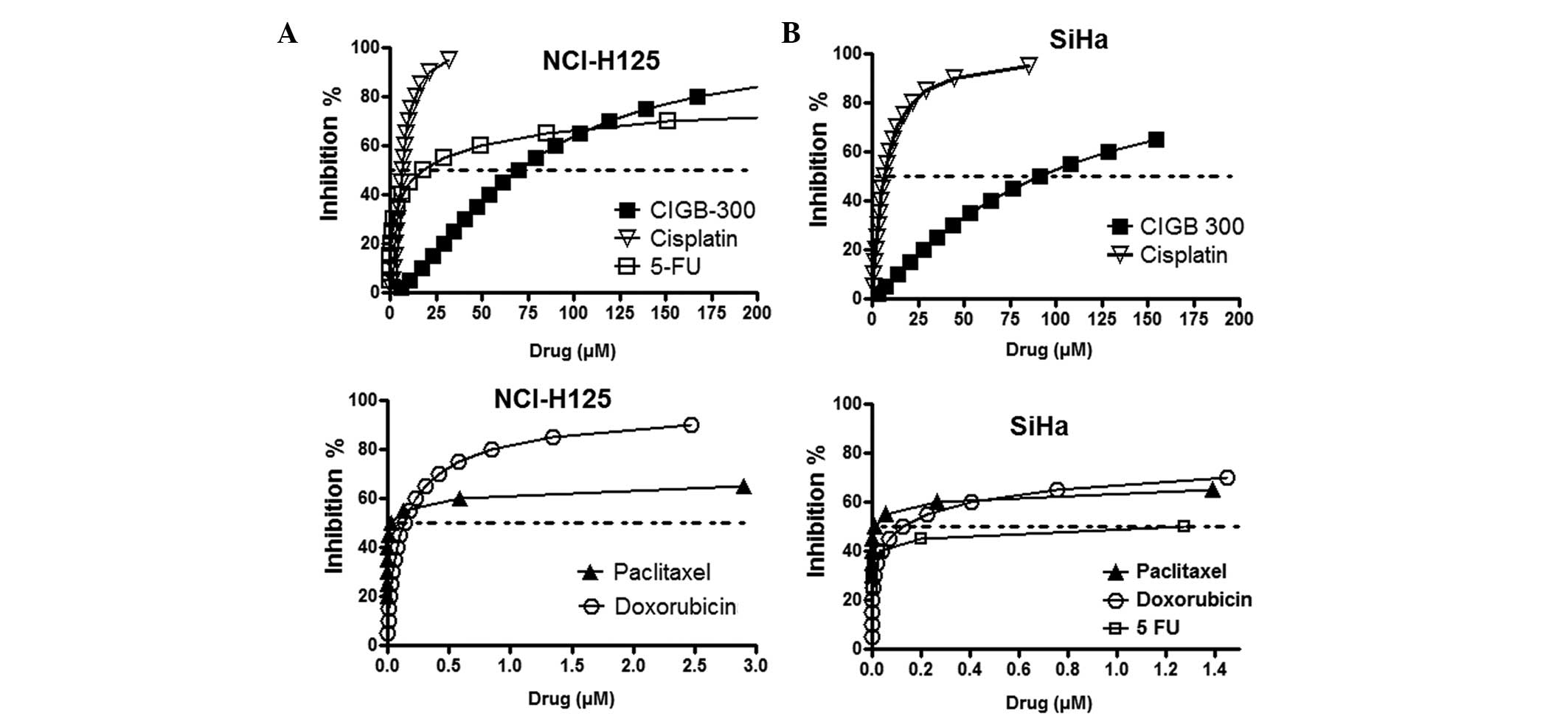
In order to select a suitable range of drug
concentrations for combination experiments, we first evaluated the
antiproliferative effect of CIGB-300 and four different
chemotherapeutics drugs used in the treatment of lung (paclitaxel
and cisplatin) and cervical cancer (paclitaxel, cisplatin,
doxorubicin and 5-fluorouracil) with the SRB-based assay (Fig. 1).
The fitting of each curve to the median effect
equation allowed us to estimate the half maximal inhibitory drug
concentration (IC50), also referred to as drug potency,
and the steepness or shape of the curve for each individual drug
(m) (Table I). As expected, due to
the diverse nature and mechanism of action of the tested compounds,
the results demonstrated that these drugs differ in both
parameters. While CIGB-300 exerts a major effect at >50 μM in
both cell lines, the majority of the chemotherapeutic drugs exert a
significant effect on cell proliferation at <5 μM. The flat
sigmoidal shape (m<1) of certain drugs, such as paclitaxel,
doxorubicin and 5-fluorouracil, prompted us to use serial 10-fold
dilutions in the combination experiments to cover an extensive
range of cytotoxic effects.
 | Table IDrug potency (IC50) and
steepness (m) of the dose-response curves obtained after fitting
the SRB-based assay data to the median effect equation using the
CalcuSyn software. |
Table I
Drug potency (IC50) and
steepness (m) of the dose-response curves obtained after fitting
the SRB-based assay data to the median effect equation using the
CalcuSyn software.
| | NCI-H125 cells | SiHa cells |
|---|
| |
|
|
|---|
| Compound | Class | IC50
(μM) | m | Drug range
(μM) | IC50
(μM) | m | Drug range
(μM) |
|---|
| CIGB-300 | CK2-targeted
therapy | 69.80 | 1.60 | 6.25–200 | 91.10 | 1.17 | 12.5–400 |
| Paclitaxel | Antimitotic | 0.03 | 0.13 | 0.00002–2 | 0.01 | 0.13 | 0.0002–2 |
| Cisplatin | Alkylating | 9.43 | 2.38 | 0.31–10 | 6.61 | 0.96 | 1.56–25 |
| Doxorubicin | Antitopoisomerase
II | 0.14 | 0.76 | 0.00025–25 | 0.13 | 0.35 | 0.00025–25 |
| 5-Fluorouracil | RNA/DNA
antimetabolite | 17.83 | 0.40 | 0.02–2000 | 1.28 | 0.11 | 0.02–200 |
Drug combination experiments
To determine the type of interaction (i.e.,
synergistic, additive or antagonistic) between the CIGB-300 peptide
and conventional chemotherapeutic drugs in the two tumor types, we
used the CalcuSyn software. A Latin square design was selected to
investigate such interactions across a range of different drug
concentrations, which included clinically achievable drug levels.
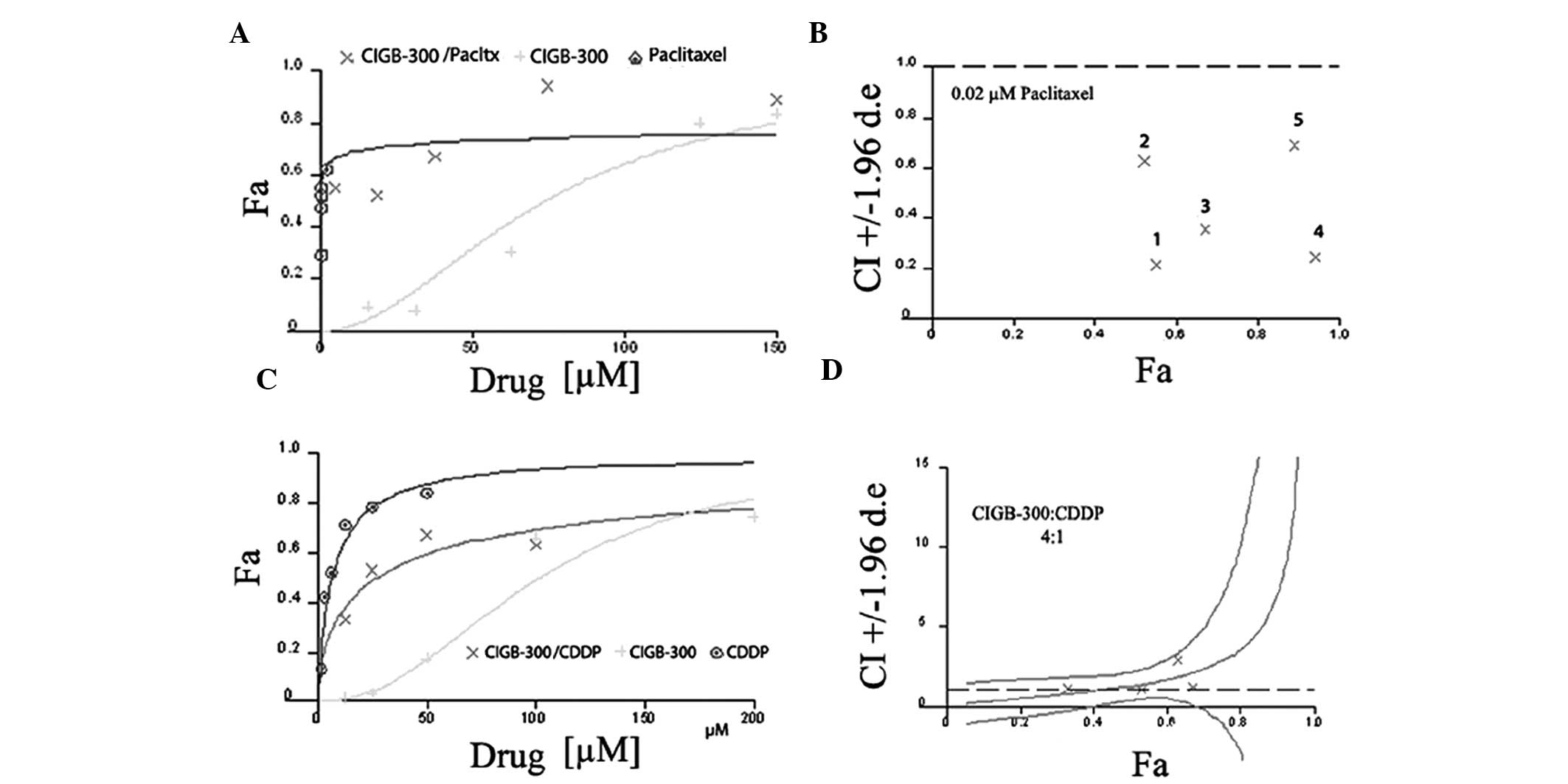
Representative examples of the graphs generated from the analysis
of two particular drug combinations, i.e., CIGB-300 plus paclitaxel
in NCI-H125 cells (non-constant ratio combinations) and CIGB-300
plus cisplatin in SiHa cells (constant-ratio combinations), are
shown in Fig. 2.
To visualize the results obtained from all 8
combination experiments (i.e., CIGB-300 pairwise combined with 4
drugs in 2 cell lines), an interaction surface was built by
representing in a color code the estimated CI for each particular
pair of drug combinations (i.e., 6×6 concentration matrix)
(Figs. 3 and 4). Moreover, in order to assess the
actual relevance of the observed interaction pattern, we also
represented the Fa by the combinations over this 2D graphical
display of interaction data.
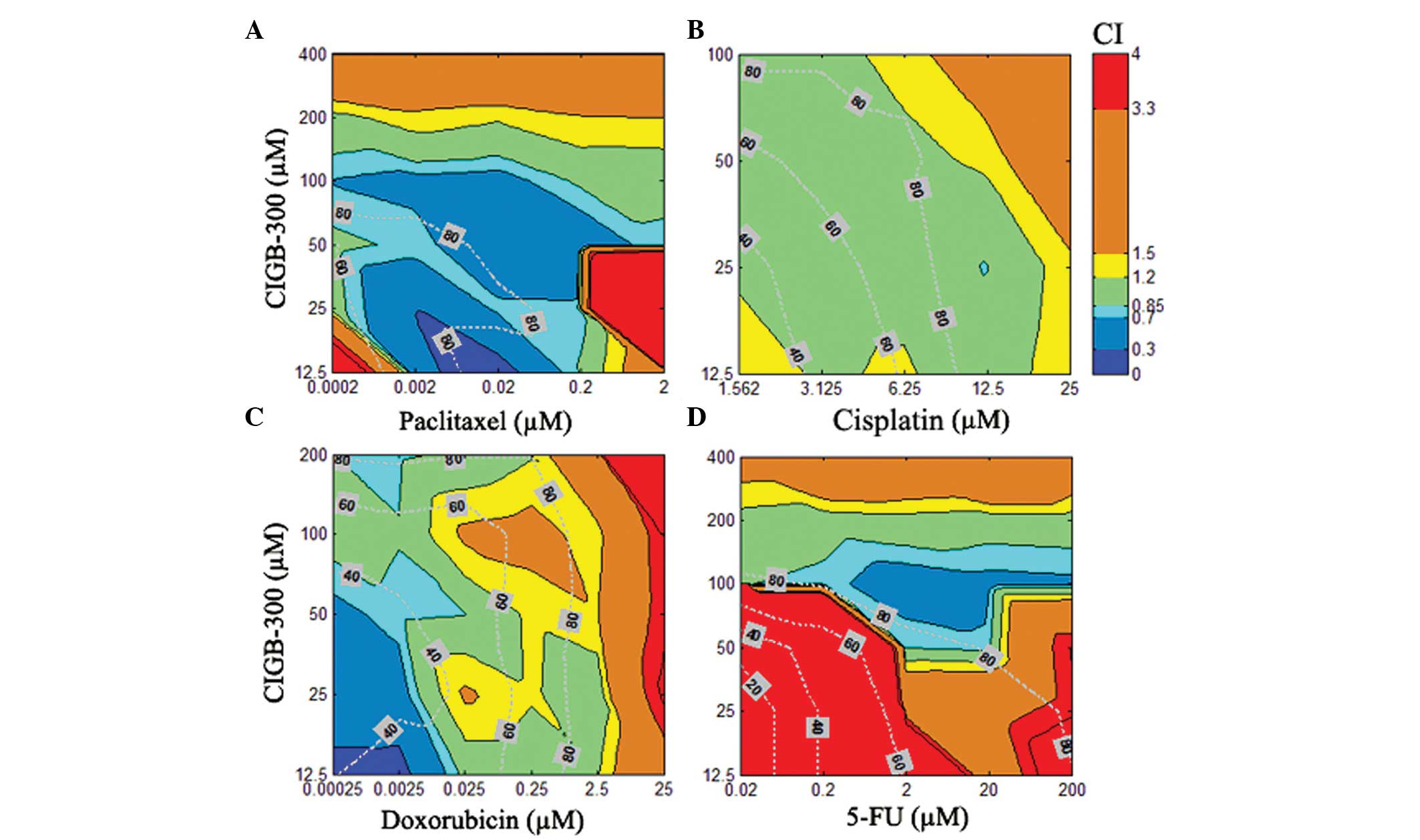
The results of the combination experiments in the
SiHa cervical cancer cell line demonstrated that CIGB-300 interacts
synergistically with paclitaxel in >50% of the interaction
surface, with effect levels or Fa>60% of the cell population,
while only small areas of antagonism were registered at extreme
concentrations (i.e., low for both drugs or very high for
paclitaxel) (Fig. 3A). A similar
but less promising interaction pattern was observed for the
combination of CIGB-300 plus doxorubicin, where regions of additive
or slight antagonism between the two drugs were found to be more
predominant at higher effect levels (>50%) when the
concentration of doxorubicin increased (Fig. 3C). An intermediary scenario was
observed for the combination of CIGB-300 plus cisplatin, with
>70% of the interaction surface exhibiting an additive pattern
even at high effect levels (40–85%) (Fig. 3B). The worse combination scenario
was clearly observed for the CIGB-300 plus 5-fluorouracil
combination, where a strong antagonism was observed in
approximately half of the interaction surface, with synergistic
interaction areas only seen at very high concentrations of both
drugs (Fig. 3D).
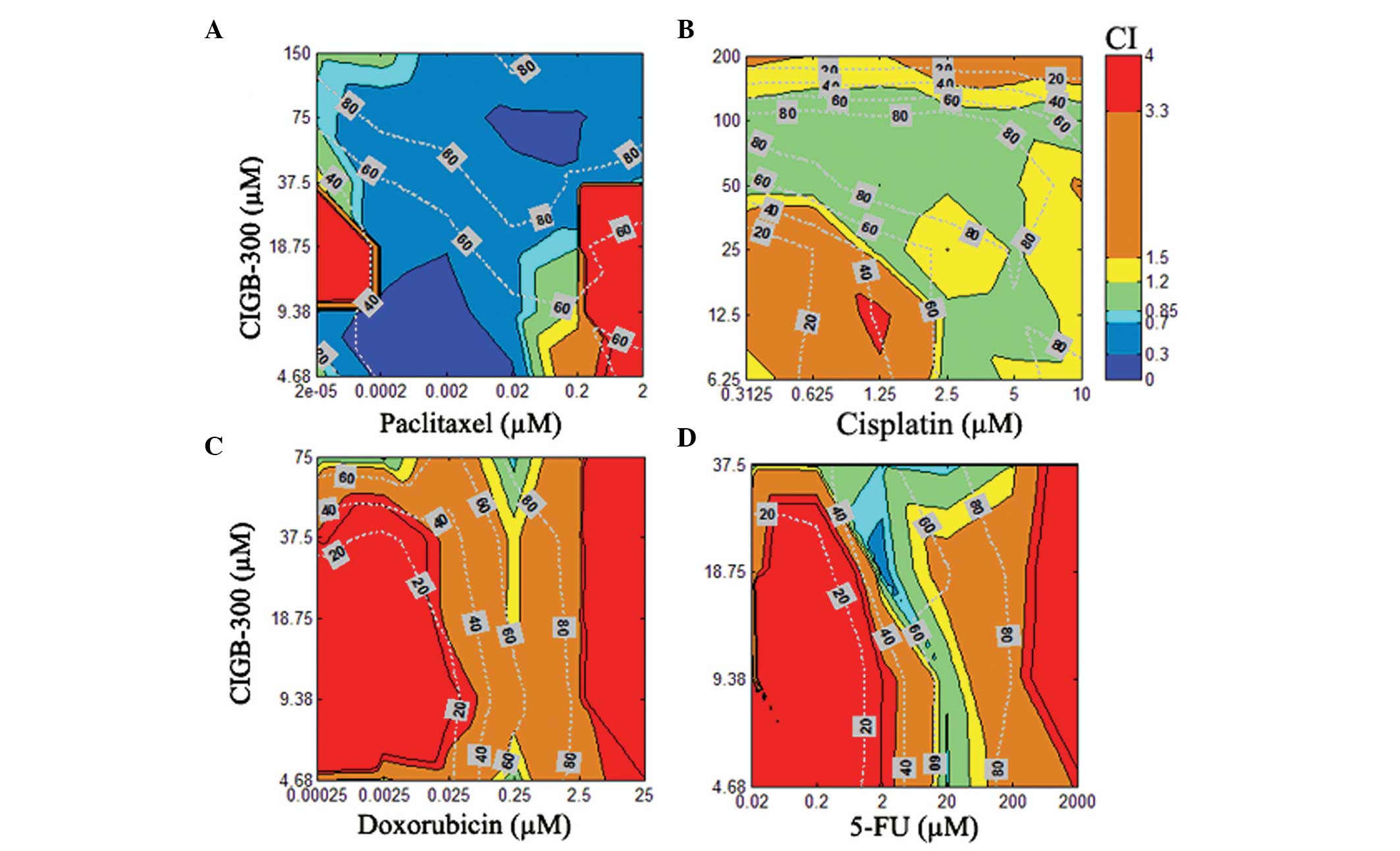
In the NCI-H125 lung cancer cell line, the
combination of CIGB-300 plus paclitaxel exhibited the best
synergistic profile, with >75% of the interaction surface
displaying a CI<0.7 and effect levels >50% (Fig. 4A). Of note, a similar interaction
pattern in lung and cervical cancer cells was observed for the
combination of CIGB-300 plus cisplatin, with significant areas of
the interaction surface exhibiting an additive pattern at effect
levels >60% (Fig. 4B). Finally,
a moderate (CI>1.20) to strong antagonism (CI>3.3) was
observed practically across the entire interaction surface when the
combinations of CIGB-300 plus doxorubicin and CIGB-300 plus
5-fluorouracil were evaluated in NCI-H125 cells (Fig. 4C and D).
Another important output derived from the analysis
of drug interaction data using CalcuSyn is the dose reduction index
(DRI). This parameter is inversely associated with CI and
represents the number of times each single drug may be reduced in a
combination setting without compromising the final therapeutic
effect (23). The estimated DRI
from the combination of CIGB-300 plus paclitaxel indicated that, in
a synergistic scenario like those exemplified in Table II, 5-fold less peptide (DRI=5.3)
is required to achieve an antiproliferative effect level of 94% in
NCI-H125 cells (Fa=0.94). Furthermore, in the same combination
setting, the concentration of paclitaxel may be reduced 3,250 times
without compromising the final antiproliferative effect in lung
cancer cells (Table II). A
significant DRI was also achieved in the combination of CIGB-300
plus cisplatin for the peptide (DRI=8.5) but not for cisplatin,
where only a small DRI was observed (DRI=1.2) (Table II). As expected, in the majority
of the combination experiments where a synergistic pattern was
observed, important DRIs were registered for all the drugs (data
not shown).
| Table IIParameters estimated using the
CalcuSyn software for two selected combinations of CIGB-300 plus
chemotherapy in NCI-H125 and SiHa cells. |
Table II
Parameters estimated using the
CalcuSyn software for two selected combinations of CIGB-300 plus
chemotherapy in NCI-H125 and SiHa cells.
| Combination | Parameters | | DRI |
|---|
|
|
| |
|
|---|
| Cell line | Drugs | (μM) | IC50
(μM) | CI | Fa | Interaction
type | Drug 1 | Drug 2 |
|---|
| NCI-H125 | CIGB-300 +
paclitaxel | 75+0.02 | 0.03 | 0.30 | 0.94 | Strong
synergism | 5.3 | 3250 |
| SiHa | CIGB-300 +
CDDP | 25+12.5 | 6.61 | 0.83 | 0.73 | Moderate
synergism | 8.5 | 1.2 |
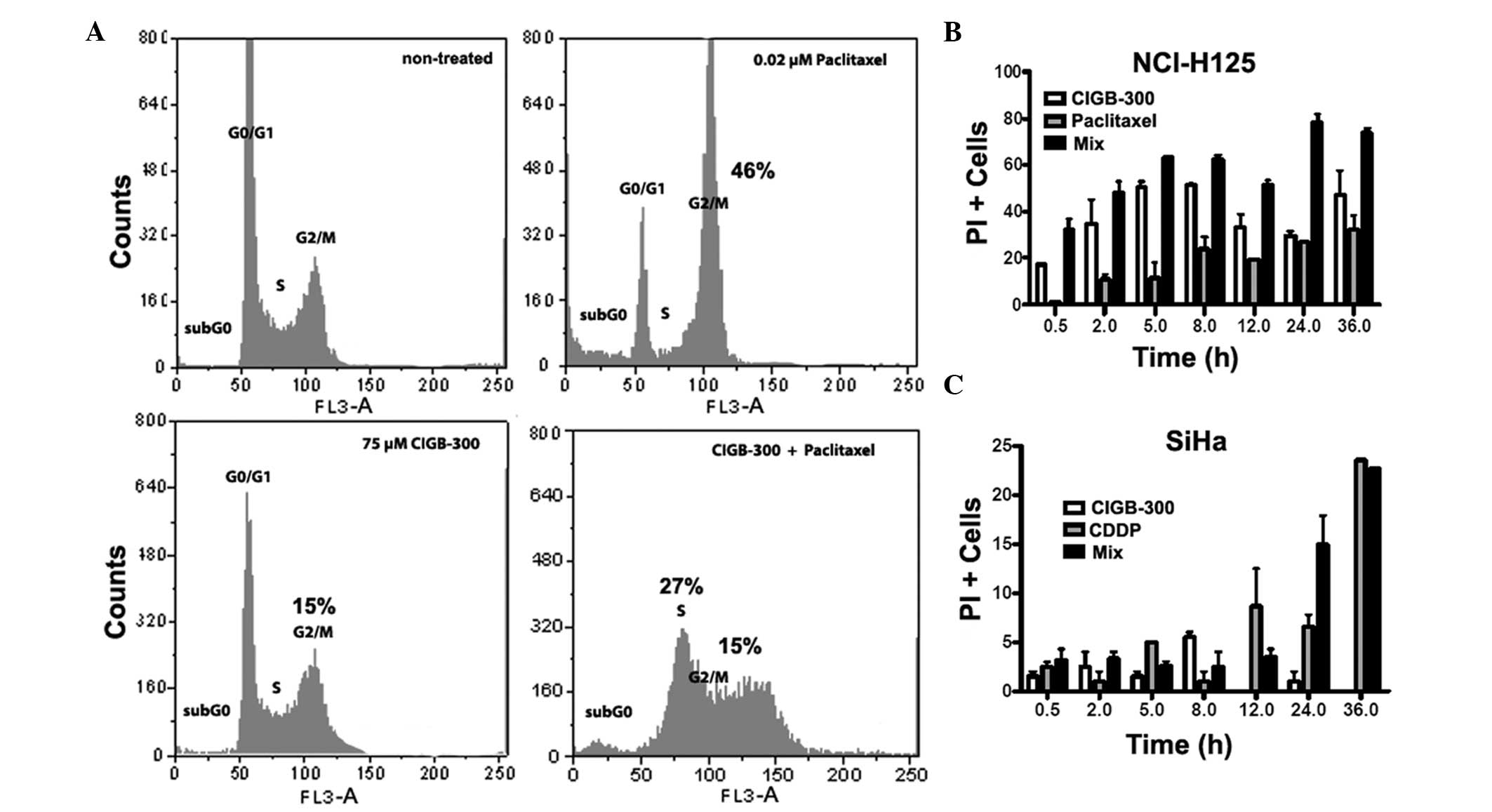
Effect of drug combinations on cell cycle
and viability
The characterization of the type of interaction
performed thus far was based on a global proliferative readout
(i.e., cell mass staining) from the SRB-based assay. To evaluate
how selected drug combinations affect cell proliferation and
viability, we measured the DNA content and the loss of cytoplasmic
membrane integrity as indicators of cell cycle progression and cell
cytotoxicity, respectively. Two particular synergistic drug
combinations, namely CIGB-300 plus paclitaxel in lung cancer cells
and CIGB-300 plus cisplatin in cervical cancer cells, were selected
for the analysis (Table II,
Figs. 3 and 4).
The effect of the combination of CIGB-300 plus
paclitaxel on cell cycle progression in lung cancer cells was
determined using PI to stain genomic DNA. After gating out cellular
debris and excluding cell doublets, the DNA distribution was fitted
to a diploid DNA content for cell cycle profiling. The analysis
demonstrated that both CIGB-300 and paclitaxel cause a cell cycle
arrest in the G2/M phase of the cell cycle (15 and 46%,
respectively), while their combination caused cell cycle arrest at
the S (27%) and G2/M (15%) phases (Fig. 5A). Of note, in SiHa cells, CIGB-300
at 25 μM did not affect the cell cycle distribution, whereas
cisplatin and the combination caused only a mild arrest at the S
phase (10%) (data not shown).
As regards the cytotoxicity of the selected drug
combinations, the results obtained with the combination of CIGB-300
plus paclitaxel in lung cancer cells corroborated the previously
observed synergistic interaction. We observed a stronger cytotoxic
effect for the combination compared to the one registered for each
drug alone, which was more evident after 24 and 36 h of incubation
(Fig. 5B). This kinetic experiment
also revealed that, while paclitaxel-induced cytotoxicity increases
gradually from 2 to 36 h, the effect of CIGB-300 may be seen as
early as 0.5 h, reaching a maximum effect between 5 and 8 h and
after 36 h. Finally, the cytotoxic effect of CIGB-300 alone at 25
μM was found to be negligible in the SiHa cervical cancer cell line
compared to that of cisplatin alone or its combination with
CIGB-300 (Fig. 5C).
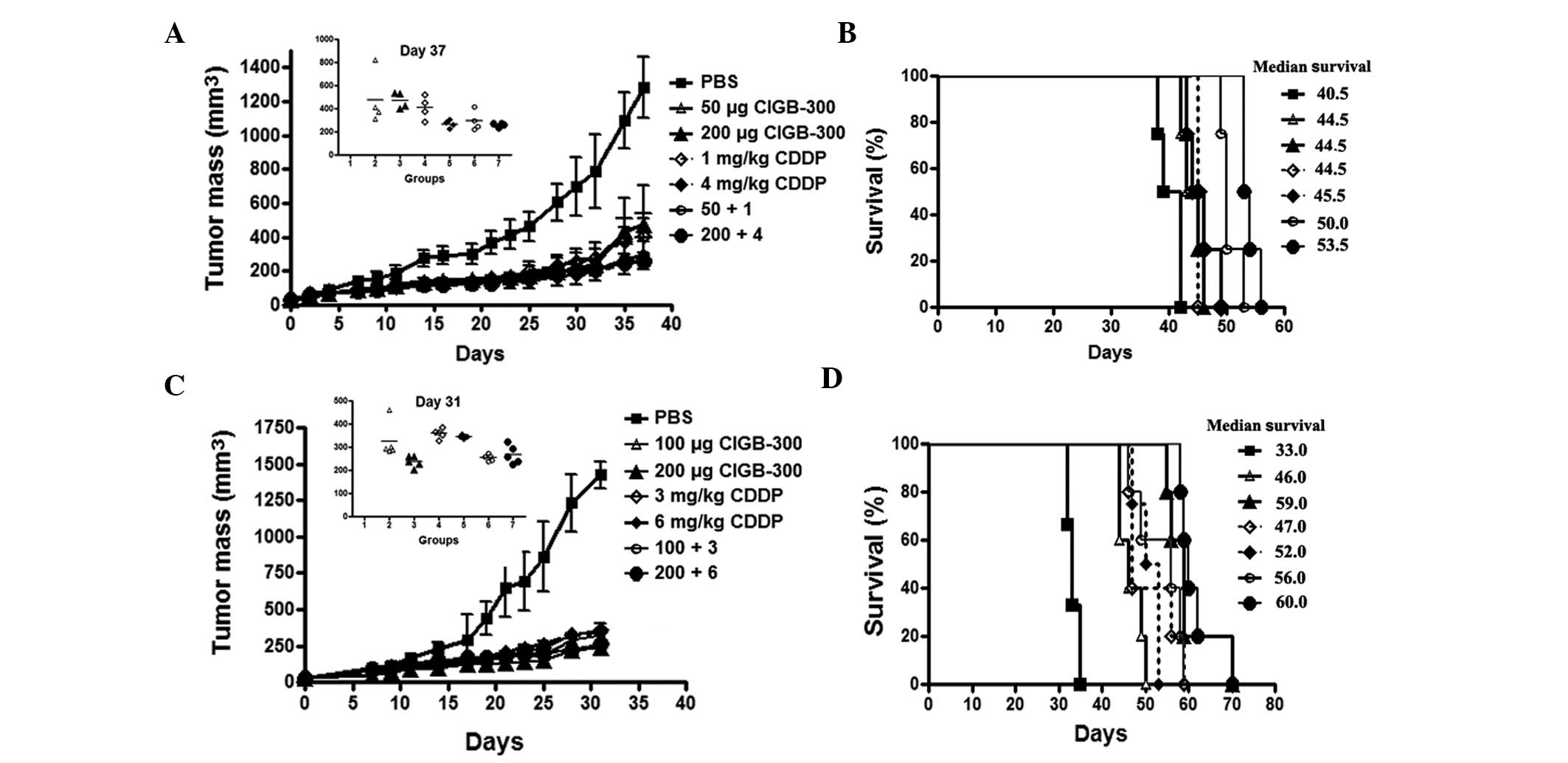
In vivo drug combination in a cervical
cancer animal model
To corroborate the potential benefit of combining
the CIGB-300 with chemotherapy in vivo, we evaluated the
effect of simultaneous administration of the peptide and cisplatin
on tumor growth and survival following the inoculation of SiHa
cells in nude mice. In both experiments (schemes A and B), the
doses for each single agent and its combinations were selected to
maintain a fixed ratio among the drugs to further evaluate the type
of interaction by CalcuSyn. In scheme A (CIGB-300:cisplatin,
2.5:1), the single agents as well as their combination
significantly reduced tumor growth (>60% inhibition, T/C
0.4-0.2, day 37) and increased the survival of treated mice (4–13
days) in this cervical cancer model (Fig. 6A and B)
Of note, in this experimental setting, the lower
dose combination of CIGB-300 plus cisplatin achieved a more
significant tumor growth delay and survival compared to each
monotherapy alone (Fig. 6A insert
and Fig. 6B). At day 37, the group
treated with this combination (CIGB-300 50 μg plus cisplatin 1
mg/kg) resulted in 77% tumor growth inhibition (T/C=0.23), while
the corresponding monotherapies achieved a less significant effect
(CIGB-300: 63% inhibition, T/C=0.37; and cisplatin: 68% inhibition,
T/C=0.32) (Fig. 6A insert).
However, no significant differences were observed between such
groups (one-way ANOVA, Tukey’s post hoc test). The superiority
regarding the tumor growth inhibition of combining the higher doses
of each compound (CIGB-300 200 μg plus cisplatin 4 mg/kg) was
rather subtle, as a consequence of the strong effect of cisplatin
alone (80% vs. 79%, respectively) (Fig. 6A). By contrast, the survival
analysis indicated that the combination of the lower doses of
CIGB-300 and cisplatin significantly increased the survival (5.5
days) of tumor-bearing mice compared to each monotherapy alone
(combination vs. CIGB-300, P<0.05; combination vs. cisplatin,
P<0.01), whereas the higher dose combination prolonged survival
up to 8 days over each single agent (combination vs. CIGB-300,
P<0.01; combination vs. cisplatin, P<0.01) (log-rank test)
(Fig. 6B).
In scheme B (CIGB-300:cisplatin, 1.7:1), although
both single agents and their combinations exerted a potent
antitumor effect (>70% inhibition, T/C 0.25–0.17, day 31), the
differences in tumor growth inhibition for monotherapies compared
to combination treatments did not reach statistical significance
(Fig. 6C insert). However, the
survival analysis indicated that the lower dose combination
(CIGB-300 100 μg plus cisplatin 3 mg/kg) prolonged mouse survival
at least by 9 days compared to each monotherapy alone (P<0.05)
(Fig. 6D).
Discussion
CIGB-300 is a novel clinical stage molecule designed
to impair CK2-mediated phosphorylation by binding to the conserved
phosphoaceptor domain on its substrates (11). The antineoplastic effect of this
peptide has been well documented in the preclinical setting in
vitro and in vivo (13,17,18).
In 2006, the peptide entered a phase I clinical trial on patients
with HSILs, where safety and efficacy signs were registered
(19). Moreover, in patients with
stage 1B2/II cervical cancer, the MTD, biodistribution and
modulation of a response biomarker were investigated following
local administration of CIGB-300 (20). Finally, evidence of the antitumor
activity of CIGB-300 in humans was recently collected from case
studies (21).
Although preclinical and clinical findings support
the use of the CIGB-300 peptide as a monotherapy, it is widely
accepted that the combination of drugs in the field of oncology are
more likely to cope with tumor complexity compared to single-agent
treatment (22). Such a view is
sustained by decades of clinical practice and by cumulative
knowledge on tumor biology (26).
Drug combination therapies are commonly used in order to increase
therapeutic efficacy, reduce toxicity and decrease the incidence of
drug resistance. In this scenario, the combination of targeted
therapies plus conventional chemotherapy has emerged as an
alternative therapeutic option that requires further investigation
(27).
To the best of our knowledge, the antineoplastic
effect of the CIGB-300 peptide when combined with different
chemotherapeutic drugs currently used in the treatment of lung and
cervical cancer was first investigated in this study. The drugs
used included antimitotic and DNA alkylating agents, topoisomerase
II inhibitors and DNA/RNA antimetabolites, hence targeting
different global cellular processes. In order to avoid false claims
of synergism commonly found in the literature, we performed our
interaction studies using the CalcuSyn software, which is based on
the median effect equation described by Chou (23). One of the main advantages of this
analysis over the classical isobologram is that the type of
interaction can be scored through an entire range of drug
combinations and effect levels once a Latin square design is
selected. Of note, in order to visualize all interaction data at
the same time, a simple script was built to draw an interaction
surface. On such an interaction surface, additional representation
of the effect level allowed us to better assess the relevance of
the observed interaction pattern ‘at a first glance’. Indeed,
synergistic interactions at low effect levels (e.g., Fa<0.4) may
be less relevant compared to slight synergism or even additive
interaction at higher effect levels (e.g., Fa>80), since the
complete eradication of cancer cells is the final therapeutic goal
in the clinical oncology setting.
Our in vitro interaction studies suggested
that the anti-mitotic drug paclitaxel may be a suitable partner to
combine with CIGB-300 in lung and cervical malignancies. A plain
synergistic interaction was observed in most of the interaction
surface from NCI-H125 and SiHa cells. Importantly, such synergistic
interaction resulted in >70% proliferation inhibition
(Fa>0.70) in both cell lines. Moreover, since CI and DRI are
inversely associated, such a ‘positive’ interaction suggests that,
each drug in the combination may be significantly reduced without
compromising the final therapeutic outcome, hence limiting the
chances of unwanted side effects. This issue is of paramount
importance in the case of paclitaxel, a drug known to be associated
with significant toxicities, such as myelosuppression and
peripheral neuropathy (28).
The combination of CIGB-300 with the alkylating drug
cisplatin displayed an additive interaction pattern across most of
the interaction surface in SiHa cells. Of note, for paclitaxel and
cisplatin, further combination experiments demonstrated that the
simultaneous addition of CIGB-300 and the chemotherapeutic agent is
more favorable compared to their sequential addition, according to
schedule-dependent experiments (data not shown). Altogether, while
the combination of the CIGB-300 peptide with the antimitotic drug
paclitaxel appears to be more favorable for lung cancer treatment,
the combination of CIGB-300 with cisplatin may be more promissory
in cervical cancer. Considering that paclitaxel and cisplatin are
chemotherapeutic drugs currently used for the treatment of lung and
cervical cancer, respectively, the evaluation of these particular
drug combinations in their already approved clinical niches may be
more expedite. However, a quite different profile was obtained from
combining CIGB-300 with doxorubicin in lung and cervical cancer
cells. In lung cancer cells, the combination of CIGB-300 with the
topoisomerase II inhibitor and with DNA/RNA antimetabolites,
resulted in antagonistic surfaces with rather scarce areas of
additive or synergistic effects under these experimental
conditions.
A further layer of analysis in drug combination
experiments is whether drug concentrations resulting in a
synergistic interaction are actually achievable in vivo. The
pharmacokinetic analysis of CIGB-300 in animal models and humans
indicated that this peptide may reach concentrations up to 30 μM in
the plasma, whereas after a 3-h infusion of paclitaxel at doses of
135–225 mg/m2 the estimated Cmax ranged between 3.3 and
7.6 μM. (unpublished results; 29). According to such reported
values, a significant area of the synergistic interaction between
these two drugs occurs in the range of clinically achievable
concentrations. Importantly, a similar analysis also suggested that
the predominant additive profile obtained from the combination of
CIGB-300 plus cisplatin in cervical cancer cells occurs in a
concentration range achievable in vivo for CIGB-300 (<30
μM) and cisplatin (<15 μM) (30,31).
Considering that in the in vivo setting
precise drug concentrations or ratios are difficult to achieve
(32), broad favorable interaction
surfaces in vitro (i.e., synergistic or additive) may
suggest a higher chance of obtaining an antitumor effect at the
organism level, both in mouse models and hopefully the clinical
setting. Likewise, such favorable profiles provide us with more
confidence to exclude potential antagonism arising from drug
combinations at particular ratios or concentrations. Following this
rationale and taking into account that cervical cancer is one of
the clinical niches where CIGB-300 is currently being evaluated as
a monotherapy (20), we attempted
to corroborate the potential therapeutic benefit of combining
CIGB-300 with cisplatin in vivo after inoculating human
cervical cancer cells in nude mice. Although such experiments were
initially designed to be analyzed by CalcuSyn, the reduction of
tumor growth with monotherapies and drug combinations were far
above the IC50 in all cases (T/C<0.5), thus
precluding the generation of reliable dose-response curves for CI
determination (23). However, our
results demonstrated that two different CIGB-300 plus cisplatin
combinations (i.e., different doses and drug ratios) significantly
increased animal survival when compared to each monotherapy alone.
Such in vivo results are in complete accordance with the
broad additive interaction surface observed with the concentration
range and drug ratios evaluated in vitro in the
antiproliferative setting.
Although drug combination experiments may be
performed over days or weeks, the elucidation of the cellular and
molecular basis underlying the observed interactions typically
requires years of research. The analysis at the cellular level for
one particular drug combination (i.e., CIGB-300 plus paclitaxel) in
lung cancer cells indicated that the observed synergistic
interaction arose from their effect on cell cycle progression and
viability. However, only cytotoxicity appeared to contribute to the
observed synergism regarding the combination of CIGB-300 plus
cisplatin in cervical cancer cells, suggesting that particular drug
combinations affect cell proliferation at different levels.
The first molecular clues that may explain the
synergistic interaction with anticancer drugs in lung cancer cells
were previously revealed by proteomics analysis of CIGB-300-treated
NCI-H125 cells (15). In such
studies, the peptide modulated an array of proteins associated with
drug resistance and survival, which may favor the cytotoxic effect
of several chemotherapeutic drugs. One of the downregulated
proteins was found to be the nuclease-sensitive element-binding
protein 1 (YB-1), a transcriptional factor that activates the
expression of the multidrug resistance 1 protein (MDR1) in response
to genotoxic stress (33). MDR1
pumps out a myriad of cytotoxic compounds, including paclitaxel;
therefore, a putative decrease in MDR1 expression via YB-1 may
explain the increased sensitivity of tumor cells to paclitaxel
(34). Furthermore, YB-1
participates in the nucleotide excision repair pathway and plays a
role in cisplatin-DNA adduct reparation (35); therefore, its downregulation may
also account for the observed additive pattern in the combination
of CIGB-300 plus cisplatin. Intriguingly, although YB-1 is a
validated substrate for CK2 (36),
this protein does not interact with CIGB-300 according to previous
pull-down experiments conducted on lung cancer cells (12).
CIGB-300 was also shown to decrease the levels of
glutathione S-transferase Pi (GST-Pi), a cytoplasmic detoxifying
enzyme, whose increased levels correlate with resistance to a wide
range of drugs (37). Although
paclitaxel and cisplatin are not direct substrates for this enzyme,
both drugs require the activation of the MAP kinase pathway to
exert their maximal cytotoxicity, which is prevented by high levels
of GST-Pi. Moreover, at least three other proteins associated with
drug resistance were clearly downregulated by CIGB-300 in NCI-H125
cells (15). Whether the
synergistic/additive interaction patterns observed with the
combination of CIGB-300 with cisplatin or paclitaxel may be
explained by these molecular events require elucidation by further
studies.
Despite the availability of a significant number of
studies on CK2, the number of reports on the use of CK2 inhibitors
in combination with anticancer compounds remains limited (38). Such pioneering studies mainly rely
on the genetic manipulation of the catalytic subunit CK2α or the
use of the first -generation semi-selective CK2 inhibitors DRB and
TBB (38). Although these former
studies provided valuable preclinical data, only with the advent of
clinical-grade inhibitors the true potential of combining anti-CK2
approaches with conventional or emerging therapies may be
translated into clinical grounds. Apart from the CIGB-300 peptide,
only the ATP-competitive inhibitor CX-4945 has been evaluated in
humans as an anti-CK2 therapy (9,10). A
synergistic pattern for particular drug concentrations or ratios
was reported when CX-4945 was combined with different compounds
targeting the phosphatidylinositol 3 kinase (PI3K)/Akt/mammalian
target of rapamycin (mTOR) signaling axis in breast or lung
preclinical cancer models (39,40).
Considering that CK2 phosphorylates critical substrates in this
pathway (e.g., Akt, phosphatase and tensin homolog, p70SK6), the
rationale behind such combination experiments was to reinforce the
inhibitory effect of compounds such as erlotinib (epithelial growth
factor receptor), LY294002 (PI3K) or the dual inhibitor PI-103
(PI3K and mTOR). Moreover, the combination of CX-4945 with
cisplatin also resulted in a synergistic drug interaction in two
ovarian cancer cell lines according to the Bliss additivity method
(41,42). Beyond the fact that the methods of
drug interaction analysis differ, those studies only evaluated
particular drug concentrations in vitro, without providing
an explanation for its selection or investigating a wide range of
concentrations for each of the compounds (39,41).
As a novel approach to impair the CK2-mediated
signaling, we investigated the potential therapeutic benefit of
combining the CIGB-300 peptide with different cytotoxic compounds
targeting global cellular processes, which are firmly established
in clinical oncology. Although we are currently focused on the
elucidation of the molecular and cellular basis of observed
synergistic/additive interactions, the preclinical data provided in
this study may pave the way for near-future clinical trials
investigating the combination of the CIGB-300 peptide with
conventional chemotherapeutic agents, such as paclitaxel and
cisplatin in two frequent human cancers.
Acknowledgements
This study was supported by C.I.G.B. and Biorec.
Grant CIGB-300.
References
|
1
|
Guerra B and Issinger OG: Protein kinase
CK2 in human diseases. Curr Med Chem. 15:1870–1886. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ruzzene M and Pinna LA: Addiction to
protein kinase CK2: a common denominator of diverse cancer cells?
Biochim Biophys Acta. 1804.499–504. 2010.PubMed/NCBI
|
|
3
|
Zandomeni R, Zandomeni MC, Shugar D and
Weinmann R: Casein kinase type II is involved in the inhibition by
5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole of specific RNA
polymerase II transcription. J Biol Chem. 261:3414–3419.
1986.PubMed/NCBI
|
|
4
|
Szyszka R, Grankowski N, Felczak K and
Shugar D: Halogenated benzimidazoles and benzotriazoles as
selective inhibitors of protein kinases CK I and CK II from
Saccharomyces cerevisiae and other sources. Biochem Biophys
Res Commun. 208:418–424. 1995.PubMed/NCBI
|
|
5
|
Sarno S, Reddy H, Meggio F, et al:
Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP
site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’).
FEBS Lett. 496:44–48. 2001.
|
|
6
|
Pagano MA, Meggio F, Ruzzene M,
Andrzejewska M, Kazimierczuk Z and Pinna LA:
2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: a novel
powerful and selective inhibitor of protein kinase CK2. Biochem
Biophys Res Commun. 321:1040–1044. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Duncan JS, Gyenis L, Lenehan J, Bretner M,
Graves LM, Haystead TA and Litchfield DW: An unbiased evaluation of
CK2 inhibitors by chemoproteomics: characterization of inhibitor
effects on CK2 and identification of novel inhibitor targets. Mol
Cell Proteomics. 7:1077–1088. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pagano MA, Bain J, Kazimierczuk Z, et al:
The selectivity of inhibitors of protein kinase CK2: an update.
Biochem J. 415:353–365. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marschke RF, Andreopoulou E, Von Hoff DD,
Lim JK, Padgett CS and Northfelt DW: Phase I clinical trial of
CX-4945: A first-in-class orally administered small molecule
inhibitor of protein kinase CK2. Mol Cancer Ther. 8:C392009.
View Article : Google Scholar
|
|
10
|
Siddiqui-Jain A, Drygin D, Streiner N, et
al: CX-4945, an orally bioavailable selective inhibitor of protein
kinase CK2, inhibits prosurvival and angiogenic signaling and
exhibits antitumor efficacy. Cancer Res. 70:10288–10298. 2010.
View Article : Google Scholar
|
|
11
|
Perea SE, Reyes O, Puchades Y, et al:
Antitumor effect of a novel proapoptotic peptide that impairs the
phosphorylation by the protein kinase 2 (casein kinase 2). Cancer
Res. 64:7127–7129. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perera Y, Farina HG, Gil J, et al:
Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its
CK2-mediated phosphorylation, and leads to apoptosis through its
nucleolar disassembly activity. Mol Cancer Ther. 8:1189–1196. 2009.
View Article : Google Scholar
|
|
13
|
Perera Y, Costales HC, Diaz Y, et al:
Sensitivity of tumor cells towards CIGB-300 anticancer peptide
relies on its nucleolar localization. J Pept Sci. 18:215–223. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meggio F and Pinna LA:
One-thousand-and-one substrates of protein kinase CK2? FASEB J.
17:349–368. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rodriguez-Ulloa A, Ramos Y, Gil J, et al:
Proteomic profile regulated by the anticancer peptide CIGB-300 in
non-small cell lung cancer (NSCLC) cells. J Proteome Res.
9:5473–5483. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Farina HG, Benavent Acero F, Perera Y, et
al: CIGB-300, a proapoptotic peptide, inhibits angiogenesis in
vitro and in vivo. Exp Cell Res. 317:1677–1688. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perera Y, Farina HG, Hernandez I, et al:
Systemic administration of a peptide that impairs the protein
kinase (CK2) phosphorylation reduces solid tumor growth in mice.
Int J Cancer. 122:57–62. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Perea SE, Reyes O, Baladron I, et al:
CIGB-300, a novel proapoptotic peptide that impairs the CK2
phosphorylation and exhibits anticancer properties both in vitro
and in vivo. Mol Cell Biochem. 316:163–167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Solares AM, Santana A, Baladron I, et al:
Safety and preliminary efficacy data of a novel casein kinase 2
(CK2) peptide inhibitor administered intralesionally at four dose
levels in patients with cervical malignancies. BMC Cancer.
9:1462009. View Article : Google Scholar
|
|
20
|
Soriano-García JL, López-Díaz A,
Solares-Asteasuainzarra M, et al: Pharmacological and safety
evaluation of CIGB-300, a casein kinase 2 inhibitor peptide,
administered intralesionally to patients with cervical cancer stage
IB2/II. J Cancer Res Ther. 1:163–173. 2013.
|
|
21
|
Perea SE, Baladron I, Garcia Y, et al:
CIGB-300, a synthetic peptide-based drug that targets the CK2
phosphoaceptor domain. Translational and clinical research. Mol
Cell Biochem. 356:45–50. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Al-Lazikani B, Banerji U and Workman P:
Combinatorial drug therapy for cancer in the post-genomic era. Nat
Biotechnol. 30:679–692. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boyd M: The NCI In Vitro Anticancer Drug
Discovery Screen. Concept, Implementation, and Operation,
1985–1995. Anticancer Drug Development Guide: Preclinical
Screening, Clinical Trials, and Approval. Humana Press; Totowa, NJ,
USA: 1997
|
|
25
|
Chou TC and Hayball MP: CalcuSyn for
Windows: multiple-drug dose effect analyzer and manual. Biosoft;
Cambridge (UK): 1997
|
|
26
|
Kitano H: Cancer as a robust system:
implications for anticancer therapy. Nat Rev Cancer. 4:227–235.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dancey JE and Chen HX: Strategies for
optimizing combinations of molecularly targeted anticancer agents.
Nat Rev Drug Discov. 5:649–659. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Marupudi NI, Han JE, Li KW, Renard VM,
Tyler BM and Brem H: Paclitaxel: a review of adverse toxicities and
novel delivery strategies. Expert Opin Drug Saf. 6:609–621. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gianni L, Kearns CM, Giani A, et al:
Nonlinear pharmacokinetics and metabolism of paclitaxel and its
pharmacokinetic/pharmacodynamic relationships in humans. J Clin
Oncol. 13:180–190. 1995.PubMed/NCBI
|
|
30
|
Andersson A, Fagerberg J, Lewensohn R and
Ehrsson H: Pharmacokinetics of cisplatin and its monohydrated
complex in humans. J Pharm Sci. 85:824–827. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuenen BC, Rosen L, Smit EF, et al:
Dose-finding and pharmacokinetic study of cisplatin, gemcitabine,
and SU5416 in patients with solid tumors. J Clin Oncol.
20:1657–1667. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mayer LD and Janoff AS: Optimizing
combination chemotherapy by controlling drug ratios. Mol Interv.
7:216–223. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ohga T, Uchiumi T, Makino Y, Koike K, Wada
M, Kuwano M and Kohno K: Direct involvement of the Y-box binding
protein YB-1 in genotoxic stress-induced activation of the human
multidrug resistance 1 gene. J Biol Chem. 273:5997–6000. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hennessy M and Spiers JP: A primer on the
mechanics of P-glycoprotein the multidrug transporter. Pharmacol
Res. 55:1–15. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gaudreault I, Guay D and Lebel M: YB-1
promotes strand separation in vitro of duplex DNA containing either
mispaired bases or cisplatin modifications, exhibits
endonucleolytic activities and binds several DNA repair proteins.
Nucleic Acids Res. 32:316–327. 2004. View Article : Google Scholar
|
|
36
|
Skabkin MA, Evdokimova V, Thomas AA and
Ovchinnikov LP: The major messenger ribonucleoprotein particle
protein p50 (YB-1) promotes nucleic acid strand annealing. J Biol
Chem. 276:44841–44847. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Townsend DM and Tew KD: The role of
glutathione-S-transferase in anti-cancer drug resistance. Oncogene.
22:7369–7375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Pinna LA: Protein Kinase CK2.
Wiley-Blackwell, . John Wiley & Sons, Inc; 2013, View Article : Google Scholar
|
|
39
|
Drygin D, Bliesath J, Ho C, et al:
CX-4945, a novel small molecule inhibitor of CK2 protein kinase,
reduces hyperactivated Akt signaling and synergizes with Akt
inhibitors in breast cancer cells. In: 21st AACR-NCI-EORTC
Symposium on Molecular Targets and Cancer Therapeutics; Boston, MA.
Mol Cancer Ther. 8. pp. C1982009, View Article : Google Scholar
|
|
40
|
Bliesath J, Huser N, Omori M, et al:
Combined inhibition of EGFR and CK2 augments the attenuation of
PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer
Lett. 322:113–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Siddiqui-Jain A, Bliesath J, Macalino D,
et al: CK2 inhibitor CX-4945 suppresses DNA repair response
triggered by DNA-targeted anticancer drugs and augments efficacy:
mechanistic rationale for drug combination therapy. Mol Cancer
Ther. 11:994–1005. 2012. View Article : Google Scholar
|
|
42
|
Siddiqui-Jain A, Streiner N, Bliesath J,
et al: CX-4945, an inhibitor of protein kinase CK2, potentiates the
antitumor activity of platinum chemotherapy in models of ovarian
cancer by preventing phosphorylation of XRCC1 and MDC1 and
disrupting DNA damage repair. Cancer Res. 71:54942011. View Article : Google Scholar
|