Introduction
Sialoblastoma is a rare salivary gland tumor that
recapitulates the primitive salivary gland anlage. Sialoblastoma
was first described in 1966 by Vawter and Tefftas and referred to
as embryoma (1). In 1988, Taylor
suggested the term ‘sialoblastoma’, as it describes both the
dysontogenic characteristics and the origin of this tumor from the
salivary gland (2–4). The various appellations include
congenital basal cell adenoma, basal cell adenoma, basaloid
adenocarcinoma, congenital hybrid basal cell adenoma-adenoid cystic
carcinoma, and embryoma. Sialoblastoma most commonly affects the
major salivary glands and is histologically characterized by
variably arranged, tight clusters or clumps of basaloid cells and
partially formed ductal and pseudo-ductal spaces separated by thin
fibrous bands. The overall prognosis of this type of tumor remains
controversial. Sialoblastoma has a tendency to progress to local
invasion, local recurrence and occasional metastasis. In 1996,
according to the third series of the Armed Forces Institute of
Pathology (AFIP) classification of salivary gland tumors,
sialoblastoma was classified as a benign epithelial neoplasm.
However, according to World Health Organization (WHO)
classification of head and neck tumor and the fourth series of the
AFIP classification of salivary gland tumors, sialoblastoma was
reclassified as a malignant epithelial tumor (5). The authors herein report the clinical,
histopathological and immunohistochemical characteristics of
sialoblastoma.
Case report
Clinical summary
A 1-year-old female infant presented with a palpable
mass of the right cheek, which had originally appeared and
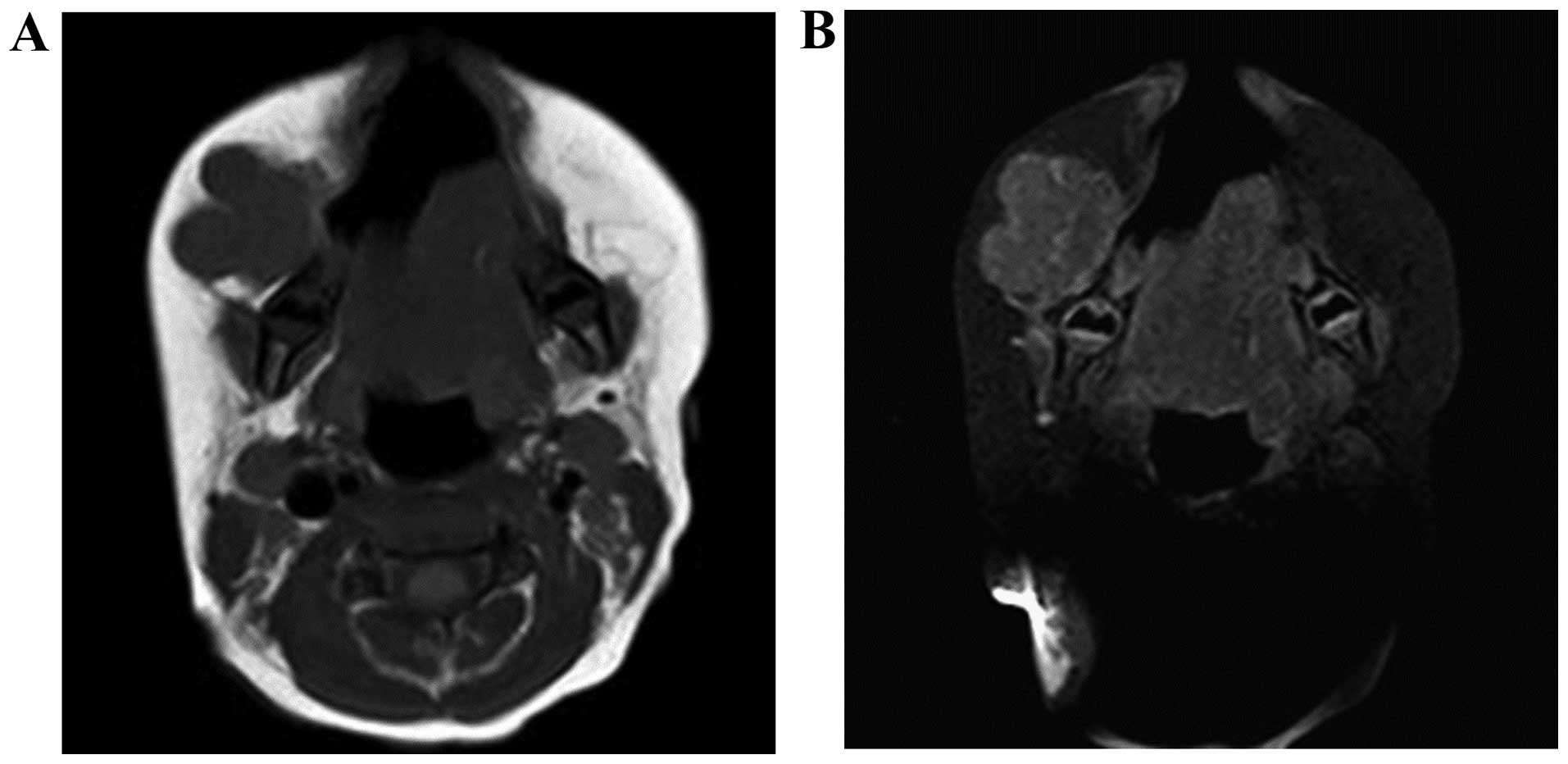
progressively enlarged since the age of 2 months. The magnetic
resonance axial T1-weighted (T1W) images revealed a lobulated soft
tissue lesion anterior to the right mandible (Fig. 1A) and the axial contrast-enhanced T1W
images showed minimal enhancement (Fig.
1B). An incisional biopsy was performed and histological
examination revealed basaloid tumor cells with focal ductal
differentiation, suggestive of sialoblastoma. The post-incisional
serum α-fetoprotein (AFP) level was 99 ng/ml. Subsequently,
complete excision was performed. The final pathological diagnosis
was high-grade sialoblastoma with unfavorable histology, T2N0M0.
The postoperative course was uneventful. Following complete
surgical removal, the serum AFP normalized and radiographic disease
disappeared. The patient remains free from recurrence 2 years after
complete excision.
Pathological findings
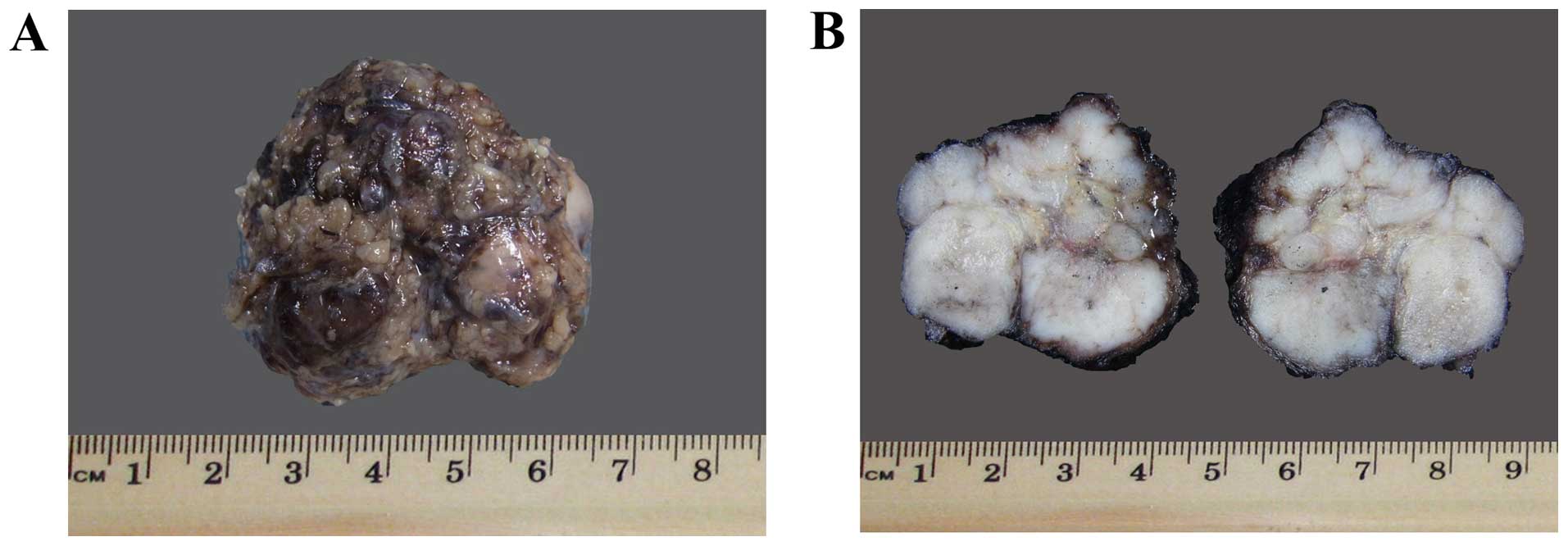
On gross examination, a lobulated soft tissue mass,
measuring 3.8×3×2.5 cm and weighing 12 g, was excised (Fig. 2A). The cut sections displayed a
homogeneous solid gray-tan surface (Fig.
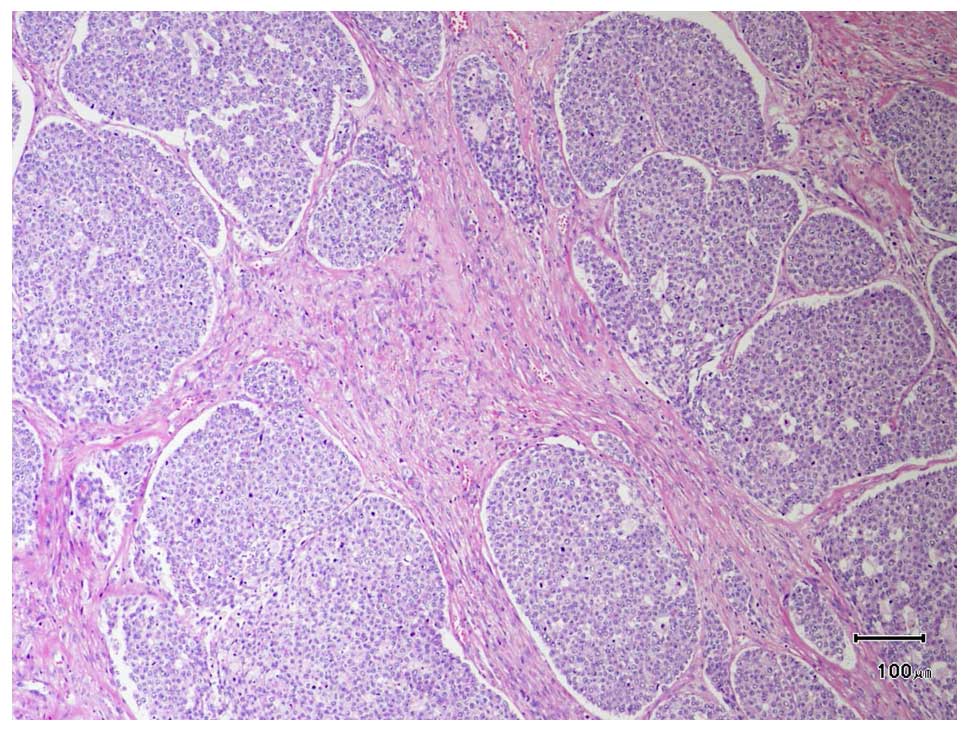
2B). The microscopic findings included solid nests of basaloid
cells and partially formed ductal and pseudo-ductal spaces
separated by thin fibrous bands (Fig.
3). The basaloid cells were characterized by high
nuclear:cytoplasmic ratio, round to oval nuclei, fine nuclear
chromatin, small nucleoli and scant to moderate pale eosinophilic
cytoplasm. The number of mitotic figures was 50 per 10 high-power
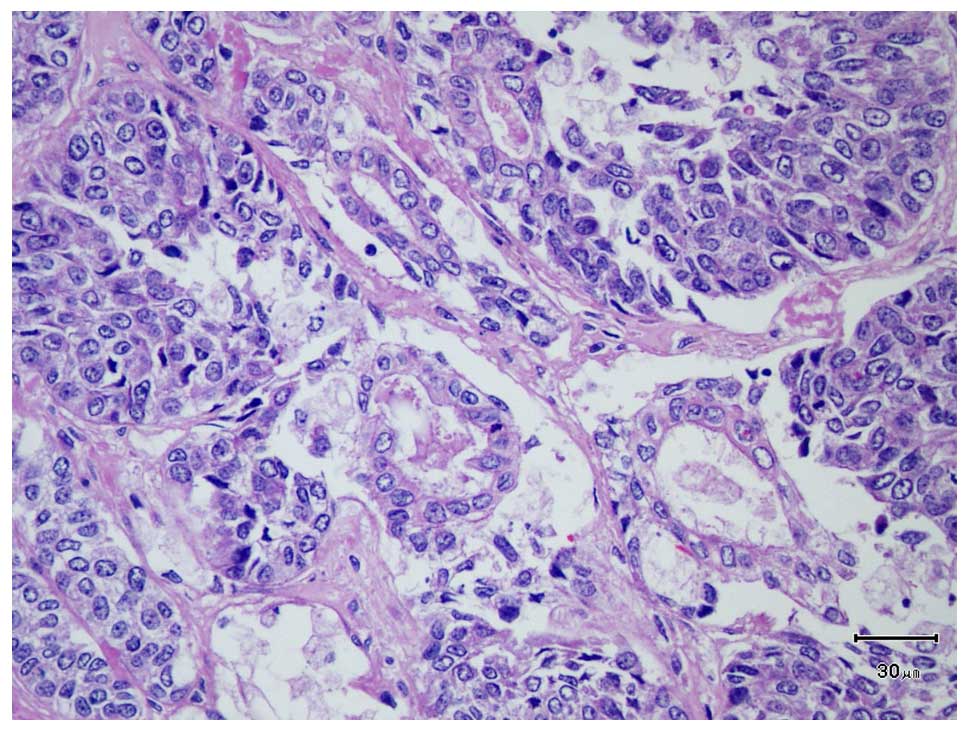
fields (HPFs). Primitive duct formations were observed (Fig. 4). Vascular, perineural and skeletal
muscular invasion was also observed, as was 10% tumor cell
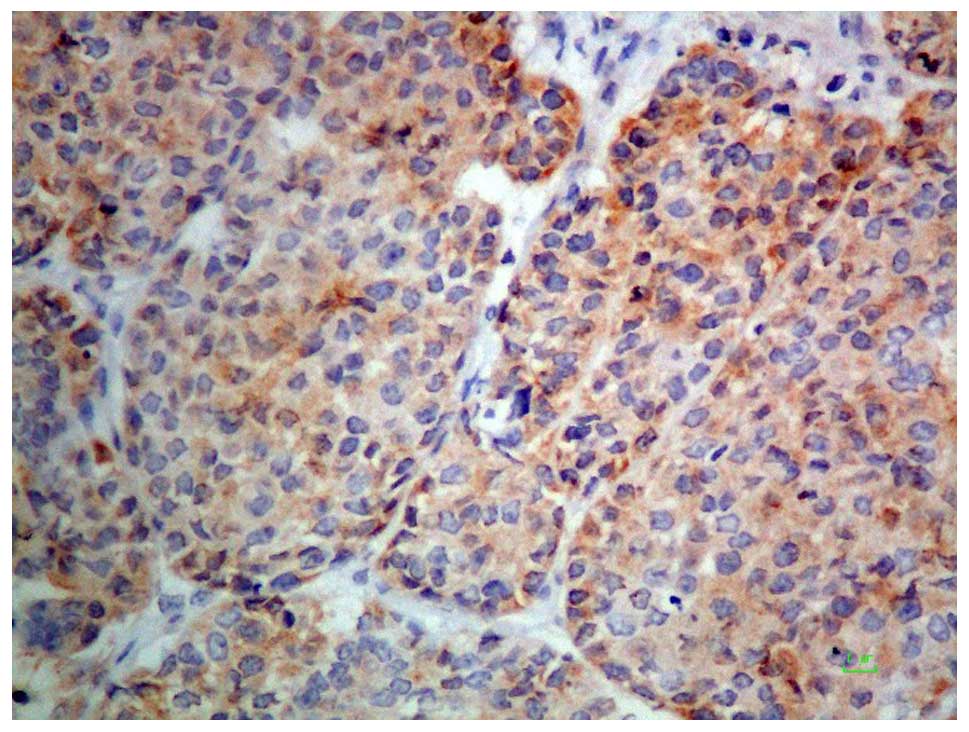
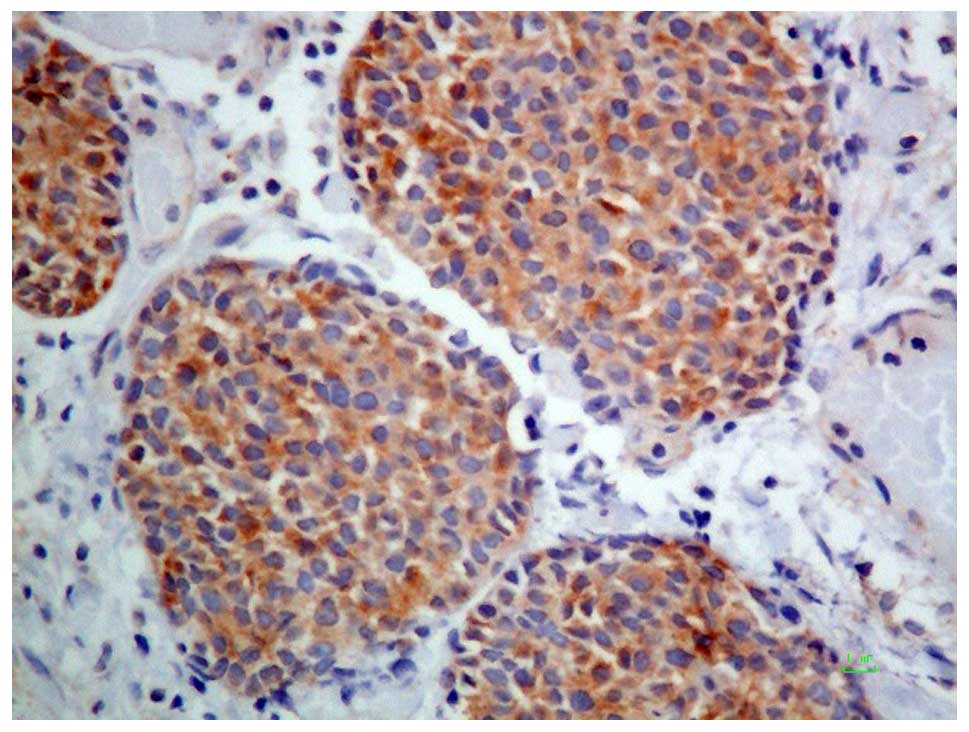
necrosis. Immunohistochemical staining showed the tumor cells to be
positive for cytokeratin AE1/AE3. Some tumor cells were also
positive for epithelial membrane antigen, cytokeratin 5/6, smooth
muscle actin, p63, CD99, S100 protein, AFP, and Hep Par-1 (Figs. 5 and 6),
while immunostaining for cytokeratins 7 and 8/18 was only positive
in the ductal structures. Vimentin immunostaining was positive in
both tumor cells and fibroblastic stromal cells. Finally,
immunostaining was negative for cytokeratin 20, neuron-specific
enolase, chromogranin A, synaptophysin, CD56, CD57, CD31, CD34,
leukocyte common antigen, placental alkaline phosphatase, human
melanoma black 45, Wilms tumor 1, glial fibrillary acidic protein
and desmin. The Ki67 proliferative index was 70%. The tumor cells
exhibited a negative signal for Epstein-Barr virus-encoded small
RNAs on in situ hybridization.
Discussion
Primary salivary gland tumors are rare in infancy
and childhood (1). The incidence of
salivary gland tumors in children aged <5 years is extremely
rare (1), particularly during infancy
(1). In the first decade of life, the
incidence of salivary gland tumors is <0.25% (2). The majority of salivary gland tumors in
children are non-epithelial neoplasms (8). Hemangioma is the most common salivary
gland tumor during the first year of life (6,7).
Sialoblastoma is a rare epithelial neoplasm of
salivary gland origin in children (3), with ~50 cases reported in the English
literature (5). This tumor may occur
during the congenital, neonatal, or childhood periods (9,10).
However, the vast majority of the cases are identified at or
shortly after birth (5), with a
median age at diagnosis of 9.8 months (9). Sialoblastoma exhibits a male
predilection of ~2:1. These tumors are mostly found in the parotid
and submandibular glands, although involvement of the minor
salivary glands has also been reported. The size of tumor is
ranging from 1.5 to 15 cm in greatest dimension (5). The majority of the patients clinically
present with a slow-growing, painless, subcutaneous mass (8).
Regarding the macroscopic findings, most tumors are
multilobulated, firm, tan-pink to yellow masses (8,10).
Microscopically, sialoblastomas exhibit the primitive embryonic
histological characteristics of salivary gland tissue, together
with basaloid epithelial components and a variable amount of stroma
(1,10). This tumor consists of variably
arranged, tight clusters or clumps of basaloid-like cells in a
background of dispersed epithelial and myoepithelial cells, and
partially formed ductal and pseudo-ductal spaces separated by thin
fibrous bands (8,11). These microscopic characteristics
reflect the pre-acinar stage of salivary gland morphogenesis,
together with primitive cell masses, forming ducts and
pseudo-ductular spaces, without acinar differentiation (3,11).
The differential diagnosis includes adenoid cystic
carcinoma and basal cell adenoma. One-third of sialoblastomas
exhibit a cribriform growth pattern, which is different from
adenoid cystic carcinoma regarding various histological
characteristics, including prominent, elongated and branching
ductal structures within basaloid cells bulbs, thickened tubules
with basaloid cells, and larger basaloid cells with more obvious
cytoplasm (12). In contrast to basal
cell adenoma, sialoblastoma is composed of more primitive cells
that exhibit less prominent peripheral palisading of nuclei,
significant cytological atypia and higher mitotic activity, and
often infiltrate the surrounding tissue, including vascular,
perineural and skeletal muscular invasion.
Immunohistochemical studies have reported that
sialoblastoma stains positive for cytokeratin in the ductal cells
and for vimentin, smooth muscle actin and S100 in the outer ductal
parts (5,11). Solid nests of basaloid cells are
focally positive for S100 and vimentin (11). The immunohistochemical results in our
case were compatible with previous reports. In addition, our case
was also immunohistologically and serologically positive for AFP
(11,13). Moreover, immunohistochemical
investigation demonstrated positivity for Hep Par-1 in a
considerable number of tumor cells.
The presence of anaplasia, neurovascular invasion
and necrosis were suggested as adverse prognostic factors (2,6). Other
adverse histological characteristics include infiltration of the
capsule (12). Sialoblastomas may be
histomorphologically divided into two groups, namely favorable and
unfavorable groups (10). The
characteristics of the favorable group include circumscription and
infrequent mitoses (<3–4/10 HPFs) (10), whereas the characteristics of the
unfavorable group include anaplasia, frequent mitoses (≥4/10 HPFs),
apoptosis, necrosis and a high Ki67 index (10). According to this classification, the
pathological findings of our patient were those of the unfavorable
group, namely frequent mitoses (50/10 HPFs) and high Ki67
proliferation index (70%).
The pathogenesis of sialoblastoma has not been fully
elucidated. It has been suggested that sialoblastoma originates
from retained blastema cells in the salivary gland, rather than
basal reserve cells. Dysembryonic salivary gland changes have been
described adjacent to the tumor, with proliferation of the terminal
ductal epithelial bulbs. In tumor chromosome study, the cytogenetic
karyotype of sialoblastoma was 47, XX, del(3)(q13),inv(9)(p11q12)c,+?r [5]/46, XX, inv(9)(p11q12)c[41] (14). The aberrations detected in the
sialoblastoma (deletion of the long arm of chromosome 3 and ring
chromosome formation) are different from those detected in other
salivary gland tumors, suggesting that the pathogenetic mechanisms
differ between sialoblastoma and other salivary gland neoplasms
(14).
A case with a concurrent sialoblastoma associated
with hepatoblastoma has been reported (9). Furthermore, there are few reports in the
literature documenting the focal expression of AFP in tumor cells.
Our patient also demonstrated co-expression of AFP and Hep Par-1 in
tumor cells. AFP is an oncofetal antigen that is produced by the
fetal liver. These data suggest that there may be an association
between hepatic tumorigenesis and the development of sialoblastoma.
However, since studies on the association between sialoblastoma and
hepatic markers remain scarce, the pathogenesis of sialoblastoma
should be further investigated.
The prognosis of sialoblastoma is controversial.
Sialoblastoma has a tendency to progress further with local
invasion, local recurrence and occasional metastasis (5). In general, in infants and children,
sialoblastoma is usually of low-grade malignancy and most cases
have a favorable outcome (12).
However, although they generally behave benignly, these tumors may
be associated with lymphatic metastases (12). Approximately one-third of
sialoblastomas develop local recurrence and metastasis (12). Recurrences have been reported as late
as 4 years after excision (7) and
distant metastases usually involve the lung and lymph nodes
(5,12).
Due to the limited number of the cases, definitive
treatment guidelines have not yet been established (7,9). Early
complete surgical excision remains the cornerstone of the surgical
management of sialoblastoma (2,7,9,15), whereas
radiotherapy and chemotherapy remain controversial (15). Radiation may play a role in cases with
incomplete tumor resection (8).
However, since the disease mostly occurs in childhood, the use of
radiotherapy may be limited, due to the radiation-related side
effects, such as impaired bone growth, abnormal facial structure
and mutagenic effects (8,9). Sialoblastoma is likely sensitive to
chemotherapy (9), which may be useful
in patients with extensive or metastatic tumors, relapsed cases, or
inadequate surgical excision of a primary tumor (1,7,9,15). The
protocols are usually based on the chemosensitivity, as with other
mesenchymal tumors (7). The use of
neoadjuvant chemotherapy for locally invasive tumors, rather than
extensive, mutilating surgery, has also been suggested (9). The follow-up should be frequent and
prolonged. AFP may be a useful marker of tumor response in patients
with sialoblastoma.
References
|
1
|
Brandwein M, Said-Al-Naeif N, Manwani D,
Som P, Goldfeder L, Rothschild M and Granowetter L: Sialoblastoma:
Clinicopathological/immunohistochemical study. Am J Surg Pathol.
23:342–348. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Vidyadhar M, Amanda C, Thuan Q and
Prabhakaran K: Sialoblastoma. J Pediatr Surg. 43:e11–e13. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marucci DD, Lawson K, Harper J, Sebire NJ
and Dunaway DJ: Sialoblastoma arising in ectopic salivary gland
tissue. J Plast Reconst Aesthet Surg. 62:e241–e246. 2009.
View Article : Google Scholar
|
|
4
|
Taylor GP: Congenital epithelial tumor of
the parotid - sialoblastoma. Pediatr Pathol. 8:447–452. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ellis GL: What's new in the AFIP fascicle
on salivary gland tumors: A few highlights from the 4th Series
Atlas. Head Neck Pathol. 3:225–230. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Eken M, Altin G, Aydin S, Hardal U and
Sanli A: Sialoblastoma of the parotid gland. Int J Pediatr
Otorhinolaryngol Extra. 5:63–65. 2010. View Article : Google Scholar
|
|
7
|
Mostafapour SP, Folz B, Barlow D and
Manning S: Sialoblastoma of the submandibular gland: Report of a
case and review of the literature. Int J Pediatr Otorhinolaryngol.
53:157–161. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fuchsmann C, Viremouneix L,
Collardeau-Frachon S, Ayari-Khalfallah S, Bouvier R, Guibaud L, et
al: Management and treatment of a sialoblastoma of the
submandibular gland in a neonate. Int J Pediatr Otorhinolaryngol
Extra. 6:9–12. 2011. View Article : Google Scholar
|
|
9
|
Prigent M, Teissier N, Peuchmaur M, El
Maleh-Berges M, Philippe-Chomette P, Cardin P and Orbach D:
Sialoblastoma of salivary glands in children: Chemotherapy should
be discussed as an alternative to mutilating surgery. Int J Pediatr
Otorhinolaryngol. 74:942–945. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Williams SB, Ellis GL and Warnock GR:
Sialoblastoma: A clinicopathologic and immunohistochemical study of
7 cases. Ann Diagn Pathol. 10:320–326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ersoz S, Turgutalp H, Cobanoglu U, Bektas
D and Yaris N: Sialoblastoma in the parotid gland: A case report.
Pediatr Int. 52:670–672. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dardick I, Daley TD and McComb RJ:
Sialoblastoma in adults: Distinction from adenoid cystic carcinoma.
Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 109:109–116.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ozdemir I, Simsek E, Silan F and Demirci
F: Congenital sialoblastoma (embryoma) associated with premature
centromere division and high level of alpha-fetoprotein. Prenat
Diagn. 25:687–689. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mertens F, Wahlberg P and Domanski HA:
Clonal chromosome aberrations in a sialoblastoma. Cancer Genet
Cytogenet. 189:68–69. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karaman E, Duman C, Cansız H, Yildiz I,
Dervişoglu S and Ozdemir G: Sialoblastoma in cheek region: Report
of a case. Int J Pediatr Otorhinolaryngol Extra. 5:47–49. 2010.
View Article : Google Scholar
|